

Abstract
The phyla Planctomycetes, Verrucomicrobia, Chlamydiae, Lentisphaerae, and “Candidatus Omnitrophica (OP3)” comprise bacteria that share an ancestor but show highly diverse biological and ecological features. Together, they constitute the PVC superphylum. Using large-scale comparative genome sequence analysis, we identified a protein uniquely shared among all of the known members of the PVC superphylum. We provide evidence that this signature protein is expressed by representative members of the PVC superphylum. Its predicted structure, physicochemical characteristics, and overexpression in Escherichia coli and gel retardation assays with purified signature protein suggest a housekeeping function with unspecific DNA/RNA binding activity. Phylogenetic analysis demonstrated that the signature protein is a suitable phylogenetic marker for members of the PVC superphylum, and the screening of published metagenome data indicated the existence of additional PVC members. This study provides further evidence of a common evolutionary history of the PVC superphylum and presents a unique case in which a single protein serves as an evolutionary link among otherwise highly diverse members of major bacterial groups.
INTRODUCTION
The bacterial phyla Planctomycetes, Verrucomicrobia, Chlamydiae, Lentisphaerae, “Candidatus Omnitrophica (OP3),” and “Candidatus Poribacteria” were proposed to share an ancestor on the basis of their monophyletic grouping in 16S rRNA-based phylogenetic trees (1). This diverse assemblage of phyla was termed the PVC superphylum and later received additional support from genomic and phylogenetic analysis of conserved proteins (2–4). Most recently, 16 housekeeping and ribosomal proteins were used to infer evolutionary relationships among the members of the PVC superphylum (5). This further established the common evolutionary origin of the members of the PVC superphylum.
Despite their common origin, the members of the PVC superphylum differ greatly with respect to life-style, physiology, and ecology (1). Each phylum includes members that attracted significant research interest because of their importance in carbon and nitrogen cycling (e.g., Rhodopirellula and “Candidatus Kuenenia” species [6, 7]), as pathogens or symbionts (e.g., Chlamydia and Protochlamydia species [8–10]), or as environmental microbes in aquatic and soil habitats (e.g., Verrucomicrobia [11, 12]). In addition to their ecological, biotechnological, and medical relevance, some members of the PVC superphylum show genetic and cellular features that are unusual for bacteria but reminiscent of eukaryotes or archaea (13–15). Because of these similarities, members of the PVC superphylum have been implicated in the emergence and evolution of eukaryotes, a hypothesis that is controversially discussed (14, 16–20).
In this study, we performed an extensive comparative genomic analysis in order to identify unifying links among the diverse members of the PVC superphylum. We describe the analysis and characterization of a protein, independently identified very recently (5), that is shared by all of the members of the superphylum but absent from all other bacteria. Computational analysis and functional assays provided evidence of a putative housekeeping function for this protein. Because of its conservation among the members of the PVC superphylum, we were able to use this protein to extract information about the occurrence and diversity of the members of the PVC superphylum from the available environmental metagenomes.
MATERIALS AND METHODS
Identification of the signature protein (SP).
Predicted coding sequences from completely sequenced PVC and representative non-PVC genomes were obtained from the INSDC (21) and NCBI RefSeq (22) databases. All-versus-all pairwise sequence similarities were precalculated by the SIMAP database (23). From SIMAP we obtained all of the bidirectionally best-matching protein pairs (BBH) between all of the genomes, in which the alignment covered at least 50% of both protein sequences and the E value was not higher than 1e-04. The score of each BBH was additionally used as the threshold to determine inparalogs from the respective genomes. In order to cluster BBHs from the PVC superphylum into clusters of orthologous groups (COGs), we first determined all of the three-cliques (triangles) formed by PVC BBHs. Triangles were grouped into COGs if they shared a BBH. The remaining PVC BBHs were added to COGs if one of the proteins was already a member of a COG and the other was not. All of the other PVC BBHs were considered individual COGs. Inparalogs associated with BBH proteins were added to the respective COGs in all of the clustering steps mentioned above.
For each COG, we determined the presence or absence of the proteins encoded in PVC genomes. For COGs occurring in all of the PVC genomes, we determined their presence or absence in the representative non-PVC genomes from BBHs between PVC and non-PVC genomes. Only one COG, the PVC SP, was present in all of the PVC genomes and absent from all of the non-PVC genomes.
COG-based presence-or-absence analysis.
The COGs of all of the bacterial genomes were obtained from the eggNOG (24) database. The BBHs between the PVC and non-PVC genomes described above were used to determine the presence or absence of each COG in the PVC genomes not yet contained in eggNOG. A matrix was then created with all of the COGs in the first column and the organism names in the top row. Then the table was filled with a 1 or a 0 for each COG for each genome on the basis of its presence or absence, respectively, allowing a quick overview of COG conservation across PVC and non-PVC bacteria as selective sums.
For each COG without representatives in the PVC superphylum, the Escherichia coli representative was found and used as the query in searches against the NCBI Refseq database (22) with BLAST (25). The first 10 proteins of nonredundant origin (different organisms) were collected. With these sets, the average protein size and isoelectric point (pI) were calculated for each COG. The pI was calculated by solving the Henderson-Hasselbach equation by a local Perl script.
Screening of metagenome data.
All of the assembled metagenomes available at the JGI Genome Portal (26) were downloaded and organized into BLAST databases with makeblastdb (included in the BLAST+ suit) according to their originating environments. The nucleotide sequence databases were searched for the presence of the SP by using tBLASTx (25) with default settings and all of the known SP sequences as queries. The output files were then merged, and the matching translated sequences were collected. All of the redundant sequences (exact or substring match) and those that contained stop codons or were shorter than 45 amino acids were removed. The remaining sequences were submitted to the Conserved Domains Database (27), and the presence of the SP domain was verified in all of them.
Phylogenetic analysis.
Amino acid sequences from sequenced members of the PVC superphylum with or without metagenomic proteins were aligned by using MUSCLE (28) in MEGA5 (29), and their evolutionary history was inferred by the unweighted-pair group method using average linkages (UPGMA) (30) or FastTree (31). The evolutionary distances were computed with the JTT (32) for UPGMA and the WAG model (33) for FastTree, while a gamma value of 20 was used for both. Phylogenetic trees were visualized with iTOL (34).
Reverse transcriptase PCR.
Verrucomicrobium spinosum DSM 4136 and Rhodopirellula baltica SH1 were inoculated from colonies grown on agar plates to flasks containing 100 ml of the appropriate media described by Schlesner (35, 36), respectively, and grown while shaking at 22°C. Initially, growth characteristics were determined by measuring optical density at 600 nm (OD600) in a spectrophotometer. Cultures were harvested after 3 days (exponential growth phase) and 5 to 6 days (stationary phase), respectively. Cells were lysed by bead beating (FastPrep FP120, Savant), and total RNA was extracted with TRIzol (Molecular Research Center, Inc.) according to the manufacturer's instructions. Primers were designed to target the genes encoding the V. spinosum and R. baltica SPs, respectively (VssignF, 5′-TCCCAGCATCGTAGTCTCAA-3′; VssignR, 5′-TAAGCTTCCGGCTTGGTCT-3′; RbsignF, 5′-TAAGAGTCGCAACGTCCTGA-3′; RbsignR, 5′-TTCTTCTTGTCGTCGGCTTC-3′). The housekeeping gene coding for glyceraldehyde 3-phosphate dehydrogenase from V. spinosum was used as a positive control (37) (VsqapdhF, 5′-CGGTCTCTTTACCGAAGCTG-3′; VsqapdhR, 5′-CGTTGGAGATGATGTTGTGG-3′). Reverse transcriptase PCR was performed with Moloney murine leukemia virus polymerase (Invitrogen) and an annealing temperature of 55°C for 35 cycles.
Cloning, expression, and purification of recombinant proteins.
The genes coding for the SP of R. baltica (GenBank/EMBL/DDBJ accession number KF733603) and Protochlamydia amoebophila (YP_008052) were synthesized (GenScript Corp.) flanked by restriction sites for EcoRI (Thermo Scientific) and XhoI (Thermo Scientific) that were used for subsequent cloning into the pGEX 4T-1 vector (GE Healthcare) containing an N-terminal glutathione S-transferase (GST) tag at the multiple cloning site. The final constructs were then transformed into electrocompetent E. coli strain BL21 (λDE3). Transformed E. coli cells were grown overnight in 5 ml of Luria-Bertani (LB) medium containing 50 μg/ml ampicillin (LB-Amp) at 37°C on a shaker (120 rpm), and the next day, 1 ml of each culture was used to inoculate flasks containing 100 ml of LB-Amp. The cells were incubated for 2 h (OD600 of ∼0.4), and then the expression of the proteins was induced by 100 μM (final concentration) isopropyl-β-d-thiogalactopyranoside. After 2 h of induction, cells expressing the GST signature fusion protein were collected by centrifugation in 50-ml tubes at 6,000 rpm for 10 min at 4°C. The supernatant was discarded, and the tubes containing the cell pellets were stored at −20°C.
For protein purification, the collected cell pellets were resuspended by vortexing in 4.5 ml binding buffer (125 mM Tris, 150 mM NaCl, 1% Triton X [pH 8], protease inhibitors [Roche Diagnostics]) plus 0.5 ml lysozyme from a 2-mg/ml stock solution. The tubes were incubated horizontally on a rocking platform for 15 min at room temperature and then placed on ice. The final cell disruption was performed with three rounds of sonication for 30 s at 70% strength (Bandelin Electronic) with intervals of cooling. The lysates were centrifuged at 12,000 rpm for 10 min, and the supernatant was transferred to new tubes. After three rounds of washing in 20 ml of binding buffer, 2 ml of a glutathione-coated magnetic bead slurry (Pierce) was mixed with the lysate and kept shaking horizontally for 1 h. With an appropriate magnetic stand, the beads were washed three times with washing buffer (125 mM Tris, 500 mM NaCl, 1% Triton X [pH 8]). Finally, 4 ml of elution buffer (125 mM Tris, 500 mM NaCl, 50 mM reduced glutathione [pH 9], protease inhibitors) was added and the beads were incubated for an additional 15 min before elution, three times, keeping the eluates separated. The purity and quantity of purified proteins were determined by 12.5% SDS-PAGE and staining with colloidal Coomassie blue (Invitrogen).
For desalting, 2-ml volumes of pooled protein purifications were placed in an Ultracell 10K spin column (Millipore) and phosphate-buffered saline (PBS; pH 7.4) was used to fill the column to 15 ml. The column was centrifuged for 30 min at 5,000 × g. The desalting was repeated with another 15 ml of PBS, resulting in 200 μl of desalted and concentrated protein.
Electrophoretic mobility shift assay.
To evaluate the effect of SP on the mobility of nucleic acids, purified proteins were mixed with DNA or RNA samples and gel loading dye (New England BioLabs). The mixtures were then loaded onto 1% agarose gels, run for 1 h at 120 V, and visualized by staining with ethidium bromide. When cleaved protein was used, 3 μl of thrombin (GE Healthcare Life Sciences) was added to 30 μl of a desalted and concentrated stock of fusion protein and left overnight at room temperature. Complete cleavage was then verified by SDS-PAGE.
Nucleotide sequence accession number.
The gene sequence coding for the SP of R. baltica was deposited in GenBank/EMBL/DDBJ under accession number KF733603.
RESULTS AND DISCUSSION
To investigate the evolutionary history of the PVC superphylum, early after the original proposal, we performed a comparative genome analysis to identify orthologous genes conserved among all of the PVC members. We discovered a single protein-coding gene of unknown function that is uniquely shared among all of the members of the superphylum that we refer to as the SP of the PVC superphylum (I. Lagkouvardos, T. Rattei, and M. Horn, 8th German Chlamydia Workshop, Munich, Germany, 24 to 26 February 2010). In the following, we verified its presence in all of the further sequenced PVC genomes published since with PSI-BLAST (25) and found the SP in all of the 55 available genome sequences. The only exceptions were (i) missing gene predictions (e.g., for R. baltica SH1) that we identified only with tblastn and (ii) incomplete genome sequences (e.g., the Poribacteria draft genome that has been estimated to represent 75% of the complete genome [38]) that we did not consider suitable for presence-or-absence analysis (see Table S1 in the supplemental material). Recently, 16 housekeeping and ribosomal proteins were used to infer evolutionary relationships among the members of the PVC superphylum (5), which further established the common evolutionary origin of the PVC superphylum. By searching for conserved signature insertions or deletions, the same study independently recovered the SP to be encoded in all of the known members (except for Poribacteria) (5). The presence of a protein in all of the PVC members that does not show any sequence similarity to other known proteins serves as a unifying link among the members of this diverse assemblage of microbes and suggests a conserved function.
Asking whether the SP is expressed, we searched available transcriptomic and proteomic data on members of the PVC superphylum. Members of the phylum Chlamydiae are represented the best in such studies, with few reports on Planctomycetes. We found evidence of its expression only in members of the phylum Chlamydiae, where the SP seems to be expressed constitutively in small amounts similar to those of some housekeeping proteins (see Table S2 in the supplemental material). To compensate for the lack of evidence of transcription in Planctomycetes and Verrucomicrobia, we performed reverse transcriptase PCR assays with RNA from R. baltica SH1 and V. spinosum DSM 4136 isolated in the logarithmic and stationary growth phases, respectively. This demonstrated that the SP is also expressed in these organisms (see Fig. S1 in the supplemental material). Taken together, these findings are evidence of the expression of SP by representatives of all of the major phyla within the PVC superphylum.
The SP is a small, 50- to 60-amino-acid protein exhibiting considerable conservation of its sequence (55% average amino acid sequence similarity among all of the representatives; Fig. 1A) and physicochemical properties. In silico prediction of its localization, isoelectric point, and secondary structure revealed a highly basic cytosolic protein (pI 10 to 11) (39) consisting of an alpha helix followed by a putative second alpha helix, depending on the prediction software (Fig. 1B), which is reminiscent of the DNA binding helix-turn-helix motif (40). Structure prediction and physicochemical characteristics thus point toward a nucleic acid-associated protein such as histone-like proteins, transcription factors, or ribosomal proteins. Consistent with this observation, the SP has been recognized as a protein family in TIGRFAM (TIGR04137 [41]), where a possible rRNA interaction is proposed.
FIG 1.

Features of the PVC superphylum SP. (A) Conservation of the SP amino acid sequence. A sequence logo based on a MUSCLE alignment of all of the known SPs generated by WebLogo 3 is shown (28, 48). The overall height of the alignment positions indicates sequence conservation, while the height of each symbol indicates the relative frequency of each amino acid at the respective position. Symbol colors reflect amino acid chemical properties. Highly conserved positions can be observed along the complete length of the alignment, with a longer conserved region in the middle, corresponding to a predicted α-helix. (B) Predicted secondary and tertiary structures of representative SPs compared to those of small DNA/RNA binding proteins of E. coli. Predictions were performed with I-TASSER (49) (i to iii) and the QUARK server (50) (iv to vi). Structures: i and iv, SP of Protochlamydia amoebophila. UWE25 (GenBank/EMBL/DDBJ accession number YP_008052); ii and v, SP of V. spinosum (WP_009960041); iii and vi, SP of R. baltica (KF733603); vii, E. coli ribosomal protein L30 (Protein Data Bank accession number 2AW4); viii, E. coli DNA binding protein H-NS (Protein Data Bank accession number 1HNS); ix, E. coli histone like protein HU (Protein Data Bank accession number 1MUL). Pink, alpha-helix; yellow, beta-sheet; blue, turn; gray, unstructured. Independently of the software, a central alpha-helix is predicted for all of the SPs. The SP of all of the Planctomycetes shows a C-terminal lysine-rich extension that forms additional secondary-structure elements. (C) Nucleic acid mobility retardation by SP of R. baltica and P. amoebophila. Agarose gel i, retardation assay with sheared genomic DNA. Lanes: 1, molecular size markers; 2, empty; 3, genomic DNA with GST-tagged SP of R. baltica, 4: GST-tagged SP of R. baltica without DNA; 5, genomic DNA only. Agarose gel ii, retardation assay with purified total RNA. Lanes: 1 and 5, molecular size markers; 2, RNA only; 3, RNA with GST-tagged SP of R. baltica; 4, RNA with GST-tagged SP of P. amoebophila; 6, RNA with GST only. Arrows indicate bands representing the 16S and 23S rRNAs, respectively. (Bottom agarose gel) retardation assay with PCR products. Lanes: 1, molecular size markers; 2, only PCR product; 3 to 7, PCR product with increasing concentrations of GST-tagged SP of P. amoebophila; 8, PCR product with GST only. The same molecular size marker was used in all of the experiments, and fragment sizes in base pairs are shown on the left. The retardation assays suggest unspecific binding of SP to DNA and RNA.
To verify the in silico prediction and to investigate the in vitro activity of the SP, we heterologously expressed the SPs of P. amoebophila (as a representative of Chlamydiae) and R. baltica (Planctomycetes) as glutathione S-transferase (GST) fusion proteins in E. coli. The expressed proteins were purified with glutathione-coated magnetic beads and subsequently used for gel retardation assays. When the fusion proteins were incubated with various DNA and RNA products (sheared genomic DNA, total RNA, or PCR products), the mobility of the nucleic acids in agarose gels was retarded (Fig. 1C). This was also observed after the removal of the GST tag by protease treatment but never when only the GST tag was used (Fig. 1C; see Fig. S2 in the supplemental material). Dose-dependent retardation was observed when increasing amounts of SP were added to PCR products (Fig. 1C). Together, these findings demonstrate an unspecific and concentration-dependent DNA and RNA binding activity of the SP from R. baltica and P. amoebophila in vitro. This mode of nucleic acid interaction seems to rule out a role for the SP as a transcription factor, which typically shows highly specific DNA binding activity.
To investigate whether the SP could function as a histone-like protein, we analyzed E. coli cells overexpressing R. baltica or P. amoebophila SP. Overexpression of histones generally leads to nucleation of chromatin, which can be detected by staining with DNA-specific dyes (40). However, no nucleation was observed during the overexpression of both SPs in E. coli (see Fig. S3 in the supplemental material). Although we cannot exclude a histone function for the SP in vivo in R. baltica or P. amoebophila, expression in the heterologous host does not support such a role. In addition, P. amoebophila, showing a condensed nucleoid in the elementary body stage, encodes other histone-like proteins that are likely to be involved in chromatin condensation (42, 43).
The occurrence and documented expression of the SP in all of the members of the PVC superphylum point toward a highly conserved function. This function could be unique to the superphylum, or the SP could substitute for the role of an otherwise conserved and essential protein in non-PVC organisms. To search for proteins that are well conserved in most other organisms but do not occur in PVC members, we conducted a COG-based comparative analysis of all of the available PVC genomes and a representative set of non-PVC genomes. This analysis revealed several highly conserved bacterial functions with no representation by a protein homolog in the PVC superphylum (Table 1). Of those bacterial homologs missing from PVC bacteria, the ribosomal protein L30, which is also present in archaea and eukaryotes, shows a striking physicochemical similarity to the SP of the PVC superphylum. Despite the absence of any amino acid sequence similarity, the two proteins have similar size, pI, and expression profiles (Table 1). Together with its observed nucleic acid binding activity, this suggests the possibility that the SP is a functional analog of ribosomal protein L30, which is missing from all of the members of the superphylum. Further experimental investigation is needed to verify the presence and function of the SP in the ribosome of PVC members.
TABLE 1.
Functional categories conserved among bacterial genomes absent from members of the PVC superphyluma
| COG | No. found in: |
Function | Avg length (amino acids) | Avg pI | |
|---|---|---|---|---|---|
| PVCb | Other bacteriac | ||||
| COG0806 | 0 | 466 | 16S rRNA processing protein RimM | 182 | 4.8 |
| COG0779 | 0 | 439 | Ribosome maturation factor RimP | 154 | 4.5 |
| COG1559 | 0 | 405 | Aminodeoxychorismate lyase | 339 | 8.9 |
| COG1841 | 0 | 375 | Ribosomal protein L30/L7E | 60 | 11.0 |
| COG1660 | 0 | 334 | Predicted P loop-containing kinase | 294 | 5.8 |
| COG2884 | 0 | 327 | Cell division ATP-binding protein FtsE | 224 | 9.5 |
| COG2177 | 0 | 318 | Cell division protein FtsX | 304 | 7.6 |
| COG0595 | 0 | 300 | mRNA degradation RNase J1/J2 | 537 | 5.4 |
| SP | 56 | 0 | DNA/RNA binding | 60 | 11.6 |
COGs absent from all members of the PVC superphylum but conserved in at least 60% of all non-PVC bacteria analyzed are listed together with basic physicochemical properties. The SP of the PVC superphylum is shown for comparison.
Total n = 56.
Total n = 490.
The high sequence conservation and exclusive presence of the SP in all of the members of the PVC superphylum suggest that it may serve as an additional phylogenetic marker for the superphylum. In fact, the topology of amino acid-based phylogenetic trees resembles that of the 16S rRNA gene (see Fig. S4 in the supplemental material). Simple clustering by UPGMA recovered all of the PVC phyla with good bootstrap support, and the structures within the different phyla are largely similar (see Fig. S4A). The 16S rRNA tree topology was less well recovered in approximately maximum-likelihood SP trees with FastTree (31). Here the Verrucomicrobia SPs were not monophyletic but included the Lentisphaerae sequences (see Fig. S4B). Still, the overall congruence between 16S rRNA gene- and SP-based trees allowed us to exploit the SP for the analysis of metagenomic data sets from various environmental samples to obtain insights into the diversity of the PVC superphylum. To this end, metagenomic data sets available in IMG/m (44) and SIMAP (45) were first screened with PSI-BLAST (46). In addition, tblastx was used to detect the SP even in the absence of correctly predicted coding sequences. A total of 233 nonredundant SP sequences were detected, mainly in metagenomes originating from freshwater (36%), soil (34%), and marine (21%) samples (see Table S1). Phylogenetic analysis of these sequences showed that the majority of the metagenomic SPs are related to one of the known phyla within the PVC superphylum (Fig. 2). Within the different phyla, however, several novel evolutionary lineages could be observed, significantly expanding the known diversity of the PVC superphylum as inferred from SP phylogeny. Lentisphaerae was the least diverse phylum, followed by Chlamydiae and “Candidatus Omnitrophica (OP3)”; Planctomycetes and Verrucomicrobia were the most diverse. Interestingly, the majority of the Verrucomicrobia sequences originated from soil metagenomes, while most of the “Candidatus Omnitrophica (OP3)” and Chlamydiae sequences originated from freshwater samples (including sediments); no trend was observed for the other phyla. Although this analysis cannot be used to quantitatively assess the abundance of PVC microbes in the different habitats, the observed ecological patterns are consistent with those of known members of the superphylum. For example, the relatively low number of metagenomic SPs branching with known members of the Chlamydiae phylum is consistent with the generally low abundance of chlamydial protein sequences detected in metagenomes in a recent study (47). An explanation for this could be the low abundance of members of the phylum Chlamydiae (which are typically associated with eukaryotic hosts) in environmental samples, which would result in a low coverage of chlamydial genomes in metagenomic data sets. Overall, the suitability of the SP as a phylogenetic marker allows the identification of genomic fragments containing the SP as originating from a PVC member and thus helps in the binning of metagenomic data and in the estimation of the overall presence of PVC members in such data sets. In addition, concatenation of the SP with other conserved proteins should help in the construction of robust phylogenetic trees to analyze the diversity and evolutionary history of the PVC superphylum (5).
FIG 2.
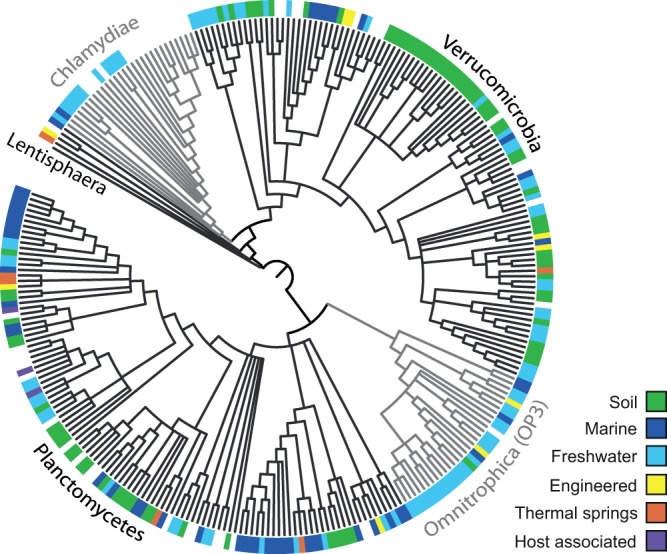
Evolutionary relationships of all of the known PVC superphylum SPs and their metagenomic homologs. The environmental origin of SPs is color coded at the tips of the tree for metagenomic sequences but not for SPs originating from complete genome sequences. An approximate maximum-likelihood tree is shown; nodes with less than 70% support are collapsed.
In summary, all of the known members of the PVC superphylum produce a small, conserved SP with nucleic acid binding activity. There is evidence of the expression of this protein by some PVC members, and its physicochemical properties, predictions of its structure, and the absence of ribosomal protein L30 from all of the members of the superphylum suggest that the SP has a conserved function and is possibly associated with the ribosome. We demonstrated that the SP is a useful marker for the analysis of metagenomic data and that it may serve to investigate the diversity and ecology of bacteria related to this medically and biotechnologically important superphylum.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Michael Wagner for helpful discussions.
This work was funded by Austrian Science Fund (FWF) grant Y277-B03 and the University of Vienna (Graduate School Symbiotic Interactions). Matthias Horn acknowledges support from the European Research Council (ERC StG EvoChlamy).
Footnotes
Published ahead of print 1 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02655-13.
REFERENCES
- 1.Wagner M, Horn M. 2006. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr. Opin. Biotechnol. 17:241–249. 10.1016/j.copbio.2006.05.005 [DOI] [PubMed] [Google Scholar]
- 2.Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH, Petroni G. 2008. Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J. Bacteriol. 190:3192–3202. 10.1128/JB.01797-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths E, Gupta RS. 2007. Phylogeny and shared conserved inserts in proteins provide evidence that Verrucomicrobia are the closest known free-living relatives of chlamydiae. Microbiology 153:2648–2654. 10.1099/mic.0.2007/009118-0 [DOI] [PubMed] [Google Scholar]
- 4.Kamneva OK, Knight SJ, Liberles DA, Ward NL. 2012. Analysis of genome content evolution in PVC bacterial super-phylum: assessment of candidate genes associated with cellular organization and lifestyle. Genome Biol. Evol. 4:1375–1390. 10.1093/gbe/evs113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta RS, Bhandari V, Naushad HS. 2012. Molecular signatures for the PVC clade (Planctomycetes, Verrucomicrobia, Chlamydiae, and Lentisphaerae) of bacteria provide insights into their evolutionary relationships. Front. Microbiol. 3:327. 10.3389/fmicb.2012.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shu QL, Jiao NZ. 2008. Different Planctomycetes diversity patterns in latitudinal surface seawater of the open sea and in sediment. J. Microbiol. 46:154–159. 10.1007/s12275-008-0002-9 [DOI] [PubMed] [Google Scholar]
- 7.Strous M, Pelletier E, Mangenot S, Rattei T, Lehner A, Taylor MW, Horn M, Daims H, Bartol-Mavel D, Wincker P, Barbe V, Fonknechten N, Vallenet D, Segurens B, Schenowitz-Truong C, Medigue C, Collingro A, Snel B, Dutilh BE, Op den Camp HJM, van der Drift C, Cirpus I, van de Pas-Schoonen KT, Harhangi HR, van Niftrik L, Schmid M, Keltjens J, van de Vossenberg J, Kartal B, Meier H, Frishman D, Huynen MA, Mewes HW, Weissenbach J, Jetten MSM, Wagner M, Le Paslier D. 2006. Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440:790–794. 10.1038/nature04647 [DOI] [PubMed] [Google Scholar]
- 8.Bebear C, de Barbeyrac B. 2009. Genital Chlamydia trachomatis infections. Clin. Microbiol. Infect. 15:4–10. 10.1111/j.1469-0691.2008.02647.x [DOI] [PubMed] [Google Scholar]
- 9.Corsaro D, Greub G. 2006. Pathogenic potential of novel chlamydiae and diagnostic approaches to infections due to these obligate intracellular bacteria. Clin. Microbiol. Rev. 19:283–297. 10.1128/CMR.19.2.283-297.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horn M. 2008. Chlamydiae as symbionts in eukaryotes. Annu. Rev. Microbiol. 62:113–131. 10.1146/annurev.micro.62.081307.162818 [DOI] [PubMed] [Google Scholar]
- 11.Zwart G, Crump BC, Agterveld MPKV, Hagen F, Han SK. 2002. Typical freshwater bacteria: an analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 28:141–155. 10.3354/ame028141 [DOI] [Google Scholar]
- 12.Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, Knight R, Fierer N. 2011. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 43:1450–1455. 10.1016/j.soilbio.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santarella-Mellwig R, Franke J, Jaedicke A, Gorjanacz M, Bauer U, Budd A, Mattaj IW, Devos DP. 2010. The compartmentalized bacteria of the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum have membrane coat-like proteins. PLoS Biol. 8:e1000281. 10.1371/journal.pbio.1000281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devos DP, Reynaud EG. 2010. Evolution. Intermediate steps. Science 330:1187–1188. 10.1126/science.1196720 [DOI] [PubMed] [Google Scholar]
- 15.Fuerst JA, Sagulenko E. 2011. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 9:403–413. 10.1038/nrmicro2578 [DOI] [PubMed] [Google Scholar]
- 16.Fuchsman CA, Rocap G. 2006. Whole-genome reciprocal BLAST analysis reveals that Planctomycetes do not share an unusually large number of genes with Eukarya and Archaea. Appl. Environ. Microbiol. 72:6841–6844. 10.1128/AEM.00429-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McInerney JO, Martin WF, Koonin EV, Allen JF, Galperin MY, Lane N, Archibald JM, Embley TM. 2011. Planctomycetes and eukaryotes: a case of analogy not homology. Bioessays 33:810–817. 10.1002/bies.201100045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budd A, Devos DP. 2012. Evaluating the evolutionary origins of unexpected character distributions within the bacterial Planctomycetes-Verrucomicrobia-Chlamydiae superphylum. Front. Microbiol. 3:401. 10.3389/fmicb.2012.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forterre P. 2011. A new fusion hypothesis for the origin of Eukarya: better than previous ones, but probably also wrong. Res. Microbiol. 162:77–91. 10.1016/j.resmic.2010.10.005 [DOI] [PubMed] [Google Scholar]
- 20.Fuerst JA, Sagulenko E. 2012. Keys to eukaryality: planctomycetes and ancestral evolution of cellular complexity. Front. Microbiol. 3:167. 10.3389/fmicb.2012.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura Y, Cochrane G, Karsch-Mizrachi I. 2013. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 41:D21–24. 10.1093/nar/gks1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35:D61–D65. 10.1093/nar/gkl842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rattei T, Tischler P, Gotz S, Jehl MA, Hoser J, Arnold R, Conesa A, Mewes HW. 2010. SIMAP—a comprehensive database of pre-calculated protein sequence similarities, domains, annotations and clusters. Nucleic Acids Res. 38:D223–226. 10.1093/nar/gkp949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, Jensen LJ, von Mering C, Bork P. 2012. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40:D284–289. 10.1093/nar/gkr1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, Otillar R, Poliakov A, Ratnere I, Riley R, Smirnova T, Rokhsar D, Dubchak I. 2012. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 40:D26–32. 10.1093/nar/gkr947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Zheng CJ, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu SN, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang DC, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D348–D352. 10.1093/nar/gks1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sneath PHA, Sokal RR. 1973. Numerical taxonomy; the principles and practice of numerical classification. W. H. Freeman, San Francisco, CA [Google Scholar]
- 31.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282 [DOI] [PubMed] [Google Scholar]
- 33.Whelan S, Goldman N. 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18:691–699. 10.1093/oxfordjournals.molbev.a003851 [DOI] [PubMed] [Google Scholar]
- 34.Letunic I, Bork P. 2007. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128. 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
- 35.Schlesner H. 1987. Verrucomicrobium spinosum gen. nov., sp. nov.—a fimbriated prosthecate bacterium. Syst. Appl. Microbiol. 10:54–56. 10.1016/S0723-2020(87)80010-3 [DOI] [Google Scholar]
- 36.Schlesner H. 1994. The development of media suitable for the microorganisms morphologically resembling Planctomyces spp., Pirellula spp., and other Planctomycetales from various aquatic habitats using dilute media. Syst. Appl. Microbiol. 17:135–145. 10.1016/S0723-2020(11)80042-1 [DOI] [Google Scholar]
- 37.Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E. 1999. Housekeeping genes as internal standards: use and limits. J. Biotechnol. 75:291–295. 10.1016/S0168-1656(99)00163-7 [DOI] [PubMed] [Google Scholar]
- 38.Siegl A, Kamke J, Hochmuth T, Piel J, Richter M, Liang CG, Dandekar T, Hentschel U. 2011. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5:61–70. 10.1038/ismej.2010.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FSL. 2010. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615. 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison SC. 1991. A structural taxonomy of DNA-binding domains. Nature 353:715–719. 10.1038/353715a0 [DOI] [PubMed] [Google Scholar]
- 41.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJA, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. 2009. InterPro: the integrative protein signature database. Nucleic Acids Res. 37:D211–D215. 10.1093/nar/gkn785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes HW, Wagner M. 2004. Illuminating the evolutionary history of chlamydiae. Science 304:728–730. 10.1126/science.1096330 [DOI] [PubMed] [Google Scholar]
- 43.Sixt BS, Heinz C, Pichler P, Heinz E, Montanaro J, Op den Camp HJM, Ammerer G, Mechtler K, Wagner M, Horn M. 2011. Proteomic analysis reveals a virtually complete set of proteins for translation and energy generation in elementary bodies of the amoeba symbiont Protochlamydia amoebophila. Proteomics 11:1868–1892. 10.1002/pmic.201000510 [DOI] [PubMed] [Google Scholar]
- 44.Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D, Chen IM, Grechkin Y, Dubchak I, Anderson I, Lykidis A, Mavromatis K, Hugenholtz P, Kyrpides NC. 2008. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 36:D534–D538. 10.1093/nar/gkm869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rattei T, Tischler P, Arnold R, Hamberger F, Krebs J, Krumsiek J, Wachinger B, Stumpflen V, Mewes W. 2008. SIMAP—structuring the network of protein similarities. Nucleic Acids Res. 36:D289–D292. 10.1093/nar/gkm963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 47.Lagkouvardos I, Weinmaier T, Lauro FM, Cavicchioli R, Rattei T, Horn M. 15 August 2013. Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the chlamydiae. ISME J. (Epub ahead of print.) 10.1038/ismej.2013.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: A sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu D, Zhang Y. 2012. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80:1715–1735. 10.1002/prot.24065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.