

Abstract
Cytochrome P450 monooxygenases (P450s), which constitute a superfamily of heme-containing proteins, catalyze the direct oxidation of a variety of compounds in a regio- and stereospecific manner; therefore, they are promising catalysts for use in the oxyfunctionalization of chemicals. In the course of our comprehensive substrate screening for all 27 putative P450s encoded by the Streptomyces griseus genome, we found that Escherichia coli cells producing an S. griseus P450 (CYP154C3), which was fused C terminally with the P450 reductase domain (RED) of a self-sufficient P450 from Rhodococcus sp., could transform various steroids (testosterone, progesterone, Δ4-androstene-3,17-dione, adrenosterone, 1,4-androstadiene-3,17-dione, dehydroepiandrosterone, 4-pregnane-3,11,20-trione, and deoxycorticosterone) into their 16α-hydroxy derivatives as determined by nuclear magnetic resonance and high-resolution mass spectrometry analyses. The purified CYP154C3, which was not fused with RED, also catalyzed the regio- and stereospecific hydroxylation of these steroids at the same position with the aid of ferredoxin and ferredoxin reductase from spinach. The apparent equilibrium dissociation constant (Kd) values of the binding between CYP154C3 and these steroids were less than 8 μM as determined by the heme spectral change, indicating that CYP154C3 strongly binds to these steroids. Furthermore, kinetic parameters of the CYP154C3-catalyzed hydroxylation of Δ4-androstene-3,17-dione were determined (Km, 31.9 ± 9.1 μM; kcat, 181 ± 4.5 s−1). We concluded that CYP154C3 is a steroid D-ring 16α-specific hydroxylase which has considerable potential for industrial applications. This is the first detailed enzymatic characterization of a P450 enzyme that has a steroid D-ring 16α-specific hydroxylation activity.
INTRODUCTION
Cytochrome P450 monooxygenases (P450s or CYPs) are found in all three domains of life, i.e., archaea, bacteria, and eukarya, and are classified into more than 1,000 families by their amino acid sequence homology (1). They have the ability to hydroxylate inactive carbons of hydrocarbons or aromatic compounds regio- and stereospecifically (2, 3). In animals, P450s are mainly involved in xenobiotic metabolism and steroid biosynthesis (4, 5). In plants, many P450s are involved in the biosynthesis of plant hormones and secondary metabolites (6). In contrast, the functions of bacterial P450s are largely unknown, although some bacterial P450s are required for secondary metabolite biosynthesis and assimilation of terpenoids, such as cineol or terpineol (7–10). In spite of their unknown physiological functions, however, bacterial P450s have attracted much attention as a rich resource for new biocatalysts for use in the oxyfunctionalization of chemicals (11, 12). For example, microbial conversion of steroid precursors is widely used for large-scale synthesis of steroid drugs (13–17).
CYP154 family members are widely found in actinomycetes, and many putative CYP154 genes have been found in the genomes of Streptomyces species. The structure of two CYP154 enzymes from Streptomyces coelicolor A3(2), CYP154C1 and CYP154A1, has already been elucidated by X-ray crystallography (18, 19). CYP154C1 has been shown to convert the 12- and 14-membered ring macrolides, narbomycin and YC-17, into pikromycin and neomethymycin, respectively, in vitro (18); however, not enough substrate screening of CYP154C1 has been performed. A metabolomics approach revealed an endogenous substrate of CYP154A1; unexpectedly, CYP154A1 has been shown to catalyze intramolecular cyclization of a novel dipentaenone to a Paternò-Büchi-like product without oxidation/reduction (20). To date, there is no other report on the function of CYP154 enzymes of Streptomyces species, while several studies on the CYP154 enzymes of Thermobifida fusca and Nocardia francinica have been reported. CYP154H1 of T. fusca hydroxylates ethylbenzene, n-propylbenzene, styrene, and indole and oxidizes sulfide compounds into sulfoxides and sulfones in vitro (21). In contrast, CYP154C5 of N. francinica has been shown to hydroxylate testosterone at the 16α position of the D ring by a bioconversion experiment (13). Very recently Bracco et al. showed that this enzyme can also hydroxylate five other steroids (pregnenolone, dehydroepiandrosterone, progesterone, Δ4-androstene-3,17-dione, and nandrolone) at the 16α position of the D ring (22). It is also noteworthy that in 1957, Streptomyces roseochromogenus was reported to hydroxylate two steroids (9α-fluorohydrocortisone and 9α-fluoroprednisolone) at the 16α position of the D ring (23), suggesting that S. roseochromogenus has a P450 that catalyzes a reaction similar to that of CYP154C5 of N. francinica. In fact, Berrie et al. purified and characterized this P450 enzyme (24). Very recently, Von et al. reported on the substrate screening of two CYP154 enzymes, CYP154E1 of T. fusca and CYP154A8 of N. francinica (25). CYP154E1 and CYP154A8 oxidize many compounds, including saturated fatty acids, primary alcohols, and terpenoids. Both enzymes convert neither anthraquinones nor steroids.
The streptomycin producer Streptomyces griseus IFO13350 has 27 putative P450 genes in its genome (26). Among them, only SGR6619 (P450mel, CYP107F1) has already been studied. CYP107F1 is involved in the hexahydroxyperylenequinone (HPQ) melanin biosynthesis and catalyzes oxidative biaryl coupling of 1,3,6,8-tetrahydroxynaphthalene to produce HPQ (27). We were interested in the functions of other P450s, including three CYP154 family members, CYP154M2 (SGR622), CYP154C3 (SGR1085), and CYP154C4 (SGR3108); hence, we carried out a comprehensive screening of their substrates using E. coli cells producing each P450 as a fusion protein with the P450 reductase domain of a self-sufficient P450 from Rhodococcus sp. and a low-molecular-weight compound library composed of 57 substrate candidates (Fig. 1). In the course of this screening, we found that one of the three CYP154 enzymes (CYP154C3) catalyzed regio- and stereospecific hydroxylation of various steroids at the 16α position of the D ring, indicating that this enzyme has considerable potential for industrial applications. Here, we describe the detailed enzymatic characterization of CYP154C3, including its substrate specificity, reaction specificity, and kinetic parameters.
FIG 1.

Compounds used for the screening of substrates of S. griseus P450s.
MATERIALS AND METHODS
Recombinant DNA techniques.
All recombinant DNA techniques were conducted by a previously described standard molecular biology technique (28) or by following the instructions provided by the suppliers.
Construction of expression plasmids.
The SGR1085 gene was amplified by PCR using two primers, SGR1085_F (5′-TACCATATGAACTGCCCGCACACTGC-3′; the NdeI site is underlined, and the start codon is in boldface) and SGR1085_R (5′-TACGAATTCGCCCAGGAGGACCG-3′; the EcoRI site is underlined), with S. griseus chromosomal DNA (25 ng) as the template. The 1.2-kb fragment amplified was digested with NdeI and EcoRI and cloned between the NdeI and EcoRI sites of pRED (29) to construct pCYP154C3-RED. In this plasmid, the original stop codon (TAG) of the SGR1085 gene was removed, and the gene was fused in frame with the RhF gene of Rhodococcus species. To construct a nonfused pCYP154C3, the SGR1085 gene was amplified by PCR using two primers, SGR1085_F2 (5′-TACCATATGAACTGCCCGCACACTGCC-3′; the NdeI site is underlined, and the start codon is in boldface) and SGR1085_R2 (5′-TACCTCGAGGCCCAGGAGGACCG-3′; the XhoI site is underlined), with pCYP154C3-RED (10 ng) as the template. The 1.2-kb fragment amplified was digested with NdeI and XhoI and cloned between the NdeI and XhoI sites of pET26b(+) (Merck KGaA, Darmstadt, Germany) to construct pCYP154C3. In this plasmid, the original stop codon (TAG) of the SGR1085 gene was removed, and the gene was fused in frame with a His6 tag sequence.
Expression of the CYP154C3-RED fusion gene and in vivo substrate screening assay.
E. coli BLR(DE3) carrying pCYP154C3-RED was incubated in 2×YT medium (2 ml) containing 100 μg/ml carbenicillin at 37°C for 4 h with reciprocal shaking. After the optical density at 600 nm (OD600) reached 0.8, the culture was transferred into fresh 2×YT medium (500 ml) containing 80 μg/ml 5-aminolevulinic acid hydrochloride (5-ALA), 100 μg/ml carbenicillin, 0.1 mM Fe(NH4)2(SO4)2, and 200 μl/liter trace element solution [27.0 g/liter FeCl3 · 6H2O, 2.0 g/liter CoCl3 · 6H2O, 2.0 g/liter (NH4)2MoO4 · 2H2O, 1.0 g/liter ZnCl2, 1.0 g/liter CaCl · 2H2O, 1.0 g/liter CuCl2, 0.5 g/liter MnCl2 · 4H2O, 0.5 g/liter H3BO3, and 20 ml of concentrated hydrochloric acid]. Cultivation was continued at 37°C until the OD600 reached 0.8. Isopropyl-β-d-thiogalactopyranoside (IPTG; 0.01 mM) was added to the culture, and incubation was continued for another 20 h at 20°C on a rotary shaker. The concentration of CYP154C3-RED was examined by the CO difference spectrum analysis (30).
The cells were collected by centrifugation and suspended in 100 ml of 50 mM potassium phosphate buffer (pH 7.2) containing 10% glycerol. An aliquot (500 μl) of this cell suspension was dispensed into each well of a 96-well sterile plate (PR-Master Block 2ML; Greiner Bio-One, Frickenhausen, Germany), which contained each substrate candidate (final concentration, 1 mM) dissolved in dimethylsulfoxide (DMSO; final concentration, 1%). Chemical compounds used as substrate candidates and authentic samples were purchased from Tokyo Chemical Industry Co. (Tokyo, Japan), Sigma-Aldrich Co. (St. Louis, MO, USA), Wako Pure Chemical Industries (Osaka, Japan), or Nacalai Tesque (Kyoto, Japan). Bioconversion was performed at 25°C for 24 h with shaking at 1,500 rpm on a rotary shaker (MS 3 Basic; IKA, Staufen im Breisgau, Germany). To stop the reaction, 200 μl of saturated sodium chloride solution and 600 μl of ethyl acetate were added, in this order, to the reaction mixture. The mixture then was shaken for 5 min. After centrifugation, the organic phase (500 μl) was collected and dried by evaporation, and the materials were dissolved in methanol (200 μl) for high-performance liquid chromatography (HPLC) analysis using a Waters e2695 separation module (Waters Corp., MA, USA) equipped with an on-line photodiode array detector (Waters 2998). An aliquot (6 μl) of the organic phase was applied to HPLC and separated using a MonoBis ODS column (inner diameter, 3.2 mm by 50 mm; Kyoto Monotech, Kyoto, Japan). The compounds were eluted with an acetonitrile-water gradient (both containing 0.1% trifluoroacetic acid) at a flow rate of 1 ml/min. The details of HPLC conditions are the following: solvent A (5% acetonitrile in 0.1% trifluoroacetic acid) for 4 min, and then a linear gradient from solvent A to solvent B (100% CH3CN–0.1% trifluoroacetic acid) for 18 min, and finally solvent B for 3 min. Maximum absorbance was monitored in the range from 200 to 500 nm (maximum plot).
Purification of recombinant CYP154C3.
For the production of C-terminally 6× histidine-tagged CYP154C3, the E. coli BLR(DE3) strain harboring pCYP154C3 was inoculated into 2×YT medium (2 ml) containing 100 μg/ml of carbenicillin and grown at 37°C for 4 h. After the OD600 reached 0.8, the cell culture was transferred into terrific broth (1 liter) containing 80 μg/ml 5-ALA, 1% glucose, 100 μl/ml carbenicillin, 0.1 mM Fe(NH4)2(SO4)2, and 200 μl/liter trace element solution. Cultivation was continued at 37°C until the OD600 reached 0.8. IPTG (0.01 mM) was added to the culture, and incubation was continued for another 20 h at 20°C on a rotary shaker. Cells were harvested by centrifugation and resuspended in 200 ml of buffer A (50 mM sodium phosphate buffer, pH 7.4, containing 500 mM NaCl, 0.1 mM dithiothreitol [DTT], and 20% [vol/vol] glycerol) and then disrupted by sonication. The crude cell extract was prepared by removal of cell debris by centrifugation at 10,000 × g for 30 min. The recombinant protein fused with a histidine tag was purified using a nickel-nitrilotriacetic acid superflow (Qiagen, Venlo, Netherlands) according to the manufacturer's instructions. CYP154C3-containing fractions were dialyzed against 5 liters of buffer B (50 mM sodium phosphate buffer, pH 7.4, containing 0.1 mM EDTA, 0.1 mM DTT, and 20% [vol/vol] glycerol), and applied to anion-exchange chromatography. Chromatography was performed using a ResourceQ column (0.64 by 3 cm) preequilibrated in buffer B. Proteins were eluted in a linear gradient of buffer B to buffer B containing 500 mM NaCl. CYP154C3-containing fractions were dialyzed against 5 liters of buffer B and concentrated by ultrafiltration (Amicon Ultra-4; Millipore, MA, USA). The concentration of CYP154C3 was examined by the CO difference spectrum analysis (30).
Measurement of the shift in the spin state of the heme accompanied by the substrate binding of CYP154C3.
Five microliters of substrate solution (in DMSO; final substrate concentrations were 25.0, 12.5, 6.25, 3.13, 1.56, 0.781, 0.391, 0.195, and 0 μM) was added to 0.1 nM CYP154C3 in a buffer (95 μl; 50 mM sodium phosphate, pH 7.4, containing 0.1 mM EDTA, 0.1 mM DTT, and 20% [vol/vol] glycerol). The mixture was incubated at 25°C for 2 min. Thereafter, the absorbance spectrum (from 340 to 490 nm) was recorded to show the shift in the spin state of the heme. The absorbance difference spectrum was determined by subtracting the spectrum of CYP154C3 solution containing DMSO from the spectrum of CYP154C3 solution containing the substrate. Equilibrium dissociation constants (apparent Kd values) were calculated according to a reported method (31).
In vitro steroid conversion assay.
An in vitro steroid conversion assay was performed with reaction solution (250 μl) containing 50 mM sodium phosphate, pH 7.4, 0.1 mM EDTA, 0.1 mM DTT, and 20% (vol/vol) glycerol at 20°C for 3 h. The reaction mixture contained 0.8 μM ferredoxin (from spinach; Sigma-Aldrich), ferredoxin reductase (0.01 U [at pH 7.5 and 25°C]; from spinach; Sigma-Aldrich), 0.2 nM CYP154C3, and 0.5 mM steroid. Steroids were dissolved in DMSO prior to addition to the reaction mixture (the final DMSO concentration in the reaction mixture was 1%). The reaction was started by the addition of NADPH at a final concentration of 0.5 mM. The products were analyzed by HPLC with the same method as that used for the bioconversion experiments described above.
Kinetic analysis of CYP154C3.
To determine the kinetic parameters of the Δ4-androstene-3,17-dione conversion, the reactions were performed with reaction solution (250 μl) containing 50 mM sodium phosphate buffer, pH 7.4, 0.1 mM EDTA, 0.1 mM DTT, and 20% (vol/vol) glycerol at 20°C for 20 min. The reaction mixture contained 2 μM ferredoxin, ferredoxin reductase (0.025 U), 0.65 nM CYP154C3, and Δ4-androstene-3,17-dione, which was dissolved in DMSO prior to addition to the reaction mixture (the final DMSO concentration in the reaction mixture was 1%). The concentrations of Δ4-androstene-3,17-dione ranged from 0 to 750 μM. The reaction was started by the addition of NADPH at a final concentration of 1 mM. The products were analyzed by HPLC with the same method as that used for the bioconversion experiments described above. To show the initial rate of the reaction, we measured the area of product peak in the UV chromatogram and calculated the amount of product from an analytical curve, which was created by the authentic 16α-hydroxy-Δ4-androstene-3,17-dione sample.
Large-scale production and purification of the product.
For structural determination of products, large-scale cultivation was carried out by inoculation of the preculture (2 ml) into 2×YT medium (1 liter) containing 80 μg/ml 5-ALA, 1% glucose, 100 μl/ml carbenicillin, 0.1 mM Fe(NH4)2(SO4)2, and 300 μl/liter trace element solution in a baffled Erlenmeyer flask at 37°C. When the OD600 reached 0.8, 0.01 mM IPTG was added to the culture and incubation was continued for another 20 h at 20°C on a rotary shaker at 160 rpm. Cells were collected by centrifugation and resuspended in 50 mM potassium phosphate (pH 7.2; 200 ml) containing 10% (vol/vol) glycerol. Each substrate dissolved in DMSO was added at a final concentration of 1 mM to the cell suspension (the final DMSO concentration in the reaction mixture was 1%), and bioconversion was performed by incubation at 26°C for 48 h with shaking (150 rpm). Saturated sodium chloride solution (200 ml) and ethyl acetate (600 ml) then were added, in this order, to the reaction mixture (200 ml), followed by shaking for 30 min. After centrifugation, the compound was purified from the organic phase by SiO2 column chromatography and HPLC equipped with a reverse-phase column, an 5C18-AR-II column (20 mm [inner diameter] by 250 mm; Nacalai Tesque) or DOCOSIL-B column (20 mm [inner diameter] by 250 mm; Senshu Scientific Co., Ltd., Tokyo, Japan).
Structure analysis by NMR spectrometry and high-resolution mass spectrometry.
Nuclear magnetic resonance (NMR) spectra were recorded in CD3OD or CDCl3 on a JNM-A500 spectrometer (JEOL, Tokyo, Japan). High-resolution mass spectrometry (HR-MS) was recorded in methanol on a JMS-T100LC device (JEOL, Tokyo, Japan).
RESULTS
Production of the functional CYP154C3 enzyme in E. coli as a fusion protein with the P450 reductase domain of P450Rhf.
We constructed an expression plasmid (pCYP154C3-RED) for the production of CYP154C3 that was C terminally fused with the P450 reductase domain (RED) of the self-sufficient P450, P450Rhf, of Rhodococcus species. The CYP154C3-RED fusion protein was produced in E. coli BLR(DE3) harboring pCYP154C3-RED, as described in Materials and Methods. The amount of active CYP154C3-RED was 11.5 ± 2.2 nmol per 1 liter of culture, as determined by the CO difference spectrum analysis (Fig. 2). The production level of CYP154C3-RED was sufficient for the ensuing bioconversion experiments.
FIG 2.
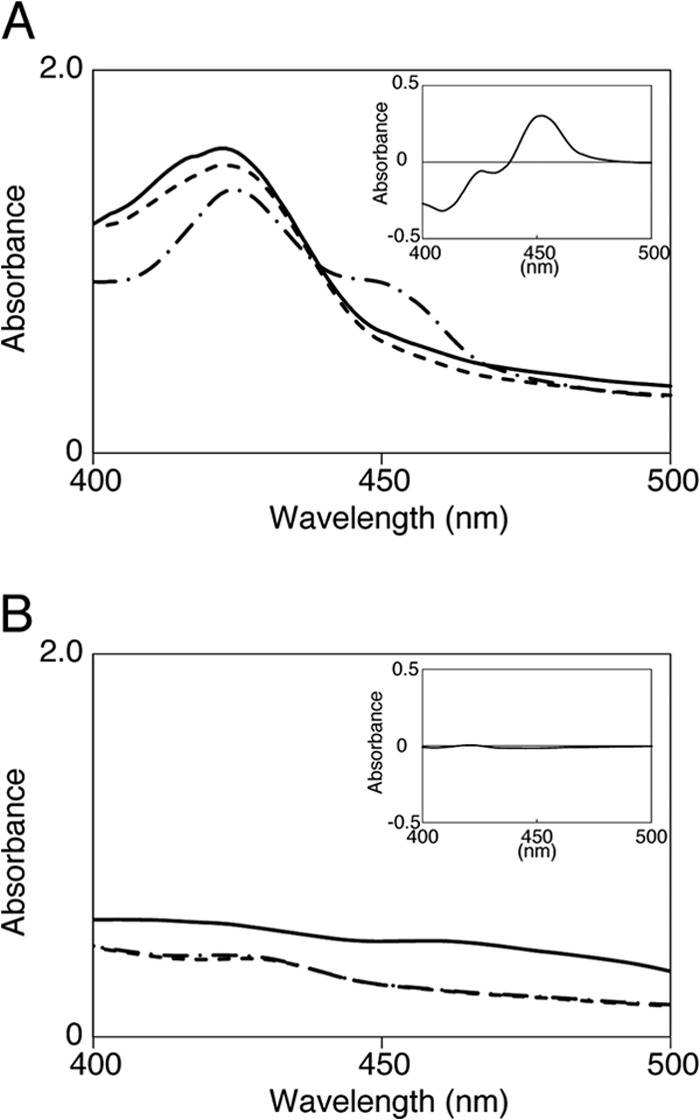
Spectral analysis of CYP154-RED. (A) Spectral features of the recombinant CYP154C3 fused C terminally with the P450 reductase domain of P450Rhf (CYP154-RED) in the cell extract. Spectra are shown for the oxidized (solid line), sodium-hydrosulfite-reduced (dashed line), and CO-bound (chain line) forms of the enzyme. The CO difference spectrum is shown in the inset. (B) Negative-control experiment using the cell extract of E. coli harboring pRED.
Substrate screening of CYP154C3 using E. coli BLR(DE3) harboring pCYP154C3-RED.
We carried out a systematic screening for substrate(s) of CYP154C3 using E. coli BLR(DE3) harboring pCYP154C3-RED and a low-molecular-weight compound library, which contained 57 substrate candidates, including steroids, sesquiterpenes, naphthalene derivatives, biphenyl derivatives, flavonoids, and other chemicals with aromatic rings (Fig. 1). E. coli BLR(DE3) cells, which produced active CYP154C3-RED, were incubated with each substrate candidate and possible bioconversion products were analyzed by HPLC, as described in Materials and Methods. Consequently, we found that testosterone (compound 1) and progesterone (compound 3) were efficiently converted into compounds 2 and 4, respectively (Fig. 3A and B). β-Estradiol (compound 5; Fig. 4 depicts its structure) was also converted into a different compound with a low efficiency (data not shown). Conversion of the other 53 compounds was not observed. The conversion rate of testosterone (compound 1), progesterone (compound 3), and β-estradiol (compound 5) was 74.0% ± 15.6%, 85.0% ± 18.2%, and 9.9% ± 8.7%, respectively, when estimated from the amounts of residual substrates in the bioconversion experiments, in which E. coli BLR(DE3) cells harboring pRED (which directed the production of RED only) were used as a negative control.
FIG 3.
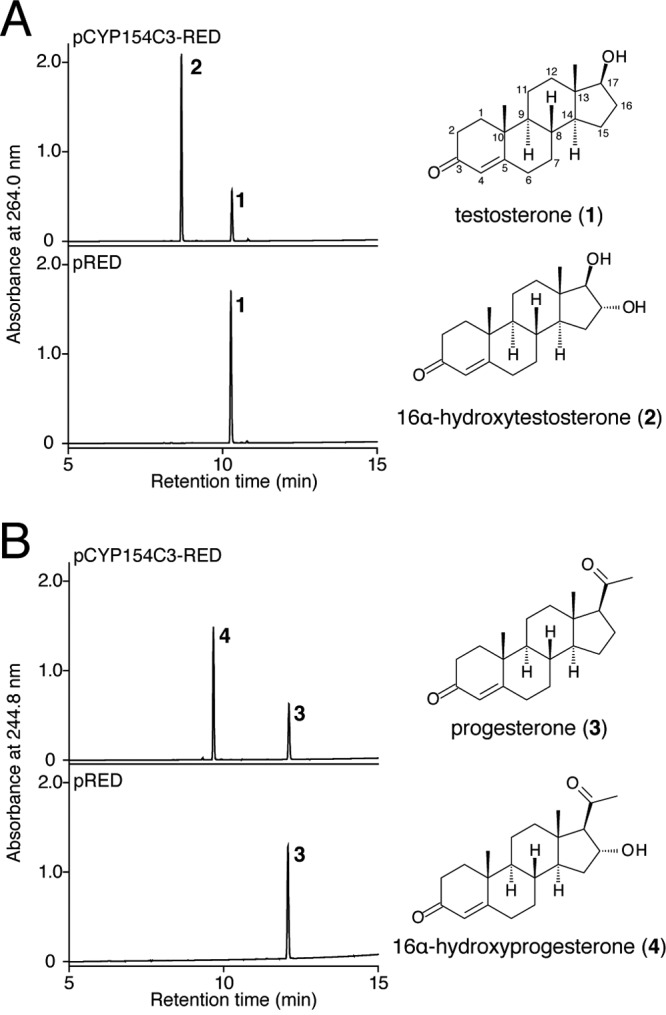
HPLC analysis of extracts of the bioconversion reactions using E. coli cells harboring pCYP154-RED and pRED. Bioconversion of testosterone (A) and progesterone (B) by E. coli cells harboring pCYP154-RED (upper) and pRED (lower). Chemical structures of testosterone (1), 16α-hydroxytestosterone (2), progesterone (3), and 16α-hydroxyprogesterone (4) are also shown.
FIG 4.

Steroid compounds used for the screening of substrates of CYP154C3.
Structure determination of compounds 2 and 4.
To elucidate the structure of compounds 2 and 4, we tried to obtain large amounts of these compounds by a large-scale bioconversion of testosterone (compound 1) and progesterone (compound 3), as described in Materials and Methods. Consequently, from the 1-liter reaction mixture, sufficient amounts of compound 2 (36.7 mg) and compound 4 (67.1 mg) were purified by SiO2 column chromatography and HPLC equipped with a reverse-phase column. We then analyzed the structure of compounds 2 and 4 by NMR (1H NMR, 13C NMR, correlation spectroscopy [COSY], heteronuclear multiple quantum coherence [HMQC], heteronuclear multiple-bond coherence [HMBC], and nuclear Overhauser effect spectroscopy [NOESY]) and HR-MS analyses. Compounds 2 and 4 gave an [M-H]− molecular ion at m/z 303.19684 and m/z 329.21189, respectively. According to this observation, the molecular formulas of compounds 2 and 4 were determined as C19H28O3 and C21H30O3, respectively. This result indicated that one OH group was introduced into each substrate. The position of the OH group was determined to be C-16α by the observation of 1H-1H vicinal spin networks between H-13, H-14, H-15, and H-17 and an NOE signal observed between H-16β, H-15α, and H-18 (see Tables S1 and S2 in the supplemental material). Thus, compounds 2 and 4 were determined to be 16α-hydroxytestosterone and 16α-hydroxyprogesterone, respectively (Fig. 3 depicts their structures).
Bioconversion of various steroids by E. coli BLR(DE3) harboring pCYP154C3-RED.
To obtain further insight into the substrate specificity and reaction specificity of CYP154C3, we examined the bioconversion of another 23 steroid compounds, shown in Fig. 4, by E. coli BLR(DE3) cells harboring pCYP154C3-RED. Fourteen of the 23 steroids were converted into different compounds with various conversion rates (Fig. 5A). Using the same method as that for the structure determination of compounds 2 and 4, we determined the structure of six products (from Δ4-androstene-3,17-dione [compound 6], 1,4-androstadiene-3,17-dione [compound 7], adrenosterone [compound 8], deoxycorticosterone [compound 9], 4-pregnane-3,11,20-trione [compound 10], and dehydroepiandrosterone [compound 11]), which were obtained with high yields in the reactions (see Tables S3 to S8 in the supplemental material). Similar to compounds 2 and 4, all six bioconversion products were found to have one OH group at the 16α position of the D ring. Three of the six bioconversion products, 16α-hydroxyadrenosterone (from compound 7), 16α-hydroxydeoxycorticosterone (from compound 9), and 16α-hydroxy-4-pregnene-3,11,20-trione (from compound 10), are novel compounds. This result indicates that CYP154C3 catalyzes regio- and stereospecific hydroxylation of various steroids at the D ring in the 16α position. Therefore, it was reasonable that dexamethasone (compound 21), which has a methyl group at C-16α, and estriol (compound 22), which has an OH group at C-16α, were not transformed. Many of the other compounds that were not transformed have a bulky substitute group at the D ring. We speculate that these bulky substitute groups are a structural hindrance for the substrates to enter the catalytic pocket of CYP154C3. Although we did not determine the structure of the bioconversion products from compounds 5 and 12 to 20, we speculated that they all have one OH group at the D ring in the 16α position, similar to the other steroids described above.
FIG 5.
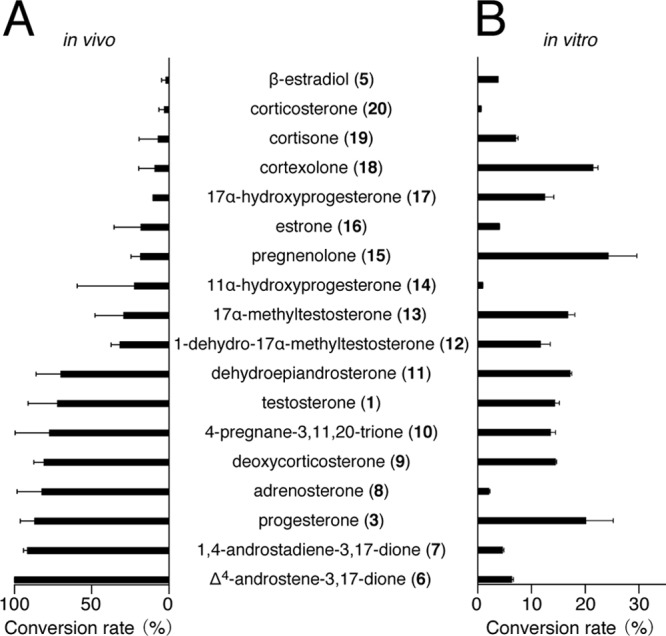
Conversion rates of each substrate in the bioconversion using E. coli cells harboring pCYP154-RED (A) and in the enzymatic conversion using purified CYP154C3 with the aid of ferredoxin and ferredoxin reductase from spinach (B). Values indicate the means from four (A) and three (B) independent experiments with standard deviations.
Equilibrium dissociation constants between CYP154C3 and the substrates.
We next investigated the equilibrium dissociation constants between CYP154C3 and the substrates. For this purpose, we prepared purified CYP154C3 that was not fused with RED, as described in Materials and Methods (Fig. 6A). The CO difference spectrum indicated that the purified CYP154C3 enzyme has an active form (Fig. 6B). Generally, when a substrate associates with P450, a spectrum change is observed; a peak at about 385 nm and a trough at about 420 nm should appear. This spectral change is caused by the substrate binding at a site very close to the heme, which leads to a transition from the hexacoordinate state of native low-spin P450 (Fe3+ heme; spin quantum number [S] = 1/2) to its pentacoordinate high-spin P450 (Fe3+ heme; S = 5/2) after the loss of H2O near the iron ligand (32). In other words, a compound that causes such a spectral change in the heme has a high possibility of being a substrate of the examined P450. Furthermore, by analyzing this spectral change, equilibrium dissociation constants (apparent Kd values) between a P450 and its substrate can be estimated. We examined the binding between CYP154C3 and all eight preferred steroid substrates (testosterone [compound 1], progesterone [compound 3], Δ4-androstene-3,17-dione [compound 6], adrenosterone [compound 7], 1,4-androstadiene-3,17-dione [compound 8], dehydroepiandrosterone [compound 9], 4-pregnane-3,11,20-trione [compound 10], and deoxycorticosterone [compound 11]). The spectral change in the heme caused by the binding between CYP154C3 and Δ4-androstene-3,17-dione is shown in Fig. 7A as a representative (see Fig. S1 in the supplemental material for all results). The apparent Kd value of the binding between CYP154C3 and Δ4-androstene-3,17-dione was calculated to be 1.51 ± 0.01 μM (see Fig. S1). Similarly, we estimated the apparent Kd values for the other steroids (Fig. 7B). The apparent Kd values for all eight preferred steroid substrates were less than 8 μM, indicating that CYP154C3 strongly binds to them.
FIG 6.
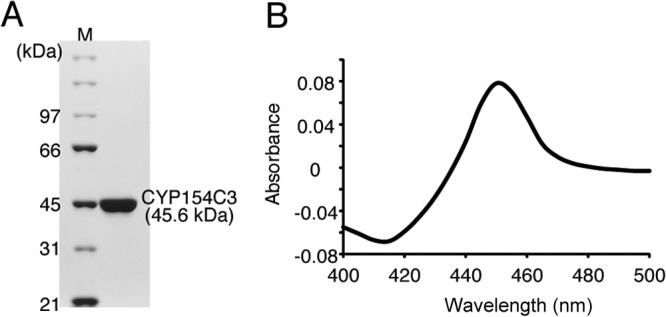
Production of the recombinant CYP154C3 enzyme. (A) SDS-PAGE analysis of purified CYP154C3 (45.6 kDa). (B) Dithionite-reduced CO difference spectrum of purified CYP154C3.
FIG 7.
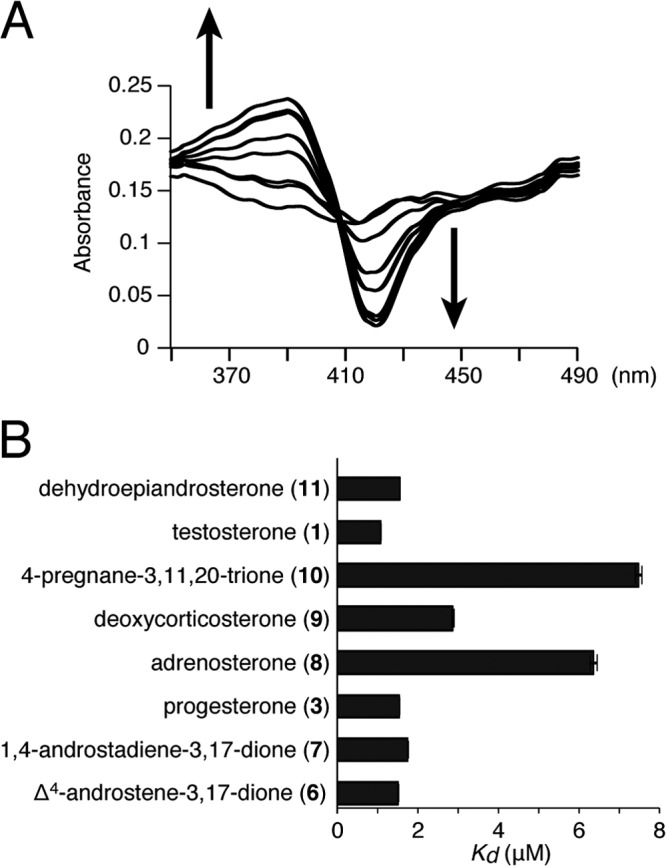
Equilibrium dissociation constants between CYP154C3 and the substrates. (A) Shift of the spin state of the heme (type I spectral shift) induced by the binding between CYP154C3 and Δ4-androstene-3,17-dione (final concentrations are 0, 0.195, 0.391, 0.781, 1.56, 3.13, 6.25, 12.5, and 25.0 μM). The shift accompanied by the increasing substrate concentrations is indicated by arrows. (B) Apparent Kd values of CYP154C3 and each substrate. Values indicate the means from three independent experiments with standard deviations.
Enzymatic conversion of various steroids in vitro.
To obtain further insight into the catalytic function of CYP154C3, we examined the enzymatic conversion of 18 steroids which showed bioconversion, using purified CYP154C3, spinach ferredoxin, and spinach ferredoxin reductase. We detected enzymatic conversion of all 18 examined steroids (Fig. 5B). All of the conversion products were the same compounds as the products in the respective bioconversion experiment described above, as determined by HPLC, indicating that CYP154C3 catalyzed the regio- and stereospecific hydroxylation of various steroids at the 16α position independently of the redox partners. Interestingly, there was no correlation between the conversion rates of the bioconversion using E. coli cells producing CYP154C3-RED and the enzymatic conversion (Fig. 5A and B).
Kinetic parameters of the CYP154C3-catalyzed hydroxylation of Δ4-androstene-3,17-dione.
We determined the kinetic parameters of the CYP154C3-catalyzed hydroxylation of Δ4-androstene-3,17-dione, which was the most preferable substrate in the bioconversion but a relatively poor substrate in the enzymatic conversion. Ferredoxin and ferredoxin reductase from spinach were used as redox partners. The Km value for Δ4-androstene-3,17-dione was estimated to be 31.9 ± 9.1 μM (see Fig. S2 in the supplemental material). The kcat value of the reaction was estimated to be 181 ± 4.5 s−1 (see Fig. S2).
DISCUSSION
In this study, we identified CYP154C3 of S. griseus as a steroid D-ring 16α-specific hydroxylase and analyzed its substrate specificity, reaction specificity, and kinetic parameters. The CYP154C5 enzyme of N. francinica has been shown to hydroxylate six steroids, testosterone (compound 1), pregnenolone (compound 15), dehydroepiandrosterone (compound 11), progesterone (compound 3), Δ4-androstene-3,17-dione (compound 6), and nandrolone, at the D ring in the 16α position, similar to CYP154C3 (14, 22). These two CYP154C enzymes are phylogenetically close to each other (both show 66% identity in the amino acid sequence). Thus, steroid D-ring 16α-specific hydroxylases seem to be conserved across the genera Streptomyces and Nocardia, although their physiological function remains unknown, as is the case for many other bacterial P450s. Interestingly, the substrate specificity of CYP154C3 seems to be different from that of CYP154C5. Progesterone is preferable to testosterone as the substrate for CYP154C5 (22). In contrast, CYP154C3 hydroxylates both progesterone and testosterone with similar efficiency.
It has been reported that CYP154H1 of T. fusca hydroxylates ethylbenzene, n-propylbenzene, styrene, and indole, and it oxidizes sulfide compounds, including thioanisole, into sulfoxides and sulfones in vitro (21). Ethylbenzene, n-propylbenzene, indole, and thioanisole were included in our substrate candidate library (Fig. 1), but no conversion of these compounds was observed in our bioconversion experiment, indicating that these compounds are not CYP154C3 substrates. This result showed that the substrate specificities are very different between CYP154H1 and CYP154C3. Taking the substrate specificities of other characterized CYP154 enzymes (CYP154C5, CYP154C1, CYP154A1, CYP154E1, and CYP154A8) into consideration, the CYP154 family seems to have a very broad substrate spectrum.
One of the important features of CYP154C3 is its strict substrate selectivity. For example, cyanobacterial CYP110E1 (33) uses various compounds with diverse molecular sizes and structures as substrates, and it catalyzes hydroxylation and deprotonation at various positions. CYP153A13a (P450balk) isolated from the marine bacterium Alcanivorax borkumensis SK2 hydroxylates various compounds with diverse structures, such as n-octane, cyclohexane, and n-butylbenzene (34). The CYP116 enzyme of Rhodococcus ruber DSM 44319 also hydroxylates various aromatic compounds, including 7-ethoxycoumarin, naphthalene, and toluene (35). In contrast to these enzymes, CYP154C3 accepts only steroids, and the hydroxylation occurs specifically at the D ring in the 16α position. We speculate that CYP154C3 has a catalytic pocket that is very suitable for the accommodation of steroid substrates. The low apparent Kd values (less than 8 μM) of the binding between CYP154C3 and steroid substrates support this speculation.
In the bioconversion experiment, we used E. coli BLR(DE3) harboring pCYP154C3-RED, in which CYP154C3 was produced as a fusion protein with the P450 reductase domain of P450Rhf (RED). We think that the CYP154C3 enzyme was active with the aid of the redox partner fused to it. However, we note that intrinsic ferredoxin and ferredoxin reductase of E. coli also have the potential to act as redox partner proteins of CYP154C3. Interestingly, E. coli BLR(DE3) producing only native CYP154C3 was able to transform testosterone into 16α-hydroxytestosterone with high efficiency; the conversion rate was approximately 60% (for 24 h at 25°C) when 5 mM testosterone was used as a substrate (data not shown). Therefore, we cannot eliminate the possibility that the E. coli ferredoxin and ferredoxin reductase function as redox partner proteins of CYP154C3, even when it is fused with RED. Nevertheless, we postulate that RED is the main redox partner of CYP154C3 in E. coli BLR(DE3) harboring pCYP154C3-RED, because CYP154C3 and RED are physically close to each other.
In this study, we performed bioconversion using E. coli cells producing the CYP154C3-RED fusion protein as well as enzymatic conversion using purified CYP154C3 with the aid of ferredoxin and ferredoxin reductase from spinach. To the best of our knowledge, this is the first example of a detailed comparison between the conversion rates of these two different conversion experiments. Interestingly, there was no correlation between the conversion rates of these experiments (Fig. 5A and B). For example, Δ4-androstene-3,17-dione (compound 6) was the most preferable substrate in the bioconversion, but it was a rather poor substrate in the enzymatic conversion. Conversely, pregnenolone (compound 15) was the most preferable substrate in the enzymatic conversion, but it was a rather poor substrate in the bioconversion. We speculated that the cytoplasmic membrane permeability of each steroid greatly affects the conversion rate in the bioconversion experiment. If this speculation is correct, then we cannot estimate correct substrate preference of an enzyme by a bioconversion experiment; an in vitro enzyme assay is required for this. However, another possibility is that the difference in conversion rates comes from the difference in redox partner proteins. In the bioconversion using E. coli BLR(DE3) harboring pCYP154C3-RED, the P450 reductase domain of P450Rhf from Rhodococcus species was used as the main redox partner. In contrast, in the in vitro enzyme assay, ferredoxin and ferredoxin reductase from spinach were the redox partner proteins. Several studies have indicated that in vitro P450 activity depends on the redox partner proteins used for the reaction (36, 37). Therefore, the difference in conversion rates between bioconversion and in vitro enzyme assay can be ascribed to the difference in redox partner. Furthermore, if the effect of the redox partner on P450 activity varies among respective substrates, the difference in redox partner should be considered for the difference in substrate preference between two conversion experiments.
The kcat/Km value of the hydroxylation of Δ4-androstene-3,17-dione by CYP154C3 was estimated to be 5.6 μM−1 s−1. There are only a limited number of reports on kcat/Km values for P450s. Examples are CYP105D7 (0.0006 μM−1 s−1) from Streptomyces avermitilis (38), which is involved in the biosynthesis of pentalenolactone, and CYP107L (2.3 μM−1 s−1) from Streptomyces platensis (39), which hydroxylates terfenadine, a kind of antihistamine agent. Compared to these values, the kcat/Km value (5.6 μM−1 s−1) of the CYP154C3-catalyzed hydroxylation of Δ4-androstene-3,17-dione seems to be in an appropriate range.
16α-Hydroxysteroids have various uses. Maternal urine steroid measurements are effective for detecting fetal steroid sulfatase deficiency, and 16α-hydroxydehydroepiandrosterone (16α-OH-DHA) sulfate is an important biomarker; 16α-OH-DHA sulfate is the precursor of estriol (compound 22), which is an estrogen (40). After hydrolysis of 16α-OH-DHA sulfate by a sulfatase, 16α-OH-DHA is detected by gas chromatography-mass spectrometry (GC-MS) (40). Therefore, 16α-OH-DHA is used for an important authentic standard in the diagnostic test. Meanwhile, 16α-hydroxyprogesterone seems to be a useful precursor for the synthesis of allopregnane-3,6,20-trione and pregnane-3,11,20-trion, which are metabolites of progesterone and could be important authentic standards in a diagnostic test. To the best of our knowledge, there is no efficient method for synthesizing 16α-hydroxyprogesterone with a high yield. Some 16α-hydroxylated steroids may also be important intermediates in the synthesis of steroid drugs in a wide range of therapeutic applications. Chemical synthesis of 16α-hydroxysteroids is generally difficult because of their complicated structure and many stereocenters. In contrast, using E. coli BLR(DE3) cells harboring pCYP154C3-RED, many steroids can be easily hydroxylated to produce 16α-hydroxysteroids with a high yield. Accordingly, the regio- and stereospecific hydroxylation of steroids at the D ring in the 16α position by CYP154C3 is very useful for the synthesis of 16α-hydroxysteroids. Thus, CYP154 has considerable potential for industrial applications.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kazuo Furihata for help with the NMR analysis.
This research was supported by a funding program for next-generation world-leading researchers from the Bureau of Science, Technology, and Innovation Policy, Cabinet Office, Government of Japan.
Footnotes
Published ahead of print 13 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03504-13.
REFERENCES
- 1.Nelson DR. 2011. Progress in tracing the evolutionary paths of cytochrome P450. Biochim. Biophys. Acta 1814:14–18. 10.1016/j.bbapap.2010.08.008 [DOI] [PubMed] [Google Scholar]
- 2.Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT. 2013. Reactive intermediates in cytochrome P450 catalysis. J. Biol. Chem. 288:17074–17081. 10.1074/jbc.R113.473108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortiz de Montellan PR. (ed). 2005. Cytochrome P450: structure, mechanism, and biochemistry, 3rd ed. Kluwer Academic/Plenum Publishers, New York, NY [Google Scholar]
- 4.Uno T, Ishizuka M, Itakura T. 2012. Cytochrome P450 (CYP) in fish. Environ. Toxicol. Pharmacol. 34:1–13. 10.1016/j.etap.2012.02.004 [DOI] [PubMed] [Google Scholar]
- 5.Pikuleva IA, Waterman MR. 2013. Cytochromes P450: roles in diseases. J. Biol. Chem. 288:17091–17098. 10.1074/jbc.R112.431916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizutani M, Ohta D. 2010. Diversification of P450 genes during land plant evolution. Annu. Rev. Plant Biol. 61:291–314. 10.1146/annurev-arplant-042809-112305 [DOI] [PubMed] [Google Scholar]
- 7.Podust LM, Sherman DH. 2012. Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep. 29:1251–1266. 10.1039/c2np20020a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawkes DB, Adams GW, Burlingame Ortiz de Montellano ALPR, De Voss JJ. 2002. Cytochrome P450cin (CYP176A), isolation, expression, and characterization. J. Biol. Chem. 277:27725–27732. 10.1074/jbc.M203382200 [DOI] [PubMed] [Google Scholar]
- 9.Peterson JA, Lu JY, Geisselsoder J, Graham-Lorence S, Carmona C, Witney F, Lorence MC. 1992. Cytochrome P-450terp. Isolation and purification of the protein and cloning and sequencing of its operon. J. Biol. Chem. 267:14193–14203 [PubMed] [Google Scholar]
- 10.Manivasagan P, Venkatesan J, Sivakumar K, Kim SK. 2013. Pharmaceutically active secondary metabolites of marine actinobacteria. Microbiol. Res. 2013:S0944-5013(13)00125-0. 10.1016/j.micres.2013.07.014 [DOI] [PubMed] [Google Scholar]
- 11.Kyoung-Rok K, Deok-kun O. 2013. Production of hydroxy fatty acids by microbial fatty acid-hydroxylation enzymes. Biotechnol. Adv. 31:1473–1485. 10.1016/j.biotechadv.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 12.Hayashi K, Yasuda K, Sugimoto H, Ikushiro S, Kamakura M, Kittaka A, Horst RL, Chen TC, Ohta M, Shiro Y, Sakaki T. 2010. Three-step hydroxylation of vitamin D3 by a genetically engineered CYP105A1: enzymes and catalysis. FEBS J. 277:3999–4009. 10.1111/j.1742-4658.2010.07791.x [DOI] [PubMed] [Google Scholar]
- 13.Agematsu H, Matsumoto N, Fujii Y, Kabumoto H, Doi S, Machida K, Ishikawa J, Arisawa A. 2006. Hydroxylation of testosterone by bacterial cytochromes P450 using the Escherichia coli expression system. Biosci. Biotechnol. Biochem. 70:307-311. 10.1271/bbb.70.307 [DOI] [PubMed] [Google Scholar]
- 14.Mahato SB, Mukherjee A. 1984. Microbial transformation of testosterone by Aspergillus fumigatus. J. Steroid Biochem. 21:341–342. 10.1016/0022-4731(84)90289-9 [DOI] [PubMed] [Google Scholar]
- 15.Smith KE, Latif S, Kirk DN. 1989. Microbial transformation of steroids-II. Transformations of progesterone, testosterone and androstenedione by Phycomyces blakesleeanus. J. Steroid Biochem. 32:445–451 [DOI] [PubMed] [Google Scholar]
- 16.Smith KE, Latif S, Kirk DN. 1990. Microbial transformations of steroids–VI. Transformation of testosterone and androsterone by Botryosphaerica obtusa. J. Steroid Biochem. 35:115–120 [DOI] [PubMed] [Google Scholar]
- 17.Kolek T, Swizdor A. 1998. Biotransformation XLV: transformations of 4-ene-3-oxo steroids in Fusarium culmorum culture. J. Steroid Biochem. Mol. Biol. 67:63–69. 10.1016/S0960-0760(98)00073-9 [DOI] [PubMed] [Google Scholar]
- 18.Podust LM, Kim Y, Arase M, Neely BA, Beck BJ, Bach H, Sherman DH, Lamb DC, Kelly SL, Waterman MR. 2003. The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1: a new monooxygenase that functionalizes macrolide ring systems. J. Biol. Chem. 278:12214–12221. 10.1074/jbc.M212210200 [DOI] [PubMed] [Google Scholar]
- 19.Podust LM, Bach H, Kim Y, Lamb DC, Arase M, Sherman DH, Kelly SL, Waterman MR. 2004. Comparison of the 1.85 Å structure of CYP154A1 from Streptomyces coelicolor A3(2) with the closely related CYP154C1 and CYPs from antibiotic biosynthetic pathways. Protein Sci. 13:255–268. 10.1110/ps.03384804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Q, Lamb DC, Kelly SL, Lei L, Guengerich FP. 2010. Cyclization of a cellular dipentaenone by Streptomyces coelicolor cytochrome P450 154A1 without oxidation/reduction. J. Am. Chem. Soc. 132:15173–15175. 10.1021/ja107801v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schallmey A, den Besten G, Teune IG, Kembaren RF, Janssen DB. 2011. Characterization of cytochrome P450 monooxygenase CYP154H1 from the thermophilic soil bacterium Thermobifida fusca. Appl. Microbiol. Biotechnol. 89:1475–1485. 10.1007/s00253-010-2965-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bracco P, Janssen DB, Schallmey A. 2013. Selective steroid oxyfunctionalisation by CYP154C5, a bacterial cytochrome P450. Microb. Cell Fact. 12:95. 10.1186/1475-2859-12-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thoma RW, Fried J, Bonanno S, Grabowich P. 1957. Oxidation of steroids by microorganisms. IV. 16α-Hydroxylation of 9α-fluorohydrocortisone and 9α-fluoroprednisolone by Streptomyces roseochromogenus. J. Am. Chem. Soc. 79:4818 [Google Scholar]
- 24.Berrie JR, Williams RAD, Smith KE. 1999. Microbial transformations of steroids-XI. Progesterone transformation by Streptomyces roseochromogenes—purification and characterisation of the 16α-hydroxylase system. J. Steroid Biochem. 71:153–165 [DOI] [PubMed] [Google Scholar]
- 25.Von BC, Le-Huu P, Urlacher VB. 2013. Cluster screening: an effective approach for probing the substrate space of uncharacterized cytochrome P450s. ChemBioChem [Epub ahead of print.] 10.1002/cbic.201300271 [DOI] [PubMed] [Google Scholar]
- 26.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S. 2008. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 190:4050–4060. 10.1128/JB.00204-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funa N, Funabashi M, Ohnishi Y, Horinouchi S. 2005. Biosynthesis of hexahydroxyperylenequinone melanin via oxidative aryl coupling by cytochrome P-450 in Streptomyces griseus. J. Bacteriol. 187:8149–8155. 10.1128/JB.187.23.8149-8155.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29.Nodate M, Kubota M, Misawa N. 2006. Functional expression system for cytochrome P450 genes using the reductase domain of self-sufficient P450RhF from Rhodococcus sp. NCIMB 9784. Appl. Microbiol. Biotechnol. 71:455–462. 10.1007/s00253-005-0147-y [DOI] [PubMed] [Google Scholar]
- 30.Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239:2370–2378 [PubMed] [Google Scholar]
- 31.Brill E, Hannemann F, Zapp J, Brüning G, Jauch J, Bernhardt R. 2013. A new cytochrome P450 system from Bacillus megaterium DSM319 for the hydroxylation of 11-keto-β-boswellic acid (KBA). Appl. Microbiol. Biotechnol. [Epub ahead of print.] 10.1007/s00253-013-5029-0 [DOI] [PubMed] [Google Scholar]
- 32.Jefcoate CR. 1978. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 52:258–279. 10.1016/S0076-6879(78)52029-6 [DOI] [PubMed] [Google Scholar]
- 33.Makino T, Otomatsu T, Shindo K, Kitamura E, Sandmann G, Harada H, Misawa N. 2012. Biocatalytic synthesis of flavones and hydroxyl-small molecules by recombinant Escherichia coli cells expressing the cyanobacterial CYP110E1 gene. Microb. Cell Fact. 11:95. 10.1186/1475-2859-11-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita N, Sumisa F, Shindo K, Kabumoto H, Arisawa A, Ikenaga H, Misawa N. 2009. Comparison of two vectors for functional expression of a bacterial cytochrome P450 gene in Escherichia coli using CYP153 genes. Biosci. Biotechnol. Biochem. 73:1825–1830. 10.1271/bbb.90199 [DOI] [PubMed] [Google Scholar]
- 35.Liu L, Schmid RD, Urlacher VB. 2006. Cloning, expression, and characterization of a self-sufficient cytochrome P450 monooxygenase from Rhodococcus ruber DSM 44319. Appl. Microbiol. Biotechnol. 72:876–882. 10.1007/s00253-006-0355-0 [DOI] [PubMed] [Google Scholar]
- 36.Chun YJ, Shimada T, Sanchez-Ponce R, Martin MV, Lei L, Zhao B, Kelly SL, Waterman MR, Lamb DC, Guengerich FP. 2007. Electron transport pathway for a Streptomyces cytochrome P450: cytochrome P450 105D5-catalyzed fatty acid hydroxylation in Streptomyces coelicolor A3(2). J. Biol. Chem. 282:17486–17500. 10.1074/jbc.M700863200 [DOI] [PubMed] [Google Scholar]
- 37.Tyson CA, Lipscomb JD, Gunsalus IC. 1972. The roles of putidaredoxin and P450cam in methylene hydroxylation. J. Biol. Chem. 247:5777–5784 [PubMed] [Google Scholar]
- 38.Takamatsu S, Xu LH, Fushinobu S, Shoun H, Komatsu M, Cane DE, Ikeda H. 2011. Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis. J. Antibiot. 64:65–71. 10.1038/ja.2010.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lombard M, Salard I, Sari MA, Mansuy D, Buisson D. 2011. A new cytochrome P450 belonging to the 107L subfamily is responsible for the efficient hydroxylation of the drug terfenadine by Streptomyces platensis. Arch. Biochem. Biophys. 508:54–63. 10.1016/j.abb.2011.01.008 [DOI] [PubMed] [Google Scholar]
- 40.Marcos J, Craig WY, Palomaki GE, Kloza EM, Haddow JE, Roberson M, Bradley LA, Shackleton CHL. 2009. Maternal urine and serum steroid measurements to identify steroid sulfatase deficiency (STSD) in second trimester. Prenat. Diagn. 29:771–780. 10.1002/pd.2284 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.