

Abstract
Iron is essential for most living organisms but iron excess can be toxic. Cellular and systemic iron balance is therefore tightly controlled. Iron homeostasis is dysregulated in chronic kidney disease (CKD) and contributes to the anemia that is prevalent in this patient population. Iron supplementation is one cornerstone of anemia management in CKD patients, but has not been rigorously studied in large prospective randomized controlled trials. This review highlights important advances from genetic studies and animal models that have provided key insights into the molecular mechanisms governing iron homeostasis and its disturbance in CKD, and summarizes how these findings may yield advances in the care of this patient population.
Keywords: anemia, chronic kidney disease, hepcidin, iron, review
SYSTEMIC IRON BALANCE
As a transition metal that can donate and accept electrons, iron has a critical role in fundamental biological processes including oxygen and electron transport, cellular respiration and DNA synthesis. However, excess iron can lead to the production of toxic free radicals and cell death. Disturbances of iron homeostasis lead to many common diseases such as anemia and the iron overload disorder hemochromatosis that in aggregate affect over 1 billion people worldwide [1]. Iron is therefore tightly controlled via a network of proteins involved in the import, storage, export and transport of iron, at both cellular and systemic levels.
Humans have a daily requirement of ∼25 mg of iron, nearly 80% of which is used for erythropoiesis [1]. A small fraction of this iron is provided by dietary absorption (∼1–2 mg), while the majority is provided by recycling iron from senescent erythrocytes via macrophages in liver, spleen and bone marrow. The circulating pool of iron contains only ∼10% (∼3 mg) of the daily requirement for erythropoiesis, and therefore must be turned over every 2–3 h. Iron loss is an unregulated process that occurs primarily through cell shedding and blood loss (Figure 1).
FIGURE 1:
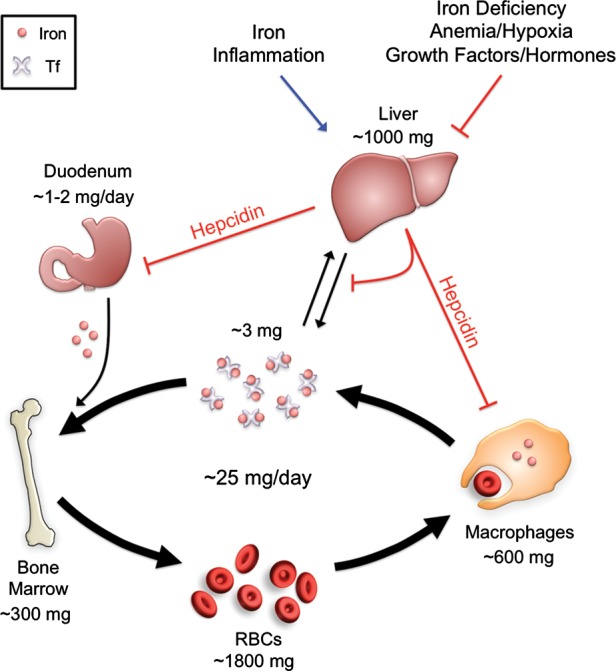
Systemic iron regulation. Iron is absorbed by the duodenum where it is released into the circulation via the iron exporter ferroportin to be loaded onto transferrin (Tf). The majority of iron is utilized by red blood cells (RBCs) for the synthesis of the hemoglobin, requiring ∼25 mg of iron per day. The daily requirements for intestinal iron uptake are only 1–2 mg per day due to efficient recycling of iron from RBCs. Iron recycling is performed primarily by reticuloendothelial macrophages which phagocytize senescent RBCs and then export iron via ferroportin back into the circulating pool of Tf-bound iron. Excess iron is also stored within hepatocytes. Hepcidin regulates systemic iron balance by inducing ferroportin degradation to inhibit iron absorption from the duodenum and iron release from macrophage and hepatocyte stores. Hepcidin production in the liver is stimulated by iron and inflammation to limit iron availability, while hepcidin production is inhibited by iron deficiency, anemia and hypoxia to increase iron availability. Several other growth factors and steroid hormones have recently been demonstrated to suppress hepcidin expression in the liver, including EGF, HGF, testosterone and estrogen.
DIETARY IRON ABSORPTION
Dietary iron absorption occurs primarily in the duodenum (Figure 2). Dietary iron exists in both heme and non-heme forms, but the molecular mechanisms underlying heme absorption are poorly understood. The liberation of non-heme iron from food and its solubilization is aided by the acidic pH of the stomach [2]. Soluble iron is reduced from the ferric (Fe3+) to the ferrous form (Fe2+) in a process that is thought to involve ferrireductases located on the intestinal apical cell membrane and ascorbic acid [3]. One candidate ferrireductase is DCYTB [3], but Dcytb null mice do not appear to have a significant iron phenotype suggesting that additional ferrireductases may play a redundant role in iron reduction [4]. Ferrous iron is then transported across the apical surface of the duodenal enterocyte via divalent metal transporter 1 (DMT1), mutations of which lead to iron deficiency anemia [5–7]. A H+/Fe2+ symporter, DMT1-mediated iron uptake is also aided by an acidic microenvironment [6]. Although the mechanism of heme uptake by the enterocyte remains obscure, it has been suggested that subsequent intracellular metabolism by heme oxygenase 1 releases iron into a common pathway shared with non-heme iron [8].
FIGURE 2:
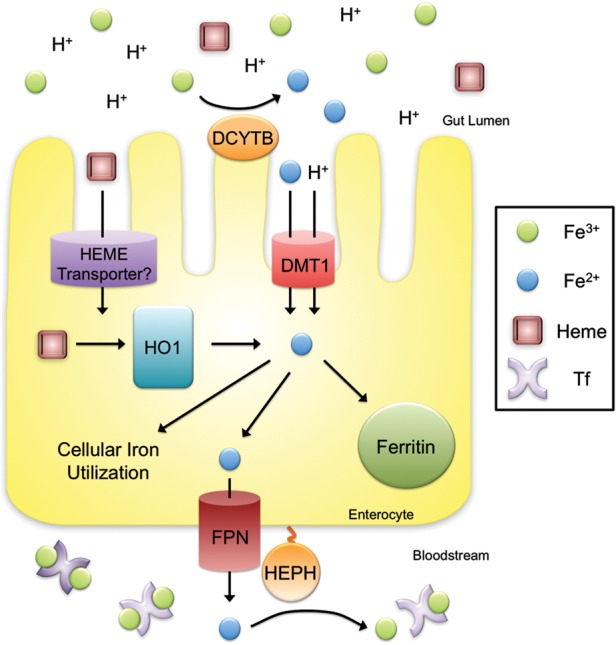
Enterocyte iron uptake. Dietary iron absorption occurs via the reduction of ferric (Fe3+) iron to ferrous (Fe2+) iron by ferrireductases such as DCYTB. Ferrous iron is then transported across the apical membrane of duodenal enterocytes by the symporter DMT1. Heme is also an important source of dietary iron, although the mechanism for heme uptake is unclear. Heme oxygenase 1 (HO1) is thought to facilitate the degradation of heme into iron, biliverdin and carbon monoxide. Cytoplasmic iron can be stored by the ferritin complex, utilized by various molecular enzymes or exported into the bloodstream by ferroportin (FPN). The multicopper ferroxidase hephaestin (HEPH) works in conjunction with ferroportin to facilitate iron export coupled with oxidization of Fe2+ to Fe3+ and loading onto Tf.
Once taken up into duodenal enterocytes, iron is either utilized by the cell, stored or exported across the basolateral membrane into circulation for systemic use. Iron is stored in an inactive Fe3+ form in ferritin, a multimeric protein comprising 24 light and heavy chain subunits surrounding a core of up to 4500 iron atoms [1]. In the absence of export across the basolateral membrane, this stored iron is lost as enterocytes are sloughed off into the gut lumen every few days.
Iron export across the enterocyte basolateral membrane into circulation is mediated by ferroportin. Importantly, ferroportin is also expressed in iron recycling macrophages and hepatocytes, and is the only known mammalian iron export protein responsible for iron entry into the bloodstream [9–11]. Iron export by ferroportin is coupled with ferroxidases including hephaestin in the intestine and ceruloplasmin that convert Fe2+ back to Fe3+ and facilitate iron loading onto the plasma iron carrying protein transferrin (Tf [12–15]).
THE HEPCIDIN–FERROPORTIN AXIS REGULATES SYSTEMIC IRON BALANCE
Ferroportin and its ligand hepcidin are key regulators of systemic iron balance, coordinating communication between tissues and cells that acquire, store and utilize iron. Discovered in 2000–01, hepcidin is a 25 amino acid peptide hormone primarily secreted by the liver that resembles other proteins involved in innate immunity [16–18]. Hepcidin was soon recognized to have an important role in iron homeostasis regulation since hepcidin null mice and human patients with hepcidin mutations develop a severe juvenile-onset form of hemochromatosis [19–21]. In contrast, hepcidin transgenic mice and human patients with hepcidin-expressing adenomas develop profound iron deficiency anemia [22, 23]. In 2004, it was demonstrated that ferroportin was the receptor for hepcidin, and that hepcidin binding caused ferroportin to be internalized and degraded [24]. The hepcidin–ferroportin axis therefore controls iron entry into circulation from dietary sources, iron recycling macrophages and hepatocyte stores (Figure 1).
Although the hepcidin–ferroportin axis has a central role in regulating body iron balance, there are many additional levels of regulation. For example, the enterocyte exerts local control over iron absorption through the regulation of proteins involved in iron transport (DMT1 and ferroportin) and sequestration (ferritin) via both transcriptional and post-transcriptional mechanisms involving hypoxia inducible factors (particularly HIF-2α) and iron regulatory proteins (reviewed in ref. [25]).
HEPCIDIN REGULATION
Hepcidin expression in the liver is regulated by a number of factors (Figure 1). Iron increases hepcidin expression as a homeostatic mechanism to limit further iron entry into the bloodstream [17, 26, 27]. Inflammation also stimulates hepcidin expression [17, 26–30], which is hypothesized to function as a protective mechanism to sequester iron from infectious organisms. However, in chronic inflammatory states, this results in macrophage iron sequestration, hypoferremia and iron restricted erythropoiesis that contributes to anemia of chronic disease [31]. Iron deficiency, hypoxia and anemia inhibit hepcidin expression to increase iron availability for erythropoiesis [26]. Recently, several growth factors, steroid hormones and other endocrine signals have also been identified to have a role in hepcidin regulation [32–36].
Hepcidin regulation by iron
Key insights into the iron-mediated hepcidin regulatory pathways came from studying the genetic iron overload disorder hereditary hemochromatosis. This is a heterogeneous disorder caused by mutations in any of several genes that ultimately result in impaired regulation of the hepcidin–ferroportin axis, leading to increased dietary iron absorption, increased iron release from macrophage stores, progressive tissue iron deposition and consequent multiorgan damage and disease [37]. Hereditary hemochromatosis can be caused by mutations in hepcidin itself, mutations in ferroportin that interfere with hepcidin binding or hepcidin-mediated internalization or mutations in one of three other genes that are involved in the iron-mediated regulation of hepcidin expression: hemojuvelin (HJV, also known as HFE2), HFE and transferrin receptor 2 (TFR2) [37]. Among these genes, HJV has the most critical role in hepcidin regulation since HJV mutations lead to the more severe juvenile onset form of hemochromatosis that is similar to the phenotype seen with mutations in hepcidin itself [21, 38].
HJV functions as a co-receptor for the bone morphogenetic protein (BMP)-SMAD signaling pathway [39], which is central to hepcidin transcriptional regulation in response to iron [40, 41] (Figure 3). A subfamily of the transforming growth factor beta (TGF-β) superfamily of signaling molecules, BMPs have an important role in a number of biologic functions, particularly during development [42]. Moreover, there is redundancy in the system with a number of BMP ligands and several BMP type I and type II receptors that can lead to the same intracellular SMAD signaling cascade [42]. Nevertheless, HJV mediates a crucial and unique function of BMP-SMAD signaling in the liver to regulate hepcidin expression and systemic iron balance, since mice and patients with HJV mutations have hepcidin deficiency and hemochromatosis but no other obvious phenotype [38, 43, 44]. It is hypothesized that HJV expression sensitizes hepatocytes to respond to low levels of BMP ligand, which would not otherwise generate a response in the absence of the co-receptor [39]. By enhancing the affinity of the binding interaction, HJV may also help cells to selectively respond to a certain subset of BMP ligands using a certain subset of BMP type I and type II receptors that are required to specifically regulate hepcidin in liver cells, in particular the ligand BMP6 [45–47], the BMP type I receptors ALK3 and ALK2 [48, 49], and the BMP type II receptor ACTRIIA [48] (Figure 3).
FIGURE 3:

Molecular regulation of hepcidin by iron and inflammation. Increased systemic iron stimulates the production of the ligand bone morphogenetic protein 6 (BMP6), which binds to the BMP Type I (ALK2/ALK3) and II (ACTRIIA) receptors, and the co-receptor HJV to stimulate phosphorylation of the SMAD1/5/8 intracellular signaling molecules. Phosphorylated SMAD 1/5/8 binds to SMAD4 and translocates to the nuclease to activate hepcidin transcription. The mechanism by which the hemochromatosis protein HFE and/or TFR2 regulate hepcidin expression is unknown but appears to involve an interaction with the BMP-SMAD signaling pathway. It has been proposed that an interaction between HFE and TFR1 is reduced under high iron conditions due to competitive binding of holotransferrin to TFR1. Displaced HFE could then associate with TFR2 and possibly the HJV-BMP receptor complex to regulate hepcidin. Inflammation also stimulates hepcidin production, in part via a canonical janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway in which inflammation increases interleukin 6 (IL6) binding to the IL6-receptor (IL6R) and thereby stimulating phosphorylation of JAKs and STAT3. Phosphorylated-STAT3 homodimers translocate to the nuclease and bind to the hepcidin promoter to stimulate hepcidin expression. Other mediators of inflammation and infection can also regulate hepcidin expression in this context (not shown). A mechanism of crosstalk between inflammatory signals and BMP signaling has been proposed in which inflammation induces activin B, which binds to BMP receptors to stimulate SMAD1/5/8 phosphorylation. SMADs and STAT3 may also interact at the level of the hepcidin promoter.
HJV may also connect the BMP-SMAD signaling response to molecules involved in iron sensing, but the molecular mechanisms for this remain to be fully elucidated. It has been hypothesized that HFE and TFR2 sense circulating iron levels in the form of iron-bound transferrin, since TFR2 can bind to transferrin [50, 51] and HFE competes for transferrin binding to transferrin receptor 1 (TFR1) [52–55] (Figure 3). There is some evidence from in vitro overexpression systems that HFE, TFR2 and HJV can interact with each other [56–58], but it is uncertain whether this occurs in vivo. HFE and TFR2 do appear to intersect with the BMP-SMAD pathway at some level, since mice and human patients with HFE and TFR2 mutations exhibit impairment in liver SMAD signaling [59–64]. However, the functions of HFE and TFR2 in regulating hepcidin are not entirely overlapping given the differential severity of the iron overload phenotype in mice and patients with HFE mutations alone, TFR2 mutations alone and double HFE/TFR2 mutations [61, 64, 65].
Hepcidin regulation by inflammation
Another well-characterized hepcidin regulatory pathway is the IL6-JAK-STAT3 pathway, which mediates, at least in part, hepcidin transcriptional induction in response to inflammation [27, 28, 66–68] (Figure 3). Other mediators of inflammation and infection including IL-22, type I interferon, tumor necrosis factor alpha and endoplasmic reticulum stress have also been implicated in hepcidin regulation [27, 29, 30]. Notably, liver SMAD signaling is also induced in many inflammatory models [69, 70], and hepcidin induction by inflammation is reduced when the BMP-SMAD signaling pathway is inhibited, indicating crosstalk between these regulatory pathways [41, 71–75]. Hypothesized mechanisms for this crosstalk are an interaction between STAT3 and SMADs at the level of the hepcidin promoter, and the TGF-β superfamily member activin B [70, 73] (Figure 3).
Hepcidin regulation by erythropoietic activity and hypoxia
Increased erythropoietic activity, for example in response to anemia or erythropoiesis-stimulating agent (ESA) administration, is a potent suppressor of hepcidin expression. This appears to be mediated by a secreted factor from proliferating red blood cell (RBC) precursors in the bone marrow, since inhibition of erythropoiesis by chemotherapy, irradiation or an erythropoietin blocking antibody prevents hepcidin suppression by anemia or ESAs [76, 77]. TGF-β/BMP superfamily modulators GDF15 and TWSG1 have been proposed to mediate hepcidin suppression in iron loading anemias with ineffective erythropoiesis such as β-thalassemia [78, 79], but may not mediate hepcidin suppression in other contexts [80, 81]. Recent data in genetic mouse models suggest that hypoxia-mediated hepcidin suppression occurs indirectly by stimulating erythropoiesis [82, 83], although other mechanisms for hypoxia-mediated hepcidin suppression have also been proposed [84, 85].
Hepcidin regulation by growth factors, steroid hormones and other endocrine factors
Recently, several growth factors and steroid hormones have been demonstrated to suppress hepcidin expression in the liver including hepatocyte growth factor (HGF, 32), epidermal growth factor (EGF, 32), estrogen [33, 34] and testosterone [35, 36] (Figure 1). HGF, EGF and testosterone are proposed to intersect with BMP-SMAD signaling in the regulation of hepcidin [32, 35, 36], while estrogen is suggested to act via an estrogen response element in the hepcidin promoter [33, 34]. The effects of steroid hormones on hepcidin regulation may help explain gender differences in iron homeostasis that have been observed [86]. Recent data presented in abstract form suggests that vitamin D administration may also suppress circulating hepcidin levels, and that vitamin D inhibits hepcidin transcription in mononuclear cells [87]. In contrast, prolonged fasting [88] and glucose [89] have been shown to increase circulating hepcidin levels, and the glucose-mediated hepcidin increase was associated with a decrease in serum iron levels [89]. The mechanism of hepcidin regulation by glucose and fasting is still undetermined, but interestingly, while glucose did not affect hepcidin secretion in hepatoma-derived cell cultures, it did induce hepcidin secretion by insulinoma-derived cell cultures [89]. These findings suggest intriguing links between iron metabolism and multiple endocrine systems, and raise the possibility that hepcidin production in non-hepatic tissues may functionally contribute to circulating hepcidin levels and systemic iron balance in some contexts, although this will need to be validated by future studies.
DISORDERED IRON BALANCE IN CHRONIC KIDNEY DISEASE
Iron deficiency-limiting erythropoiesis is an important cause of anemia and resistance to ESAs in chronic kidney disease (CKD) patients [90–93]. Iron administration is therefore a vital part of CKD anemia management. Moreover, the use of iron agents appears to be increasing [94] in the wake of recent large clinical trials that raised safety concerns for ESAs [95–97], and clinical practice guidelines that have liberalized recommendations regarding iron use in CKD patients [98]. However, current diagnostic tests to evaluate iron status are limited, the targets of iron therapy are largely opinion based, and the safety of iron has not been rigorously evaluated in large prospective randomized controlled trials in this patient population [98]. Our increasing understanding about the molecular mechanisms governing iron homeostasis regulation and its disturbance in CKD may lead to improved diagnostic and therapeutic strategies for managing this patient population.
The causes of iron deficiency in CKD patients are multifactorial (Figure 4). Some patients have true iron deficiency, characterized by decreases in both circulating iron levels and total body iron stores. Other patients have functional iron deficiency, characterized by a decrease in circulating iron that limits erythropoiesis, which can occur even in the context of normal or adequate body iron stores. A combination of these features may also be present. Factors predisposing CKD patients to iron deficiency include increased blood loss, increased iron utilization from ESA therapy, impaired dietary iron absorption and impaired iron release from body storage sites [84] (Figure 4). Blood loss can arise from frequent phlebotomy, blood trapping in the dialysis apparatus and gastrointestinal or other bleeding as a result of uremic platelet dysfunction. Dietary iron absorption can be impaired by antacid medications or phosphate binders that block enterocyte iron uptake. It is now apparent that hepcidin excess also contributes to the impaired dietary iron absorption and impaired iron release from body storage sites in CKD patients by downregulating ferroportin expression to block iron entry into the circulation [99–101]. Mechanisms leading to hepcidin excess in these patients are thought to include reduced renal clearance of this small peptide hormone, and increased inflammatory-mediated hepcidin transcription caused by the dialysis procedure itself and/or the underlying disease process [84]. Hepcidin levels in CKD patients are also influenced by iron and ESA administration (Figure 4) [84, 100].
FIGURE 4:
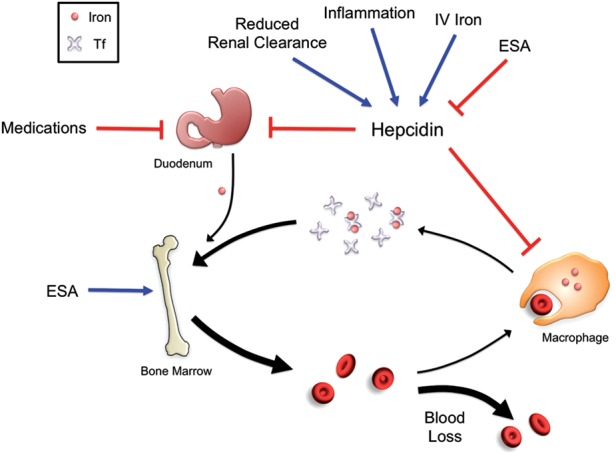
Disordered iron balance in CKD. Chronic inflammation and reduced renal clearance in patients with CKD lead to increased levels of hepcidin, which reduces duodenal iron uptake and iron release from cellular iron stores. Intestinal iron uptake is also inhibited by medications such as phosphate binders and antacids. ESAs stimulate increased iron usage for erythropoiesis, while blood loss due to frequent phlebotomy, blood trapping in the dialysis apparatus and gastrointestinal bleeding further deplete the circulating iron pool. Iron administration stimulates hepcidin expression, which can paradoxically worsen the iron restriction, while ESAs have an inhibitory effect on hepcidin expression.
IRON STATUS EVALUATION IN CKD
Current Kidney Disease: Improving Global Outcomes clinical practice guidelines regarding the use of iron agents to manage anemia of CKD [98] revolve around two diagnostic tests: serum Tf saturation and serum ferritin levels. Serum Tf saturation measures circulating iron that is immediately available for erythropoiesis, while serum ferritin serves as a surrogate measure of body iron levels. A major limitation of these diagnostic tools is that they are not reliable for estimating body iron stores or predicting which patients will respond well to iron therapy [98, 102–105]. Indeed, ferritin is also an acute phase reactant, and so must be interpreted with caution in the setting of inflammation. While there is general agreement that patients with total body iron deficiency as indicated by low Tf saturation and low ferritin should be treated with iron therapy, there are limited data on how to manage patients as ferritin levels rise [98, 106]. There is therefore a need for new diagnostic tests to understand the iron status of CKD patients and to help determine which patients will benefit from iron therapy.
ALTERNATIVE AND NOVEL DIAGNOSTIC TOOLS FOR IRON AND ANEMIA MANAGEMENT IN CKD
Reticulocyte hemoglobin content
By evaluating the hemoglobin content of reticulocytes, which are early RBC forms, reticulocyte hemoglobin content (CHr) provides an indication of iron availability for erythropoiesis within the last few days. Several studies have suggested that CHr may also be helpful to predict responsiveness to iron in hemodialysis patients [107–112], although it is less well studied and may not be as widely available as Tf sat and ferritin.
Percentage of hypochromic RBCs
Percentage of hypochromic RBCs measures the concentration of hemoglobin in RBCs, which reflects both the absolute amount of hemoglobin and the RBC size. This test has also shown utility in predicting iron responsiveness in hemodialysis patients [110, 113], but can be impacted by blood storage time, which leads to artificial RBC expansion, thereby limiting utility in dialysis centers that use national laboratories [114].
Soluble transferrin receptor
Transferrin receptor 1 mediates uptake of iron into developing RBCs. Its expression and release into circulation as soluble transferrin receptor (sTFR) is increased in the setting of iron deficiency and increased erythroid activity. Although the literature on sTFR is limited, a few studies have suggested that sTFR may be helpful to predict iron responsiveness [110, 113]. However, interpretation of this test in patients on ESAs is complicated by the fact that erythropoiesis itself increases sTFR levels [115]. The use of this assay is also limited by lack of widespread availability and cost.
Hepcidin
The understanding that hepcidin excess contributes to disordered iron homeostasis in CKD patients has garnered interest in measuring hepcidin levels as a marker of iron status, iron responsiveness and/or ESA responsiveness in CKD patients. There are two general types of assays now available to the research community to measure circulating hepcidin levels: immunologic and mass spectrometry-based assays. Both types of assays have their inherent strengths and weaknesses and give an overall large variation in the absolute values of hepcidin levels, but do show overall good correlation in relative hepcidin levels with each other [116]. Older assays that also recognize the precursor form of hepcidin (prohepcidin) are not useful because prohepcidin levels do not correlate with hepcidin biological activity [117, 118]. Using the more recent assays, many studies have now confirmed that circulating hepcidin levels are increased in CKD patients, with the highest levels in patients on hemodialysis [99–101]. Hepcidin levels in CKD patients have the strongest correlation with serum ferritin [100, 101, 119], but are also influenced, at least in some studies, by inflammation, iron administration, estimated glomerular filtration rate, dialysis clearance, ESA dose and hemoglobin [100, 101, 119–121]. One important limitation for the use of hepcidin levels as a diagnostic tool in CKD patients is the large intra-individual variability of both immunologic and mass spectrometry-based assays [122, 123]. Notably, hepcidin levels have not been shown to consistently predict responsiveness or resistance to iron therapy or ESAs [120, 124]. Thus, for the time being, there is no convincing evidence that hepcidin assays offer any advantage or additional information compared with currently available diagnostic tests with regard to CKD iron and anemia management, but this remains an area of active investigation.
Soluble HJV
Recent studies have explored the utility of measuring circulating levels of endogenous soluble HJV (sHJV) as a measure of iron status in human patients both without and with CKD [125–129]. sHJV release from cells can be mediated by the proprotein convertase furin, the transmembrane serine protease TMPRSS6 and phospholipase C [130–134], and sHJV has been detected in the conditioned media of transfected cells and in the bloodstream of animals and humans [125–130, 135–137]. While cell-surface, GPI-anchored HJV functions as a BMP co-receptor to stimulate hepcidin expression (Figure 3) [39], sHJV can function as an inhibitor of BMP signaling and hepcidin expression, presumably by sequestering BMP ligands from interacting with cell surface signaling receptors [45, 72, 135]. Interestingly, some studies have suggested that sHJV may be decreased by iron treatment and increased by iron deficiency [125, 130, 135–137], suggesting that (i) sHJV could be useful as a diagnostic tool to indicate iron status and (ii) the generation sHJV could have a functional role to inhibit hepcidin expression in the context of iron deficiency. However, one important concern regarding these early human studies quantitating sHJV levels is assay validity. Indeed, one commercial ELISA assay used in studies focusing on CKD patients [128, 129] has subsequently been shown not to recognize HJV [138]. Future studies will be needed using well-validated assays and larger patient populations to determine if sHJV could have value as a diagnostic marker to guide iron therapy in CKD patients.
Other markers
The putative role of GDF15 hepcidin regulation by erythropoietic drive has generated interest in investigating this molecule as a novel diagnostic tool for iron and anemia management in CKD patients [139]. However, currently available clinical data are very limited [139]. Moreover, while one study suggested that GDF15 may be increased by iron deficiency [140], this was not robustly supported by another study [141], and GDF15 levels may also be influenced by inflammation [141, 142], malnutrition [142] and kidney disease [142], which may complicate its usefulness in this setting.
IRON THERAPY FOR CKD PATIENTS
Iron administration remains one of the cornerstones of anemia management in CKD patients to improve hemoglobin levels and ESA responsiveness [98]. Iron supplementation is currently given in two general forms: oral or parenteral. Oral iron supplementation is the easiest and cheapest. However, oral iron agents can have gastrointestinal side effects that limit adherence, due to the formation of local reactive oxygen species and oxidative damage in the gut mucosa [143]. Moreover, several studies have suggested that oral iron is less effective than parenteral iron, particularly in hemodialysis patients, for improving or preventing iron deficiency, ameliorating anemia or reducing ESA dose [98, 144–146]. The limited effectiveness of oral iron supplements in this patient population is likely due to medications such as antacids and phosphate binders that inhibit iron entry into duodenal enterocytes, and hepcidin excess that decreases ferroportin expression to limit iron release from duodenal enterocytes into the bloodstream (Figure 4)
There are several intravenous (IV) iron preparations that can be used to treat iron-restricted erythropoiesis in CKD patients, including iron dextrans, iron sucrose, ferric gluconate, ferric carboxymaltose, iron isomaltoside 1000 and ferumoxytol. These preparations are generally comprising an iron core shielded by a carbohydrate shell with different molecular weights and physiochemical properties yielding differential degradation kinetics and ability to release ‘free’ iron into the circulation [143]. This determines the maximal single dose for each preparation, with the newer, higher molecular weight, more stable complexes enabling larger doses over shorter time frames [143]. Iron dextrans (particularly high molecular weight dextrans) have been limited by dextran-induced anaphylactic reactions in ∼0.6–0.7% of patients [98]. There is some limited data suggesting that various iron preparations may have different effects on markers of oxidative stress and inflammation, but this did not necessarily correlate with the compounds' molecular weight, stability or ability to release free iron into circulation [147, 148]. Comparative safety of these IV iron preparations in CKD patients remains largely unknown due to the lack of direct head-to-head clinical trials.
Understanding the physiology of systemic iron balance and its pathophysiology in CKD and other iron disorders raises several potential limitations shared by all IV iron preparations. Regardless of the iron preparation, once the iron is taken up into erythrocytes, macrophages or other body storage sites, hepcidin excess and ferroportin downregulation will limit the availability of the iron for recycling and subsequent use. Moreover, iron itself stimulates hepcidin expression and therefore can paradoxically worsen the iron restriction (Figure 4). Additional concerns, particularly with regard to repetitive iron administration as ferritin levels rise, are the potential for oxidant-mediated tissue injury from excess iron deposition as seen in iron overload disorders such as hemochromatosis. Iron deposition has also been associated with the pathogenesis of many other common disorders including neurodegenerative diseases, diabetes mellitus and atherosclerosis [1, 149, 150]. Additionally, withholding iron from invading pathogens is an important function of the immune system, and iron loading is associated with worse outcomes in several infectious diseases including malaria, tuberculosis and HIV [151–153]. Large prospective randomized trials in the CKD population are long overdue to evaluate the efficacy of repetitive IV iron administration with regard to hard clinical outcomes and long-term safety, to further characterize which patients will benefit from iron therapy and to determine treatment targets of iron therapy.
NOVEL TREATMENT STRATEGIES FOR IRON-RESTRICTED ERYTHROPOIESIS IN CKD PATIENTS
The understanding that hepcidin excess contributes to iron-restricted erythropoiesis in CKD patients has generated interest in developing new therapies that target the hepcidin–ferroportin axis to more directly address the underlying pathophysiology of this disease. Such therapies would be expected to increase iron availability from the diet and from the patients own body iron stores, and are a particularly attractive option for patients with higher ferritin levels.
Several categories of hepcidin/ferroportin-based therapeutics are currently in development (reviewed in [31]). One category is direct hepcidin antagonists, including anti-hepcidin antibodies, other hepcidin-binding proteins (anticalins), hepcidin-binding spiegelmers and hepcidin siRNAs and antisense oligonucleotides [31]. Dialysis itself also reduces hepcidin levels [121, 154], but the levels quickly rebound [154], potentially due in part to the induction of inflammatory cytokines by the dialysis procedure, as well as the high basal turnover rate of hepcidin [155]. Another category is agents that inhibit hepcidin production by targeting either the BMP-SMAD signaling pathway or the IL6-STAT3 pathway [31]. BMP-SMAD pathway inhibitors include anti-BMP6 antibodies, sHJV linked to the constant region of IgG1 (HJV.Fc), small molecule BMP type I receptor antagonists (LDN-193189) and heparin (which has been shown to sequester BMP ligands) [31, 41, 45, 72, 74, 75, 156]. IL6-STAT3 pathway inhibitors include anti-IL6 antibodies (Siltuximab), anti-IL6 receptor antibodies (Tocilizumab), JAK2 inhibitors (AG490) and STAT3 inhibitors (PpYLKTK) [31]. ESAs and other stimulators of ESA production such as prolyl hydroxylase inhibitors also fall in this category since they inhibit hepcidin production. A third category is ferroportin agonists/stabilizers, including anti-ferroportin antibodies and thiol-reactive compounds that interfere with hepcidin binding to ferroportin, as well as agents that interfere with ferroportin internalization or potentiate ferroportin synthesis [31]. Notably, many of these agents have shown efficacy for treating iron-restricted erythropoiesis and anemia in animal models with anemia of chronic disease [74, 75, 157–160], and several are currently in human clinical trials [161–164]. The safety and efficacy of these agents in human CKD patients compared with current treatment strategies remains to be determined.
CONCLUSIONS
The last 13 years have yielded significant advances in understanding the molecular mechanisms underlying systemic iron balance and its dysregulation in CKD patients. These studies hold the promise for developing new, rationally designed diagnostic and therapeutic tools to improve anemia management in CKD patients. Novel therapies targeting hepcidin have shown particular promise, and several have already entered human clinical trials. More research is needed to better understand the efficacy, long-term safety and targets of current iron therapies as well as novel hepcidin-lowering approaches in large prospective randomized controlled trials.
CONFLICT OF INTEREST STATEMENT
J.L.B. has ownership interest in a start-up company FerruMax Pharmaceuticals, which has licensed technology from the Massachusetts General Hospital based on the work cited here and in prior publications.
ACKNOWLEDGEMENTS
J.L.B. was supported in part by NIH grant RO1-DK087727 and a Howard Goodman Fellowship Award from the Massachusetts General Hospital.
REFERENCES
- 1.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 2.Kovac S, Anderson GJ, Baldwin GS. Gastrins, iron homeostasis and colorectal cancer. Biochim Biophys Acta. 2011;1813:889–895. doi: 10.1016/j.bbamcr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 4.Gunshin H, Starr CN, Direnzo C, et al. Cybrd1(duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood. 2005;16:16. doi: 10.1182/blood-2005-02-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleming MD, Trenor CC, III, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 6.Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 7.Gunshin H, Fujiwara Y, Custodio AO, et al. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weintraub LR, Weinstein MB, Huser HJ, et al. Absorption of hemoglobin iron: the role of a heme-splitting substance in the intestinal mucosa. J Clin Invest. 1968;47:531–539. doi: 10.1172/JCI105749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 10.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 11.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 12.Osaki S, Johnson DA. Mobilization of liver iron by ferroxidase (ceruloplasmin) J Biol Chem. 1969;244:5757–5758. [PubMed] [Google Scholar]
- 13.Osaki S, Johnson DA, Frieden E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem. 1966;241:2746–2751. [PubMed] [Google Scholar]
- 14.Roeser HP, Lee GR, Nacht S, et al. The role of ceruloplasmin in iron metabolism. J Clin Invest. 1970;49:2408–2417. doi: 10.1172/JCI106460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 16.Krause A, Neitz S, Mägert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 17.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 18.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 19.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesbordes-Brion JC, Viatte L, Bennoun M, et al. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood. 2006;108:1402–1405. doi: 10.1182/blood-2006-02-003376. [DOI] [PubMed] [Google Scholar]
- 21.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 22.Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinstein DA, Roy CN, Fleming MD, et al. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100:3776–3781. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 24.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 25.Mastrogiannaki M, Matak P, Peyssonnaux C. The gut in iron homeostasis: role of HIF-2 under normal and pathological conditions. Blood. 2013 doi: 10.1182/blood-2012-11-427765. doi:10.1182/blood-2012-11-427765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 29.Armitage AE, Eddowes LA, Gileadi U, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118:4129–4139. doi: 10.1182/blood-2011-04-351957. [DOI] [PubMed] [Google Scholar]
- 30.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768–772. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 31.Sun CC, Vaja V, Babitt JL, et al. Targeting the hepcidin–ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol. 2012;87:392–400. doi: 10.1002/ajh.23110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodnough JB, Ramos E, Nemeth E, et al. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56:291–299. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Q, Jian J, Katz S, et al. 17β-estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology. 2012;153:3170–3178. doi: 10.1210/en.2011-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou Y, Zhang S, Wang L, et al. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene. 2012;511:398–403. doi: 10.1016/j.gene.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 35.Guo W, Bachman E, Li M, et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell. 2013;12:280–291. doi: 10.1111/acel.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Latour C, Kautz L, Besson-Fournier C, et al. Testosterone perturbs systemic iron balance through activation of EGFR signaling in the liver and repression of hepcidin. Hepatology. 2013 doi: 10.1002/hep.26648. doi:10.1002/hep.26648. [DOI] [PubMed] [Google Scholar]
- 37.Babitt JL, Lin HY. The molecular pathogenesis of hereditary hemochromatosis. Semin Liver Dis. 2011;31:280–292. doi: 10.1055/s-0031-1286059. [DOI] [PubMed] [Google Scholar]
- 38.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 39.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 40.Corradini E, Meynard D, Wu Q, et al. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology. 2011;54:273–284. doi: 10.1002/hep.24359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu PB, Hong CC, Sachidanandan C, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corradini E, Babitt JL, Lin HY. The RGM/DRAGON family of BMP co-receptors. Cytokine Growth Factor Rev. 2009;20:389–398. doi: 10.1016/j.cytogfr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang FW, Pinkus JL, Pinkus GS, et al. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–2186. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andriopoulos B, Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–487. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41:478–481. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 47.Wu Q, Sun CC, Lin HY, et al. Repulsive guidance molecule (RGM) family proteins exhibit differential binding kinetics for bone morphogenetic proteins (BMPs) PLoS One. 2012;7:e46307. doi: 10.1371/journal.pone.0046307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia Y, Babitt JL, Sidis Y, et al. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008;111:5195–5204. doi: 10.1182/blood-2007-09-111567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinbicker AU, Bartnikas TB, Lohmeyer LK, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118:4224–4230. doi: 10.1182/blood-2011-03-339952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 51.West AP, Jr, Bennett MJ, Sellers VM, et al. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem. 2000;275:38135–38138. doi: 10.1074/jbc.C000664200. [DOI] [PubMed] [Google Scholar]
- 52.Feder JN, Penny DM, Irrinki A, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lebrón JA, West AP, Jr, Bjorkman PJ. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J Mol Biol. 1999;294:239–245. doi: 10.1006/jmbi.1999.3252. [DOI] [PubMed] [Google Scholar]
- 54.West AP, Jr, Giannetti AM, Herr AB, et al. Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites. J Mol Biol. 2001;313:385–397. doi: 10.1006/jmbi.2001.5048. [DOI] [PubMed] [Google Scholar]
- 55.Giannetti AM, Björkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279:25866–25875. doi: 10.1074/jbc.M401467200. [DOI] [PubMed] [Google Scholar]
- 56.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 57.Gao J, Chen J, Kramer M, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9:217–227. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D'Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012;57:1052–1060. doi: 10.1016/j.jhep.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 59.Corradini E, Garuti C, Montosi G, et al. Bone morphogenetic protein signaling is impaired in a Hfe knockout mouse model of hemochromatosis. Gastroenterology. 2009;137:1489–1497. doi: 10.1053/j.gastro.2009.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kautz L, Meynard D, Besson-Fournier C, et al. BMP/Smad signaling is not enhanced in Hfe-deficient mice despite increased Bmp6 expression. Blood. 2009;114:2515–2520. doi: 10.1182/blood-2009-02-206771. [DOI] [PubMed] [Google Scholar]
- 61.Wallace DF, Summerville L, Crampton EM, et al. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology. 2009;50:1992–2000. doi: 10.1002/hep.23198. [DOI] [PubMed] [Google Scholar]
- 62.Bolondi G, Garuti C, Corradini E, et al. Altered hepatic BMP signaling pathway in human HFE hemochromatosis. Blood Cells Mol Dis. 2010;45:308–312. doi: 10.1016/j.bcmd.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryan JD, Ryan E, Fabre A, et al. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology. 2010;52:1266–1273. doi: 10.1002/hep.23814. [DOI] [PubMed] [Google Scholar]
- 64.Corradini E, Rozier M, Meynard D, et al. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology. 2011;141:1907–1914. doi: 10.1053/j.gastro.2011.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pietrangelo A, Caleffi A, Henrion J, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology. 2005;128:470–479. doi: 10.1053/j.gastro.2004.11.057. [DOI] [PubMed] [Google Scholar]
- 66.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verga Falzacappa MV, Vujic Spasic M, Kessler R, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109:353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 68.Pietrangelo A, Dierssen U, Valli L, et al. STAT3 is required for IL-6-gp130 dependent activation of hepcidin in vivo. Gastroenterology. 2007;132:294–300. doi: 10.1053/j.gastro.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 69.Theurl I, Schroll A, Nairz M, et al. Pathways for the regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica. 2011;96:1761–1769. doi: 10.3324/haematol.2011.048926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120:431–439. doi: 10.1182/blood-2012-02-411470. [DOI] [PubMed] [Google Scholar]
- 71.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 72.Babitt JL, Huang FW, Xia Y, et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–1939. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verga Falzacappa MV, Casanovas G, Hentze MW, et al. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J Mol Med. 2008;86:531–540. doi: 10.1007/s00109-008-0313-7. [DOI] [PubMed] [Google Scholar]
- 74.Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118:4977–4984. doi: 10.1182/blood-2011-03-345066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood. 2011;117:4915–4923. doi: 10.1182/blood-2010-10-313064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pak M, Lopez MA, Gabayan V, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vokurka M, Krijt J, Sulc K, et al. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006;55:667–674. doi: 10.33549/physiolres.930841. [DOI] [PubMed] [Google Scholar]
- 78.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 79.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181–186. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casanovas G, Spasic MV, Casu C, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444–447. doi: 10.3324/haematol.2012.069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95:505–508. doi: 10.3324/haematol.2009.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Q, Davidoff O, Niss K, et al. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122:4635–4644. doi: 10.1172/JCI63924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mastrogiannaki M, Matak P, Mathieu JR, et al. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica. 2012;97:827–834. doi: 10.3324/haematol.2011.056119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis. 2010;55:726–741. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finberg KE. Regulation of systemic iron homeostasis. Curr Opin Hematol. 2013;20:208–214. doi: 10.1097/MOH.0b013e32835f5a47. [DOI] [PubMed] [Google Scholar]
- 86.Harrison-Findik DD. Gender-related variations in iron metabolism and liver diseases. World J Hepatol. 2010;2:302–310. doi: 10.4254/wjh.v2.i8.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bacchetta J, Zaritsky J, Lisse TS, et al. Poster #FR-PO1560. Vitamin D as a new regulator of iron metabolism: vitamin D suppresses hepcidin in vitro and in vivo. J Am Soc Nephrol. 2011;22:474A. [Google Scholar]
- 88.Troutt JS, Rudling M, Persson L, et al. Circulating human hepcidin-25 concentrations display a diurnal rhythm, increase with prolonged fasting, and are reduced by growth hormone administration. Clin Chem. 2012;58:1225–1232. doi: 10.1373/clinchem.2012.186866. [DOI] [PubMed] [Google Scholar]
- 89.Aigner E, Felder TK, Oberkofler H, et al. Glucose acts as a regulator of serum iron by increasing serum hepcidin concentrations. J Nutr Biochem. 2013;24:112–117. doi: 10.1016/j.jnutbio.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 90.Mircescu G, Garneata L, Capusa C, et al. Intravenous iron supplementation for the treatment of anaemia in pre-dialyzed chronic renal failure patients. Nephrol Dial Transplant. 2006;21:120–124. doi: 10.1093/ndt/gfi087. [DOI] [PubMed] [Google Scholar]
- 91.Silverberg DS, Iaina A, Peer G, et al. Intravenous iron supplementation for the treatment of the anemia of moderate to severe chronic renal failure patients not receiving dialysis. Am J Kidney Dis. 1996;27:234–238. doi: 10.1016/s0272-6386(96)90546-6. [DOI] [PubMed] [Google Scholar]
- 92.Fishbane S, Frei GL, Maesaka J. Reduction in recombinant human erythropoietin doses by the use of chronic intravenous iron supplementation. Am J Kidney Dis. 1995;26:41–46. doi: 10.1016/0272-6386(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 93.Sunder-Plassmann G, Horl WH. Importance of iron supply for erythropoietin therapy. Nephrol Dial Transplant. 1995;10:2070–2076. [PubMed] [Google Scholar]
- 94.Pisoni RL, Fuller DS, Bieber BA, et al. The DOPPS practice monitor for US dialysis care: trends through August 2011. Am J Kidney Dis. 2012;60:160–165. doi: 10.1053/j.ajkd.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 95.Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584–590. doi: 10.1056/NEJM199808273390903. [DOI] [PubMed] [Google Scholar]
- 96.Singh AK, Szczech L, Tang KL, et al. CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–2098. doi: 10.1056/NEJMoa065485. [DOI] [PubMed] [Google Scholar]
- 97.Pfeffer MA, Burdmann EA, Chen CY, et al. TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019–2032. doi: 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- 98.Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl. 2012;2:279–335. [Google Scholar]
- 99.Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood. 2008;112:4292–4297. doi: 10.1182/blood-2008-02-139915. [DOI] [PubMed] [Google Scholar]
- 100.Ashby DR, Gale DP, Busbridge M, et al. Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease. Kidney Int. 2009;75:976–981. doi: 10.1038/ki.2009.21. [DOI] [PubMed] [Google Scholar]
- 101.Zaritsky J, Young B, Wang HJ, et al. Hepcidin—a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4:1051–1056. doi: 10.2215/CJN.05931108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coyne DW, Kapoian T, Suki W, et al. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV iron with elevated ferritin (DRIVE) Study. J Am Soc Nephrol. 2007;18:975–984. doi: 10.1681/ASN.2006091034. [DOI] [PubMed] [Google Scholar]
- 103.Singh AK, Coyne DW, Shapiro W, et al. Predictors of the response to treatment in anemic hemodialysis patients with high serum ferritin and low transferrin saturation. Kidney Int. 2007;71:1163–1171. doi: 10.1038/sj.ki.5002223. [DOI] [PubMed] [Google Scholar]
- 104.Stancu S, Barsan L, Stanciu A, et al. Can the response to iron therapy be predicted in anemic nondialysis patients with chronic kidney disease? Clin J Am Soc Nephrol. 2010;5:409–416. doi: 10.2215/CJN.04280609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fishbane S, Kowalski EA, Imbriano LJ, et al. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol. 1996;7:2654–2657. doi: 10.1681/ASN.V7122654. [DOI] [PubMed] [Google Scholar]
- 106.Locatelli F, Bárány P, Covic A, et al. ERA-EDTA ERBP Advisory Board. Kidney disease: improving Global Outcomes guidelines on anaemia management in chronic kidney disease: a European Renal Best Practice position statement. Nephrol Dial Transplant. 2013;28:1346–1359. doi: 10.1093/ndt/gft033. [DOI] [PubMed] [Google Scholar]
- 107.Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis. 1997;30:912–922. doi: 10.1016/s0272-6386(97)90104-9. [DOI] [PubMed] [Google Scholar]
- 108.Fishbane S, Galgano C, Langley RC, Jr, et al. Reticulocyte hemoglobin content in the evaluation of iron status of hemodialysis patients. Kidney Int. 1997;52:217–222. doi: 10.1038/ki.1997.323. [DOI] [PubMed] [Google Scholar]
- 109.Fishbane S, Shapiro W, Dutka P, et al. A randomized trial of iron deficiency testing strategies in hemodialysis patients. Kidney Int. 2001;60:2406–2411. doi: 10.1046/j.1523-1755.2001.00077.x. [DOI] [PubMed] [Google Scholar]
- 110.Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant. 2001;16:1416–1423. doi: 10.1093/ndt/16.7.1416. [DOI] [PubMed] [Google Scholar]
- 111.Mitsuiki K, Harada A, Miyata Y. Assessment of iron deficiency in chronic hemodialysis patients: investigation of cutoff values for reticulocyte hemoglobin content. Clin Exp Nephrol. 2003;7:52–57. doi: 10.1007/s101570300007. [DOI] [PubMed] [Google Scholar]
- 112.Chuang CL, Liu RS, Wei YH, et al. Early prediction of response to intravenous iron supplementation by reticulocyte haemoglobin content and high-fluorescence reticulocyte count in haemodialysis patients. Nephrol Dial Transplant. 2003;18:370–377. doi: 10.1093/ndt/18.2.370. [DOI] [PubMed] [Google Scholar]
- 113.Bovy C, Gothot A, Delanaye P, et al. Mature erythrocyte parameters as new markers of functional iron deficiency in haemodialysis: sensitivity and specificity. Nephrol Dial Transplant. 2007;22:1156–1162. doi: 10.1093/ndt/gfl765. [DOI] [PubMed] [Google Scholar]
- 114.Wish JB. Assessing iron status: beyond serum ferritin and transferrin saturation. Clin J Am Soc Nephrol. 2006;1(Suppl. 1):S4–S8. doi: 10.2215/CJN.01490506. [DOI] [PubMed] [Google Scholar]
- 115.Ahluwalia N, Skikne BS, Savin V, et al. Markers of masked iron deficiency and effectiveness of EPO therapy in chronic renal failure. Am J Kidney Dis. 1997;30:532–541. doi: 10.1016/s0272-6386(97)90313-9. [DOI] [PubMed] [Google Scholar]
- 116.Kroot JJ, Kemna EH, Bansal SS, et al. Results of the first international round robin for the quantification of urinary and plasma hepcidin assays: need for standardization. Haematologica. 2009;94:1748–1752. doi: 10.3324/haematol.2009.010322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roe MA, Spinks C, Heath AL, et al. Serum prohepcidin concentration: no association with iron absorption in healthy men; and no relationship with iron status in men carrying HFE mutations, hereditary haemochromatosis patients undergoing phlebotomy treatment, or pregnant women. Br J Nutr. 2007;97:544–549. doi: 10.1017/S0007114507336829. [DOI] [PubMed] [Google Scholar]
- 118.Kemna E, Pickkers P, Nemeth E, et al. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood. 2005;106:1864–1866. doi: 10.1182/blood-2005-03-1159. [DOI] [PubMed] [Google Scholar]
- 119.van der Weerd NC, Grooteman MP, Bots ML, et al. CONTRAST Investigators. Hepcidin-25 in chronic hemodialysis patients is related to residual kidney function and not to treatment with erythropoiesis stimulating agents. PLoS One. 2012;7:e39783. doi: 10.1371/journal.pone.0039783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kato A, Tsuji T, Luo J, et al. Association of prohepcidin and hepcidin-25 with erythropoietin response and ferritin in hemodialysis patients. Am J Nephrol. 2008;28:115–121. doi: 10.1159/000109968. [DOI] [PubMed] [Google Scholar]
- 121.Zaritsky J, Young B, Gales B, et al. Reduction of serum hepcidin by hemodialysis in pediatric and adult patients. Clin J Am Soc Nephrol. 2010;5:1010–1014. doi: 10.2215/CJN.08161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ford BA, Eby CS, Scott MG, et al. Intra-individual variability in serum hepcidin precludes its use as a marker of iron status in hemodialysis patients. Kidney Int. 2010;78:769–773. doi: 10.1038/ki.2010.254. [DOI] [PubMed] [Google Scholar]
- 123.Peters HP, Rumjon A, Bansal SS, et al. Intra-individual variability of serum hepcidin-25 in haemodialysis patients using mass spectrometry and ELISA. Nephrol Dial Transplant. 2012;27:3923–3929. doi: 10.1093/ndt/gfs164. [DOI] [PubMed] [Google Scholar]
- 124.Tessitore N, Girelli D, Campostrini N, et al. Hepcidin is not useful as a biomarker for iron needs in haemodialysis patients on maintenance erythropoiesis-stimulating agents. Nephrol Dial Transplant. 2010;25:3996–4002. doi: 10.1093/ndt/gfq321. [DOI] [PubMed] [Google Scholar]
- 125.Brasse-Lagnel CG, Poli M, Lesueur C, et al. Immunoassay for human serum hemojuvelin. Haematologica. 2010;95:2031–2037. doi: 10.3324/haematol.2010.022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Finkenstedt A, Widschwendter A, Brasse-Lagnel CG, et al. Hepcidin is correlated to soluble hemojuvelin but not to increased GDF15 during pregnancy. Blood Cells Mol Dis. 2012;48:233–237. doi: 10.1016/j.bcmd.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 127.Luciani N, Brasse-Lagnel C, Poli M, et al. Hemojuvelin: a new link between obesity and iron homeostasis. Obesity (Silver Spring) 2010;19:1545–1551. doi: 10.1038/oby.2011.12. [DOI] [PubMed] [Google Scholar]
- 128.Rumjon A, Sarafidis P, Brincat S, et al. Serum hemojuvelin and hepcidin levels in chronic kidney disease. Am J Nephrol. 2012;35:295–304. doi: 10.1159/000336528. [DOI] [PubMed] [Google Scholar]
- 129.Malyszko J, Malyszko JS, Levin-Iaina N, et al. Is hemojuvelin a possible new player in iron metabolism in hemodialysis patients? Int Urol Nephrol. 2011;44:1805–1811. doi: 10.1007/s11255-011-0084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111:924–931. doi: 10.1182/blood-2007-07-100677. [DOI] [PubMed] [Google Scholar]
- 131.Kuninger D, Kuns-Hashimoto R, Nili M, et al. Pro-protein convertases control the maturation and processing of the iron-regulatory protein, RGMc/hemojuvelin. BMC Biochem. 2008;9:9. doi: 10.1186/1471-2091-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lin L, Nemeth E, Goodnough JB, et al. Soluble hemojuvelin is released by proprotein convertase-mediated cleavage at a conserved polybasic RNRR site. Blood Cells Mol Dis. 2008;40:122–131. doi: 10.1016/j.bcmd.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–511. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang AS, West AP, Jr, Wyman AE, et al. Interaction of hemojuvelin with neogenin results in iron accumulation in human embryonic kidney 293 cells. J Biol Chem. 2005;280:33885–33894. doi: 10.1074/jbc.M506207200. [DOI] [PubMed] [Google Scholar]
- 135.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106:2884–2889. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 136.Zhang AS, Anderson SA, Meyers KR, et al. Evidence that inhibition of hemojuvelin shedding in response to iron is mediated through neogenin. J Biol Chem. 2007;282:12547–12556. doi: 10.1074/jbc.M608788200. [DOI] [PubMed] [Google Scholar]
- 137.Chen W, Sun CC, Chen S, et al. A novel validated enzyme-linked immunosorbent assay to quantify soluble hemojuvelin in mouse serum. Haematologica. 2013;98:296–304. doi: 10.3324/haematol.2012.070136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gutiérrez OM, Sun CC, Chen W, et al. Statement of concern about a commercial assay used to measure soluble hemojuvelin in humans. Am J Nephrol. 2012;36:332–333. doi: 10.1159/000342519. [DOI] [PubMed] [Google Scholar]
- 139.Małyszko J, Koc-Żórawska E, Levin-Iaina N, et al. New parameters in iron metabolism and functional iron deficiency in patients on maintenance hemodialysis. Pol Arch Med Wewn. 2012;122:537–542. [PubMed] [Google Scholar]
- 140.Lakhal S, Talbot NP, Crosby A, et al. Regulation of growth differentiation factor 15 expression by intracellular iron. Blood. 2009;113:1555–1563. doi: 10.1182/blood-2008-07-170431. [DOI] [PubMed] [Google Scholar]
- 141.Theurl I, Finkenstedt A, Schroll A, et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. 2010;148:449–455. doi: 10.1111/j.1365-2141.2009.07961.x. [DOI] [PubMed] [Google Scholar]
- 142.Breit SN, Carrero JJ, Tsai VW, et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15) and mortality in end-stage renal disease. Nephrol Dial Transplant. 2012;27:70–75. doi: 10.1093/ndt/gfr575. [DOI] [PubMed] [Google Scholar]
- 143.Macdougall IC, Geisser P. Use of intravenous iron supplementation in chronic kidney disease: an update. Iran J Kidney Dis. 2013;7:9–22. [PubMed] [Google Scholar]
- 144.Fudin R, Jaichenko J, Shostak A, et al. Correction of uremic iron deficiency anemia in hemodialyzed patients: a prospective study. Nephron. 1998;79:299–305. doi: 10.1159/000045053. [DOI] [PubMed] [Google Scholar]
- 145.Macdougall IC, Tucker B, Thompson J, et al. A randomized controlled study of iron supplementation in patients treated with erythropoietin. Kidney Int. 1996;50:1694–1699. doi: 10.1038/ki.1996.487. [DOI] [PubMed] [Google Scholar]
- 146.Markowitz GS, Kahn GA, Feingold RE, et al. An evaluation of the effectiveness of oral iron therapy in hemodialysis patients receiving recombinant human erythropoietin. Clin Nephrol. 1997;48:34–40. [PubMed] [Google Scholar]
- 147.Toblli JE, Cao G, Olivieri L, et al. Comparison of the renal, cardiovascular and hepatic toxicity data of original intravenous iron compounds. Nephrol Dial Transplant. 2010;25:3631–3640. doi: 10.1093/ndt/gfq260. [DOI] [PubMed] [Google Scholar]
- 148.Martin-Malo A, Merino A, Carracedo J, et al. Effects of intravenous iron on mononuclear cells during the haemodialysis session. Nephrol Dial Transplant. 2012;27:2465–2471. doi: 10.1093/ndt/gfr711. [DOI] [PubMed] [Google Scholar]
- 149.Wilson JG. Iron and glucose homeostasis: new lessons from hereditary haemochromatosis. Diabetologia. 2006;49:1459–1461. doi: 10.1007/s00125-006-0289-1. [DOI] [PubMed] [Google Scholar]
- 150.Saeed O, Otsuka F, Polavarapu R, et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:299–307. doi: 10.1161/ATVBAHA.111.240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367:133–143. doi: 10.1016/S0140-6736(06)67962-2. [DOI] [PubMed] [Google Scholar]
- 152.Boelaert JR, Vandecasteele SJ, Appelberg R, et al. The effect of the host's iron status on tuberculosis. J Infect Dis. 2007;195:1745–1753. doi: 10.1086/518040. [DOI] [PubMed] [Google Scholar]
- 153.McDermid JM, Jaye A, Schim van der Loeff MF, et al. Elevated iron status strongly predicts mortality in West African adults with HIV infection. J Acquir Immune Defic Syndr. 2007;46:498–507. doi: 10.1097/qai.0b013e31815b2d4b. [DOI] [PubMed] [Google Scholar]
- 154.Kuragano T, Shimonaka Y, Kida A, et al. Determinants of hepcidin in patients on maintenance hemodialysis: role of inflammation. Am J Nephrol. 2010;31:534–540. doi: 10.1159/000312381. [DOI] [PubMed] [Google Scholar]
- 155.Xiao JJ, Krzyzanski W, Wang YM, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J. 2010;12:646–657. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011;117:997–1004. doi: 10.1182/blood-2010-06-289082. [DOI] [PubMed] [Google Scholar]
- 157.Sasu BJ, Cooke KS, Arvedson TL. Anti-hepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation induced anemia. Blood. 2010;115:3616–3624. doi: 10.1182/blood-2009-09-245977. [DOI] [PubMed] [Google Scholar]
- 158.Hashizume M, Uchiyama Y, Horai N. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int. 2010;30:917–923. doi: 10.1007/s00296-009-1075-4. [DOI] [PubMed] [Google Scholar]
- 159.Sun CC, Vaja V, Chen S, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant. 2013;28:1733–1743. doi: 10.1093/ndt/gfs584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schwoebel F, van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013;121:2311–2315. doi: 10.1182/blood-2012-09-456756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Eli Lilly and Company. A phase 1 study of LY2787106 in cancer and anemia. http://clinicaltrials.gov/ct2/show/NCT01340976. (27 June 2013, date last accessed)
- 162.Eli Lilly and Company. A First Human Study of a Ferroportin Antibody. http://clinicaltrials.gov/ct2/show/NCT01330953. (27 June 2013, date last accessed)
- 163.Noxxon Pharma AG. Efficacy of NOX-H94 on anemia of chronic disease in patients with cancer. http://clinicaltrials.gov/ct2/show/NCT01691040. (27 June 2013, date last accessed)
- 164.FerruMax Pharmaceuticals, Inc. A phase 2A trial of FMX-8 treatment for anemia in patients with ESRD on hemodialysis HD. http://clinicaltrials.gov/ct2/show/NCT01873534. (27 June 2013, date last accessed)