

Abstract
There is growing interest in using cannabinoid receptor 2 (CB2) agonists for the treatment of neuropathic pain and other indications. In continuation of our ongoing program aiming for the development of new small molecule cannabinoid ligands, we have synthesized a novel series of carbazole and γ-carboline derivatives. The affinities of the newly synthesized compounds were determined by a competitive radioligand displacement assay for human CB2 cannabinoid receptor and rat CB1 cannabinoid receptor. Functional activity and selectivity at human CB1 and CB2 receptors were characterized using receptor internalization and [35S]GTP-γ-S assays. The structure-activity relationship and optimization studies of the carbazole series have led to the discovery of a non-selective CB1 and CB2 agonist, compound 4. Our subsequent research efforts to increase CB2 selectivity of this lead compound have led to the discovery of CB2 selective compound 64, which robustly internalized CB2 receptors. Compound 64 had potent inhibitory effects on pain hypersensitivity in a rat model of neuropathic pain. Other potent and CB2 receptor–selective compounds, including compounds 63 and 68, and a selective CB1 agonist, compound 74 were also discovered. In addition, we identified the CB2 ligand 35 which failed to promote CB2 receptor internalization and inhibited compound CP55,940-induced CB2 internalization despite a high CB2 receptor affinity. The present study provides novel tricyclic series as a starting point for further investigations of CB2 pharmacology and pain treatment.
Keywords: Cannabinoid, CB2, GPCR, neuropathic pain, carbazole, CB1
1. Introduction
Two cannabinoid receptors, CB1 and CB2, have been characterized and cloned[1, 2]. The CB1 receptor is found predominantly in the brain[3]. Impairment of cognitive functions and psychoactivity induced by cannabinoids ligands are mediated by CB1 receptors activation[4]. Selective activation of CB2 receptors has been proposed as a strategy to curtail the negative central side effects seen with nonselective CB1/CB2 agonists. Several CB2-selective agonists have been described previously[5–11]. Although multiple preclinical studies suggest that the CB2 receptor is a viable target to decrease both acute and neuropathic pain responses[12, 13], synthetic CB2 agonists have not advanced through clinical trials. In part, this is due to a lack of a thorough understanding of CB2-mediated analgesic mechanisms[14]. CB2 modulation is also implicated in immunomodulation and neuroprotection but the functional profile of the CB2 ligands inducing these effects has not been clearly defined[15]. Activation of CB2 receptor induces its coupling to the Gi/o class of G proteins. The dissociation of the α and βγ subunits resulting from the CB2 activation can influence multiple effector systems including adenylyl cyclase, p42/44 MAPK (ERK1/2) or ion channels[16]. Currently, the diversity of cannabinoid agonists is very broad and continues to expand rapidly (figure 1). In our previous communications, the SARs of potent and selective CB2- receptor agonists[17] and antagonists[6] based on the isatin scaffold were described (MDA19 in Figure 1).
Figure 1.
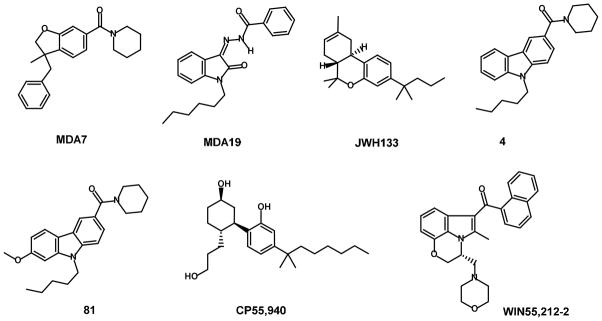
Chemical structures of CB2 modulators (1-7). MDA7[12]; MDA19[17]; JWH133[62]; (4) NMP7[7]; (81) NMP4[7]; CP55,940[63]; WIN55,212-2[64].
Although the isatin core was identified as a good scaffold for designing CB2-selective cannabinoid agonists, we hypothesized that the intramolecular hydrogen bond pattern in the isatin scaffold can be replaced with a covalent C-C bond. Also considering the high pharmacological potential of carbazole-based natural products[18], we focused our attention on this scaffold for use of studying CB1/CB2 receptor pharmacology. Thus, we first identified compound 4 as a potent non-selective CB1/CB2 agonist[19]. We recently reported the cannabinoid and T-type calcium channel activities for three compounds from this novel series of cannabinoid ligands[7]. In this article, we optimized the CB2 selectivity of this series. We also explored the effect on CB2 activity of the introduction of quaternary ammonium moieties or introduction of heteroatoms increasing the polar surface area to help future identification of peripherally-restricted CB2 agonists in our tricyclic series. This research effort led to the discovery of a selective CB2 agonists with a polar surface area > 70 Å2 and expected to have a low blood-brain barrier (BBB) permeability. Functional activities of our most active compounds were determined using [35S]GTP-γ-S assays and by characterizing their ability to internalize human CB1 and CB2 receptors. The ability of these compounds to interact with the orthosteric site on human CB2 receptor was assessed by determining their ability to block CP55940-induced hCB2 internalization. Moreover, the binding mode prediction through ligand-steered modeling highlighted a potential H-bond interaction between the alkylsulfonamide moiety in the N-1 position bored by compound 64 and N7.45 of the CB2 receptor. Similarly to what was previously observed within the isatin series, the presence of the OMe fragment in the carbazole scaffold turned out to be a key substituent responsible for the agonist-to-antagonist functionality switch.
2. Results and discussion
2.1. Chemistry
The pathway of achieving the synthesis of the first series of compounds (4-11 and 14-23) is outlined in Scheme 1. We found that desirable analogs 4-11 can be conveniently prepared from commercially available carbazole via consecutive substitution with n-pentyl bromide under alkaline conditions followed by electrophilic formylation using standard Vilsmeier–Haack conditions, oxidation, and, finally, amidation under standard peptide coupling conditions. Later, a more convenient method was developed for the synthesis of compound 4 that utilized a direct Friedel-Crafts reaction using piperidinecarbonyl chloride. While our conditions for the Friedel-Crafts reaction (compounds 4, 18, and 19) were not expected to be optimal, this strategy did provide a rapid access to the desired compounds. Compound 19 was subjected to nucleophilic attack by the corresponding amines to give analogs 20-22. Deprotection of the Boc protecting group in compounds 12 and 13 furnished compounds 14–15. Analogs 16 and 17 were prepared by coupling of corresponding bromomethylpyridyl derivatives and acid 3 in the presence of TBAF under basic conditions [20]. The tertiary base 9 was converted to the quaternary form 23 by treatment with methyl iodide in diethyl ether at room temperature.
Scheme 1.

Synthesis of 3-carboxamide 9-pentyl-9H-carbazole derivatives 4-17 and 20-23.a
a Reagents and conditions: (a) Method A: n-pentyl bromide, Cs2CO3, DMF, μw at 140°C, 1 h. Method B: n-pentyl bromide, NaOH, acetone, reflux, 16 h; (b) DMF, POCl3, 0°C → μw at 100°C, 1h; (c) KMnO4, acetone-water, 0°C → reflux, 3h; (d) DIEA, DMAP, corresponding amine, EDAC·HCl, DCM, 0°C → rt, 16 h; (f) corresponding bromomethyl pyridines, Et3N, Na2CO3,TBAF, THF, 16 h at rt → μw at 60°C, 1h; (g) AlCl3, bromoacetyl bromide or benzoyl chloride, benzene, 0°C → rt, 19 h; (h) corresponding amine, Et3N, DMF, μw at 90°C, 5 min; (i) MeI, Et2O, rt, 18 h; (j) HCl gas, EtOAc, 0°C, 10 min.
Next, we focused on modifications (Scheme 2) that led to a series of compounds in which the original 3-carbonyl group in the carbazole framework was either derivatized or entirely eliminated. Compounds 24-26, 28 and 30 were synthesized following protocols as outlined in Scheme 2. Nitrile 24 was prepared by a one-pot solvent-free procedure from aldehyde 2 and hydroxylamine. Thioamide 25 was prepared by heating amide 4 with Lawesson’s reagent under microwave conditions. Tertiary amine 26 was obtained upon treatment of 4 with LAH. The preparation of amines 28 and 30 was achieved by palladium-catalyzed coupling in analogy to known literature methods (e.g.[21]).
Scheme 2.

Synthetic modifications of the carbonyl group of 3-carboxamide 9-pentyl-9H-carbazole derivatives 25-26 and 28-30.a
a Reagents and conditions: (a) NH2-OH·HCl, p-TSA, 235°C → μw at 105°C, 5 min → 235°C; (b) LR, toluene, 140°C, 3 h; (c) LAH, THF, 0°C → reflux, 10 min; (d) n-pentyl bromide, Cs2CO3, DMF, μw at 140°C, 1 h; (e) corresponding potassium BF3-methylpiperazinium salts, K2CO3, Pd(OAc)2, (2-biphenyl)-di-tert-butylphosphine, DMF, μw at 120°C, 20 min; (f) TFA, DCM, rt, 6 h.
The pyrido[3,4-b]indole-based analogs 33-35 were prepared as depicted in Scheme 3. Commercially available ethyl 9H-pyrido[3,4-b]indole-3-carboxylate was alkylated with n-pentyl bromide under microwave conditions. Hydrolysis of ester 31 gave the corresponding carboxylic acid 31. Activation of acid 32, followed by coupling with the corresponding amines provided the corresponding target carboxamides 33-35.
Scheme 3.
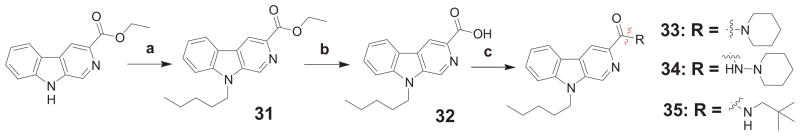
Synthesis of 3-carboxamide 9-pentyl-9H-pyrido[3,4-b]indole derivatives 33-35.a
a Reagents and conditions: (a) n-pentyl bromide, Cs2CO3, DMF, μw at 140°C, 1 h; (b) KOH, ethanol/water, reflux, 16h; (c) DIEA, DMAP, corresponding amine, EDAC·HCl, DCM, 0°C → rt, 16 h.
A series of N-substituted analogues were prepared according to Scheme 4. In an analogous fashion as previously described for compound 4, N-alkylations of carbazole (compounds 36-39) followed by Friedel–Crafts reaction using piperidinecarbonyl chloride onto the resultant substrates led to analogs 40-42. N-alkyl derivatives 43-44 were prepared according to the literature[22]. Intermediate 44 was prepared from carbazole and ethylene oxide under basic conditions. Conversion of alkyl carbazoles 37, 44, and 45 to the corresponding aldehydes 46-49 was achieved using standard Vilsmeier–Haack conditions as described above. Vilsmeier–Haack reaction allowed simultaneous installation of an aldehyde group into the aromatic core and a halogen atom into the N-alkyl chain. Similarly, the obtained aldehydes were then oxidized to the corresponding carboxylic acids 50-53. After activation, these acids were used for coupling with the corresponding amines under standard peptide coupling conditions to furnish compounds 54-57. The N-alkylated chloride derivatives 56 and 57 were converted to the corresponding iodides 58 and 59. Taking advantage of the ease of nucleophilic displacement of the iodide substituent, compounds 60-64 were obtained by base-mediated aminolysis of compounds 58 and 59. Although some of the targets from this series could still be prepared starting from carbazole, we realized that it would more expedient to achieve the same goal by using Buchwald–Hartwig amination and subsequent palladium-mediated oxidative cyclization of the diarylamines [23]. Following this strategy, the diaryl amine 65 was formed and then oxidized with Pd(OAc)2 to give the carbazole ester 66. Subsequent ester hydrolysis of 66 delivered the corresponding acid which was amidated with piperidine to produce the generic compound 67. Alkylation of carbazole 67 afforded the target compounds 68-76. Deprotection of the Boc group within 72 afforded compound 73 after aqueous work-up with sodium hydroxide.
Scheme 4.

Synthesis of (9-substituted-9H-carbazol-3-yl)(piperidin-1-yl)methanone derivatives 54-57, 60-64 and 68-76.
a Reagents and conditions: (a) n-alkyl bromide or iodide, NaOH, acetone, reflux, 16h; (b) AlCl3, piperidinecarbonyl chloride, benzene, 0°C → μw at 100°C, 1h; (c) DMF, POCl3, 0°C → μw at 100°C, 1h; (d) ethyl acrylate, K2CO3, DMF, rt, 19h; (e) ethylene oxide, KOH, methyl vinyl ketone, μw at 50°C, 1h; (f) LAH, THF, 0°C → rt, 1h; (g) KMnO4, acetone-water/acetic acid, 0°C → reflux, 3h; (h) DIEA, DMAP, piperidine, EDAC·HCl, DCM, 0°C → rt, 16h; (i) NaI, acetonitrile, reflux, 72h; (j) corresponding amine, Cs2CO3, acetone, rt → 80°C, 1 h; (k) NaH, DMF, 0°C → rt, 16h; (l) aniline, 5 mol% Pd(OAc)2, 5 mol% rac-BINAP, K2CO3, toluene, μW at 160°C, 1h; (m) Pd(OAc)2, AcOH, reflux, 1 h; (n) KOH, EtOH/H2O, reflux, 16 h; (o) DIEA, DMAP, piperidine, EDAC·HCl, DCM, 16h; (p) corresponding substituted alkyl bromide, TBAI, Cs2CO3, DMF, μW at 140°C, 2 h, or KOt-Bu, rt, 16 h; (q) gaseous HCl, EtOAc, 0°C → rt.
Following a procedure analogous to that of Scheme 4 for compound 67, compounds 81, 84, and 85 were prepared as depicted in Scheme 5. Alternatively, further modification of the carboxamide part of analog 55 was enabled starting from carboxylic acid 51, which was subjected to amide coupling conditions to afford the desired amides 82 and 83.
Scheme 5.

Synthesis of 3-carboxamide-9-substituted-9H-carbazol derivatives 81-83 and 85.a
a Reagents and conditions: (a) methyl 4-bromobenzoate, 5 mol% Pd(OAc)2, 5 mol% rac-BINAP, K2CO3, toluene, μW at 160°C, 2h; (b) Pd(OAc)2, AcOH, reflux, 1 h; (c) n-pentyl bromide, Cs2CO3, DMF, μw at 140°C, 2 h; (d) KOH, EtOH/H2O, reflux, 16 h; (e) DIEA, DMAP, corresponding amine, EDAC·HCl, DCM, 16h; (f) tert-butyl N-(3-bromo-propyl)carbamate, NaOH, acetone, μW at 80°C, 1h.
2.2. Binding affinities for cannabinoid receptors and modeling: structure-activity relationship
We previously identified a series of isatin acylhydrazone CB2 modulators[6, 17]. These compounds showed potent CB2 agonist or antagonist activities. The lead compound among the isatin series possessed potent antiallodynic effects in a rat model of neuropathic pain without affecting the rat locomotor activity[13] at the therapeutic dose. We next turned our attention to substituting the isatin acylhydrazone core with a carbazole scaffold, which is devoid of the potential hydrazone bond isomerization and would have improved potency due to its rigid structural template that may ensure the precise spatial orientation of important functional groups.
Starting with the lead compound 4 we carried out structural modifications in order to expand the SARs and optimize binding affinity, selectivity, and functional activity associated with the CB2 receptor. The CB1 and CB2 receptors binding affinities were determined by performing [3H]CP55,940 radioligand competition binding experiments (Table 1, 2, 3, 4 and 5). Previous studies have shown that binding affinities of CB1 ligands were in the same range for human and rat CB1 receptors[12, 24].
Table 1.
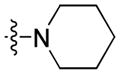
Radioligand competitive binding assays (mean ± SEM) for 9H-carbazole-3-carboxy-based analogues.
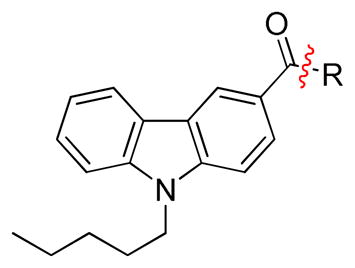
| |||||
|---|---|---|---|---|---|
| Compound | R | TPSAa | rCB1 Kib (nM) | hCB2 Kib (nM) | CB1/CB2 ratioc |
| WIN55,212-2 | – | 43.70 | 36.0 ± 8.3 | 13.0 ± 2.1 | 2.77 |
| 4 |
|
25.24 | 77.0 ± 35.5 | 73.0 ± 15.1 | 1.05 |
| 5 |
|
25.24 | 105 ± 14.6 | 51.8 ± 5.6 | 2.03 |
| 6 |
|
59.38 | 440.0 ± 101.3 | 225.0 ± 51.8 | 1.96 |
| 7 |
|
37.27 | n.b. | 1609.0 ± 279.1 | – |
| 8 |
|
34.47 | 60.0 ± 12.4 | 84.5 ± 9.7 | 0.71 |
| 9 |
|
28.48 | 123.0 ± 19.8 | 35.4 ± 4.4 | 3.47 |
| 10 |
|
34.03 | 1451 ± 273.2 | 288.5 ± 39.3 | 5.03 |
| 11 |
|
34.03 | n.b. | 3594.0 ± 424.1 | – |
| 14 |
|
50.64 | n.b. | n.b. | – |
| 15 |
|
41.85 | 283.0 ± 65.2 | 2833.0 ± 1304.6 | 0.10 |
| 16 |
|
44.12 | 108 ± 24.9 | 1392 ± 320.5 | 0.08 |
| 17 |
|
44.12 | 679 ± 156.3 | 1927 ± 443.7 | 0.35 |
| 18 |
|
22 | 291.0 ± 67.0 | 101.0 ± 23.3 | 2.88 |
| 20 |
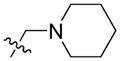
|
26.44 | 3330.0 ± 766.8 | 1093.0 ± 201.3 | 3.05 |
| 21 |
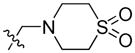
|
59.38 | 675.0 ± 155.4 | 502.0 ± 80.9 | 1.34 |
| 22 |
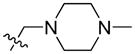
|
29.68 | 1504.0 ± 346.3 | 398.0 ± 64.2 | 3.78 |
| 23 |
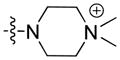
|
25.24 | n.b. | n.b. | – |
Topological polar surface area[25].
Values are means of three experiments run in triplicates with standard deviation.
Ki of CB1/Ki of CB2;
n.b. no binding.
Table 2.
Radioligand competitive binding assays for 9H-carbazole analogues: focusing on the role of the carbonyl group with concomitant variation in basicity.
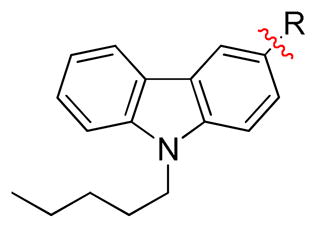
| |||||
|---|---|---|---|---|---|
| Compound | R | TPSAa | rCB1 Kib (nM) | hCB2 Kib (nM) | CB1/CB2 Ratioc |
| 24 |
|
28.72 | n.b. | n.b. | |
| 25 |
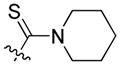
|
8.17 | 40.6 ± 4.2 | 49.6 ± 4.4 | 0.82 |
| 26 |
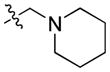
|
9.37 | 122.0 ± 28.1 | 370 ± 85.2 | 0.33 |
| 28 |
|
12.61 | 244.0 ± 56.2 | 740.0 ± 119.3 | 0.33 |
| 30 |
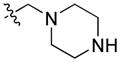
|
24.78 | n.b. | n.b. | – |
Topological polar surface area[25].
Values are means of three experiments run in triplicates with standard deviation.
Ki of CB1/Ki of CB2;
n.b. no binding.
Table 3.
Radioligand competitive binding assays (mean ± SEM) for the 9H-pyrido[3,4-b]indole-3-carboxy-based analogues: a tactical approach to assess the impact of potential hydrogen bonding in the 3-carboxy region.
| |||||
|---|---|---|---|---|---|
| Compound | R | TPSAa | rCB1 Kia (nM) | hCB2 Kia (nM) | CB1/CB2 ratiob |
| 31 |
|
44.12 | 1859.0 ± 363.2 | 1367.0 ± 194.6 | 1.36 |
| 33 |
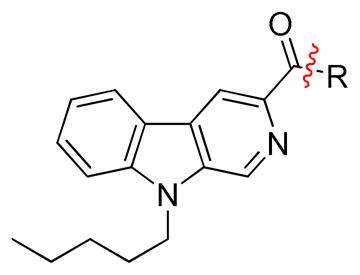
|
38.13 | 136.2 ± 19.9 | 35.3 ± 5.5 | 3.86 |
| 34 |
|
50.16 | 354.5 ± 54.0 | 171.1 ± 17.7 | 2.07 |
| 35 |
|
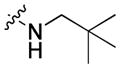
46.92 | 43.6 ± 6.4 | 26.5 ± 4.1 | 1.65 |
Topological polar surface area[25].
Values are means of three experiments run in triplicates with standard deviation.
Ki of CB1/Ki of CB2.
Table 4.
Radioligand competitive binding assays (mean ± SEM) for carbazole-based analogues: systematic variation in lipophilicity and basicity in the 9H-region.
| |||||
|---|---|---|---|---|---|
| Compound | R | TPSAa | rCB1 Kib (nM) | hCB2 Kib (nM) | CB1/CB2 Ratioc |
| 40 |
|
25.24 | 600.0 ± 138.2 | 521.0 ± 108.0 | 1.15 |
| 41 |
|
25.24 | 241.0 ± 111.0 | 96.0 ± 15.5 | 2.51 |
| 42 |
|
25.24 | 942.5 ± 433.8 | 827.0 ± 171.4 | 1.14 |
| 54 |
|
25.24 | 63.0 ± 14.5 | 264 ± 48.6 | 0.24 |
| 55 |
|
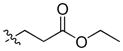
51.54 | 4976 ± 2291 | 1313 ± 242 | 3.79 |
| 56 |
|
25.24 | 2360 ± 543 | 519.0 ± 119 | 4.55 |
| 57 |
|
25.24 | 123.5 ± 28.3 | 88.0 ± 18.2 | 1.40 |
| 60 |
|
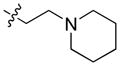
29.68 | n.b. | n.b. | – |
| 61 |
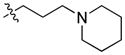
|
29.68 | n.b. | 2169 ± 449 | – |
| 62 |
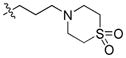
|
62.62 | 2376 ± 1094 | 249.0 ± 40.1 | 9.54 |
| 63 |
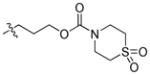
|
88.92 | 2256 ± 519 | 73.0 ± 13.4 | 30.90 |
| 64 |
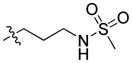
|
71.41 | 1471 ± 677 | 62.0 ± 12.8 | 23.73 |
| 67 | H | 36.10 | n.b. | n.b. | – |
| 68 |
|
34.47 | 1442 ± 664 | 83.0 ± 19.1 | 8.88 |
| 69 |
|
34.47 | 19.5 ± 4.5 | 20.0 ±4.6 | 0.98 |
| 70 |
|
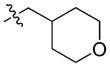
51.54 | 1848 ± 425 | 208.0 ± 47.9 | 8.88 |
| 71 |
|
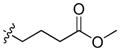
29.68 | 2387 ± 403 | 403.0 ± 92.8 | 5.92 |
| 72 |
|
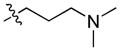
63.57 | n.b. | 2690 ± 619 | – |
| 73 |
|
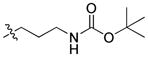
52.88 | n.b. | n.b. | – |
| 74 |
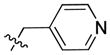
|
38.13 | 393.0 ± 90.5 | 91.0 ± 21.0 | 4.32 |
| 75 |
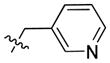
|
38.13 | 1024 ± 236 | 600.0 ± 138.2 | 1.71 |
| 76 |
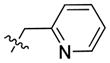
|
38.13 | 403.0 ± 92.8 | 215.0 ± 49.5 | 1.87 |
Topological polar surface area[25].
Values are means of three experiments run in triplicates with standard deviation.
Ki of CB1/Ki of CB2;
n.b. no binding.
Table 5.
Radioligand competitive binding assays (mean ± SEM) for carbazole-based analogues with extra and simultaneous modification at two variation points.
The structural model of 4 complexed with the CB2 receptor is shown in Fig. 2. The ligand occupies a cavity defined by helices 3, 5, 6 and 7, with the alkyl chain buried within the binding site, and the piperidine moiety facing the extracellular side. In agreement with this predicted binding mode, deletion of the chain born by the endocyclic nitrogen in 67 was detrimental to its binding. Replacement of the piperidine ring by a nitrile moiety in 24 or a methyl ester moiety in 31 had a strong negative effect on CB2 affinity, probably due to the lack of ligand-receptor van der Waals contacts. In order to further improve the physicochemical properties and metabolic stability of compound 4, we decided to incorporate polar groups at position 4 of the piperidine ring[26]. However, such a chemical modification did not produce significant improvement in affinity towards the CB2 receptor for analogues 6 and 8. Alternatively, replacing the piperidine ring by the more polar N-methylpiperazine ring (compound 9), decreased affinity at CB1 but increased toward CB2, resulting in a 3-fold improvement of CB2 selectivity compared to the piperidine analogue 4. It is notable that the removal of the methyl moiety (compound 15) from the N-methylpiperazine analogue, dramatically decreased the CB2 binding affinity indicating that a hydrophobic moiety in this part of the molecule may be essential for the CB2 affinity. In contrast, the CB1 affinity in this case decreased by only 4-fold. Next, the quaternary ammonium derivative 23 was prepared in order to probe electrostatic interactions with CB2 and investigate if this strategy can be used to design CB2 agonists that do not cross the blood-brain barrier. Unfortunately, compound 23 turned out to be devoid of CB1 or CB2 affinity. This result lends further support to our view that lipophilicity is a required feature on the carboxamide part of the molecule. Since there was compelling evidence that a lipophilic moiety is required for a good CB2 affinity, we decided to add a methylene moiety or a nitrogen spacer in between the carbonyl and the lipophilic piperidine moiety to test whether the biological profile can be further improved. However, as compared to the piperidine analogue 4, the N-aminopiperidine derivative 7, the N-methylpiperidine derivative 20 and the methylpiperidine derivative 14 did not show relevant CB1 or CB2 affinities, thus suggesting a scarce adaptability of the lipophilic pocket in the CB2 binding site. One can notice that among common features present in these three compounds is not only a longer substituent, but also a tertiary amine offering a site of N-cation interactions. Therefore, it may be speculated that perhaps either the increased basicity of the tertiary amine or the subsequent cation interactions are responsible for destabilizing the complex. The thiomorpholine 1,1-dioxide analogue 21 exhibited moderate affinities for both CB1 and CB2. Our attempts to increase the conformational flexibility in the carboxamide region by opening the piperidine cycle, as exemplified by compound 5 slightly increased CB2 affinity and the selectivity toward CB2. Replacement of the piperidine by the planar phenyl ring, as in compound 18, resulted in a slight decrease of CB1 affinity, while the CB2 affinity was in the range of the parent compound. These observations support the binding mode proposed for 4 (Figure 2). On the other hand, an increase of lipophilicity in compound 10, resulted in an improvement in CB2 selectivity. In this case the adamantyl group is probably too bulky to fit properly in the CB1 receptor-binding pocket. One of the unique benefits of “soft drugs” such as esters for treating inflammatory disorders lies in the susceptibility of their ester functionality to hydrolysis, which can limit their CNS penetration. Therefore, such molecules should lack the undesired CNS-induced side effects associated with CB1 receptor activation. The pyridine ester analogues 16 and 17 were synthesized to build aqueous solubility required for bioavailabilty via the hydrolysable ester bonds. Surprisingly, both compounds exhibited higher affinity at the CB1 rather than at the CB2 receptor. This decrease in CB2 affinity is in good agreement with what is reported above regarding the longer substituents. Further exploration of the SARs around the carbazole series led us to examine the impact of the amide toward CB2 affinity. Replacement of the carbonyl (compound 4) by a methylene in compound 26 resulted in a decrease of CB2 affinity while the CB1 affinity was in the same range, what could be explained in terms of the lost hydrogen-bond with the OH group of S7.39 (cf. Figure 2). On the other hand, the replacement of the carbonyl by the thiocarbonyl in 25 resulted in the CB2 affinity being restored. Replacement of amide bonds by thio-amide bonds have been shown to destabilize hydrogen bond by the higher steric demands imposed by the larger sulfur atom, which leads to non-optimal angles required to form hydrogen bonding[27]. In addition, the sulfur atom has lower electronegativity compared to oxygen atom. However, C=S bond in 25 may induce the required conformation and with the correct position of the piperidine ring needed for high CB2 affinity[28]. Nevertheless, thioamide derivatives cannot be considered as viable alternatives to design potent CB2 agonists since it was shown that thioamides behave as amide prodrugs in vivo. As expected, the piperazine analogues 28 and 30 did not exhibit potent CB2 affinity even though the CB1 affinity was slightly lower for 28 compared 9. Introduction of an extra nitrogen atom in the carbazole scaffold was expected to impact both water solubility and positioning of the lipophilic moiety attached to the carbonyl. As a result, CB2 affinity was slightly increased for the carboline analogue 33 when compared to the respective carbazole analogue. For the former, the CB1 affinity was in the same range as for its carbazole counterpart 4. Notably, the CB2 affinity was substantially higher for the N-aminopiperidine carboline derivative 34 as compared to the carbazole derivative 7. This higher affinity could potentially result from a hydrogen bond between the secondary amino group and the nitrogen on the carboline scaffold, thus providing some critical assistance in accommodating the respective lipophilic moiety in the CB2 binding site. Introduction of the neopentyl moiety in 35 resulted in increased affinity at both CB1 and CB2 but the selectivity toward CB2 receptor was rather low (ca. 1.65).
Figure 2.

CB2 receptor complexed with compound 4. The ligand is displayed with yellow carbon atoms, overlaid to a transparent CPK representation. Key receptor residues are displayed as sticks, with light grey carbons. Hydrogen bonds are shown as small spheres.
In the course of our initial exploratory work on the structure–activity relationship for this novel series of CB2 modulators, we decided to determine the optimal chain length attached to the carbazole’s endocyclic nitrogen (compounds 40 to 54). Among the linear N-1 alkyl chains, a pentyl chain seemed to be the most optimal for occupying the CB2 lipophilic cavity, because systematically increasing and decreasing the length from n-pentyl in 4 negatively impacted the respective CB2 affinities. Introduction of halogen atoms was shown to increase the affinity of small molecules toward proteins even though halogen bond interactions are significantly weaker than hydrogen bond interactions. Previous results from literature suggest that interactions between C-Cl moieties and carbonyl groups or other H-bond acceptors increase affinity of corresponding ligands to biological targets[29]. Introduction of a chlorine atom into the ethyl or propyl chain of 56 and 57 did not increase CB2 affinity. The longer chloropropyl chain in 57 mimicked the pentyl chain in 4, while the use of a shorter one in 56 suffered a dramatic loss of CB2 affinity. Hydrogen bond interactions were shown to provide specificity to the process of receptor recognition although not always did they add much of free binding energy[30]. Since we achieved only modest improvements in terms of CB2 selectivity, we next sought to expand our library by targeting potential hydrogen bond interactions between the chain carried by the endocyclic nitrogen and CB2 receptor. First, we turned our attention on to alkyl ethers, despite the fact that ethers are known to be weak hydrogen bond acceptors. However, an interesting feature of ethers in this regard is that they exhibit a somewhat higher tolerance for angular requirement between the hydrogen-donor and acceptor[30]. In order to explore the SARs within this context, we synthesized acyclic (compound 68) and cyclic (compound 69) ethers. The cyclic ethers are known to be stronger hydrogen bond acceptors than the acyclic ones[30]. Consistent with our predictions (Fig. 2), 69 demonstrated higher CB2 affinity as compared to 4 although the CB2 selectivity remained poor (Fig. 2). A replacement of the 4-methyltetrahydropyran fragment by the 4-methylpyridine moiety as exemplified by compound 74 resulted in a 4-fold improvement in CB2 selectivity, while the respective CB2 affinities were similar. The binding mode of 74 is shown in Fig. 3, exhibiting a similar pose when compared to 4 with hydrogen bonding occurring between the amide carbonyl of 74 and S7.39. In this case, the only difference is that an additional hydrogen bond is present between the nitrogen of the 4-methylpypridine and the side chain of W6.48. Consistent with this model, the 2- and 3-methylpyridine analogues (compounds 75 and 76) exhibited low CB2 affinities. In contrast replacement of the pentyl chain by the acyclic ether in compound 68 resulted in a significant increase of selectivity due to a specific decrease CB1 affinity. Acyclic esters functionality such as in compound 70 resulted in a dramatic improvement in selectivity due to significantly lower affinity of this compound at the CB1 receptor. Interestingly, this effect was observed for both the ester analogue and the dimethyl amine analogue 71. A significant loss of CB2 binding affinity in these cases may be attributed to three obstacles such as higher degree of angular preferences, higher steric demands, or difference in electronic properties. In comparison, the ethyl ester counterpart 55, which has the ester functionality closer to the carbazole scaffold did not exhibit significant affinity at CB1, and its affinity at CB2 was not promising either. In part, this might be explained by poor ability of 55 to promote hydrogen bonding with the corresponding receptors. When the nitrogen atom was free of substituents as in 73 or carried a bulky Boc moiety as in 72, a significant loss of CB2 affinity was observed. In the former case, the charged terminal amine group was likely to destabilize the complex, which is consistent with our previous observations. Employment of alkyl chains featuring the sulfonyl functional group is typical in cases when a further increase in the PSA values of the corresponding analogs is desirable. The dual character of the weakly polar sulfonyl group which allows it to form hydrogen bonds and at the same time participate in nonpolar hydrophobic interactions through van der Waals contacts drawn our attention to the possibility of applying this strategy to improve CB2 affinity[30, 31]. As expected, the methyl sulfonamide derivative 64 demonstrated the most specific CB2 affinity.
Figure 3.

CB2 receptor complexed with compound 74. The ligand is displayed with yellow carbon atoms, overlaid to a transparent CPK representation. Key receptor residues are displayed as sticks, with light grey carbons. Hydrogen bonds are shown as small spheres.
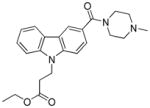
The binding mode of compound 64 is shown in Figure 4; the pose is very similar to that of compound 4 (Fig. 2). The first thing one can notice in this model is a weak hydrogen bond between the oxygen of the sulfonyl group and the side chain of N7.45 in addition to a hydrogen bond between the amide carbonyl of 64 and S7.39. Introducing a basic residue such as a piperidine ring into the N-aliphatic side chain resulted in the inactive compounds (61 and 60). A possible explanation for this might be that the charged amine in this case is buried within the potential hydrophobic binding site. In contrast, the dioxothiomorpholine counterpart 62 regained both good selectivity and CB2 affinity. A similar trend was observed for analogue 63 which offered a 30-fold selectivity over CB1. For comparative purposes, we next decided to carry out structure-activity relationship studies looking at the effects of ring substitution to determine if they would parallel those in our previous isatin series. Indeed, we observed a similar result when a methoxy group was introduced in position 6 of the carbazole scaffold resulting in compound 81 which displayed a higher CB2 affinity and was endowed with inverse agonist instead of agonist functional activity.
Figure 4.

CB2 receptor complexed with compound 64. The ligand is displayed with yellow carbon atoms, overlaid to a transparent CPK representation. Key receptor residues are displayed as sticks, with light grey carbons. Hydrogen bonds are shown as small spheres.
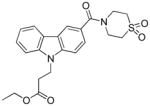
In the carbazole series, we also briefly explored whether it is possible to impact the spatial position of the N-alkyl chain by introducing substituents in position 4 of the piperidine ring. As shown in Table 5, both the methylpiperazine analogue 82 and the dioxothiomorpholine analogue 83 turned out to be lacking CB2 affinity. This result may be explained by the fact that the methylpiperazine and dioxothiomorpholine rings did not allow the chain containing the ester moiety to be set further down into the lipophilic pocket in order to restore the CB2 affinity within the same range of 70. As expected (compare with 72), the dioxothiomorpholine analogue 85 bearing a distal Boc moiety on the N-propyl chain showed no measurable activity against both the CB1 and CB2 receptors.
2.3. Cannabinoid receptors functional activity
Functional activity for compound 64 was evaluated by using a [35S] guanosine-5′-triphosphate (GTP)-γ-S assay in Chinese hamster ovarian cell membrane extracts expressing recombinant hCB1 or hCB2 receptor (Table 6). In this system, agonists stimulate [35S]GTP-γ-S binding, whereas antagonists have no effect and inverse agonists decrease [35S]GTP-γ-S basal binding. Efficacies (Emax) for CB1 or CB2 were expressed as a percentage of the efficacy of compound CP55,940. Compound 64 behaved as a selective CB2 agonist since it did not show any agonistic or antagonistic activities at human CB1 receptors in the [35S] GTPγS assay at 1 μM whereas the EC50 value for the CB2 receptor was 7.3 nM, with an efficacy of almost 60%. Thus, compound 64 appears to be a selective CB2 agonist.
Table 6.
Determination of potency (EC50) and maximal stimulation (Emax) on hCB1 and hCB2 receptors of selected compoundsa
| Compound | GTPγ[35S] functional assaysa | |||
|---|---|---|---|---|
| Human CB1 | Human CB2 | |||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |
| CP55,940 | N.D. | N.D. | 4.13 | 100 |
| 4 | 96.9 ± 11.9b | 73.6b | 10.5 ± 1.8b | 30.8b |
| 64 | N.A. | N.A. | 7.3± 6.7 | 59.6 |
| 81 | 118.3 ± 4b | 30.4b | 9.8 ± 0.3b | −76.4b |
CB1 and CB2 assay data are presented as the mean of two determinations. Assay reproducibility was monitored by the use of the reference compound CP55,940. For replicate determinations, the maximum variability tolerated in the test was 20% around the average of the replicates. Efficacies (Emax) for CB1 or CB2 are expressed as a percentage relative to the efficacy of compound CP55,940. N.D. = not determined; N.A.: not active at 1 μM;
from ref [7].
Receptor internalization and functional selectivity are important emerging pharmacological concepts that increase the complexity of the biological profiling of G protein-coupled receptor (GPCR) ligands. Recently, it has been shown that WIN55,212-2 and other aminoalkylindoles failed to promote CB2 receptor internalization, whereas compound CP55,940 robustly internalized CB2 receptors[14]. Despite these differences in term of CB2 induced-internalization, both compounds activated CB2 receptors mediated ERK 1/2 phosphorylation and recruited β-arrestin2 to the membrane. In contrast, whereas CP55,940 inhibited voltage-gated calcium channels via CB2 receptor activation, WIN55,212-2 was ineffective on its own and antagonized the effects of CP55,940. These differences in terms of functional activity and the polypharmacology associated with cannabinoids ligands[7, 32] can possibly explain the variability observed in preclinical models of neuropathic pain and the difficulties of translating results from the preclinical models to human pain states. As a way for rapidly screening for orthosteric interactions with CB1 and CB2 receptors, internalizations of these receptors induced by our most active compounds was determined (Table 7). In our series of compounds both CB1 and CB2 receptor internalization correlated well with the GTPγ[35S] functional assays since 81, a partial CB1 agonist and CB2 inverse agonist in the GTPγ[35S] functional assays only promoted 33% of CB1 internalization and failed to internalize CB2 at 10 μM. In contrast, 4 which was a full CB1 agonist and a partial CB2 agonist in the GTPγ[35S] functional assay, promoted internalization of both hCB1 and hCB2 to similar extent. Despite high CB2 affinities, compounds 5, 9, 18 and 63 promoted only partial CB2 internalization. These data suggest that these compounds behave as partial agonist at CB2, at least with respect to the internalization. Similarly, 6 and 35 behaved as CB1 partial agonists as they partially induced CB1 internalization despite the high CB1 affinity. Interestingly, compound 26 failed to induce CB1 internalization despite a CB1 affinity of around 100 nM. This result may be explained by the fact that the carbonyl moiety between the carbazole scaffold and the piperidine is essential for CB1 internalization as exemplified by compound 4, which induced CB1 internalization. Low CB1 affinity is associated with low CB1 induced internalization as illustrated by compounds 70 or 20. The lack of CB2 internalization observed with 35 could potentially result from the hydrogen bond between the secondary amino group and the nitrogen on the carboline scaffold. This hydrogen bond may compromise the conformational flexibility of 35 required by the CB2 receptor lipophilic pocket to accommodate the neopentyl moiety essential to induce internalization. Compound 35 produced little CB1 internalization despite a good affinity, behaving as a partial CB1 agonist.
Table 7.
Determination of hCB1 and hCB2 receptor internalization by selected compoundsa.
| Compound | Receptor internalization | |
|---|---|---|
| hCB1 (%) | hCB2 (%) | |
| CP55,940 | 100 | 100 |
| 4 | 140.4 | 117.9 |
| 5 | 101.9 | 53.1 |
| 6 | 23.9 | 91.5 |
| 8 | 115.3 | 78.7 |
| 9 | 84.7 | 43.9 |
| 18 | 77.0 | 39.3 |
| 20 | −20.4 | 102.6 |
| 25 | 124.4 | 63.0 |
| 26 | −11.1 | 79.6 |
| 33 | 127.8 | 101.1 |
| 35 | 34.7 | 0.1 |
| 41 | 121.8 | 77.0 |
| 54 | 84.8 | 67.9 |
| 57 | 106.3 | 110.7 |
| 63 | 65.6 | 27.9 |
| 64 | 54.1 | 116.2 |
| 68 | 63.2 | 68.1 |
| 69 | 141.8 | 86.2 |
| 70 | 9.4 | 37.3 |
| 74 | 74.9 | 66.3 |
| 81 | 33.4 | −1.1 |
CB1 and CB2 assay data are presented as the mean of two determinations.
To further explore the concentration dependence of this effect, we performed complementary experiments with hCB2 cells. First, we applied a constant 10 nM compound CP55,940 with cotreatments of 3 uM of selected compounds. As expected, the CB2 inverse agonist compound 81 reduced compound CP55,940-induced internalization in hCB2 cells (Fig. 5). Unexpectedly, compound 18 and 35 reduced by more than 50% hCB2 internalization induced by CP55,940. These compounds may behave as inverse agonists or despite their effect on internalization, they may activate CB2 receptors without inducing receptor internalization as has recently been shown for compound WIN55,212-2. A more detailed investigation into the CB2 functional activity profiles of these compounds is required to confirm these hypotheses.
Figure 5.

Effects of selected compounds on CP55,940-induced CB2 internalization. hCB2 cells were cotreated with 10 nM of compound CP55,940 and 3 μM selected compounds.
2.4. In vivo studies
Peripheral nerve injury can cause clinically relevant chronic neuropathic pain. The L5 and L6 spinal nerve ligation produced long-lasting mechanical hypersensitivity (tactile allodynia) on the ipsilateral hind paw in rats. Since compound 64 exhibited a favorable CB2 selectivity profile and induced CB2 internalization, we were interested in characterizing its activity in the SNL in vivo model of neuropathic pain. Compound 64 administered intraperitoneally (5 – 20 mg/kg) significantly attenuated tactile allodynia in a dose-dependent manner. The higher doses (20 mg/kg and 10 mg/kg) produced a longer duration of the antiallodynic effect than that observed with the 5 mg/kg of compound 64 (Figure 6).
Figure 6.

Effect of compound 64 (administered intraperitoneally) on to the paw withdrawal threshold, tested with von Frey filaments, in a neuropathic pain model in rats (seven rats per group). Repeated measures ANOVA with Dunnet’s post hoc test were used to determine the statistical difference in each group. * P < 0.05 compared with the baseline control (time 0). Data are expressed as mean ± SEM.
3. Conclusion
In this investigation, we presented a broad range of experimental data on the novel series of carbazole-based cannabinoid ligands. Within this series, sulfonamide analogue 64 was identified as a selective CB2 agonist. Our structure modeling and docking studies for compound 64 based on the ligand-steered approach highlighted a potential H-bond interaction in a burrow-like site between the alkylsulfonamide moiety at the N-1 position and N7.45 of the CB2 receptor. This selective CB2 ligand exhibited functional agonist activity as assessed by using [35S]GTP-γ-S assay and the CB2 internalization study. Similarly to our isatin series, the presence of a methoxy group in position 7 of the carbazole scaffold in compound 81 turned out to be a key substituent responsible for the agonist-to-antagonist functionality switch. Compound 81 exhibited [35S]GTP-γ-S inverse agonist activity and inhibited compound CP55,940-induced CB2 internalization. On the basis of our results, compound 35 has a high CB2 affinity, inhibits compound CP55,940-mediated internalization, and merits further pharmacology characterization. Compound 64, which induced CB2 internalization, attenuated tactile allodynia in a dose-dependent manner in the SNL in vivo model of neuropathic pain. In summary, we have identified a novel series of tricyclic CB2 selective agonists with a well-defined CB2 functional activity that can be used as a platform for the future development of specific CB2 agonists as treatments of pain. The present study also provides an additional insight into the internalization of CB2 receptors induced by CB2 agonist, which should further facilitate optimization of this novel class of tricyclic CB2 modulators for the treatment of pain.
4. Experimental Section
4.1. Synthesis
Unless otherwise stated, all reactions were carried out under a nitrogen or argon atmosphere, using commercially available reagents and solvents. Anhydrous THF and Et2O were obtained by distillation from sodium and benzophenone followed by distillation from LAH. All other solvents are reagent grade and were used without further purification. All procedures were carried out at room temperature unless otherwise stated. Magnesium sulfate was used as the drying agent. The crude products were purified by flash chromatography using prepacked Biotage® cartridges on a Biotage® Isolera separation system. Analytical thin-layer chromatography (TLC) was performed on precoated, aluminum-backed silica gel (200 μm thick, Sorbent Technologies, UV254). Melting points were obtained on a Start SMP3 melting point apparatus and are uncorrected. The microwave irradiation was effected using a Biotage® Initiator microwave synthesizer. High Resolution mass spectrometry (HRMS) analyses were performed on a Waters/Micromass LCT-TOF instrument. The HPLC systems used to analyze the target compounds were either: (i) Waters 2790 high-performance liquid chromatograph with an autosampler connected to a Waters 2487 dual absorbance UV detector and Waters Micromass LCT KC290 mass spectrometer or (ii) Waters Alliance e2695 high-performance liquid chromatograph with an autosampler connected to a Waters 2998 photodiode array detector and Waters Micromass LCT Premier mass spectrometer. NMR spectra were obtained either on Varian Inova-500 or Bruker Ascend™-400 instruments. Chemical shifts for 13C NMR spectra are recorded in parts per million using either the central peak of deuterated chloroform (77.23 ppm) or deuterated DMSO (39.51 ppm) as the internal standards. Characteristic splitting patterns due to spin-spin coupling are expressed as follows: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet. All coupling constants are measured in hertz. All synthesized piperidine-amides, except for thioamide analogs, displayed a characteristic broadening effect in their 13C NMR spectra for α- and β-CH2 carbons of the piperidine ring, reflecting a restricted rotation around the peptide bond. For this reason, the corresponding chemical shifts values for these analogs were reported only when possible. 2-Hydroxypropyl-β-cyclodextrin with an average degree of molar substitution of 4.4 was purchased from CTD Holdings Inc (Alachua, FL, USA)
4.1.1. 9-Pentyl-9H-carbazole (1)
Method A
Under argon atmosphere, a solution of carbazole (2.5 g, 14.95 mmol), 1-bromopentane (2.225 mL, 17.94 mmol), and Cs2CO3 (7.3 g, 22.41 mmol) in DMF (10 mL) was subjected to microwave irradiation at 140°C for 1 h. The reaction mixture was cooled, diluted with EtOAc (50 mL), and filtered. The organic solvents were evaporated in vacuo. The resultant dark oil was distilled under reduced pressure (bp 125°C, 2 mmHg) to afford the title compound as light yellow oil (3.169 g, 89%). 1H NMR (500 MHz, CDCl3) δ 8.08 (dd, J = 7.6, 0.8 Hz, 2H), 7.47 – 7.41 (m, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.21 (td, J = 7.5, 1.0 Hz, 2H), 4.24 (t, J = 7.3 Hz, 2H), 1.89 – 1.77 (m, 2H), 1.40 – 1.26 (m, 4H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 140.64 (C), 125.75 (CH), 123.02 (C), 120.52 (CH), 118.88 (CH), 108.84 (CH), 43.23 (CH2), 29.62 (CH2), 28.87 (CH2), 22.69 (CH2), 14.15 (CH3).
Method B
A mixture of carbazole (10 g, 59.81 mmol), n-pentyl bromide (11 mL, 88.92 mmol), and finely ground NaOH (4 g, 100 mmol) in dry acetone (100 mL) was refluxed for 16 h under nitrogen. After all volatile components were removed by rotary evaporation in vacuo, the residue was extracted with tert-butyl methyl ether (150 mL). The organic phase was washed with water, brine, dried (MgSO4), filtered, and evaporated in vacuo. The obtained residue was crystallized from ice-cold ethanol. Yield: 12.72 g (90%); mp 51°C. NMR spectra are identical with those by Method A.
4.1.2. 9-Pentyl-9H-carbazole-3-carbaldehyde (2)
The title compound was prepared according to a modified literature procedure[33]. POC13 (2.6 mL, 28.40 mmol) was added, over a period of 10 min, to an ice-cooled, stirred DMF (7.43 mL, 95.96 mmol) under nitrogen. The reddish solution was allowed to stir at room temperature for 1 h. 9-Pentyl-9H-carbazole (1) (3.169 g, 13.35 mmol) was added over 10 min, and the obtained mixture was subjected to microwave irradiation at 100°C for 1 h. The reaction mixture was cooled and then poured into crushed ice. After warming to room temperature, the resultant product was extracted with EtOAc. The organic phase was washed with water, brine, dried (MgSO4), filtered, and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a white solid (3.49 g, 99%); mp 63–64°C. 1H NMR (500 MHz, CDCl3) δ 10.07 (s, 1H), 8.56 (s, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 8.5, 1.1 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.30 (t, J = 7.4 Hz, 1H), 4.27 (t, J = 7.3 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H), 1.34 (d, J = 3.7 Hz, 4H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 191.86 (C=O), 144.21 (C), 141.33 (C), 128.66 (C), 127.27 (CH), 126.85 (CH), 124.09 (CH), 123.21 (C), 123.15 (C), 120.87 (CH), 120.43 (CH), 109.55 (CH), 109.07 (CH), 43.54 (CH2), 29.50 (CH2), 28.78 (CH2), 22.59 (CH2), 14.08 (CH3).
4.1.3. 9-Pentyl-9H-carbazole-3-carboxylic acid (3)
To an ice-cold solution of 9-pentyl-3-formylcarbazole (2) (2.96 g, 11.16 mmol) in water/acetone (100 mL, 1:1. v/v) was added dropwise under stirring a solution of potassium permanganate (1.8 g, 11.39 mmol) in acetone (50 mL). The mixture was heated 3 h at reflux and then allowed to cool to room temperature. After that the reaction mixture was quenched with ethanol (20 mL), and then stirred for 30 min at reflux. After cooling to room temperature, the mixture was filtered through a pad of Celite® and concentrated in vacuo. The concentrated solution was diluted with water (100 mL), basified with NaOH to pH ca. 10, and extracted with heptane/ether (4:1, v/v, 50 mL × 3) to remove the unreacted starting material. The aqueous solution was cooled on an ice-water bath and then acidified with ice-cold solution of sulfuric acid (20%) to pH ca. 2. The resultant bulky precipitate was extracted with EtOAc (150 mL). The organic layer was washed with brine (30 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The precipitated product was collected by filtration, washed with heptanes (20 mL), and dried overnight to produce the title compound 3 (1.743 g, 55%) as a light yellow-to-greenish solid; mp 147°C. 1H NMR (500 MHz, CDCl3) δ 12.66 (s, 1H), 8.91 (d, J = 1.5 Hz, 1H), 8.25 (dd, J = 8.6, 1.6 Hz, 1H), 8.15 (d, J = 7.7 Hz, 1H), 7.54 – 7.47 (m, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.34 – 7.26 (m, 1H), 4.27 (t, J = 7.3 Hz, 2H), 1.86 (p, J = 7.4 Hz, 2H), 1.40 – 1.29 (m, 4H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 173.58 (C=O), 143.80 (C), 141.22 (C), 128.04 (CH), 126.60 (CH), 123.91 (CH), 123.15 (C), 122.85 (C), 120.92 (CH), 120.17 (CH), 119.84 (C), 109.35 (CH), 108.43 (CH), 43.48 (CH2), 29.55 (CH2), 28.82 (CH2), 22.65 (CH2), 14.15 (CH3).
4.1.4. (9-Pentyl-9H-carbazol-3-yl)(piperidin-1-yl)methanone (4)
Method A: amide coupling
Carboxylic acid 3 (300 mg, 1.07 mmol), piperidine (215 mg, 2.53 mmol), DIPEA (363 μL, 2.14 mmol), and DMAP (156 mg, 1.28 mmol) were added to DCM (30 mL) under nitrogen. The obtained solution was cooled down on an ice-water bath. EDC (350 mg, 1.83 mmol) was added to the solution, and the reaction mixture was stirred for 16 h while warming at room temperature. The solvent was removed in vacuo, and the obtained residue was extracted with EtOAc (100 mL). The organic layer was washed consecutively with 5% citric acid solution (50 mL × 3), concentrated sodium bicarbonate (50 mL × 3), brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light yellow gum (345 mg, 93%). 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 1.2 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.53 (dd, J = 8.4, 1.6 Hz, 1H), 7.49 – 7.43 (m, 1H), 7.41 – 7.35 (m, 2H), 7.26 – 7.20 (m, 1H), 4.25 (t, J = 7.2 Hz, 2H), 3.73 (br.m, 4H), 1.83 (p, J = 7.3 Hz, 2H), 1.72 – 1.65 (m, 3H), 1.61 (br.s, 3H), 1.36 – 1.29 (m, 4H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.53 (C=O), 140.99 (C), 140.90 (C), 126.74 (C), 126.13 (CH), 125.08 (CH), 122.74 (C), 122.43 (C), 120.54 (CH), 119.88 (CH), 119.28 (CH), 109.01 (CH), 108.40 (CH), 43.20 (CH2), 29.41 (CH2), 28.71 (CH2), 24.79 (CH2), 22.53 (CH2), 14.03 (CH3). ESI: m/z 349.5 (M + H)+. HRMS calcd for C23H29N2O (M + H)+ 349.2280, found 349.2311.
4.1.5. N,N-diethyl-9-pentyl-9H-carbazole-3-carboxamide (5)
Using carboxylic acid 3 (112 mg, 0.40 mmol) and diethylamine (74 μL, 0.71 mmol) as starting compounds, the title compound was prepared as a clear, colorless viscous oil according to the procedure described above for 4. Yield: 44 mg (33%). 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 1.5 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.52 (dd, J = 8.4, 1.6 Hz, 1H), 7.50 – 7.46 (m, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.26 – 7.22 (m, 1H), 4.30 (t, J = 7.2 Hz, 2H), 3.83 – 3.16 (br. m, 4H), 1.87 (p, J = 7.4 Hz, 2H), 1.35 (dq, J = 7.2, 3.6 Hz, 4H), 1.31 – 1.07 (m, 6H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 172.59 (C=O), 141.00 (C), 140.94 (C), 127.71 (C), 126.23 (CH), 124.61 (CH), 122.87 (C), 122.53 (C), 120.67 (CH), 119.36 (CH), 119.28 (CH), 109.11 (CH), 108.58 (CH), 43.36 (CH2), 29.57 (CH2), 28.85 (CH2), 22.67 (CH2), 14.16 (CH3). ESI: m/z 337.2 (M + H)+. HRMS calcd for C22H29N2O (M + H)+ 337.2280, found 337.2309.
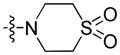
4.1.6. (9-Pentyl-9H-carbazol-3-yl)(1,1-dioxo-thiomorpholino)methanone (6)
Using carboxylic acid 3 (115 mg, 0.41 mmol) and 1,1-dioxo-thiomorpholine (80 mg, 0.59 mmol) as starting compounds, the title compound was prepared as a clear, colorless gum according to the procedure described above for 4. The obtained product solidified upon refrigeration to a white solid Yield: 83 mg (51%); mp 115°C. 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 1.5 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.55 (dd, J = 8.4, 1.7 Hz, 1H), 7.51 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.43 (t, J = 8.3 Hz, 2H), 7.30 – 7.24 (m, 1H), 4.30 (t, J = 7.3 Hz, 2H), 4.17 (s, 4H), 3.10 (s, 4H), 1.86 (p, J = 7.4 Hz, 2H), 1.35 (dq, J = 7.2, 3.7 Hz, 4H), 0.91 – 0.83 (m, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 172.50 (C=O), 141.69 (C), 141.08 (C), 126.72 (CH), 125.12 (CH), 123.87 (C), 122.79 (C), 122.49 (C), 120.72 (CH), 120.47 (CH), 119.83 (CH), 109.31 (CH), 108.91 (CH), 52.18 (CH2), 43.38 (CH2), 29.46 (CH2), 28.76 (CH2), 22.57 (CH2), 14.09 (CH3). ESI: m/z 399.1 (M + H)+. HRMS calcd for C22H27N2O3S (M + H)+ 399.1742, found 399.1718.
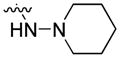
4.1.7. 9-Pentyl-N-(piperidin-1-yl)-9H-carbazole-3-carboxamide (7)
Using carboxylic acid 3 (100 mg, 0.36 mmol) and 1-aminopiperidine (39 μL, 0.36 mmol) as starting compounds, the title compound was prepared as a light yellow solid according to the procedure described above for 4 with the exception of not using 5% citric acid wash during the workup. Yield: 109 mg (84%); mp 158°C. 1H NMR (500 MHz, CDCl3) δ 8.51 (s, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.49 (ddd, J = 8.2, 7.1, 1.0 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.26 (dd, J = 8.1, 6.8 Hz, 1H), 6.96 (s, 1H), 4.28 (t, J = 7.2 Hz, 2H), 2.93 (br. s, 4H), 1.85 (p, J = 7.5 Hz, 2H), 1.82 – 1.73 (m, 4H), 1.46 (br. s, 2H), 1.38 – 1.30 (m, 4H), 0.86 (t, J = 6.8 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 166.22 (C=O), 142.29 (C), 141.06 (C), 126.35 (CH), 124.90 (CH), 124.62 (C), 122.92 (C), 122.64 (C), 120.75 (CH), 120.02 (CH), 119.63 (CH), 109.16 (CH), 108.49 (CH), 57.45 (CH2), 43.31 (CH2), 29.47 (CH2), 28.75 (CH2), 25.57 (CH2), 23.50 (CH2), 22.58 (CH2), 14.09 (CH3). ESI: m/z 364.2 (M + H)+. HRMS calcd for C23H30N3O (M + H)+ 364.2389, found 364.2356.
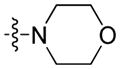
4.1.8. Morpholino(9-pentyl-9H-carbazol-3-yl)methanone (8)
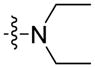
Using carboxylic acid 3 (100 mg, 0.36 mmol) and morpholine (62 μL, 0.71 mmol) as starting compounds, the title compound was prepared as a white solid according to the procedure described above for 4. Yield: 109 mg (84%); mp 98°C. 1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 1.6 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 8.4, 1.7 Hz, 1H), 7.48 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.40 (t, J = 8.5 Hz, 2H), 7.25 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 4.28 (t, J = 7.2 Hz, 2H), 3.73 (s, 8H), 1.90 – 1.79 (m, 2H), 1.38 – 1.28 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.72 (C=O), 141.26 (C), 140.98(C), 126.36 (CH), 125.45 (C), 125.29(CH), 122.66 (C), 122.57 (C), 120.63 (CH), 120.29 (CH), 119.50 (CH), 109.14 (CH), 108.59 (CH), 67.11 (CH2), 43.28 (CH2), 29.45 (CH2), 28.74 (CH2), 22.57 (CH2), 14.08 (CH3). ESI: m/z 351.2 (M + H)+. HRMS calcd for C22H27N2O2 (M + H)+ 351.2073, found 351.2071.
4.1.9. (4-Methylpiperazin-1-yl)(9-pentyl-9H-carbazol-3-yl)methanone (9)
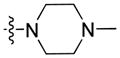
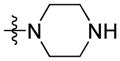
Using carboxylic acid 3 (110 mg, 0.39 mmol) and 1-methylpiperazine (71 mg, 0.71 mmol) as starting compounds, the title compound was prepared as a clear, orange viscous oil according to the procedure described above for 4 with the exception of not using 5% citric acid during the workup. Yield: 134 mg (94%). 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 1.3 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.55 (dd, J = 8.4, 1.6 Hz, 1H), 7.48 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.27 – 7.22 (m, 1H), 4.28 (t, J = 7.2 Hz, 2H), 3.78 (br. s, 4H), 2.46 (br. s, 4H), 2.33 (s, 3H), 1.85 (p, J = 7.3 Hz, 2H), 1.37 – 1.32 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.63 (C=O), 141.19 (C), 140.97 (C), 126.30 (CH), 125.97 (C), 125.31 (CH), 122.72 (C), 122.51 (C), 120.64 (CH), 120.21 (CH), 119.44 (CH), 109.11 (CH), 108.52 (CH), 46.21 (CH3), 43.29 (CH2), 29.47 (CH2), 28.76 (CH2), 22.58 (CH2), 14.09 (CH3). ESI: m/z 364.2 (M + H)+. HRMS calcd for C23H30N3O (M + H)+ 364.2389, found 364.2356.
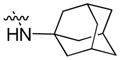
4.1.10. N-(1-adamantyl)-9-pentyl-9H-carbazole-3-carboxamide (10)
Using carboxylic acid 3 (112 mg, 0.40 mmol) and 1-adamantylamine (74 μL, 0.71 mmol) as starting compounds, the title compound was prepared as a light yellow glass according to the procedure described above for compound 4. Yield: 91 mg (62%). 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 1.7 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.85 (dd, J = 8.6, 1.8 Hz, 1H), 7.48 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.29 – 7.22 (m, 1H), 5.97 (s, 1H), 4.26 (t, J = 7.2 Hz, 2H), 2.20 (d, J = 2.6 Hz, 6H), 2.15 (br. s, 3H), 1.88 – 1.80 (m, 2H), 1.79 – 1.69 (m, 6H), 1.37 – 1.28 (m, 4H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 167.51 (C=O), 142.14 (C), 141.13 (C), 126.79 (C), 126.31 (CH), 124.69 (CH), 123.03 (C), 122.62 (C), 120.70 (CH), 119.59 (CH), 119.56 (CH), 109.19 (CH), 108.41 (CH), 52.36 (C), 43.36 (CH2), 42.01 (CH2), 36.64 (CH2), 29.74 (CH), 29.51 (CH2), 28.80 (CH2), 22.62 (CH2), 14.13 (CH3). ESI: m/z 415.2 (M + H)+. HRMS calcd for C28H35N2O (M + H)+ 415.2749, found 415.2762.
4.1.11. N-(4-chlorophenethyl)-9-pentyl-9H-carbazole-3-carboxamide (11)
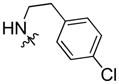
Using carboxylic acid 3 (105 mg, 0.37 mmol) and 2-(4-chlorophenyl)ethanamine (90 mg, 0.58 mmol) as starting compounds, the title compound was prepared as a white solid according to the procedure described above for 4. Yield: 38 mg (23%); mp 153°C. 1H NMR (500 MHz, CDCl3) δ 8.57 – 8.43 (m, 1H), 8.07 (d, J = 7.6 Hz, 1H), 7.81 (ddd, J = 8.6, 1.7, 0.8 Hz, 1H), 7.49 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.30 – 7.22 (m, 3H), 7.17 (d, J = 8.4 Hz, 2H), 4.26 (t, J = 7.3 Hz, 2H), 3.76 – 3.70 (m, 2H), 2.94 (t, J = 7.0 Hz, 2H), 1.84 (p, J = 7.4 Hz, 2H), 1.37 – 1.31 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 168.39 (C=O), 168.32 (C=O), 142.37 (C), 141.17 (C), 137.85 (C), 132.46 (C), 130.42 (CH), 128.92 (CH), 126.49 (CH), 125.15 (C), 125.12 (C), 124.65 (CH), 122.96 (C), 122.77 (C), 120.74 (CH), 119.90 (CH), 119.76 (CH), 109.25 (CH), 108.58 (CH), 43.42 (CH2), 41.38 (CH2), 41.26 (CH2), 35.47 (CH2), 35.46 (CH2), 29.54 (CH2), 28.82 (CH2), 22.63 (CH2), 14.14 (CH3). ESI: m/z 419.4 (M + H)+. HRMS calcd for C26H28N2OCl (M + H)+ 419.1890, found 419.1870.
4.1.12. Tert-butyl 4-((9-pentyl-9H-carbazole-3-carboxamido)methyl)piperidine-1-carboxylate (12)
Carboxylic acid 3 (700 mg, 2.49 mmol), HOBt (404 mg, 2.99 mmol), DIPEA (847 μL, 4.98 mmol), DMAP (365 mg, 2.99 mmol), and EDC (573 mg, 2,99 mmol) were added upon stirring to DCM (50 mL) under nitrogen. The obtained solution was cooled down on an ice-water bath. Tert-butyl 4-(aminomethyl)piperidine-1-carboxylate (749 mg, 2.99 mmol) was added in one portion, and the resulting reaction mixture was then allowed to warm to room temperature and stirred for 16 h. The solvent was removed in vacuo, and the obtained residue was extracted with EtOAc (100 mL). The organic layer was washed consecutively with 5% citric acid solution (50 mL × 3), concentrated sodium bicarbonate (50 mL × 3), and brine (50 mL). The organic layer was then separated, dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography eluting with heptane/EtOAc (gradient elution) to give 1.096 g (92%) of 14 as a yellow glass. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 1.1 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.90 (dd, J = 8.6, 1.4 Hz, 1H), 7.47 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.24 (t, J = 7.5 Hz, 1H), 6.64 (br s, 1H), 4.26 (t, J = 7.2 Hz, 2H), 4.07 (br m, 2H), 3.39 (br s, 2H), 2.68 (t, J = 12.1 Hz, 2H), 1.91 – 1.79 (m, 3H), 1.75 (br d, J = 13.2 Hz, 2H), 1.45 (s, 9H), 1.37 – 1.29 (m, J = 8.9, 5.2 Hz, 4H), 1.20 (qd, J = 12.5, 4.3 Hz, 2H), 0.86 (t, J = 6.8 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 168.49 (C=O), 154.76 (C), 142.05 (C), 140.86 (C), 126.12 (CH), 125.05 (C), 124.86 (CH), 122.76 (C), 122.43 (C), 120.43 (CH), 119.80 (CH), 119.38 (CH), 108.96 (CH), 108.19 (CH), 79.24 (C), 45.45 (CH2), 43.00 (CH2), 36.48 (CH), 29.91 (CH2), 29.20 (CH2), 28.50 (CH2), 28.41 (CH3), 22.33 (CH2), 13.87 (CH3). ESI: m/z 478.2 (M + H)+. HRMS calcd for C29H40N3O3 (M + H)+ 478.3070, found 478.3007.
4.1.13. Tert-butyl 4-(9-pentyl-9H-carbazole-3-carbonyl)piperazine-1-carboxylate (13)
Using carboxylic acid 3 (700 mg, 2.49 mmol) and tert-butyl piperazine-1-carboxylate (556 mg, 2.99 mmol) as starting compounds, the title compound was prepared as a light yellow glass according to the procedure described above for compound 14. Yield: 1.03 g (92%). 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 1.2 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 8.4, 1.5 Hz, 1H), 7.48 (J = 8.3, 7.0, 1.2 Hz, ddd, 1H), 7.41 (t, J = 8.1 Hz, 2H), 7.26 (ddd, J = 7.0, 5.9, 1.1 Hz, 1H), 4.30 (t, J = 7.2 Hz, 2H), 3.68 (br s, 4H), 3.50 (br s, 4H), 1.93 – 1.82 (m, 2H), 1.48 (s, 9H), 1.39 – 1.31 (m, 4H), 0.87 (t, J = 7.0 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 171.98 (C=O), 154.83 (C), 141.38 (C), 141.09 (C), 126.45 (CH), 125.71 (C), 125.32 (CH), 122.77 (C), 122.69 (C), 120.70 (CH), 120.35 (CH), 119.59 (CH), 109.20 (CH), 108.66 (CH), 80.42 (C), 43.39 (CH2), 29.53 (CH2), 28.81 (CH2), 28.56 (CH3), 22.62 (CH2), 14.11 (CH3). ESI: m/z 450.1 (M + H)+. HRMS calcd for C27H36N3O3 (M + H)+ 450.2757, found 450.2777.
4.1.14. 9-Pentyl-N-(piperidin-4-ylmethyl)-9H-carbazole-3-carboxamide (14)
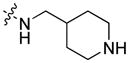
Dry hydrogen chloride gas was passed through a solution containing 760 mg (1.59 mmol) of 12 dissolved in 30 mL of EtOAc for about ten minutes. The solution was stirred overnight, and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 1N NaOH. The organic layer was washed with brine (30 mL), dried (MgSO4), and filtered. The solvent was evaporated under reduced pressure, and the residue self-crystallized. The white crystalline solid was collected by filtration, washed with heptanes (20 mL) and dried in vacuo. Yield (as a free base): 527 mg (88%); mp 163°C. 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 1.4 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.90 (dd, J = 8.6, 1.7 Hz, 1H), 7.48 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.24 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.57 (t, J = 5.7 Hz, 1H), 4.26 (t, J = 7.2 Hz, 2H), 3.39 (t, J = 6.2 Hz, 2H), 3.09 (d, J = 12.1 Hz, 2H), 2.60 (td, J = 12.2, 2.5 Hz, 2H), 1.98 (s, 1H), 1.91 – 1.80 (m, 2H), 1.80 – 1.71 (m, 3H), 1.40 – 1.29 (m, 4H), 1.23 (qd, J = 12.8, 3.7 Hz, 2H), 0.86 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 168.51 (C=O), 142.32 (C), 141.15 (C), 126.40 (CH), 125.41 (C), 124.79 (CH), 122.99 (C), 122.73 (C), 120.73 (CH), 119.82 (CH), 119.67 (CH), 109.21 (CH), 108.50 (CH), 46.47 (CH2), 46.20 (CH2), 43.37 (CH2), 36.91 (CH), 31.37 (CH2), 29.49 (CH2), 28.77 (CH2), 22.59 (CH2), 14.09 (CH3). ESI: m/z 378.2 (M + H)+. HRMS calcd for C24H32N3O (M + H)+ 378.2545, found 378.2583.
4.1.15. (9-Pentyl-9H-carbazol-3-yl)(piperazin-1-yl)methanone (15)
Starting with compound 13 (973 mg, 2.16 mmol), the title compound was prepared as a light yellow glass according to the procedure described above for compound 14. Yield (as a free base): 732 mg (97%). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 1.1 Hz, 1H), 8.13 (d, J = 7.7 Hz, 1H), 7.57 (dd, J = 8.4, 1.5 Hz, 1H), 7.52 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.44 (t, J = 8.6 Hz, 2H), 7.29 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 4.33 (t, J = 7.2 Hz, 2H), 3.72 (br s, 4H), 2.95 (br s, 4H), 2.02 (s, 1H), 1.90 (p, J = 7.3 Hz, 2H), 1.45 – 1.31 (m, 4H), 0.90 (t, J = 7.0 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 171.74 (C=O), 141.20 (C), 141.04 (C), 126.32 (CH), 126.18 (C), 125.31 (CH), 122.80 (C), 122.60 (C), 120.67 (CH), 120.21 (CH), 119.47 (CH), 109.13 (CH), 108.55 (CH), 46.47 (CH2), 43.34 (CH2), 29.50 (CH2), 28.78 (CH2), 22.60 (CH2), 14.09 (CH3). ESI: m/z 350.1 (M + H)+. HRMS calcd for C22H28N3O (M + H)+ 350.2232, found 350.2235.
4.1.16. Pyridin-4-yl-methyl 9-pentyl-9H-carbazole-3-carboxylate (16)
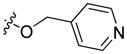
To a stirred mixture of carboxylic acid 3 (200 mg, 0.71 mmol), 4-(bromomethyl)pyridine (147 mg, 0.85 mmol), triethylamine (178 μL, 1.27 mmol), and sodium carbonate (1 g, 9.43 mmol) in THF (3 mL) in a 20 mL microwave vessel was added 1 M TBAF in THF (853 μL, 0.853 mmol). The mixture was stirred at room temperature for 16 h under a nitrogen atmosphere, and then subjected to microwave irradiation at 60°C for 1 h. After cooling to room temperature, the reaction mixture was diluted with DCM (30 mL) and transferred to a round-bottomed flask. The volatiles were removed in vacuo, and the obtained residue was dissolved in DCM (150 mL). The organic layer was washed with 0.05 M NaOH (50 mL × 2), dried over magnesium sulfate, filtered, and concentrated in vacuo. The obtained residue was purified by flash chromatography eluting with heptane/EtOAc (gradient elution) on a Biotage® KP-NH cartridge yielding compound 12 as a yellowish oil that solidified on standing to a white solid. Yield: 203 mg, (77%); mp 104°C. 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 8.68 (br. s, 2H), 8.22 (d, J = 8.6 Hz, 1H), 8.16 (d, J = 7.7 Hz, 1H), 7.58 – 7.48 (m, 3H), 7.44 (d, J = 7.5 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.30 (t, J = 7.4 Hz, 1H), 5.48 (s, 2H), 4.32 (t, J = 7.2 Hz, 2H), 1.98 – 1.80 (m, 2H), 1.45 – 1.29 (m, 4H), 0.88 (t, J = 6.7 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 166.97 (C=O), 148.37 (CH), 148.27 (C), 143.63 (CH), 141.28 (C), 127.56 (CH), 126.75 (CH), 123.34 (CH), 123.06 (C), 122.95 (C), 122.61 (CH), 120.88 (CH), 120.22 (CH), 119.73 (C), 109.43 (CH), 108.60 (CH), 64.37 (CH2), 43.53 (CH2), 29.53 (CH2), 28.80 (CH2), 22.61 (CH2), 14.10 (CH3). ESI: m/z 373.2 (M + H)+. HRMS calcd for C24H25N2O2 (M + H)+ 373.1916, found 373.1890.
4.1.17. Pyridin-2-ylmethyl 9-pentyl-9H-carbazole-3-carboxylate (17)
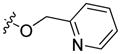
The title compound was prepared from carboxylic acid 3 (316 mg, 1.12 mmol) as a light amber viscous oil by the same procedure as described for 16. Yield: 392 mg, (99%). 1H NMR (500 MHz, CDCl3) δ 8.86 (d, J = 1.5 Hz, 1H), 8.58 (ddd, J = 4.8, 1.1, 0.7 Hz, 1H), 8.20 (dd, J = 8.6, 1.7 Hz, 1H), 8.07 (d, J = 7.7 Hz, 1H), 7.62 (td, J = 7.7, 1.8 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.43 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.31 (d, J = 8.2 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.22 (ddd, J = 7.8, 7.1, 0.7 Hz, 1H), 7.14 (ddd, J = 7.5, 4.9, 0.9 Hz, 1H), 5.55 (s, 2H), 4.11 (t, J = 7.2 Hz, 2H), 1.74 (p, J = 7.3 Hz, 2H), 1.33 – 1.17 (m, 4H), 0.80 (t, J = 6.9 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 166.77 (C=O), 156.23 (C), 149.00 (CH), 143.01 (C), 140.79 (C), 136.80 (CH), 127.24 (CH), 126.24 (CH), 122.90 (CH), 122.69 (C), 122.64 (CH), 122.40 (C), 121.59 (CH), 120.45 (CH), 119.91 (C), 119.71 (CH), 109.01 (CH), 108.09 (CH), 66.65 (CH2), 42.93 (CH2), 29.11 (CH2), 28.41 (CH2), 22.26 (CH2), 13.81 (CH3). ESI: m/z 373.1 (M + H)+. HRMS calcd for C24H25N2O2 (M + H)+ 373.1916, found 373.1890.
4.1.18. (9-Pentyl-9H-carbazol-3-yl)(phenyl)methanone (18)
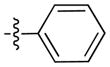
Under argon atmosphere, AlCl3 (309 mg, 2.32 mmol) was added to a solution of 9-pentyl-9H-carbazole (1) (500 mg, 2.11 mmol) in anhydrous benzene (30 mL), and the obtained solution was cooled by an ice bath for 20 min. Benzoyl chloride (282 μL, 2.43 mmol) was added dropwise via a syringe to the solution, and the reaction mixture was stirred for 16 h while warming at room temperature. The reaction mixture was cooled on an ice-water bath then poured onto a mixture of ice and 4 M NaOH solution (50 mL) and extracted with diethyl ether (150 mL). The organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to give the target product (514 mg, 71%) as a light yellow solid: mp 116°C. 1H NMR (500 MHz, CDCl3) δ 8.61 (d, J = 1.0 Hz, 1H), 8.10 (d, J = 7.7 Hz, 1H), 8.03 (dd, J = 8.5, 1.3 Hz, 1H), 7.85 (d, J = 7.1 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.56 – 7.47 (m, 3H), 7.44 (d, J = 2.2 Hz, 1H), 7.43 (d, J = 2.9 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H), 4.32 (t, J = 7.2 Hz, 2H), 1.89 (p, J = 7.0 Hz, 2H), 1.46 – 1.29 (m, 4H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 196.79 (C=O), 143.21 (C), 141.29 (C), 139.22 (C), 131.82 (CH), 130.08 (CH), 128.61 (CH), 128.59 (C), 128.35 (CH), 126.60 (CH), 124.22 (CH), 123.28 (C), 122.59 (C), 120.89 (CH), 120.08 (CH), 109.41 (CH), 108.45 (CH), 43.51 (CH2), 29.55 (CH2), 28.85 (CH2), 22.65 (CH2), 14.14 (CH3). ESI: m/z 342.1 (M + H)+. HRMS calcd for C24H24NO (M + H)+ 342.1858, found 342.1859.
4.1.19. 2-Bromo-1-(9-pentyl-9H-carbazol-3-yl)ethanone (19)
Under argon atmosphere, anhydrous AlCl3 (658 mg, 4.93 mmol) was added to a solution of 9-pentyl-9H-carbazole (1) (1.172 g, 4.94 mmol) in anhydrous benzene (20 mL), and the obtained solution was cooled by an ice bath for 20 min. 1-Bromoacetyl bromide (429 μL, 4.94 mmol) was added dropwise to the solution, and the reaction mixture was stirred for 19 h while warming at room temperature. The reaction mixture was quenched with 3 mL of concentrated HCl solution, and then extracted with diethyl ether (150 mL). The organic phase was washed with saturated solution of ascorbic acid (3 × 30 mL), water (30 mL), saturated aqueous sodium bicarbonate (2 × 30 mL), brine, dried (MgSO4), filtered and evaporated in vacuo. The residue was purified on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light yellow gum (522 mg, 67%) which self-crystallized shortly after standing: mp 91°C. 1H NMR (500 MHz, CDCl3) δ 8.75 (d, J = 1.7 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.12 (dd, J = 8.7, 1.8 Hz, 1H), 7.53 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.44 (d, J = 8.2 Hz, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.32 (ddd, J = 7.8, 7.0, 0.7 Hz, 1H), 4.58 (s, 2H), 4.30 (t, J = 7.3 Hz, 2H), 1.88 (p, J = 7.4 Hz, 2H), 1.36 (dq, J = 7.2, 3.6 Hz, 4H), 0.88 (t, J = 7.1 Hz, 1H). 13C and APT NMR (126 MHz, CDCl3) δ 190.94 (C=O), 143.74 (C), 141.33 (C), 127.11 (CH), 126.89 (CH), 125.32 (C), 123.17 (C), 122.99 (C), 122.75 (CH), 120.89 (CH), 120.43 (CH), 109.55 (CH), 108.80 (CH), 43.54 (CH2), 31.43 (CH2), 29.52 (CH2), 28.82 (CH2), 22.62 (CH2), 14.13 (CH3).
4.1.20. 1-(9-Pentyl-9H-carbazol-3-yl)-2-(piperidin-1-yl)ethanone (20)
Under nitrogen atmosphere, a mixture of bromide 19 (208 mg, 0.58 mmol), piperidine (165 μL, 1.67 mmol), and triethylamine (234 μL, 1.68 mmol) in DMF (3 mL) was subjected to microwave irradiation at 90°C for 5 min. The mixture was allowed to cool to room temperature, and the organic solvents were evaporated in vacuo. The residue was purified on a Biotage® KP-NH cartridge (amino-modified silica gel) using cyclohexane/EtOAc in different proportions to afford to give the title compound as a clear yellowish oil (202 mg, 100%), which darkened on standing. 1H NMR (500 MHz, CDCl3) δ 8.80 (d, J = 0.8 Hz, 1H), 8.15 (dd, J = 8.7, 1.0 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.45 (dd, J = 8.0, 7.3 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 4.16 (t, J = 7.2 Hz, 2H), 3.88 (s, 2H), 2.59 (br. s, 4H), 1.84 – 1.73 (m, 2H), 1.72 – 1.61 (m, 4H), 1.51 – 1.40 (m, 2H), 1.34 – 1.23 (m, 4H), 0.83 (t, J = 6.8 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 196.04 (C=O), 143.06 (C), 140.97 (C), 127.63 (C), 126.30 (CH), 126.23 (CH), 123.10 (C), 122.38 (C), 121.55 (CH), 120.49 (CH), 119.82 (CH), 109.15 (CH), 108.12 (CH), 65.40 (CH2), 54.95 (CH2), 43.09 (CH2), 29.25 (CH2), 28.56 (CH2), 25.87 (CH2), 24.08 (CH2), 22.39 (CH2), 13.91 (CH3). ESI: m/z 363.1 (M + H)+. HRMS calcd for C24H31N2O (M + H)+ 363.2436, found 363.2408.
4.1.21. 1-(9-Pentyl-9H-carbazol-3-yl)-2-(1,1-dioxo-thiomorpholino)ethanone (21)
Using bromide 19 (200 mg, 0.56 mmol), 1,1-dioxo-thiomorpholine (226 mg, 1.67 mmol), and triethylamine (234 μL, 1.68 mmol) as starting compounds, the title compound was prepared as a beige glass according to the procedure described above for 20. Yield: 207 mg (90%). 1H NMR (500 MHz, CDCl3) δ 8.68 (d, J = 1.5 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 8.05 (dd, J = 8.7, 1.6 Hz, 1H), 7.52 – 7.47 (m, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 4.22 (t, J = 7.2 Hz, 2H), 4.12 (s, 2H), 3.25 – 3.17 (m, 4H), 3.17 – 3.09 (m, 4H), 1.89 – 1.75 (m, 2H), 1.38 – 1.26 (m, 4H), 0.85 (t, J = 6.9 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 194.76 (C=O), 143.24 (C), 140.98 (C), 126.63 (C), 126.58 (CH), 125.80 (CH), 122.83 (C), 122.45 (C), 121.25 (CH), 120.51 (CH), 120.04 (CH), 109.33 (CH), 108.46 (CH), 62.20 (CH2), 51.46 (CH2), 51.10 (CH2), 43.14 (CH2), 29.18 (CH2), 28.50 (CH2), 22.32 (CH2), 13.90 (CH3). ESI: m/z 413.1 (M + H)+. HRMS calcd for C23H29N2O3S (M + H)+ 413.1899, found 413.1874.
4.1.22. 2-(4-Methylpiperazin-1-yl)-1-(9-pentyl-9H-carbazol-3-yl)ethanone (22)
Using bromide 19 (265 mg, 0.74 mmol), 1-methylpiperazine (214 μL, 1.93 mmol), and triethylamine (269 μL, 1.93 mmol) as starting compounds, the title compound was prepared as a clear amber gum according to the procedure described above for 20. Yield: 247 mg (89%). 1H NMR (500 MHz, CDCl3) δ 8.73 (d, J = 1.5 Hz, 1H), 8.11 (d, J = 7.4 Hz, 1H), 8.09 (dd, J = 8.6, 1.6 Hz, 1H), 7.47 (ddd, J = 8.1, 6.9, 0.9 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.30 (d, J = 8.7 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 4.18 (t, J = 7.2 Hz, 2H), 3.98 (s, 2H), 3.08 – 2.62 (br. m, 8H), 2.48 (s, 3H), 1.84 – 1.74 (m, 2H), 1.35 – 1.23 (m, 4H), 0.84 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 194.89 (C=O), 142.97 (C), 140.79 (C), 126.99 (C), 126.26 (CH), 125.81 (CH), 122.79 (C), 122.24 (C), 121.21 (CH), 120.34 (CH), 119.76 (CH), 109.04 (CH), 108.11 (CH), 63.60 (CH2), 54.26 (CH2), 52.11 (CH2), 44.88 (CH3), 42.93 (CH2), 29.02 (CH2), 28.33 (CH2), 22.15 (CH2), 13.71 (CH3). ESI: m/z 378.2 (M + H)+. HRMS calcd for C24H32N3O (M + H)+ 378.2545, found 378.2565.
4.1.23. 1,1-Dimethyl-4-[(9-pentyl-9H-carbazol-3-yl)carbonyl]piperazin-1-ium iodide (23)
Methyl iodide (764 μL, 12.32 mmol) was added to a stirred solution of the tertiary amine 9 (320 mg, 0.88 mmol) in anhydrous diethyl ether (10 mL). A precipitate immediately started forming, and stirring was continued for 18 h at room temperature. The precipitated solid was isolated by filtration, washed with diethyl ether (ca. 50 mL), and dried under high vacuum to provide the title compound (186 mg, 42%) as a light yellow solid: mp 119°C (with decomposition). 1H NMR (500 MHz, CDCl3) δ 8.37 (s, 1H), 8.14 (d, J = 7.7 Hz, 1H), 7.55 (d, J = 8.6 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 9.4 Hz, 1H), 7.15 (t, J = 7.4 Hz, 1H), 4.13 (t, J = 6.7 Hz, 2H), 3.97 (s, 4H), 3.78 (s, 4H), 3.49 (s, 6H), 1.73 (dt, J = 14.6, 7.4 Hz, 2H), 1.30 – 1.21 (m, 4H), 0.81 (t, J = 6.8 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 171.73 (C=O), 141.39 (C), 140.91 (C), 126.56 (CH), 125.51 (CH), 123.77 (C), 122.54 (C), 122.49 (C), 121.25 (CH), 120.92 (CH), 119.71 (CH), 109.15 (CH), 109.04 (CH), 61.31 (CH2), 52.26 (CH3), 43.28 (CH2), 29.35 (CH2), 28.67 (CH2), 22.50 (CH2), 14.09 (CH3). ESI: m/z 378.1 (M + H)+. HRMS calcd for C24H32N3O (M + H)+ 378.2545, found 378.2523.
4.1.24. 9-Pentyl-9H-carbazole-3-carbonitrile (24)
A mixture of 9-pentyl-9H-carbazole-3-carbaldehyde (500 mg, 1.88 mmol), hydroxylamine hydrochloride (270 mg, 4.19 mmol), and p-toluenesulfonic acid (73 mg, 0.42 mmol) was stirred at 235°C in a 20 mL microwave vessel under a stream of nitrogen until foaming and evolution of gas ceased. The mixture was then subjected to microwave irradiation at 105°C for 5 min. The reaction mixture was allowed to cool to room temperature, and then heated again at 235°C under a stream of nitrogen for 20 min. The obtained residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light orange solid. Yield: 336 mg (68%), mp 71°C. 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 1.3 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.62 (dd, J = 8.5, 1.5 Hz, 1H), 7.51 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H),7.40 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 4.21 (t, J = 7.3 Hz, 2H), 1.81 (p, J = 7.3 Hz, 2H), 1.42 – 1.18 (m, 4H), 0.85 (t, J = 6.9 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 142.09 (C), 141.02 (C), 128.86 (CH), 127.19 (CH), 125.18 (CH), 123.01 (C), 121.94 (C), 120.74 (CH), 120.36 (CH), 109.44 (CH), 109.37 (CH), 101.43 (C), 43.35 (CH2), 29.38 (CH2), 28.64 (CH2), 22.48 (CH2), 13.99 (CH3). ESI: m/z 263.2 (M + H)+. HRMS calcd for C18H19N2 (M + H)+ 263.1548, found 263.1522.
4.1.25. 9-Pentyl-9H-carbazol-3-yl)(piperidin-1-yl)methanethione (25)
Under argon atmosphere, a solution of 4 (60 mg, 0.17 mmol) and LR (49 mg, 0.12 mmol) in toluene (3 mL) was tightly capped in a 5 mL microwave vessel. The mixture was subjected to microwave irradiation at 140°C for 3 h and then cooled to room temperature. The organic solvent was evaporated in vacuo, and the residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to yield the target product as a yellow glass. Yield: 48 mg (76%). 1H NMR (500 MHz, CDCl3) δ 8.12 – 8.00 (m, 2H), 7.47 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.44 (dd, J = 8.4, 1.7 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.25 – 7.20 (m, 1H), 4.42 (br. s, 2H), 4.28 (t, J = 7.2 Hz, 2H), 3.68 – 3.60 (m, 2H), 1.90 – 1.82 (m, 4H), 1.76 (dt, J = 11.8, 6.1 Hz, 2H), 1.62 – 1.56 (m, 2H), 1.35 (dq, J = 7.2, 3.7 Hz, 4H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 201.31 (C=S), 141.09 (C), 140.55 (C), 134.30 (C), 126.23 (CH), 124.23 (CH), 122.84 (C), 122.47 (C), 120.71 (CH), 119.33 (CH), 118.53 (CH), 109.09 (CH), 108.45 (CH), 53.76 (CH2), 51.46 (CH2), 43.34 (CH2), 29.51 (CH2), 28.83 (CH2), 27.16 (CH2), 25.76 (CH2), 24.44 (CH2), 22.64 (CH2), 14.13 (CH3). ESI: m/z 366.1 (M + H)+. HRMS calcd for C23H29N2S (M + H)+ 365.2051, found 365.2019.
4.1.26. 9-Pentyl-3-(piperidin-1-ylmethyl)-9H-carbazole (26)
A solution of 4 (495mg, 1.42 mmol) in anhydrous tetrahydrofuran (5 mL) was added dropwise to a 0°C cooled suspension of LAH (108 mg, 2.85 mmol) in anhydrous THF (50 mL). The reaction was then heated under reflux for 10 min. The reaction mixture was cooled to 0°C, and then water (3 mL) followed by 3.6 M NaOH solution (70 mL) were carefully added dropwise to the reaction to destroy the excess of LAH. The reaction mixture was then extracted with methyl tert-butyl ether (3 × 50 mL). The combined organic layers were washed with brine, dried (MgSO4) and evaporated. The obtained residue was purified by flash chromatography eluting with heptane/EtOAc (gradient elution) on a Biotage® KP-NH cartridge yielding compound 26 as a light orange oil (263 mg, 55%). 1H NMR of free base (500 MHz, CDCl3) δ 8.23 (d, J = 7.7 Hz, 1H), 8.17 (s, 1H), 7.59 – 7.51 (m, 2H), 7.46 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.33 (t, J = 7.4 Hz, 1H), 4.29 (t, J = 7.2 Hz, 2H), 3.78 (s, 2H), 2.57 (br. s, 4H), 1.92 (p, J = 7.2 Hz, 2H), 1.80 – 1.67 (m, 4H), 1.62 – 1.52 (br. m, 2H), 1.49 – 1.36 (m, 4H), 0.98 (t, J = 7.0 Hz, 3H). 13C and APT NMR of free base (101 MHz, CDCl3) δ 140.71 (C), 139.76 (C), 128.58 (C), 127.39 (CH), 125.49 (CH), 122.79 (C), 122.71 (C), 121.16 (CH), 120.40 (CH), 118.64 (CH), 108.62 (CH), 108.20 (CH), 64.29 (CH2), 54.47 (CH2), 43.02 (CH2), 29.40 (CH2), 28.71 (CH2), 25.99 (CH2), 24.51 (CH2), 22.50 (CH2), 13.99 (CH3). ESI: m/z 335.2 (M + H)+. HRMS calcd for C23H31N2 (M + H)+ 335.2487, found 335.2495.
4.1.27. 3-Bromo-9-pentyl-9H-carbazole (27)
Under an argon atmosphere, a mixture of 3-bromo-9H-carbazole (400 mg, 1.63 mmol), 1-bromopentane (0.3 mL, 2.45 mmol) and Cs2CO3 (1 g, 3.25 mmol) in DMF (5 mL) was subjected to microwave irradiation at 140°C for 1 h. After cooling to room temperature, the reaction was diluted with EtOAc and filtered. The organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with ethyl acetate/heptanes in different proportions to give 390.4 g (75.9%) of the target product in the form of a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 7.77 Hz, 1H) 8.01(s, 1H) 7.49-7.36 (m, 3H) 7.33 (d, J = 7.83 Hz, 1H) 7.21 (t, J = 7.78 Hz, 1H) 4.29 (q, J = 7.3 Hz, 2H), 1.95-1.84 (m, 2H) 1.42-1.31 (m, 4H) 0.87 (t, J = 6.85 Hz, 3H) 13C NMR (126 MHz, CDCl3) δ 140.72, 139.83, 128.16, 127.25, 125.63, 122.72, 121.25, 120.44, 118.74, 118.60, 108.78, 108.40, 55.15, 29.46, 28.75, 22.55, 13.99.
4.1.28. 3-((4-Methylpiperazin-1-yl)methyl)-9-pentyl-9H-carbazole (28)
Under an argon atmosphere, a mixture of bromide 27 (100 mg, 0.31 mmol), potassium methyl-4-trifluoroboratomethyl-piperizine (75.1 mg, 0.34), K2CO3 (131 mg, 0.95 mmol), Pd(OAc)2 (4.6 mg, 0.015 mmol), and 2-biphenyl-di-tert-butylphosphine (12.2 mg, 0.031 mmol) in DMF (2 mL) was subjected to microwave irradiation at 120°C for 20 minutes. The reaction was then cooled to room temperature, diluted with EtOAc, and filtered through a pad of Celite. The filtrate was then washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with ethyl DCM/methanol in different proportions to give 30 mg (28.5%) of the target product in the form of a yellow oil. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.83 Hz, 1H) 8. 02(s, 1H) 7. 47-7.38 (m, 3) 7. 34 (d, J = 7.82 Hz, 1H) 7.23 (t, J = 7.83 Hz, 1H) 4.28 (q, J = 7.34 Hz, 2H) 3.74 (s, 2H) 2.65-2. 58(m, 4H), 2.56-47 (m, 4H), 1. 92-1.85 (m, 2H), 1.62 (s, 9H), 1.42-1.35 (m, 4H), 0.88 (t, J = 6.85 Hz, 3H). 13C NMR and DEPT NMR (126 MHz, CDCl3) δ 140.68 (C), 139.79 (C), 128.12 (CH), 127.21 (C), 125.59 (CH), 122.68 (C), 121.21 (C), 120.40 (C), 118.70 (CH), 118.56 (C), 108.74 (CH), 108. 36 (CH), 63.48 (CH2), 56.04 (CH2), 55.11 (CH2), 49.64 (CH2), 46.61 (CH2), 29.42 (CH3), 28.70 (CH2), 22.50 (CH2), 13.95 (CH3). HRMS calcd for C23H32N3 (M + H)+ 350.2596, found 350.2609.
4.1.29. Tert-butyl 4-((9-pentyl-9H-carbazol-3-yl)methyl)piperazine-1-carboxylate (29)
Under an argon atmosphere, bromide 27 (100 mg, 0.31), potassium trifluoroboratomethyl -4-N-Boc-piperazine (104.1 mg, 0.341 mmol), K2CO3 (131 mg, 0.95 mmol), Pd(OAc)2 (4.6 mg, 0.015 mmol), (2-biphenyl)-di-tert-butylphosphine (12.2 mg, 0.031 mmol), and DMF (2 mL) were added to a microwave vessel (10 mL). The reaction was heated in a microwave at 120°C for 20 minutes. The reaction was then cooled to room temperature, diluted with EtOAc, and filtered through a pad of Celite. The filtrate was then washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with DCM/methanol in different proportions to give 85.7 mg (63.5%) of the target product in the form of a yellow oil. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.83 Hz, 1H), 8.02(s, 1H), 7. 47-7.38 (m, 3H), 7.34 (d, J = 7.82 Hz, 1H), 7.23 (t, j = 7.77 Hz, 1H), 4.28 (q, J = 7.34 Hz, 2H), 3.74 (s, 2H), 2.65-2.58 (m, 4H), 2. 56-47 (m, 4H), 1. 92-1.85 (m, 2H), 1.62 (s, 9H) 1. 42-1.35 (m, 4H), 0.88 (t, J = 6.85 Hz, 3H). 13C NMR and DEPT NMR (126 MHz, CDCl3) δ 156.16 (C), 140. 40 (C), 139. 59 (C), 128.12 (CH), 127.31 (CH), 125.79 (C), 122.68 (CH), 121.21 (C), 120.58 (CH), 118.70 (CH), 118.56 (C), 108.71 (CH), 108.24 (CH), 79.01 (C), 63.49 (CH2), 55.11 (CH2), 53.56 (CH2), 50.52 (CH2), 29.45 (CH2), 28.71 (CH3), 28.46 (CH2), 22.53 (CH2), 13.97 (CH3).
4.1.30. 9-Pentyl-3-(piperazin-1-ylmethyl)-9H-carbazole (30)
Compound 29 (60 mg, 0.16 mmol) was added to a solution of DCM (2 mL) and TFA (2 mL). The reaction mixture was stirred for 6 hours at room temperature. The reaction mixture was then diluted with DCM (30 mL), and the organic phase was washed consequtively with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with ethyl acetate/heptanes in different proportions to give 51.2 mg (94.9%) of the target product in the form of a yellow oil. 1H NMR 8.09 (d, J = 7.83 Hz, 1H), 8.02(s, 1H), 7.46(t, J = 7.34 Hz, 1H), 7.39 (d, J = 7.83 Hz, 2H), 7.34 (d, J = 8.31 Hz, 1H), 7.22 (t, J = 7.34 Hz, 1H), 4.28 (q, J = 7.35 Hz, 2H), 3.72 (s, 2H), 2.89-2.80 (m, 4H), 2.47-2.39 (m, 4H), 1.92-84 (m, 2H), 1.42-1.34 (m, 4H), 0.88 (t, J = 6.85 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 140.41 (C), 139.68 (C), 128.12 (CH), 127.31 (C), 125.21 (C), 122.68 (CH), 121.24 (C), 120.44 (CH), 118.77 (C), 118.57 (CH), 108.74 (CH), 108.36 (CH), 63.45 (CH2), 57.53 (CH2), 55.11 (CH2), 45.92 (CH2), 29.37 (CH2), 28.72 (CH2), 22.51 (CH2), 13.94 (CH3). HRMS calcd for C22H30N3 (M + H)+ 336.2440, found 336.2466.
4.1.31. Ethyl 9-pentyl-9H-pyrido[3,4-b]indole-3-carboxylate (31)
Using ethyl 9H-pyrido[3,4-b]indole-3-carboxylate (1 g, 4.16 mmol) and n-bromopentane (772 μL, 6.24 mmol) as starting compounds, the title compound was prepared as a white solid according to the procedure described above for compound 1 using method A. The target product was purified using heptanes/EtOAc in different proportions to afford the title compound as a white solid (1.08 g, 84%); mp 92°C. 1H NMR (500 MHz, CDCl3) δ 8.91 (s, 1H), 8.82 (s, 1H), 8.13 (d, J = 7.8 Hz, 1H), 7.59 (ddd, J = 8.4, 7.1, 1.2 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 4.32 (t, J = 7.1 Hz, 2H), 1.97 – 1.76 (m, 2H), 1.51 (t, J = 7.1 Hz, 3H), 1.39 – 1.21 (m, 4H), 0.83 (t, J = 6.7 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 166.07 (C=O), 141.31 (C), 137.59 (C), 137.39 (C), 131.54 (CH), 128.64 (CH), 128.07 (C), 121.90 (CH), 121.16 (C), 120.40 (CH), 117.49 (CH), 109.77 (CH), 61.40 (CH2), 43.41 (CH2), 29.16 (CH2), 28.75 (CH2), 22.25 (CH2), 14.48 (CH3), 13.77 (CH3). ESI: m/z 311.1 (M + H)+. HRMS calcd for C19H23N2O2 (M + H)+ 311.1760, found 311.1784.
4.1.32. 9-Pentyl-9H-pyrido[3,4-b]indole-3-carboxylic acid (32)
Potassium hydroxide (3 g, 53.57 mmol) was added to a stirred solution of ester 31 (715 mg, 2.30 mmol) in a mixture of ethanol (40 mL) and water (10 mL). The reaction mixture was stirred at reflux for 16 hrs and then cooled to room temperature. The solvents were evaporated under reduced pressure, and the residue was diluted with DI water. Using external cooling (ice-bath), the solution was acidified to pH ca. 2 by dropwise addition of 1M aqueous HCl. The precipitated product was extracted with EtOAc (150 mL). The organic layer was washed with brine under acidic pH, dried over MgSO4, and filtered. The solvent was then evaporated under reduced pressure, and the resulting residue was purified by silica chromatography using EtOAc/heptane in different proportions to yield the title compound as a pinkish solid. Yield: 647 mg (100%). 1H NMR (400 MHz, DMSO-d6) δ 14.56 (br. s, 1H), 9.49 (s, 1H), 9.33 (s, 1H), 8.66 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 4.72 (t, J = 7.1 Hz, 2H), 1.88 – 1.71 (m, 2H), 1.34 – 1.21 (m, 4H), 0.79 (t, J = 6.9 Hz, 3H). 13C and APT NMR (101 MHz, DMSO-d6) δ 162.53 (C=O), 143.52 (C), 136.37 (C), 131.58 (CH), 131.36 (C), 130.41 (C), 127.97 (CH), 123.83 (CH), 122.05 (CH), 120.08 (C), 118.89 (CH), 111.57 (CH), 43.63 (CH2), 28.58 (CH2), 28.35 (CH2), 21.85 (CH2), 13.81 (CH3).
4.1.33. 1-((9-Pentyl-9H-pyrido[3,4-b]indol-3-yl}carbonyl)piperidine (33)
Using acid 32 (241 mg, 0.85 mmol) and piperidine (102 mg, 1.20 mmol) as starting compounds, the title compound was prepared as an off-white solid according to the analogous procedure described above for 4 by the amide coupling protocol. Yield: 122 mg (41%); mp 126°C. 1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.38 (s, 1H), 8.13 (d, J = 7.8 Hz, 1H), 7.59 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 4.36 (t, J = 7.2 Hz, 2H), 3.81 (br. s, 2H), 3.63 (br. s, 2H), 1.89 (p, J = 7.1 Hz, 2H), 1.79 – 1.66 (m, 4H), 1.60 (br. s, 2H), 1.39 – 1.28 (m, 4H), 0.85 (t, J = 6.9 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 168.86 (C=O), 143.75 (C), 141.56 (C), 136.45 (C), 129.95 (CH), 128.80 (C), 128.68 (CH), 122.09 (CH), 121.22 (C), 120.06 (CH), 115.60 (CH), 109.72 (CH), 48.71 (CH2), 43.77 (CH2), 43.54 (CH2), 29.31 (CH2), 28.87 (CH2), 26.70 (CH2), 25.73 (CH2), 24.75 (CH2), 22.40 (CH2), 13.92 (CH3). ESI: m/z 350.2 (M + H)+. HRMS calcd for C22H28N3O (M + H)+ 350.2232, found 350.2249.
4.1.34. 9-Pentyl-N-(piperidin-1-yl)-9H-pyrido[3,4-b]indole-3-carboxamide (34)
Using acid 32 (100 mg, 0.35 mmol) and 1-aminopiperidine (36 mg, 0.36 mmol) as starting compounds, the title compound was prepared as a light yellow glass according to the analogous procedure described above for 4 by the amide coupling protocol with the exception of not using 5% citric acid solution during the workup. Yield: 71 mg (56%). 1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 8.81 (s, 1H), 8.71 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 4.36 (t, J = 7.1 Hz, 2H), 2.93 (br. s, 4H), 1.94 – 1.85 (m, 2H), 1.82 (dt, J = 10.9, 5.4 Hz, 4H), 1.49 (br. s, 2H), 1.39 – 1.28 (m, 4H), 0.86 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 162.31 (C=O), 141.59 (C), 139.89 (C), 137.77 (C), 129.82 (CH), 128.96 (C), 128.81 (CH), 122.46 (CH), 121.58 (C), 120.48 (CH), 114.90 (CH), 109.82 (CH), 57.49 (CH2), 43.73 (CH2), 29.42 (CH2), 29.00 (CH2), 25.41 (CH2), 23.55 (CH2), 22.51 (CH2), 14.02 (CH3). ESI: m/z 365.1 (M + H)+. HRMS calcd for C22H29N4O (M + H)+ 365.2341, found 365.2316.
4.1.35. N-(2,2-dimethylpropyl)-9-pentyl-9H-pyrido[3,4-b]indole-3-carboxamide (35)
Using acid 32 (100 mg, 0.35 mmol) and neopentylamine (37 mg, 0.42 mmol) as starting compounds, the title compound was prepared as a white solid according to the analogous procedure described above for 4 by the amide coupling protocol. Yield: 67 mg (54%); mp 91°C. 1H NMR (500 MHz, CDCl3) δ 8.93 (d, J = 0.9 Hz, 1H), 8.74 (d, J = 0.9 Hz, 1H), 8.33 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.60 (td, J = 7.2, 3.6 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.32 (ddd, J = 8.0, 7.1, 0.9 Hz, 1H), 4.36 (t, J = 7.2 Hz, 2H), 3.36 (d, J = 6.6 Hz, 2H), 2.02 – 1.78 (m, 2H), 1.45 – 1.27 (m, 4H), 1.04 (s, 9H), 0.87 (t, J = 7.0 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 165.53 (C=O), 141.53 (C), 140.07 (C), 137.61 (C), 129.84 (CH), 128.91 (C), 128.61 (CH), 122.24 (CH), 121.52 (C), 120.28 (CH), 114.38 (CH), 109.68 (CH), 50.67 (CH2), 43.60 (CH2), 32.32 (C), 29.31 (CH2), 28.89 (CH2), 27.39 (CH3), 22.39 (CH2), 13.91 (CH3). ESI: m/z 352.2 (M + H)+. HRMS calcd for C22H30N3O (M + H)+ 352.2389, found 352.2386.
4.1.36. 9-Propyl-9H-carbazole (36)
From carbazole (2 g, 11.96 mmol), n-propyl iodide (1.75 mL, 17.91 mmol), and NaOH (718 mg, 17.95 mmol) a similar procedure as that described compound 1 (Method B) afforded the title product as a white waxy solid. Yield: 2.07 g (83%). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 2H), 7.44 (ddd, J = 8.2, 6.9, 1.2 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 7.21 (ddd, J = 8.0, 6.8, 1.1 Hz, 2H), 4.24 (t, J = 7.2 Hz, 2H), 2.01 – 1.78 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 140.71 (C), 125.75 (CH), 123.00 (C), 120.53 (CH), 118.90 (CH), 108.89 (CH), 44.80 (CH2), 22.51 (CH2), 12.02 (CH3).
4.1.37. 9-Butyl-9H-carbazole (37)
From carbazole (10 g, 59.81 mmol) and n-butyl iodide (10 mL, 87.87 mmol), a similar procedure as that described for compound 1 (Method B) afforded the title product as a white solid. Yield: 10.53 g (79%); mp 62°C. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.7 Hz, 2H), 7.45 (ddd, J = 8.2, 7.1, 1.2 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.21 (J = 7.02, 6.83, 1.1 Hz, 1H), 4.27 (t, J = 7.2 Hz, 2H), 1.89 – 1.76 (m, 2H), 1.44 – 1.32 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 140.65 (C), 125.75 (CH), 123.02 (C), 120.53 (CH), 118.89 (CH), 108.85 (CH), 43.02 (CH2), 31.33 (CH2), 20.78 (CH2), 14.09 (CH3).
4.1.38. 9-Hexyl-9H-carbazole (38)
A mixture of carbazole (1.5 g, 8.97 mmol), n-hexyl bromide (1.89 mL, 13.46 mmol), and powdered NaOH (538 mg, 13.45 mmol) in dry acetone (50 mL) was refluxed for 16 h under nitrogen. After all volatile components were removed by rotary evaporation in vacuo, the residue was extracted with tert-butyl methyl ether (100 mL). The organic phase was washed with water, brine, dried (MgSO4), filtered, and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a colorless oil which crystallized on standing (2.03 g, 90%); mp 66°C. 1H NMR (500 MHz, CDCl3) δ 8.10 (d, J = 7.7 Hz, 2H), 7.45 (t, J = 7.3 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.22 (t, J = 7.6 Hz, 2H), 4.28 (t, J = 7.3 Hz, 2H), 1.86 (dt, J = 15.0, 7.5 Hz, 2H), 1.43 – 1.34 (m, 2H), 1.34 – 1.21 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 140.61 (C), 125.75 (CH), 122.99 (C), 120.53 (CH), 118.87 (CH), 108.84 (CH), 43.28 (CH2), 31.81 (CH2), 29.15 (CH2), 27.20 (CH2), 22.77 (CH2), 14.25 (CH3).
4.1.39. 9-Heptyl-9H-carbazole (39)
From carbazole (2 g, 11.96 mmol), n-heptyl bromide (3.21 g, 17.94 mmol), and NaOH (4 g, 100 mmol), an analogous procedure as that described for compound 38 afforded the target product as a colorless oil. Yield 2.88 g (91%). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.8 Hz, 2H), 7.42 (ddd, J = 8.3, 7.0, 1.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 7.19 (ddd, J = 7.9, 6.9, 1.0 Hz, 2H), 4.19 (t, J = 7.3 Hz, 2H), 1.86 – 1.73 (m, 2H), 1.38 – 1.13 (m, 8H), 0.84 (t, J = 6.9 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 140.62 (C), 125.73 (CH), 123.01 (C), 120.51 (CH), 118.87 (CH), 108.83 (CH), 43.20 (CH2), 31.92 (CH2), 29.28 (CH2), 29.16 (CH2), 27.46 (CH2), 22.79 (CH2), 14.25 (CH3).
4.1.40. Piperidin-1-yl(9-propyl-9H-carbazol-3-yl)methanone (40)
Using 9-propyl-9H-carbazole (1.484 g, 7.09 mmol), piperidine-1-carbonyl chloride (1.064 mL, 8.51 mmol), and anhydrous AlCl3 (1.04 g, 7.80 mmol) as starting compounds, the title compound was prepared as a white crystalline solid according to the procedure described above for 4 (Method B: Friedel–Crafts acylation). Yield: 1.720 g (76%); mp 149°C. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.53 (dd, J = 8.4, 1.5 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 8.6 Hz, 2H), 7.23 (t, J = 7.3 Hz, 1H), 4.25 (t, J = 7.0 Hz, 2H), 3.62 (br. s, 4H), 1.98 – 1.82 (m, 2H), 1.77 – 1.48 (br. m, 6H), 0.94 (t, J = 7.4 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 171.60 (C=O), 141.15 (C), 141.06 (C), 126.87 (C), 126.19 (CH), 125.13 (CH), 122.82 (C), 122.51 (C), 120.60 (CH), 119.93 (CH), 119.36 (CH), 109.11 (CH), 108.50 (CH), 44.83 (CH2), 26.33 (CH2), 24.86 (CH2), 22.40 (CH2), 11.89 (CH3). ESI: m/z 321.1 (M + H)+. HRMS calcd for C21H25N2O (M + H)+ 321.1967, found 321.1934.
4.1.41. (9-Hexyl-9H-carbazol-3-yl)(piperidin-1-yl)methanone (41)
From 9-hexyl-9H-carbazole (1.651 g, 6.57 mmol), piperidine-1-carbonyl chloride (986 μL, 7.88 mmol), and anhydrous AlCl3 (963 mg, 7.22 mmol) an analogous microwave procedure as that described for 4 (Method B: Friedel–Crafts acylation) afforded the title product as a light yellow gum. Yield 1.77 g (74%). 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 1.2 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.53 (dd, J = 8.4, 1.6 Hz, 1H), 7.47 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.24 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 4.28 (t, J = 7.3 Hz, 2H), 3.62 (br. s, 4H), 1.85 (dt, J = 15.0, 7.4 Hz, 2H), 1.76 – 1.67 (br. m, 3H), 1.63 (br. s, 3H), 1.41 – 1.32 (m, 2H), 1.32 – 1.21 (m, 4H), 0.85 (t, J = 7.1 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 171.53 (C=O), 140.99 (C), 140.90 (C), 126.76 (C), 126.11 (CH), 125.06 (CH), 122.75 (C), 122.43 (C), 120.53 (CH), 119.86 (CH), 119.26 (CH), 108.98 (CH), 108.37 (CH), 43.24 (CH2), 31.59 (CH2), 28.94 (CH2), 26.96 (CH2), 26.25 (CH2), 24.79 (CH2), 22.56 (CH2), 14.04 (CH3). ESI: m/z 363.2 (M + H)+. HRMS calcd for C24H30N2O (M + H)+ 363.2436, found 363.2423.
4.1.42. 9-Heptyl-9H-carbazol-3-yl)(piperidin-1-yl)methanone (42)
Using 9-heptyl-9H-carbazole (1.743 g, 6.57 mmol), piperidine-1-carbonyl chloride (986 μL, 7.88 mmol), and anhydrous AlCl3 (963 mg, 7.22 mmol) as starting compounds, the title compound was prepared as a light yellow gum according to the procedure described above for for 4 (Method B: Friedel–Crafts acylation). Yield: 1.737 g (70%). 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 1.2 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.53 (dd, J = 8.4, 1.6 Hz, 1H), 7.48 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.24 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 4.29 (t, J = 7.2 Hz, 2H), 3.62 (br. s, 4H), 1.92 – 1.79 (m, 2H), 1.76 – 1.66 (br. m, 3H), 1.63 (br. s, 3H), 1.41 – 1.28 (m, 4H), 1.28 – 1.17 (m, 4H), 0.85 (t, J = 6.9 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 171.67 (C=O), 141.13 (C), 141.04 (C), 126.89 (C), 126.23 (CH), 125.20 (CH), 122.89 (C), 122.57 (C), 120.67 (CH), 119.99 (CH), 119.40 (CH), 109.11 (CH), 108.50 (CH), 43.38 (CH2), 31.86 (CH2), 29.21 (CH2), 29.12 (CH2), 27.40 (CH2), 26.37 (CH2), 24.92 (CH2), 22.73 (CH2), 14.20 (CH3). ESI: m/z 377.3 (M + H)+. HRMS calcd for C25H32N2O (M + H)+ 377.2593, found 377.2554.
4.1.43. Ethyl 3-(9H-carbazol-9-yl)propanoate (43)
The title compound was prepared according to a procedure already described. (J. Med. Chem. 50, 4648-4655, 2007). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.8 Hz, 2H), 7.48 – 7.39 (m, 4H), 7.22 (ddd, J = 8.0, 6.4, 1.8 Hz, 2H), 4.61 (t, J = 7.2 Hz, 2H), 4.06 (q, J = 7.2 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 171.59 (C=O), 140.17 (C), 125.95 (CH), 123.26 (C), 120.56 (CH), 119.35 (CH), 108.80 (CH), 61.06 (CH2), 38.90 (CH2), 33.73 (CH2), 14.19 (CH3).
4.1.44. 2-(9H-carbazol-9-yl)ethanol (44)
To a stirred solution of carbazole (3 g, 17.94 mmol) in methyl ethyl ketone (10 mL) in a 20 mL microwave vessel, powdered KOH (1 g, 17.82 mmol) was added under nitrogen, and the solution was cooled to −60°C. Ice-cold ethylene oxide (8 mL, 2.5 M solution in THF) was added dropwise to the obtained solution, and the reaction vessel was tightly capped. The reaction mixture was then subjected to microwave irradiation at 50°C for 1 h with stirring and then cooled to room temperature. The solution was extracted with tert-butyl methyl ether (150 mL). The organic phase was washed with water, hydrochloric acid (aq, 2 M), brine, dried (MgSO4), and filtered. The organic solvents were evaporated in vacuo, and the obtained residue self-crystallized. The crystalline product was removed by vacuum filtration, washed with hexanes (20 mL), and then dried under high vacuum for 12 h to give the title product as a white solid, which was used without further purification. Yield: 3.136 g (83%); mp 83°C. 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 7.7 Hz, 2H), 7.37 (ddd, J = 8.2, 7.2, 1.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.16 (ddd, J = 7.9, 7.0, 0.9 Hz, 2H), 4.19 (t, J = 5.4 Hz, 2H), 3.71 (t, J = 5.4 Hz, 2H), 1.82 (s, 1H). 13C and APT NMR (101 MHz, CDCl3) δ 140.77 (C), 125.93 (CH), 123.01 (C), 120.46 (CH), 119.30 (CH), 108.95 (CH), 61.38 (CH2), 45.46 (CH2).
4.1.45. 3-(9H-carbazol-9-yl)propan-1-ol (45)
The title compound was prepared according to a procedure already described[22]. 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 7.8 Hz, 2H), 7.46 – 7.35 (m, 4H), 7.20 (ddd, J = 7.9, 6.6, 1.5 Hz, 2H), 4.34 (t, J = 6.6 Hz, 2H), 3.47 (t, J = 5.9 Hz, 2H), 1.99 (p, J = 6.3 Hz, 2H), 1.72 (br. s, 1H). 13C NMR (126 MHz, CDCl3) δ 140.57 (C), 125.83 (CH), 122.94 (C), 120.49 (CH), 119.02 (CH), 108.79 (CH), 59.67 (CH2), 39.29 (CH2), 31.50 (CH2).
4.1.46. 9-Butyl-9H-carbazole-3-carbaldehyde (46)
Using 9-butyl-9H-carbazole (2.982 g, 13.35 mmol), DMF (7.43 mL, 95.96 mmol), and POC13 (2.80 mL, 30.04 mmol) as starting compounds, the title compound was prepared as yellowish crystals following the procedures described in preparation of compound 2. Yield: 3.185 g (quantitative); mp 58°C. 1H NMR (400 MHz, CDCl3) δ 10.05 (s, 1H), 8.55 (s, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.30 (t, J = 7.4 Hz, 1H), 4.26 (t, J = 7.1 Hz, 2H), 1.91 – 1.73 (m, 2H), 1.46 – 1.28 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.88 (C=O), 144.16 (C), 141.28 (C), 128.58 (C), 127.22 (CH), 126.82 (CH), 124.06 (CH), 123.14 (C), 123.08 (C), 120.84 (CH), 120.40 (CH), 109.53 (CH), 109.05 (CH), 43.26 (CH2), 31.16 (CH2), 20.63 (CH2), 13.97 (CH3).
4.1.47. Ethyl 3-(3-formyl-9H-carbazol-9-yl)propanoate (47)
Using ethyl 3-(9H-carbazol-9-yl)propanoate (5.012 g, 18.75 mmol), DMF (10 mL, 129.16 mmol), and POC13 (3.5 mL, 37.44 mmol) as starting compounds, the title compound was prepared as white crystals following the procedures described in preparation of compound 2. Yield: 3.642 g (66%); mp 79°C. 1H NMR (500 MHz, CDCl3) δ 10.06 (s, 1H), 8.55 (d, J = 1.0 Hz, 1H), 8.11 (d, J = 7.8 Hz, 1H), 7.98 (dd, J = 8.5, 1.5 Hz, 1H), 7.52 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.32 (ddd, J = 7.9. 6.8, 1.1 Hz, 1H), 4.63 (t, J = 7.0 Hz, 2H), 4.07 (q, J = 7.2 Hz, 2H), 2.85 (t, J = 7.0 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 191.88 (HC=O), 171.17 (C=O), 143.79 (C), 140.79 (C), 128.93 (C), 127.46 (CH), 127.01 (CH), 123.89 (CH), 123.38 (C), 123.25 (C), 120.91 (CH), 120.78 (CH), 109.46 (CH), 109.17 (CH), 61.23 (CH2), 39.12 (CH2), 33.60 (CH2), 14.15 (CH3).
4.1.48. 9-(2-Chloroethyl)-9H-carbazole-3-carbaldehyde (48)
Using compound 44 (2.86 g, 13.54 mmol), DMF (9 mL, 116.24 mmol), and POC13 (3.37 mL, 36.16 mmol) as starting compounds, the title compound was prepared as a white-tan solid following the procedures described in preparation of compound 2. Yield: 2.826 g (81%); mp 167°C. 1H NMR (400 MHz, CDCl3) δ 10.08 (s, 1H), 8.58 (s, 1H), 8.14 (d, J = 7.8 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.35 (t, J = 7.4 Hz, 1H), 4.66 (t, J = 6.7 Hz, 2H), 3.89 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 191.86 (CHO), 144.10 (C), 141.00 (C), 129.32 (C), 127.65 (CH), 127.21 (CH), 124.02 (CH), 123.56 (C), 123.36 (C), 121.13 (CH), 121.10 (CH), 109.33 (CH), 109.14 (CH), 45.13 (CH2), 41.29 (CH2).
4.1.49. 9-(3-Chloropropyl)-9H-carbazole-3-carbaldehyde (49)
The title compound was prepared according to a modified literature procedure[34]. POC13 (2.8 mL, 30.04 mmol) was added, over a period of 10 min, to an ice-cooled, stirred DMF (7.43 mL, 95.96 mmol) under nitrogen. The reddish solution was allowed to stir at room temperature for 1 h. Compound 45 (2.447 g, 10.86 mmol) was added over 10 min, and the obtained mixture was subjected to microwave irradiation at 100°C for 1 h. The reaction mixture was cooled and then poured into crushed ice. After warming to room temperature, the resultant product was extracted with DCM. The organic phase was washed with water, brine, dried (MgSO4), filtered, and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light yellow solid (2.436 g, 83%); mp 88°C. 1H NMR (500 MHz, CDCl3) δ 10.05 (s, 1H), 8.54 (s, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.61 – 7.41 (m, 3H), 7.32 (t, J = 7.3 Hz, 1H), 4.48 (t, J = 6.5 Hz, 2H), 3.49 (t, J = 5.8 Hz, 2H), 2.45 – 2.19 (m, 2H). 13C and APT NMR (126 MHz, CDCl3) δ 191.84 (C=O), 144.07 (C), 141.11 (C), 128.86 (C), 127.52 (CH), 127.06 (CH), 123.90 (CH), 123.26 (C), 123.15 (C), 120.92 (CH), 120.74 (CH), 109.39 (CH), 108.96 (CH), 42.15 (CH2), 40.15 (CH2), 31.68 (CH2).
4.1.50. 9-Butyl-9H-carbazole-3-carboxylic acid (50)
Powdered potassium permanganate (3 g, 18.98 mmol) was added in one portion to a solution of 9-butyl-9H-carbazole-3-carbaldehyde (2.936 g, 11.68 mmol) in a mixture of acetone (200 mL) and water (3 mL) at room temperature. The resulting solution was stirred for 5 min at this temperature, and then, the mixture was heated 16 h at reflux under stirring. The mixture was quenched with ethanol (20 mL), and then stirred for 30 min at reflux. After cooling, the solid precipitate (MnO2) was filtered off through a pad of Celite®; the filtrate was concentrated in vacuo to remove organic solvents. The obtained syrup was diluted with water (100 mL), basified with NaOH to pH ca. 10, and extracted with ether (50 mL × 3) to remove the unreacted starting material. The aqueous solution was cooled on an ice-water bath and acidified with ice-cold solution of sulfuric acid (20%) to pH ca. 2. The resultant bulky precipitate was extracted with EtOAc and the extract was washed with brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. The precipitated product was collected by filtration, washed with heptanes (20 mL × 2), and dried overnight to produce the title compound (1.993 g, 64%) as a white solid; mp 159°C. 1H NMR (500 MHz, CDCl3) δ 12.82 (s, 1H), 8.91 (d, J = 1.6 Hz, 1H), 8.25 (dd, J = 8.6, 1.6 Hz, 1H), 8.15 (d, J = 7.7 Hz, 1H), 7.50 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.29 (ddd, 7.9, 6.9, 1 Hz, 1H), 4.27 (t, J = 7.2 Hz, 2H), 1.92 – 1.77 (m, 2H), 1.46 – 1.31 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 173.60 (C=O), 143.80 (C), 141.22 (C), 128.03 (CH), 126.59 (CH), 123.90 (CH), 123.14 (C), 122.84 (C), 120.91 (CH), 120.17 (CH), 119.83 (C), 109.36 (CH), 108.44 (CH), 43.25 (CH2), 31.22 (CH2), 20.72 (CH2), 14.05 (CH3).
4.1.51. 9-(3-Ethoxy-3-oxopropyl)-9H-carbazole-3-carboxylic acid (51)
Powdered potassium permanganate (736 mg, 4.66 mmol) was added in portions under stirring at room temperature to a solution of 47 (747 mg, 2.53 mmol) in a mixture of acetone (50 mL) and acetic acid (805 μL). After the addition, the mixture was heated 5 h at reflux under stirring and then allowed to cool to room temperature. The mixture was filtered through a pad of Celite® and concentrated in vacuo to remove organic solvents. The obtained residue was purified by column chromatography on silica gel, eluenting with EtOAc/heptanes in different proportions to give the title compound (434 mg, 55%) as yellow crystalls; mp 153°C. 1H NMR (500 MHz, CDCl3) δ 12.69 (br. s, 1H), 8.88 (d, J = 1.4 Hz, 1H), 8.26 (dd, J = 8.6, 1.6 Hz, 1H), 8.14 (d, J = 7.7 Hz, 1H), 7.52 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.47 (d, J = 8.4 Hz, 2H), 7.33 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 4.63 (t, J = 7.1 Hz, 2H), 4.08 (q, J = 7.2 Hz, 2H), 2.86 (t, J = 7.1 Hz, 2H), 1.14 (t, J = 7.2 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 173.18 (C=O), 171.35 (C=O), 143.44 (C), 140.80 (C), 128.22 (CH), 126.82 (CH), 123.87 (CH), 123.38 (C), 123.15 (C), 120.99 (CH), 120.60 (CH), 120.41 (C), 109.31 (CH), 108.54 (CH), 61.27 (CH2), 39.16 (CH2), 33.72 (CH2), 14.21 (CH3).
4.1.52. 9-(2-Chloroethyl)-9H-carbazole-3-carboxylic acid (52)
From compound 48 (2.826 g, 10.97 mmol), potassium permanganate (2.08 g, 13.16 mmol), DI water (2 mL, 111.11 mmol), and acetone (100 mL), a similar procedure as that described for compound 50 afforded the title product as a yellow crystalline solid. Yield 2.422 g (81%); mp 204°C. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.19 (d, J = 7.7 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.23 (t, J = 7.4 Hz, 1H), 4.78 (t, J = 5.6 Hz, 2H), 4.06 (t, J = 5.7 Hz, 2H), 1.87 (s, 1H). 13C and APT NMR (101 MHz, DMSO-d6) δ 169.70 (C=O), 141.27 (C), 140.54 (C), 129.48 (C), 127.51 (CH), 125.64 (CH), 122.77 (C), 121.51 (C and CH), 120.20 (CH), 119.33 (CH), 109.65 (CH), 108.13 (CH), 44.19 (CH2), 43.06 (CH2).
4.1.53. 9-(3-Chloropropyl)-9H-carbazole-3-carboxylic acid (53)
From 9-(3-chloropropyl)-9H-carbazole-3-carbaldehyde (2.436 g, 8.96 mmol), potassium permanganate (2 g, 12.66 mmol), DI water (2 mL, 111.11 mmol), and acetone (100 mL), a similar procedure as that described for compound 50 afforded the title product as a yellowish solid. Yield 1.605 g (62%); mp 218°C. 1H NMR (500 MHz, DMSO-d6) δ 12.68 (s, 1H), 8.82 (d, J = 1.5 Hz, 1H), 8.28 (d, J = 7.7 Hz, 1H), 8.09 (dd, J = 8.6, 1.7 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.65 (d, J = 8.3 Hz, 1H), 7.51 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.27 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 4.54 (t, J = 6.9 Hz, 2H), 3.63 (t, J = 6.4 Hz, 2H), 2.22 (p, J = 6.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 168.02 (C=O), 142.48 (C), 140.57 (C), 127.19 (CH), 126.54 (CH), 122.57 (CH), 122.31 (C), 121.99 (C), 121.42 (C), 120.82 (CH), 119.91 (CH), 109.56 (CH), 108.83 (CH), 42.63 (CH2), 39.90 (CH2), 31.46 (CH2).
4.1.54. (9-Butyl-9H-carbazol-3-yl)(piperidin-1-yl)methanone (54)
Using carboxylic acid 50 (400 mg, 1.50 mmol) and piperidine (177 μL, 1.79 mmol) as starting compounds, the title compound was prepared as a colorless viscous gum according to the procedure described above for compound 4. Yield: 325 mg (65%). 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 1.2 Hz, 1H), 7.97 (d, J = 7.7 Hz, 1H), 7.42 (dd, J = 8.4, 1.6 Hz, 1H), 7.35 (ddd, J = 8.3, 7.0, 1.3, 1H), 7.27 (t, J = 8.6 Hz, 2H), 7.11 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 4.14 (t, J = 7.1 Hz, 2H), 3.51 (br. s, 4H), 1.70 (dt, J = 15.0, 7.3 Hz, 2H), 1.62 – 1.53 (br. m, 3H), 1.50 (br. s, 3H), 1.30 – 1.18 (m, 2H), 0.80 (t, J = 7.4 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 171.48 (C=O), 140.94 (C), 140.86 (C), 126.69 (C), 126.08 (CH), 125.02 (CH), 122.68 (C), 122.38 (C), 120.48 (CH), 119.83 (CH), 119.24 (CH), 108.99 (CH), 108.37 (CH), 42.91 (CH2), 31.08 (CH2), 24.74 (CH2), 20.52 (CH2), 13.90 (CH3). ESI: m/z 335.3 (M + H)+. HRMS calcd for C22H27N2O (M + H)+ 335.2123, found 335.2086.
4.1.55. Ethyl 3-(3-(piperidine-1-carbonyl)-9H-carbazol-9-yl)propanoate (55)
Using carboxylic acid 51 (200 mg, 0.64 mmol) and piperidine (76 μL, 0.78 mmol) as starting compounds, the title compound was prepared as a colorless gum according to the procedure described above for compound 4. Yield: 175 mg (72%). 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 1.2 Hz, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.53 (dd, J = 8.4, 1.5 Hz, 1H), 7.48 – 7.39 (m, 3H), 7.23 (ddd, J = 8.0, 6.5, 1.5 Hz, 1H), 4.60 (t, J = 7.0 Hz, 2H), 4.04 (q, J = 7.2 Hz, 2H), 3.66 (br. s, 2H), 3.55 (br. s, 2H), 2.81 (t, J = 7.0 Hz, 2H), 1.77 – 1.64 (m, 3H), 1.61 (br. s, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 171.24 (C=O), 171.18 (C=O), 140.45 (C), 140.37 (C), 127.16 (C), 126.23 (CH), 125.06 (CH), 122.86 (C), 122.65 (C), 120.48 (CH), 119.80 (CH), 119.63 (CH), 108.91 (CH), 108.35 (CH), 60.87 (CH2), 38.79 (CH2), 33.48 (CH2), 24.66 (CH2), 13.97 (CH3). ESI: m/z 379.3 (M + H)+. HRMS calcd for C23H27N2O3 (M + H)+ 379.2022, found 379.2017.
4.1.56. (9-(2-Chloroethyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (56)
From carboxylic acid 52 (2.2 g, 8.04 mmol) and piperidine (953 μL, 9.64 mmol), a similar procedure as that described for compound 4 gave the title compound (2.144 g, 78%) as a light yellow crystalline solid; mp 172°C. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.40 (dd, J = 8.2, 4.2 Hz, 2H), 7.26 (t, J = 7.3 Hz, 1H), 4.60 (t, J = 6.8 Hz, 2H), 3.83 (t, J = 6.8 Hz, 2H), 3.77 – 3.22 (m, 4H), 1.83 – 1.65 (m, 3H), 1.65 – 1.42 (br. m, 3H). 13C and APT NMR (101 MHz, CDCl3) δ 171.28 (C=O), 140.79 (C), 140.68 (C), 127.75 (C), 126.53 (CH), 125.35 (CH), 123.07 (C), 122.88 (C), 120.78 (CH), 120.11 (CH), 120.01 (CH), 108.89 (CH), 108.38 (CH), 44.90 (CH2), 41.28 (CH2), 26.28 (CH2), 24.82 (CH2). ESI: m/z 341.1 (M + H)+. HRMS calcd for C20H22N2OCl (M + H)+ 341.1421, found 341.1382.
4.1.57. (9-(3-Chloropropyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (57)
Using carboxylic acid 53 (800 mg, 2.78 mmol) and piperidine (604 μL, 6.11 mmol) as starting compounds, the title compound was prepared as a colorless glass according to the procedure described above for compound 4. Yield: 540 mg (55%). 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 1.1 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.54 (dd, J = 8.4, 1.6 Hz, 1H), 7.52 – 7.43 (m, 3H), 7.26 (ddd, J = 7.9, 6.1, 2.0 Hz, 1H), 4.51 (t, J = 6.5 Hz, 2H), 3.94 – 3.30 (m, 4H), 3.50 (t, J = 6 Hz, 2H), 2.42 – 2.26 (m, 2H), 1.76 – 1.67 (br. m, 3H), 1.63 (br. s, 3H). 13C and APT NMR (126 MHz, CDCl3) δ 171.46 (C=O), 141.01 (C), 140.90 (C), 127.34 (C), 126.49 (CH), 125.35 (CH), 122.98 (C), 122.72 (C), 120.75 (CH), 120.02 (CH), 119.80 (CH), 109.00 (CH), 108.41 (CH), 42.34 (CH2), 39.99 (CH2), 31.84 (CH2), 24.88 (CH2). ESI: m/z 355.1 (M + H)+. HRMS calcd for C21H24N2OCl (M + H)+ 355.1577, found 355.1579.
4.1.58. (9-(2-Iodoethyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (58)
Powdered NaI (5.78 g, 38.56 mmole) was added to a solution of compound 56 (1.95 g, 5.71 mmole) in acetonitrile (100 mL) under nitrogen. After stirring at reflux for 72 h, the reaction mixture was filtered, and the solvent was removed under reduced pressure. The obtained residue was extracted with EtOAc (150 mL). The extract was washed consecutively with 5% aqueous sodium dithionite (50 mL, 2 times), brine, dried over MgSO4, and filtered. The solvent was evaporated under reduced pressure, and the residue self-crystallized. The white crystalline solid was collected by filtration, washed with heptanes (20 mL) and dried in vacuo. Yield: 2.22 g (90%); mp 161°C. According to the NMR and mass-spec analyses the final product represented a 4:1 mixture of iodide and the unreacted chloride 56. 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 1H), 7.42 (dd, J = 12.8, 6.3 Hz, 2H), 7.28 (t, J = 7.8 Hz, 1H), 4.69 (t, J = 7.9 Hz, 2H), 3.62 (br. s, 4H), 3.43 (t, J = 7.9 Hz, 2H), 1.70 (br. m, 3H), 1.63 (br. s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.31 (C=O), 140.34 (C), 140.29 (C), 127.89 (C), 126.62 (CH), 125.43 (CH), 123.15 (C), 122.97 (C), 120.92 (CH), 120.25 (CH), 120.16 (CH), 108.85 (CH), 108.31 (CH), 45.91 (CH2), 26.38 (CH2), 24.88 (CH2), −0.06 (CH2).
4.1.59. (9-(3-Iodopropyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (59)
Powdered NaI (3.7 g, 24.68 mmole) was added to a solution of compound 57 (1.247 g, 3.51 mmole) in acetonitrile (100 mL) under nitrogen. After stirring at reflux for 72 h, the reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography on silica gel using EtOAc/heptane in different proportions. The resulting colorless oil was dried in high vacuo to yield the target compound as a colorless glass. Yield: 1.328 g (85%). 1H NMR (500 MHz, CDCl3) δ 8.09 (s, 1H), 7.98 (d, J = 7.7 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.41 – 7.31 (m, 3H), 7.16 (t, J = 6.5 Hz, 1H), 4.29 (t, J = 6.5 Hz, 2H), 3.92 – 3.17 (br. m, 4H), 3.02 (t, J = 6.4 Hz, 2H), 2.36 – 2.17 (m, 2H), 1.59 (br. s, 3H), 1.53 (br. s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.33 (C=O), 140.85 (C), 140.76 (C), 127.17 (C), 126.39 (CH), 125.23 (CH), 122.85 (C), 122.62 (C), 120.66 (CH), 119.94 (CH), 119.71 (CH), 109.03 (CH), 108.46 (CH), 43.22 (CH2), 32.71 (CH2), 24.77 (CH2), 3.06 (CH2).
4.1.60. Piperidin-1-yl(9-(2-(piperidin-1-yl)ethyl)-9H-carbazol-3-yl)methanone (60)
Under nitrogen atmosphere, a solution of compound 58 (600 mg, 1.39 mmol), piperidine (411 μL, 4.17 mmol), and Cs2CO3 (904 mg, 2.77 mmol) in acetone (5 mL) was subjected to microwave irradiation at 80°C for 1 h. The reaction mixture was cooled, diluted with ethyl acetate (50 mL), and filtered. The organic solvents were evaporated in vacuo, and the obtained residue was dissolved in DCM (150 mL). The organic layer was washed with 1 N NaOH (50 mL × 2), dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified on a Biotage® KP-NH cartridge using EtOAc/heptane in different proportions to give the title compound as a pale yellow glass. Yield: 203 mg (37%). 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.33 – 7.19 (m, 3H), 7.06 (t, J = 7.3 Hz, 1H), 4.20 (t, J = 7.4 Hz, 2H), 3.76 – 3.13 (m, 4H), 2.49 (t, J = 7.4 Hz, 2H), 2.28 (br. s, 4H), 1.60 – 1.31 (m, 10H), 1.30 – 1.19 (m, 2H). 13C and APT NMR (101 MHz, CDCl3) δ 171.07 (C=O), 140.62 (C), 140.53 (C), 126.64 (C), 125.87 (CH), 124.78 (CH), 122.48 (C), 122.22 (C), 120.18 (CH), 119.58 (CH), 119.12 (CH), 108.65 (CH), 108.05 (CH), 56.61 (CH2), 54.74 (CH2), 40.93 (CH2), 25.91 (CH2), 25.74 (CH2), 24.45 (CH2), 23.98 (CH2). ESI: m/z 390.2 (M + H)+. HRMS calcd for C25H32N3O (M + H)+ 390.2545, found 390.2591.
4.1.61. Piperidin-1-yl(9-(3-(piperidin-1-yl)propyl)-9H-carbazol-3-yl)methanone (61)
Using iodide 59 (263 mg, 0.59 mmol), piperidine (166 μL, 1.68 mmol), and Cs2CO3 (365 mg, 1.12 mmol) in acetone (5 mL) as starting compounds, the title compound was prepared as a light-orange gum according to the procedure described above for compound 60. Yield: 202 mg (85%). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 8.3 Hz, 1H), 7.21 (t, J = 6.9 Hz, 1H), 4.34 (t, J = 6.5 Hz, 2H), 3.60 (br. s, 4H), 2.26 (br. s, 4H), 2.19 (t, J = 6.7 Hz, 2H), 1.98 (p, J = 6.4 Hz, 2H), 1.65 (br. s, 3H), 1.63 – 1.51 (m, 7H), 1.41 (br. s, 2H). 13C and DEPT NMR (101 MHz, CDCl3) δ 171.42 (C=O), 141.03 (C), 140.89 (C), 126.66 (C), 125.95 (CH), 124.82 (CH), 122.64 (C), 122.33 (C), 120.30 (CH), 119.66 (CH), 119.17 (CH), 109.15 (CH), 108.56 (CH), 55.49 (CH2), 54.35 (CH2), 40.59 (CH2), 26.00 (CH2), 25.92 (CH2), 24.66 (CH2), 24.44 (CH2). ESI: m/z 404.1 (M + H)+. HRMS calcd for C26H34N3O (M + H)+ 404.2702, found 404.2675.
4.1.62. Piperidin-1-yl(9-(3-(1,1-dioxo-thiomorpholino))-9H-carbazol-3-yl)methanone (62)
Using iodide 59 (290 mg, 0.65 mmol), 1,1-dioxo-thiomorpholine (227 mg, 1.68 mmol), and Cs2CO3 (365 mg, 1.12 mmol) in acetone (5 mL) as starting compounds, the title compound was prepared as a colorless gum according to the procedure described above for compound 60. Yield: 112 mg (38%). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.07 (d, J = 7.7, 1H), 7.51 (d, J = 8.3, 1H), 7.48 – 7.34 (m, 3H), 7.23 (t, J = 7.1, 1H), 4.34 (t, J = 6.6 Hz, 2H), 3.61 (br. s, 4H), 2.83 (br. s, 4H), 2.73 (br. s, 4H), 2.32 (t, J = 6.6 Hz, 2H), 2.02 – 1.90 (m, 2H), 1.68 (br. s, 3H), 1.62 (br. s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.05 (C=O), 140.71 (C), 126.89 (C), 126.01 (CH), 124.87 (CH), 122.51 (C), 122.25 (C), 120.41 (CH), 119.66 (CH), 119.37 (CH), 108.90 (CH), 108.28 (CH), 53.31 (CH2), 50.89 (CH2), 50.46 (CH2), 40.24 (CH2), 25.51 (CH2), 24.53 (CH2). ESI: m/z 454.2 (M + H)+. HRMS calcd for C25H32N3O3S (M + H)+ 454.2164, found 454.2132.
4.1.63. 3-(3-(Piperidine-1-carbonyl)-9H-carbazol-9-yl)propyl (1,1-dioxo-thiomorpholino)-4-carboxylate (63)
The title compound was obtained as a by-product in the form of a light yellow glass from the synthesis of compound 62. Yield: 75 mg (23%). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 8.5 Hz, 2H), 7.27 (dd, J = 8.6, 6.5 Hz, 1H), 4.43 (t, J = 6.1 Hz, 2H), 4.18 (t, J = 5.6 Hz, 2H), 3.86 (br. s, 2H), 3.77 – 3.25 (br. m, 6H), 2.92 (br. s, 2H), 2.76 (br. s, 2H), 2.41 – 2.17 (m, 2H), 1.70 (br. s, 3H), 1.64 (br. s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.23 (C=O), 154.26 (C=O), 140.75 (C), 127.45 (C), 126.46 (CH), 125.38 (CH), 122.88 (C), 122.59 (C), 120.81 (CH), 119.96 (CH), 119.84 (CH), 108.82 (CH), 108.23 (CH), 64.56 (CH2), 51.75 (CH2), 42.52 (CH2), 40.54 (CH2), 28.28 (CH2), 24.81 (CH2). ESI: m/z 498.2 (M + H)+. HRMS calcd for C26H32N3O5S (M + H)+ 498.2063, found 498.2085.
4.1.64. N-(3-(3-(Piperidine-1-carbonyl)-9H-carbazol-9-yl)propyl)methanesulfonamide (64)
Sodium hydride (60% dispersion in mineral oil, 108 mg, 2.82 mmol) was added portionwise to a solution of compound 59 (247 mg, 0.55 mmol) in dry DMF (3 mL) under nitrogen atmosphere, followed by methanesulfonamide (257 mg, 2.70 mmol) at 0°C. The reaction mixture was stirred for 16 h while warming at room temperature. After quenching with water, the reaction mixture was extracted with EtOAc (150 mL), washed with water (5×50 mL), then brine, dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified using EtOAc/heptane in different proportions to give the title compound as a colorless gum. Yield: 206 mg (90.6%). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 8.02 (d, J = 7.7, 1H), 7.38 (t, J = 7.5, 1H), 7.30 (d, J = 8.5, 2H), 7.19 (t, J = 6.8, 2H), 6.11 (s, 1H), 4.16 (t, J = 6.7, 2H), 3.68 (br. s, 2H), 3.45 (br. s, 2H), 2.98 (t, J = 7.1 Hz, 2H), 2.79 (s, 3H), 1.97 – 1.84 (m, 2H), 1.66 (br. s, 3H), 1.58 (br. s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.53 (C=O), 140.62 (C), 140.55 (C), 126.61 (C), 126.38 (CH), 124.84 (CH), 122.53 (CH), 122.45 (C), 120.49 (C), 119.72 (CH), 119.58 (CH), 108.97 (CH), 108.36 (CH), 40.77 (CH2), 40.13 (CH2), 39.49 (CH3), 29.01 (CH2), 24.60 (CH2). ESI: m/z 414.1 (M + H)+. HRMS calcd for C22H28N3O3S (M + H)+ 414.1851, found 414.1853.
4.1.65. Methyl 4-(phenylamino)benzoate (65)
Under argon atmosphere, a solution of methyl 4-bromobenzoate (3.5 g, 16.28 mmol), aniline (1.819 g, 19.53 mmol), palladium (II) acetate (218 mg, 0.97 mmol), rac-BINAP (506 mg, 0.81 mmol), and potassium carbonate (6.72 g, 48.62 mmol) in toluene (ca. 10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 160°C for 2 h and then cooled to rt. The reaction mixture was diluted with DCM and filtered. The organic solvents were evaporated in vacuo, and the residue was suspended in methyl tert-butyl ether (150 mL). The organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography (gradient elution starting with 5% EtOAc in heptane to 60% EtOAc in heptane) to afford title compound 65 (3.55 g, 96% yield) as a pale green solid: mp 121°C. 1H NMR (500 MHz, CDCl3) δ 7.94 – 7.87 (m, 2H), 7.35 – 7.29 (m, 2H), 7.16 (d, J = 7.5 Hz, 2H), 7.05 (t, J = 7.4 Hz, 1H), 7.00 – 6.95 (m, 2H), 6.15 (s, 1H), 3.86 (s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 167.19 (C=O), 148.27 (C), 140.99 (C), 131.63 (CH), 129.64 (CH), 123.21 (CH), 121.10 (C), 120.55 (CH), 114.69 (CH), 51.91 (CH3).
4.1.66. Methyl 9H-carbazole-3-carboxylate (66)
In a 100 mL round-bottom flask, a mixture of palladium acetate (1.821 g, 8.11 mmol) and diphenylamine 36 (1.676 g, 7.37 mmol) in glacial acetic acid (40 mL) was stirred under reflux for 1 hr. The organic solvent was removed by distillation. The precipitated metallic palladium was separated by transferring the obtained black residue into a folded paper filter and continuous extraction with acetone in a Soxhlet extractor until the condensing solvent turned colorless. The extract was concentrated in vacuo, and the resultant solid was sonicated for 10 min in a bath sonicator with 1 M hydrochloric acid (100 mL), filtered, rinsed with distilled water (50 mL × 3), and then dried in vacuo. The dry precipitate was sublimed under vacuum to afford the title compound as a light yellow solid. Yield: 1.081 g (65%); mp 180°C. 1H NMR (500 MHz, DMSO-d6) δ 11.73 (s, 1H), 8.81 (d, J = 1.6 Hz, 1H), 8.25 (d, J = 7.8 Hz, 1H), 8.03 (dd, J = 8.5, 1.7 Hz, 1H), 7.56 (t, J = 8.3 Hz, 2H), 7.45 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.25 – 7.20 (m, 1H), 3.89 (s, 3H). 1H NMR (500 MHz, CDCl3) δ 8.84 – 8.79 (m, 1H), 8.50 (s, 1H), 8.12 (dd, J = 8.5, 1.7 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.47 – 7.40 (m, 2H), 7.38 (d, J = 8.6 Hz, 1H), 7.27 (ddd, J = 8.0, 6.6, 1.5 Hz, 1H), 3.97 (s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 168.24 (C=O), 142.52 (C), 140.14 (C), 127.59 (CH), 126.74 (CH), 123.43 (C), 123.26 (C), 123.07 (CH), 121.39 (C), 120.78 (CH), 120.47 (CH), 111.13 (CH), 110.38 (CH), 52.21 (CH3).
4.1.67. (9H-carbazol-3-yl)(piperidin-1-yl)methanone (67)
Potassium hydroxide (3 g, 53.47 mmol) was added to a stirred solution of methyl ester 37 (818 mg, 3.63 mmol) in a mixture of ethanol (40 mL) and water (10 mL). The reaction mixture was stirred at reflux for 16 hrs and then cooled to room temperature. The solvents were evaporated under reduced pressure, and the residue was diluted with DI water. Using external cooling (ice-bath), the solution was acidified to pH ca. 2 by dropwise addition of 1M aqueous HCl. The precipitated product was extracted with EtOAc (150 mL). The organic layer was washed with brine under acidic pH, dried over MgSO4, and filtered. The solvent was then evaporated under reduced pressure, and the resulting residue was purified by silica chromatography using 70% EtOAc in heptane to yield the title compound as a beige solid. Yield: 737 mg (96%); mp 271°C. The obtained 9H-carbazole-3-carboxylic acid (742 mg, 3.51 mmol), piperidine (416 μL, 4.21 mmol), DIPEA (966 μL, 5.68 mmol), and DMAP (43 mg, 0.35 mmol) were added to DCM (30 mL) under nitrogen. The obtained solution was cooled down on an ice-water bath. EDAC (808 mg, 4.21 mmol) was added to the solution, and the reaction mixture was stirred for 16 h while warming at room temperature. The solvent was removed in vacuo, and the obtained residue was extracted with EtOAc (150 mL). The organic layer was washed consecutively with 5% citric acid solution (50 mL × 3), concentrated sodium bicarbonate (50 mL × 3), and brine (50 mL). The organic phase was then dried (MgSO4), filtered, and concentrated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to give the title compound as a beige solid. Yield: 839 mg (86%); mp 221°C.1H NMR (500 MHz, DMSO-d6) δ 11.47 (s, 1H), 8.18 (d, J = 8.0 Hz, 2H), 7.51 (dd, J = 8.2, 1.8 Hz, 2H), 7.42 (dd, J = 4.4, 1.3 Hz, 1H), 7.41 – 7.38 (m, 1H), 7.18 (t, J = 7.4 Hz, 1H), 3.50 (s, 4H), 1.67 – 1.57 (m, 2H), 1.52 (s, 4H). 13C NMR and DEPT (126 MHz, DMSO-d6) δ 170.10 (C=O), 140.16 (C), 140.08 (C), 126.56 (C), 126.00 (CH), 124.69 (CH), 122.31 (C), 121.91 (C), 120.53 (CH), 119.50 (CH), 118.92 (CH), 111.16 (CH), 110.48 (CH), 25.72 (CH2), 24.19 (CH2). ESI: m/z 279.1 (M + H)+. HRMS calcd for C18H19N2O (M + H)+ 279.1497, found 279.1514.
4.1.68. 9-(3-Methoxypropyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (68)
Under argon atmosphere, a solution of the carbazole amide 67 (100 mg, 0.36 mmol), 1-bromo-3-methoxypropane (61 μL, 0.54 mmol), and Cs2CO3 (234 mg, 0.72 mmol) in DMF (10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 140°C for 1 h and then cooled to room temperature. The reaction mixture was diluted with EtOAc and filtered. The organic solvents were evaporated in vacuo. The residue was suspended in methyl tert-butyl ether (150 mL), and the organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to give the title product (119 mg, 95%) as a clear, colorless gum. 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.50 – 7.40 (m, 3H), 7.24 (ddd, J = 8.0, 6.6, 1.3 Hz, 1H), 4.41 (t, J = 6.5 Hz, 2H), 3.64 (s, 4H), 3.29 (s, 3H), 3.23 (t, J = 5.6 Hz, 2H), 2.08 (p, J = 6.04 Hz, 2H), 1.73 – 1.66 (m, 2H), 1.62 (s, 4H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.48 (C=O), 141.09 (C), 140.95 (C), 126.87 (C), 126.19 (CH), 125.08 (CH), 122.73 (C), 122.43 (C), 120.48 (CH), 119.81 (CH), 119.38 (CH), 109.01 (CH), 108.39 (CH), 68.91 (CH2), 58.60 (CH3), 39.57 (CH2), 29.10 (CH2), 24.76 (CH2). ESI: m/z 351.1 (M + H)+. HRMS calcd for C22H27N2O2 (M + H)+ 351.2073, found 351.2048.
4.1.69. Piperidin-1-yl(9-((tetrahydro-2H-pyran-4-yl)methyl)-9H-carbazol-3-yl)methanone (69)
Using carbazole amide 67 (100 mg, 0.36 mmol) and 4-(bromomethyl)tetrahydro-2H-pyran (97 mg, 0.54 mmol) as starting compounds, the title compound was prepared as a colorless glass according to the procedure described above for compound 68. Yield: 120 mg (89%). 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 1.2 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.53 (dd, J = 8.4, 1.6 Hz, 1H), 7.47 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.38 (t, J = 8.7 Hz, 2H), 7.27 – 7.21 (m, 1H), 4.15 (d, J = 7.3 Hz, 2H), 3.93 (t, J = 2.8 Hz, 1H), 3.91 (t, J = 2.9 Hz, 1H), 3.49 (br. m, 4H), 3.30 – 3.21 (m, 2H), 2.23 (dp, J = 15.1, 7.4 Hz, 1H), 1.68 (br. s, 2H), 1.62 (br. s, 4H), 1.51 – 1.45 (m, 4H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.35 (C=O), 141.21 (C), 141.14 (C), 126.99 (C), 126.20 (CH), 125.12 (CH), 122.66 (C), 122.38 (C), 120.54 (CH), 119.80 (CH), 119.47 (CH), 109.18 (CH), 108.64 (CH), 67.49 (CH2), 48.98 (CH2), 35.60(CH), 31.18 (CH2), 24.75 (CH2). ESI: m/z 377.1 (M + H)+. HRMS calcd for C24H29N2O2 (M + H)+ 377.2229, found 377.2192.
4.1.70. Methyl 4-(3-(piperidine-1-carbonyl)-9H-carbazol-9-yl)butanoate (70)
Using carbazole amide 67 (100 mg, 0.36 mmol) and methyl 4-bromobutanoate (54 μL, 0.43 mmol) as starting compounds, the title compound was prepared as a colorless glass according to the procedure described above for compound 68. Yield: 127 mg (93%). 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 8.07 (d, J = 7.7, 1H), 7.52 (d, J = 8.4, 1H), 7.49 – 7.44 (m, 1H), 7.40 (t, J = 8.0, 2H), 7.24 (t, J = 7.4, 1H), 4.36 (t, J = 7.0, 2H), 3.64 (s, 3H), 3.60 (br.s, 4H), 2.31 (t, J = 7.0, 2H), 2.21 – 2.14 (m, 2H), 1.68 (br.s, 3H), 1.62 (br.s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 173.28 (C=O), 171.38 (C=O), 140.83 (C), 140.74 (C), 127.01 (C), 126.26 (CH), 125.12 (CH), 122.76 (C), 122.51 (C), 120.56 (CH), 119.88 (CH), 119.50 (CH), 108.92 (CH), 108.30 (CH), 51.72 (CH3), 41.96 (CH2), 30.75 (CH2), 24.74 (CH2), 23.87 (CH2). ESI: m/z 379.1 (M + H)+. HRMS calcd for C23H27N2O3 (M + H)+ 379.2022, found 379.1993.
4.1.71. (9-(3-(Dimethylamino)propyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (71)
Under argon atmosphere, a solution of carbazole amide 67 (248 mg, 0.89 mmol), 3-chloropropyldimethylamine hydrochloride (311 mg, 1.97 mmol), TBAI (128 mg, 0.35 mmol), and Cs2CO3 (886 mg, 2.72 mmol) in DMF (10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 140°C for 2 h and then cooled to room temperature. The reaction mixture was diluted with EtOAc (50 mL) and filtered. The organic solvents were evaporated in vacuo. The residue was suspended in methyl tert-butyl ether (150 mL), and the organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on a Biotage® KP-NH (amino-modified silica gel) cartridge using heptanes/EtOAc in different proportions to afford the title compound as a light yellow viscous gum. Yield: 180 mg, 56%. 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 1.5 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.53 (dd, J = 8.4, 1.6 Hz, 1H), 7.48 – 7.42 (m, 3H), 7.23 (ddd, J = 8.0, 4.7, 3.4 Hz, 1H), 4.36 (t, J = 6.8 Hz, 2H), 3.62 (br.s, 4H), 2.22 (t, J = 6.7 Hz, 2H), 2.19 (s, 6H), 1.98 (p, J = 6.7 Hz, 2H), 1.67 (br.s, 3H), 1.61 (br.s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.47 (C=O), 141.02 (C), 140.90 (C), 126.74 (C), 126.11 (CH), 125.00 (CH), 122.68 (C), 122.39 (C), 120.42 (CH), 119.75 (CH), 119.27 (CH), 109.08 (CH), 108.49 (CH), 56.38 (CH2), 45.42 (CH3), 40.66 (CH2), 26.89 (CH2), 24.72 (CH2). ESI: m/z 364.1 (M + H)+. HRMS calcd for C23H30N3O (M + H)+ 364.2389, found 364.2359.
4.1.72. Tert-butyl 3-(3-(piperidine-1-carbonyl)-9H-carbazol-9-yl)propylcarbamate (72)
Using amide 67 (435 mg, 1.56 mmol) and tert-butyl N-(3-bromopropyl)-carbamate (558 mg, 2.34 mmol) as starting compounds, the title compound was prepared as a light yellow glass according to the procedure described above for compound 71. Yield: 617 mg (91 %). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.46 (d, J = 7.5 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.28 – 7.21 (m, 1H), 4.68 (br. s, 1H), 4.34 (t, J = 6.4 Hz, 2H), 3.63 (br. s, 4H), 3.22 – 3.04 (m, 2H), 2.14 – 1.98 (m, 2H), 1.70 (br. s, 3H), 1.63 (br. s, 3H), 1.43 (s, 9H). 13C and DEPT NMR (101 MHz, CDCl3) δ 171.54 (C=O), 156.20 (C=O), 140.88 (C), 140.82 (C), 127.17 (C), 126.43 (CH), 125.29 (CH), 122.96 (C), 122.71 (C), 120.77 (CH), 120.06 (CH), 119.66 (CH), 108.94 (CH), 108.31 (CH), 79.59 (C), 40.75 (CH2), 38.60 (CH2), 29.36 (CH2), 28.55 (CH3), 24.90 (CH2). ESI: m/z 436.3 (M + H)+. HRMS calcd for C26H34N3O3 (M + H)+ 436.2600, found 436.2644.
4.1.73. 9-(3-Aminopropyl)-9H-carbazol-3-yl)(piperidin-1-yl)methanone (73)
Starting with compound 72 (354 mg, 0.81 mmol), the title compound was prepared as a light yellow glass according to the analogous procedure described above for compound 29. The obtained residue was purified by flash chromatography eluting with EtOAc/EtOH (gradient elution) on a Biotage® KP-NH cartridge to give 230 mg (84 %) of the target product in the form of a light amber glass. 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 1.5 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.47 (dd, J = 8.4, 1.6 Hz, 1H), 7.44 – 7.39 (m, 1H), 7.39 – 7.34 (m, 2H), 7.21 – 7.17 (m, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.82 – 3.32 (br. m, 4H), 2.61 (t, J = 6.8 Hz, 2H), 1.90 (p, J = 6.8 Hz, 2H), 1.70 – 1.60 (m, 3H), 1.57 (br. s, 3H), 1.47 (s, 2H). 13C and APT NMR (126 MHz, CDCl3) δ 171.30 (C=O), 140.81 (C), 140.71 (C), 126.67 (C), 126.05 (CH), 124.90 (CH), 122.54 (C), 122.27 (C), 120.36 (CH), 119.64 (CH), 119.23 (CH), 108.91 (CH), 108.32 (CH), 49.00 (CH2), 43.46 (CH2), 40.39 (CH2), 39.27 (CH2), 32.16 (CH2), 26.02 (CH2), 24.59 (CH2). ESI: m/z 336.2 (M + H)+. HRMS calcd for C21H26N3O (M + H)+ 336.2076, found 336.2069.
4.1.74. Piperidin-1-yl(9-(pyridin-4-ylmethyl)-9H-carbazol-3-yl)methanone (74)
Amide 67 (100 mg, 0.36 mmol) and potassium tert-butoxide (305 mg, 2.71 mmol) were added to DMF (5 mL) in a 25 mL round bottom flask. 4-(Bromomethyl)pyridine hydrobromide (136 mg, 0.54 mmol) was added to the reaction mixture in one portion. The reaction mixture was stirred at room temperature for 2 days, diluted with EtOAc (25 mL) and filtered. The solvents were evaporated in vacuo, and the obtained syrup was extracted with EtOAc. The organic layer was washed with an aqueous sodium bicarbonate, brine, dried (MgSO4) and filtered. The volatiles were removed in vacuo, and the obtained syrup was purified on a Biotage® KP-NH cartridge eluting with EtOAc/heptanes in different proportions to afford the target product as a light yellow glass. Yield: 118 mg (89%). 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 5.0 Hz, 2H), 8.23 (s, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.29 (t, J = 8.0 Hz, 2H), 7.27 – 7.23 (m, 1H), 6.97 (d, J = 5.4 Hz, 2H), 5.48 (s, 2H), 3.58 (br.m, 4H), 1.79 – 1.67 (m, 3H), 1.63 (br.s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.22 (C=O), 150.35 (CH), 145.93 (C), 140.90 (C), 140.86 (C), 127.88 (C), 126.69 (CH), 125.43 (CH), 123.04 (C), 122.91 (C), 121.40 (CH), 120.85 (CH), 120.26 (CH), 120.07 (CH), 108.96 (CH), 108.41 (CH), 45.66 (CH2), 24.78 (CH2). ESI: m/z 370.2 (M + H)+. HRMS calcd for C24H24N3O (M + H)+ 370.1919, found 370.1886.
4.1.75. Piperidin-1-yl(9-(pyridin-3-ylmethyl)-9H-carbazol-3-yl)methanone (75)
Using amide 67 (100 mg, 0.36 mmol) potassium tert-butoxide (160 mg, 1.42 mmol) and 3-(bromomethyl)pyridine hydrobromide (136 mg, 0.54 mmol) as starting compounds, the title compound was prepared as a colorless glass according to the procedure described above for compound 26. Yield: 45 mg (34%). 1H NMR (500 MHz, CDCl3) δ 8.56 (s, 1H), 8.48 (s, 1H), 8.21 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.51 – 7.47 (m, 1H), 7.47 – 7.41 (m, 1H), 7.37 – 7.30 (m, 2H), 7.30 – 7.23 (m, 2H), 7.15 – 7.09 (m, 1H), 5.49 (d, J = 4.1 Hz, 2H), 3.55 (br.m, 4H), 1.68 (br.s, 3H), 1.61 (br.s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.27 (C=O), 149.31 (CH), 148.36 (CH), 140.89 (C), 140.85 (C), 134.23 (CH), 132.53 (C), 127.79 (C), 126.67 (CH), 125.43 (CH), 123.85 (CH), 123.10 (C), 122.95 (C), 120.84 (CH), 120.17 (CH), 120.07 (CH), 109.03 (CH), 108.48 (CH), 44.34 (CH2), 24.82 (CH2). ESI: m/z 370.1 (M + H)+. HRMS calcd for C24H24N3O (M + H)+ 370.1919, found 370.1895.
4.1.76. Piperidin-1-yl(9-(pyridin-2-ylmethyl)-9H-carbazol-3-yl)methanone (76)
Using carbazole amide 67 (100 mg, 0.36 mmol) and potassium tert-butoxide (305 mg, 2.72 mmol) were added to DMF (5 mL) in a 25 mL round bottom flask. 2-(Bromomethyl)pyridine hydrobromide (340 mg, 1.34 mmol) was added to the reaction mixture in one portion. The reaction mixture was stirred at room temperature for 2 days, diluted with EtOAc (25 mL) and filtered. The solvents were evaporated in vacuo, and the obtained syrup was extracted with EtOAc. The organic layer was washed with an aqueous sodium bicarbonate, brine, dried (MgSO4) and filtered. The volatiles were removed in vacuo, and the obtained syrup was purified on a Biotage® KP-NH cartridge eluting with EtOAc/heptanes in different proportions to afford the target product as a light yellow glass. Yield 106 mg (80%). 1H NMR (500 MHz, CDCl3) δ 8.61 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 8.23 (d, J = 1.1 Hz, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.50 (dd, J = 8.4, 1.6 Hz, 1H), 7.47 – 7.42 (m, 1H), 7.41 (dd, J = 7.8, 1.7 Hz, 1H), 7.39 – 7.34 (m, 2H), 7.29 – 7.25 (m, 1H), 7.13 (dd, J = 7.1, 5.3 Hz, 1H), 6.62 (d, J = 7.9 Hz, 1H), 5.62 (s, 2H), 3.69 (br.m, 4H), 1.78 – 1.66 (m, 3H), 1.62 (br.s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.31 (C=O), 156.79 (C), 149.63 (CH), 141.05(C), 140.99 (CH), 137.15 (CH), 127.60 (C), 126.54 (CH), 125.33 (CH), 123.01 (C), 122.85 (C), 122.64 (CH), 120.69 (C), 120.48 (CH), 120.00 (CH), 119.95 (CH), 109.19 (CH), 108.67 (CH), 48.69 (CH2), 24.77 (CH2). ESI: m/z 370.2 (M + H)+. HRMS calcd for C24H24N3O (M + H)+ 370.1919, found 370.1901.
4.1.77. Methyl 4-(3-methoxyphenylamino)benzoate (77)
Under argon atmosphere, a solution of methyl 4-bromobenzoate (3.5 g, 16.28 mmol), 3-methoxyaniline (2 g, 16.24 mmol), palladium (II) acetate (218 mg, 0.97 mmol), rac-BINAP (506 mg, 0.81 mmol), and potassium carbonate (6.72 g, 48.62 mmol) in toluene (ca. 10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 160°C for 2 h. The reaction mixture was allowed to cool down to room temperature, diluted with DCM (50 mL) and filtered. The solvents were removed under reduced pressure, and the obtained residue was distilled in vacuo to afford a light yellow oil: b.p. 160–165°C at 0.2 mm Hg. Yield: 3.348 g (80%). 1H NMR (500 MHz, CDCl3) δ 7.93 – 7.88 (m, 2H), 7.22 (t, J = 8.1 Hz, 1H), 7.02 – 6.98 (m, 2H), 6.75 (ddd, J = 8.0, 2.1, 0.9 Hz, 1H), 6.72 (t, J = 2.2 Hz, 1H), 6.60 (ddd, J = 8.3, 2.4, 0.7 Hz, 1H), 6.18 (s, 1H), 3.87 (s, 3H), 3.78 (s, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 167.17 (C=O), 160.78 (C), 147.99 (C), 142.36 (C), 131.60 (CH), 130.39 (CH), 121.27 (C), 115.09 (CH), 112.68 (CH), 108.35 (CH), 106.07 (CH), 55.42 (CH3), 51.92 (CH3).
4.1.78. Methyl 7-methoxy-9H-carbazole-3-carboxylate (Clausine C or clauszoline-L, 78)
Using benzoate 77 (1.897 g, 7.37 mmol) and palladium acetate (1.987 g, 8.85 mmol) as starting compounds, the title compound was prepared following the procedures described in preparation of compound 66 as a pale yellow solid. Yield: 1.6 g (85%). 1H NMR (500 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.66 (d, J = 1.6 Hz, 1H), 8.11 (d, J = 8.6 Hz, 1H), 7.93 (dd, J = 8.5, 1.7 Hz, 1H), 7.49 (dd, J = 8.5, 0.5 Hz, 1H), 7.02 (d, J = 2.2 Hz, 1H), 6.83 (dd, J = 8.6, 2.3 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H). 13C NMR and DEPT (126 MHz, DMSO-d6) δ 167.06 (C=O), 159.06 (C), 142.67 (C), 141.83 (C), 125.53 (CH), 122.55 (C), 121.48 (CH), 121.19 (CH), 119.89 (C), 116.05 (C), 110.45 (CH), 108.75 (CH), 94.86 (CH), 55.32 (CH3), 51.73 (CH3).
4.1.79. Methyl 7-methoxy-9-pentyl-9H-carbazole-3-carboxylate (79)
Under argon atmosphere, a solution of carbazole 78 (462 mg, 1.81 mmol), 1-bromopentane (320 μL, 2.59 mmol), and Cs2CO3 (1.123 g, 3.45 mmol) in DMF (10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 140°C for 2 h and then cooled to rt. The reaction mixture was diluted with EtOAc and filtered. The organic solvents were evaporated in vacuo. The residue was suspended in methyl tert-butyl ether (150 mL), and the organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on silica gel, eluent: EtOAc–heptanes (1/99, v/v) → EtOAc–heptanes (2/3, v/v) to give the title compound (480 mg, 82%) as a light yellow viscous oil. 1H NMR (500 MHz, CDCl3) δ 8.70 (d, J = 1.2 Hz, 1H), 8.08 (dd, J = 8.6, 1.7 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.31 (d, J = 8.6 Hz, 1H), 6.88 (dd, J = 8.5, 2.2 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 4.20 (t, J = 7.3 Hz, 2H), 3.96 (s, 3H), 3.92 (s, 3H), 1.84 (p, J = 7.3 Hz, 2H), 1.38 – 1.30 (m, 4H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 168.17 (C=O), 159.61 (C), 143.48 (C), 142.56 (C), 126.25 (CH), 122.92 (C), 122.01 (CH), 121.54 (CH), 120.84 (C), 116.95 (C), 108.06 (CH), 108.04 (CH), 93.88 (CH), 55.88 (CH3), 52.06 (CH3), 43.37 (CH2), 29.50 (CH2), 28.64 (CH2), 22.62 (CH2), 14.12 (CH3).
4.1.80. 7-Methoxy-9-pentyl-9H-carbazole-3-carboxylic acid (80)
Potassium hydroxide (3 g, 53.47 mmol) was added to a stirred solution of methyl ester 79 (626 mg, 1.92 mmol) in a mixture of ethanol (40 mL) and water (10 mL). The reaction mixture was stirred at reflux for 16 h and then cooled to room temperature. The solvents were evaporated under reduced pressure, and the residue was diluted with DI water. The solution was placed in an ice-water bath, and acidified to pH ca. 2 by dropwise addition of 1M aqueous HCl. The precipitated product was extracted with EtOAc, washed with brine under acidic pH (ca. 2), and dried over MgSO4. After evaporation of the solvent under reduced pressure, the residue was chromatographed on silica gel with 70% EtOAc in heptanes to yield the title compound as a beige solid. Yield: 480 mg (80%); mp 171°C. 1H NMR (600 MHz, CDCl3) δ 8.79 (d, J = 1.3 Hz, 1H), 8.18 (dd, J = 8.6, 1.6 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.34 (d, J = 8.6 Hz, 1H), 6.91 (dd, J = 8.5, 2.1 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 4.22 (t, J = 7.3 Hz, 2H), 3.94 (s, 3H), 1.86 (p, J = 7.21 Hz, 2H), 1.42 – 1.31 (m, 4H), 0.88 (t, J = 7.1 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 173.33 (C=O), 159.70 (C), 144.05 (C), 142.65 (C), 126.95 (CH), 123.06 (C), 122.86 (CH), 121.68 (CH), 119.94 (C), 116.98 (C), 108.22 (CH), 108.19 (CH), 94.01 (CH), 55.95 (CH3), 43.47 (CH2), 29.54 (CH2), 28.68 (CH2), 22.66 (CH2), 14.16 (CH3).
4.1.81. (7-Methoxy-9-pentyl-9H-carbazol-3-yl)(piperidin-1-yl)methanone (81)
Carboxylic acid 80 (1 g, 3.21 mmol), piperidine (636 μL, 6.42 mmol), DIPEA (1.1 mL, 6.42 mmol), and DMAP (785 mg, 6.42 mmol) were added to DCM (100 mL) under nitrogen. The obtained solution was cooled down on an ice-water bath. EDC (1231 mg, 6.42 mmol) was added to the solution, and the reaction mixture was stirred for 16 h while warming at room temperature. The solvent was removed in vacuo, and the obtained residue was extracted with EtOAc (150 mL). The organic layer was washed consecutively with 5% citric acid solution (50 mL × 3), concentrated sodium bicarbonate (50 mL × 3), brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified on silica gel using heptanes/EtOAc (gradient elution) to afford the title compound as a light yellow gum that self-solidified upon standing in the refrigerator to a light yellow solid. Yield: 1109 mg, (91%); mp 96°C. 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 1.3 Hz, 1H), 7.94 (d, 1H), 7.45 (dd, J = 8.3, 1.6 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 6.86 (dq, J = 4.3, 2.2 Hz, 2H), 4.22 (t, J = 7.2 Hz, 2H), 3.93 (s, 3H), 3.63 (br.s, 4H), 1.85 (p, J = 7.3 Hz, 2H), 1.74 – 1.66 (m, 3H), 1.62 (br. s, 3H), 1.38 – 1.31 (m, 4H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR and DEPT (126 MHz, CDCl3) δ 171.73 (C=O), 159.45 (C), 142.37 (C), 141.25 (C), 126.97 (C), 123.86 (CH), 122.75 (C), 121.38 (CH), 119.03 (CH), 116.73 (C), 108.18 (CH), 107.62 (CH), 93.57 (CH), 55.84 (CH3), 43.26 (CH2), 29.48 (CH2), 28.64 (CH2), 26.37 (CH2), 24.88 (CH2), 22.62 (CH2), 14.11 (CH3). ESI: m/z 379.3 (M + H)+. HRMS calcd for C24H31N2O2 (M + H)+ 379.2386, found 379.2390.
4.1.82. Ethyl 3-(3-(4-methylpiperazine-1-carbonyl)-9H-carbazol-9-yl)propanoate (82)
Using carboxylic acid 51 (240 mg, 0.77 mmol) and 1-methylpiperazine (103 μL, 0.93 mmol) as starting compounds, the title compound was prepared as a light amber gum according to the procedure described above for compound 4 with the exception of not using 5% citric acid solution during the workup. Yield: 268 mg (88%). 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 1.3 Hz, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 8.4, 1.6 Hz, 1H), 7.50 – 7.37 (m, 3H), 7.23 (ddd, J = 7.9, 6.8, 1.3 Hz, 1H), 4.59 (t, J = 7.0 Hz, 2H), 4.04 (q, J = 7.2 Hz, 2H), 3.71 (br. s, 4H), 2.81 (t, J = 7.0 Hz, 2H), 2.44 (br. s, 4H), 2.31 (s, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 171.17 (C=O), 171.08 (C=O), 140.51 (C), 140.31 (C), 126.33 (C), 126.26 (CH), 125.18 (CH), 122.71 (C), 122.60 (C), 120.45 (CH), 120.01 (CH), 119.64 (CH), 108.89 (CH), 108.37 (CH), 60.80 (CH2), 45.98 (CH3), 38.71 (CH2), 33.39 (CH2), 13.92 (CH3). ESI: m/z 394.3 (M + H)+. HRMS calcd for C23H28N3O3 (M + H)+ 393.2131, found 394.2091.
4.1.83. Ethyl 3-(3-((1,1-dioxo-thiomorpholino)-4-carbonyl)-9H-carbazol-9-yl)propanoate (83)
Using 9-(3-ethoxy-3-oxopropyl)-9H-carbazole-3-carboxylic acid (400 mg, 1.28 mmol) and 1,1-dioxo-thiomorpholine (209 mg, 1.55 mmol) as starting compounds, the title compound was prepared as a colorless glass following the procedures described in preparation of compound 4. Yield: 289 mg, (52%). 1H NMR (500 MHz, CDCl3) δ 8.14 (d, J = 1.0 Hz, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.48 (dd, J = 8.4, 1.6 Hz, 1H), 7.46 – 7.39 (m, 3H), 7.21 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 4.58 (t, J = 7.0 Hz, 2H), 4.09 (br. s, 4H), 4.00 (q, J = 7.2 Hz, 2H), 3.02 (br. s, 4H), 2.77 (t, J = 7.0 Hz, 2H), 1.07 (t, J = 7.2 Hz, 3H). 13C and DEPT NMR (126 MHz, CDCl3) δ 172.35 (C=O), 171.28 (C=O), 141.32 (C), 140.66 (C), 126.94 (CH), 125.21 (CH), 124.42 (C), 123.13 (C), 122.73 (C), 120.80 (CH), 120.51 (CH), 120.29 (CH), 109.29 (CH), 109.03 (CH), 61.20 (CH2), 52.18 (CH2), 39.04 (CH2), 33.63 (CH2), 14.17 (CH3). ESI: m/z 429.3 (M + H)+. HRMS calcd for C22H25N2O5S (M + H)+ 429.1484, found 429.1476.
4.1.84. (9H-carbazol-3-yl)(1,1-dioxothiomorpholino)methanone (84)
Using 9H-carbazole-3-carboxylic acid (204 mg, 0.97 mmol), thiomorpholine 1,1-dioxide (154 mg, 1.14 mmol), DIPEA (330 μL, 1.89 mmol), DMAP (12 mg, 0.1 mmol), and EDAC (218 mg, 1.14 mmol), the title compound was prepared as an off-white solid following the procedures described in preparation of compound 67. Yield: 119 mg (38%); mp 326°C. 1H NMR (500 MHz, DMSO-d6) δ 11.54 (s, 1H), 8.34 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.59 – 7.50 (m, 3H), 7.43 (t, J = 7.5 Hz, 1H), 7.20 (t, J = 7.4 Hz, 1H), 3.96 (br. s, 4H), 3.31 (br. s, 4H). 13C and DEPT NMR (126 MHz, DMSO-d6) δ 170.79 (C=O), 140.46 (C), 140.21 (C), 126.17 (CH), 125.02 (CH), 124.88 (C), 122.28 (C), 121.91 (C), 120.57 (CH), 119.93 (CH), 119.08 (CH), 111.26 (CH), 110.70 (CH), 50.97 (CH2).
4.1.85. Tert-butyl 3-(3-((1,1-dioxo-thiomorpholino)-1-carbonyl)-9H-carbazol-9-yl)propyl-carbamate (85)
Under argon atmosphere, a solution of carbazole amide 84 (114 mg, 0.35 mmol), tert-butyl N-(3-bromopropyl)-carbamate (161 mg, 0.68 mmol), and powdered NaOH (21 mg, 0.53 mmol) in acetone (10 mL) was tightly capped in a 20 mL microwave vessel. The mixture was subjected to microwave irradiation at 80°C for 1 h and then cooled to room temperature. The reaction mixture was diluted with EtOAc (50 mL) and filtered. The organic solvents were evaporated in vacuo. The residue was suspended in methyl tert-butyl ether (150 mL), and the organic phase was washed with saturated aqueous sodium bicarbonate, brine, dried (MgSO4), filtered and evaporated in vacuo. The obtained residue was purified by column chromatography on a Biotage® KP-NH (amino-modified silica gel) cartridge using heptanes/EtOAc in different proportions to afford the title compound as a colorless glass. Yield: 143 mg (84%). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.49 (t, J = 10.0 Hz, 2H), 7.43 – 7.33 (m, 2H), 7.30 – 7.21 (m, 1H), 4.83 (s, 1H), 4.32 (t, J = 6.8 Hz, 2H), 4.14 (br. s, 4H), 3.21 – 2.98 (m, 6H), 2.03 (p, J = 6.2 Hz, 2H), 1.43 (s, 9H). 13C and APT NMR (101 MHz, CDCl3) δ 172.26 (C=O), 156.09 (C=O), 141.38 (C), 140.79 (C), 126.78 (CH), 125.11 (CH), 124.13 (C), 122.81 (C), 122.48 (C), 120.71 (CH), 120.41 (CH), 119.95 (CH), 109.07 (CH), 108.68 (CH), 79.47 (C), 52.04 (CH2), 40.68 (CH2), 38.45 (CH2), 29.24 (CH2), 28.45 (CH3). ESI: m/z 486.0 (M + H)+. HRMS calcd for C25H32N3O5S (M + H)+ 486.2063, found 486.2076.
4.2. CB2-agonist structural characterization through ligand-steered modeling
The crude model of the active-state CB2 receptor was based on a multi-template approach [5, 35], using the crytal structures of A2A adenosine receptor (A2AR) in the active state bound to agonist UK-432097[36] (PDB 3QAK), and the sphingosine 1-phosphate receptor 1 (S1P1R) bound to an antagonist sphingolipid mimic[37] (PDB 3V2Y). A multiple alignment of CB2, S1P1R, A2AR, together with the sequences of CB1, S1P2R, S1P3R, S1P4R, S1P5RL, lysophosphatidic acid receptor 1 (LPAR1), LPAR2 and LPAR3 was built using BLAST[34] and taking into account the information of the key conserved residues within class-A GPCRs[38]. The extracted alignment of CB2, S1P1R and A2AR is shown in Fig. S1. The two-residue gap in helix 5 of CB2 and S1P1R was introduced according to the structural superposition of GPCR crystal structures[39]. The nomenclature of Ballesteros and Weinstein[40] is used throughout the text, whereby X.50 denotes the most conserved residue in helix X. In agreement with Gonzalez et al.[39], residue L201 was considered as 5.50 in CB2. The aligment was used as input in ICM[41] to build a preliminary model of CB2 using the A2AR in the active state as template (3QAK), with the exception of the E2-loop and helix 5, which taking into account both sequence similarity and the smaller number of gaps, were built using S1P1R (3V2Y) as template. The N-term and C-term CB2 residues (1-29 and 316-360, respectively), and the TL4 residues conecting helices 4 and 5 in the A2AR structure were deleted. The disulfide bond within the E2-loop between C174 and C179 was inherited from the S1P1R template, and is in agreement with the work of others[42, 43]. This crude model was subjected to a restraint energy minimization to relieve structural strain stemming from the non-conservative substitutions. The refinement of the binding site was performed using the ligand-steered modeling method[6, 44–46]. Starting from the crude model developed through the multitemplate approach described above, agonist WIN55,212-2 was extracted from the GPCR Ligand Library (GLL)[47] (http://cavasotto-lab.net/Databases/GDD/), and seeded into the pocket, and a structural set of 50 structures was generated by randomizing the position and orientation of the ligand. Then, a multistep energy minimization was performed, in which the van der Waals interaction was gradually switched on, as already described[48, 49]. The ligand and receptor were held flexible in this stage without restraints. The structures in the set were subjected to a full flexible ligand-flexible side chain Monte Carlo-based global energy optimization in ICM[50–53]. The top-ranking structures were then visually inspected for ligand-receptor interactions, and a CB2-WIN55,212-2 complex was thus selected. It is observed that the interaction between the ligand and W5.43 revealed by mutagenesis analysis[54] was present in our model. The CB2 structural model thus obtained was used model complexes between compounds and CB2 using the ligand-steered method outlined above.
4.3. In vitro receptor radioligand binding studies
CB1 and CB2 radioligand binding data were obtained using National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) resources as described earlier[7, 55–57]. Compounds were screened in a competitive binding experiment using, respectively, membrane fractions prepared from rat brain homogenate expressing CB1 receptor and HEK293 cells expressing the human CB2 receptor. The competition binding experiment for CB1 and CB2 was performed in 96 well plates containing Standard Binding Buffer (50 mM Tris HCl, 1 mM EDTA, 3 mM MgCl2, 5 mg/ml fatty acid-free BSA, pH 7.4). The radioligand was [3H]CP55,940, and the reference compound was compound CP55,940. A solution of the compound to be tested was prepared as a 1 mg/ml stock in DMSO and then diluted in Standard Binding Buffer by serial dilution. Radioligand was diluted to five times the assay concentration in Standard Binding Buffer. Aliquots (50 μl) of radioligand were dispensed into the wells of a 96-well plate containing 100 μl of Standard Binding Buffer. Then, duplicate 50-μl aliquots of the test and reference compound dilutions were added. Finally, crude membrane fractions of cells were resuspended in 3 ml of chilled Standard Binding Buffer and homogenized by several passages through a 26 gauge needle, then 50 μl are dispensed into each well. The 250-μl reactions were incubated at room temperature for 1.5 hours, and then harvested by rapid filtration onto Whatman GF/B glass fiber filters pre-soaked with 0.3% polyethyleneimine using a 96-well Brandel harverster. Four rapid 500-μl washes were performed. Filters were placed in 6-ml scintillation tubes and allowed to dry overnight. Bound radioactivity was harvested onto 0.3% polyethyleneimine-treated, 96-well filter mats using a 96-well Filtermate harvester. The filter mats were dried, then scintillant was melted onto the filters and the radioactivity retained on the filters counted in a Microbeta scintillation counter. Raw data (dpm) representing total radioligand binding (i.e., specific + non-specific binding) were plotted as a function of the logarithm of the molar concentration of the competitor (i.e., test or reference compound). Non-linear regression of the normalized (i.e., percent radioligand binding compared to that observed in the absence of test or reference compound) raw data was performed in Prism 4.0 (GraphPad Software) using the built-in three parameter logistic model describing ligand competition binding to radioligand-labeled sites: y = bottom + [(top-bottom)/(1 + 10x-logIC50)] where bottom equals the residual radioligand binding measured in the presence of 10 μM reference compound (i.e., non-specific binding) and top equals the total radioligand binding observed in the absence of competitor. The log IC50 (i.e., the log of the ligand concentration that reduces radioligand binding by 50%) is thus estimated from the data and used to obtain the Ki by applying the Cheng-Prusoff approximation: Ki = IC50/(1 + [ligand]/KD) where [ligand] equals the assay radioligand concentration and KD equals the affinity constant of the radioligand for the target receptor.
4.4. [35S]GTP-γ-S functional assays
Functional activity was evaluated using GTP-γ-[35S] assay in Chinese hamster ovarian cell membrane extracts expressing recombinant hCB1 receptors or hCB2 receptors. The assay relies on the binding of GTP-γ-[35S], a radiolabeled nonhydrolyzable GTP analogue, to the G protein upon binding of an agonist of the G-protein-coupled receptor. In this system, agonists stimulate GTP-γ-[35S] binding whereas antagonists have no effect and inverse agonists decrease GTP-γ-[35S] basal binding. Compounds were solubilized in 100% DMSO at a concentration of 10 mM within 4 h of the first testing session (master solution). A predilution for the dose-response curve was performed in 100% DMSO and then diluted 100-fold in assay buffer at a concentration fourfold higher than the concentration to be tested. Compounds were tested for agonist activity at eight concentrations in duplicate: 10, 3, 1, 0.3, 0.1, 0.03, 0.01, and 0.001 μM, with compound CP55,940 (Tocris, 0949) as the reference agonist. For GTP-γ-[35S], membranes (Euroscreen s.a., Gosselies, Belgium) were mixed with GDP diluted in assay buffer to give 30μM solution (volume:volume) and incubated for at least 15 min on ice. In parallel, GTP-γ-[35S] (Amersham, SJ1308) was mixed with the beads PVT-WGA (Amersham, RPNQ001) diluted in assay buffer at 50 mg/ml (0.5 mg/10 μl) (volume:volume) just before starting the reaction. The following reagents were successively added in the wells of an Optiplate (Perkin Elmer): 50 μl of ligand, 20 μl of the membranes:GDP mix, 10 μl of assay buffer for agonist testing, and 20 μl of the GTP-γ-[35S]:beads mix. The plates were covered with a topseal, shaken on an orbital shaker for 2 min, and then incubated for 1 h at room temperature. Then the plates were centrifuged for 10 min at 2000 rpm, incubated at room temperature for 1 h, and counted for 1 min/well with a PerkinElmer TopCount reader. Assay reproducibility was monitored by the use of a reference compound CP55,940. For replicate determinations, the maximum variability tolerated in the test was of ± 20% around the average of the replicates. Efficacies (Emax) for CB1 and CB2 are expressed as a percentage relative to the efficacy of compound CP55,940.
4.5. CB1 and CB2 receptor internalization
The ability of the synthesized compounds to orthosterically interact with CB1 or CB2 receptors was assessed by determining if the compounds either caused internalization on their own (i.e., were agonists), prevented internalization by CP55,940 (i.e., were antagonists), or had no effect (i.e., were not orthosteric ligands). Receptor internalization was performed as described previously using cells stably expressing either human CB1 receptors or human CB2 receptors[58]. Data are presented as percent of internalization induced by 10 nM CP55,940 by 3 μM of each test compound. Thus, no internalization would be 0% and internalization equal to CP55,940 would be 100%. To determine if the compound was acting as an antagonist, cells were exposed to 10 nM of CP55,940 and 3 μM of each test compound. The reversal of internalization by 10 nM of compound CP55,940 by each test compound was calculated with the following formula: reversal = ((internalization in the presence of test compound with 10 nM CP55,940) − (internalization by 10 nM CP55,940))/(1− (internalization by 10 nM CP55,940))
4.6. In Vivo Evaluation
4.6.1. Animals
Adult male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) weighing 120–150 g were used in experimental procedures approved by the Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center. Animals were housed three per cage on a 12-h light/12-h dark cycle with water and food pellets available ad libitum.
4.6.2. Chronic neuropathic pain model
All surgical procedures were performed under 2–3% isoflurane anesthesia in 100% O2. The spinal nerve ligation was performed as described previously[59]. Briefly, a midline incision was made in the lumbar spinal region. The left L5 and L6 spinal nerves were isolated under a surgical microscope, and both nerves were tightly ligated with 6-0 silk. A prophylactic antibiotic (5 mg/kg of norfloxacin subcutaneously) and a prophylactic analgesic (0.2–0.5 mg/kg of buprenorphine subcutaneously) were administered once daily for 3 days. All of the behavioral tests were conducted three weeks after spinal nerve ligation.
4.6.3. Drug administration
All dosing solutions were prepared within 1 h prior to injection and stored at room temperature until use. Compound 64 and hydroxypropyl-β-cyclodextrins (30%) were mixed in appropriate ratios in PBS under constant magnetic stirring (200 rpm) at room temperature. 64 dosing solution was administrated i.p. as a single bolus injection.
4.6.4. Assessment of mechanical withdrawal thresholds
For assessment of pain hypersensitivity (tactile allodynia), rats were placed in a compartment on a mesh floor and allowed to acclimate for at least 30 min before testing. Mechanical sensitivity was assessed by using a series of von Frey filaments with logarithmic incremental stiffness. A series of calibrated von Frey filaments was used (Stoelting, Wood Dale, IL), as previously described[60], and 50% probability withdrawal thresholds were calculated with the up-down method[61]. In brief, filaments were applied one by one to the plantar surface of the hind paw for 6 s. If no withdrawal response was observed, the next stiffer filament was applied; if there was a withdrawal response, the next less stiff filament was applied. Six consecutive responses from the first change in the response were used to calculate the withdrawal threshold (in grams).
Supplementary Material
Discovery of a CB2 selective compound 64 based on a carbazole scaffold
64 internalized CB2 receptors and activity was confirmed using [35S]GTP-γ-S assays
64 had inhibitory effects on pain hypersensitivity in a rat model of neuropathic pain
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant P30-NS055022 (RRP, FD, SWM and PD), NS073935 (HLP), and DA011322 and DA035068 (JWM, LK, KM). This work was also supported by a grant from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2011-2778 to CNC). CNC thanks Molsoft LLC for providing an academic license for the ICM program. Radioligand binding assays were performed by the National Institute of Mental Health’s Psychoactive Drug Screening Program Contract # HHSN-271-2008-00025-C (NIMH/PDSP). NIMH/PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. We also thank Dr. Leonid Kalachev for helping us to calculate the affinity values. Marvin was used for calculating TPSA values, Marvin 5.11.5, 2013, ChemAxon (http://www.chemaxon.com).
ABBREVIATIONS
- BSA
bovine serum albumin
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- EC50
half maximal effective concentration
- EDTA
ethylenediaminetetraacetic acid
- GTP
guanosine-5′-triphosphate
- hCB1
human CB1
- hCB2
human CB2
- IC50
median inhibition concentration
Appendix A. Supplementary data
Supplementary data related to this article can be found at
Footnotes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ameri A. The effects of cannabinoids on the brain. Progress in Neurobiology. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 5.Diaz P, Phatak SS, Xu J, Fronczek FR, Astruc-Diaz F, Thompson CM, Cavasotto CN, Naguib M. 2,3-Dihydro-1-Benzofuran Derivatives as a Series of Potent Selective Cannabinoid Receptor 2 Agonists: Design, Synthesis, and Binding Mode Prediction through Ligand-Steered Modeling. ChemMedChem. 2009;4:1615–1629. doi: 10.1002/cmdc.200900226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz P, Phatak SS, Xu J, Astruc-Diaz F, Cavasotto CN, Naguib M. 6-Methoxy-N-alkyl Isatin Acylhydrazone Derivatives as a Novel Series of Potent Selective Cannabinoid Receptor 2 Inverse Agonists: Design, Synthesis, and Binding Mode Prediction. J Med Chem. 2009;52:433–444. doi: 10.1021/jm801353p. [DOI] [PubMed] [Google Scholar]
- 7.You H, Gadotti V, Petrov R, Zamponi G, Diaz P. Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol Pain. 2011;7:89. doi: 10.1186/1744-8069-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasquini S, De Rosa M, Ligresti A, Mugnaini C, Brizzi A, Caradonna NP, Cascio MG, Bolognini D, Pertwee RG, Di Marzo V, Corelli F. Investigations on the 4-quinolone-3-carboxylic acid motif. 6. Synthesis and pharmacological evaluation of 7-substituted quinolone-3-carboxamide derivatives as high affinity ligands for cannabinoid receptors. Eur J Med Chem. 2012;58:30–43. doi: 10.1016/j.ejmech.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 9.Yogeeswari P, Sharma M, Samala G, Gangadhar M, Karthick S, Mallipeddi S, Semwal A, Sriram D. Discovery of novel tetrahydro-pyrazolo [4,3-c] pyridines for the treatment of neuropathic pain: Synthesis and neuropharmacology. Eur J Med Chem. 2013;66:211–220. doi: 10.1016/j.ejmech.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Tourteau A, Andrzejak V, Body-Malapel M, Lemaire L, Lemoine A, Mansouri R, Djouina M, Renault N, Bakali JE, Desreumaux P, Muccioli GG, Lambert DM, Chavatte P, Rigo B, Leleu-Chavain N, Millet R. 3-Carboxamido-5-aryl-isoxazoles as new CB2 agonists for the treatment of colitis. Bioorg Med Chem. 2013;21:5383–5394. doi: 10.1016/j.bmc.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Madadi NR, Penthala NR, Brents LK, Ford BM, Prather PL, Crooks PA. Evaluation of (Z)-2-((1-benzyl-1H-indol-3-yl)methylene)-quinuclidin-3-one analogues as novel, high affinity ligands for CB1 and CB2 cannabinoid receptors. Bioorg Med Chem Lett. 2013;23:2019–2021. doi: 10.1016/j.bmcl.2013.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naguib M, Diaz P, Xu JJ, Astruc-Diaz F, Craig S, Vivas-Mejia P, Brown DL. MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol. 2008;155:1104–1116. doi: 10.1038/bjp.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu JJ, Diaz P, Astruc-Diaz F, Craig S, Munoz E, Naguib M. Pharmacological Characterization of a Novel Cannabinoid Ligand, MDA19, for Treatment of Neuropathic Pain. Anest Analg. 2010;111:99–109. doi: 10.1213/ANE.0b013e3181e0cdaf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K. Functional Selectivity in CB2 Cannabinoid Receptor Signaling and Regulation: Implications for the Therapeutic Potential of CB2 Ligands. Mol Pharmacol. 2012;81:250–263. doi: 10.1124/mol.111.074013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller AM, Stella N. CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz-Laviada I, Ruiz-Llorente L. Signal Transduction Activated by Cannabinoid Receptors. Mini Rev Med Chem. 2005;5:619–630. doi: 10.2174/1389557054368808. [DOI] [PubMed] [Google Scholar]
- 17.Diaz P, Xu J, Astruc-Diaz F, Pan HM, Brown DL, Naguib M. Design and Synthesis of a Novel Series of N-Alkyl Isatin Acylhydrazone Derivatives that Act as Selective Cannabinoid Receptor 2 Agonists for the Treatment of Neuropathic Pain. J Med Chem. 2008;51:4932–4947. doi: 10.1021/jm8002203. [DOI] [PubMed] [Google Scholar]
- 18.Knölker HJ, Reddy KR. Isolation and Synthesis of Biologically Active Carbazole Alkaloids. Chem Rev. 2002;102:4303–4428. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]
- 19.Petrov RR, Astruc-Diaz F, Cavasotto CN, Diaz P. Novel series of CB2 selective agonists for the treatment of neuropathic pain. 241st ACS National Meeting & Exposition; Anaheim, CA, United States. 2011. p. MEDI-227. [Google Scholar]
- 20.Wu CY, Brik A, Wang SK, Chen YH, Wong CH. Tetrabutylammonium Fluoride-Mediated Rapid Alkylation Reaction in Microtiter Plates for the Discovery of Enzyme Inhibitors in Situ. ChemBioChem. 2005;6:2176–2180. doi: 10.1002/cbic.200500295. [DOI] [PubMed] [Google Scholar]
- 21.Blangetti M, Rosso Hn, Prandi C, Deagostino A, Venturello P. Suzuki-Miyaura Cross-Coupling in Acylation Reactions, Scope and Recent Developments. Molecules. 2013;18:1188–1213. doi: 10.3390/molecules18011188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferorelli S, Abate C, Colabufo NA, Niso M, Inglese C, Berardi F, Perrone R. Design and evaluation of naphthol- and carbazole-containing fluorescent sigma ligands as potential probes for receptor binding studies. J Med Chem. 2007;50:4648–4655. doi: 10.1021/jm070373b. [DOI] [PubMed] [Google Scholar]
- 23.Akermark B, Eberson L, Jonsson E, Pettersson E. Palladium-promoted cyclization of diphenyl ether, diphenylamine, and related compounds. J Org Chem. 1975;40:1365–1367. [Google Scholar]
- 24.Gonsiorek W, Lunn CA, Fan X, Deno G, Kozlowski J, Hipkin RW. Sch35966 is a potent, selective agonist at the peripheral cannabinoid receptor (CB2) in rodents and primates. Br J Pharmacol. 2007;151:1262–1271. doi: 10.1038/sj.bjp.0707336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ertl P, Rohde B, Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 26.St Jean DJ, Fotsch C. Mitigating Heterocycle Metabolism in Drug Discovery. J Med Chem. 2012;55:6002–6020. doi: 10.1021/jm300343m. [DOI] [PubMed] [Google Scholar]
- 27.Bachmann A, Wildemann D, Praetorius F, Fischer G, Kiefhaber T. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc Natl Acad Sci. 2011;108:3952–3957. doi: 10.1073/pnas.1012668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Jahreis G, Fischer G, Lücke C. Atomic Polarizability Dominates the Electronic Properties of Peptide Bonds upon Thioxo or Selenoxo Substitution. Chem Eur J. 2012;18:9841–9848. doi: 10.1002/chem.201200863. [DOI] [PubMed] [Google Scholar]
- 29.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 30.Bissantz C, Kuhn B, Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostermann N, Ruedisser S, Ehrhardt C, Breitenstein W, Marzinzik A, Jacoby E, Vangrevelinghe E, Ottl J, Klumpp M, Hartwieg JCD, Cumin F, Hassiepen U, Trappe J, Sedrani R, Geisse S, Gerhartz B, Richert P, Francotte E, Wagner T, Krömer M, Kosaka T, Webb RL, Rigel DF, Maibaum J, Baeschlin DK. A Novel Class of Oral Direct Renin Inhibitors: Highly Potent 3,5-Disubstituted Piperidines Bearing a Tricyclic P3-P1 Pharmacophore. J Med Chem. 2013;56:2196–2206. doi: 10.1021/jm301706j. [DOI] [PubMed] [Google Scholar]
- 32.Pertwee RG. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem. 2010;17:1360–1381. doi: 10.2174/092986710790980050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pajda MB, Dariusz Fast preparation method of aromatic aldehydes from carbazole and hydroxyaromatic derivatives under microwave irradiation. Modern polym mater environ appl. 2006:129–132. [Google Scholar]
- 34.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsson P, Wallner B, Lindahl E, Elofsson A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 2008;17:990–1002. doi: 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez A, Cordomi A, Caltabiano G, Pardo L. Impact of helix irregularities on sequence alignment and homology modeling of G protein-coupled receptors. Chembiochem. 2012;13:1393–1399. doi: 10.1002/cbic.201200189. [DOI] [PubMed] [Google Scholar]
- 40.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 41.ICM. MolSoft, LLC; La Jolla, CA: 2012. [Google Scholar]
- 42.Gouldson P, Calandra B, Legoux P, Kerneis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Mutational analysis and molecular modelling of the antagonist SR 144528 binding site on the human cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 43.Montero C, Campillo NE, Goya P, Paez JA. Homology models of the cannabinoid CB1 and CB2 receptors. A docking analysis study. Eur J Med Chem. 2005;40:75–83. doi: 10.1016/j.ejmech.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Cavasotto CN. Homology models in docking and high-throughput docking. Curr Top Med Chem. 2011;11:1528–1534. doi: 10.2174/156802611795860951. [DOI] [PubMed] [Google Scholar]
- 45.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O’Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 46.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Gatica EA, Cavasotto CN. Ligand and Decoy Sets for Docking to G Protein-Coupled Receptors. J Chem Inf Model. 2012;52:1–6. doi: 10.1021/ci200412p. [DOI] [PubMed] [Google Scholar]
- 48.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 49.Phatak SS, Gatica EA, Cavasotto CN. Ligand-steered modeling and docking: A benchmarking study in Class A G-Protein-Coupled Receptors. J Chem Inf Model. 2010;50:2119–2128. doi: 10.1021/ci100285f. [DOI] [PubMed] [Google Scholar]
- 50.Monti MC, Casapullo A, Cavasotto CN, Napolitano A, Riccio R. Scalaradial, a Dialdehyde-Containing Marine Metabolite That Causes an Unexpected Noncovalent PLA(2) Inactivation. Chembiochem. 2007;8:1585–1591. doi: 10.1002/cbic.200700217. [DOI] [PubMed] [Google Scholar]
- 51.Monti MC, Casapullo A, Cavasotto CN, Tosco A, Dal Piaz F, Ziemys A, Margarucci L, Riccio R. The binding mode of petrosaspongiolide M to the human group IIA phospholipase A(2): exploring the role of covalent and noncovalent interactions in the inhibition process. Chem Eur J. 2009;15:1155–1163. doi: 10.1002/chem.200801512. [DOI] [PubMed] [Google Scholar]
- 52.Domenech R, Hernandez-Cifre JG, Bacarizo J, Diez-Pena AI, Martinez-Rodriguez S, Cavasotto CN, de la Torre JG, Camara-Artigas A, Velazquez-Campoy A, Neira JL. The Histidine-Phosphocarrier Protein of the Phosphoenolpyruvate: Sugar Phosphotransferase System of Bacillus sphaericus Self-Associates. PLoS One. 2013;8:e69307. doi: 10.1371/journal.pone.0069307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovacs JA, Cavasotto CN, Abagyan RA. Conformational Sampling of Protein Flexibility in Generalized Coordinates: Application to ligand docking. J Comp Theor Nanosci. 2005;2:354–361. [Google Scholar]
- 54.Zhang Y, Xie Z, Wang L, Schreiter B, Lazo JS, Gertsch J, Xie XQ. Mutagenesis and computer modeling studies of a GPCR conserved residue W5.43(194) in ligand recognition and signal transduction for CB2 receptor. Int Immunopharmacol. 2011;11:1303–1310. doi: 10.1016/j.intimp.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armbruster BN, Roth BL. Mining the Receptorome. J Biol Chem. 2005;280:5129–5132. doi: 10.1074/jbc.R400030200. [DOI] [PubMed] [Google Scholar]
- 56.Jensen NH, Roth BL. Massively parallel screening of the receptorome. Comb Chem High Throughput Screen. 2008;11:420–426. doi: 10.2174/138620708784911483. [DOI] [PubMed] [Google Scholar]
- 57.Strachan RT, Ferrara G, Roth BL. Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discov Today. 2006;11:708–716. doi: 10.1016/j.drudis.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 58.Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim SHCJ. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 60.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 61.Dixon W. The up-and-down method for small samples. J Am Stat Assoc. 1965;60:967–978. [Google Scholar]
- 62.Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR. 3-(1′,1′-Dimethylbutyl)-1-deoxy-[Delta]8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 63.MRM, Johnson LS. Cannabinoids as Therapeutic Agents. Boca Raton, FL: 1986. [Google Scholar]
- 64.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.