

Abstract
Technological development led to an increased interest in systems biological approaches in plants to characterize developmental mechanism and candidate genes relevant to specific tissue or cell morphology. AUX-IAA proteins are important plant-specific putative transcription factors. There are several reports on physiological response of this family in Arabidopsis but in cotton fiber the transcriptional network through which AUX-IAA regulated its target genes is still unknown. in-silico modelling of cotton fiber development specific gene expression data (108 microarrays and 22,737 genes) using Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) reveals 3690 putative AUX-IAA target genes of which 139 genes were known to be AUX-IAA co-regulated within Arabidopsis. Further AUX-IAA targeted gene regulatory network (GRN) had substantial impact on the transcriptional dynamics of cotton fiber, as showed by, altered TF networks, and Gene Ontology (GO) biological processes and metabolic pathway associated with its target genes. Analysis of the AUX-IAA-correlated gene network reveals multiple functions for AUX-IAA target genes such as unidimensional cell growth, cellular nitrogen compound metabolic process, nucleosome organization, DNA-protein complex and process related to cell wall. These candidate networks/pathways have a variety of profound impacts on such cellular functions as stress response, cell proliferation, and cell differentiation. While these functions are fairly broad, their underlying TF networks may provide a global view of AUX-IAA regulated gene expression and a GRN that guides future studies in understanding role of AUX-IAA box protein and its targets regulating fiber development.
Keywords: GRN gene regulatory network, AUX-IAA auxin-indole acetic acid, ARF Auxin response factor, TF's Transcription Factor's
Background
AUX/IAA genes were first isolated as members of a family of genes that were rapidly induced in response to auxin (indole-3-acetic acid, IAA). Aux/IAA genes have been found in dicots (pea, soybean, Medicago truncatula, Arabidopsis, tomato, tobacco and cotton), grasses (maize, rice) and pine trees. They have not been found in animal, bacterial or fungal genomes and are therefore unique to plants. The AUX-IAA family proteins play crucial roles in diverse aspects of plant development; this has become evident in recent years and has been documented in an increasing number of publications. Auxin plays important roles in many aspects of plant growth and developmental processes, such as apical dominance, tropism, and lateral root and flower formation [1]. This wide range of effects is attributed to the role of auxin as a signal factor that activates a series of downstream pathways mainly related to cell wall loosening, cell proliferation, cell expansion (Figure 1). Auxin can rapidly and specifically alter the transcription levels of most of these genes without protein synthesis [2]. Moreover, molecular, genetic, and biochemical studies have shown that AUX/IAA proteins play central roles in auxin signal transduction [3, 4] .These genes have been grouped into three major classes: auxin/indole-3- acetic acid (AUX/IAA), growth hagen 3 (GH3), and small auxin-up RNA (SAUR) gene families [3]. Recently it has been identify that IAA8 involved in lateral root formation and interacts with the TIR1 auxin receptor and ARF transcription factors in Arabidopsis [5]. Further tomato (Solanum lycopersicum) SlIAA15 is involved in trichome formation and axillary shoot development.
Figure 1.
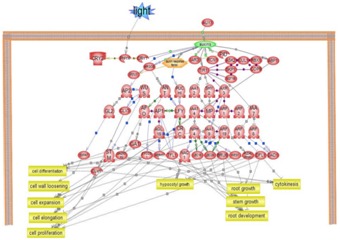
Summarization of general Biological and Cellular process governing through AUX-IAA Transcription factors in Arabidopsis thaliana.
Cotton is one of the most important economic crops in the world whose fibers are highly elongated single-celled seed trichomes that initiate from the seed coat, and they serve as a good experimental model for cell elongation. Various studies on cotton fiber cell development have identified plant hormones as critical regulators of fiber development. Auxin is known to play an important role in fiber development. However, little information about the Aux/IAA superfamily has been reported in cotton. Auxin promotes the fiber cell development of in vitro cultured ovules, and a positive correlation between final fiber length and IAA levels has also been observed [6]. The mRNA transcripts of GhAux1, GhAux2, and GhAux3 accumulated in the root, stem, and leaf, implying that these genes might be related to the regulation of vegetative growth. Where as, mRNA transcripts of GhAux4, GhAux5, GhAux6, and GhAux7 showed higher expression in ovules at 0 DPA when compared with other tissues. The expression levels of GhAux4 in stems; GhAux6 in stems, leaves, and fibers at 10 DPA and 15 DPA; and GhAux7 in stems and leaves were also relatively high. These results indicated that although the four genes played significant roles in fiber initiation, they also showed functional diversity in cotton vegetative growth and fiber elongation. GhAux8 had a higher expression level in stems and fibers at 2 to 5 DPA and 23 DPA, which may indicate a role in stem development, early fiber elongation, and secondary cell wall thickening [7]. Interestingly GhAux9 was fiber-specific and had no detectable expression in roots, stems, or leaves. The high level expression of GhAux9 was detected in fibers at 10 DPA when rapid fiber elongation occurred. This was observed that expression began to decline at 15 DPA, and became high again at 23 DPA. This indicated that GhAux9 may play an important role at the fiber elongation stage and the secondary cell wall thickening stage.
GhIAA16 had its expression peak at −3 DPA in the fiber initiation stage, consistent with Suo et al. (2002), and high expression levels in fiber at 20 DPA and 23 DPA. Here, we provide the first identification of putative targets of eight Aux/IAA family members in cotton. The identification and characterization of these AUX-IAA targets in elongating cotton fiber cells might promote the further study of fiber development regulation mechanisms.
Methodology
Gene expression profile dataset:
We used 108 expression profile where 99 gene expression profiles previously generated by our labs GEO (Series GSE36228) for six different fiber devopment stages of two superior and three inferior genotype using the Affymetrix cotton Gene Chip System. We also used nine other gene expression profiles available in GEO (Series GSE36021 and GSE29810) Probe sets with expression mean µ < 50 and S.D. σ < 0.3µ was considered uninformative and was therefore excluded, leaving 18,892 probe sets for the analysis. Supplementary File 1 (Available with authors) summarizes 108 samples included in this study.
Data analysis and bioinformatics:
Network analyses:
Co-regulatory gene networks were analysed using Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe). Raw data from 108 samples, form GEO database were first normalized (through RMA) and log2 transformed and median centered. Normalize expression values of 18,892 genes were used as input files to infer global regulatory networks.
ARACNE
ARACNE uses mutual information [8] and it uses the Data Processing Inequality (DPI) [9] to retain only those regulatory relationships that are direct (rather than indirect) [10]. In other words, if genes g1 and g3 interact only through a third gene, g2, then DPI indicates:
I(g1,g3)≥min[I(g1,g2);I(g2,g3)]
Thus, the edge with the least value gets eliminated. The “DPI tolerance” used for ranking of I values, to minimize the impact of I value variance was set at 0.15 in this case study. DPI tolerance values of greater than 0.2 have been determined to yield high false positive edges. Furthermore, the threshold p-value for establishing that the mutual information between gene pairs was significant in this study was set at 5.0 × 10−11.
Gene Ontology Analyses:
In silico identified candidate targets of AUX-IAA transcription factor within the fiber transcriptome were further analysed using agriGO tool (http://bioinfo.cau.edu.cn/agriGO/) [11]. Enrichment of certain GO terms was determined based on Fisher's exact test. During analysis a multiple correction control (permutation to control false discovery rate, FDR) was implemented to set up the threshold to obtain the lists of significantly over-represented GO terms.
Hierarchical Clustering of AUX-IAA target genes:
Candidate AUX-IAA targeted genes were further used for hierarchical cluster analyses through dChip (http://www.hsph.harvard.edu /cli/complab/dchip /) [12] using ecludiean matrix for gene expression data of six developmental time point for 5 genotype (two superior and three inferior) for identifying their expression trend. Two different cluster were formed on the basis of their expression pattern in different fiber development time point.
Correlation Analyses:
Correlation analyses for AUX-IAA target genes with homologues Arabidopsis genes were performed with the help of ATTED tool (http://atted.jp/). There were 139 correlated genes were found and 128 genes were overlapped with homologues cotton genes which needs further validation for understanding their potential role in developing fiber cells.
Results
Overview of the analytic procedure:
Our analytic procedure followed that of Xiaofei Yu et al.2011. In the study, a global regulatory network was inferred for cotton fiber development stages using oligonucleotide microarray data from GEO database. Here we focused on modelling gene regulatory networks only for the 8 auxin genes. From the use of the network model, we then extracted their downstream targets. Therefore, our study is more suitable to evaluate whether a regulatory model can successfully identify key upstream regulators (e.g. markers) for fiber development purely based on expression profiles without depending on external knowledge.
Regulatory gene networks:
Using a combined stringent cut off of an error tolerance ε = 0.2 and a P-value threshold of mutual information (MI) at 1e-7, ARACNE inferred global gene networks with direct interactions in the cotton fiber development. Eight AUX-IAA genes (Ghi.3606.1.A1 _at; Ghi.4821.1.S1_s _at; Ghi.9984.1.S1_ s_at; GraAffx.24472.1.S1_s_at; GhiAffx.1868.2.A1_at; GhiAffx.18267.1.S1_s_at; Gra.1987.1.S1_s_at; GhiAffx.34525.1. S1_s_at; GhiAffx.1868.2.S1_a_at) controlling more than 50 targets of all direct interactions considered as hubs or master regulators (Supplementary File 2 (Available with authors), Figure 2). If the AUX-IAA TFs indeed function to mediate auxin responses, we should be able to observe some expression correlation among them, which can be demonstrated by reconstruction of a gene regulatory network (GRN). For this purpose, we employed ARACNe (Algorithm for the Reconstruction of Accurate Cellular Networks), which was developed and previously used to infer a GRN in human B cells [13]. ARACNe calculates expression correlations between genes based on mutual information and picks out statistically significant correlation. ARACNE inferred direct interactions (1st neighbours) and 2nd neighbours for selected hub genes.
Figure 2.
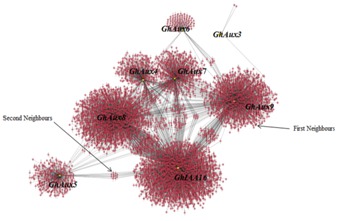
A transcriptional network for AUX-IAA regulated gene expression. GRN inferred by ARACNe. Red circles represent AUX-IAA targets. Yellow circles represent Gossypium AUX-IAA transcription factors (as a hub genes) in developing fibre cells of Gossypium hirsutum.
Expression correlation and gene ontology (GO) analyses:
GO terms significantly enriched in the fiber cells were identified using agriGO tool (Supplementary File 3 (Available with authors). In the first step of the analyses, we extracted and ranked the 128 genes out of 135 genes, whose expression most tightly correlates with that of AUX-IAA genes (Supplementary File 4 (Available with authors), Figure 3). We took advantage of a visualization tool called Cytoscape to view expression data and analysis results. Using the available data regarding correlations between different Arabidopsis genes, we constructed a network composed of the AUX-IAA genes and their co-expressed genes in developing cotton fiber cells.
Figure 3.
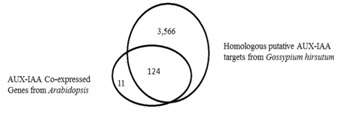
Venn analysis showing overlapping genes in between AUX-IAA co expressed genes of Arabidopsis and identified homologous putative AUX-IAA targets within developing fibre cells of Gossypium hirsutum.
To gain an insight over AUX-IAA correlated gene -percent distribution for transcription factor and cell signalling related genes we performed group categorization using Pathway studio 5.9 software with a parameter of p-value ≤ 0.05. We found that there were 136 transcription factor, 22 types of phosphatase, 11 receptor, and 119 protein kinase Table 1 (see supplementary material). In order to identify a functional role for these AUX-IAA genes, the correlated genes were further analysed using enrichment analysis module to identify any bias in GO functional annotation terms in the correlated using homologues A. thaliana genes. Most of these related to various development process such as pollen ovule, anther, root and shoot development, biotic-abiotic stress response, and transcription factor activity. Other biological processes identified were associated with extracellular activities, and enzyme activity. Thus, given previous work suggesting that AUX-IAA genes play a role in controlling architecture [14, 15]. We can infer that AUX-IAA genes may not only do this but also take part in many other biological processes.
Metabolic Interactome analysis:
To interpret functional associations between co-expressed proteins, we mapped the cotton genes whose expression was correlated with AUX-IAA genes (124 genes) using the Arabidopsis thaliana locus ids. Interactome analysis of this GRN suggested that many genes and proteins co-expressed with AUX-IAA genes supports their role in a diversity of physiology processes within fiber cell (Figure 4). Analysis of the connected genes identified functional modules for cell differentiation, trichome differentiation and development, flower leaf and shoot development, leaf morphogenesis and initiation, defence response as well as disease resistance. These observations support the theory that the network modelled here constitutes a framework that can guide in-depth experimental study of genes and proteins related to AUX-IAA gene function within developing fiber of Gossypium spp.
Figure 4.
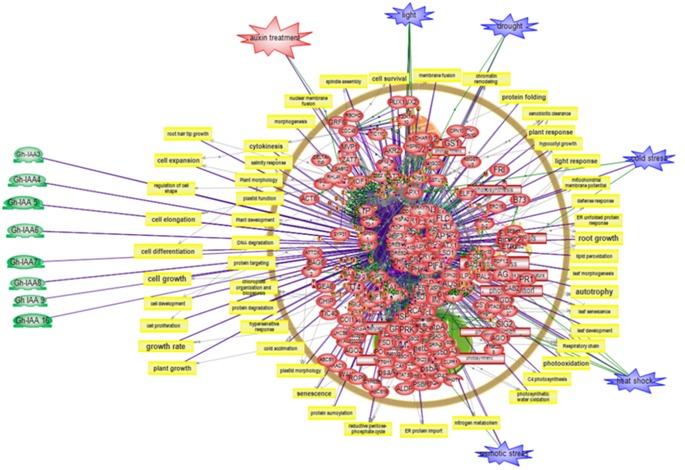
Metabolic network analysis showing probable biological functions of AUX-IAA targets genes identified in developing fibre cells of Gossypium hirsutum.
Differential expression pattern of AUX-IAA target genes reveals their putative role in fiber quality:
Two contrasting pattern identified here in genotype subgroup from cluster analysis reveals that AUX-IAA target genes has significant role in determining fiber quality. We categorized genes as early and late inducible on the basis of their expression in early (i.e. 0dpa) and late phase (19 and 25 dpa) of fiber development in superior and inferior genotypes respectively (Figure 5).
Figure 5.

Hierarchical clustering (using Ecludian distance matrix) of putative AUX-IAA targets genes in developing fibre cells (at six time point i.e. 0, 6, 9, 12, 19, 25) of five Genotypes of Gossypium hirsutum. Genotype Dependent: Early inducible differential putative AUX-IAA targets, Genotype Independent: Late inducible differential putative AUX-IAA targets.
Discussion
Although transcription factors are generally expressed at basic level and their half-life are short, microarray has been widely used for transcription factors expression research [16, 17] for function analysis of transcription factors their co-expression, new areas related with system biology and co-expression network biology are emerging day by day. In our analysis, we used microarray data to generate and classify co-expression network of AUX-IAA genes. We think that modelled gene regulatory network may help us to infer the function of putative AUX-IAA target genes in cotton fiber biology.
AUX-IAA target genes are involved in multiple aspects of auxin pathway:
AUX-IAA directly targets genes of different functional groups, including signalling molecules, enzymes, and many with unknown functions (Supplementary File 1). Some BR-responsive AUX-IAA target genes contributed to cell elongation and cellular growth, such as cell wall modifying enzymes. AUX-IAA also tightly connects the auxin pathway to othe r hormone responses in Arabidopsis. Genes involved, response to auxin stimulus (p value = 0.000064), auxin transport (p value = 0.000086), auxin polar transport (p value = 0.000086), auxin metabolic process (p value = 0.000046), auxin mediated signalling pathway (p value = 0.000068) auxin homeostasis, (p value = 0.000085), auxin binding (p value = 0.000021) were over-represented in AUX-IAA target genes according to gene ontology analysis (Supplementary file1).
AUX-IAA target genes may functions in determining fiber quality:
From our mining analysis it can be inferred that the gene that responsible for trichome development and initiation may also play role in fiber quality determination, as we previously shown that their contrasting expression pattern in two contrasting genotypes group may be responsible for fiber quality differ. It is well known that some genes related to auxin signaling pathway and pectin modification such as ARF2 (auxin reponse factor 2) antiauxin-resistant 3 (AAR3) preferentially up-regulated at the fast elongation stage in G. barbadense compared with G. hirsutum at 25 DPA [18]. Thus there may be differential regulation of AUX-IAA targeted gene during fiber development in superior and inferior genotypes which may need further investigation. This is first report on correlation between AUX-IAA and cotton fiber quality (Figure 4).
AUX-IAA target genes may play a key role in development of different organ of Arabidopsis:
Transcriptional control of the expression of organ development related genes is a crucial part until whole organogenesis. Nevertheless, our GO analysis revealed that some genes correlated with AUX-IAA genes were involved in the pollen development, carpel development xylem development, root development, shoot development showing its ubiquitous role within plant development e.g. MBA10.13, WUS, AUX-IAA, JAG (anther development), STM, AG, SPT, SHP2, SHP1, CRC, KAN, STK, JAG, KAN2, SEP4 (carpel development), STM, AG, SPT, SHP2, SHP1, CRC, KAN, STK, JAG, KAN2, SEP4 (cotyledon development), AGL63,SPT,RPL,AGL8,AFO,YAB3,ARF10 (fruit development) (Supplementary File 1, Figure 4). Previously it has been shown plants that Aux/IAAs and ARFs influence apical dominance, vascular development, tropic movements, and various tissue development with ubiquitous expression along with strong expression of some genes in xylem and phloem specifically [19, 20].
A interaction network between AUX-IAA target genes and their correlated TF's:
Our GO analysis shows transcription activity to be an important function for co-expressed genes. AUX-IAA genes can activate other transcription factor families, including RABC2b, MYH9.12, LIP1, F5A8.11, LOC100284549, Sb01g041370, GA3OX1, UFO, RBR1, AGL20, CKB3, AGL44, AGL16, VSP1, AGL21, RABC2a, FLR1. We believe that AUX-IAA genes function by controlling the expression of these transcription families. We therefore hypothesize a complex control network linking AUX-IAA genes with other transcription families.
AUX-IAA target genes and their interaction with other hormones:
AUX-IAA interacting genes showed to involve in various plant hormone signalling pathway related with abscisic acid mediated signalling pathway, auxin mediated signalling pathway, brassinosteroid homeostasis, ethylene mediated signaling pathway, jasmonic acid mediated signalling pathway, response to salicylic acid stimulus. One interesting acitvity within AUX-IAA correlated gene that we observed was jasmonic acid and ethylene-dependent systemic resistance. It is well reported that auxin interact with brassinosteroid during vegetative growth [21]. Recently researches have proposed that auxin regulated growth-promoting genes during hypocotyl growth follows GA-dependent and -independent pathways. Different genes related with each of these category were mentioned in (Supplementary File 1, Figure 4).
AUX-IAA target genes and its role in stress stimuli:
Our analysis showed that AUX-IAA genes helps in perceiving stimuli of different biotic and abiotic stress, i.e. cellular response to drought, osmotic stress, cold stress, heat stress etc. A number of genes that comes within these AUX-IAA correlated categories show their role in diverse role of stress stimuli and response (supplementary File 1). Recently a Genome-wide analysis have been performed for Aux/IAA gene family in different plant focusing on evolution and expression patterns in various tissues, developmental process and in response to auxin or abiotic stresses [22, 23, 24] (Supplementary File 1, Figure 4).
AUX-IAA target genes have varied functions:
The AUX-IAA gene family is a group of plant-specific genes that encodes plant-specific transcription factor. AUX-IAA transcription factors may have a variety of physiological functions. To date, several important and divergent biological processes regulated by AUX-IAA genes have been reported. Our co-expression analysis has revealed that AUX-IAA genes may play a role in diverse developmental processes. Moreover, several auxin-responsive genes were differentially expressed under various abiotic stress conditions, indicating probable crosstalk between auxin and abiotic stress signaling [25]. Network analysis suggests that AUX-IAA genes function by controlling other transcription factor families. The interactome of the correlated genes has shown that AUX-IAA genes may also take part in sucrose stimuli, ion -transport inorganic salt, and ATP production.
Conclusion
Cotton fiber biology in itself is a vast area where multiple transcription factors are major players influencing its trait. Among them AUX-IAA box gene family represent to a group of plant-specific zinc finger protein encoding genes. The expression of AUX-IAA genes are significantly correlated with that of genes involved in the cell wall, genes related with unidimensional cell morphogenesis. The expression of AUX-IAA genes are correlated with other transcription factors gene and some integral membrane proteins. Additionally some AUX-IAA genes are correlated with themselves. All of these analyses suggest that AUX-IAA genes may play an important role in developing fiber cell.
Accession codes
The gene expression profiles used in this study were Series GSE36228 (in house) and two Series GSE36021 and GSE29810 (publically available)
Funding sources
DN thanks to CSIR India for supporting fellowship as SRF.
Conflict of Interest
The authors declare that there is no conflict of interest.
Supplementary material
Acknowledgments
DN is grateful to the Council of Scientific and Industrial Research, Government of India, New Millennium Technology Leadership Initiative (NMITLI) program for the financial support. DN, is thankful to CSIR for senior research fellowship.
Footnotes
Citation:Nigam & Sawant, Bioinformation 9(20): 996-1002 (2013)
References
- 1.Friml J. Curr Opin Plant Biol. 2003;6:7. doi: 10.1016/s1369526602000031. [DOI] [PubMed] [Google Scholar]
- 2.Abel S, Theologis A. Plant Physiol. 1996;111:9. doi: 10.1104/pp.111.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guilfoyle T, et al. Plant Physiol. 1998;118:341. doi: 10.1104/pp.118.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leyser O. Annu Rev Plant Biol. 2002;53:377. doi: 10.1146/annurev.arplant.53.100301.135227. [DOI] [PubMed] [Google Scholar]
- 5.Arase F, et al. PLoS One. 2012;7:e43414. doi: 10.1371/journal.pone.0043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gokani SJ, Thaker VS. Field crops research. 2002;77:127. [Google Scholar]
- 7.Han X, et al. Gene. 2012;503:83. doi: 10.1016/j.gene.2012.03.069. [DOI] [PubMed] [Google Scholar]
- 8.Margolin AA, et al. BMC Bioinformatics. 2006;7:S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaudry NJ, Renner R. Quantum Information & Computation. 2012;12:432. [Google Scholar]
- 10.Cover TM, Thomas JA. Elements of Information Theory 2nd Edition. 2006 [Google Scholar]
- 11.Nigam D, et al. Plant Biotechnology J. 2013 [Google Scholar]
- 12.Li C, Wong WH. DNA-chip analyzer (dChip) 2003;120 [Google Scholar]
- 13.Basso K, et al. Nat Genet. 2005;37:382. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 14.Notaguchi M, et al. J Integr Plant Biol. 2012;54:760. doi: 10.1111/j.1744-7909.2012.01155.x. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Gera H, Ori N. Plant Signal Behav. 2012;7:1255. doi: 10.4161/psb.21550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arora R, et al. BMC Genomics. 2007;8:242. doi: 10.1186/1471-2164-8-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nijhawan A, et al. Plant Physiol. 2008;146:333. doi: 10.1104/pp.107.112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, et al. PLoS One. 2012;7:e30056. doi: 10.1371/journal.pone.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalluri UC, et al. BMC Plant Biol. 2007;7:59. doi: 10.1186/1471-2229-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodward AW, Bartel B. Ann Bot. 2005;95:707. doi: 10.1093/aob/mci083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura A, et al. Plant J. 2005;45:193. [Google Scholar]
- 22.Audran-Delalande C, et al. Plant Cell Physiol. 2012;53:659. doi: 10.1093/pcp/pcs022. [DOI] [PubMed] [Google Scholar]
- 23.Scherer GFE, et al. Trends Plant Sci. 2010;15:693. doi: 10.1016/j.tplants.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Song Y, et al. Planta. 2009;229:577. doi: 10.1007/s00425-008-0853-7. [DOI] [PubMed] [Google Scholar]
- 25.Jain M, Khurana JP. FEBS J. 2009;276:3148. doi: 10.1111/j.1742-4658.2009.07033.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.