

Abstract
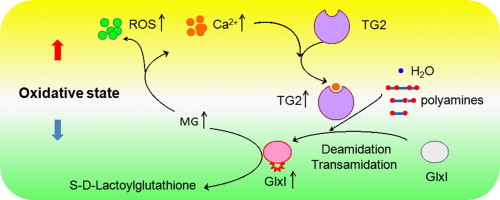
Glyoxalase 1 (GlxI) is the key enzyme that converts the highly reactive α-oxo-aldehydes into the corresponding α-hydroxy acids using l-glutathione as a cofactor. In our preliminary data, GlxI was identified as a substrate of transglutaminase 2 (TG2), a ubiquitous enzyme with multiple functions. According to the catalytic properties of TG2, protein cross-linking, polyamine conjugation, and/or deamidation are potential post-translational modifications. In this article, we have demonstrated that TG2 catalyzes either polyamine conjugation or deamidation to GlxI depending on the presence of polyamines or not. Deamidation leads to activation of GlxI while polyamine conjugation results in activation of GlxI as well as stabilization of GlxI against denaturation treatment. In cultured HeLa cells, methylglyoxal challenge causes increase in intracellular levels of reactive oxygen species (ROS) and calcium leading to TG2 activation and subsequent transamidation and activation of GlxI. The inhibition of TG2 significantly weakens the cell resistance to the methylglyoxal challenge. Thus, GlxI is a novel substrate of TG2 and is activated by TG2 in vitro and in cellulo. Exposure to methylglyoxal elicits a negative feedback loop entailing ROS, calcium, TG2 and GlxI, thus leading to attenuation of the increase in the methylglyoxal level. The results imply that cancer cells highly express TG2 or GlxI can endure the oxidative stress derived from higher glycolytic flux and may gain extra growth advantage from the aerobic glycolysis.
Keywords: Transglutaminase 2, Glyoxalase 1, Methylglyoxal, Oxidative stress
Graphical abstract
Highlights
-
•
We have demonstrated novel modifications of glyoxalase I by transglutaminase 2.
-
•
The modifications mediated by transglutaminse 2 modulate the glyoxalase I activities.
-
•
Methylglyoxal treatment in cells induces increases in the levels of endogenous reactive oxygen species and activation transglutaminase 2 and glyoxalase I.
-
•
Cells dispose the accumulated intracellular methylglyoxal by a negative feedback loop consisting of reactive oxygen species, calcium, transglutaminase 2 and glyoxalase I.
Introduction
Transglutaminases (EC 2.3.2.13) are encoded by a family of nine structurally related genes, eight of which encode functional enzymes [1], [2]. Among them, transglutaminase 2 (TG2), also called tissue type transglutaminase, is expressed ubiquitously in many different cell types. Transglutaminases, in the presence of high concentrations of calcium, catalyze the formation of isopeptide linkages between the γ-carboxamide group of the glutamine residue and the ε-amino group of the lysine residue resulting in the formation of soluble protein dimer or stable, insoluble macromolecular complexes. In addition, low-molecular-mass amines such as polyamines, can replace lysine residues in the transamidating reactions resulting in the formation of N-mono(γ-glutamyl) polyamines [3], [4]. The transglutaminase-catalyzed polyamine conjugation possibly alters structure and function of the substrates since polyamines are polycationic in structure in contrast to the uncharged property of glutamine side chain. Furthermore, several noncanonical activities associated with TG2 have been reported such as deamidation of glutamine residues [5], [6], GTP-binding and hydrolysis [7], [8], protein disulfide isomerase [9], protein kinase [10], [11] and binding proteins for signaling molecules [12], [13], [14], [15].
Methylglyoxal, a highly reactive dicarbonyl compound toward the amino groups of proteins and nucleic acids, is formed spontaneously from glyceraldehyde-3-phosphate and dihydroxyacetone phosphate generated in the glycolytic pathway [16]. It is estimated that up to 0.1–2% of arginine residues of cellular proteins and 1 in 105 nucleobases of cellular DNA is modified by methylglyoxal [17]. Glyoxalase 1 (EC 4.4.1.5; GlxI), a dimeric Zn (II) metalloenzyme of molecular mass 42 kDa, acts on the hemithioacetal formed spontaneously from methylglyoxal and glutathione to produce S-d-lactoylglutathione. Glyoxalase 2 subsequently converts S-d-lactoylglutathione to d-lactate returning glutathione consumed in the reaction catalyzed by GlxI [18], [19]. Excessive accumulation of methylgloxal may induce modifications of mitochondrial proteins leading to dysfunction of the mitochondria [20] and increases in the production of reactive oxygen species (ROS) [21], [22]. Furthermore, GlxI overexpression attenuates hyperglycemia-induced accumulation of methylglyoxal-modified mitochondrial proteins and synthesis of ROS and thus enhances the lifespan in Caenorhabditis elegans [23]. In contrast, inhibition of GlxI reduces mean and maximum life span of the worms. Similarly, overexpression of GlxI reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats [24]. Since accumulation of methylglyoxal leads to increases in ROS [21], [22] and increases in intracellular ROS activate TG2 [25], [26], we wish to investigate whether treatment of cultured cells with methylglyoxal induces activation of TG2 or not. In addition, we wish to study whether GlxI is regulated by TG2. Our data indicate that TG2 acts on GlxI leading to polyamine–conjugation or deamidation modification depending on the presence of polyamines; deamidation leading to activation and polyamine–conjugation to activation and stabilization of GlxI. Moreover, activity of TG2 and GlxI increase in HeLa cells in response to challenged methylglyoxal and the increase in GlxI activity requires the transamidating activity of TG2. We propose that exposure of cells to methylglyoxal elicits a negative feedback loop entailing ROS, calcium, TG2 and GlxI leading to decrease in the levels of methylglyoxal.
Materials and methods
Affinity purification of TG2 substrates
HeLa cells were lysed with 20 mM Tris–HCl, pH 8.0, containing 2 mM CaCl2, 0.1 mM dithiothreitol, 2% Triton X-100 and 1 mM biotinamidopentylamine (bPA, EZ-Link® Pentylamine-Biotin, Pierce), and the lysate was incubated at 25 °C for 30 min. After repeated protein precipitation with 80% ammonium sulfate at 4 °C, the protein pellets were dissolved in TE buffer (20 mM Tris–HCl, pH 8.0, 5 mM EDTA). The protein samples were then applied to a streptavidin–agarose (Fluka) column equilibrated with TE buffer. After washing with 10 bed volumes of TE buffer and 10 bed volumes of 0.1% SDS in TE buffer, bound proteins were obtained by boiling in the SDS sample buffer for 5 min.
Constructing plasmids
The first-stranded cDNA library was prepared using mRNA isolated from HeLa cells. Target DNA was amplified from 50 ng of cDNA by the Advantage 2 PCR kit (Promega) with the 5' primer hGlxIF, 5'–GTC GAC ATG GCA GAA CCG CA-3', and the 3' primer hGlxIR, 5'–GCG GCC GCC ATT AAG GTT GCC-3', each with endonuclease recognition sites (underlined) for SalI and NotI, respectively. The DNA product was restriction-digested and then ligated with pET21b expression vector (Novagen) and transformed into Escherichia coli BL21. With the same procedures, the 5' primer hGlxIF, 5'–AGG CTT ATG GCA GAA CCG CA-3', and the 3' primer hGlxIR, 5'–GAA TTC GCC ATT AAG GTT GCC-3', each with the endonuclease recognition site (underlined) for HindIII and EcoRI, respectively, were used for constructing the insert into p3XFLAG-CMVTM-14 expression vector (Sigma-Aldrich) for expressing FLAG-GlxI in cells. Although, the deduced amino acid sequence differs from other isolated sequences with glutamic acid residue replacing alanine at position 111, the kinetic properties of recombinant human glyoxalase I (rhGlxI) were indistinguishable from those of native enzyme [27]. In addition, our rhGlxI has extra 21 amino acid residues to the N-terminus; MASMTGGQQMGKGSEFELKKM- due to cloning artifacts.
Preparation and purification of anti-spermine antibody
The conjugation of spermine onto bovine serum albumin (BSA) was carried out by electrolytic reduction and followed by Schiff base reaction [28]. Ten milligrams of BSA was electrolyzed in 5 ml of 1% acetic acid containing 0.1 M NaCl at 100 V for 4 h, and the solution were then adjusted to pH 7.0–8.0 by NaOH prior to reaction with spermine. The reduced BSA solution was added by 5 ml of 0.1 M spermine and allowed to incubate at 25 °C for 30 min for Schiff reaction. The spermineconjugated BSA was separated by preparative SDS-PAGE and the gel band was sliced for preparing antigens. Anti-spermine antibody was obtained from guinea pig by subcutaneous injection of spermineconjugated BSA with adjuvant. The harvested serum was further purified with spermineconjugated agarose. Two milliliter of NHS-activated agarose (Thermo scientific) was incubated with 2 ml of 0.5 M spermine in PBS for 3 h and quenched by incubation with 4 ml of 1 M Tris–HCl, pH 8.0. The IgGs were pulled down by Hitrap Protein A HP column (GE Healthcare Life Science) in PBS and then eluted by 0.1 M glycine-HCl, pH 2.8. The eluted IgGs were reconstituted in PBS and subsequently purified by spermineconjugated agarose. The affinity bound IgG by spermineconjugated agarose was eluted by 0.1 M glycine-HCl, pH 2.8 and reconstituted in PBS as anti-spermine antibody.
In vitro transamidation
Reaction was carried out by adding TG2 (Sigma-Aldrich), to a final concentration of 50 mU/ml, to a substrate mixture in the TG2 reaction buffer (20 mM Tris–HCl, pH 8.0, 0.15 M NaCl, 0.1 mM DTE, and 5 mM CaCl2) in the presence of 1 μg/μl bPA at 25 °C for 30 min incubation. For inhibition of in vitro transamidation, inhibitors such as EGTA, cysteamine (Fluka), cystamine (Fluka), or monodansylcadeverine (Fluka), each 10 mM at a final concentration, was included in the reaction. Biotin-tagged proteins were resolved by SDS-PAGE, immunoblotting and streptavidin-peroxidase overlay assay. For immunoblotting, the protein was recognized by the specific antibody, anti-glyoxalase I (FL-184, Santa Gruz biotechnology), TG2 (Ab-1, NeoMarkers), or anti-spermine antibody (prepared in house) and magnified by the horseradish peroxidase-conjugated second antibody (Jackson ImmunoResearch Inc.).
Streptavidin-peroxidase blot overlay
The PVDF membrane was blocked with PBS containing 0.05% Tween 20 (PBS-T) and 3% skim milk for 1 h and then washed with PBS-T three times, each for 5 min. For streptavidin-peroxidase blot overlay, proteins were probed with 1 μg/ml horseradish peroxidase-conjugated streptavidin (Thermo Scientific) in PBS-T containing 3 mg/ml of BSA for 30 min. After washed with PBS-T three times, each for 5 min, horseradish peroxidase catalyzed signals were detected with the standard ECL protocol (Millipore).
Detection of TG2-catalyzed deamidation of rhGlxI
Detection of deamidation of GlxI was carried out by two consecutive reactions with TG2, an initial deamidation and a latter transamidation in the absence and presence of 1 μg/μl bPA, respectively. The deamidation reaction was catalyzed by 0 mU to 6 mU of recombinant human TG2 (Zedira, 0.59 U/mg) at 25 °C for 18 h in the TG2 reaction buffer. For the transamidation reaction, to each sample was then added another 2.5 mU of TG2 and bPA to a final concentration of 1 μg/μl for further incubation at 25 °C for 30 min. The samples were then subjected to streptavidin-peroxidase overlay assay. The extent of deamidation was revealed by the decreases in bPA incorporation into rhGlxI. Alternatively, endoproteinase Glu-C cleavage was used for detection of the newly generated glutamyl amino acid residues catalyzed by TG2 [29]. The rhGlxI was incubated without or with 2 mU TG2 (Zedira) at 25 °C for 18 h in the TG2 reaction buffer and then added with Glu-C (Sigma-Aldrich) to 0.1 mU/μl for additional 2 h incubation at 37 °C. The product was resolved by 10% SDS-PAGE, transferred to a PVDF membrane, and stained with Amido Black. N-terminal amino acid sequences (Applied Biosystems Model 477 A) were obtained from the excised protein bands.
GlxI activity assay
The procedure was modified from the previous description [30]. GlxI activity was determined spectrophotometrically in a reaction buffer containing 8 mM methylglyoxal (Sigma-Aldrich), 1 mM glutathione, 15 mM MgSO4 and 0.2 M imidazole-HCl, pH 7.0 at 25 °C. For 200 μl of reaction, 20 μl of 0.1 μg/μl rhGlxI solution or cells lysate was added to 180 μl of reaction buffer. The formation of S-d-lactoylglutathione was monitored by absorbance at 240 nm. GlxI activity was obtained from each catalytic velocity curve in the first 3 min with 15 s intervals by extrapolating the slope of absorbance over time.
Cell culture
Cells were obtained originally from American Type Culture Collection. Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), high glucose medium containing 10% FBS within 5% CO2 atmosphere at 37 °C.
In situ transamidation assay
Cells at 80% confluence were primed with fresh serum-free DMEM medium containing 1 mM bPA for 1–2 h. The cells were then treated with 0–5 mM methylglyoxal or with 2 mM methylglyoxal in the absence or presence of 1 mM cystamine, 0.1 mM N1-N11-diethylnorspermine (DENSPM, Tocris), or 0.1 mM monodansylcadaverine (Sigma-Aldrich) for 1–2 h and then harvested by scraping in a lysis buffer formed by 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1% SB 3–12, 1% CHAPS after PBS wash three times. Each cell lysate was subjected to the streptavidin-peroxidase blot overlay assay, and the TG2 activity was evaluated by densitometry analysis of the blots. Endogenous biotin signals obtained from control cells in the absence of bPA were subtracted from the calculation. The expression of TG2 activity was normalized to that of the bPA primed and untreated cells.
Measurement of intracellular methylglyoxal
Methylglyoxal derivatization
The procedures were modified from the previous description except that we used 2,3-diaminopyridine (Sigma-Aldrich) instead of o-phenylenediamine in the derivatization reaction [31]. The HeLa cells were extracted with 66% methanol in water at a ratio of 600 μl to 1.2×106 cells. After centrifugation, 400 μl of supernatant was vacuum-dried, reconstituted in 20 μl of 20 mM 2,3-diaminopyridine (Sigma) in 2.6 mM formic acid and incubated for 24 h at 25 °C. The derivatized samples were filled up to 100 μl with ultrapure water for LC-ESI-Q-TOF analysis. Methylglyoxal standard (1–100 pmol) was used to establish the linear range of reaction.
LC-ESI-MS analysis
The LC-ESI-MS system consisted of an ultra-performance liquid chromatography (UPLC) system (Ultimate 3000 RSLC, Dionex) and an electrospray ionization (ESI) source of quadrupole time-of-flight (TOF) mass spectrometer (maXis HUR-QToF system, Bruker Daltonics). The autosampler was kept at 4 °C. Separation was performed with reversed-phase liquid chromatography (RPLC) on a BEH C18 column (2.1×100 mm, Walters). The elution started from 99% mobile phase A (0.1% formic acid in ultrapure water) and 1% mobile phase B (0.1% formic acid in acetonitrile), raised to 90% B in 1 min, held at 90% B for 2 min, and then lowered to 1% B in 0.5 min. The column was equilibrated by pumping 1% B for 3.5 min. The flow rate was set at 0.4 ml/min with per injection volume 10 μl. LC-ESI-MS chromatogram were acquired under the following conditions: capillary voltage of 4500 V in positive ion mode, dry temperature at 190 °C, dry gas flow maintained at 8 l/min, nebulizer gas at 1.4 bar, and acquisition range of m/z 100–1000. Reaction products of methylglyoxal and 2,3-diaminopyridine gave rise to signals of at least 6-fold intensity as compared with that of methylglyoxal and o-phenylenediamine in mass spectrometry detection.
Measurement of intracellular ROS and calcium concentration
Cells were treated with 5 μM 2',7'-dichlorofluorescin diacetate (DCFH-DA, Sigma-Aldrich) or Fura-2 (Invitrogen) in serum free DMEM medium for 30 min. The cells were then challenged with 2 mM methylglyoxal in the absence or presence of 1 mM cystamine or 0.1 mM DENSPM in fresh serum free DMEM medium for 2 h. Next, cells were washed with PBS three times and trypsinized for the flow cytometry analysis with a FACSCaliburflow cytometer (BD Bioscience). The geometric mean of DCF and Fura-2 fluorescent intensity was obtained and used for indicating the intracellular ROS levels and [Ca2+], respectively.
MTT assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Sigma-Aldrich] in PBS solution (5 mg/ml) was infused to the culture medium by the ratio 1:10. After 2–5 h, the purple formazan was dissolved by DMSO and the solution was applied to measure the absorbance at 560 nm and 670 nm, the latter for background subtraction. The amount of viable cells was evaluated by absorbance readout and graphed as a histogram.
Statistics
Statistical comparisons were performed using Student's unpaired, two-tailed t test with results expressed as the mean±standard deviation (S.D.). A p value of<0.05 was considered statistically significant.
Cell transfection
Reverse transfection was performed according to the published procedures [32]. In brief, cells were trypsinized and suspended in DMEM medium containing 10% FBS. After centrifugation, the cell pellet was dispersed at the density of 8×105 cells in 1 ml of serum free DMEM medium. DNA or RNA was delivered by using Lipofectamine 2000 reagent (Invitrogen). After 12 h incubation within 5% CO2 at 37 °C, the cells were refreshed with DMEM medium containing 10% FBS for further 36 h culture.
Short interfering RNA
TGM2, ID: s14087 siRNA(Ambion) was used to knock down TG2 protein expression, GLO1, ID: s5824 or GLO1, ID: s5825 siRNA (Ambion) was used to knock down GlxI, and negative control #1 siRNA (Ambion) was used for negative control.
Results
Identification of GlxI as a TG2 substrate
From our previous data, GlxI was identified as one of TG2 substrates from a proteomic survey. For further confirmation, HeLa cell lysate was incubated with 1 μg/μl biotinamidopentylamine (bPA) in the presence of 5 mM calcium and 0.1 mM dithiothreitol for creating TG2-catalyzed transamidation reactions. As a result, endogenous TG2 would be activated and bPA would be incorporated into endogenous TG2 substrates. Potential TG2 substrates were purified by the streptavidin affinity column chromatography, biotin-labeled proteins were probed with streptavidin-peroxidase (Fig. 1A, upper panel) and GlxI was detected by immunoblotting as a protein band of 24 kD among the biotin-labeled proteins (Fig. 1A, lower panel). Notice that endogenous biotin-labeled proteins were purified from the control cell lysate and those proteins were no longer purified from the cell lysate treated with bPA possibly due to the limited capacity of streptavidin-agarose in the assay. Next, recombinant human GlxI (rhGlxI) was analyzed by an in vitro transamidation assay. Recombinant rhGlxI was susceptible to bPA incorporation catalyzed by TG2 and bPA incorporation was inhibited by TG2 inhibitors; cysteamine, cystamine, EGTA, and monodansylcadaverine (MDC). There was little change in the levels of monomeric rhGlxI in the presence of TG2 suggesting that TG2 did not catalyze significantly the crosslinking of rhGlxI (Fig. 1B). Therefore, transamidation with small amine-containing ligands or deamidation would be the potential modifications of GlxI catalyzed by TG2.
Fig. 1.
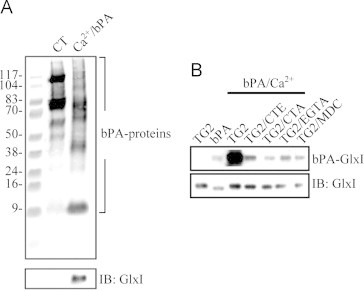
Confirmation of glyoxalase 1 (GlxI) as a transglutaminase 2 (TG2) substrate. (A) HeLa cell lysate was subjected to the in vitro transamidation reaction with endogenous TG2 and substrates in the presence of 1 μg/μl bPA. The bPA labeled proteins were pulled down using streptavidin-agarose and applied to the biotin overlay assay (upper panel) and immunoblotting (lower panel). (B) Purified rhGlxI was subjected to the in vitro transamidation reaction and applied to the biotin overlay assay (upper panel) and immunoblotting (lower panel). Lane 1: TG2 only; lane 2: bPA only; lane 3–7: TG2, bPA and various reagents. CT: control, CTE: cysteamine, CTA: cystamine, MDC: monodansylcadaverine.
Polyamine incorporation and deamidation of Glx1 catalyzed by TG2
Polyamines including putrescine, spermidine and spermine are the most important amine-containing ligands used by TG2 in the amine conjugation into TG2 substrates. We then assayed the incorporation of spermine into GlxI by the in vitro transamidation assay in the absence or presence of various TG2 inhibitors (Fig. 2A). Incorporation of spermine into the recombinant GlxI or proteins in the cell lysate was detected by immunoblotting using an in-house antiserum against spermine. All known inhibitors of TG2 potently suppressed the incorporation of spermine into rhGlxI. Interestingly, rhGlxI seemed to bind spermine strongly enough to resist SDS-PAGE because the anti-spermine immunoblotting recognized the rhGlxI pre-incubated with spermine. Further, the level of bPA incorporation into rhGlxI was suppressed by the addition of spermine in the in vitro transamidation catalyzed by TG2 (Fig. 2B). Along with the decrease in bPA incorporation, there was an increase in spermine conjugation into rhGlxI in a dose-dependent manner. Similarly, bPA incorporation into rhGlxI was also competed by putrescine and spermidine in the in vitro transamidation, albeit not as potent as spermine (Fig. 2C). Therefore, all polyamines and bPA can be used by TG2 in the amine incorporation into TG2 substrates and compete with each other. Besides transamidation, TG2 can cause deamidation, hydrolysis of the amide group on glutamine side chain, of its substrates. Besides elaborated analysis using mass spectrometry, Gln deamidation can be examined by differential cleavage by Glu-C protease (also called V8 protease) specific for Glu residues [29]. However, deamidation also occurs during sample preparation steps in mass spectrometry prior to the trypsin digestion [33]. To investigate the deamidation of rhGlxI, the in vitro transamidation assay was modified as follows. By pretreating rhGlxI with the activated TG2 in the absence of primary amines, the amide group of some glutaminyl residues of rhGlxI would be hydrolyzed by TG2 resulting in conversion to glutamyl residues, which would then be resistant to further transamidation with the addition of primary amines. Indeed, pretreatment of rhGlxI with the activated TG2 significantly decreased the extent of bPA incorporation at the TG2 doses of 3–6 mU (Fig. 2D). Alternatively, the TG2-treated rhGlxI either denatured in SDS (right panel) or not (left panel) was subjected to proteolysis by the endoproteinase Glu-C which is specific for glutamate and aspartate residues, but not for glutamine residues. As a result, extra peptide bands were resolved by SDS-PAGE from which two amino-termini were determined as the following sequence C-terminal to Gln9 (designated as M10 in the left panel) and to Gln54 (designated as T55 in the right panel) (Fig. 2E). The results suggest that TG2 tends to catalyze amine incorporation into GlxI or deamidation, depending on the presence or absence of primary amines, instead of protein cross-linking of GlxI.
Fig. 2.
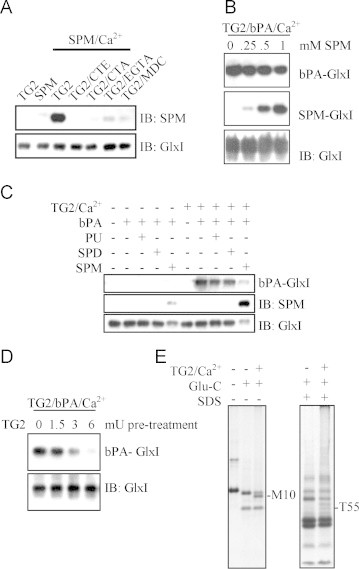
Polyamination and deamidation of GlxI catalyzed by TG2. (A) Spermine (SPM) was applied to the in vitro transamidation reaction, in the presence or absence of TG2 inhibitors, for determination of GlxI polyamination. The products were analyzed by immunoblotting with an antiserum against spermine. Lane 1: TG2 only; lane 2: spermine only; lane 3–7: TG2, spermine and various reagents. (B) Spermine competed with bPA for incorporation into rhGlxI catalyzed by TG2. (C) Polyamines competed with bPA for incorporation into rhGlxI catalyzed by TG2. (D) Deamidation of rhGlxI catalyzed by TG2 in the absence of primary amine. In the absence of primary amine, rhGlxI was incubated with activated TG2 and the level of deamidation was evaluated by the decrease in incorporation of bPA after further catalysis of TG2 in the presence of bPA. (E) Deamidation of rhGlxI catalyzed by TG2 in the absence of primary amines. The newly generated glutamic acid residues catalyzed by TG2 generated new Glu-C cleavage sites. TG2: transglutaminase 2, CTE: cysteamine, CTA: cystamine, MDC: monodansylcadaverine, PU: putrescine, SPD: spermidine.
Polyamine binding, conjugation and deamidation modulating GlxI activity and stability
We next examined whether polyamine binding, conjugation and deamidation of GlxI altered the protein stability or affected enzyme activity. After treatment with one polyamine or one polyamine in combination with TG2 for 30 min, the treated rhGlxI was immediately assayed for its enzyme activity or assayed after exposure to 25 mM sodium hydroxide treatment for 30 min. Because a small portion of the enzyme solution was used in the final assay, sodium hydroxide did not change the pH value of the final assay solution. Without TG2 catalyzed reaction, the rhGlxI activity slightly increased in the presence of putrescine and spermine. After preincubation with any one of the polyamines, rhGlxI became resistant to the sodium hydroxide treatment (Fig. 3A). In contrast, control rhGlxI was prone to denaturation upon the alkaline treatment rendering it less active later in the assay. After TG2 catalysis, the activity of rhGlxI either polyamine conjugated or deamidated increased by more than 80% as compared to that of untreated rhGlxI (Fig. 3B, left panel). No significant difference between polyamine-conjugated and deamidated rhGlxI in the enzyme activities was found. Polyamine-conjugated rhGlxI appeared to be resistant to the sodium hydroxide treatment with only a slight decrease in enzyme activity (Fig. 3B, right panel). Unexpectedly, deamidated rhGlxI lost its activity to a similar extent as compared to that of untreated rhGlxI after the sodium hydroxide treatment. Although noncovalent binding of spermidine did not stimulate rhGlxI, the enzyme activity of rhGlxI increased in the presence of spermidine and TG2 reaching to about 2 folds of that of untreated rhGlxI (Fig. 3C). Overall, the results indicate that GlxI activity is upregulated by TG2-catalyzed polyamine conjugation and deamidation. In addition, TG2-catalyzed polyamine conjugation causes conformational change of TG2 rendering it more resistant to alkaline denaturation.
Fig. 3.
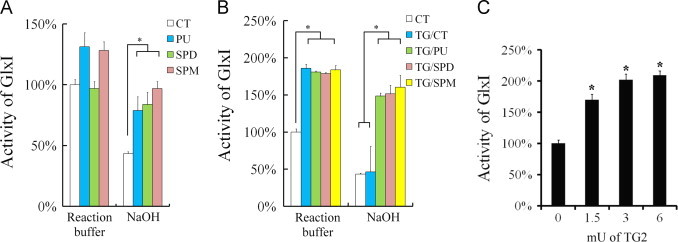
Modulation of GlxI activity by polyamine binding and TG2-catalyzed modifications. (A) Effects of polyamines binding on GlxI activity and stability. GlxI was incubated with one of the polyamines; putrescine (PU), spermidine or spermine, or with an equal volume of TG2 reaction buffer (CT). Two sets of such samples were prepared and one of them was then mixed with 25 mM NaOH solution (NaOH) and another with an equal volume of TG2 reaction buffer. After 30 min incubation, 20 μl of the sample was taken into 180 μl of the assay solution for GlxI activity. The activities of all treated samples were compared with that of CT in the reaction buffer. (B) Effects of TG2 catalyzed modifications on GlxI activity and stability. The duplicate samples in Fig. 4A were incubated in the presence of 150 mU/ml TG2. Two sets of such samples were prepared and one of them was then mixed with 25 mM NaOH solution (NaOH) and another with an equal volume of TG2 reaction buffer. The activities of all treated samples were compared with that of CT sample in TG2 reaction buffer. (C) Effects of TG2-catalyzed spermidine incorporation on the activity of GlxI. The activity of GlxI was measured after GlxI was treated with TG2 in the presence of spermidine as compared with that of buffer-treated control. Results were presented as mean of three independent experiments plus standard error. Results significantly different from control at p<0.05 are indicated by ⁎.
Increases in the levels of ROS and calcium concentration in HeLa cells in response to methylglyoxal treatment
We next determined intracellular [Ca2+] in HeLa cells following methylglyoxal treatment since calcium is required for the activation of TG2. As a result, intracellular [Ca2+] significantly increased in methylglyoxal-treated HeLa cells. DENSPM was used to deplete intracellular polyamines by activating spermidine/spermine acetyl transferase (SSAT) which would promote the catabolism of endogenous spermidine and spermine. The presence of cystamine or DENSPM did not affect the rise in intracellular calcium induced by methylglyoxal, but slightly decrease the basal levels of calcium in HeLa cells (Fig. 4A). Intracellular ROS level in all methylglyoxal-treated HeLa cells was higher than that in the control cells. Treatment of both methylglyoxal and cystamine even induced more ROS in HeLa cells. On the other hand, treatment of cystamine or DENAPM alone decreased the level of ROS in HeLa cells slightly (Fig. 4B). The results indicate that following methylglyoxal treatment, levels of intracellular ROS and [Ca2+] are elevated.
Fig. 4.
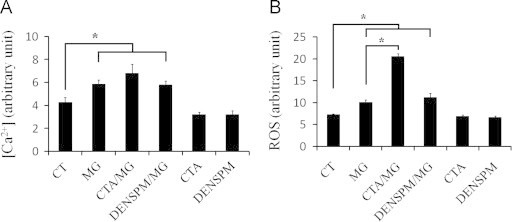
Methylglyoxal treatment caused the increases in intracellular [Ca2+] and ROS. HeLa cells were incubated in media containing 2 mM methylglyoxal in the absence or presence of 1 mM cystamine or 0.1 mM DENSPM for 2 h. (A) Intracellular calcium level was determined with Fura-2 by flow cytometry. (B) Intracellular ROS was determined with DCFH-DA fluorescence by flow cytometry. Results represent an average of three independent experiments+S.E. Results significantly different from control at p<0.05 are indicated by ⁎.
Methylglyoxal treatment activating endogenous TG2 in HeLa cells
Since methylglyoxal is capable of inducing the increase of intracellular [Ca2+], TG2 may be activated when cells are challenged by exposure to methylglyoxal. To confirm this notion, in situ transamidation assay was performed by incubating the cells with 1 mM bPA in the media. As shown by the extent of bPA incorporation into the endogenous protein substrates, the endogenous TG2 activity was evaluated after methylglyoxal treatment for 1 h and 2 h. The results showed that endogenous TG2 was activated as revealed by the incorporation of bPA into cellular proteins after methylglyoxal treatment. The activity TG2 was increased up to 2 folds after 2 mM methylglyoxal treatment for 2 h as compared to that of the untreated cells. In addition, further activation of TG2 was observed at 3, 4, and 5 mM methylglyoxal for either 1 h or 2 h treatment, with activity increasing up to 4 to 12 folds (Fig. 5A). Then, HeLa cells were treated with 2 mM methylglyoxal in the presence TG2 inhibitors. The TG2 activity induced by methylglyoxal treatment was reduced by at least 50% and 85% with cystamine or by 18% and 60% with MDC as compared to the methylglyoxal treated cells at 1 h and at 2 h, respectively (Fig. 5B). In the presence of DENSPM, the TG2 activity remained elevated at about 2.5 folds as compared to the untreated cells at 1 h and 2 h. The increases in TG2 activity did not result from the increases in TG2 protein levels (Fig. 5A). In addition, the protein level of GlxI did not change significantly following methylglyoxal treatment. The data indicate that cystamine and MDC work well in HeLa cells as TG2 inhibitor but not DENSPM probably because bPA is used in the assay as the amine donor. Depletion of endogenous spermidine and spermine would favor the use of bPA as the amine donor in the amine incorporation reaction. The data indicate that HeLa cells respond to the methylglyoxal treatment with TG2 activated in a dose-dependent manner.
Fig. 5.
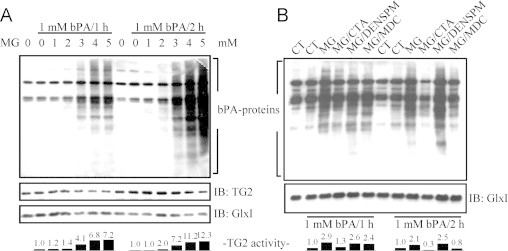
Increases in TG2 activity following methylglyoxal treatment in HeLa cells. HeLa cells were incubated in media containing 1 mM bPA and (A) various concentrations of methylglyoxal or (B) 2 mM methylglyoxal in the presence of 1 mM cystamine, 0.1 mM DENSPM, or 0.1 mM MDC, and then the cell lysate was subjected to the biotin overlay assay.
Transamidation and activation of GlxI in HeLa cells treated with methylglyoxal
To find out whether transamidation of GlxI occurs in HeLa cells, Flag-rhGlxI was overexpressed in HeLa cells and analyzed for transamidation by in situ bPA incorporation. Both Flag-rhGlxI and endogenous GlxI were resolved and appeared to be transamidated in methylglyoxal-treated HeLa cells (Fig. 6A) since they were both tagged with biotin. In addition, the dimer of Flag-rhGlxI and GlxI was formed in HeLa cells as indicated by the presence of GlxI in the anti-Flag immunoprecipitates. S-d-lactoylglutathione is the product of methylglyoxal and glutathione catalyzed by GlxI. The cell lysate from methylglyoxal-treated cells was used to evaluate the enzyme activity of GlxI by measuring the turnover of methylglyoxal into the nontoxic S-d-lactoylglutathione in the assay. With various concentrations of methylglyoxal treatment, GlxI enzyme activity reached 150% compared to the untreated cells with the increase of methylglyoxal concentration from 1 to 5 mM (Fig. 6B). When cells were treated with cystamine and methylglyoxal together, the turnover rate of S-d-lactolyglutathione dropped at least by 50% as compared to that treated with methylglyoxal alone. However, DENSPM only slightly suppressed the methylglyoxal-induced increases in GlxI activity (Fig. 6C). The data indicate that methylglyoxal treatment causes transamidation of GlxI catalyzed by TG2 resulting in activation of GlxI.
Fig. 6.
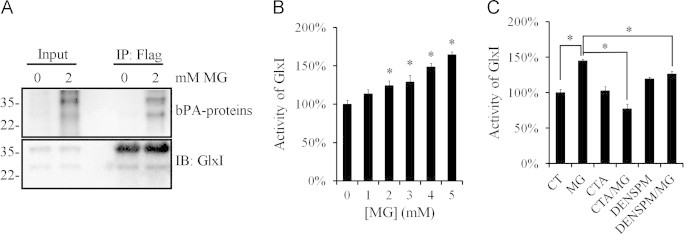
Methylglyoxal treatment inducing transamidation and activation of GlxI in HeLa cells. (A) HeLa cells expressing Flag-tagged GlxI (Flag-rhGlxI) were incubated in media containing 1 mM bPA in the presence or absence of 2 mM methylglyoxal for 2 h. Flag-rhGlxI was immunoprecipitated with anti-Flag-agarose and analyzed by the biotin overlay assay. HeLa cells were incubated with (B) various concentrations of methylglyoxal or (C) 2 mM methylglyoxal in the presence of 1 mM cystamine or 0.1 mM DENSPM for 2 h, and then GlxI activity in the lysate was monitored by measuring the rate of S-d-lactoylgutathione formation. Results represent an average of three independent experiments +S.E. Results significantly different from control at p<0.05 are indicated by ⁎.
The roles of TG2 and polyamines in HeLa cells in response to oxidative stress
Suppression of TG2 activity leads to decreases in the turnover of intracellular methylglyoxal which may result in ROS accumulation. Therefore, we wish to learn whether TG2 activation is required for HeLa cells to resist oxidative damage induced by methylglyoxal treatment. HeLa cells were treated with methylglyoxal accompanying TG2 inhibition or polyamine depletion and then the cell viability was evaluated by the MTT assay. HeLa cells were treated with methylglyoxal at various concentrations for 24 h in DMEM medium containing 10% FBS, and the cell death occurred in a biphasic manner as a function of methylglyoxal concentration. Methylglyoxal treatment caused maximal cell death at 2 mM, but viable cells unexpectedly increased under the treatment of methylglyoxal at 3–5 mM as compared to the treatment of methylglyoxal at 1–2 mM. According to the results, cystamine alone did not induce significant cell death up to the highest concentration tested, 1 mM. In combination with cystamine treatment, the same methylgloxal treatment caused more cell death in a dose-dependent manner, and the unexpected increase in cell viability at higher concentrations of methylglyoxal treatment was suppressed by the presence of 0.2, 0.5 and 1 mM cystamine (Fig. 7A). Thus, HeLa cells treated with cystamine appears more vulnerable to methylglyoxal treatment, and the biphasic response to methylglyoxal is suppressed by the cystamine treatment. However, one should be cautious that cystamine is not a specific inhibitor of TG2 [34], [35]. The data suggest that treatment of higher concentrations of methylglyoxal causes activation of TG2 rendering cells more resistant to methylglyoxal treatment. Similarly, HeLa cells treated with DENSPM appeared more vulnerable to methylglyoxal treatment. However, the biphasic response to methylglyoxal was only slightly suppressed by DENSPM treatment (Fig. 7B). In comparison, we also tested the role of TG2 in HeLa cells in response to staurosporine, an inducer of cell apoptosis. Cystamine at 1 mM enhanced the effects of staurosporine in inducing cell death when staurosporine concentration was higher than 30 μM (Fig. 7C). By contrast, depletion of polyamines in staurosporine-treated cells caused no extra effect on cell viability (Fig. 7D). It appears that the role of TG2 in HeLa cell resistance to staurosporine treatment is less important than to methylglyoxal treatment. The data indicate that TG2 activation is required for HeLa cells to resist damage induced by methylglyoxal treatment. In addition, the effect of GlxI is only partially mediated by polyamine conjugation.
Fig. 7.
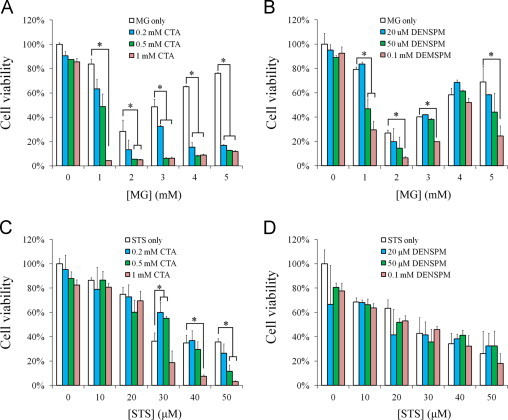
Resistance of cultured cells to methylglyoxal treatment or staurosporin (STS) under TG2 inhibition or polyamine depletion. HeLa cells were treated with various concentrations of methylglyoxal (A, B) or STS (C, D) in the presence of varying concentrations of cystamine or DANSPM for 24 h, and the cell viability was evaluated by MTT assay. Results represent an average of three independent experiments +S.E. Results significantly different from control at p<0.05 are indicated by ⁎.
Expression of TG2 and GlxI are critical for the tolerance against methylglyoxal assaults
For specific characterization of TG2 in response to methylglyoxal, siRNA was used to suppress the expression of TG2. TG2-knockdown cells were exposed to methylglyoxal treatment for 12 h in serum-free DMEM medium. In HeLa cells, 90% of cells survived after 2 mM methylglyoxal treatment as compared with that of the control cells, and only 70% of TG2-knockdown cells survived from 2 mM methylglyoxal treatment. Along with the knockdown of GlxI, 65% of cells survived after 2 mM methylglyoxal treatment as compared with that of the control cells (Fig. 8A). Moreover, 65% of HepG2 cells survived from 2 mM methylglyoxal treatment. Along with the knockdown of TG2, 50% of HepG2 cells survived after 2 mM methylglyoxal treatment as compared with that of the control cells. Meanwhile, along with the knockdown of GlxI, the cell viability was only 35% as compared with that of the control cells after 2 mM methylglyoxal treatment (Fig. 8B). Therefore, expression of both GlxI and TG2 are beneficial for cell resistance to the methylglyoxal insults.
Fig. 8.
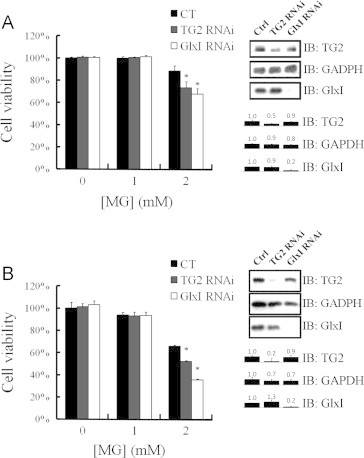
Resistance of cultured cells to methylglyoxal treatment under TG2 or GlxI gene knockdown. TG2- or GlxI-knocked down (A) HeLa cells or (B) HepG2 cells were treated with various concentrations of methylglyoxal and the cell viability was evaluated by MTT assay. Results represent an average of three independent experiments +S.E. Results significantly different from control at p<0.05 are indicated by ⁎.
Next, we measured the expression levels of TG2 and GlxI in three different cells and also their tolerance against MG assaults. The protein expression levels of TG2, GAPDH, and GlxI were evaluated in a fixed amount of cellular protein. HeLa cells expressed the highest level of all three enzymes and 3T3 cells expressed the lowest level of these enzymes. However, COS-1 cells expressed a similar level of TG2 as compared to that of HeLa cells (Fig. 9A). 3T3 cells appeared to be the most vulnerable, COS-1 next and HeLa cells the least, to the methylglyoxal treatment (Fig. 9B). At last, cellular levels of methylglyoxal were determined after 2 h treatment of methylglyoxal in three cells. Under normal culture conditions, intracellular levels of methylglyoxal were between 14 and 25 μM. Exposure to methylglyoxal resulted in dramatic increases in intracellular levels of methylglyoxal to 42, 118, and 116 μM in response to 200, 500, and 1000 μM methylglyoxal exposure, respectively, in 3T3 cells. However, no significant change of intracellular methlglyoxal levels was observed in COS-1 cells, and methylglyoxal levels in HeLa cells were slightly elevated up to 24, 28, and 35 μM in response to 200, 500, and 1000 μM methylglyoxal exposure, respectively (Fig. 9C). The results indicate that the expression levels of both GlxI and TG2 are highly correlated with cell resistance to the methylglyoxal insults. Altogether, the data suggest that GlxI is required for the disposal of excess methylglyoxal and shortage of TG2 activity significantly impairs the process.
Fig. 9.
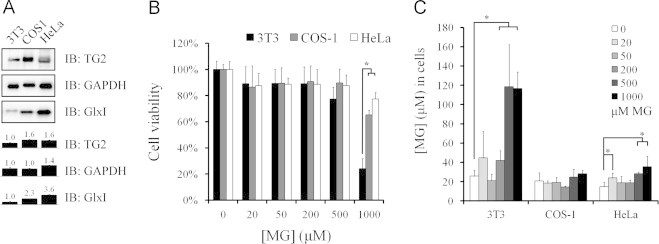
Protein expression levels of GlxI and TG2 in cells and cell resistance to methylglyoxal challenge. (A) The protein expression of TG2, GAPDH, and GlxI were evaluated by immunoblotting in a fixed amount of protein applied for SDS-PAGE. (B) Cell viability was measured in cells treated with various concentrations of methylglyoxal for 24 h. (C) Intracellular concentrations of methylglyoxal were determined in cells treated with various concentrations of methylglyoxal for 2 h. Results represent an average of three independent experiments+S.E. Results significantly different from control at p<0.05 are indicated by ⁎.
Discussion
In this communication, we have provided evidence indicating GlxI is a novel substrate of TG2 whose action elicits Gln deamidation or polyamine conjugation to GlxI. Nonenzymatic deamidation of Asn and Gln residues is a well-known artifact generated by analytical steps especially under mildly alkaline pH and prolonged incubations at 37 °C [36]. Deamidation of Asn at neutral pH usually proceeds through a cyclic imide reaction mechanism [37] whereas deamidation of Gln is thermodynamically less favorable in forming a six-member imide intermediate [38]. Besides elaborated analysis using mass spectrometry, Gln deamidation can be examined by differential cleavage by Glu-C protease specific for Glu residues [29]. Here we have demonstrated a novel method, called TG2 tandem reaction, for detecting Gln deamidation catalyzed by TG2. In this practice, TG2 substrates are treated with TG2 twice, first for deamidation and then for primary amine incorporation. Decrease in the levels of primary amine incorporation reveals the occurrence of prior Gln deamidation. In this communication, GlxI deamidation catalyzed by TG2 has been shown by both the Glu-C differential cleavage and TG2 tandem reaction methods. Our results suggest that TG2 tends to catalyze amine incorporation into GlxI or deamidation, depending on the presence or absence of primary amines, instead of protein cross-linking of GlxI. In addition, both deamidation and polyamination greatly enhance the enzyme activity of GlxI and polyamination renders GlxI more resistant to alkaline denaturation. The identified modification site Q54 of the recombinant GlxI corresponds to Q33 of the active site formed by Q33, E99, H126, E172 and zinc ion in native enzyme [39]. Q33E mutant was demonstrated to lose zinc binding ability [39]. Our data are not consistent with the previous results. We speculate that Q33 deamidation or transamidation would not be the only modification caused by TG2. Deamidation or transamidation on sites other than Q33 may increase the stability of GlxI dimer on the facts that transamidation with spermine provides opportunities of hydrogen bonding, salt bridges and hydrophobic interaction and glutamic acid side chain provides salt bridges and hydrogen bonding, rather than directly engage in catalysis.
TG2 up-regulation has been observed in cells under oxidative stress [25], [26], [40], [41]. However, its roles in cell response to oxidative stress remain elusive, depending on the cell type and the kind of stresses encountered [42], [43]. It has been demonstrated that the transamidation activity of TG2 is required for the survival of cultured cells in response to various stresses [44], [45], [46]. However, only limited transamidated substrates of TG2 have been shown to underscore the anti-apoptotic function of TG2; crosslinking of caspase 3 leading to inhibition [47] and crosslinking of cathepsin d leading to depletion [48]. In this report, we have added one more TG2 substrate, GlxI, to this short list whose transamidation results in stabilization and activation of the enzyme in cells in response to high concentrations of methylglyoxal. In addition, activation of GlxI renders cells more resistant to methylglyoxal insult.
Among many altered phenotypes, sustained aerobic glycolysis (also known as the Warburg effect) is notable for providing important advantages in tumor growth and survival. Aerobic glycolysis is preferred over oxidative phosphorylation also in rapidly proliferating cells such as embryonic stem cells and lymphocytes. In addition to generating ATP, increases in glycolytic flux also provides precursors used for the synthesis of nucleic acids, proteins, and lipids [49]. However, maintaining high levels of glycolytic intermediates may inevitably lead to production and accumulation of methylglyoxal. It is conceivable that many tumor cells overexpress GlxI to dispose the hazardous byproduct of aerobic glycolysis [50]. In fact, GlxI gene is amplified in many primary tumors and cancer cell lines [51] and enzymes involved in cellular redox balance including GlxI are induced by oncogenic transformation [52]. Interestingly, expression of TG2 is also elevated in many types of cancer cells resulting in increased cell invasiveness and cell survival [53], [54], [55]. In particular, gene amplification of GlxI has been observed in high percentage in breast cancer [51] and overexpression of TG2 in breast cancer is associated with higher drug resistance and metastatic potential [56], [57], [58]. Our results indicate that the expression levels of both GlxI and TG2 in cells are correlated with cell resistance to the methylglyoxal insults, GlxI is required for the disposal of excess methyglyoxal and shortage of TG2 activity significantly impairs the process. We speculate that overexpression of both TG2 and GlxI confers tumor cells greater advantage in cell survival, drug resistance and metastasis since transamidation of GlxI stabilizes and activates the enzyme. Thus, tumor cells overexpressing both TG2 and GlxI can endure higher glycolytic flux and may benefit from aerobic glycolysis without accumulation of methylglyoxal.
Summary
In this article, we have described that (1) GlxI is a novel substrate of TG2 and TG2 catalyzes either polyamine conjugation or deamidation depending the presence of polyamines, (2) deamidation or polyamine conjugation leads to activation of GlxI or stabilization and activation of GlxI, respectively, (3) methylglyoxal challenge causes increases in intracellular levels of ROS and calcium leading to TG2 activation and transamidation and activation of GlxI in HeLa cells, (4) both TG2 and GlxI are involved in cells resistance to methylglyoxal challenge. The data suggest that exposure to methylglyoxal elicits a negative feedback loop entailing ROS, calcium, TG2 and GlxI leading to decrease in the levels of methylglyoxal.
Acknowledgments
This research was supported by grants NTU-CESRP-102R7602B1 and NTU-CESRP-101R7602B1 from the National Taiwan University. Metabolomic mass spectrometry analyses were performed at the Technology Commons in the College of Life Science, National Taiwan University.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Griffin M., Casadio R., Bergamini C.M. Transglutaminases: nature's biological glues. Biochem. J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorand L., Graham R.M. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nature reviews. Mol. Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 3.Beninati S., Piacentini M. The transglutaminase family: an overview: minireview article. Amino Acids. 2004;26:367–372. doi: 10.1007/s00726-004-0091-7. [DOI] [PubMed] [Google Scholar]
- 4.Folk J.E., Park M.H., Chung S.I., Schrode J., Lester E.P., Cooper H.L. Polyamines as physiological substrates for transglutaminases. J. Biol. Chem. 1980;255:3695–3700. [PubMed] [Google Scholar]
- 5.Molberg O., McAdam S.N., Korner R., Quarsten H., Kristiansen C., Madsen L., Fugger L., Scott H., Noren O., Roepstorff P., Lundin K.E., Sjostrom H., Sollid L.M. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 6.van de Wal Y., Kooy Y., van Veelen P., Pena S., Mearin L., Papadopoulos G., Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J. Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- 7.Nakaoka H., Perez D.M., Baek K.J., Das T., Husain A., Misono K., Im M.J., Graham R.M. Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 8.Achyuthan K.E., Greenberg C.S. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J. Biol. Chem. 1987;262:1901–1906. [PubMed] [Google Scholar]
- 9.Hasegawa G., Suwa M., Ichikawa Y., Ohtsuka T., Kumagai S., Kikuchi M., Sato Y., Saito Y. A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem. J. 2003;373:793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra S., Murphy L.J. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 2004;279:23863–23868. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- 11.Mishra S., Saleh A., Espino P.S., Davie J.R., Murphy L.J. Phosphorylation of histones by tissue transglutaminase. J. Biol. Chem. 2006;281:5532–5538. doi: 10.1074/jbc.M506864200. [DOI] [PubMed] [Google Scholar]
- 12.Akimov S.S., Krylov D., Fleischman L.F., Belkin A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentile V., Porta R., Chiosi E., Spina A., Valente F., Pezone R., Davies P.J., Alaadik A., Illiano G. tTGase/G alpha h protein expression inhibits adenylate cyclase activity in Balb-C 3T3 fibroblasts membranes. Biochim. Biophys. Acta. 1997;1357:115–122. doi: 10.1016/s0167-4889(97)00024-4. [DOI] [PubMed] [Google Scholar]
- 14.Kumar S., Mehta K. Tissue transglutaminase constitutively activates HIF-1alpha promoter and nuclear factor-kappaB via a non-canonical pathway. PloS one. 2012;7:e49321. doi: 10.1371/journal.pone.0049321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucholski J., Johnson G.V. Tissue transglutaminase directly regulates adenylyl cyclase resulting in enhanced cAMP-response element-binding protein (CREB) activation. J. Biol. Chem. 2003;278:26838–26843. doi: 10.1074/jbc.M303683200. [DOI] [PubMed] [Google Scholar]
- 16.Phillips S.A., Thornalley P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. (FEBS) 1993;212:101–105. doi: 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- 17.Thornalley P.J., Battah S., Ahmed N., Karachalias N., Agalou S., Babaei-Jadidi R., Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abordo E.A., Minhas H.S., Thornalley P.J. Accumulation of alpha-oxoaldehydes during oxidative stress: a role in cytotoxicity. Biochem. Pharmacol. 1999;58:641–648. doi: 10.1016/s0006-2952(99)00132-x. [DOI] [PubMed] [Google Scholar]
- 19.Thornalley P.J., Edwards L.G., Kang Y., Wyatt C., Davies N., Ladan M.J., Double J. Antitumour activity of S-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996;51:1365–1372. doi: 10.1016/0006-2952(96)00059-7. [DOI] [PubMed] [Google Scholar]
- 20.Biswas S., Ray M., Misra S., Dutta D.P., Ray S. Selective inhibition of mitochondrial respiration and glycolysis in human leukaemic leucocytes by methylglyoxal. Biochem. J. 1997;323(Pt 2):343–348. doi: 10.1042/bj3230343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang T., Wang R., Wu L. Methylglyoxal-induced nitric oxide and peroxynitrite production in vascular smooth muscle cells. Free Radic. Biol. Med. 2005;38:286–293. doi: 10.1016/j.freeradbiomed.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 22.Rosca M.G., Mustata T.G., Kinter M.T., Ozdemir A.M., Kern T.S., Szweda L.I., Brownlee M., Monnier V.M., Weiss M.F. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. American journal of physiology. Renal Physiol. 2005;289:F420–F430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 23.Schlotterer A., Kukudov G., Bozorgmehr F., Hutter H., Du X., Oikonomou D., Ibrahim Y., Pfisterer F., Rabbani N., Thornalley P., Sayed A., Fleming T., Humpert P., Schwenger V., Zeier M., Hamann A., Stern D., Brownlee M., Bierhaus A., Nawroth P., Morcos M. C. elegans as model for the study of high glucose-mediated life span reduction. Diabetes. 2009;58:2450–2456. doi: 10.2337/db09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brouwers O., Niessen P.M., Ferreira I., Miyata T., Scheffer P.G., Teerlink T., Schrauwen P., Brownlee M., Stehouwer C.D., Schalkwijk C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011;286:1374–1380. doi: 10.1074/jbc.M110.144097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Z.W., Kwon S.M., Kim S.W., Yi S.J., Kim Y.M., Ha K.S. Activation of in situ tissue transglutaminase by intracellular reactive oxygen species. Biochem. Biophys. Res. Commun. 2003;305:633–640. doi: 10.1016/s0006-291x(03)00835-0. [DOI] [PubMed] [Google Scholar]
- 26.Yi S.J., Choi H.J., Yoo J.O., Yuk J.S., Jung H.I., Lee S.H., Han J.A., Kim Y.M., Ha K.S. Arachidonic acid activates tissue transglutaminase and stress fiber formation via intracellular reactive oxygen species. Biochem. Biophys. Res. Commun. 2004;325:819–826. doi: 10.1016/j.bbrc.2004.10.122. [DOI] [PubMed] [Google Scholar]
- 27.Ridderstrom M., Mannervik B. Optimized heterologous expression of the human zinc enzyme glyoxalase I. Biochem. J. 1996;314(Pt 2):463–467. doi: 10.1042/bj3140463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee D.Y., Chang G.D. Electrolytic reduction: modification of proteins occurring in isoelectric focusing electrophoresis and in electrolytic reactions in the presence of high salts. Anal. Chem. 2009;81:3957–3964. doi: 10.1021/ac900281n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae N., Yang J.W., Sitte H., Pollak A., Marquez J., Lubec G. An electrophoretic approach to screen for glutamine deamidation. Anal. Biochem. 2012;428:1–3. doi: 10.1016/j.ab.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 30.Kellum M.W., Oray B., Norton S.J. A convenient quantitative synthesis of methylglyoxal for glyoxalase I assays. Anal. Biochem. 1978;85:586–590. doi: 10.1016/0003-2697(78)90258-0. [DOI] [PubMed] [Google Scholar]
- 31.Mittelmaier S., Funfrocken M., Fenn D., Berlich R., Pischetsrieder M. Quantification of the six major alpha-dicarbonyl contaminants in peritoneal dialysis fluids by UHPLC/DAD/MSMS. Anal. Bioanal. Chem. 2011;401:1183–1193. doi: 10.1007/s00216-011-5195-9. [DOI] [PubMed] [Google Scholar]
- 32.Amarzguioui M. Improved siRNA-mediated silencing in refractory adherent cell lines by detachment and transfection in suspension. BioTechniques. 2004;36(766–768):770. doi: 10.2144/04365BM03. [DOI] [PubMed] [Google Scholar]
- 33.Du Y., Wang F., May K., Xu W., Liu H. Determination of deamidation artifacts introduced by sample preparation using 18O-labeling and tandem mass spectrometry analysis. Anal. Chem. 2012;84:6355–6360. doi: 10.1021/ac3013362. [DOI] [PubMed] [Google Scholar]
- 34.Lesort M., Lee M., Tucholski J., Johnson G.V. Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J. Biol. Chem. 2003;278:3825–3830. doi: 10.1074/jbc.M205812200. [DOI] [PubMed] [Google Scholar]
- 35.Ientile R., Campisi A., Raciti G., Caccamo D., Currò M., Cannavò G., Li Volti G., Macaione S., Vanella A. Cystamine inhibits transglutaminase and caspase-3 cleavage in glutamate-exposed astroglial cells. J. Neurosci. Res. 2003;74:52–59. doi: 10.1002/jnr.10702. [DOI] [PubMed] [Google Scholar]
- 36.Hao P., Ren Y., Alpert A.J., Sze S.K. Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol. Cell. Proteomics: (MCP) 2011;10(O111):009381. doi: 10.1074/mcp.O111.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meinwald Y.C., Stimson E.R., Scheraga H.A. Deamidation of the asparaginyl-glycyl sequence. Int. J. Pept. Protein Res. 1986;28:79–84. doi: 10.1111/j.1399-3011.1986.tb03231.x. [DOI] [PubMed] [Google Scholar]
- 38.Robinson N.E., Robinson A.B. Deamidation of human proteins. Proc. Natl. Acad. Sci. USA. 2001;98:12409–12413. doi: 10.1073/pnas.221463198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ridderstrom M., Cameron A.D., Jones T.A., Mannervik B. Involvement of an active-site Zn2+ ligand in the catalytic mechanism of human glyoxalase I. J. Biol. Chem. 1998;273:21623–21628. doi: 10.1074/jbc.273.34.21623. [DOI] [PubMed] [Google Scholar]
- 40.Groenen P.J., Seccia M., Smulders R.H., Gravela E., Cheeseman K.H., Bloemendal H., de Jong W.W. Exposure of beta H-crystallin to hydroxyl radicals enhances the transglutaminase-susceptibility of its existing amine-donor and amine-acceptor sites. Biochem. J. 1993;295(Pt 2):399–404. doi: 10.1042/bj2950399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin D.M., Jeon J.H., Kim C.W., Cho S.Y., Kwon J.C., Lee H.J., Choi K.H., Park S.C., Kim I.G. Cell type-specific activation of intracellular transglutaminase 2 by oxidative stress or ultraviolet irradiation: implications of transglutaminase 2 in age-related cataractogenesis. J. Biol. Chem. 2004;279:15032–15039. doi: 10.1074/jbc.M308734200. [DOI] [PubMed] [Google Scholar]
- 42.Caccamo D., Curro M., Ferlazzo N., Condello S., Ientile R. Monitoring of transglutaminase 2 under different oxidative stress conditions. Amino Acids. 2012;42:1037–1043. doi: 10.1007/s00726-011-1018-8. [DOI] [PubMed] [Google Scholar]
- 43.Fésüs L., Szondy Z. Transglutaminase 2 in the balance of cell death and survival. FEBS Lett. 2005;579:3297–3302. doi: 10.1016/j.febslet.2005.03.063. [DOI] [PubMed] [Google Scholar]
- 44.Antonyak M.A., Miller A.M., Jansen J.M., Boehm J.E., Balkman C.E., Wakshlag J.J., Page R.L., Cerione R.A. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J. Biol. Chem. 2004;279:41461–41467. doi: 10.1074/jbc.M404976200. [DOI] [PubMed] [Google Scholar]
- 45.Datta S., Antonyak M.A., Cerione R.A. Importance of Ca(2+)-dependent transamidation activity in the protection afforded by tissue transglutaminase against doxorubicin-induced apoptosis. Biochemistry. 2006;45:13163–13174. doi: 10.1021/bi0606795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan L., Choi K., Khosla C., Zheng X., Higashikubo R., Chicoine M.R., Rich K.M. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol. Cancer Ther. 2005;4:1293–1302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi H., Wang H.G. Tissue transglutaminase serves as an inhibitor of apoptosis by cross-linking caspase 3 in thapsigargin-treated cells. Mol. Cell. Biol. 2006;26:569–579. doi: 10.1128/MCB.26.2.569-579.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S.J., Kim K.H., Ahn E.R., Yoo B.C., Kim S.Y. Depletion of cathepsin D by transglutaminase 2 through protein cross-linking promotes cell survival. Amino Acids. 2013;44:73–80. doi: 10.1007/s00726-011-1089-6. [DOI] [PubMed] [Google Scholar]
- 49.Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 50.Thornalley P.J., Rabbani N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011;22:318–325. doi: 10.1016/j.semcdb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 51.Santarius T., Bignell G.R., Greenman C.D., Widaa S., Chen L., Mahoney C.L., Butler A., Edkins S., Waris S., Thornalley P.J., Futreal P.A., Stratton M.R. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer. 2010;49:711–725. doi: 10.1002/gcc.20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young T.W., Mei F.C., Yang G., Thompson-Lanza J.A., Liu J., Cheng X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004;64:4577–4584. doi: 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- 53.Lin C.Y., Tsai P.H., Kandaswami C.C., Chang G.D., Cheng C.H., Huang C.J., Lee P.P., Hwang J.J., Lee M.T. Role of tissue transglutaminase 2 in the acquisition of a mesenchymal-like phenotype in highly invasive A431 tumor cells. Mol. Cancer. 2011;10:87. doi: 10.1186/1476-4598-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mehta K., Kumar A., Kim H.I. Transglutaminase 2: a multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem. Pharmacol. 2010;80:1921–1929. doi: 10.1016/j.bcp.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 55.Shao M., Cao L., Shen C., Satpathy M., Chelladurai B., Bigsby R.M., Nakshatri H., Matei D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201. doi: 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- 56.Mehta K. High levels of transglutaminase expression in doxorubicin-resistant human breast carcinoma cells. International journal of cancer. J. Int. Cancer. 1994;58:400–406. doi: 10.1002/ijc.2910580316. [DOI] [PubMed] [Google Scholar]
- 57.Herman J.F., Mangala L.S., Mehta K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene. 2006;25:3049–3058. doi: 10.1038/sj.onc.1209324. [DOI] [PubMed] [Google Scholar]
- 58.Singer C.F., Hudelist G., Walter I., Rueckliniger E., Czerwenka K., Kubista E., Huber A.V. Tissue array-based expression of transglutaminase-2 in human breast and ovarian cancer. Clin. Exp. Metastasis. 2006;23:33–39. doi: 10.1007/s10585-006-9015-0. [DOI] [PubMed] [Google Scholar]