

Abstract
Peptide deformylase (PDF) is an essential bacterial metalloprotease involved in deformylation of N-formyl group from nascent polypeptide chains during protein synthesis. Iron-containing variants of this enzyme from Salmonella enterica serovar Typhimurium (sPDF) and Mycobacterium tuberculosis (mPDF), although inhibited by oxidizing agents like H2O2, exhibited strikingly different 50% inhibitory concentrations (IC50s) that ranged from nanomolar (sPDF) to millimolar (mPDF) levels. Furthermore, the metal dissociation rate was higher in sPDF than mPDF. We hypothesized that a restriction in entry of environmental oxygen or oxidizing agents into the active site of mPDF might be the cause for such discrepancies between two enzymes. Since the active-site residues of the two proteins are similar, we evaluated the role of the oxidation-prone noncatalytic residue(s) in the process. Amino acid sequence analysis revealed that Cys-130 of sPDF corresponds to Met-145 of mPDF and that they are away from the active sites. Swapping methionine with cysteine in mPDF, the M145C protein displayed a drastic decrease in the IC50 for H2O2 and an increased metal dissociation rate compared to the wild type. Matrix-assisted laser desorption ionization (MALDI) analysis of a trypsin-digested fragment containing Cys-145 of the M145C protein also indicated its increased susceptibility to oxidation. To incorporate residues identical to those of mPDF, we created a double mutant of sPDF (C130M-V63C) that showed increased IC50 for H2O2 compared to the wild type. Interestingly, the oxidation state of cysteines in C130M-V63C was unaffected during H2O2 treatment. Taken together, our results unambiguously established the critical role of noncatalytic cysteine/methionine for enzymatic sensitivity to H2O2 and, thus, for conferring behavioral distinction of bacterial PDFs under oxidative stress conditions.
INTRODUCTION
Translation initiation involves an additional step of formylation of methionyl-tRNA, which is a distinctive feature in protein synthesis of prokaryotes and eukaryotic organelles, such as mitochondria and chloroplasts (1–3). As a consequence, throughout the eubacterial lineage, N-formylated methionine is the first amino acid to incorporate at the amino terminal end of nascent polypeptide chains (4). The formylated methionine is often not retained and is subsequently excised from nascent polypeptide chains by a specialized hydrolytic process, known as N-terminal methionine excision. The enzyme, peptide deformylase (PDF), is responsible for the removal of the N-formyl group of methionine by hydrolyzing its amide bond (5, 6). In fact, removal of the formyl group of the first methionine is mandatory for its subsequent excision in the process of maturation of the polypeptide chain (7); therefore, PDFs are known to be essential for bacterial survival (8–10). Also, in pathogenic bacteria, PDF is a promising and well-explored drug target (11–13).
The def gene in bacteria encodes PDF. X-ray and nuclear magnetic resonance (NMR) structures of different bacterial PDFs have been determined in recent years (14–18). They are categorized in two groups; type I has a long C-terminal tail such as in Gram-negative bacteria, while type II contains insertion sequences such as have been noted in Gram-positive bacteria (18). In spite of these differences, bacterial PDF proteins have three highly conserved motifs, motif I (GXGXAAXQ), motif II (EGCLS), and motif III (QHEXXH) (where X is any hydrophobic residue), forming an active site. Since it is a metalloprotease, the metal atom has been reported to be tetra- or pentahedrally coordinated (19, 20) with the cysteine in motif II and two histidines in motif III within the conserved enzyme active site (21–24). Any mutation in these residues led to inactivation of the enzymatic activity of the protein (21, 25).
In its naturally occurring form, either iron (Fe2+) or zinc (Zn2+) is present in different bacterial PDFs (5, 15, 25–28). Iron-containing Escherichia coli PDF was found to be enzymatically active but very labile because of conversion of Fe2+ to Fe3+ by atmospheric oxygen, and the conserved coordinating cysteine of the enzyme is reported to be oxidized in the process (16, 29). On the other hand, PDFs from Staphylococcus aureus and Mycobacterium tuberculosis did not show such oxygen sensitivity despite having iron as a metal ion (9, 15, 25, 26). These contradictory observations are really puzzling since bacterial PDFs exhibit considerable amino acid sequence homology and they are structurally similar to each other (9, 15, 16). Therefore, the data argue that if the iron oxidation status leading to modification of the conserved active site cysteine were the sole factor in governing the activities of bacterial PDFs against oxidative stress, it would have affected the functionality of any bacterial PDF protein in a similar fashion. However, there has been no detailed investigation to date explaining such distinctions in the behavior of bacterial peptide deformylases in response to oxidative stress. This is an important aspect to elucidate in the context of intracellular bacteria like M. tuberculosis, which as a successful pathogen often adopts varied survival tactics to cope with such stress.
In this study, we assessed the enzymatic activity of recombinant PDF proteins from Salmonella enterica serovar Typhimurium (here referred as S. Typhimurium; sPDF) and M. tuberculosis (mPDF) following exposure to oxidative stress utilizing H2O2, a well-known oxidizing agent. In spite of iron-containing proteins having a conserved metal-ion-coordinating cysteine (Cys-90 in sPDF and Cys-106 in mPDF) at the active site, we found that sPDF and mPDF displayed differences in enzymatic activity in response to H2O2 exposure. The IC50 (the concentration causing 50% inhibition in enzyme activity) for H2O2 was considerably higher (i.e., in the millimolar range) in mPDF than in sPDF (nanomolar range). To account for such a discrepancy in the enzymatic activities, our results of mutational analyses of both proteins unambiguously established the crucial role of oxidation-prone amino acids in mPDF. Interestingly, these residues are neither in the enzyme active site nor participating in metal ion coordination but are involved in conferring such a behavioral distinction in mPDF.
MATERIALS AND METHODS
Materials.
Restriction/modifying enzymes were procured from New England Biolabs. Different kits (PCR DNA/gel band purification, plasmid DNA preparation, Western blotting detection), dithiothreitol (DTT), and protein molecular weight markers were obtained from GE Healthcare. H2O2 (Merck), Herculase fusion DNA polymerase (Stratagene), sucrose, and HEPES buffer (MP Biomedicals) were commercially available. Other chemicals and reagents, including ammonium bicarbonate, acetonitrile, CelLytic B cell lysis reagent, α-cyno-4-hydroxy cinnamic acid, diethylenetriaminepentaacetic acid (DTPA), EDTA, ferrous sulfate, N-formyl-Met-Ala, trinitrobenzene sulfonic acid, and trypsin were from the Sigma Chemical Company. Oligonucleotides used in this study were custom synthesized (IDT/Sigma). Different media for bacterial cultures were either from Himedia or from Difco.
Site-directed mutagenesis and recombinant constructs.
Genomic DNA from S. Typhimurium (30) was used for PCR amplification of the 510-bp def gene (STM3406) using sequence-specific primers (CK1 [5′-GGAATTCCATATGTCAGTTTTGC AAGTGTTACATATT-3′] and CK2 [5′-CCCAAGCTTTTAAGCCCGGGCGTTCAG-3′]) incorporating restriction sites (NdeI in CK1 and HindIII in CK2) and Herculase fusion DNA polymerase per a standardized protocol (denaturation at 95°C for 5 min followed by reaction at 95°C for 1 min, 59°C for 30 s, and 72°C for 30 s for 29 cycles and, finally, extension at 72°C for 10 min). The amplified fragment following NdeI-HindIII digestion was ligated to the corresponding sites of pET28c. The construction of pET-mPDF (isolation of the M. tuberculosis def gene, Rv0429c, and its subsequent cloning in expression vector) is described elsewhere (25). Two external primers (CK1 and CK2 for sPDF and CR26 and CR27 for mPDF) and two internal (incorporating mutation) primers (see Table S1 in the supplemental material) were designed for creating each site-directed mutant of sPDF (V63C, C130M, and C130M-V63C) or mPDF (C59S, C68S, M145C, and M145S) employing a PCR-based overlap extension method (31). PCR products containing desired mutations following cloning in pET28c and wild-type constructs (pET-sPDF or pET-mPDF) were individually transformed into E. coli strain DH5α to build up the DNA and also into BL21(DE3) for expression and purification of recombinant proteins. All mutations were confirmed by sequencing using an automated DNA sequencer.
Expression and purification of recombinant proteins.
A fresh culture of BL21(DE3) cells harboring pET-sPDF or pET-mPDF or different mutant constructs was set up in LB broth containing 50 μg/ml kanamycin at 37°C in a shaker (shaking speed, 200 rpm) with ∼1% inoculum (from the overnight cultures), grown until an optical density at 600 nm (OD600) of ∼0.85 was reached, induced with 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and incubated a further 12 to 14 h at 18°C (except the M145C mutant, which was grown for 2.5 h at 37°C). It is worth mentioning here that, unlike urea solubilization as reported previously (25), a slight alteration of the culture conditions (as mentioned above) yielded recombinant mPDF or its mutants (except M145C) as a soluble protein. Following harvesting, cells were resuspended (1 gm cell pellet/5 ml) in lysis buffer (50 mM HEPES [pH 7.0], 100 mM NaCl, 5 mM DTT, 0.1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml pepstatin, 1 μg/ml leupeptin) for sPDF or its mutants. Cells containing mPDF or its mutants (except for M145C), on the other hand, were resuspended (1.5 g/10 ml) in lysis buffer supplemented with 6% sucrose. This was followed by sonication (each cycle of 10 s on/10 s off over ice for 15 min). BL21(DE3) cells with the M145C mutant of mPDF were solubilized in CelLytic B cell lysis reagent (1.5 g cell/20 ml) and incubated for 30 min at 25°C. The supernatant fraction was loaded in a nickel-nitrilotriacetic acid (Ni-NTA) column and washed in 10 bed volumes with 50 mM HEPES (pH 7.0) containing 100 mM NaCl and either 20 mM imidazole for sPDF or 50 mM imidazole and 6% sucrose for mPDF. The protein was eluted with 50 mM HEPES (pH 7.0) containing 100 mM NaCl and 200 mM imidazole with (mPDF) or without (sPDF) 6% sucrose. The protein concentration (32), at this step was ∼1.5 mg/ml for mPDF or ∼11 mg/ml for sPDF. To remove imidazole (carried out if it was an experimental need), the purified protein(s) was dialyzed (4°C for ∼14 h in dialysis buffer containing 50 mM HEPES (pH 7.0) and 100 mM NaCl for sPDF or the same buffer supplemented with 6% sucrose for mPDF (protein/buffer ratio of 1:100 [vol/vol]; buffer changed four times). Recombinant proteins were stored in aliquots (protein concentration of ∼1.5 mg/ml for mPDF or ∼11 mg/ml for sPDF) at −80°C until used. They were utilized in assays following dilution in the same buffer. In experiments where apo-enzyme was used, starting from cell lysis and proceeding to protein purification, 1 mM EDTA was added in buffers. Apo-enzymes were always made imidazole-free by dialyzing in buffers (see above) supplemented with 1 mM EDTA and used on the same day as the day of preparation. HEPES was replaced with Tris-HCl as the buffer in protein preparations used for circular dichroism (CD) experiments.
Enzyme assay.
The ability of the enzyme (sPDF or mPDF) to deformylate N-formyl-Met-Ala was assessed through a spectrophotometric assay as described earlier (25) with slight modification for carrying it out in a microtiter plate. Briefly, PDF protein (50 ng of mPDF or 125 ng of sPDF, 50 mM HEPES buffer, pH 7.0, containing 100 mM NaCl) was incubated with the substrate (5 mM for the single-point assay or 0 to 20 mM for enzyme kinetics; total reaction volume = 10 μl) at 30°C for 30 min. The reaction was terminated by addition of 4% HClO4 (10 μl), and the reaction mixture was further incubated (37°C for 2 h) with trinitrobenzene sulfonic acid (TNBSA) reagent (0.01% in 0.1 M NaHCO3 buffer, pH 8.4; 130 μl). This was followed by addition of 10% SDS (50 μl)–1 M HCl (25 μl), and absorption of the highly chromogenic derivative generated due to the reaction of primary amine with TNBSA was monitored at 340 nm in an enzyme-linked immunosorbent assay (ELISA) reader (Molecular Devices). The readings were corrected by subtracting the blank (all ingredients except enzyme) values, and the specific enzyme activity (nmol of free amino group produced/min/mg of protein) was calculated from standard curves prepared with l-methionine (0 to 21.4 nmol). Km and Vmax values were obtained from non-linear-fit plots, and for calculating kcat, the molecular mass of recombinant enzyme was considered 21 kDa (for sPDF) or 23 kDa (for mPDF). We have noted that irrespective of the methods used for cell lysis (sonication and use of CelLytic B cell lysis reagent), there was no difference in the specific activity values determined for the mPDF enzyme. Unless mentioned otherwise, data for enzyme activity represent means ± standard deviations (SD) of the values from at least three independent experiments.
Atomic absorption spectroscopy.
Recombinant His-tagged dialyzed protein (mPDF or sPDF or their mutants; 500 μg protein/assay) was digested with 5% HNO3 (150°C for 2 h), filtered (0.45-μm-pore-size filter), and injected into an atomic absorption spectrophotometer (Shimadzu) to determine the metal content. The readings were corrected by subtracting the blank (buffer containing 5% HNO3), and, finally, the metal ion concentration of samples was calculated from a standard curve calculated with known metals (nickel, iron, and zinc). Under our experimental conditions, the metal content of different purified preparations (mPDF and sPDF and their mutants) revealed iron as the coordinating ion (∼1 mol of iron/mol of polypeptide) with no detectable amount of nickel or zinc contamination, even when recombinant His-tagged proteins were purified in Ni-NTA columns.
Western blotting.
Purified proteins (1 μg of protein/slot) were resolved in 12% SDS-PAGE and transferred to nitrocellulose membranes (0.45-μm pore size) using a Mini Trans-Blot apparatus (Bio-Rad). Membranes were stained with Ponceau S to ensure transfer, processed using anti-His antibody, and detected through an ECL detection system following the recommended protocol of the manufacturer (GE Healthcare).
CD spectroscopy.
CD spectra of wild-type and mutant proteins were compared using a Jasco J-810 spectropolarimeter. Measurements in the far UV region (250 to 190 nm) were carried out on protein solutions (0.25 mg/ml) employing a cell with a path length of 0.1 cm at 25°C. The mean residue ellipticity, (θ), was calculated using a mean residue molecular mass of 110 Da. Each spectrum reported represents an average of 5 scans.
Matrix-assisted laser desorption ionization (MALDI)–MS.
Purified recombinant protein (mPDF or M145C; 1 μg/assay) was incubated (4°C for 15 min) with or without H2O2 (10 mM) and diluted 10 times with buffer (20 mM Tris buffer [pH 7.0] containing 75 mM NaCl). Samples were then concentrated (6,500 × g for 30 min) in a Microcon filter (YM-10), washed twice in same buffer, mixed (1:1) with 40 mM ammonium bicarbonate (pH 8.2) containing 9% acetonitrile, and digested with trypsin (37°C for 18 h). Following mixing of trypsin-digested samples with an equal volume of the matrix (α-cyno-4-hydroxy cinnamic acid), mass spectra for peptide mass fingerprint analysis and, subsequently, amino acid sequences of the fragments (tandem mass spectrometry [MS/MS]; carried out under collision-induced dissociation [CID] “off” conditions in 1-kV positive mode) were obtained (AB Sciex tandem time of flight [TOF/TOF] 5800 spectrometer). The isotope abundance for peptides was calculated using Protein Prospector (http://prospector.ucsf.edu), and the peptide sequences were identified utilizing the Mascot server.
Assessment of thiol modification.
7-Chloro-4-nitrobenz-2-oxa-1,3-diazole chloride (NBD-Cl; 20-fold molar excess of protein) was added to dialyzed protein (500 μg of sPDF or C130M-V63C in 20 mM Tris [pH 7.0] containing 75 mM NaCl) following incubation (25°C for 15 min) with or without H2O2 (100-fold molar excess), and a thiol modification reaction (total volume = 600 μl) was allowed to proceed in the dark for 45 min. This was followed by removal of unbound NBD by washing through a Microcon (YM-10) centrifugal filter (three washes with 20 mM Tris [pH 7.0] containing 75 mM NaCl), and the labeling was detected by a spectral scan (260 to 600 nm) using a spectrophotometer (Shimadzu).
Bioinformatics and structural analysis.
Multiple sequence alignment was carried out using the Clustal X program (33) with default input parameters for gap opening and extension penalties. We observed ∼100% sequence identity (except for three amino acids) in the nucleotide-derived amino acid sequences of PDFs from E. coli and S. Typhimurium. Therefore, the crystal structure of PDF from E. coli (Protein Data Bank identification no. [PDB ID] 3K6L) was considered a model for S. Typhimurium and compared to that of the M. tuberculosis (PDB ID 3E3U). Analysis was performed by displaying the structures in the PYMOL (www.pymol.org) graphics program.
RESULTS
Assessment of the H2O2 sensitivity profiles of sPDF and mPDF.
Iron-containing PDFs are known to be sensitive to treatment with oxidizing agents because of oxidation of Fe2+ to Fe3+ at the metal binding core (29, 34). To have an insight on this aspect, deformylation abilities of sPDF and mPDF were compared following preincubation (15 min at 4°C) of proteins (50 ng for mPDF or 125 ng for sPDF) with different concentrations of H2O2 (0 to 100 mM). As with other bacterial PDFs, the enzyme activities of sPDF and mPDF were affected. Surprisingly, while 10 μM H2O2 completely inhibited the deformylase activity of sPDF, ≥100 mM was required for mPDF (Fig. 1). The IC50s of H2O2 for sPDF and mPDF were ∼46 nM and ∼16.1 mM, respectively (Table 1). Since the buffers used may contaminate with traces of metal ions and considering the fact that bacterial PDFs are able to confer enzymatic activity in the presence of divalent cations such as Ni2+, Co2+, and Zn2+ (35, 36), we compared the deformylation abilities of mPDF following H2O2 treatment in the presence or absence of DTPA, a metal ion chelator that entraps dissociated metals without actively extracting the prosthetic group of an enzyme (34). Interestingly, no significant alteration in mPDF activity due to addition of DTPA was noticed and its (200 μM) presence did not affect H2O2 sensitivity (inset, Fig. 1A).
FIG 1.
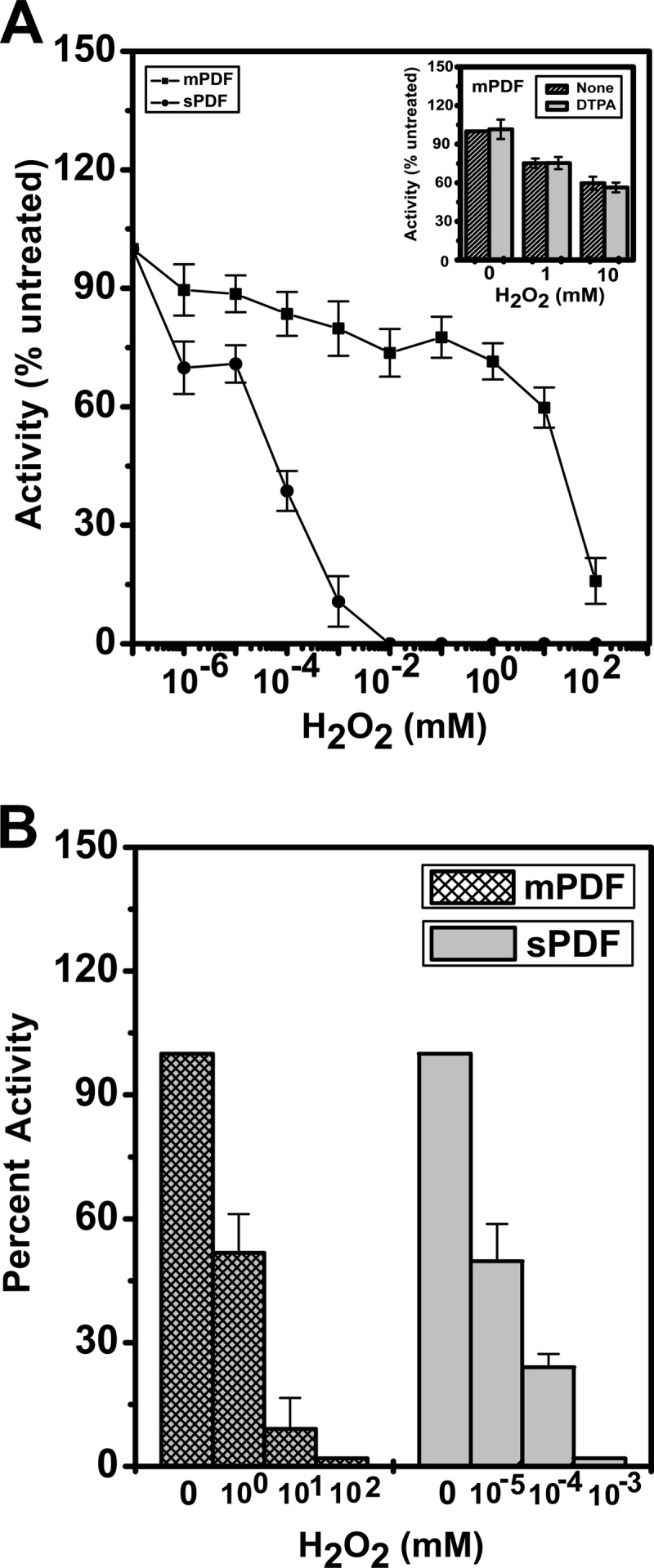
Deformylation abilities of mPDF and sPDF in response to exposure to H2O2. (A) The enzyme assay was carried out with mPDF (50 ng/assay) or sPDF (125 ng/assay) following incubation with H2O2 (15 min at 4°C). The inset shows the peptide deformylase activities of mPDF in the presence of DTPA following incubation with H2O2. (B) Effect of H2O2 on peptide deformylase activity of apo-enzyme (mPDF or sPDF) following reconstitution with iron (Fe2+). Apo-proteins were prepared by inclusion of 1 mM EDTA in cell lysis buffer, purified, dialyzed, and used for reconstitution of enzyme subjected to metallation as mentioned in the text.
TABLE 1.
IC50s for H2O2 exhibited by mPDF and sPDF mutants
| Protein | IC50 (mM) ± SD | No. of expts |
|---|---|---|
| sPDF | 4.6 ± 1.1 × 10−5 | 5 |
| C90S | NAa | 3 |
| C130S | 4.9 ± 0.5 × 10−5 | 3 |
| C130 M | 16 ± 2.7 × 10−5 | 3 |
| V63C | 15 ± 3.0 × 10−5 | 3 |
| C130 M-V63C | 144 ± 29 × 10−5 | 5 |
| mPDF | 16.1 ± 3.1 | 5 |
| C59S | 13.6 ± 0.8 | 3 |
| C68S | 0.5 ± 0.05 | 3 |
| C106S | NA | 3 |
| M145S | 0.07 ± 0.01 | 3 |
| M145C | 0.06 ± 0.01 | 5 |
NA, no activity.
To ensure that iron occupied the active site of the enzyme throughout the experimental procedure, we prepared apo-proteins of both mPDF and sPDF by inclusion of 1 mM EDTA in the lysis buffer (see Materials and Methods). The apo-enzyme was purified in Ni-NTA columns and dialyzed to remove imidazole in the buffer containing EDTA. The apo-enzyme prepared in this way was found to be enzymatically inactive. The enzyme was then reconstituted with the Fe2+ ion utilizing a method described elsewhere (34). Briefly, the apo-protein was diluted 1:1 with buffer containing 3 mM Fe2SO4 such that the Fe2+ concentration was 600 μM, which exceeds the carryover EDTA concentration. The reconstituted enzyme was further diluted to 1:100 in assay buffer (50 mM HEPES buffer [pH 7.0], 100 mM NaCl) containing 100 μM Fe2+. This was followed by addition of 200 μM DTPA, and the reaction mixture was utilized either for assessing deformylation ability directly or for monitoring the effect of H2O2. The reaction following preincubation with or without H2O2 was terminated by adding catalase (∼550 units/ml; see reference 34), and the reaction mixture was incubated with the substrate for determining enzyme activity. We found that the inactive apo-enzymes became active and exhibited behavior patterns in response to H2O2 treatment (Fig. 1B) similar to those observed with recombinant mPDF or sPDF (the IC50 for H2O2 was in the nanomolar range for sPDF, whereas it was in the millimolar range for mPDF).
The striking difference between sPDF and mPDF in their levels of sensitivity to oxidative stress might be the consequence of alteration in their rates of iron dissociation from the enzyme active site (37). Therefore, the deformylation abilities of mPDF and sPDF were compared following incubation with DTPA for 0 to 8 h at 25°C. Iron (Fe2+) oxidation by environmental oxygen would lead to dissociation of the metal (Fe3+) from the enzyme active site. The free metal released in this way would be trapped by the DTPA and thus result in a decrease in the enzymatic activity of both proteins, as the passage of time would reflect their rates of metal dissociation. Interestingly, the values corresponding to the half-life (t1/2) of enzyme inactivation in the presence of DTPA were ∼47 min and ∼330 min for sPDF and mPDF, respectively (Table 2). Thus, these results showed that the rate of metal dissociation from the enzyme was higher in sPDF than in mPDF.
Table 2.
Half-life of the enzyme activity at 25°C in the presence of DTPA
| Protein | t1/2 (min) ± SD | No of expts |
|---|---|---|
| sPDF | 47 ± 6 | 3 |
| mPDF | 330 ± 21 | 3 |
| M145C | 0.5 ± 0.01 | 7 |
The low rate of metal dissociation compared to that of sPDF suggested a restriction in entry of environmental oxygen or oxidizing agent into the active site of the mPDF enzyme. Since the active-site residues of the two proteins are similar, we further evaluated the role of oxidation-prone noncatalytic residue(s) in the process. Generally, cysteines in proteins are known to be vulnerable to oxidation (16, 29, 34, 38–40). In amino acid sequence comparisons, we observed that sPDF and mPDF have two (Cys-90 and Cys-130) and three (Cys-59, Cys-68, and Cys-106) cysteines, respectively (Fig. 2A). Interestingly, except for the metal-ion-coordinating cysteines in the EGCLS motif (Cys-90 in sPDF and Cys-106 in mPDF), the cysteines are not conserved and are located away from the active site (Fig. 2A).
FIG 2.

Sequence alignment and structural analysis of mPDF and sPDF. (A) Pairwise alignment of nucleotide-derived amino acid sequences of mPDF and sPDF. Cysteines are highlighted in boxes. Notations used: st, S. Typhimurium; mt, M. tuberculosis; double dots, conserved substitutions; single dots, semiconserved substitutions; stars, identical residues. (B) Positioning of different cysteines in protein structures. A superposition of the mPDF structure (pink; PDB ID 3E3U) with the E. coli PDF (EcPDF) structure (gray; PDB ID 3K6L) is shown. Important residues are labeled for EcPDF, and the corresponding residues in mPDF are indicated within parentheses. The metal ion from EcPDF is represented as a sphere (red).
To assess the positioning of cysteines in mPDF and sPDF, we analyzed their protein structures. The mPDF structure was already available (17); for sPDF, however, we used the E. coli structure since the two proteins exhibited 100% similarity in their primary sequences (data not shown). Superimposition of the two structures revealed that the positioning of the conserved metal ion coordinating Cys-106 of mPDF was identical to that of the Cys-90 of sPDF. It is well established that oxidation of Fe2+ leading to modification of this active site cysteine in E. coli PDF affected enzymatic activity (29, 34). Among other cysteines in mPDF, Cys-59 is surface exposed in the structure and Cys-68 is in a loop in close proximity (4.5 Å) to Met-145. Interestingly, Met-145 and Cys-68 of mPDF can be superimposed on Cys-130 and Val-63 of sPDF, respectively (Fig. 2B).
Amino acid sequence as well as structural analyses of sPDF and mPDF led us to generate mutants of both proteins by replacing cysteines with serines (one at a time), to address the question of the distinction in their sensitivities for H2O2. Substitution of cysteines with serine in the PDF from S. Typhimurium yielded proteins that were either defective (C90S; no activity) or without any significant alteration (C130S; 1.3 ± 0.1 μmol/min/mg protein versus 2.0 ± 0.6 μmol/min/mg protein for the wild type), and the H2O2 sensitivity profile was indistinguishable from that of the wild-type sPDF (Table 1). In sequence comparisons, Cys-130 of sPDF aligned with Met-145 of mPDF (Fig. 2A). Interestingly, the C130M mutant protein was found to have decreased sensitivity for H2O2 (Table 1). Similarly, the V63C mutant protein, where Val-63 of sPDF corresponded to Cys-68 of mPDF (Fig. 2A), displayed a decrease in sensitivity for H2O2 (Table 1). Therefore, it is tempting to speculate that the decrease in sensitivity for H2O2 exhibited by these mutant proteins of sPDF might be an amino acid- or position-specific effect.
In mPDF, consistent with our earlier reports, mutation at Cys-106 (C106S protein) showed hardly any deformylase activity (Table 1; see also references 25 and 26). To our surprise, the C68S but not the C59S mutant protein exhibited altered IC50s (∼32-fold decrease) for H2O2 compared to wild-type mPDF results (Table 1). Thus, it is apparent that in addition to conserved iron-coordinating cysteine, mutation at nonconserved residues might affect the deformylation abilities of both sPDF and mPDF in response to H2O2 exposure.
Effect of mutation at Met-145 of mPDF on its H2O2 sensitivity.
Comparisons of amino acid sequences and structures indicated the alignment of Cys-130 of sPDF with Met-145 of mPDF (Fig. 2). This observation further led us to create the M145C mutant of mPDF, and we monitored the effect of this substitution on the enzyme activity. Compared to the wild type, the mutant protein (M145C) was found to be deficient in deformylation ability, as indicated in its kcat/Km values (Table 3). In fact, calculation of enzymatic activity as a function of protein concentrations yielded curves which emphasized the need to use increased amounts of protein (∼3-fold more than the wild-type level) to obtain activity in a comparable range (Fig. 3A). The authenticity of the mutant protein was established by Western blotting using anti-His antibody (inset, Fig. 3A). Interestingly, M145C exhibited a significant increase in sensitivity to H2O2 (the IC50 of M145C was ∼6.0 × 10−2 mM as opposed to ∼16.1 mM for the wild type; Table 1) compared to that of the mPDF (Fig. 3B). We also noticed that replacement of Met with Ser (M145S) resulted in a deficient mutant (1.3 ± 0.4 μmol/min/mg protein for the M145S mutant compared to 3.9 ± 0.9 μmol/min/mg protein for the wild type) and exhibited an IC50 for H2O2 that was similar to that observed with M145C (Table 1). To gain an insight into the rate of iron dissociation from the active site of the M145C protein, the culture was incubated at 25°C with DTPA (200 μM). Interestingly, we observed a drastic decrease in the half-life of inactivation (t1/2 = 0.5 min) of the enzymatic activity of M145C in the presence of DTPA compared to that of the mPDF (Table 2).
Table 3.
Kinetics of deformylase activities of mPDF and sPDF proteins
| Protein | Km (mM) ± SD | kcat (s−1) ± SD | kcat/Km (M−1 s−1) ± SD | No of expts |
|---|---|---|---|---|
| sPDF | 8.0 ± 0.5 | 4.1 ± 0.5 | 0.5 ± 0.01 × 103 | 3 |
| C130 M-V63C | 7.7 ± 0.8 | 0.8 ± 0.05 | 0.1 ± 0.02 × 103 | 3 |
| mPDF | 2.9 ± 0.5 | 8.8 ± 0.8 | 0.3 ± 0.03 × 104 | 3 |
| M145C | 4.7 ± 0.5 | 3.1 ± 0.5 | 0.7 ± 0.1 × 103 | 3 |
FIG 3.

Peptide deformylase activities of M145C protein. (A) Effect of increasing protein concentrations on the deformylase activity of M145C. The enzyme assay was carried out with the indicated amounts of mutant protein with formylated Met-Ala as the substrate (5 mM/assay). The inset shows Western blotting of M145C using anti-His antibody. (B) Effect of H2O2 on the M145C enzyme activity. Following preincubation (15 min at 4°C) with the indicated concentrations of H2O2, the deformylation activity of the mutant (150 ng protein/assay) was compared with that of the wild type (50 ng protein/assay).
To elucidate the oxidation status of Met-145 in mPDF as well as Cys-145 in M145C, both proteins were digested with trypsin following treatment with or without H2O2 and analyzed by MALDI. The expected trypsin cleavage patterns for both proteins (mPDF and M145C) were generated using PAWS (http://www.softpedia.com/progDownload/Genomic-PAWS-Download-178463.html) and Protein Prospector (http://prospector.ucsf.edu) software for predicting the isotope abundance of the peptides. The peptide mass fingerprinting results determined for mPDF within the m/z range of 2,040 to 2,070 exhibited ion patterns centered on m/z values of 2,045 and 2,062 (Fig. 4A). Among them, the Met-145-containing fragment (spanning amino acids 145 to 161) and another tryptic fragment (spanning amino acids 124 to 144) exhibited the expected m/z values of 2,045 and 2,062 (Fig. 4A). Interestingly, upon H2O2 treatment the trypsin-digested fragment of mPDF at m/z of 2,045 disappeared and slight changes in ion patterns around the m/z of 2,062 were noticed (Fig. 4B; see appearance at the m/z of 2,061 and the increased intensity of the m/z 2,062 fragment). MS/MS spectra of m/z 2,061 and 2,062 (Fig. 4C) revealed a mixture of populations (amino acid residues spanning 124 to 144 and an oxidized methionine-containing fragment of 145 to 161). In M145C protein, on the other hand, irrespective of H2O2 treatment, trypsin digestion exhibited ion patterns of very low intensity (∼5%) at m/z 2,017, which is expected for a Cys-145-containing unoxidized fragment. The majority of the tryptic fragment was around m/z of 2,062 to 2,065 (Fig. 4D and E). Since the expected mass of the hyperoxidized form of cysteine (Cys-SO3) in the Cys-145-containing tryptic fragment of M145C is 2,065 Da, we carried out MS/MS analyses of peaks of 2,062 to 2,065. Interestingly, irrespective of H2O2 exposure, the MS/MS spectra at m/z 2,062 to 2,065 indicated the presence of a Cys-145-containing tryptic fragment of M145C protein within the mixture of isotopic peaks (Fig. 4F). All these lines of evidence indicated that replacement of Met-145 with Cys (M145C) in mPDF increased the susceptibility of the protein to oxidation.
FIG 4.

Mass spectroscopy of M145C protein. (A, B, D, and E) MALDI analysis following trypsin digestion of mPDF (A and B) and M145C (D and E) proteins before and after H2O2 treatment, respectively. Fragment m/z values and corresponding amino acid sequences are indicated. (C and F) MS/MS spectra of oxidized methionine (C)- and cysteine (F)-containing peptides. b and y ions are indicated by arrows.
H2O2 sensitivity pattern following alteration of nonconserved cysteine residues of sPDF.
Met-145 and Cys-68 of mPDF, in sequence alignment as well as in structural superimposition, corresponded to Cys-130 and Val-63 of sPDF, respectively (Fig. 2). To incorporate residues identical to those of mPDF, we generated a double mutant, C130M-V63C, of sPDF. CD analysis of C130M-V63C did not show any significant alteration in the secondary structure of the mutant protein (Fig. 5A), and it was also recognized by an anti-His antibody in Western blotting (inset in Fig. 5A). However, determinations of enzyme activity as a function of increasing protein concentrations illustrated that an excess amount of C130M-V63C protein was necessary to exhibit deformylation ability in a range comparable to that seen with sPDF (Fig. 5B and its inset). Although there was no significant alteration in its Km value (Table 3), C130M-V63C exhibited an ∼5-fold decrease in its kcat/Km value compared to that of sPDF (Table 3 and Fig. 5C). As with mPDF (25), the deformylation ability of sPDF and C130M-V63C proteins was found to be inhibitory at substrate concentrations beyond 20 mM (insets in Fig. 5C). However, the enzyme activity of this mutant protein showed a decrease in its sensitivity to H2O2 compared to that of sPDF (Fig. 5D). Interestingly, the calculated IC50 for H2O2 also showed a significant increase for C130M-V63C (1.4 ± 0.3 μM) compared to that of the sPDF (Table 1).
FIG 5.

H2O2 sensitivity profile of C130M-V63C protein. (A) Effect of mutations on the secondary structure of sPDF. Ni-NTA-purified protein samples were dialyzed (overnight at 4°C with 4 changes of buffer) to remove imidazole and were used (at 0.25 mg/ml) for spectroscopic studies. Far-UV CD spectra of the wild type and the double mutant in 20 mM Tris buffer (pH 7.0) were obtained between wave lengths of 250 and 190 nm. [θ]degcm2dm−1, molar ellipticity. (B) Enzyme activities as a function of the amounts of proteins. The inset shows the deformylase activity of the wild-type sPDF with increasing amounts of protein. (C) Kinetic analysis of the deformylase activity of the double mutant. The inset shows the deformylase activity of sPDF and C130M-V63C, revealing their identical behavior patterns in response to substrate at concentrations beyond 20 mM. (D) Effect of H2O2 on the deformylase activity of C130M-V63C. Following preincubation (15 min at 4°C) with the indicated concentrations of H2O2, the enzyme activity of the mutant (1,600 ng protein/assay) was monitored.
NBD-Cl is known to conjugate with sulfenic acid (Cys-SO-NBD), an intermediate oxidation product of cysteine, and reduced thiol (Cys-S-NBD), which have UV-visible absorption properties at 347 and 420 nm, respectively (38–40). Capitalizing on this aspect, we treated sPDF and C130M-V63C mutant proteins with NBD-Cl following incubation with or without H2O2 and compared their behavior patterns. As shown in Fig. 6A, NBD conjugated with both the Cys-SO and Cys-SH was present in sPDF protein and exhibited two populations. However, after the H2O2 treatment, due to oxidation of cysteine(s) in sPDF, the Cys-SH population mostly disappeared and absorption of the NBD-labeled conjugate (the sPDF-SO-NBD adduct) shifted from 420 to 347 nm. In the C130M-V63C mutant protein, as with the wild-type sPDF, both populations were evident upon the reaction of NBD-Cl. Interestingly, C130M-V63C protein did not show any alteration in the distribution of Cys-SO and Cys-SH populations on exposure to H2O2 (Fig. 6B). Thus, our spectral data were also consistent with our findings about the role of Met, which is in the place of Cys-130 in the double-mutant protein, as a determinant for its H2O2 sensitivity.
Fig 6.

UV-visible spectra of NBD-Cl-modified sPDF (A) and C130M-V63C (B) proteins in response to H2O2 exposure. NBD-Cl (20-fold molar excess of protein) was added to dialyzed protein following incubation with or without a 100-fold molar excess of H2O2. Unbound NBD-Cl was removed by washing, and the labeling was detected by a spectral scan (260 to 600 nm) as described in Materials and Methods.
DISCUSSION
PDF, responsible for deformylation of formylated initiator methionine during bacterial protein synthesis, is an essential enzyme throughout the bacterial lineage. Although our knowledge on this iron-containing metalloenzyme from E. coli dates back to 1968 (41), its so-called extreme oxygen sensitivity resulting in conversion of active Fe2+ to the inactive Fe3+ form and, ultimately, oxidation of active site cysteine prevented its isolation as well as characterization for more than 2 decades (41, 42). However, in subsequent years, with the advancement of recombinant DNA technology and, recently, in the postgenomic era, studies on iron-containing PDFs from different bacterial species, particularly S. aureus (15) and M. tuberculosis (25), revealed that this enzyme is not that short-lived. Despite having conserved active-site residues, including a cysteine, these iron-containing PDFs exhibited discrepancies in their responses to oxidative stress. To address this issue in greater detail, we initiated this study with the PDFs from S. Typhimurium and M. tuberculosis.
We observed that the sPDF has 100% amino acid sequence similarity to that of the E. coli protein. It was catalytically active and displayed biochemical characteristics similar to those reported earlier for the mPDF (25). However, comparisons of enzymatic activities showed a disparity between these two proteins in their sensitivities to H2O2 that was reflected in their IC50s (Fig. 1A; see also Table 1). Since Fe2+ easily dissociates from many enzymes, particularly when oxygen is available to oxidize it, one may infer that the decreased H2O2 sensitivity of mPDF was the effect of “metallation” (i.e., metal addition) with a metal other than iron. To rule out such a possibility, we prepared apo-proteins and reconstituted the enzyme activity with Fe2+. The reconstituted enzymes, when exposed to H2O2, behaved similarly to what was observed with wild-type mPDF (Fig. 1B).
We further compared the rates of metal dissociation for mPDF and sPDF. Interestingly, as reflected in the half-life of enzyme inactivation in the presence of DTPA (Table 2), mPDF protein exhibited a low metal dissociation rate compared to sPDF. Such an observation suggested a restriction in the entry of environmental oxygen or oxidizing agents into the active site of the mPDF enzyme. This is a possibility, if we presume an enclosed active site of the mPDF enzyme. Such a proposition does not seem to be unrealistic since a surface-exposed “RRR” sequence within the insertion sequence (absent in type I bacterial PDFs), together with glycine in its conserved motif III, was ascribed to modulation of the active site of the mPDF enzyme in the “action-at-a-distance” mode (9, 43).
In studies of amino acid sequence alignment as well as structural superimposition of mPDF and sPDF, we noted that the amino acid corresponding to Cys-130 of sPDF is methionine (Met-145) in mPDF (Fig. 2). Although swapping cysteine for methionine (M145C) in mPDF resulted in a deficient mutant, the results showed a drastic increase (>250-fold) in sensitivity to H2O2 as indicated by IC50s (Fig. 3B and Table 1). Interestingly, the metal dissociation rate of M145C protein was remarkably high compared to that of the wild-type enzyme (Table 2). Thus, a common attribute of the M145C enzyme underlies the increased H2O2 sensitivity as well as rate of metal dissociation from the active site due to the replacement of noncatalytic residue Met-145 with Cys. We further monitored the oxidation status of the M145C protein. Under our experimental conditions, MALDI analysis of the mPDF protein following trypsin digestion revealed that the fragment containing Met-145 had the ability to be oxidized (16 Da added to form methionine sulfoxide) in response to exposure to H2O2 (Fig. 4B). However, the M145C protein had increased susceptibility to oxidation as evident by peptide mass fingerprinting as well as MS/MS data, where in our experimental setup cysteine was converted to cysteine sulfonic acid (Cys-SO3) even without H2O2 treatment (Fig. 4D). These observations, therefore, strongly argued the association of Met-145 in mPDF with exhibiting resistance to oxidative stress.
To have a second line of evidence in support of our findings with M145C protein, we generated a double mutant of sPDF, C130M-V63C, by incorporating residues identical to those of mPDF. Compared to the wild type, the double mutant, although deficient (∼5-fold) in its enzyme turnover rate, showed that its affinity for the substrate or its secondary structure was not significantly altered (Fig. 5A and C and Table 3). Compared to the wild type, C130M-V63C exhibited an ∼30-fold increase in IC50 for H2O2 (Table 1; see also Fig. 5D). Under our experimental conditions, E. coli cultures harboring a pET-sPDF or pET-C130M-V63C construct were grown in aerobic conditions as reported elsewhere (5, 16, 29). As a result, the recombinant proteins (sPDF and C130M-V63C) exhibited two populations with different oxidation states of cysteines (Cys-SO-NBD and Cys-S-NBD) when treated with NBD-Cl. Interestingly, the oxidation state of cysteines in sPDF indicated the predominance of the Cys-SO-NBD population of the protein in response to H2O2 treatment (Fig. 6A). On the other hand, H2O2 was unable to affect Cys-SO-NBD and Cys-S-NBD populations in C130M-V63C mutant protein (Fig. 6B). Since the sPDF and the C130M-V63C variant have equal numbers of cysteines (Cys-90/Cys-130 in sPDF and Cys-90/Cys-63 in C130M-V63C), this observation strongly insinuates the involvement of Met-130 (replacement of Cys-130) in regulating the deformylation ability of the double-mutant protein in response to H2O2 treatment.
In iron-containing E. coli PDF, the metal atom in the active site of this metalloprotease is exposed in order to bind substrate, and therefore, when treated with H2O2, it is vulnerable to inactivation by molecular oxygen through two different mechanisms (29, 34, 44). First, oxygen can oxidize the ferrous atom to the ferric form, causing it to dissociate. This event is reversible, and enzyme activity is restored when ferrous ion is added back (34). Second, oxygen can oxidize the metal-coordinating Cys residue when the enzyme is in the apo-protein form (34). sPDF is governed by a mechanism similar to that seen with the E. coli PDF, yielding an IC50 for H2O2 in the nanomolar range (Table 1), and the enzyme is completely inactivated at 10 μM (Fig. 1). This is very likely to be the case in all type I PDFs, since a BLAST search using sPDF as the query sequence with the nonredundant database revealed that they have two cysteines aligning with the Cys-90 and Cys-130 positions (of sPDF).
A BLAST search performed with mPDF as the query sequence identified all type II PDFs, the majority of them having 2 cysteines (ranging from 3 to 5) in alignment with Cys-68 and Cys-106 (of mPDF); Met-145 is also highly conserved. Interestingly, the positioning of these residues (Cys-68/Cys-106/Met-145) is similar in the PDF from the minimal genome of M. leprae (45). Thus, based on our experimental evidence determined with M145C (Figs. 3 and 4 and Table 2), it is tempting to speculate that besides the conserved metal-coordinating Cys-106, Met-145 in mPDF was crafted in such a way that it exhibited characteristics enabling it to withstand a high level of stress upon treatment with H2O2. This is at least reflected in the M. tuberculosis physiology, where the bacterium efficiently copes with oxidative stress to be a successful pathogen.
Finally, it is evident from our results that type I and type II PDFs have distinct means to cope with oxidative stress. Given the experimental data provided here, with respect to the mPDF or a type II PDF, it seems logical to postulate that the low metal dissociation rate compared to sPDF or a type I PDF is definitely a factor (Table 2) contributing to H2O2 sensitivity. Such an observation is very likely indicative of an enclosed enzyme active site of mPDF that might prevent Fe2+ being affected by H2O2 through a gating mechanism. In such a corollary, treatment with H2O2 would oxidize available oxidation-prone noncatalytic amino acids like Met-145 and Cys-68 in mPDF (Table 1). Since a role of methionine in protection against oxidation and in endogenous antioxidant defense of proteins has already been reported (46–49), Met-145 would shield mPDF from oxidative stress to some extent. This corroborates well with our findings determined with M145C protein, which exhibited faster metal dissociation as well as an increased sensitivity to H2O2 (Tables 2 and 3; see also Fig. 3B). Intriguingly, our peptide mass fingerprinting studies also indicated considerable change in the oxidation status of fragments containing Cys-145 in M145C protein compared to Met-145 in the wild type even without treatment with H2O2 (Fig. 4; compare panels A and D). Further studies in this direction would unravel how oxidation/reduction of methionine, which is a reversible process (49), could regulate the event. Nonetheless, we present here for the first time firm evidence that bacterial PDFs might display disparity in their sensitivities to H2O2 because of the involvement of noncatalytic amino acids such as cysteine/methionine in mPDF or in type II PDFs.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to Rahul Saxena, Department of Biochemistry and Cell Biology, Georgetown University Medical Center, Washington, DC, for his help during the initial phase of this study. We are thankful to J. Prasad for providing us with excellent technical assistance.
Senior Research Fellowships awarded to Sanjay Kumar by the Council of Scientific and Industrial Research (CSIR), New Delhi, India, and to Pavitra Kanudia by the Department of Biotechnology (DBT), New Delhi, India, are gratefully acknowledged. This study was supported by research grants from CSIR (Supra institutional project) and DBT.
Footnotes
Published ahead of print 18 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00916-13.
REFERENCES
- 1.Halbreich A, Rabinowitz M. 1971. Isolation of Saccharomyces cerevisiae mitochondrial formyltetrahydrofolic acid:methionyl-tRNA transformylase and the hybridization of mitochondrial fMet-tRNA with mitochondrial DNA. Proc. Natl. Acad. Sci. U. S. A. 68:294–298. 10.1073/pnas.68.2.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meinnel T, Mechulam Y, Blanquet S. 1993. Methionine as translation start signal: a review of the enzymes of the pathway in Escherichia coli. Biochimie 75:1061–1075. 10.1016/0300-9084(93)90005-D [DOI] [PubMed] [Google Scholar]
- 3.Giglione C, Serero A, Pierre M, Boisson B, Meinnel T. 2000. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J. 19:5916–5929. 10.1093/emboj/19.21.5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitt E, Guillon JM, Meinnel T, Mechulam Y, Dardel F, Blanquet S. 1996. Molecular recognition governing the initiation of translation in Escherichia coli. A review. Biochimie 78:543–554. 10.1016/S0300-9084(96)80001-0 [DOI] [PubMed] [Google Scholar]
- 5.Rajagopalan PT, Datta A, Pei D. 1997. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry 36:13910–13918. 10.1021/bi971155v [DOI] [PubMed] [Google Scholar]
- 6.Groche D, Becker A, Schlichting I, Kabsch W, Schultz S, Wagner AF. 1998. Isolation and crystallization of functionally competent Escherichia coli peptide deformylase forms containing either iron or nickel in the active site. Biochem. Biophys. Res. Commun. 246:342–346. 10.1006/bbrc.1998.8616 [DOI] [PubMed] [Google Scholar]
- 7.Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan JE., Jr 1999. Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J. Mol. Biol. 290:607–614. 10.1006/jmbi.1999.2913 [DOI] [PubMed] [Google Scholar]
- 8.Mazel D, Pochet S, Marliere P. 1994. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. 13:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxena R, Kanudia P, Datt M, Dar HH, Karthikeyan S, Singh B, Chakraborti PK. 2008. Three consecutive arginines are important for the mycobacterial peptide deformylase enzyme activity. J. Biol. Chem. 283:23754–23764. 10.1074/jbc.M709672200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84. 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 11.Jones RN, Fritsche TR, Sader HS. 2004. Antimicrobial spectrum and activity of NVP PDF-713, a novel peptide deformylase inhibitor, tested against 1,837 recent Gram-positive clinical isolates. Diagn. Microbiol. Infect. Dis. 49:63–65. 10.1016/j.diagmicrobio.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 12.Fonseca-Aten M, Salvatore CM, Mejias A, Rios AM, Chavez-Bueno S, Katz K, Gomez AM, McCracken GH, Jr, Hardy RD. 2005. Evaluation of LBM415 (NVP PDF-713), a novel peptide deformylase inhibitor, for treatment of experimental Mycoplasma pneumoniae pneumonia. Antimicrob. Agents Chemother. 49:4128–4136. 10.1128/AAC.49.10.4128-4136.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui K, Lu W, Zhu L, Shen X, Huang J. 2013. Caffeic acid phenethyl ester (CAPE), an active component of propolis, inhibits Helicobacter pylori peptide deformylase activity. Biochem. Biophys. Res. Commun. 435:289–294. 10.1016/j.bbrc.2013.04.026 [DOI] [PubMed] [Google Scholar]
- 14.Meinnel T, Blanquet S, Dardel F. 1996. A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase. J. Mol. Biol. 262:375–386. 10.1006/jmbi.1996.0521 [DOI] [PubMed] [Google Scholar]
- 15.Baldwin ET, Harris MS, Yem AW, Wolfe CL, Vosters AF, Curry KA, Murray RW, Bock JH, Marshall VP, Cialdella JI, Merchant MH, Choi G, Deibel MR., Jr 2002. Crystal structure of type II peptide deformylase from Staphylococcus aureus. J. Biol. Chem. 277:31163–31171. 10.1074/jbc.M202750200 [DOI] [PubMed] [Google Scholar]
- 16.Kreusch A, Spraggon G, Lee CC, Klock H, McMullan D, Ng K, Shin T, Vincent J, Warner I, Ericson C, Lesley SA. 2003. Structure analysis of peptide deformylases from Streptococcus pneumoniae, Staphylococcus aureus, Thermotoga maritima and Pseudomonas aeruginosa: snapshots of the oxygen sensitivity of peptide deformylase. J. Mol. Biol. 330:309–321. 10.1016/S0022-2836(03)00596-5 [DOI] [PubMed] [Google Scholar]
- 17.Pichota A, Duraiswamy J, Yin Z, Keller TH, Alam J, Liung S, Lee G, Ding M, Wang G, Chan WL, Schreiber M, Ma I, Beer D, Ngew X, Mukherjee K, Nanjundappa M, Teo JW, Thayalan P, Yap A, Dick T, Meng W, Xu M, Koehn J, Pan SH, Clark K, Xie X, Shoen C, Cynamon M. 2008. Peptide deformylase inhibitors of Mycobacterium tuberculosis: synthesis, structural investigations, and biological results. Bioorg. Med. Chem. Lett. 18:6568–6572. 10.1016/j.bmcl.2008.10.040 [DOI] [PubMed] [Google Scholar]
- 18.Guilloteau JP, Mathieu M, Giglione C, Blanc V, Dupuy A, Chevrier M, Gil P, Famechon A, Meinnel T, Mikol V. 2002. The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types: a platform for the structure-based design of antibacterial agents. J. Mol. Biol. 320:951–962. 10.1016/S0022-2836(02)00549-1 [DOI] [PubMed] [Google Scholar]
- 19.Karambelkar VV, Xiao C, Zhang Y, Sarjeant AA, Goldberg DP. 2006. Geometric preferences in iron(II) and zinc(II) model complexes of peptide deformylase. Inorg. Chem. 45:1409–1411. 10.1021/ic050995s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu XH, Quan JM, Wu YD. 2007. Theoretical study of the catalytic mechanism and metal-ion dependence of peptide deformylase. J. Phys. Chem. B 111:6236–6244. 10.1021/jp068611m [DOI] [PubMed] [Google Scholar]
- 21.Meinnel T, Lazennec C, Villoing S, Blanquet S. 1997. Structure-function relationships within the peptide deformylase family. Evidence for a conserved architecture of the active site involving three conserved motifs and a metal ion. J. Mol. Biol. 267:749–761. 10.1006/jmbi.1997.0904 [DOI] [PubMed] [Google Scholar]
- 22.Han C, Wang Q, Dong L, Sun H, Peng S, Chen J, Yang Y, Yue J, Shen X, Jiang H. 2004. Molecular cloning and characterization of a new peptide deformylase from human pathogenic bacterium Helicobacter pylori. Biochem. Biophys. Res. Commun. 319:1292–1298. 10.1016/j.bbrc.2004.05.120 [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z, Song X, Li Y, Gong W. 2004. Unique structural characteristics of peptide deformylase from pathogenic bacterium Leptospira interrogans. J. Mol. Biol. 339:207–215. 10.1016/j.jmb.2004.03.045 [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Van Aller GS, Taylor AN, Kerrigan JJ, Liu WS, Trulli JM, Lai Z, Holmes D, Aubart KM, Brown JR, Zalacain M. 2006. Phylogenomic and biochemical characterization of three Legionella pneumophila polypeptide deformylases. J. Bacteriol. 188:5249–5257. 10.1128/JB.00866-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxena R, Chakraborti PK. 2005. The carboxy-terminal end of the peptide deformylase from Mycobacterium tuberculosis is indispensable for its enzymatic activity. Biochem. Biophys. Res. Commun. 332:418–425. 10.1016/j.bbrc.2005.04.142 [DOI] [PubMed] [Google Scholar]
- 26.Saxena R, Chakraborti PK. 2005. Identification of regions involved in enzymatic stability of peptide deformylase of Mycobacterium tuberculosis. J. Bacteriol. 187:8216–8220. 10.1128/JB.187.23.8216-8220.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Chen Z, Gong W. 2002. Enzymatic properties of a new peptide deformylase from pathogenic bacterium Leptospira interrogans. Biochem. Biophys. Res. Commun. 295:884–889. 10.1016/S0006-291X(02)00777-5 [DOI] [PubMed] [Google Scholar]
- 28.Nguyen KT, Wu JC, Boylan JA, Gherardini FC, Pei D. 2007. Zinc is the metal cofactor of Borrelia burgdorferi peptide deformylase. Arch. Biochem. Biophys. 468:217–225. 10.1016/j.abb.2007.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajagopalan PT, Pei D. 1998. Oxygen-mediated inactivation of peptide deformylase. J. Biol. Chem. 273:22305–22310. 10.1074/jbc.273.35.22305 [DOI] [PubMed] [Google Scholar]
- 30.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856. 10.1038/35101614 [DOI] [PubMed] [Google Scholar]
- 31.Shirley K, Sheng YC, John JS, Alice W. 1995. Design and use of mismatched and degenerate primers, p 143–155 In Dieffenbach CW, Dveksler GS. (ed), PCR primer: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 32.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 33.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anjem A, Imlay JA. 2012. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J. Biol. Chem. 287:15544–15556. 10.1074/jbc.M111.330365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Che X, Hu J, Wang L, Zhu Z, Xu Q, Lv J, Fu Z, Sun Y, Sun J, Lin G, Lu R, Yao Z. 2011. Expression, purification, and activity assay of peptide deformylase from Escherichia coli and Staphylococcus aureus. Mol. Cell. Biochem. 357:47–54. 10.1007/s11010-011-0874-6 [DOI] [PubMed] [Google Scholar]
- 36.Rajagopalan PT, Grimme S, Pei D. 2000. Characterization of cobalt(II)-substituted peptide deformylase: function of the metal ion and the catalytic residue Glu-133. Biochemistry 39:779–790. 10.1021/bi9919899 [DOI] [PubMed] [Google Scholar]
- 37.Gu M, Imlay JA. 2013. Superoxide poisons mononuclear iron enzymes by causing mismetallation. Mol. Microbiol. 89:123–134. 10.1111/mmi.12263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole LB, Ellis HR. 2002. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 348:122–136. 10.1016/S0076-6879(02)48632-6 [DOI] [PubMed] [Google Scholar]
- 39.Ellis HR, Poole LB. 1997. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry 36:15013–15018. 10.1021/bi972191x [DOI] [PubMed] [Google Scholar]
- 40.Griffiths SW, King J, Cooney CL. 2002. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J. Biol. Chem. 277:25486–25492. 10.1074/jbc.M203089200 [DOI] [PubMed] [Google Scholar]
- 41.Adams JM. 1968. On the release of the formyl group from nascent protein. J. Mol. Biol. 33:571–589. 10.1016/0022-2836(68)90307-0 [DOI] [PubMed] [Google Scholar]
- 42.Meinnel T, Blanquet S. 1995. Enzymatic properties of Escherichia coli peptide deformylase. J. Bacteriol. 177:1883–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narayanan SS, Sokkar P, Ramachandran M, Nampoothiri KM. 2011. Glycine in the conserved motif III modulates the thermostability and oxidative stress resistance of peptide deformylase in Mycobacterium tuberculosis. FEMS Microbiol. Lett. 320:40–47. 10.1111/j.1574-6968.2011.02289.x [DOI] [PubMed] [Google Scholar]
- 44.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11:443–454. 10.1038/nrmicro3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011. 10.1038/35059006 [DOI] [PubMed] [Google Scholar]
- 46.Levine RL, Mosoni L, Berlett BS, Stadtman ER. 1996. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. U. S. A. 93:15036–15040. 10.1073/pnas.93.26.15036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, DalleDonne I. 2000. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 9:1774–1782. 10.1110/ps.9.9.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melkani GC, Kestetter J, Sielaff R, Zardeneta G, Mendoza JA. 2006. Protection of GroEL by its methionine residues against oxidation by hydrogen peroxide. Biochem. Biophys. Res. Commun. 347:534–539. 10.1016/j.bbrc.2006.06.136 [DOI] [PubMed] [Google Scholar]
- 49.Luo S, Levine RL. 2009. Methionine in proteins defends against oxidative stress. FASEB J. 23:464–472. 10.1096/fj.08-118414 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.