

Abstract
The impact of substratum surface property change on biofilm community structure was investigated using laboratory biological aerated filter (BAF) reactors and molecular microbial community analysis. Two substratum surfaces that differed in surface properties were created via surface coating and used to develop biofilms in test (modified surface) and control (original surface) BAF reactors. Microbial community analysis by 16S rRNA gene-based PCR-denaturing gradient gel electrophoresis (DGGE) showed that the surface property change consistently resulted in distinct profiles of microbial populations during replicate reactor start-ups. Pyrosequencing of the bar-coded 16S rRNA gene amplicons surveyed more than 90% of the microbial diversity in the microbial communities and identified 72 unique bacterial species within 19 bacterial orders. Among the 19 orders of bacteria detected, Burkholderiales and Rhodocyclales of the Betaproteobacteria class were numerically dominant and accounted for 90.5 to 97.4% of the sequence reads, and their relative abundances in the test and control BAF reactors were different in consistent patterns during the two reactor start-ups. Three of the five dominant bacterial species also showed consistent relative abundance changes between the test and control BAF reactors. The different biofilm microbial communities led to different treatment efficiencies, with consistently higher total organic carbon (TOC) removal in the test reactor than in the control reactor. Further understanding of how surface properties affect biofilm microbial communities and functional performance would enable the rational design of new generations of substrata for the improvement of biofilm-based biological treatment processes.
INTRODUCTION
Fixed-growth (or biofilm) processes are important environmental biotechnologies for wastewater treatment. The biofilm processes possess numerous advantageous features, including low energy consumption, smaller footprint, and shock load resistance, which are largely attributable to their excellent biomass retention and heterogeneous microbial community structures (1, 2). Traditionally, the substratum surface for biofilm growth has been primarily viewed as a biomass carrier (3–5), while recent studies have shown that the substratum surface properties, such as surface roughness (6, 7), surface hydrophobicity (7, 8), and surface charge (9, 10), could affect bacterial cell attachment. Different cell attachment strengths not only can lead to preferential colonization by some bacterial populations over others at the beginning of biofilm formation but also may create different biofilm depths that favor different microbial populations due to diffusion-limited substrate mass transfer and continuous microbial metabolism along the biofilm depth (2, 11).
Recent advancements in molecular biology and next-generation sequencing technologies have made it possible to investigate how substratum surface properties affect microbial community structures. PCR amplification of environmental 16S rRNA genes and subsequent sequencing and database comparison have enabled the detection of microbial populations without cultivation, greatly expanding the coverage of microbial diversity (12). The 16S rRNA gene-based microbial community fingerprinting techniques, such as denaturing gradient gel electrophoresis (DGGE) (13), provide a rapid tool for comparative analysis of major populations among multiple microbial communities. The recently developed bar-coded pyrosequencing of 16S rRNA gene PCR amplicons has further deepened the coverage of microbial community analysis because of its ability to generate a large number of sequence reads, revealing both phylogenetic and abundance information of individual microbial populations (14). The use of bar-coded pyrosequencing in microbial community analysis has been demonstrated in studies on human gut microbiota (15, 16), soil microbiomes (17, 18), and microbial communities in wastewater treatment plants (19, 20).
In this study, we aimed to use microbial community fingerprinting and pyrosequencing techniques to investigate the impact of substratum surface property changes on biofilm microbial community structure and subsequently treatment performance of laboratory biological aerated filter (BAF) reactors. Two substratum surfaces with different surface roughness and similar surface hydrophobicity properties were created via surface coating and used to develop biofilms in the test BAF reactor (modified surface) and in the control BAF reactor (original surface). The functional performance of the test and the control BAF reactors was compared based on the total organic carbon (TOC) removal efficiency during independent reactor start-ups and under various organic loading regimes. Microbial communities of the BAF reactors under steady-state operational conditions were determined using 16S rRNA gene-based DGGE analysis to compare community profiles. Bar-coded pyrosequencing was subsequently performed to characterize the microbial communities (i.e., determining the phylogenetic information and relative abundance of bacterial populations), which were then compared to further assess the impact of surface modification on microbial community structures.
MATERIALS AND METHODS
Surface modification and characterization.
Surface properties of spherical acrylic plastic beads were modified by coating them with a conductive paint (Electrodag 502; Ted Pella, Redding, CA), which was selected to change surface roughness without significantly changing surface hydrophobicity. The coating process involved submerging the beads in butanone-diluted paint (1:1 dilution ratio) and subsequent curing in the air, which was repeated three times. Surface roughness was quantified using atomic force microscopy (AFM) using the following procedure. Topographical images were first obtained in air with the contact mode using an Innova AFM (Bruker, Santa Barbara, CA). A silicon nitride cantilever with a spring constant of 0.12 N/m and a nominal tip radius of 2 nm was used. Each sample was characterized at multiple scan areas to obtain a good representation of the surface structures (see Fig. S1 in the supplemental material for example AFM images). Surface roughness was calculated using Nanoscope software and is expressed as the arithmetic mean (roughness average, Ra) over a scan area of 5 by 5 μm based on a procedure previously described by Zuo et al. (21). Surface hydrophobicity was determined by measuring the static contact angle of sessile drops of ultrapure water. The contact angle was determined from the sessile drop image using axisymmetric drop shape analysis (ADSA) (22). The surface modification introduced 10 times higher surface roughness to the modified surface than the original, while surface hydrophobicity was not significantly altered (Table 1).
TABLE 1.
Surface properties of the original and modified acrylic plastic beads
| Surface property | Original | Modified |
|---|---|---|
| Water contact angle (°) | 88 ± 1 | 91 ± 1 |
| Surface roughness (nm) | 4.2 ± 0.1 | 44.6 ± 3.8 |
BAF reactor setup and start-up.
The BAF reactors consisted of a cylindrical container, four air diffusers for aeration, a peristaltic pump for fluid handling, and the acrylic beads as substratum for biofilm growth (Fig. 1). The cylindrical reactor (inner diameter, 12.7 cm; height, 20.3 cm; volume, 2.32 liters) consisted of a lower compartment housing the influent inlet and air diffusers and an upper compartment housing the beads and an effluent outlet. Four stone air diffusers were placed at the bottom of the reactor to eject compressed air for aeration and agitation. The influent was delivered from a storage tank using a peristaltic pump. Plastic beads (diameter, 8 mm; 1.2 liters), either the original ones (control) or the ones with modified surface properties (test), were used as the substratum for biofilm growth in two otherwise identical BAF reactors.
FIG 1.
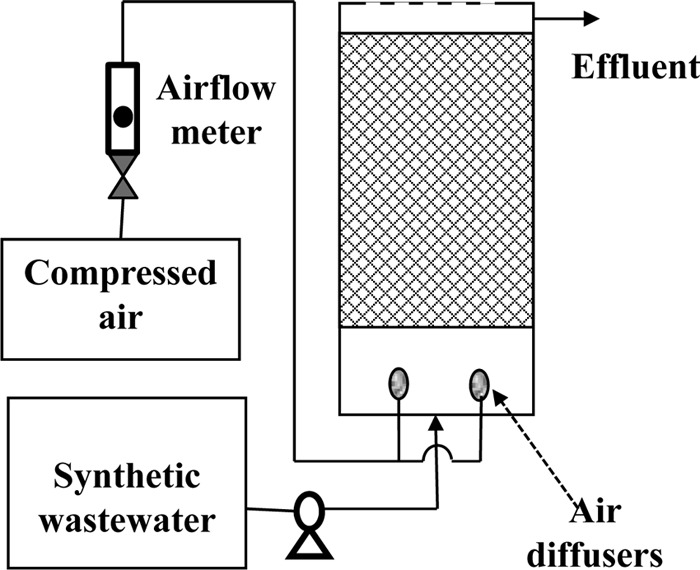
Schematic illustration of the BAF reactor setup.
The test and control BAF reactors were started up independently three times and operated to steady state at room temperature (ca. 22°C). The reactors were inoculated with 5 ml of activated sludge freshly collected from the Honouliuli wastewater treatment plant at Honolulu, HI. The same inocula were used for the test and control BAF reactors, but different inocula were used in the independent reactor start-ups. Between different reactor start-ups, the beads were thoroughly cleaned with sterile deionized (DI) water, air dried, and reused in subsequent experiments. A constant airflow (1.0 liters/min) was provided in both reactors throughout the experiments. Artificial wastewater at pH 7.5 (23) was amended with sucrose as the single carbon source to make different influent TOC concentrations. The first reactor start-up used a flow rate of 0.24 liters/h and an influent TOC concentration of 82 mg/liter, while the second reactor start-up used a flow rate of 0.58 liters/h and an influent TOC concentration of 190 mg/liter, resulting in a higher organic loading rate. A third reactor start-up was performed specifically to quantify biofilm biomass quantity, and this used the same organic loading as the second reactor start-up. Steady-state operation was indicated by the maintenance of a stable effluent TOC concentration for multiple days.
Biofilm biomass quantification.
The amount of biofilm biomass was estimated using both culture-dependent heterotrophic plate counts (HPC) and culture-independent 16S rRNA gene-based quantitative PCR (qPCR) quantification. Three beads were randomly collected from each BAF reactor daily as replicates during the third reactor start-up. The beads were placed in individual microcentrifuge tubes with 0.8 ml of sterile deionized (DI) water and 0.2 g of glass beads (ca. 0.25 mm in diameter) and vigorously shaken using a bead beater (Bio-Rad, Hercules, CA) for 1 min to dislodge biomass from the bead surface into water. For HPC determination, the biomass extracts underwent 10-fold serial dilutions before being plate counted on tryptic soy agar (TSA) plates, which were incubated at 37°C for 2 days before colony enumeration. The biomass was expressed as HPC per bead.
For 16S rRNA gene-based qPCR quantification, the extracted biomasses were first centrifuged at 13,000 × g for 10 min to pellet the biomass, which was then subjected to total genomic DNA extraction using a PowerSoil DNA isolation kit (MO-Bio, Carlsbad, CA) according to the manufacturer's instructions. The DNA extracts were quantified in triplicate by qPCR to determine bacterial concentration. The qPCRs were performed in a total volume of 20 μl using iTaqMan Universal PCR Master Mix (Applied Biosystem, Foster City, California) containing a 0.25 μM concentration of each of the universal forward (5′-TCCTACGGGAGGCAGCAGT-3′) and reverse primers (5′-GGACTACCAGGGTATCTAATCCTGTT-3′) and the 0.125 μM fluorogenic probe (6FAM-5′-CGTATTACCGCGGCTGCTGGCAC-3′-TAMRA, where 6FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine) (24) in an ABI 7300 real-time PCR system (Applied Biosystem). The thermocycler conditions were 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Exponential-stage Escherichia coli cells were used as qPCR calibration standards, and the bacterial biomass densities were expressed as calibrator cell equivalents (CCE) per bead.
BAF reactor performance assessment.
The BAF reactors during the second reactor start-up were operated under five different hydraulic retention times ([HRT] 1.6, 2.3, 3.5, 4.7, and 5.8 h) and five different influent TOC concentrations (126, 168, 252, 337, and 505 mg/liter). The HRT values and TOC concentrations were selected based on typical HRT values used in BAF operation and the range of TOC concentrations typically encountered in municipal wastewater and high-strength industrial wastewater, and the values reported were calculated by measuring the actual flow rate and influent TOC concentration. The combinations of HRT and influent TOC concentration produced 25 different organic loading rates (0.05 to 0.66 kg m−3 · h−1). The experiments started with the lowest influent TOC concentration and proceeded stepwise toward the highest influent TOC concentration. For each influent TOC concentration, the five different HRT were tested before backwashing was conducted by increasing the airflow rate to 5 liters/min, which was necessary to remove excessive biomass buildup in the BAF reactors. Each organic loading rate was operated for 2 days before samples were taken for subsequent analysis. For each organic loading level, the influent and effluent of the BAF reactors were simultaneously sampled to determine TOC removal efficiency.
PCR and DGGE.
The steady-state BAF reactors during the first two reactor start-ups were sampled by randomly collecting five beads at different depths of the reactors, which were used as biofilm sample replicates. Biofilm biomass extraction from the samples and total genomic DNA extraction followed the procedure described above. PCR-DGGE analysis of the bacterial community was based on the procedure described previously by Feng et al. (25). Briefly, the SYBR green I-stained gels were visualized on a UV transilluminator, and the gel digital image was analyzed using a GelCompar software package (Applied-Maths, Sint-Martens-Latem, Belgium). The software carries out a density profile analysis for each lane with background subtraction and least square filtering. The lanes were auto-detected, and the distortion of the lanes was modified manually. Subsequently, the lanes were normalized with a reference position and auto-detected to identify the bands. The areas of the peaks for each band were then used to calculate a similarity matrix using Pearson's correlation. Similarities between the bacterial community profiles were determined by drawing a dendrogram using the unweighted pair group method with arithmetic means (UPGMA) for cluster analysis.
Pyrosequencing.
DNA extracts from the five replicate samples for each BAF reactor during each start-up were pooled to make four composite DNA samples. The 454 pyrosequencing of the bar-coded 16S rRNA gene amplicons of the four DNA samples was performed using bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) with 16S rRNA gene primers 104F and 530R for bacteria (26). The sequence data were first denoised by removing reads without a similar or exact match on the region and by constructing consensus sequences of the clustered sequences to correct base pair errors using the USEARCH program (27). Potential chimeras in the denoised sequences were detected and removed using the de novo method of UCHIME with the entire sequence reads of the region as reference. The resulting sequence reads were then quality checked to remove sequences with low-quality tags, primers, or ends and lengths of less than 250 bp. The resulting high-quality sequence reads were then queried against a 16S rRNA gene database derived from NCBI using BLASTN+ (KrakenBLAST). The BLAST queries generated identity scores to well-characterized 16S rRNA gene sequences, and different threshold identity scores were used for the resolution at different taxonomic levels (the species level, >97% identity; the genus level, 95% to 97% identity; the family level, 90% to 95% identity; the order level, 85% to 90% identity; the class level, 80 to 85% identity; and the phylum level, 77% to 80% identity).
Chemical measurement and data processing.
The water samples were first filtered through glass fiber membranes, and the filtrates were analyzed using a TOC analyzer (Shimadzu, Columbia, MD) to determine TOC concentration according to the manufacturer's instructions. Biofilm biomass quantities of samples collected from the test and control BAF reactors were compared using Student's t test. The default significance level is a P value of <0.05 unless stated otherwise.
RESULTS
BAF reactor start-ups and steady-state operation.
During the independent start-ups, the test reactor (with a modified substratum surface) and the control reactor (with the original substratum surface) took similar times (between 1 and 2 days) to reach steady-state operational conditions, which was indicated by the maintenance of a stable effluent TOC concentration for more than 7 days (data not shown). During the first reactor start-up, which used an organic loading of 0.008 kg · m−3 · h−1, the TOC removal efficiencies of the test reactor (93.4% ± 0.2%) and the control reactor (91.8% ± 2.2%) were very similar at the steady state. During the second reactor start-up, which used a high organic loading of 0.05 kg · m−3 · h−1, higher average TOC removal rates were observed in the test reactor (89.1% ± 5.0%) than in the control reactor (76.3% ± 8.6%). The third reactor start-up used the same organic loading rate as the second one, and a similar TOC removal pattern between the test BAF reactor (93.5% ± 2.8%) and the control BAF reactor (84.7% ± 14.8%) was observed. During the third reactor start-up, biofilm biomass in the test and control reactors was quantified (see Fig. S2 in the supplemental material); samples from the test BAF reactor contained 7.76 ± 0.34 log CFU (by HPC) and 7.64 ± 0.38 log CCE (by qPCR) per bead, while samples from the control reactor contained 7.72 ± 0.26 log CFU and 7.74 ± 0.32 log CCE per bead, which indicated no significant difference.
The impact of substratum surface properties and of the resulting biofilm communities on BAF reactor performance was investigated by comparing their respective treatment efficiencies under various organic loading regimes (Fig. 2). TOC removal efficiencies in both the test and control BAF reactors decreased as the organic loading increased. Higher TOC removal was consistently observed in the test reactor than in the control reactor under all organic loadings tested. Among the 25 different organic loadings, the test BAF reactor showed 0 to 10% improvement in TOC removal in 14 loadings (loading range, 0.06 to 0.66 kg · m−3 · h−1), 10 to 20% improvement in 8 loadings (loading range, 0.10 to 0.31 kg · m−3 · h−1), and 20 to 30% improvement in 3 loadings (loading range, 0.09 to 0.16 kg · m−3 · h−1).
FIG 2.
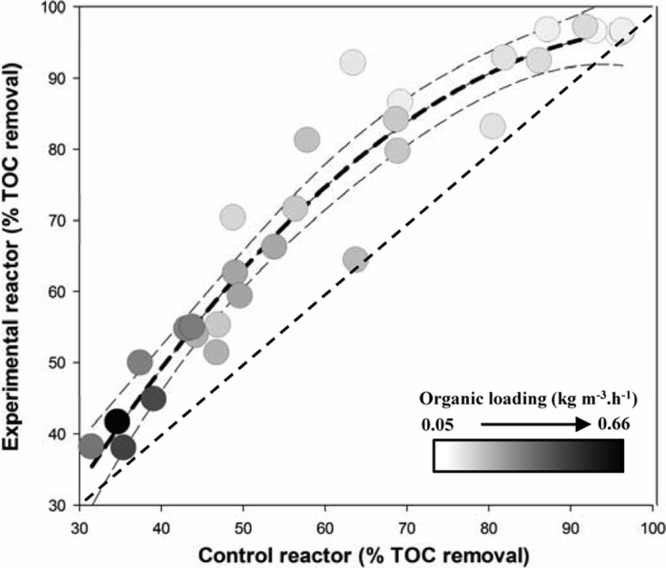
Comparison of TOC removal efficiencies of the test reactor (y axis) and control reactor (x axis) with the same organic loadings (according to the color gradient). A total of 25 different organic loadings were tested, and the organic loadings ranged from 0.05 to 0.66 kg m−3 h−1. The black dotted line is the quadratic regression curve of the data, and the dotted gray lines are the 95% confidence bands of regression. The gray gradient within the symbol indicates organic loading rates.
Steady-state biofilm communities.
The impact of substratum surface properties on the biofilm microbial community in the BAF reactors under steady-state operational conditions was first examined by community fingerprinting using 16S rRNA gene-based PCR-DGGE. Considerable differences in the microbial community structures emerged between the test reactor and the control reactor, and the communities clustered separately (Fig. 3); intrareplicate similarity was 83.6% for the first reactor start-up (T1 versus C1) and 62.9% for the second reactor start-up (T2 versus C2). This contrasts with the highly similar bacterial community structures observed in replicate biofilm samples from the same reactor. Interreplicate similarity was 86. 0% (test sample 1 [T1]) and 88.3% (control sample 1 [C1]) for the first reactor start-up and 70.4% (test sample 2 [T2]) and 89.3% (control sample 2 [C2]) for the second reactor start-up, indicating that the community differences between the two reactors were caused by the different biofilm substratum surfaces. The difference in microbial community structures from the two different reactor start-ups were more pronounced than the difference between the test reactor and the control reactor during the same start-up, corresponding to the different microbial inocula used in the different reactor start-ups.
FIG 3.

Steady-state bacterial community structures revealed by DGGE in the test reactors (T1 and T2) and the control reactors (C1 and C2) during two independent experimental start-ups. Each group contained five replicate biofilm samples.
Pyrosequencing of bar-coded 16S rRNA gene amplicons was subsequently conducted for deep community sequencing and identification of unique phylotypes. Since the replicate samples exhibited highly similar microbial community structures based on DGGE analysis (Fig. 3), DNA extracts from the replicates were pooled to make four composite DNA samples (C1, T1, C2, and T2) for subsequent pyrosequencing. After various quality-control measures, a total of 13,154 sequence reads were obtained for the four samples, and 72 unique phylotypes were identified (Table 2). The sequencing effort appeared to have covered the majority of the bacterial diversity in the reactors, as indicated by the observed species richness (Sobs) and the richness estimator (Chao 1). The diversity indices, including Shannon's index (H′), Shannon's equitability index (EH), and Simpson's reciprocal index (1/D), indicated that bacterial species diversity in the samples was low, which indicated that the community composition was skewed toward the most dominant populations.
TABLE 2.
Pyrosequencing and biodiversity of bacterial communities of the test and control BAF reactors
| Samplea | No. of readsb | Sobsc | Chao 1 | H′d | EHe | 1/Df |
|---|---|---|---|---|---|---|
| C1 | 2,195 | 27 | 27.9 | 1.60 | 0.48 | 3.8 |
| T1 | 2,803 | 32 | 35.5 | 1.81 | 0.51 | 3.6 |
| C2 | 2,383 | 35 | 38.0 | 2.07 | 0.56 | 4.5 |
| T2 | 5,773 | 51 | 51.1 | 1.70 | 0.43 | 3.9 |
C1 and C2, control samples; T1 and T2, test samples.
Number of high-quality sequence reads.
Observed species richness.
Shannon's index.
Shannon's equitability index.
Simpson's reciprocal index.
Bacterial community composition.
The relative abundances of bacterial populations at the phylum and order levels were estimated from the detection frequencies of the corresponding sequence reads, which showed clear differences between the test reactor and the control reactor during both experimental start-ups (Fig. 4). Among the 19 orders of bacteria detected, Burkholderiales and Rhodocyclales of the Betaproteobacteria class were numerically dominant and accounted for 90.5 to 97.4% of the sequence reads. The bacterial communities in the test reactors contained more members of the Burkholderiales (70.9% for test 1 and 81.2% for test 2) than those from the control reactors (39.8% for control 1 and 44.2% for control 2). Rhodocyclales showed an opposite pattern; the test reactors contained fewer members of the Rhodocyclales (26.4% for test 1 and 9.3% for test 2) than the control reactors (54.4% for control 1 and 52.1% for control 2).
FIG 4.

Relative abundances of different orders of bacteria detected in the control reactor (open bars) and the test reactor (filled bars) during the first (A) and the second (B) independent experimental start-ups.
Twelve major bacterial species (relative abundance of >1%) and five dominant species (relative abundance of >10%) were detected, and their relative abundances differed between the test reactor and the control reactor (Fig. 5). Three of the five dominant bacterial species, Zoogloea sp., Acidovorax sp., and Comamonas sp., showed relative abundance changes between the test and control reactors that were consistent during both start-ups. Zoogloea sp. was the most dominant species in the control reactor (48.8% and 39.1% for the first and second start-ups, respectively), while its relative abundance was significantly reduced in the test reactor (24.5% and 8.1%). Acidovorax sp. showed the same pattern, being more dominant in the control reactor (13.8% and 17.8%) than in the test reactor (7.3% and 14.5%). Comamonas sp. showed the opposite pattern, being more dominant in the test reactor (23.8% and 40.1%) than in the control reactor (3.6% and 24.5%).
FIG 5.
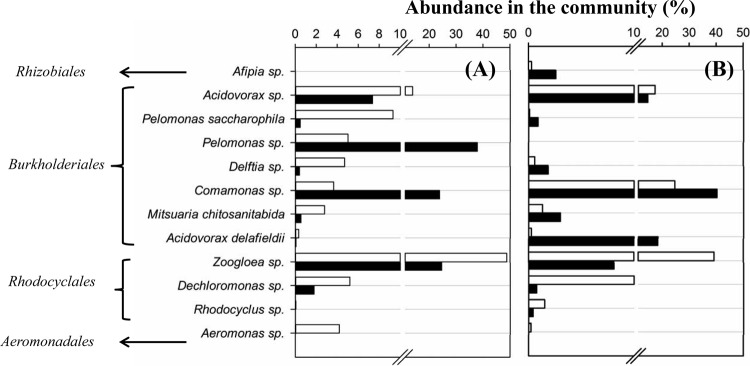
Relative abundances of major bacterial species detected in the control reactor (open bars) and the test reactor (filled bars) during the first (A) and the second (B) independent experimental start-ups.
DISCUSSION
The surface modification procedure in this study resulted in two surfaces with different surface roughness and similar hydrophobicity properties (Table 1). Surface roughness and hydrophobicity were targeted in the surface modification because they were generally considered important factors in biofilm formation. Surface roughness helps biofilm development through the provision of nearby surfaces to which detached biofilm pieces can reattach (7) and thus enhances bacterial adhesion and biomass retention (6, 7, 28), while surface hydrophobicity can affect bacterial adhesion and subsequent biofilm growth (7, 8). It is experimentally difficult to determine the contribution of individual surface properties to the observed biofilm microbial community change, primarily due to the technical difficulties in creating surfaces that differ only in one surface property. For example, the two surfaces in this study also differed in surface conductivity and potentially other surface physical and chemical characteristics that were not targeted for characterization, which may also contribute to a change in the biofilm microbial community. Nevertheless, it is clearly shown that the two different surfaces resulted in different biofilm microbial communities in the BAF reactors (Fig. 3, 4, and 5).
The impact of different surface properties on biofilm community structure was first demonstrated using 16S rRNA gene-based PCR-DGGE. One advantage of using DGGE-based community analysis is the ability to analyze multiple samples in parallel, which enabled direct comparisons of biofilm communities from replicate samples of the same reactor as well as samples from different treatments. DGGE was used in this study to determine the similarity in microbial community structure among individual replicate samples of the same experimental treatment as well samples between different treatments. The high interreplicate similarities in the community profiles (Fig. 3) highlighted the differences observed between the test reactor and the control reactor.
One limitation of DGGE, however, is that the sensitivity threshold of this method is estimated to be 1% of the total population (13), and hence the DGGE-based community analysis was limited to detecting only the major bacterial species. While the major microbial populations are often the focus of microbial community characterization because of their expected significant contribution to the process outcome, rare populations may also play important roles. Rare populations are essential for maintaining microbial diversity, which confers functional robustness and versatility against environmental perturbation and changes (31). Rare populations may also be more metabolically active than the major species, thus contributing disproportionally to ecosystem functions (32). The bar-coded pyrosequencing of 16S rRNA gene amplicons, due to its ability to generate a large number of sequence reads for each sample, provided a very useful tool to capture both major and rare species. Out of the 72 unique bacterial populations detected, 12 were identified as major populations based on a relative abundance cutoff value of 1%, while the rest (also the majority) belong to rare populations (Fig. 4).
The detection frequencies of unique sequence reads from pyrosequencing efforts are commonly used to estimate the relative abundances of bacterial populations in the community (15, 17, 18). Although this method may be biased by different 16S rRNA gene copy numbers (29), different DNA extraction efficiencies (33), and the intragenomic heterogeneity of the 16S rRNA gene (34), its use in community comparison should be acceptable, with the assumption that the same bias occurs to all samples. The detection frequencies of microbial populations in both the test reactor and the control reactor indicated that the majority of the populations belong to the Burkholderiales and Rhodocyclales orders of the Betaproteobacteria class (Fig. 4). The Betaproteobacteria were commonly found to be the most dominant class in biological wastewater treatment processes (19, 20). Previous works have shown that microbial populations belonging to Burkholderiales and Rhodocyclales can rapidly increase their relative abundances in response to labile organic substrates such as sucrose (35), which was used as the single carbon source in this study. The use of sucrose as the single carbon source can also explain the small microbial diversity observed in this study (Table 2 and Fig. 4), which is significantly less than that observed in actual wastewater treatment processes that treat complex organic wastes (19, 20).
Among the 12 major bacterial populations detected in the BAF reactors, Acidovorax and Comamonas of Burkholderiales and Zoogloea of Rhodocyclales were detected as dominant populations and showed consistent relative abundance levels between the test reactor and the control reactors during both reactor start-ups (Fig. 5). These groups are well documented in bacterial populations in wastewater biological treatment processes. Acidovorax spp. were frequently detected in wastewater (36, 37) and were credited for the degradation of various organic substrates (30). Comamonas spp. have been frequently observed in wastewater as important degraders of various organic compounds and have been shown to form biofilms using a range of organic substrates (38). Zoogloea spp. are known components of activated sludge, tend to aggregate to form flocs (39), and are commonly found in wastewater-activated sludge processes (19).
Modifying the substratum surface appeared to have altered the proportions of these microbes within the biofilm communities. Looking at the order level, 10 out of 14 orders and 11 out of 15 orders had increased relative abundances in the test reactor compared to the control reactor during the two independent experimental start-ups, respectively (Fig. 4). This is especially apparent in the abundance levels of the two most dominant orders, Burkholderiales and Rhodocyclales, where the test BAF reactor with modified surface apparently contained more bacteria belonging to the Burkholderiales and less belonging to the Rhodocyclales than the control reactor. Looking at the species level, the increase of Burkholderiales in the test reactor resulted from the interplay of multiple species, including Comamonas sp. and Pelomonas sp. (Fig. 5). Since Comamonas sp. is found in biofilm processes (38) but not as a major group in suspended growth processes, such as activated sludge processes (19, 40), its relative abundance increase in the test reactor suggests that the modified surface enhanced biofilm formation. The Pelomonas sp. showed a significant increase in relative abundance in the test reactor during the first reactor start-up but was not detected at all during the second reactor start-up, indicating the bacteria's transient and fortuitous behavior in the process, which was consistent with its infrequent detection in the environment (41).
The surface modification also resulted in reduced abundance of multiple populations in the test reactor, including Zoogloea sp. and Dechloromonas sp. of Rhodocyclales and Acidovorax sp. of Burkholderiales. Interestingly, Zoogloea sp. (19, 39, 40), Dechloromonas sp. (19, 40), and Acidovorax sp. (30, 42) were all commonly associated with activated sludge processes, where microorganisms engage primarily in suspended growth mode rather than attached growth mode. Therefore, their decreases in relative abundance in the test reactor further corroborate the notion that the modified surface enhanced biofilm formation and promoted microbial populations suited for biofilm growth.
The test BAF reactor with the modified substratum surface exhibited improved TOC removal at various organic loadings relative to the control reactor (Fig. 2). Since the modified substratum surface did not appear to increase the amount of biomass carried, this improved functional performance should be primarily attributed to the microbial community structure changes as a result of the surface modification. It is particularly interesting that the microbial community analysis showed that the surface modification reduced the abundance of some microbial populations typically associated with suspended growth, including Zoogloea sp., Dechloromonas sp., and Acidovorax sp., but increased the abundance of populations typically associated with biofilm processes, such as Comamonas sp.
To our knowledge, this is the first direct demonstration of the effects of substratum surface properties on biofilm community structures and reactor treatment performance in a biofilm process. Previous studies on clinically relevant biofilms were almost exclusively focused on the interaction between substratum surfaces and single bacterial strains, while studies on wastewater treatment processes that involved mixed microbial communities have predominantly focused on biomass density, distribution, and the resulting treatment performance. Further studies to understand how different surface properties mechanistically affect biofilm microbial communities and functional performance are needed to enable the rational design of new generations of substrata for the improvement of biofilm-based biological treatment processes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joseph Lichwa for assistance with chemical analysis.
The study was financially supported by a Sea Grant development award (NA09OAR4170060 to T.Y.). L.K. was supported by a Ford Foundation International Fellowship (no. 15076702) and a Vietnam Education Foundation Fellowship (no. F12012M).
Footnotes
Published ahead of print 18 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03001-13.
REFERENCES
- 1.Rittmann B, Manem J. 1992. Development and experimental evaluation of a steady-state, multi-species biofilm model. Biotechnol. Bioeng. 39:914–922. 10.1002/bit.260390906 [DOI] [PubMed] [Google Scholar]
- 2.Zhang T, Bishop PL. 1994. Density, porosity and pore structure of biofilms. Water Res. 28:2267–2277. 10.1016/0043-1354(94)90042-6 [DOI] [Google Scholar]
- 3.Mann A, Mendoza-Espinosa L, Stephenson T. 1998. A comparison of floating and sunken media biological aerated filters for nitrification. J. Chem. Technol. Biotechnol. 72:273–279. [DOI] [Google Scholar]
- 4.Mendoza-Espinosa L, Stephenson T. 1999. A review of biological aerated filters (BAFs) for wastewater treatment. Environ. Eng. Sci. 16:201–216. 10.1089/ees.1999.16.201 [DOI] [Google Scholar]
- 5.Moore RE, Quarmby J, Stephenson T. 2001. The effects of media size on the performance of biological aerated filters. Water Sci. Technol. 35:2514–2522. 10.1016/j.bbr.2011.03.031 [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Cheng X, Zhang X, Sun D. 2012. Influence of surface modification of polyethylene biocarriers on biofilm properties and wastewater treatment efficiency in moving-bed biofilm reactors. Water Sci. Technol. 65:1021–1026. 10.2166/wst.2012.915 [DOI] [PubMed] [Google Scholar]
- 7.Eginton PJ, Gibson H, Holah J, Handley PS, Gilbert P. 1995. The influence of substratum properties on the attachment of bacterial cells. Colloids Surf. B Biointerfaces 5:153–159. 10.1016/0927-7765(95)01219-9 [DOI] [Google Scholar]
- 8.Dexter SD, Sullivan JD, Jr, Williams J, III, Watson SW. 1975. Influence of substrate wettability on the attachment of marine bacteria to various surfaces. Appl. Microbiol. 30:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harkes G, Dankert J, Feijen J. 1992. Bacterial migration along solid surfaces. Appl. Environ. Microbiol. 58:1500–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terada A, Okuyama K, Nishikawa M, Tsuneda S, Hosomi M. 2012. The effect of surface charge property on Escherichia coli initial adhesion and subsequent biofilm formation. Biotechnol. Bioeng. 109:1745–1754. 10.1002/bit.24429 [DOI] [PubMed] [Google Scholar]
- 11.Wanner O, Gujer W. 1984. Competition in biofilms. Water Sci. Technol. 17:27–44 [Google Scholar]
- 12.Pace NR, Burgin AB. 1990. Processing and evolution of the ribosomal RNAs, p 417–425 In Hill WE, Dahlberg A, Garrett RA, Moore PB, Schlessinger D, Warner JR. (ed), The structure, function, and evolution of ribosomes. American Society for Microbiology, Washington, DC [Google Scholar]
- 13.Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. 2008. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5:235–237. 10.1038/nmeth.1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. 2008. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. 10.1371/journal.pone.0002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dethlefsen L, Huse S, Sogin ML, Relman DA. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280. 10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N. 2009. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 3:442–453. 10.1038/ismej.2008.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolton M, Meller-Harel Y, Pasternak Z, Graber ER, Elad Y, Cytryn E. 2011. Impact of biochar application to soil on the root-associated bacterial community structure of fully developed greenhouse pepper plants. Appl. Environ. Microbiol. 77:4924–4930. 10.1128/AEM.00148-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Hu M, Xia Y, Wen X, Ding K. 2012. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl. Environ. Microbiol. 78:7042–7047. 10.1128/AEM.01617-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye L, Shao MF, Zhang T, Tong AH, Lok S. 2011. Analysis of the bacterial community in a laboratory-scale nitrification reactor and a wastewater treatment plant by 454-pyrosequencing. Water Res. 45:4390–4398. 10.1016/j.watres.2011.05.028 [DOI] [PubMed] [Google Scholar]
- 21.Zuo YY, Tadayyon SM, Keating E, Zhao L, Veldhuizen RA, Petersen NO, Amrein MW, Possmayer F. 2008. Atomic force microscopy studies of functional and dysfunctional pulmonary surfactant films, II: albumin-inhibited pulmonary surfactant films and the effect of SP-A. Biophys. J. 95:2779–2791. 10.1529/biophysj.108.130732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo YY, Ding M, Bateni A, Hoorfar M, Neumann AW. 2004. Improvement of interfacial tension measurement using a captive bubble in conjunction with axisymmetric drop shape analysis (ADSA). Colloids Surf. A Physicochem. Eng. Asp. 250:233–246. 10.1016/j.colsurfa.2004.04.081 [DOI] [Google Scholar]
- 23.Fornero JJ, Rosenbaum M, Cotta MA, Angenent LT. 2008. Microbial fuel cell performance with a pressurized cathode chamber. Environ. Sci. Technol. 42:8578–8584. 10.1021/es8015292 [DOI] [PubMed] [Google Scholar]
- 24.Nadkarni MA, Martin FE, Jacques NA, Hunter N. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257–266 [DOI] [PubMed] [Google Scholar]
- 25.Feng F, Goto D, Yan T. 2010. Effects of autochthonous microbial community on the die-off of fecal indicators in tropical beach sand. FEMS Microbiol. Ecol. 74:214–225. 10.1111/j.1574-6941.2010.00921.x [DOI] [PubMed] [Google Scholar]
- 26.Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. 2008. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326. 10.1371/journal.pone.0003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 28.Tang L, Pillai S, Revsbech NP, Schramm A, Bischoff C, Meyer RL. 2011. Biofilm retention on surfaces with variable roughness and hydrophobicity. Biofouling 27:111–121. 10.1080/08927014.2010.544848 [DOI] [PubMed] [Google Scholar]
- 29.Crosby LD, Criddle CS. 2003. Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. Biotechniques 34:790–794, 796, 798 [DOI] [PubMed] [Google Scholar]
- 30.Schulze R, Spring S, Amann R, Huber I, Ludwig W, Schleifer KH, Kampfer P. 1999. Genotypic diversity of Acidovorax strains isolated from activated sludge and description of Acidovorax defluvii sp. nov. Syst. Appl. Microbiol. 22:205–214. 10.1016/S0723-2020(99)80067-8 [DOI] [PubMed] [Google Scholar]
- 31.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120. 10.1073/pnas.0605127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones SE, Lennon JT. 2010. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U. S. A. 107:5881–5886. 10.1073/pnas.0912765107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frostegard A, Courtois S, Ramisse V, Clerc S, Bernillon D, Le Gall F, Jeannin P, Nesme X, Simonet P. 1999. Quantification of bias related to the extraction of DNA directly from soils. Appl. Environ. Microbiol. 65:5409–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun DL, Jiang X, Wu QL, Zhou NY. 2013. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79:5962–5969. 10.1128/AEM.01282-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD, Brodie EL. 2011. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. 10.3389/fmicb.2011.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adav SS, Lee D-J, Lai JY. 2010. Microbial community of acetate utilizing denitrifiers in aerobic granules. Appl. Microbiol. Biotechnol. 85:753–762. 10.1007/s00253-009-2263-6 [DOI] [PubMed] [Google Scholar]
- 37.You SJ, Hsu CL, Ouyang CF. 2002. Identification of the microbial diversity of wastewater nutrient removal processes using molecular biotechnology. Biotechnol. Lett. 24:1361–1366. 10.1023/A:1019829521663 [DOI] [Google Scholar]
- 38.Andersson S, Rajarao GK, Land CJ, Dalhammar G. 2008. Biofilm formation and interactions of bacterial strains found in wastewater treatment systems. FEMS Microbiol. Lett. 283:83–90. 10.1111/j.1574-6968.2008.01149.x [DOI] [PubMed] [Google Scholar]
- 39.Shao Y, Chung BS, Lee SS, Park W, Lee SS, Jeon CO. 2009. Zoogloea caeni sp. nov., a floc-forming bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 59:526–530. 10.1099/ijs.0.65670-0 [DOI] [PubMed] [Google Scholar]
- 40.Zhang T, Shao MF, Ye L. 2012. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 6:1137–1147. 10.1038/ismej.2011.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomila M, Bowien B, Falsen E, Moore ER, Lalucat J. 2007. Description of Pelomonas aquatica sp. nov. and Pelomonas puraquae sp. nov., isolated from industrial and haemodialysis water. Int. J. Syst. Evol. Microbiol. 57:2629–2635. 10.1099/ijs.0.65149-0 [DOI] [PubMed] [Google Scholar]
- 42.Heylen K, Lebbe L, De Vos P. 2008. Acidovorax caeni sp. nov., a denitrifying species with genetically diverse isolates from activated sludge. Int. J. Syst. Evol. Microbiol. 58:73–77. 10.1099/ijs.0.65387-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.