

Abstract
Transforming growth factor β (TGF-β) inhibits myogenesis and associated gene expression. We previously reported that the TGF-β signaling effector Smad3 mediates this inhibition, by interfering with the assembly of myogenic bHLH transcription factor heterodimers on E-box sequences in the regulatory regions of muscle-specific genes. We now show that TGF-β-activated Smad3 suppresses the function of MEF2, a second class of essential myogenic factors. TGF-β signaling through Smad3 represses myogenin expression independently of E-boxes, and prevents a tethered MyoD-E47 dimer to activate transcription indirectly through MEF2-binding sites. In addition, Smad3 interacts with MEF2C, which requires its MADS domain, and disrupts its association with the SRC-family coactivator GRIP-1, thus diminishing the transcription activity of MEF2C. Consistent with this physical displacement, TGF-β signaling blocks the GRIP-1-induced redistribution of MEF2C to discrete nuclear subdomains in 10T1/2 cells, and the recruitment of GRIP-1 to the myogenin promoter in differentiating myoblasts. These findings indicate that the TGF-β/Smad3 pathway targets two critical components of the myogenic transcription machinery to inhibit terminal differentiation.
Keywords: MEF2, myogenesis, Smad, TGF-β
Introduction
The induction of the skeletal myogenic differentiation depends on the activities of two groups of transcription factors. The first is the MyoD family of basic–helix–loop–helix (bHLH) factors, also known as myogenic regulatory factors (MRFs), which include MyoD, myogenin, Myf5 and MRF4. Ectopic expression of any single MRF converts non-myogenic, mesenchymal cells into myoblasts, and germline mutations of MRFs result in defective muscle formation in mice (Molkentin and Olson, 1996; Yun and Wold, 1996). Transcriptional activation by MRFs occurs through dimerization with E proteins, another class of bHLH proteins. MRF/E protein complexes bind to conserved E-box sequences present in enhancers of muscle-specific genes (Lassar et al, 1991). The second class of transcription factors essential for muscle development is the myocyte enhancer factor 2 (MEF2) family. MEF2A–D share homology in the MADS and adjacent MEF2 domains, which mediate dimerization, DNA binding and cofactor interactions (Black and Olson, 1998). MEF2 proteins lack myogenic activity by themselves, but potentiate the activity of MRFs through combinatorial association and transcriptional cooperation (Molkentin et al, 1995). MRFs and MEF2 factors regulate each other's expression in positive feedback loops (Braun et al, 1989; Edmondson et al, 1992).
Consistent with their pivotal roles in muscle differentiation, MRF and MEF2 are targeted by signaling pathways that restrict the progression of myogenesis (Naya and Olson, 1999; Perry and Rudnicki, 2000). Notably, activation of Ca2+/camodulin-dependent kinases (CaMKs) at the onset of muscle differentiation dynamically releases class II histone deacetylases (HDACs) from MEF2 at promoters of muscle genes, resulting in association of MEF2 with coactivators with histone acetyltransferase (HAT) activity and enhanced myogenic activity (McKinsey et al, 2000, 2002). Conversely, the cdk4/cyclin D kinase activity prevents the activation of MEF2 by the coactivator GRIP-1, thus suppressing myogenic differentiation (Lazaro et al, 2002).
Members of the transforming growth factor β (TGF-β) family are potent inhibitors of terminal differentiation of cultured myoblasts (Massagué et al, 1986; Olson et al, 1986). The suppression of differentiation by TGF-β in cells that constitutively overexpress MyoD or myogenin suggests post-transcriptional repression of muscle-specific gene expression (Vaidya et al, 1989; Brennan et al, 1991). Until recently, the intracellular signaling components and mechanisms that mediate such transcriptional repression have remained obscure.
TGF-β signals are transmitted by protein complexes consisting of receptor-activated Smads, Smad2 and/or Smad3, and a common mediator Smad4 (Shi and Massagué, 2003). Since Smads regulate gene expression through physical and functional interaction with transcription factors, we investigated the role of Smads in TGF-β repression of MRF activity. We found that Smad3, but not Smad2, blocks MyoD-mediated transcriptional activation by associating with the bHLH region of MyoD. This interaction interferes with MyoD/E protein dimerization and cooperative binding to E-boxes (Liu et al, 2001). These findings revealed a mechanism whereby TGF-β-activated Smad3 inhibits E-box-dependent muscle gene expression. Nonetheless, supplying an excess of E12 only partially overcame the repression of MyoD-mediated activation of the muscle creatine kinase (MCK) promoter by TGF-β/Smad3, suggesting that additional regulatory elements are targeted for repression (Liu et al, 2001).
Here we demonstrate that TGF-β-activated Smad3 inhibits MEF2-dependent transcription. Smad3 interacts with MEF2C, and represses its transcription activity without affecting DNA binding. Our results suggest versatile roles for Smad3 in the suppression of muscle-specific gene expression through multiple mechanisms.
Results
TGF-β-activated Smad3 represses muscle-specific transcription independently of E-boxes
TGF-β, through Smad3, inhibits the formation of MyoD/E protein heterodimers on E-boxes, a prerequisite for efficient activation of the myogenic transcription program (Liu et al, 2001). This is supported by the observation that the activity of a tethered MyoD∼E47 dimer, in which MyoD was linked to E47, was refractory to Smad3-mediated repression, as determined by expression of a reporter gene under the control of E-box sites. However, the activation of the MCK promoter by MyoD∼E47 remained partially sensitive to inhibition by TGF-β/Smad3 (Liu et al, 2001). To corroborate this finding, we evaluated the capacity of MyoD∼E47 to induce myogenic conversion of C3H10T1/2 fibroblasts in the presence or absence of elevated TGF-β signaling. TGF-β treatment and Smad3 coexpression significantly impaired the expression of myogenin, a direct transcription target of MyoD, and the late differentiation marker myosin heavy chain (MHC) in cells exogenously expressing MyoD∼E47 (Figure 1). Since TGF-β/Smad3 signaling does not affect MyoD∼E47 binding and transactivation of E-boxes (Liu et al, 2001), we concluded that in addition to E-boxes, the repression by Smad3 is directed at other cis-regulatory element(s) present in complex muscle promoters/enhancers.
Figure 1.
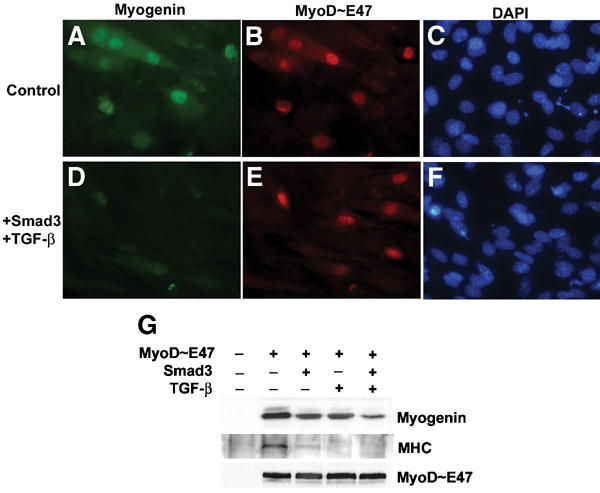
TGF-β-activated Smad3 inhibits induction of myogenin expression in C3H10T1/2 fibroblasts by the tethered MyoD∼E47 dimer. 10T1/2 cells were transfected with a plasmid encoding MyoD∼E47 (A–C), or together with a plasmid for Smad3 (D–F). After transfection, cells were cultured in DM with or without 2 ng/ml TGF-β, as indicated, for 15 h and processed for immunofluorescence staining using anti-myogenin antibody (green). Expression of exogenous MyoD∼E47 was detected using anti-HA antibody (red), and nuclei were visualized by DAPI staining (blue). The induction of myogenin or MyoD∼E47 15 h after switching to DM, and expression of skeletal MHC after 3 days in DM were evaluated by immunoblots (G).
Smad3 inhibits MEF2-dependent transcription
Like many genes associated with myogenesis, transcriptional activation of myogenin depends on the cooperation of MyoD and MEF2 proteins (Molkentin et al, 1995). A composite module of an E-box and MEF2-binding site in the myogenin promoter, located within 184 nucleotides upstream of the transcription start site, has been shown to confer its proper muscle-specific expression (Edmondson et al, 1992). As shown in Figure 2A, transient expression of MyoD∼E47, but not MEF2C alone, strongly activated the myogenin promoter in 10T1/2 cells. Deletion of the E-box did not prevent promoter activation, consistent with reports suggesting that MyoD indirectly induces myogenin expression through MEF2 (Buchberger et al, 1994). Coexpression of Smad3 repressed the reporter activity, an effect more evident in TGF-β-treated cells. On the other hand, mutation of the MEF2 site eliminated the promoter activity. These results suggest that Smad3-mediated inhibition of myogenin promoter activation is targeted at the ability of a functional MyoD/E protein complex, in cooperation with MEF2, to activate transcription through the MEF2-binding site.
Figure 2.
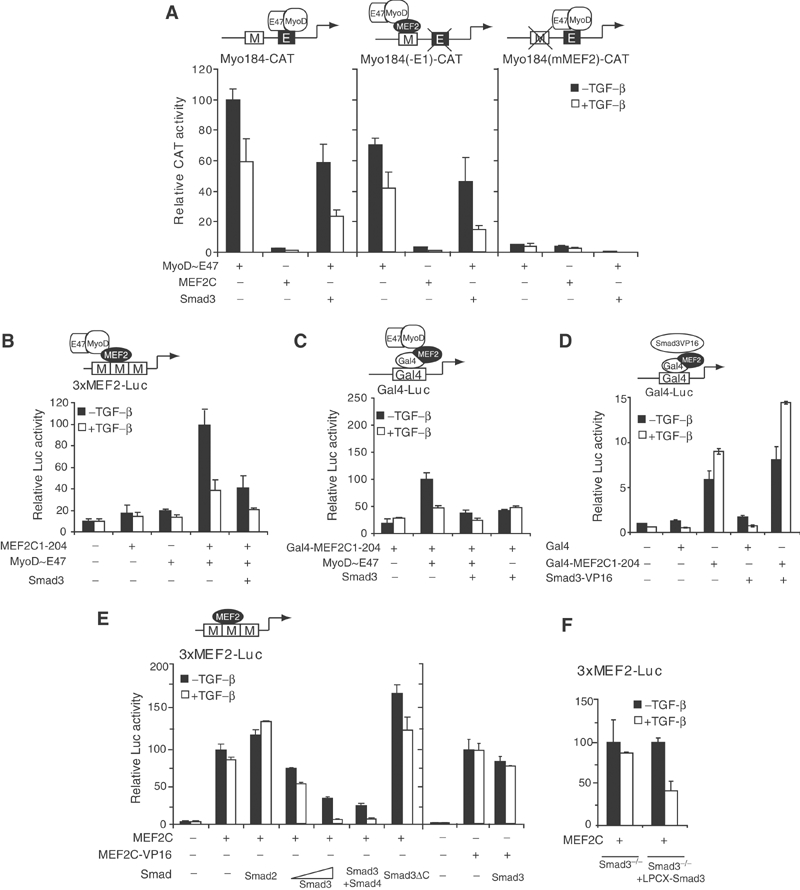
TGF-β/Smad3 represses transcription from MEF2-binding sites. (A) Effects of TGF-β and Smad3 on activation of the myogenin promoter by MyoD∼E47. 10T1/2 cells were cotransfected with MyoD∼E47 and Smad3, along with reporter plasmids driven by the myogenin promoter (Myo184-CAT), or myogenin promoter with deletion of the functional E-box site (Myo184(-E1)-CAT), or with a mutated MEF2 site (Myo184(mMEF2)-CAT). Cells were treated with or without 2 ng/ml TGF-β for 24 h before lysis to measure CAT activities. Relative CAT values were normalized against the expression of a control β-gal plasmid and are averages of replicate experiments. (B) Effects of TGF-β/Smad3 on the cooperative transcriptional activation through MyoD∼E47 and MEF2C association. 10T1/2 cells were cotransfected with MyoD∼E47, a truncated MEF2C containing amino acids 1–204, and Smad3, together with an MEF2 reporter 3xMEF2-Luc. Luciferase activities were measured in cell lysates 24 h after incubation in DM with or without 2 ng/ml TGF-β. (C) Effects of TGF-β/Smad3 on the interaction between MEF2C and MyoD∼E47. 10T1/2 cells were transfected with the indicated plasmids in Gal4 reporter assays to score transcription activity. (D) Effects of Smad3 fused to the VP16 TAD on Gal4-fused MEF2C1-204 in reporter assays as in (C), but shown on a different scale. (E) Effects of TGF-β/Smad3 on the transcriptional activity of MEF2C and MEF2C-VP16. 10T1/2 cells were cotransfected with MEF2C or MEF2C-VP16 fusion and the indicated Smads or Smad mutants, along with the 3xMEF2-Luc reporter. (F) Requirement of Smad3 for transcriptional repression of MEF2 by TGF-β. MEF2 reporter assays, as in (B), were performed in Smad3−/− mouse fibroblasts or the same cells that re-express Smad3 from a viral promoter (LPCX-Smad3).
To determine if TGF-β-activated Smad3 reduces the transcription synergy between MyoD and MEF2, we performed assays using a reporter under the control of tandem MEF2 sites (Figure 2B). Unlike the myogenin promoter that lacks an E-box, MyoD∼E47 by itself did not activate the expression of the MEF2 reporter. Neither did MEF2C1-204, a truncated MEF2C lacking the transcriptional activation domain (TAD). However, coexpression of MyoD∼E47 and MEF2C1-204 activated the reporter, presumably through MyoD TAD tethered to MEF2 sites. This activity is inhibited by Smad3, especially in the presence of TGF-β. We then determined if TGF-β/Smad3 inhibits transactivation of a heterologous promoter by MyoD TAD tethered through the MADS/MEF2 domain of MEF2C. As shown in Figure 2C, expression of Smad3, in combination with TGF-β, abolished the activation of a luciferase reporter under the control of Gal4-binding sites by MyoD∼E47, anchored through a fusion protein of the Gal4 DNA-binding domain (Gal4-DBD) and MEF2C1-204. However, fusion of Smad3 with the TAD of HSV VP16 converted Smad3 to an activator of the Gal4 reporter, presumably due to physical interaction of Smad3 with MEF2C1-204 (Figure 2D). These results demonstrate that TGF-β-activated Smad3 blocks the transcriptional activation by MyoD bound to DNA through protein–protein interaction with MEF2, without an obligatory involvement of the MEF2C transactivation function. They also raise the possibility that Smad3/MEF2C interaction interferes with the association and cooperation between MyoD∼E47 and MEF2C.
Besides serving as a cofactor for myogenic bHLH proteins during myogenesis, MEF2 can activate transcription by itself. We therefore investigated whether Smad3 could repress MEF2-dependent gene expression by interfering with the transcription activity of MEF2 itself (Figure 2E). Under conditions of excess MEF2C expression, endogenous Smad3 is inefficient in mediating repression of MEF2 activity by TGF-β signaling, whereas increasing levels of exogenous Smad3 dose-dependently inhibited the transactivation of the 3xMEF2-Luc reporter by MEF2C. This inhibition was enhanced by TGF-β, and by cotransfection of Smad4. Unlike Smad3, Smad2 mildly augmented MEF2C-dependent transcription, possibly by competing with endogenous Smad3 for Smad4 association, which has been shown to account for the antagonistic effects of Smad2 and Smad3 on goosecoid promoter activation (Labbé et al, 1998). Similarly, expression of Smad3ΔC, a dominant-negative mutant of Smad3, increased the MEF2C-activated transcription, reflecting autocrine TGF-β signaling.
In contrast to wild-type MEF2C a MEF2-VP16 fusion with the transcription activation domain of MEF2 replaced by that of VP16 activated the MEF2 reporter expression to similar levels in the presence or absence of TGF-β-activated Smad3, suggesting that repression of MEF2-dependent transcription is not due to diminished DNA binding of MEF2C (Figure 2E, last two lanes). This result also suggests that the effect of Smad3 was not due to a nonspecific transcription squelching, since a heterologous TAD from VP16 was not significantly affected.
Finally, to determine if the ability of TGF-β signaling to repress MEF2 activity requires Smad3, we assayed transcription in Smad3−/− fibroblasts. TGF-β did not significantly repress the MEF2 reporter activity. In contrast, constitutive expression of Smad3 conferred a robust inhibitory response (Figure 2F). Therefore, the repression of MEF2 activity by TGF-β is mediated through Smad3.
MH2 domain of Smad3 interacts physically with MEF2C
Considering the ability of Smads to associate with other transcription factors and modulate their activities, we evaluated the physical interaction between MEF2C and Smad proteins involved in TGF-β signaling. As shown in Figure 3A, HA-tagged MEF2C was co-immunoprecipitated with Flag-tagged Smad3 in cells cotransfected with Smad3, but not with Smad2 or Smad4. In addition, Smad3C, which contains the MH2 domain of Smad3, co-precipitated with MEF2C, whereas Smad3NL, containing the MH1 domain and linker region, did not. The lack of a Smad2/MEF2 association might explain the inability of Smad2 to inhibit the transcriptional activity of MEF2C (Quinn et al, 2001). To corroborate these results, we assessed the ability of recombinant Smads to interact with MEF2C in vitro (Figure 3B). Of the four Smad GST fusion proteins tested, Smad1–4, only Smad3 had significant affinity for in vitro-translated MEF2C. In agreement with the immunoprecipitation assays, the GST fusion of the Smad3 MH2 domain (GST-Smad3C), but not the MH1-linker region (GST-Smad3NL), bound to MEF2C.
Figure 3.
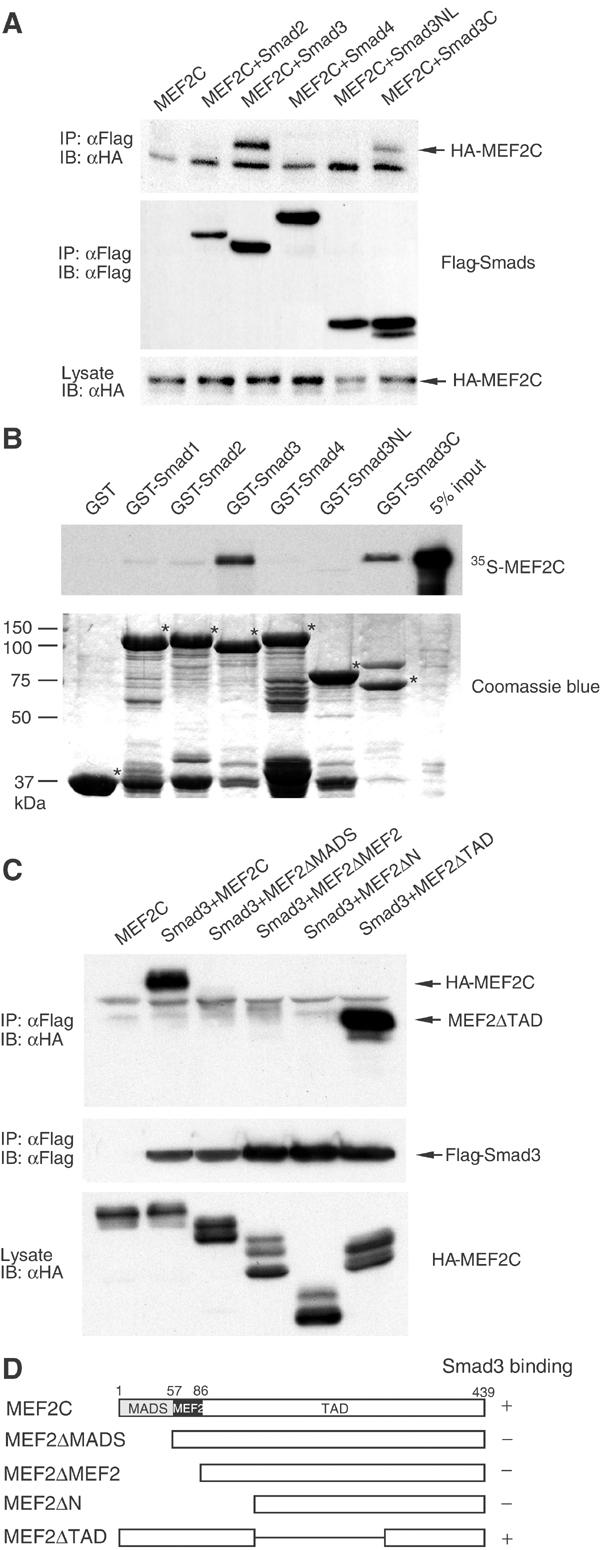
Smad3 interacts with MEF2C in vivo and in vitro. (A) Association of Smad proteins with MEF2C. COS cells were transfected with HA-tagged MEF2C and Flag-tagged Smad2, 3 or 4, or Smad3 fragments (Smad3NL and Smad3C). Smad proteins were immunoprecipitated (IP) from transfected cell lysates using anti-Flag antibody, and the Smads or MEF2C in the immune complexes were detected in immunoblots (IB) using anti-Flag or anti-HA antibody, respectively (upper and middle panels). Expression of MEF2C was assessed by immunoblotting of a portion of the cell lysates (lower panel). (B) Interaction of recombinant GST-Smad1–4 with 35S-labeled, in vitro-translated MEF2C. The upper panel shows MEF2C retained by the indicated GST-Smad proteins immobilized on agarose beads. The same gel stained with Coomassie blue, in the lower panel, shows the integrity and equal loading of the GST fusion proteins. (C) Mapping of the Smad3 interaction domain in MEF2C. HA epitope-tagged, full-length or MEF2C deletion mutants, shown in (D), were cotransfected with Flag-tagged Smad3. Flag immunoprecipitates were subjected to immunoblotting using anti-HA antibody (upper panel). Expression levels of Smad3 and MEF2C mutants in the cell lysates are shown in immunoblots (middle and lower panels).
We also tested the ability of deletion mutants of MEF2C to interact with Smad3 (Figure 3C and D). Deletion of the N-terminal MADS domain abolished the interaction. In contrast, truncating the C-terminal region did not affect the interaction of MEF2C with Smad3. Thus, the MADS domain of MEF2C is required for its interaction with Smad3.
Since the MADS domain of MEF2C also mediates DNA binding, we tested whether Smad3 could associate with MEF2C while bound to DNA. In a DNA adsorption assay, Smad3 was co-precipitated with MEF2 using a biotinylated oligonucleotide corresponding to the MEF2-binding site from the MCK enhancer (Figure 4). Smad3 did not bind to the MEF2 oligonucleotide in the absence of MEF2C, or a control mutant MEF2 oligonucleotide. This result indicates that binding of MEF2C to DNA or Smad3 is not mutually exclusive, consistent with our similar finding based on reporter assays (Figure 2C).
Figure 4.
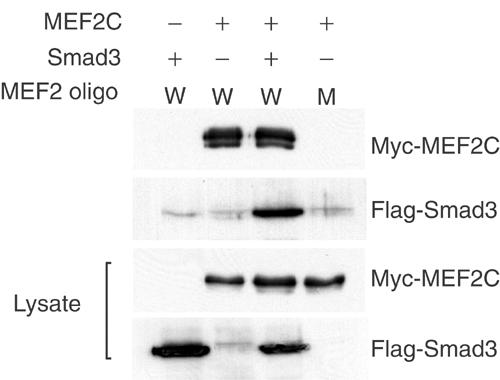
Smad3 interacts with MEF2C tethered to its target DNA sequences. Biotinylated wild-type MEF2 (W) or mutant MEF2 (M) oligonucleotide immobilized on streptavidin beads was incubated with lysate of COS cells transfected with the indicated expression plasmids. MEF2C and Smad3 proteins bound to the MEF2 cognate sequences were analyzed by immunoblotting using anti-Myc or anti-Flag antibodies, respectively. The lower panels show the expression levels of Myc-MEF2C and Flag-Smad3 proteins as analyzed in immunoblots of the cell lysates.
Smad3-mediated repression is directed at the MEF2 coactivator function of GRIP-1
The function of MEF2 family proteins is defined by interacting repressors and coactivators involved in chromatin remodeling (McKinsey et al, 2001). Because the MADS domain of MEF2 can recruit class II HDACs, which act as signal-responsive repressors (Miska et al, 1999; Lu et al, 2000), we evaluated the possibility that repression of MEF2 activity by Smad3 results from enhanced recruitment of HDACs. Our results ruled out such a mechanism, as the HDAC inhibitor trichostatin A (TSA) did not significantly affect Smad3-mediated repression in MEF2 reporter assays (Figure 5A).
Figure 5.
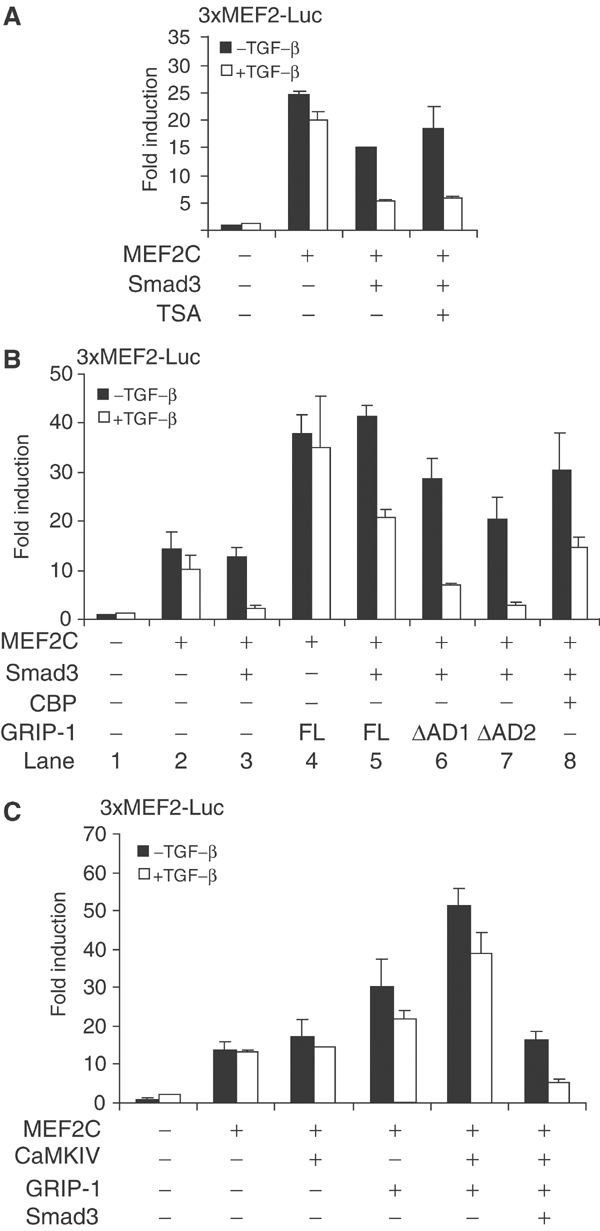
Smad3-mediated repression is targeted at a MEF2 coactivator. (A) Effect of TSA on inhibition of TGF-β/Smad3-mediated repression of MEF2 function. 10T1/2 cells were transfected with the indicated plasmids along with the 3xMEF2-Luc reporter. In lane 4, cells were treated with TSA in DM prior to reporter activity assay as in Figure 2. (B) Reversal of Smad3-mediated repression by excess GRIP-1 or CBP. 10T1/2 cells were transfected with the indicated expression plasmids for MEF2C, Smad3, GRIP-1, GRIP-1 deletion mutants (ΔAD1 and ΔAD2) or CBP, as shown. (C) Inhibition of GRIP-1 coactivation of MEF2-dependent transcription by TGF-β/Smad3. GRIP-1 and CaMKIV were coexpressed with MEF2C in MEF2 reporter assays. Smad3 overexpression and TGF-β treatment reduced the transcription synergy conferred by GRIP-1 and CaMKIV.
Conversely, the transcription activity of MEF2 is potentiated by coactivators such as CBP/p300 (Sartorelli et al, 1997) and GRIP-1 (Chen et al, 2000b) that associate with the MADS domain. GRIP-1, first identified as a nuclear receptor-interacting protein, is required for the transactivation function of MEF2 and myogenic differentiation (Chen et al, 2000b). Thus, Smad3-mediated repression of MEF2 activity may be targeted against the coactivator association. Consistent with this hypothesis, coexpression of GRIP-1 enhanced MEF2-dependent transcription and overcame the repression by TGF-β/Smad3 (Figure 5B, lane 5). A similar effect was also observed when CBP was overexpressed (Figure 5B, lane 8). Within GRIP-1, the activation domain 1 (AD1), at amino acids 1040–1120, activates transcription through recruitment of p300/CBP (Yao et al, 1996; Torchia et al, 1997). A second activation domain near the C-terminus, AD2, encodes histone acetyltransferase activity, and binds to the CARM-1 methyltransferase, which also contributes to transcriptional activation (Chen et al, 2000a; Ma et al, 2001). The GRIP-1 deletion mutants, GRIP-1-ΔAD1 and GRIP-1-ΔAD2, were less effective than full-size GRIP-1 in overcoming the repression of MEF2 reporter expression by TGF-β/Smad3 (Figure 5B, lanes 6 and 7), suggesting a role for both AD1 and AD2. This result also implies that the partial rescue of Smad3-mediated repression by CBP overexpression may relate to the compensatory HAT activity of CBP, through direct association with MEF2, or recruitment to the AD1 domain of GRIP-1.
Reciprocally, we determined the effect of increased TGF-β/Smad3 signaling on GRIP-1 coactivation of MEF2-dependent transcription. As reported (Lazaro et al, 2002), expression of GRIP-1 with activated CaMKIV enhanced the MEF2 reporter gene expression (Figure 5C). However, Smad3 overexpression in the presence of TGF-β abolished this synergistic activity. Together, these data strongly suggest that the repression of MEF2 activity by TGF-β-activated Smad3 is targeted at a coactivator function such as that of GRIP-1.
Smad3 disrupts the association of MEF2C with GRIP-1
Since both Smad3 and GRIP-1 physically interact with the MADS domain of MEF2C, and TGF-β-activated Smad3 interferes with the synergy between GRIP-1 and MEF2C, we thought that the association of MEF2C with Smad3 may preclude its association with GRIP-1. We therefore evaluated the effect of Smad3 on the GRIP-1/MEF2C interaction using mammalian two-hybrid assays (Figure 6A). In these assays, GRIP-1 fused to the Gal4 DBD (Gal4-GRIP-1) was coexpressed in 10T1/2 cells with a fusion protein of the MADS/MEF2 domain of MEF2C linked to VP16 TAD (MEF2C-VP16), along with the Gal4-luciferase reporter. The interaction between MEF2C and GRIP-1 correlates with the reporter activation through VP16 TAD (Figure 6A, lane 4). Coexpression of Smad3 largely abolished this interaction, an effect further enhanced by TGF-β (Figure 6A, lane 5). Unlike MEF2C, GRIP-1 did not interact with Smad3 (lane 6), and in contrast to its effect on GRIP-1/MEF2C association, Smad3 did not significantly affect the association of MEF2C with CBP-NS (Horvai et al, 1997; De Luca et al, 2003), a CBP fragment shown to bind MEF2 (Figure 6A, last two lanes). We conclude that increased nuclear levels of Smad3 disrupt the physical interaction of GRIP-1 and MEF2C.
Figure 6.
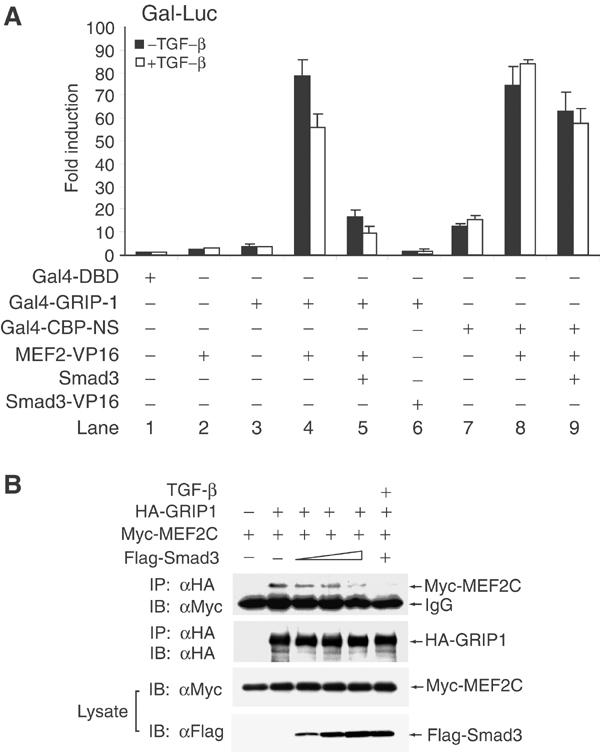
Smad3 interferes with the interaction of MEF2C with GRIP-1. (A) Effects of TGF-β/Smad3 on MEF2C/GRIP-1 or MEF2C/CBP association in mammalian two-hybrid assays. 10T1/2 cells were transfected with expression plasmids for Gal4 DNA-binding domain (Gal4-DBD), Gal4-DBD fused to GRIP-1 (Gal4-GRIP-1) or amino acids 1061–1891 of CBP (Gal4-CBP-NS), VP16 fusion of MEF2C and Smad3, and Smad3, together with the Gal4-Luc reporter. Cells were then treated with or without 2 ng/ml TGF-β and luciferase reporter assays were performed as described above. (B) Effects of TGF-β/Smad3 on co-immunoprecipitation of MEF2C and GRIP-1. HA-tagged GRIP-1 and Myc-tagged MEF2C were coexpressed with increasing amounts of Flag-tagged Smad3 in COS cells. Cell lysates were subjected to immunoprecipitation using anti-HA antibody and immunoblots for tagged GRIP-1 and MEF2C. Portions of the lysates were used to evaluate the expression of Myc-MEF2C and Flag-Smad3 by immunoblot analyses.
We also evaluated the ability of Smad3 to block the in vivo complex formation of GRIP-1 and MEF2C by co-immunoprecipitation. As shown in Figure 6B, increasing level of Smad3 resulted in dissociation of MEF2C from GRIP-1. TGF-β treatment of cells overexpressing Smad3 further depleted MEF2C from the GRIP-1 immune complexes. These results further demonstrate that competitive dissociation of the coactivator GRIP-1 from MEF2C is at the basis of the Smad3-mediated repression of MEF2C activity.
Smad3 displaces MEF2C from nuclear subdomains containing GRIP-1
GRIP-1, ectopically expressed in 10T1/2 cells, sequesters MEF2C to punctate nuclear structures (Lazaro et al, 2002). We thus investigated the effect of Smad3 on the nuclear colocalization of MEF2C and GRIP-1. As reported, GRIP-1 exhibited a punctate nuclear distribution in transfected cells (Figure 7D). When expressed alone, MEF2C was diffusely localized throughout the nucleus (Figure 7B and C). GRIP-1 coexpression localized a substantial fraction of the MEF2C protein into punctate nuclear subdomains that coincided with GRIP-1 (Figure 7E and F). Smad3 coexpression, however, conferred a less-restricted, diffuse pattern of MEF2C staining (Figure 7H–L). In contrast to the observation that Cdk4 activity excludes GRIP-1 and MEF2C from subnuclear structures (Lazaro et al, 2002), elevated TGF-β signaling did not alter the punctate GRIP-1 nuclear staining (Figure 7G and J). Therefore, while TGF-β-activated Smad3 does not influence the intranuclear distribution of GRIP-1 itself, it prevents the tethering of MEF2C by GRIP-1 to specific subnuclear structures.
Figure 7.
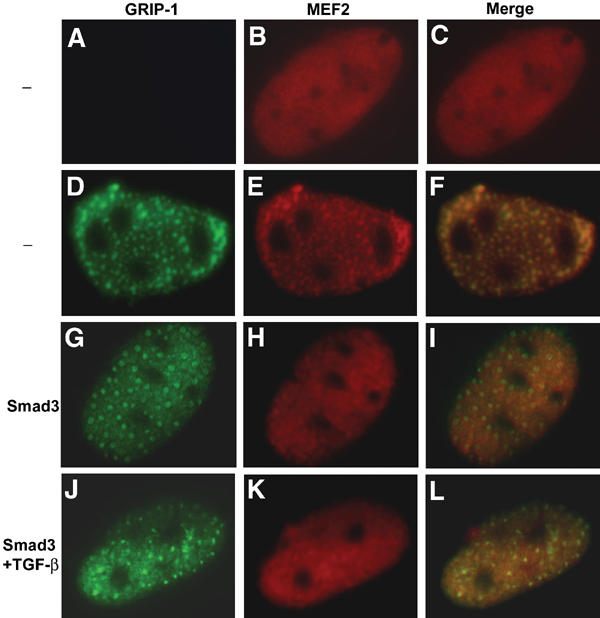
TGF-β-activated Smad3 altered the subnuclear colocalization of MEF2C and GRIP-1. 10T1/2 cells were transfected with expression plasmids for MEF2C alone (A–C), MEF2C and GRIP-1 (D–F) or MEF2C, GRIP-1 and Smad3 (G–L). Cells were cultured in the presence or absence of TGF-β, and the subnuclear distributions of MEF2C (red) and GRIP-1 (green) were determined by immunofluorescence. Colocalization of the proteins is indicated by the yellow staining in merged images.
TGF-β signaling blocks the recruitment of GRIP-1 to the myogenin promoter
During myoblast differentiation, GRIP-1 accumulates in the nucleus and assembles with MEF2 at target promoters to function as an essential coactivator for muscle-specific transcription (Chen et al, 2001; Lazaro et al, 2002). To determine if TGF-β signaling through Smad3 interferes with the recruitment of endogenous GRIP-1 to MEF2 sites on the promoters of muscle-specific genes, we carried out chromatin immunoprecipitations using PCR primers spanning the MEF2-binding sites. Thus, we isolated the MEF2/GRIP-1 transcription complexes from proliferating, undifferentiated C2C12 myoblasts and cells undergoing differentiation, as well as differentiating cells exposed to TGF-β. As shown in Figure 8A, the amount of GRIP-1 in association with the myogenin promoter segment was higher in differentiating myoblasts compared to proliferating cells. Importantly, recruitment of GRIP-1 at the myogenin promoter was reduced in TGF-β-treated cells, whereas MEF2A/C bound to chromatin was unaffected. A similarly decreased interaction of GRIP-1 also occurred at the MCK promoter (Figure 8A). In contrast, TGF-β treatment did not decrease androgen-induced recruitment of GRIP-1, through DNA binding of the activated androgen receptor, to the prostate-specific antigen (PSA) promoter (Figure 8B), which is transcriptionally repressed by TGF-β (Hayes et al, 2001). These results reinforce our conclusion that elevated TGF-β signaling results in dissociation of GRIP-1 from transcription complexes bound to the MEF2 sites, which correlates with reduced activation of MEF2-dependent transcription.
Figure 8.
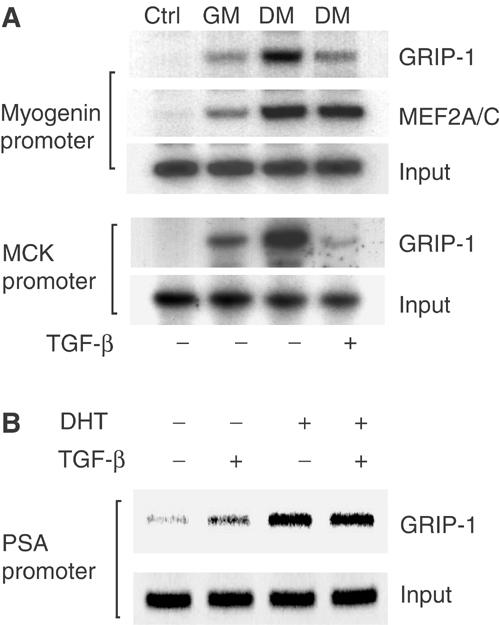
TGF-β-activated Smad3 decreased the recruitment of GRIP-1 to the myogenin and MCK promoters in C2C12 myoblasts (A) Soluble chromatin was prepared from proliferating myoblasts cultured in GM, differentiating myoblasts (DM) or cells exposed to 5 ng/ml TGF-β in DM for 6 h. Chromatin solutions were immunoprecipitated using antibodies for GRIP-1, or MEF2A/C, or control IgG (ctrl). Immune complexes were analyzed by PCR in the presence of 32P-dCTP using primers that span the MEF2-binding sites in the myogenin or MCK promoters as described (Lu et al, 2000). Equivalent amounts of input chromatin were used as shown by direct PCR of the chromatin samples. (B) TGF-β did not affect the GRIP-1 recruitment, through the androgen receptor, to the PSA promoter. Treatment of LNCaP TβRII cells with 10 nM dihydrotestosterone (DHT) for 3 h induced GRIP-1 recruitment, which was not affected by the presence of 5 ng/ml TGF-β. Immune complexes were analyzed by PCR using primers that span the androgen receptor-binding sequence of the PSA promoter.
Discussion
The data presented demonstrate that Smad3 acts as a direct effector of TGF-β signaling to antagonize MEF2 function during myogenesis through two mechanisms: (1) by disengaging the synergy between MyoD and MEF2 required for muscle-specific gene expression and (2) by displacing the GRIP-1 coactivator from MEF2 target promoters. Both mechanisms appear to result from the interaction of Smad3 with MEF2, thereby requiring its conserved MADS/MEF2 region, without affecting DNA binding of MEF2. This interaction may occur in a manner similar to the association of MEF2 with the corepressors Cabin1 or class II HDACs, in which DNA binding and corepressor recruitment are mediated by protein surfaces located on opposite sides of the MADS/MEF2 domain (Han et al, 2003). Combined with our observation that Smad3 interferes directly with MyoD function (Liu et al, 2001), these results support a model in which Smad-mediated TGF-β signaling suppresses muscle differentiation by impinging on multiple interfaces of the coordinated transcriptional circuitry that consists of MEF2 and myogenic bHLH factors.
TGF-β/Smad3 targets MEF2, in addition to MyoD, to inhibit myogenic transcription
A ‘myogenic recognition motif' (MRM) within the bHLH region of MyoD family proteins has been shown to confer both myogenic potential and susceptibility to TGF-β-induced inhibition (Martin et al, 1992). It was therefore postulated that TGF-β signaling may perturb the function of a transcriptional cofactor that depends on MRM. In agreement with this notion, we now provide evidence that TGF-β inhibits the myogenic coactivator function of MEF2. Similar mechanisms mediate the indirect inhibition of MRF activity by Notch and Raf signaling (Wilson-Rawls et al, 1999; Winter and Arnold, 2000).
The repression of MyoD-dependent transcription by TGF-β/Smad3 through mechanisms that inactivate MEF2 complements the ability of Smad3 to inhibit MyoD/E protein heterodimerization (Liu et al, 2001). At strictly E-boxes-driven promoters, MEF2C overexpression was not sufficient to overcome Smad3-mediated repression (D Liu and R Derynck, unpublished results), suggesting that in such sequence contexts, a direct effect of Smad3 on MEF2 contributes little to TGF-β-induced repression. However, many muscle-specific genes contain both E-box and MEF2 sites in their regulatory regions, which confer transcription activation through synergistic cooperation. Therefore, the inhibitory action of TGF-β/Smad3 on muscle differentiation is exerted at two restriction nodes within a mutually reinforcing myogenic regulatory circuit, in which MEF2 and MyoD family proteins, already present in myoblasts prior to differentiation, induce each other's expression to promote myogenesis (Black and Olson, 1998). Such dual roles of Smad3 allow for a broader control over the myogenic transcription program, which may explain how TGF-β signaling, through endogenous Smad3, effectively blocks the differentiation of C2 C12 myoblasts or 10T1/2 fibroblasts constitutively overexpressing MyoD or myogenin (Vaidya et al, 1989; Brennan et al, 1991).
One of the genes shown to be controlled by both MRF and MEF2 is myogenin. Its expression at the onset of differentiation depends on MEF2-mediated recruitment of MyoD or Myf5 into an active transcription complex. Thus, activation of myogenin transcription by MyoD in differentiating myoblasts, a process dependent on pre-existing MEF2, requires an MEF2-binding site, but not E-boxes (Edmondson et al, 1992; Buchberger et al, 1994). Furthermore, DNA footprinting revealed stable occupation of the MEF2 site but not the E-boxes of the myogenin promoter in differentiated myotubes, and antisense MEF2C mRNA abolished the activation of myogenin promoter by Myf5 (Johanson et al, 1999). Therefore, our observation that Smad3 repressed transcriptional activation of myogenin by MyoD∼E47 strongly supports a repression due to silencing of the coactivator function MEF2. Unlike myogenin, MRF4 expression is induced by MyoD through an E-box-dependent mechanism. The single MEF2 site in the MRF4 promoter is dispensable for its expression, although MEF2 can indirectly activate MRF4 transcription (Black et al, 1995). In this case, Smad3-mediated repression of MEF2 may seem redundant, but ensures maximal silencing of muscle gene expression.
Smad3 signaling impairs the function of GRIP-1, a MEF2 coactivator
Our observation that Smad3 regulates MEF2 coactivator association adds to previously identified signaling pathways that intersect with the myogenic transcription program at the level of cofactors involved in chromatin remodeling. TGF-β-activated Smad3 specifically perturbed the association of GRIP-1 with MEF2C and prevented the recruitment of MEF2 to subnuclear sites that accumulate GRIP-1. Consistent with these observations and supporting our conclusion that TGF-β signaling targets MEF2 function, forced expression of MEF2C in the nuclei of myoblasts confers resistance to the inhibitory action of TGF-β on differentiation (De Angelis et al, 1998), although under our conditions we did not observe the reported nuclear exclusion of MEF2C in response to TGF-β. Overexpression of CBP also partially overcame Smad3-mediated repression, albeit likely through its HAT activity, since Smad3 did not significantly affect the association of CBP with MEF2C, and the amino-acid sequences in CBP/p300 that mediate binding to MEF2 or Smad3 do not overlap (Feng et al, 1998; De Luca et al, 2003).
Diverse mechanisms lead to transcriptional repression in response to TGF-β
In contrast to the well-documented cooperation of Smads with sequence-specific transcription factors to activate transcription, the mechanisms that underlie Smad-mediated transcriptional repression are only beginning to emerge. Recent studies indicate that Smad3 forms stable complexes with transcription repressors, thereby repressing target gene transcription. Upon TGF-β stimulation, a pre-assembled protein complex containing Smad3, E2F4/5 and DP1, and the corepressor p107 translocates into the nucleus and recognizes a composite Smad-E2F site on the c-myc promoter to carry out repression (Chen et al, 2002). Likewise, Smad3 forms a complex with the transcription repressor ATF3 at the Id1 promoter and inhibits transcription (Kang et al, 2003). In both cases, binding of Smad3 to specific DNA sequences in the target promoter is required and presumably stabilizes the repressor complexes. Here we demonstrate TGF-β-induced repression of MEF2 activity through Smad3 association independently of Smad-binding sites. Also, the interference of Smad3 with MyoD function does not require Smad3 binding to MyoD-responsive promoter sequences (Liu et al, 2001). In addition, during mesenchymal differentiation, Smad3 interacts with and represses the activity of C/EBP and Runx2 (Alliston et al, 2001; Choy and Derynck, 2003), without directly contacting DNA. Thus, multiple mechanisms involving Smad3 protein complexes account for TGF-β-induced transcriptional repression.
The promoter sequence and levels of coactivators or corepressors, which may in turn depend on cell type, are determinants of the abilities of Smads to repress or activate transcription. For example, Smad3 represses Runx2-mediated activation of the osteocalcin promoter in mesenchymal cells, yet enhances this activity in epithelial cells (Alliston et al, 2001), whereas Smad3 cooperates with Runx family proteins to induce IgCα expression in both cell types (Zhang and Derynck, 2000). In response to Dpp, the Drosophila Smad homologs Mad and Medea associate with Schnurri (Shn). Binding of this complex to both Mad and Shn sites on the B-enhancer of the Ubx gene is required for synergistic activation of Ubx transcription (Dai et al, 2000; Torres-Vazquez et al, 2001), whereas Shn tethered to Brk silencer by Mad/Medea represses gene expression (Müller et al, 2003). Further studies should clarify how the architecture of the transcription factor complexes and their spatial configuration on target promoters determine the outcome of signal-dependent regulation of gene expression by Smad3.
Implications of TGF-β regulation of MEF2 in non-myogenic cells
The significance of the functional interaction between Smad3 and MEF2 may extend beyond skeletal muscle differentiation. TGF-β signaling and MEF2 have been implicated in hypertrophic cardiomyocyte growth (Brand and Schneider, 1995; Kolodziejczyk et al, 1999) and are essential for vascular development (Pepper, 1997; Lin et al, 1998). MEF2 is the major transcription factor responsible for calcium-dependent Nur77 transcription that mediates T-cell receptor (TCR)-induced apoptosis (Youn et al, 1999), whereas TGF-β is also involved in TCR-mediated thymocyte death (Wahl et al, 2000). It will be interesting to determine if control of MEF2 activity through Smad3 plays a role in regulating broader physiological responses to TGF-β family signaling.
Materials and methods
Cell culture and transfections
C3H10T1/2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). C2C12 myoblasts were maintained in growth medium (GM), or DMEM with 15% FBS. Differentiation was initiated by shifting to DMEM with 2% horse serum (differentiation medium, DM). COS cells were grown in DMEM with 10% FBS. 10T1/2 cells were transiently transfected using Effectene or Fugene, and COS cells were transfected using Lipofectamine according to the manufacturer's instructions. The quantities of transfected DNA were kept constant by adding an appropriate amount of empty vector pRK5.
Transcription reporter assays
The myogenin reporter plasmid Myo184-CAT, containing the chloramphenicol acetyl transferase (CAT) gene under the control of a 184 bp myogenin promoter, and its derivatives, Myo184(-E1)-CAT and Myo184(mMEF2)-CAT, containing inactivating mutations in the E-box or MEF2 site, respectively, have been described (Edmondson et al, 1992). The reporter for MEF2-dependent transcription, 3xMEF2-Luc, contains three tandem MEF2 sites linked to the E1b basal promoter, which controls luciferase expression (Lu et al, 2000). Mammalian two-hybrid assays were based on the interaction between Gal4 and VP16 fusion proteins, which in turn activates luciferase expression from Gal4 DNA-binding sites in Gal4-Luc (FR-Luc, Stratagene). Smad3−/− mouse fibroblasts and its derivative cell line with constitutive Smad3 expression have been described (Liu et al, 2001). CAT and luciferase activities were normalized to the β-galactosidase activity from a cotransfected pRK5-βgal control plasmid, as described (Liu et al, 2001). The results represent at least three experiments.
Immunoprecipitation and immunoblotting
10T1/2 or COS cells transfected with plasmids encoding tagged MEF2C, GRIP-1, Smads or fragments of MEF2C and Smad3 were harvested 24–48 h post-transfection, and lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40). The lysates were immunoprecipitated with antibody-coated protein A–sepharose. A fraction of the cell lysates was subjected to direct immunoblotting for protein expression levels.
GST and oligonucleotide protein binding analyses
Binding of 35S-labeled MEF2C to GST-Smad proteins was determined as described (Liu et al, 2001). In DNA precipitation assays, cells transfected with Myc-MEF2 and Flag-Smad3 were lysed by sonication in HKMG buffer (10 mM HEPES pH 7.9, 100 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT, 0.1% NP-40). Cell extracts were incubated for 2 h with 1 μg of 5′-biotinylated, double-stranded oligonucleotide corresponding to the MEF2-binding site (5′-GATCGCTCTAAAAATAACCCTGTCG-3′), or a mutant version with C–G and A–C substitutions at the underlined positions. DNA–protein complexes were precipitated with streptavidin–agarose beads for 1 h and subjected to immunoblotting.
Indirect immunofluorescence
Transfected 10T1/2 cells were fixed and permeabilized in PBS with 4% formaldehyde and 0.2% Triton X-100 for 20 min, and stained with mouse and rabbit primary antibodies. Immune complexes were detected using Oregon green-conjugated goat anti-mouse and Texas red-conjugated goat anti-rabbit IgG. Nuclei were visualized by DAPI staining. Antibodies include F5D, a monoclonal anti-myogenin antibody (Santa Cruz), anti-Flag M2 (Sigma) and anti-HA (Covance).
Chromatin immunoprecipitation analyses
C2C12 cells were grown in GM or DM, or DM followed by addition of TGF-β for 5 h. Cells were fixed in 1% formaldehyde for 40 min. Glycine was added to a concentration of 50 mM, and cells were suspended successively in PBS, lysis buffer (50 mM HEPES pH 8, 140 mM NaCl, 10% glycerol, 0.5% NP-40, 0.5% Triton X-100) with protease inhibitors, and wash buffer (10 mM Tris–HCl pH 8, 200 mM NaCl). Nuclei were pelleted and resuspended in 1 ml RIPA buffer (10 mM Tris–HCl pH 8, 140 mM NaCl, 1% Triton X-100) with protease inhibitors. Soluble chromatin was prepared by sonication and immunoprecipitated using a monoclonal antibody against GRIP-1 (Neomarker) or a polyclonal antibody against MEF2A/C (Santa Cruz). Immune complexes were collected with salmon sperm DNA/protein A/G beads, and washed with increasing stringency in RIPA buffer, RIPA buffer with 500 mM NaCl, and LiCl wash buffer (10 mM Tris–HCl pH 8, 250 mM LiCl, 1% Triton X-100, 1% Na-deoxycholate). Immune complexes were eluted in 1% SDS and 0.1M NaHCO3. Crosslinking was reversed by incubating at 65°C for 4 h, and chromatin DNA fragments in the eluent were purified using Qiaquick columns. A volume of 5 μl of DNA solution was used as a template for PCR using primers specific for the myogenin or MCK promoter (Lu et al, 2000). A portion of the chromatin preparation was included in PCR reactions as positive control.
LNCaP TβRII cells (Guo and Kyprianou, 1998) were grown in RPMI medium 1640 with 5% charcoal–dextran–stripped FBS for 3 days, and chromatin immunoprecipitation assays were performed using anti-GRIP-1 antibody (Bethyl Laboratories) as described above. For PCR amplification, primers spanning from –406 to –164 region of the PSA promoter were used: 5′-AGGGATCAGGGAGTCTCACA-3′ (forward) and 5′-GCTAGCACTTGCTGTTCTGC-3′ (reverse).
Acknowledgments
We thank B Black, A Lassar, E Olson and K Yamamoto for providing reagents, and B Black for critical reading of the manuscript. This research was supported by grants RO1-CA63101 and P60 DE-13058 (project III) to RD and an NIH postdoctoral fellowship to DL.
References
- Alliston T, Choy L, Ducy P, Karsenty G, Derynck R (2001) TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J 20: 2254–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Martin JF, Olson EN (1995) The mouse MRF4 promoter is trans-activated directly and indirectly by muscle-specific transcription factors. J Biol Chem 270: 2889–2892 [DOI] [PubMed] [Google Scholar]
- Black BL, Olson EN (1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 14: 167–196 [DOI] [PubMed] [Google Scholar]
- Brand T, Schneider MD (1995) The TGF β superfamily in myocardium: ligands, receptors, transduction, and function. J Mol Cell Cardiol 27: 5–18 [DOI] [PubMed] [Google Scholar]
- Braun T, Buschhausen-Denker G, Bober E, Tannich E, Arnold HH (1989) A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J 8: 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Edmondson DG, Li L, Olson EN (1991) Transforming growth factor β represses the actions of myogenin through a mechanism independent of DNA binding. Proc Natl Acad Sci USA 88: 3822–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Ragge K, Arnold HH (1994) The myogenin gene is activated during myocyte differentiation by pre-existing, not newly synthesized transcription factor MEF-2. J Biol Chem 269: 17289–17296 [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, Massagué J (2002) E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell 110: 19–32 [DOI] [PubMed] [Google Scholar]
- Chen D, Huang SM, Stallcup MR (2000a) Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J Biol Chem 275: 40810–40816 [DOI] [PubMed] [Google Scholar]
- Chen SL, Dowhan DH, Hosking BM, Muscat GE (2000b) The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev 14: 1209–1228 [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Wang SC, Hosking B, Muscat GE (2001) Subcellular localization of the steroid receptor coactivators (SRCs) and MEF2 in muscle and rhabdomyosarcoma cells. Mol Endocrinol 15: 783–796 [DOI] [PubMed] [Google Scholar]
- Choy L, Derynck R (2003) TGF-β inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem 278: 9609–9619 [DOI] [PubMed] [Google Scholar]
- Dai H, Hogan C, Gopalakrishnan B, Torres-Vazquez J, Nguyen M, Park S, Raftery LA, Warrior R, Arora K (2000) The zinc finger protein Schnurri acts as a Smad partner in mediating the transcriptional response to Decapentaplegic. Dev Biol 227: 373–387 [DOI] [PubMed] [Google Scholar]
- De Angelis L, Borghi S, Melchionna R, Berghella L, Baccarani-Contri M, Parise F, Ferrari S, Cossu G (1998) Inhibition of myogenesis by transforming growth factor β is density-dependent and related to the translocation of transcription factor MEF2 to the cytoplasm. Proc Natl Acad Sci USA 95: 12358–12363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Severino A, De Paolis P, Cottone G, De Luca L, De Falco M, Porcellini A, Volpe M, Condorelli G (2003) p300/cAMP-response-element-binding-protein (‘CREB')-binding protein (CBP) modulates co-operation between myocyte enhancer factor 2A (MEF2A) and thyroid hormone receptor-retinoid X receptor. Biochem J 369: 477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson DG, Cheng TC, Cserjesi P, Chakraborty T, Olson EN (1992) Analysis of the myogenin promoter reveals an indirect pathway for positive autoregulation mediated by the muscle-specific enhancer factor MEF-2. Mol Cell Biol 12: 3665–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Zhang Y, Wu RY, Derynck R (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev 12: 2153–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Kyprianou N (1998) Overexpression of transforming growth factor (TGF) β1 type II receptor restores TGF-β1 sensitivity and signaling in human prostate cancer cells. Cell Growth Differ 9: 185–193 [PubMed] [Google Scholar]
- Han A, Pan F, Stroud JC, Youn HD, Liu JO, Chen L (2003) Sequence-specific recruitment of transcriptional co-repressor Cabin1 by myocyte enhancer factor-2. Nature 422: 730–734 [DOI] [PubMed] [Google Scholar]
- Hayes SA, Zarnegar M, Sharma M, Yang F, Peehl DM, ten Dijke P, Sun Z (2001) SMAD3 represses androgen receptor-mediated transcription. Cancer Res 61: 2112–2118 [PubMed] [Google Scholar]
- Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen TM, Rose DW, Rosenfeld MG, Glass CK (1997) Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc Natl Acad Sci USA 94: 1074–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson M, Meents H, Ragge K, Buchberger A, Arnold HH, Sandmöller A (1999) Transcriptional activation of the myogenin gene by MEF2-mediated recruitment of Myf5 is inhibited by adenovirus E1A protein. Biochem Biophys Res Commun 265: 222–232 [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massagué J (2003) A self-enabling TGFβ response coupled to stress signaling. Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 11: 915–926 [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk SM, Wang L, Balazsi K, DeRepentigny Y, Kothary R, Megeney LA (1999) MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr Biol 9: 1203–1206 [DOI] [PubMed] [Google Scholar]
- Labbé E, Silvestri C, Hoodless PA, Wrana JL, Attisano L (1998) Smad2 and Smad3 positively and negatively regulate TGF β-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell 2: 109–120 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H (1991) Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 66: 305–315 [DOI] [PubMed] [Google Scholar]
- Lazaro JB, Bailey PJ, Lassar AB (2002) Cyclin D–cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev 16: 1792–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN (1998) Requirement of the MADS-box transcription factor MEF2C for vascular development. Development 125: 4565–4574 [DOI] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R (2001) TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev 15: 2950–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6: 233–244 [DOI] [PubMed] [Google Scholar]
- Ma H, Baumann CT, Li H, Strahl BD, Rice R, Jelinek MA, Aswad DW, Allis CD, Hager GL, Stallcup MR (2001) Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr Biol 11: 1981–1985 [DOI] [PubMed] [Google Scholar]
- Martin JF, Li L, Olson EN (1992) Repression of myogenin function by TGF-β1 is targeted at the basic helix–loop–helix motif and is independent of E2A products. J Biol Chem 267: 10956–10960 [PubMed] [Google Scholar]
- Massagué J, Cheifetz S, Endo T, Nadal-Ginard B (1986) Type β transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci USA 83: 8206–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408: 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2001) Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev 11: 497–504 [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci 27: 40–47 [DOI] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18: 5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN (1995) Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83: 1125–1136 [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN (1996) Combinatorial control of muscle development by basic helix–loop–helix and MADS-box transcription factors. Proc Natl Acad Sci USA 93: 9366–9373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller B, Hartmann B, Pyrowolakis G, Affolter M, Basler K (2003) Conversion of an extracellular Dpp/BMP morphogen gradient into an inverse transcriptional gradient. Cell 113: 221–233 [DOI] [PubMed] [Google Scholar]
- Naya FS, Olson E (1999) MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr Opin Cell Biol 11: 683–688 [DOI] [PubMed] [Google Scholar]
- Olson EN, Sternberg E, Hu JS, Spizz G, Wilcox C (1986) Regulation of myogenic differentiation by type β transforming growth factor. J Cell Biol 103: 1799–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS (1997) Transforming growth factor-β: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev 8: 21–43 [DOI] [PubMed] [Google Scholar]
- Perry RL, Rudnicki MA (2000) Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci 5: D750–D767 [DOI] [PubMed] [Google Scholar]
- Quinn ZA, Yang CC, Wrana JL, McDermott JC (2001) Smad proteins function as co-modulators for MEF2 transcriptional regulatory proteins. Nucleic Acids Res 29: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Huang J, Hamamori Y, Kedes L (1997) Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol 17: 1010–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massagué J (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113: 685–700 [DOI] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387: 677–684 [DOI] [PubMed] [Google Scholar]
- Torres-Vazquez J, Park S, Warrior R, Arora K (2001) The transcription factor Schnurri plays a dual role in mediating Dpp signaling during embryogenesis. Development 128: 1657–1670 [DOI] [PubMed] [Google Scholar]
- Vaidya TB, Rhodes SJ, Taparowsky EJ, Konieczny SF (1989) Fibroblast growth factor and transforming growth factor β repress transcription of the myogenic regulatory gene MyoD1. Mol Cell Biol 9: 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Orenstein JM, Chen W (2000) TGF-β influences the life and death decisions of T lymphocytes. Cytokine Growth Factor Rev 11: 71–79 [DOI] [PubMed] [Google Scholar]
- Wilson-Rawls J, Molkentin JD, Black BL, Olson EN (1999) Activated Notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol Cell Biol 19: 2853–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter B, Arnold HH (2000) Activated raf kinase inhibits muscle cell differentiation through a MEF2-dependent mechanism. J Cell Sci 113 (Part 23): 4211–4220 [DOI] [PubMed] [Google Scholar]
- Yao TP, Ku G, Zhou N, Scully R, Livingston DM (1996) The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci USA 93: 10626–10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HD, Sun L, Prywes R, Liu JO (1999) Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science 286: 790–793 [DOI] [PubMed] [Google Scholar]
- Yun K, Wold B (1996) Skeletal muscle determination and differentiation: story of a core regulatory network and its context. Curr Opin Cell Biol 8: 877–889 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Derynck R (2000) Transcriptional regulation of the transforming growth factor-α-inducible mouse germ line Igα constant region gene by functional cooperation of Smad, CREB, and AML family members. J Biol Chem 275: 16979–16985 [DOI] [PubMed] [Google Scholar]