

Abstract
Objective:
To explore the putative connection between inclusion body myopathy, Paget disease, frontotemporal dementia (IBMPFD) and motor neuron disease (MND).
Methods:
Clinical, genetic, and EMG characterization of 17 patients from 8 IBMPFD families.
Results:
Limb weakness was the most common clinical manifestation (present in 15 patients, median onset age 38 years, range 25–52), with unequivocal evidence of upper motor neuron dysfunction in 3. EMG, abnormal in all 17, was purely neurogenic in 4, purely myopathic in 6, and mixed neurogenic/myopathic in 7. Cognitive/behavioral impairment was detected in at least 8. Mutations in VCP (R155H, R159G, R155C) were identified in 6 families, and in hnRNPA2B1 (D290V) in another family. The genetic cause in the eighth family has not yet been identified.
Conclusion:
Mutations in at least 4 genes may cause IBMPFD, and its phenotypic spectrum extends beyond IBM, Paget disease, and frontotemporal dementia (FTD). Weakness, the most common and disabling manifestation, may be caused by muscle disease or MND. The acronym IBMPFD is, therefore, insufficient to describe disorders due to VCP mutations or other recently identified IBMPFD-associated genes. Instead, we favor the descriptor multisystem proteinopathy (MSP), which encompasses both the extended clinical phenotype and the previously described prominent pathologic feature of protein aggregation in affected tissues. The nomenclature MSP1, MSP2, and MSP3 may be used for VCP-, HNRNPA2B1-, and HNRNPA1-associated disease, respectively. Genetic defects in MSP implicate a range of biological mechanisms including RNA processing and protein homeostasis, both with potential relevance to the pathobiology of more common MNDs such as amyotrophic lateral sclerosis (ALS) and providing an additional link between ALS and FTD.
Inclusion body myopathy (IBM) with Paget disease and frontotemporal dementia (IBMPFD) is a rare multisystem degenerative disorder named after the organ systems originally recognized to be affected—muscle, bone, and brain. Mutations in the valosin-containing protein (VCP) gene were the first identified cause of IBMPFD,1,2 but reports of families without linkage to chromosome 9 established the genetic heterogeneity of the disorder.3,4 It has recently emerged that mutations in the HNRNPA2B1 (chromosome 7) and HNRNPA1 (chromosome 12) genes account for some families with IBMPFD.5 We first raised the possibility of a connection between IBMPFD and amyotrophic lateral sclerosis (ALS) after mutations in the VCP gene were identified in patients with familial ALS.6 Interestingly, the original report of IBMPFD7 almost 30 years ago described it as a “familial disorder of combined lower motor neuron degeneration and skeletal disorganization”; the presence of fasciculations, EMG evidence of chronic reinnervation, and muscle biopsy showing grouped atrophy all pointing toward a primarily neurogenic process. This family was subsequently found to have an R155Q mutation in the VCP gene.8 Since the original description, various reports of families with IBMPFD have made some reference to ALS, either explicitly (e.g., by the mention of “family member with ALS”) or implicitly (e.g., through clinical and electrophysiologic findings that indicate motor neuron involvement, such as fasciculations, spasticity, hyperreflexia, extensor plantar responses, chronic reinnervation changes on EMG, and prolonged central motor conduction time).8,9 Invariably, these reports overlooked the possible direct connection between VCP mutations and ALS. We have therefore conducted the present study to more clearly define the relationship between IBMPFD and motor neuron disease.
METHODS
Study population.
See appendix e-1 on the Neurology® Web site at www.neurology.org. Participants for this study were recruited from across the United States through physician referrals and patient self-identification. Seventeen individuals from 8 families, each with at least 2 family members affected by some combination of muscle weakness (presumed due to IBM or ALS), Paget disease, and cognitive/behavioral impairment, were enrolled. The institutional review board of the University of Miami approved the study protocol and all participants provided written informed consent.
Genetics.
Genetic testing (figure) established the cause of disease to be mutations in the VCP gene (10 individuals from 6 families), the HNRNPA2B1 gene (4 individuals from 1 family), or an unknown gene (3 individuals from 1 family). All 3 patients in whom the causative gene has not been identified tested negative for mutations in VCP, heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1), HNRNPA1, and C9ORF72. Mutations in other genes known to cause familial ALS were excluded through analysis of whole-exome data.
Figure. Multisystem proteinopathy pedigrees.

Pedigrees of the 6 VCP families (families 1–6), the HNRNPA2B1 family (family 7), and the gene unknown family (family 8). Arrowhead indicates the proband. Star indicates family members who were examined. Phenotype status is denoted by symbols as indicated. Pedigrees have been altered to protect privacy. ALS = amyotrophic lateral sclerosis; FTD = frontotemporal dementia.
Clinical evaluation.
The same neurologist (M.B.) examined 16 of the 17 study participants, either at the study center (n = 8) or at the patients' homes (n = 8, who were too ill to travel). The remaining subject (family 3, III-3) was examined by a second neurologist (B.O.). Evaluations included a careful history, neuromuscular examination, and bedside cognitive evaluation specifically directed at detecting signs of frontotemporal dysfunction (attention, verbal fluency, executive dysfunction, and memory). Nerve conduction studies and routine EMG was performed in all subjects and by the same neurologist (M.B.) in all but 2 cases; EMG in one of these 2 cases was performed by a coauthor (B.O.) and EMG in the other was performed by an electromyographer at the Mayo Clinic. Careful semiquantitative evaluation of motor unit action potentials (MUAPs) was performed to distinguish neurogenic from myopathic processes. Early recruitment of short-duration, low-amplitude, polyphasic MUAPs was interpreted as a sign of myopathy. Reduced recruitment of long-duration, large-amplitude, polyphasic MUAPs was interpreted to indicate a neurogenic process. The potential for late-stage development of large -amplitude, polyphasic MUAPs in chronic myopathies was recognized, and particular emphasis was placed on analysis of MUAP duration and the recruitment pattern to distinguish chronic myopathy from a primarily neurogenic process—i.e., long duration and reduced recruitment indicating a neurogenic disorder. Medical records, including alkaline phosphatase, creatine kinase, bone x-rays, radionucleotide bone scans, and muscle biopsies, were reviewed where available. The diagnosis of ALS was based on the finding of progressive upper motor neuron (UMN) and lower motor neuron (LMN) dysfunction in multiple body regions, according to the principles laid out in the El Escorial criteria.10
Neuropsychological assessment.
As mentioned above, bedside cognitive evaluations were performed on all study subjects. In addition, 4 of the participants underwent a 2- to 3-hour detailed neuropsychological test battery to assess cognitive and behavioral functioning. Logistical constraints precluded the administration of this battery to the other participants. The battery consisted of standardized measures that have been shown to be clinically and empirically sensitive to the cognitive and behavioral changes observed in patients with frontotemporal dementia. As part of the evaluation, caregivers were also asked to complete brief paper and pencil questionnaires assessing the participant's cognitive/behavioral symptoms.
Statistics.
Median age at onset or diagnosis was estimated by product-limit method when individuals who had not exhibited a particular clinical manifestation (weakness, Paget disease, or cognitive/behavioral impairment) by the time of evaluation were included in the calculation.
RESULTS
The study population comprised 17 individuals (11 male, 6 female) from 8 apparently unrelated families, including 6 families with mutations in the VCP gene (R155H in 4 families, R155C in 1 family, and R159G in 1 family); 1 family with a D290V mutation in the hnRNPA2B1 gene; and 1 family in which mutations in the VCP, hnRNPA2B1, hnRNPA1, and C9orf72 genes, as well as in other genes known to cause familial ALS, were excluded (table). Median age at appearance of first clinical manifestation was 34 years (range 25–52), with weakness being the most common initial symptom (n = 10, median age at onset 38 years, range 25–52) and Paget disease of bone the next most frequent initial diagnosis (n = 7, median age at onset 37 years, range 30–54).
Table.
Summary of clinical features
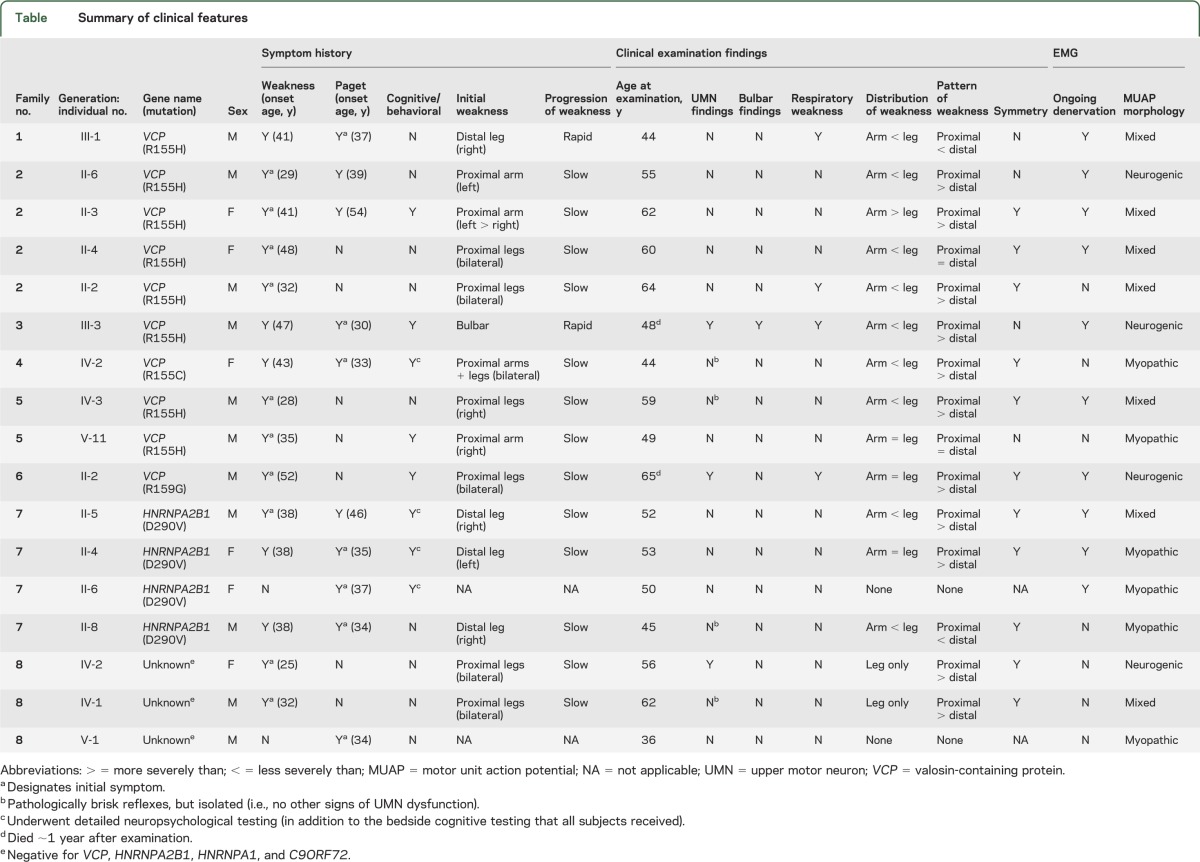
Cognitive impairment was identified in 4 subjects through bedside evaluation alone, cognitive/behavioral impairment in another 4 through both bedside cognitive evaluation as well as detailed neuropsychological testing. In this latter group, detailed testing revealed cognitive impairment in 2 and behavioral impairment in all 4. Impairments were typically of the frontotemporal variety with executive dysfunction (e.g., poor performance on the antisaccade test) and impaired verbal fluency, as well as apathy and disinhibition. Due to the limited sensitivity of bedside testing and its exclusive focus on cognitive (rather than behavioral) impairment, the prevalence of cognitive/behavioral impairment among our study subjects (8/17 = 47%) is likely an underestimate.
Among the 15 subjects who had weakness, it was asymmetric at onset in 8. Weakness remained asymmetric in 4 of these patients by the time of study evaluation, which occurred 0.5, 2, 15, and 28 years after onset of weakness. Weakness typically appeared first in the limbs (n = 14) rather than in the bulbar muscles (n = 1). Limb weakness first appeared in the arms in 3 patients (all proximal), in the legs in 10 patients (n = 6 proximal, n = 4 distal), and simultaneously in the arms and legs in 1 patient (proximal). After extended follow-up (median latency from onset of weakness to examination = 15 years), in the majority of patients weakness remained most severe in the legs, and proximal muscles were typically affected to a greater extent than distal muscles.
Manifestations of both UMN and LMN dysfunction were unequivocally present in 3 patients, with UMN dysfunction evidenced clinically by spastic dysarthria, brisk jaw jerk, limb spasticity, hyperreflexia, and extensor plantar responses (family 3, III-3); limb spasticity, hyperreflexia, and an extensor plantar response (family 6, II-2); and hyperreflexia, positive Hoffman sign, and extensor plantar responses (family 8, IV-2); pathologically brisk reflexes were evident in 4 additional patients. Among the 3 patients with both UMN and LMN dysfunction, EMG showed reduced recruitment of chronic neurogenic MUAPs in all 3 and widespread ongoing denervation in 2 of the 3 (table). In addition to these 3 patients, EMG findings were purely neurogenic in 1 other patient, whereas 6 patients evinced purely myopathic features on EMG and 7 patients demonstrated a mixed pattern of both neurogenic and myopathic findings. Overall, therefore, there was evidence for involvement of motor neurons or their axons in 11/17 (65%) study participants. Two of the 3 patients with both UMN and LMN dysfunction died of the disease 1.5 and 14 years after the onset of weakness. The third patient with both UMN and LMN dysfunction is still alive 30+ years after weakness first appeared.
Weakness generally progressed slowly over a period of decades, although 2 subjects reported periods of very rapid decline in muscle strength. One of these subjects (family 3, III-3) was already described above. The second subject (family 1, III-1) presented with a mixed neurogenic-myopathic picture without UMN signs, but weakness (including respiratory muscle involvement) progressed rapidly over a period of <2 years. There was no apparent pattern in the relationship between the site of initial weakness (arm, leg, proximal, distal, or bulbar) and the subsequent progression of disease, the underlying genotype, or physiology (neurogenic vs myopathic).
Notwithstanding the relatively long latency from onset of weakness to study evaluation and the cross-sectional nature of this study, we found evidence that either myopathic or neurogenic processes may dominate early in the course of disease. In 2 patients (family 7, II-6, and family 8, V-1) who reported no weakness and had normal strength on confrontation testing (i.e., their muscles are at most minimally affected), the EMG was myopathic. Furthermore, one individual (family 8, IV-1) complained only of weakness in the legs and confrontation testing showed normal arm strength; EMG of the legs showed chronic neurogenic changes, but EMG of the arms showed early recruitment of clearly myopathic MUAPs. Perhaps contrasting these observations is the finding of a primarily neurogenic process in patients evaluated soon (0.5–2 years) after onset of weakness (e.g., family 1, III-1, and family 3, III-3) and whose disease has since progressed rapidly. The finding of neurogenic changes early in the course of disease also mitigates the possibility that such findings are solely attributable to a longstanding and severe primary muscle disease.
There was no clear association between the mutated gene (VCP, HNRNPA2B1, unknown) or the specific mutation within the VCP gene (R155H, R155C, R159G) on the one hand, and the motor phenotype on the other. Lower motor neuron neurogenic pathology was evident in subjects with mutations in VCP (R159G), HNRNPA2B1, and the unknown gene; motor neuron disease (defined on the basis of combined UMN and LMN dysfunction) was observed in subjects with 2 different mutations in the VCP gene and a mutation in a third unknown gene.
DISCUSSION
With the goal of exploring the putative connection between IBMPFD and motor neuron disease, we have performed careful clinical and EMG evaluations of 17 consecutive patients from 8 apparently unrelated IBMPFD families. Motor neuron involvement was apparent in the majority of study participants, including 3 patients with unequivocal UMN findings and 4 additional patients with subtle UMN findings. Importantly, the neurogenic phenotype was observed across families with different genes (VCP, HNRNPA2B1, and an as yet unidentified gene) and different mutations of the same gene (e.g., VCP) (table). The rate of progression in 2 of the 3 patients with ALS was relatively slow, although atypical, similarly slow rates of progression have been reported in both sporadic11 and familial12 forms of ALS.
These observations strongly support our hypothesis that weakness in IBMPFD is not solely due to myopathy, but at least in part due to a neurogenic process that may implicate the motor neuron. The phenotypic spectrum of IBMPFD, therefore, is broader than previously recognized, extending to include motor neuron disease. As such, the term IBMPFD seems overly restrictive. IBMPFD-ALS is similarly cumbersome and probably inadequate given the rare, but well-described, involvement of other organ systems including cardiac,13 hepatic,14 visual,14 auditory,15 sensory,16 and autonomic systems,17 as well as emerging evidence that mutations in VCP may infrequently manifest as Parkinson disease,18–20 hereditary spastic paraplegia,21 or cerebellar ataxia.22 Instead, we propose to change the name of this syndrome from IBMPFD to multisystem proteinopathy (MSP), using the nomenclature of MSP1 for disease caused by mutations in VCP, MSP2 when due to mutations in HNRNPA2B1,5 MSP3 when caused by HNRNPA1 mutations,5 and MSP4 when due to mutations in the as yet unidentified gene. This nomenclature emphasizes the multisystem nature of the degenerative process (most prominently nerve, muscle, bone, and brain) as well as the conspicuous deposition of TDP-43, hnRNPA2B1, and hnRNPA1 protein in affected tissues of patients with this multisystem disease independent of genetic etiology.5,6,23,24 The neuropathology of MSP1, for example, is characterized by TDP-43 and ubiquitin-positive neuronal intracytoplasmic inclusions and dystrophic neurites in the neocortex,23 TDP-43-, hnRNPA1-, and hnRNPA2B1-positive cytoplasmic inclusions in muscle,5,24 and nuclear extrusion of TDP-43 accompanied by accumulation of TDP-43-positive cytoplasmic inclusions in the hypoglossal nucleus.6 Similarly, MSP2 and MSP3 are characterized by clearance of TDP-43 as well as hnRNPA2B1 and hnRNPA1, respectively, from myonuclei, with accumulation of large cytoplasmic inclusions that are immunoreactive against TDP-43, hnRNPA1, and hnRNPA2B1.5 Although there has been no report of TDP-43 pathology in Pagetoid bone from patients with IBMPFD (perhaps no one has yet looked), ultrastructural analysis of bone from patients with IBMPFD demonstrates tubulofilamentous inclusions similar to the inclusions identifiable in muscle tissue.1,8,13,25 The role of TDP-43, hnRNPA2B1, and hnRNPA1 in disease pathogenesis is strongly supported by the fact that missense mutations in any of the 3 genes is sufficient to cause ALS or MSP.5,26–28
The significance of defining the phenotypic overlap between IBMPFD and ALS is best understood in the context of the recent discovery that mutations in HNRNPA2B1 and HNRNPA1 are novel genetic causes of MSP.5 Along with TDP-43 and FUS (both of which are hnRNPs), hnRNPA2B1 and hnRNPA1 have conserved prion-like domains (PrLDs). Indeed, PrLDs are present in ∼250 human proteins and are particularly enriched in proteins that undergo dynamic assembly/disassembly into non-membrane-bound organelles or “granules.”29 Recently it was determined that the PrLDs in TDP-43, FUS, hnRNPA2B1, and hnRNPA1 mediate fibrillization that underlies assembly into cytoplasmic RNA granules.5,30,31 Interestingly, mutations in these genes that cause MSP tend to cluster within these PrLDs, enhance the fibrillization properties of these proteins, and show increased recruitment into cytoplasmic RNA granules with presumed impairment in metabolism of client RNAs. The discovery that mutations in HNRNPA2B1 and HNRNPA1 may also cause ALS, therefore, adds to the growing body of evidence supporting perturbed RNA metabolism as an important pathophysiologic mechanism relevant to motor neuron degeneration in ALS. Furthermore, this discovery emphasizes the importance of prion-like mechanisms in ALS pathophysiology, at least in patients with ALS due to certain genetic causes, and lends support to the idea that cell-to-cell spread through protein conformational change may underpin the “spread by contiguity” that has been reported in ALS.32 Although the specifics of how this might occur remain unclear, one speculative idea is that PrLD-containing proteins are extruded through exosomes into the cytoplasm and then engulfed by neighboring cells through a process of macropinocytosis.
Recognizing that the phenotype of MSP extends to include dysfunction of the motor neuron, including typical ALS, is important for a number of reasons. First, findings of motor neuron degeneration/dysfunction should not detract from the diagnosis of MSP. Second, patients with MSP may benefit from the infrastructure and sophistication of the multidisciplinary care that is now recommended for patients with ALS. Moreover, the genetic defects in MSP implicate a range of biological mechanisms—particularly altered protein homeostasis, PrLD-mediated self-aggregation, and RNA metabolism—that are likely relevant to the pathobiology of more common motor neuron degenerative diseases such as ALS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants in this study as well as their family members for their commitment to and engagement in the research process.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- IBM
inclusion body myopathy
- IBMPFD
inclusion body myopathy with Paget disease and frontotemporal dementia
- LMN
lower motor neuron
- MSP
multisystem proteinopathy
- MUAP
motor unit action potential
- PrLD
prion-like domain
- UMN
upper motor neuron
- VCP
valosin-containing protein
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Michael Benatar contributed to all aspects of the work described in this manuscript including study concept and design, obtaining funding, acquiring data, study supervision, and drafting/revising the manuscript. Joanne Wuu contributed to study concept and design, obtaining funding, study supervision, and drafting/revising the manuscript. Catalina Fernandez contributed to acquiring data. Conrad C. Weihl contributed to acquiring data and drafting/revising the manuscript. Heather Katzen contributed to acquiring data and drafting/revising the manuscript. Julie Steele contributed to acquiring data. Bjorn Oskarsson contributed to acquiring data and drafting/revising the manuscript. J. Paul Taylor contributed to all aspects of the work described in this manuscript including study concept and design, obtaining funding, acquiring data, and drafting/revising the manuscript.
STUDY FUNDING
ALS Association, grant 1862; ALS Recovery Fund; Robert Packard Center for ALS Research.
DISCLOSURE
Dr. Benatar receives research funding from the NIH (U10 NS077423), the Food and Drug Administration (R01 FD003517 and R01 FD003710), TKCIS (subcontract from CDC/ATSDR, 300617501), the Muscular Dystrophy Association (grants 4365, 132866, and 172123), and the ALS Association (grants 1491, 1712, and 1862). He served as a site investigator in a multicenter study funded by Alexion Pharmaceuticals and currently serves on the safety monitoring committee of a clinical trial in myasthenia gravis that is funded by Cytokinetics Inc. Dr. Benatar has received research support from CytRx Corporation, the Woodruff Foundation, and has participated in medico-legal cases. J. Wuu has received research support from the National Institute of Aging/National Institute of Health (P01 AG014449, P50 AG025688), the Food and Drug Administration (R01 FD003517), TKCIS (subcontract from CDC/ATSDR, 300617501), the Muscular Dystrophy Association (grants 4365, 172123), the ALS Association (grant 1491), the Woodruff Foundation, Consolidated Anti-Aging Foundation, and the ALS Recovery Fund. Dr. Fernandez reports no disclosures. Dr. Weihl receives research funding from the NIH (R01 AG031867) and the MDA (grant 218514). Dr. Katzen receives research funding from the NIH (K23NS045051), the National Parkinson Foundation, and The Multiple Sclerosis Society. J. Steele reports no disclosures. Dr. Oskarsson receives research funding from the NIH (UL1 TR 000002, KL2 TR 000134, U10 NS077422-01, U01 NS049640-02, R01 ES016848-01A2). He serves as a site investigator in a multicenter study funded by Knopp Pharmaceuticals and Biogen IDEC. Dr. Taylor receives research support from the NIH (R01 NS053825 and R01 AG031587) and the Muscular Dystrophy Association (186705, 63630, and 214429). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kimonis VE, Kovach MJ, Waggoner B, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2000;2:232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–381 [DOI] [PubMed] [Google Scholar]
- 3.Kottlors M, Moske-Eick O, Huebner A, et al. Late-onset autosomal dominant limb girdle muscular dystrophy and Paget's disease of bone unlinked to the VCP gene locus. J Neurol Sci 2010;291:79–85 [DOI] [PubMed] [Google Scholar]
- 4.Waggoner B, Kovach MJ, Winkelman M, et al. Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am J Med Genet 2002;108:187–191 [DOI] [PubMed] [Google Scholar]
- 5.Kim H, Kim N, Wang Y, et al. Mutations in prion-like domains found in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy. Nature Epub 2013. Mar 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker WS, Jr, Hubbard WH, Stryker TD, et al. A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans Assoc Am Physicians 1982;95:126–134 [PubMed] [Google Scholar]
- 8.Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 2008;146A:745–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar KR, Needham M, Mina K, et al. Two Australian families with inclusion-body myopathy, Paget's disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord 2010;20:330–334 [DOI] [PubMed] [Google Scholar]
- 10.Brooks B, World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299 [DOI] [PubMed] [Google Scholar]
- 11.Turner MR, Parton MJ, Shaw CE, Leigh PN, Al-Chalabi A. Prolonged survival in motor neuron disease: a descriptive study of the King's database 1990-2002. J Neurol Neurosurg Psychiatry 2003;74:995–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber M, Neuwirth C, Thierbach J, et al. ALS patients with SOD1 mutations in Switzerland show very diverse phenotypes and extremely long survival. J Neurol Neurosurg Psychiatry 2012;83:351–353 [DOI] [PubMed] [Google Scholar]
- 13.Hubbers CU, Clemen CS, Kesper K, et al. Pathological consequences of VCP mutations on human striated muscle. Brain 2007;130:381–393 [DOI] [PubMed] [Google Scholar]
- 14.Guyant-Marechal L, Laquerriere A, Duyckaerts C, et al. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 2006;67:644–651 [DOI] [PubMed] [Google Scholar]
- 15.Djamshidian A, Schaefer J, Haubenberger D, et al. A novel mutation in the VCP gene (G157R) in a German family with inclusion-body myopathy with Paget disease of bone and frontotemporal dementia. Muscle Nerve 2009;39:389–391 [DOI] [PubMed] [Google Scholar]
- 16.Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 2005;65:1304–1305 [DOI] [PubMed] [Google Scholar]
- 17.Miller TD, Jackson AP, Barresi R, et al. Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): clinical features including sphincter disturbance in a large pedigree. J Neurol Neurosurg Psychiatry 2009;80:583–584 [DOI] [PubMed] [Google Scholar]
- 18.Chan N, Le C, Shieh P, et al. Valosin-containing protein mutation and Parkinson's disease. Parkinsonism Relat Disord 2012;18:107–109 [DOI] [PubMed] [Google Scholar]
- 19.Majounie E, Traynor BJ, Chio A, et al. Mutational analysis of the VCP gene in Parkinson's disease. Neurobiol Aging 2012;33:209 e201–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spina S, Van Laar AD, Murrell JR, et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur J Neurol 2013;20:251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Bot ST, Schelhaas HJ, Kamsteeg EJ, van de Warrenburg BP. Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain 2012;135:e223; author reply e224. [DOI] [PubMed] [Google Scholar]
- 22.Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501–509 [DOI] [PubMed] [Google Scholar]
- 23.Neumann M, Mackenzie IR, Cairns NJ, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 2007;66:152–157 [DOI] [PubMed] [Google Scholar]
- 24.Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 2008;79:1186–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009;19:308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 2008;63:535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574 [DOI] [PubMed] [Google Scholar]
- 28.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gitler AD, Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011;5:179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han TW, Kato M, Xie S, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 2012;149:768–779 [DOI] [PubMed] [Google Scholar]
- 31.Kato M, Han TW, Xie S, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012;149:753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 2009;73:805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.