

Abstract
The cytochrome bc1-cytochrome aa3 complexes together comprise one of the major branches of the bacterial aerobic respiratory chain. In actinobacteria, the cytochrome bc1 complex shows a number of unusual features in comparison to other cytochrome bc1 complexes. In particular, the Rieske iron-sulfur protein component of this complex, QcrA, is a polytopic rather than a monotopic membrane protein. Bacterial Rieske proteins are usually integrated into the membrane in a folded conformation by the twin arginine protein transport (Tat) pathway. In this study, we show that the activity of the Streptomyces coelicolor M145 cytochrome bc1 complex is dependent upon an active Tat pathway. However, the polytopic Rieske protein is still integrated into the membrane in a ΔtatC mutant strain, indicating that a second protein translocation machinery also participates in its assembly. Difference spectroscopy indicated that the cytochrome c component of the complex was correctly assembled in the absence of the Tat machinery. We show that the intact cytochrome bc1 complex can be isolated from S. coelicolor M145 membranes by affinity chromatography. Surprisingly, a stable cytochrome bc1 complex containing the Rieske protein can be isolated from membranes even when the Tat system is inactive. These findings strongly suggest that the additional transmembrane segments of the S. coelicolor Rieske protein mediate hydrophobic interactions with one or both of the cytochrome subunits.
INTRODUCTION
The ability to respire oxygen is essential for the survival of almost all eukaryotes and many prokaryotes. Many members of the Gram-positive actinobacteria, including Mycobacterium tuberculosis and Streptomyces coelicolor, are obligate aerobes, requiring oxygen to support growth, although recent studies have indicated that these organisms have mechanisms to survive periods of anaerobiosis (1, 2, 3). The major route for aerobic quinol oxidation in actinobacteria is via the cytochrome bc1-cytochrome aa3 complexes, with most members of the phylum also having the ability to produce a cytochrome bd oxidase (Fig. 1A), which has a higher oxygen affinity but is less energy efficient, tending to be produced under oxygen-limiting growth conditions (1, 4, 5).
FIG 1.

The major aerobic respiratory components in S. coelicolor M145 as deduced from the genome sequence. (A) Two homologues of a Nuo-type NADH dehydrogenase are encoded by S. coelicolor. The genome also reveals two potential routes to the oxygen-dependent reoxidation of quinol, via the cytochrome bc1-cytochrome aa3 oxidase or through a higher-affinity cytochrome bd oxidase. (B) Schematic representation of the three subunits of the S. coelicolor cytochrome bc1 complex. Domains that show homology to the related components from mitochondria and Gram-negative bacteria are shown in gray. Domains that appear unique to the actinobacteria are shown in white. FeS, iron sulfur cluster; b, b-type heme; c, c-type heme; in, cytoplasm; out, cell exterior.
The cytochrome bc1-cytochrome aa3 branch of the aerobic respiratory chain that is found in actinobacteria shows unusual features when compared to the well-characterized complexes from Paracoccus denitrificans or bovine heart mitochondria (6, 7). These differences are primarily at the level of the cytochrome bc1 complex. The cytochrome b component, encoded by qcrB, is predicted to have nine transmembrane domains (TMD) instead of the eight found in the mitochondrial homologue (8), with a predicted small (approximately 100-amino-acid) globular domain at the trans side of the membrane (Fig. 1B). The last four transmembrane domains of actinobacterial cytochrome b proteins share little homology with the mitochondrial counterpart. The cytochrome c component (QcrC) is a membrane-anchored diheme cytochrome c (9, 10) rather than being a soluble protein containing a single heme (8). Unlike most other bacteria, there is no gene encoding a soluble cytochrome c in the genomes of S. coelicolor, M. tuberculosis, or Corynebacterium glutamicum (10). Finally the Rieske iron-sulfur protein (QcrA) is much more hydrophobic than previously characterized, monotopic Rieske proteins (8, 11) and has three TMD preceding the globular iron-sulfur cluster-containing domain (12) (Fig. 1B).
The biogenesis of respiratory complexes requires a high degree of coordination, since individual subunits need to be inserted into the membrane, bind redox cofactors, and associate with partner proteins in order to be active. In bacteria, most polytopic membrane proteins, such as QcrB, are inserted into the lipid bilayer by the general secretory (Sec) pathway. The Sec translocon is a heterotrimeric complex of SecYEG, which forms a narrow channel of sufficient diameter to accommodate unfolded polypeptide chains (reviewed in references 13 and 14). Cytochrome c proteins are also substrates of the Sec pathway, and the c-type heme is covalently attached to the unfolded polypeptide as it extrudes through the Sec translocon at the trans side of the membrane (15, 16).
In contrast, in bacteria and plants, the Rieske iron-sulfur protein is inserted into the membrane by the Tat machinery (17, 18, 19, 20, 21). The Tat pathway transports folded substrate proteins (see references 22 and 23 for recent reviews), and the iron-sulfur cluster is inserted into the Rieske protein in the cytoplasm prior to transport. Tat substrates are synthesized with N-terminal signal peptides that contain a conserved S-R-R-X-F-L-K twin arginine motif (24, 25). Although the twin arginine signal peptide is normally cleaved off during translocation, the Rieske proteins have uncleaved signal sequences that serve to anchor the protein in the lipid bilayer and to form contacts with the two partner proteins (11). The highly hydrophobic Rieske proteins of actinobacteria present a biosynthetic difficulty, since the Tat pathway is strictly posttranslational. Experiments examining the biosynthetic requirements of the three TMD of the S. coelicolor Rieske protein (Sco2149) expressed as a fusion protein in the heterologous host Escherichia coli indicated that only the insertion of the third TMD was Tat dependent and that the first two TMD were inserted into the membrane by the Sec pathway, most likely in a cotranslational fashion (12). It should be noted, however, that deliberate truncation, production as a fusion protein, or even small protein tags or single amino acid substitutions can interfere with the targeting route of a protein (e.g., see references 26, 27, 28, and 29). Therefore, in this study, we have examined the role of the Tat pathway in the assembly of the native S. coelicolor M145 Rieske protein. Our results are consistent with the conclusion that the protein requires the operation of at least two transport pathways for its correct assembly into the membrane. Surprisingly, the cytochrome bc1 complex can be stably isolated from a tat mutant strain.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
For cloning and intergeneric conjugation, E. coli strains were grown in LB medium (30) or superoptimal broth (SOB) (31). For growth of S. coelicolor strains in liquid culture, tryptone soya broth (32), yeast extract-malt extract (YEME) medium (32), or SOB (31) medium was used, as indicated in the figure legends. All S. coelicolor strains used in this study are derived from the M145 strain. M145 is a derivative of the wild-type strain S. coelicolor A3(2) that was used for genomic sequencing (33). For growth of S. coelicolor on solid medium, soy flour mannitol (SFM) agar or Difco nutrient agar were used (32). Spore stocks were prepared as described previously (32). Antibiotics, where required, were used at the following concentrations: ampicillin (100 μg/ml), kanamycin (50 μg/ml), chloramphenicol (30 μg/ml), apramycin (50 μg/ml), hygromycin B (50 μg/ml), and nalidixic acid (25 μg/ml). Growth rate analysis of S. coelicolor strains was performed by determining total biomass as described previously (34).
Strain and plasmid construction.
For marked deletion of qcrCAB in S. coelicolor M145, the Redirect PCR targeting approach was used (35). Briefly, the hygR-oriT cassette was amplified by PCR using pIJ10700 (Table 1) as the template, with the primer pair qcrCABFOR (5′-GCATCGACGCAGAAGATCCTGACACCGGGGTAATCCGTGATTCCGGGGATCCGTCGACC-3′) and qcrCABREV (5′-GGGGCCCTTCGACCGTGGTGTGGCGACGCTGGAGGGTCATGTAGGCTGGAGCTGCTTC-3′). The resultant product was gel purified and electroporated into E. coli strain BW25113 harboring pIJ790 and cosmid 11-D01 (which carries S. coelicolor M145 chromosomal DNA covering the qcrCAB operon; a kind gift from Paul Dyson, University of Swansea). After selection for the E. coli strain carrying the disrupted cosmid by growth on hygromycin B-containing medium, the cosmid was isolated, introduced into E. coli strain ET12567/pUZ8002, and transferred to S. coelicolor M145 by conjugation. Double-crossover mutants were selected by screening for hygromycin resistance and kanamycin sensitivity. Deletion of qcrCAB was confirmed by PCR analysis of chromosomal DNA using oligonucleotides 50-48seq For (5′-CGGCCGCACCTACGCGGCCCGGAGGTTCACCCACG) and 50-48seq Rev (5′-CGGGGCCCTTCGACCGTGGTGTGGCGACGCTGGAGGG-3′) that lie outside the deleted region and, additionally, by Western blot analysis using a QcrA polyclonal antibody (12). The ΔqcrCAB::hyg strain was subsequently designated APH2.
TABLE 1.
Strains plasmids and cosmids used in this article
| Strain, plasmid, or cosmid | Genotype or description | Reference or source |
|---|---|---|
| Escherichia coli strains | ||
| MC1061 | F− Δ(ara-leu)7697 [araD139]B/r Δ(codB-lacI)3 galK16 galE15 λ− e14− mcrA0 relA1 rpsL150(strR) spoT1 mcrB1 hsdR2(r− m+) | 52 |
| BW25113 | F− Δ(araD-araB)567 ΔlacZ4787 (::rrnB-3) λ− rpoS396(Am) rph-1 Δ(rhaD-rhaB)568 hsdR514 | 53 |
| ET12567 | dam13::Tn9 dcm6 hsdM hsdR rec143F zjj201::Tn10 galK2 galT22 ara14 lacY1 leuB1 thi1 tonA31 rpsL136 hisG4 tsx78 mtlI glnV44 F− | 54 |
| Streptomyces coelicolor strains | ||
| M145 | Wild-type derivative of S. coelicolor A3(2) lacking plasmids | 52 |
| TP4 | As M145; ΔtatC | 43 |
| APH2 | As M145; ΔqcrCAB::hyg | This work |
| APH3 | As APH2; ϕC31 (PhrdB-qcrCHis10AB) | This work |
| APH5 | As APH2; ϕC31 (PhrdB-qcrCABHis10) | This work |
| APH5-KK | As APH5 except that DNA coding for the twin arginine motif (R161 and R162) of QcrA is mutated to twin lysine codons | This work |
| APH6 | As TP4; ϕC31 (PhrdB-qcrCHis10AB) | This work |
| APH8 | As TP4; ϕC31 (PhrdB-qcrCABHis10) | This work |
| Plasmids and cosmids | ||
| pBluescript (KS+) | Cloning vector | Stratagene |
| pIJ790 | Modified λRed recombination plasmid | 35 |
| pUZ8002 | Nontransmissible oriT-mobilizing plasmid | 55 |
| pIJ10700 | pBlueScriptII (KS+) containing the hygromycin resistance gene and oriT from plasmid RP4, flanked by FLP recombination target (FRT) sites | 56 |
| Cosmid 11-D01 | SuperCos I vector containing chromosomal region 2295280 to 2330618, encoding genes sco2135 to sco2165 | Paul Dyson Swansea |
| Cosmid 11-D01(50H) | As cosmid 11-D01 except that qcrCAB (sco2150-sco2148) is deleted and replaced with FRT-oriT-hyg-FRT by homologous recombination | This work |
| pSET152 | Vector for conjugal transfer and chromosomal integration at the ϕC31 attachment site in Streptomyces | 57 |
| pAPH1 | pSET152 carrying PhrdB-qcrCHis10AB | This work |
| pAPH3 | pSET152 carrying PhrdB-qcrCABHis10 | This work |
| pAPH3KK | As APH3 except that DNA coding for the twin arginine motif of QcrA is mutated to twin lysine codons | This work |
For ectopic expression of qcrCAB, we designed a synthetic construct. This construct carried the S. coelicolor M145 hrdB (sco5820) promoter region and ribosome binding site (from position −429 to −1) joined by an in-frame NdeI restriction site to the qcrCAB operon (from the start codon of qcrA to the stop codon of qcrC). The construct also introduced decahistidine tag-coding sequences at the 3′ end of each gene, directly preceding the stop codons. In addition, the last 36 base pairs of the qcrC and qcrA genes were duplicated, in order to provide the native ribosome binding site for the subsequent gene (see Fig. S1 in the supplemental material). Pairs of silent restriction sites were included within the construct to allow the in-frame removal of the histidine tag-coding sequences, using BsiWI for qcrC, AgeI for qcrA, and NcoI for qcrB (see Fig. S1). We also silently removed nine restriction enzyme sites within the operon (numbered from position 1 of qcrC: C531G, C1601G, G1637A, G1987A, G2119C, T2365A, T2401A, G3013C, and A3145T). The whole construct was bounded by an XhoI followed directly by a BamHI restriction site at the 5′ end and an XbaI followed by a SacI site at the 3′ end. The synthetic construct was synthesized by Dundee Cell Products (Dundee, United Kingdom) and was provided in the form of a plasmid that was cloned between the XhoI and SacI sites of pBluescript.
We subsequently manipulated this construct by serially removing two of the three histidine tag-coding sequences to leave constructs coding for a synthetic operon with a decahistidine tag sequence at the 3′ end of qcrA only, qcrB only, or qcrC only. Each of these three individual operons was removed from pBluescript as a BamHI-XbaI fragment and cloned into similarly digested pSET152 to give plasmids pAPH1, pAPH2, and pAPH3 (Table 1). To introduce a twin arginine-to-twin lysine codon substitution of codons 161 and 162 of qcrA in the pBluescript clone encoding PhrdB-qcrCABHis10, whole-plasmid PCR was carried out using the partially overlapping primers RRtoKKfor (5′-CGGGAAGAAGAAGCTGATCCGCAACACGATGC-3′) and RRtoKKrev (5′-GCTTCTTCTTCCCGATCACGGACTCCTTGGC-3′) according to the method in reference 36. The construct was fully sequenced to ensure that only the desired mutations had been introduced, and then the qcrCAB-containing fragment was released as a BamHI-XbaI fragment and cloned into similarly digested pSET152 to give plasmid pAPH3KK. Each of these pSET152-based constructs was introduced into an S. coelicolor M145 strain of interest by intergeneric conjugation (using E. coli strain ET12567/pUZ8002 as the donor strain), where it integrated into the ϕC31 attachment site.
Preparation of membrane fractions.
Baffled 2-liter flasks filled with 500 ml of growth medium were inoculated with spores of the S. coelicolor M145 strain of interest and grown at 30°C and 130 rpm for 65 to 90 h. All subsequent steps were performed at 4°C. Hyphal pellets were harvested by centrifugation at 2,770 × g for 30 min and resuspended to a final volume of approximately 30 ml in TEE buffer (50 mM Tris-HCl, pH 8, 1 mM EDTA, 1 mM EGTA, and a protease inhibitor cocktail [Complete protease inhibitor; Roche]). The hyphae were lysed by sonication (Digital Sonifier 250 with microtip 102C; Branson) for 10 min (5 s on/off) at 20% power. Unbroken hyphae and cell debris were removed by a centrifugation step (6,000 × g for 30 min), and the supernatant was subjected to a high-speed spin (150,000 × g for 60 min) to pellet the membranes. The pelleted membranes were washed in TEE containing 250 mM NaCl to remove peripherally bound proteins and then again in TEE only to remove the salt. Finally, the membranes were resuspended in 50 mM Tris, pH 6.8, 10% glycerol, snap-frozen in liquid nitrogen, and stored at −80°C.
Absorbance spectroscopy and cytochrome c oxidase activity assay.
Membrane fractions were solubilized in 50 mM Tris, pH 7.0, 10% glycerol, 250 mM NaCl, 1% dodecyl maltoside (DDM) for 30 min at 4°C with stirring. The samples were diluted with 50 mM Tris, pH 7.5, 10% glycerol, 250 mM NaCl until the DDM concentration was reduced to 0.1%, and then the samples were centrifuged twice at 16,000 × g for 2 min, retaining the supernatant each time. The protein concentration was determined by Bio-Rad DC (detergent compatible) assay, and samples were adjusted to 1.0 to 3.0 mg/ml solubilized protein using the same buffer.
For absorbance spectroscopy, dithionite-reduced and air-oxidized absorbance spectra were recorded between the wavelengths of 380 and 650 nm using a lambda UV/Vis spectrophotometer (PerkinElmer). The cytochrome c oxidase activity of the cytochrome bc1 complex in solubilized membranes was measured spectrophotometrically at 550 nm using equine horse cytochrome c (Sigma). First, the menadiol substrate was prepared as a 10-fold stock solution. To do this, 50 mM Tris-HCl, pH 6.8, was supplemented with 0.05% DDM, 1 mM EDTA, 6 mM NADH, 3 mM menadione, and 6 mg/ml diaphorase in strict order, and this was incubated at 37°C for 20 min. Once prepared, the substrate stock solution was kept on ice and used within 1 h. Solubilized membrane fractions were supplemented with 2 μM equine cytochrome c and 4 μM ferricyanide, and to 90 μl of this mixture in a cuvette, 10 μl of the menadiol solution was added. The change in absorbance at 550 nm was monitored for 5 min, and the initial slope was used to calculate the activity of the sample using a molar extinction coefficient for horse heart cytochrome c at 550 nm of 29,500 M−1 cm−1 (37).
Nickel affinity purification of His-tagged cytochromes.
Membrane fractions of S. coelicolor M145 strains were solubilized in 50 mM Tris-HCl, pH 6.8, 250 mM NaCl, 20% glycerol, 1% DDM for 1 h at 4°C with gentle mixing. After the DDM concentration was diluted to 0.1% with 50 mM Tris-HCl, pH 7.5, 10% glycerol, 250 mM NaCl, 10 mM imidazole, the sample was clarified by centrifugation at 40,000 × g for 30 min at 4°C.
For small-scale batch copurification experiments, the supernatant was supplemented with 10 mM imidazole. Three hundred microliters of Ni2+-nitrilotriacetic acid (NTA) resin (Qiagen) was washed with the same buffer containing 10 mM imidazole and was incubated with the solubilized membrane for 1 h at 4°C with gentle mixing. Working at 4°C, the resin was sedimented in a disposable column and washed with 15 column volumes of 50 mM Tris-HCl, pH 7.5, 20% glycerol, 250 mM NaCl, 0.1% DDM, 20 mM imidazole. Bound protein was eluted with 50 mM Tris-HCl, pH 7.5, 20% glycerol, 250 mM NaCl, 0.1% DDM, 500 mM imidazole. The unbound, wash, and elution fractions were subsequently analyzed by SDS-PAGE and Western blotting.
For larger-scale purification experiments, the solubilized, clarified membrane sample was loaded onto a 5-ml Ni2+-charged HisTrap HP column (Qiagen) at a rate of 1 ml/min. Loosely bound proteins were washed through the column with 50 mM Tris-HCl, pH 7.5, 10% glycerol, 250 mM NaCl, 0.1% DDM, 20 mM imidazole. Bound protein was eluted with a gradient of 20 mM to 1 M imidazole in the same buffer over 1 h. Eluted fractions were concentrated and analyzed by SDS-PAGE, Western blotting, and silver staining.
ESI-TOF MS.
Peptide mass fingerprinting of the protein bands of interest was performed by the FingerPrints Proteomic Service (University of Dundee) as described previously (38). The bands were excised from Coomassie-stained acrylamide gels with a scalpel, subjected to in-gel tryptic digestion, extracted, and analyzed by electrospray ionization–time of flight mass spectrometry (ESI-TOF MS) using an Applied BioSystems DE-STR. The mass lists generated were compared to those of S. coelicolor A3(2) predicted peptides from the NCBI nr database using Mascot Daemon. The searches considered oxidation and dioxydation of methionine, pyroglutamic acid formation at the N-terminal glutamine, acetylation of the N-terminal residues, and modification of cysteine by carbamidomethylation, as well as partial cleavage, leaving a maximum of two internal sites uncleaved.
Other protein methods.
SDS-PAGE and immunoblotting were carried out according to the methods of Laemmli (39) and Towbin et al. (40), respectively. Silver staining of SDS-PAGE gels was performed using the PlusOne protein silver staining kit (GE Healthcare). Polyclonal antisera to QcrA (Sco2149) have been described previously (12). Mouse anti-His5 antibody was obtained from Qiagen. Immunodetection was performed by using the Chemiluminescent substrate (Millipore) with a peroxidase-conjugated anti-rabbit (for QcrA) or anti-mouse (for His5) IgG (Bio-Rad).
RESULTS
An active Tat transport system is required for cytochrome bc1 complex activity.
We first examined the requirement of the Tat pathway for the biogenesis and function of the cytochrome bc1 complex. It has been reported previously that tat mutant strains of S. coelicolor M145, Streptomyces lividans, and Streptomyces scabies grow more slowly than wild-type strains (41, 42, 43), which might be consistent with a defect in aerobic respiration. To assess this in more detail, we constructed a strain (strain APH2) with a marked deletion of the qcrCAB genes in S. coelicolor M145 and compared the behavior of this strain with those of a previously described tatC deletion strain (43) and the parental strain.
Initially, we examined growth on two different types of solid media, SFM and YEME agars. Previously, it has been reported that tat mutant strains of S. coelicolor M145 and S. lividans grow more slowly on SFM medium and that they struggle to grow on medium containing high sucrose, where they also fail to sporulate (42, 43). This was confirmed by the experiment whose results are shown in Fig. 2A: the ΔtatC strain produced gray colonies (due to the presence of gray-pigmented spores) which were noticeably smaller than those of the wild-type strain. On YEME plates, which contain 34% sucrose, colonies of the ΔtatC strain had a colorless “bald” appearance arising from failure to undergo the full developmental cycle. In contrast, the qcrCAB deletion strain appeared to grow almost as well as the wild type on both types of growth media, although it did appear to overproduce the dark-blue-pigmented antibiotic actinorhodin, for unknown reasons. It can be concluded that the slow growth and developmental defects displayed by the ΔtatC strain on solid growth medium do not arise from a defect in biogenesis of the cytochrome bc1 complex.
FIG 2.

Comparison of the S. coelicolor M145 and ΔqcrCAB and ΔtatC mutant strains. (A) Growth of S. coelicolor M145 (WT), APH2 (ΔqcrCAB), and TP4 (ΔtatC) on SFM or YEME agar plates. Spores of each strain were streaked and incubated at 30°C for 14 days. (B) Growth in liquid culture of the same strains used in the experiments whose results are shown in panel A. An amount of 108 spores of each strain, in triplicate, was inoculated into 100 ml TSB and grown at 30°C for 40 h with shaking at 200 rpm. Every 3 h, 1-ml samples were pelleted, cytosolic proteins released by boiling in 1 M NaOH for 10 min, and the protein content of each sample measured. (C) Absorption difference spectra of DDM-solubilized membranes (each at a protein concentration of 1.25 mg/ml) of strains M145 (WT), APH2 (ΔqcrCAB), and APH8 (ΔtatC ϕC31 [PhrdB-qcrCABHis10]). Spectra were initially collected under air oxidation, and then samples were reduced by the addition of dithionite. Note that the relevant genotype for each strain is given in the figure. (D) Expanded view of the 590- to 610-nm region of the same absorption difference spectra as shown in panel C.
We next assessed the growth of the same strains in liquid culture. It has been noted previously that in liquid medium, tat mutant strains of Streptomyces grow slowly and in a very dispersed manner, whereas the corresponding wild-type strains grow as hyphal pellets (42, 43). As shown by the results in Fig. 2B, the growth rate of the ΔtatC mutant strain was clearly significantly lower than that of the wild type. In contrast, we noted that the qcrCAB deletion strain, APH2, retained the pellet-like growth of the wild-type strain, indicating that the dispersed growth of the tat strain does not result from a defect in the cytochrome bc1 complex. However, the ΔqcrCAB strain did show a small but significant delay in entering exponential-growth phase, although this was not nearly as marked as for the ΔtatC mutant. We ascribe this small growth delay to a defect in aerobic respiration.
Analysis of reduced minus oxidized difference spectra of membranes is a sensitive way to identify the presence of individual cytochromes (44). Difference spectra of solubilized membranes of the S. coelicolor wild-type strain, M145, showed specific absorbance peaks corresponding to the presence of b-type heme at 560 nm, with a clear shoulder for c-type heme at 550 nm (Fig. 2C). A small peak for a-type heme was also visible at 600 nm (see an enlargement of this section of the spectrum in Fig. 2D, top). Membranes of the ΔqcrCAB strain, APH2, showed a broad peak of b-heme at 558 to 560 nm, potentially arising from the b-type hemes of cytochrome bd oxidase and/or succinate dehydrogenase. As expected, no shoulder of c-type heme was detected, consistent with the deletion of qcrC that encodes the only c-type cytochrome in the S. coelicolor M145 genome. There was also no obvious a-type heme detectable at 600 nm (Fig. 2D, middle), suggesting that the expression or stability of cytochrome aa3 oxidase was affected by loss of the bc1 complex.
To maximize the detection of cytochromes in the poorly growing ΔtatC mutant strain TP4, we provided the strain with an additional copy of the qcrCAB genes under the control of the constitutive housekeeping sigma factor HrdB (2) at an ectopic location (strain APH8) (Table 1). The difference spectra of membranes isolated from this strain were very similar to those for the wild-type strain (Fig. 2C). Absorbance peaks corresponding to the b-type heme at 560 nm and a c-heme shoulder at 550 nm were readily apparent (Fig. 2C). Additionally, a small peak for a-type heme was visible at 600 nm (Fig. 2D). These observations are consistent with the conclusion that the cytochrome components of the bc1 complex are properly assembled in the ΔtatC mutant strain and that the cytochrome aa3 oxidase is also present.
To confirm the lack of cytochrome bc1 activity in the ΔqcrCAB and ΔtatC strains, we developed an assay to measure the activity of the complex in dispersed membranes. Since the cytochrome bc1-cytochrome aa3 complexes are obligately coupled and have been reported to form a supercomplex in some actinobacteria (9, 45), we found it necessary to include both formate and azide to inhibit electron flow to cytochrome aa3 (46). The results in Table 2 show that membranes of wild-type S. coelicolor M145 were able to catalyze the menadiol-dependent reduction of equine cytochrome c and that this activity was abolished by deletion of the qcrCAB genes and, also, by loss of tatC function. We therefore conclude that the Tat transport system is essential for the assembly and activity of the cytochrome bc1 complex.
TABLE 2.
Cytochrome c oxidase activity in solubilized membrane fractions of S. coelicolor M145 strains
| Strain | Mean cytochrome bc1 complex activity ± SDa (nmol cyt c reduced · mg−1 protein · min−1) |
|---|---|
| M145 (WT) | 217 ± 48 |
| APH2 (ΔqcrCAB::Hygr) | 1.0 ± 20 |
| TP4 (ΔtatC) | −12 ± 14 |
| APH3 (ΔqcrCAB::Hygr ϕC31 PhrdB-qcrCHis10AB) | 44 ± 8 |
| APH5 (ΔqcrCAB::Hygr ϕC31 PhrdB-qcrCABHis10) | 66 ± 20 |
| APH5-KK (ΔqcrCAB::Hygr ϕC31 PhrdB-qcrCAR161K R162KBHis10) | 4.5 ± 4.0 |
| APH6 (ΔtatC ϕC31 PhrdB-qcrCHis10AB) | −14 ± 15 |
| APH8 (ΔtatC ϕC31 PhrdB-qcrCABHis10) | 6.9 ± 7.1 |
Values are the means ± standard deviations of 4 replicates.
The Rieske protein is membrane bound in the tat mutant strain.
It has been demonstrated previously using a reporter fusion replacement of the Rieske iron-sulfur domain in the heterologous host E. coli that the translocation of the third TMD of the S. coelicolor Rieske protein is strictly dependent upon the Tat pathway (12). Attempts to use protease accessibility studies to map the topology of QcrA in S. coelicolor were confounded by the presence of endogenous proteases. However, if we lysed hyphae in the presence of a protease inhibitor cocktail, we were able to stabilize the protein sufficiently to confirm that it was present in the membranes of both the wild-type and the ΔtatC strain (Fig. 3A). It was also found in the membrane fraction when both of the conserved arginine residues of the twin arginine motif (Arg161 and Arg162) were replaced with lysine (Fig. 3B), a mutation that has previously been shown to block Tat-dependent recognition of substrate proteins (e.g., see references 12 and 25). Membrane-bound QcrA was resistant to extraction with carbonate or urea from each of these strains, consistent with Tat-independent integration of TMD1 and TMD2 into the bilayer (12).
FIG 3.
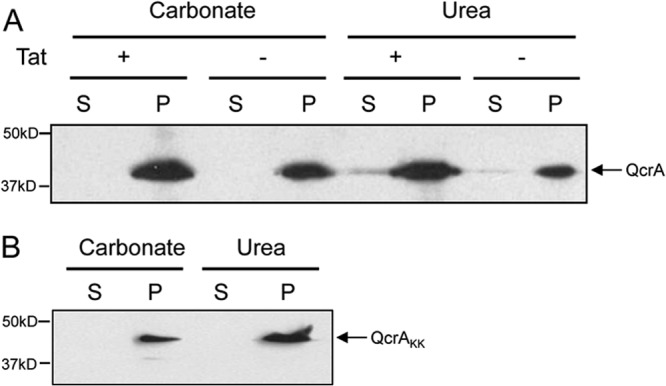
The Rieske protein, QcrA, is integrated into S. coelicolor M145 membranes in the absence of the Tat machinery. Membrane fractions were prepared from hyphae of strains M145 and TP4 (Tat + and −, respectively) (A) and strain APH5-KK (ΔqcrCAB::Hygr ϕC31 PhrdB-qcrCAR161K R162KBHis10, producing a QcrA protein with the conserved twin arginines mutated to twin lysines [QcrAKK]) (B) as described in Materials and Methods. The membranes were incubated with either 0.2 M Na2CO3 or 4 M urea, followed by recovery of the membrane pellet by ultracentrifugation. The presence of QcrA in the wash supernatant (S) and pelleted membrane (P) was analyzed by immunoblotting using anti-QcrA antiserum.
Taken together, the results described above indicate that the cytochrome components of the bc1 complex are correctly localized and assembled in the absence of a functional Tat system (since c-heme insertion occurs at the extracytoplasmic side of the membrane in bacteria) (16, 47). However, the lack of menadiol-dependent reduction of exogenous cytochrome c in the ΔtatC strain is consistent with an inability to translocate the Rieske iron-sulfur domain of QcrA across the membrane.
The cytochrome bc1 complex is still formed in the tat mutant strain.
In order to further investigate the assembly and stability of the cytochrome bc1 complex in the presence and absence of the Tat pathway, we constructed a number of strains that carry an ectopic copy of the qcrCAB genes under the expression of the constitutive hrdB promoter (48). These strains (Table 1) also encoded a decahistidine tag at the C terminus of either QcrC or QcrB. As shown by the results in Table 2, the His-tagged variants of QcrC or QcrB still supported cytochrome bc1 complex activity (which was completely abolished if the strains were also rendered ΔtatC). However, it should be noted that, in general, the overall cytochrome bc1 activity resulting from strains ectopically expressing qcrCAB was, for unknown reasons, significantly lower than that seen when the genes were expressed from their native chromosomal location.
We next investigated whether the Rieske protein, QcrA, could be copurified with the tagged QcrB in a small-scale Ni affinity pulldown experiment. To this end, membrane fractions of strain APH5 (qcrCABHis10) and the wild-type strain, M145, were solubilized with the detergent DDM, and the sample incubated with Ni-charged NTA resin. As shown by the results in Fig. 4, in the wild-type strain, there was some association between the QcrA protein and the Ni-charged resin, probably because the protein is relatively rich in His residues. This association was rather weak, since the protein could be completely washed off the resin in the presence of 50 mM imidazole. However, in the presence of His-tagged cytochrome b, the Rieske protein was bound much more tightly to the resin and did not wash off in the presence of 50 mM imidazole. Instead, QcrA was seen to coelute with the His-tagged variant of cytochrome b in the presence of 250 mM imidazole (Fig. 4), consistent with these two proteins forming a complex.
FIG 4.
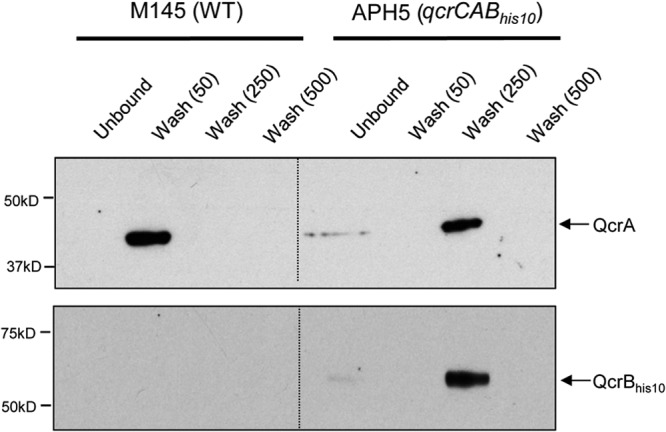
Copurification of the Rieske protein, QcrA, with decahistidine-tagged cytochrome b. Washed membrane fractions (150 mg protein) of the wild-type strain M145 or strain APH5 (ΔqcrCAB::hyg ϕC31 [PhrdB-qcrCABHis10]) were solubilized in the presence of 1% DDM. The solubilized membranes were incubated with Ni2+-charged NTA resin, and the unbound fraction was collected. Bound proteins were washed successively in buffer containing 50, 250, or 500 mM imidazole. Samples (3 μl of each indicated fraction) were separated by SDS-PAGE (14% acrylamide) and electroblotted, and the presence of QcrA or His-tagged QcrB was detected with anti-QcrA or anti-His tag antiserum, respectively.
We subsequently scaled up these experiments and investigated whether we could isolate all three of the cytochrome bc1 complex components using the affinity tag on QcrB. Solubilized membrane material was applied to a Ni-charged NTA column, and bound proteins were eluted with a gradient of imidazole. The results in Fig. 5A show that several peaks eluted from the column as the concentration of imidazole was increased. Analysis of the three main peaks by Western blotting showed that the His-tagged cytochrome b was primarily in the second peak (Fig. 5C), which eluted from the column at an imidazole concentration of approximately 250 mM. Immunoblotting the same three protein fractions with anti-QcrA antiserum indicated that the Rieske protein was also present in the major cytochrome b-containing fraction, although it was also detected in the other peaks that were examined (Fig. 5B). The fractions covering peak 2 were pooled, concentrated 50-fold, and analyzed by SDS-PAGE. As shown by the results in Fig. 5D, three major protein bands were detected in this fraction following silver staining, which were confirmed by tryptic mass fingerprinting to be QcrB, -A, and -C, with some minor degradation of cytochrome c. It should be noted that, despite previous observations that cytochrome bc1-cytochrome aa3 form a supercomplex in some actinobacteria (9, 45), we found no evidence that the cytochrome aa3 complex copurified with the cytochrome bc1 complex under these conditions.
FIG 5.

The cytochrome bc1 complex is stably assembled in both a Tat+ and ΔtatC mutant strain. Solubilized membranes from 2 g of cells of strain APH5 (ΔqcrCAB PhrdB-qcrCABHis10) (A) or strain APH8 (ΔtatC ΔqcrCAB::hyg ϕC31 [PhrdB-qcrCABHis10]) (E) were loaded onto a 5-ml Ni2+-charged NTA column and washed, and bound protein was eluted with a gradient of 30 to 500 mM imidazole (indicated by the sloping line). Fractions indicated by the arrows were analyzed by Western blotting. mAU, milli-absorbance unit. (B and C) Ten-microliter samples (from a total of 1 ml) of fractions 1 to 3 from the experiment whose results are shown in panel A. (F and G) Ten-microliter samples (from a total of 1 ml) of fractions 1 to 3 from the experiment whose results are shown in panel E. The samples were separated by SDS-PAGE (14% acrylamide), electroblotted, and incubated with anti-QcrA (B and F) or anti-His tag antibodies (C and G). The asterisk in panels B and F indicates a cross-reacting band that is recognized by the anti-QcrA antiserum, which may be His-tagged QcrB since the polyclonal QcrA antiserum was raised against a His-tagged QcrA (12). (D and H) The fractions numbered 2 in panels A and E, respectively, were concentrated from 8 ml to 150 μl, and 20 μl of each sample was analyzed by SDS-PAGE and silver staining. The identity of the indicated protein bands was confirmed by tryptic mass fingerprinting.
Finally, we examined the organization of the cytochrome bc1 complex from solubilized membranes of the ΔtatC mutant. We found that His-tagged cytochrome b from the ΔtatC strain behaved similarly to that from the tat+ strain, with the protein eluting predominantly in peak 2 (Fig. 5E and G), again corresponding to an imidazole concentration of approximately 250 mM. Interestingly, the Rieske protein, despite being incorrectly assembled in the ΔtatC background, also behaved similarly, with significant levels of protein present in the cytochrome b-containing fraction (Fig. 5F). The pooled and concentrated fractions from peak 2 were analyzed again by SDS-PAGE and silver staining (Fig. 5H). Four predominant protein bands were detected which, again, were confirmed by tryptic mass fingerprinting to be QcrB, QcrA, and two bands corresponding to cytochrome c (see Fig. S2 in the supplemental material). Our data therefore indicate that the cytochrome bc1 complex is also stably assembled in the absence of the Tat pathway.
DISCUSSION
Prior work has shown that the polytopic Rieske protein component of the S. coelicolor cytochrome bc1 complex follows an unusual pathway for membrane integration, where the first two TMD are inserted into the membrane by the action of Sec and YidC, while integration of TMD3 and translocation of the globular domain is dependent on the Tat pathway (12). However, these previous findings were for a chimeric protein comprising a reporter protein fused to the three Rieske TMDs and were undertaken in a heterologous host, E. coli. In this study, we have examined the assembly of the Rieske protein in the native organism, and the results we obtained here are consistent with these previous conclusions (12).
Our results clearly show that the cytochrome bc1 complex is inactive when the S. coelicolor M145 Tat system is absent but that at least the cytochrome c component of this complex is correctly assembled and with its heme cofactor inserted. Previous proteomic analysis has shown that the Rieske protein is a Tat substrate in S. coelicolor M145 (43), as it is in all other bacteria that have been examined (e.g., see references 17, 18, and 19). Therefore, the inactivity of the cytochrome bc1 complex in the absence of a functional Tat pathway is most likely due to the incorrect localization of the Rieske protein, QcrA.
Closer analysis of the Rieske protein in the ΔtatC strain shows that the protein is tightly integrated into the membrane. This is consistent with the insertion of the first two TMD of the protein in a Tat-independent manner, as had been observed previously when the hydrophobic portion of the protein was produced in E. coli (12). A lack of appropriate experimental tools means that it is not possible to confirm that these two TMD follow the same route of membrane integration in S. coelicolor as they do in E. coli, but it should be noted that both Sec and YidC components are conserved in Streptomyces (49), and it is highly likely that the mechanisms of assembly of TMD1 and -2 are the same. Taken together, we conclude that the Rieske protein is a dually targeted membrane protein in S. coelicolor.
The Rieske proteins of all actinobacteria analyzed to date are highly unusual because they are polytopic instead of monotopic proteins, as in other organisms. Indeed, the Rieske protein of the actinomycete Kitasatospora setae KM-6054 is predicted to have five TMD prior to the iron-sulfur containing domain (and with a consensus twin arginine motif preceding TMD5 [G. Chandra and T. Palmer, unpublished data]). What is the role of these extra TMD? Currently, it is not clear, although it is interesting to note that these highly hydrophobic Rieske proteins co-occur with membrane-bound cytochrome c and with cytochrome b proteins that also have an additional hydrophobic domain. Our results support the idea that additional Rieske TMD are involved in hydrophobic interactions with the other cytochrome bc1 components. Thus, we showed that the Rieske protein forms a stable complex with the other two cytochrome bc1 components in the ΔtatC strain. In this scenario, we anticipate that TMD1 and -2 are integrated into the membrane but that TMD3 and the iron-sulfur cluster-binding domain are localized at the cytoplasmic side of the membrane (Fig. 6). It is not clear whether the iron-sulfur cluster is inserted into the Rieske protein at this stage, since we were unable to obtain enough material from the ΔtatC strain to look for its presence by electron paramagnetic resonance (EPR). However, it should be noted that Bachmann et al. (18) showed that the cytosolic fraction of a Paracoccus denitrificans strain in which the twin arginine motif of the Rieske protein had been inactivated by substituting twin lysines gave an EPR signature of the Rieske iron-sulfur cluster, consistent with cofactor insertion in the cytoplasm prior to interaction with the Tat machinery. It is difficult to envisage how the complex we isolated from the membranes of the S. coelicolor ΔtatC strain would be stable to detergent solubilization and purification if the first two TMD of the Rieske protein were not interacting with one or both of the cytochromes. We attempted to acquire further data in support of this hypothesis by constructing truncations of QcrA producing just TMD3 and the globular domain. However, our truncates did not produce stable protein (data not shown)—it is not clear whether this resulted from poor translation of the protein or whether they lacked stability (for any of a number of reasons, including an inability to be recognized by the Tat pathway or to be stabilized through interaction with partner proteins).
FIG 6.
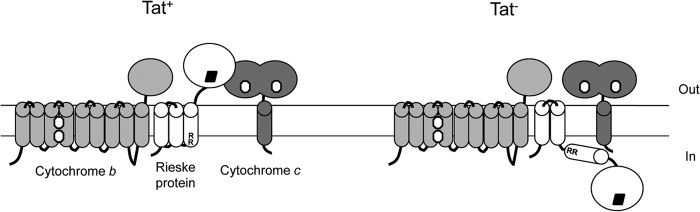
Models showing the predicted organization of the cytochrome bc1 complex components in a tat+ and a Δtat strain. White oblong, heme group; black diamond, 2Fe 2S cluster; RR, twin arginines of the Tat recognition motif; in, cytoplasm; out, cell exterior.
The results presented here have shown that the Tat system is integral to the activity of one branch of the aerobic respiratory pathway in S. coelicolor. It is interesting to note that, not only is this same branch of the pathway essential in the human actinobacterial pathogen M. tuberculosis, but the Tat pathway is also essential in this organism (50, 51), presumably because of its role in the assembly of the Rieske protein. A more-detailed understanding of cytochrome bc1 assembly in actinobacteria may eventually pave the way for novel strategies to limit the growth of pathogenic actinomycetes.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by Medical Research Council grant G0901653.
We thank Frank Sargent, Rebecca Keller, David Widdick, and Joanna Fyans for helpful discussions and advice. Grant English is thanked for his help with nickel affinity experiments.
Footnotes
Published ahead of print 18 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00776-13.
REFERENCES
- 1.Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. U. S. A. 102:15629–15634. 10.1073/pnas.0507850102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Keulen G, Alderson J, White J, Sawers RG. 2007. The obligate aerobic actinomycete Streptomyces coelicolor A3(2) survives extended periods of anaerobic stress. Environ. Microbiol. 9:3143–3149. 10.1111/j.1462-2920.2007.01433.x [DOI] [PubMed] [Google Scholar]
- 3.Watanabe S, Zimmermann M, Goodwin MB, Sauer U, Barry CE, III, Boshoff HI. 2011. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 7:e1002287. 10.1371/journal.ppat.1002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Mello R, Hill S, Poole RK. 1996. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: implications for regulation of activity in vivo by oxygen inhibition. Microbiology 142:755–763. 10.1099/00221287-142-4-755 [DOI] [PubMed] [Google Scholar]
- 5.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 187:6300–6308. 10.1128/JB.187.18.6300-6308.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker SC, Ferguson SJ, Ludwig B, Page MD, Richter OM, van Spanning RJ. 1998. Molecular genetics of the genus Paracoccus: metabolically versatile bacteria with bioenergetic flexibility. Microbiol. Mol. Biol. Rev. 62:1046–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenaz G, Genova ML. 2012. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv. Exp. Med. Biol. 748:107–144. 10.1007/978-1-4614-3573-0_5 [DOI] [PubMed] [Google Scholar]
- 8.Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. 1997. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277:60–66. 10.1126/science.277.5322.60 [DOI] [PubMed] [Google Scholar]
- 9.Niebisch A, Bott M. 2003. Purification of a cytochrome bc1-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. Identification of a fourth subunit of cytochrome aa3 oxidase and mutational analysis of diheme cytochrome c1. J. Biol. Chem. 278:4339–4346. 10.1074/jbc.M210499200 [DOI] [PubMed] [Google Scholar]
- 10.Sone N, Nagata K, Kojima H, Tajima J, Kodera Y, Kanamaru T, Noguchi S, Sakamoto J. 2001. A novel hydrophobic diheme c-type cytochrome. Purification from Corynebacterium glutamicum and analysis of the QcrCBA operon encoding three subunit proteins of a putative cytochrome reductase complex. Biochim. Biophys. Acta 1503:279–290. 10.1016/S0005-2728(00)00205-X [DOI] [PubMed] [Google Scholar]
- 11.Kurisu G, Zhang H, Smith JL, Cramer WA. 2003. Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science 302:1009–1014. 10.1126/science.1090165 [DOI] [PubMed] [Google Scholar]
- 12.Keller R, de Keyzer J, Driessen AJ, Palmer T. 2012. Co-operation between different targeting pathways during integration of a membrane protein. J. Cell Biol. 199:303–315. 10.1083/jcb.201204149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Driessen AJ, Nouwen N. 2008. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 77:643–667. 10.1146/annurev.biochem.77.061606.160747 [DOI] [PubMed] [Google Scholar]
- 14.Park E, Rapoport TA. 2012. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 41:21–40. 10.1146/annurev-biophys-050511-102312 [DOI] [PubMed] [Google Scholar]
- 15.Schulz H, Hennecke H, Thony-Meyer L. 1998. Prototype of a heme chaperone essential for cytochrome c maturation. Science 281:1197–1200. 10.1126/science.281.5380.1197 [DOI] [PubMed] [Google Scholar]
- 16.Thony-Meyer L, Kunzler P. 1997. Translocation to the periplasm and signal sequence cleavage of preapocytochrome c depend on sec and lep, but not on the ccm gene products. Eur. J. Biochem. 246:794–799. 10.1111/j.1432-1033.1997.t01-1-00794.x [DOI] [PubMed] [Google Scholar]
- 17.Aldridge C, Spence E, Kirkilionis MA, Frigerio L, Robinson C. 2008. Tat-dependent targeting of Rieske iron-sulphur proteins to both the plasma and thylakoid membranes in the cyanobacterium Synechocystis PCC6803. Mol. Microbiol. 70:140–150. 10.1111/j.1365-2958.2008.06401.x [DOI] [PubMed] [Google Scholar]
- 18.Bachmann J, Bauer B, Zwicker K, Ludwig B, Anderka O. 2006. The Rieske protein from Paracoccus denitrificans is inserted into the cytoplasmic membrane by the twin-arginine translocase. FEBS J. 273:4817–4830. 10.1111/j.1742-4658.2006.05480.x [DOI] [PubMed] [Google Scholar]
- 19.De Buck E, Vranckx L, Meyen E, Maes L, Vandersmissen L, Anne J, Lammertyn E. 2007. The twin-arginine translocation pathway is necessary for correct membrane insertion of the Rieske Fe/S protein in Legionella pneumophila. FEBS Lett. 581:259–264. 10.1016/j.febslet.2006.12.022 [DOI] [PubMed] [Google Scholar]
- 20.Meloni S, Rey L, Sidler S, Imperial J, Ruiz-Argueso T, Palacios JM. 2003. The twin-arginine translocation (Tat) system is essential for Rhizobium-legume symbiosis. Mol. Microbiol. 48:1195–1207. 10.1046/j.1365-2958.2003.03510.x [DOI] [PubMed] [Google Scholar]
- 21.Molik S, Karnauchov I, Weidlich C, Herrmann RG, Klosgen RB. 2001. The Rieske Fe/S protein of the cytochrome b6f complex in chloroplasts: missing link in the evolution of protein transport pathways in chloroplasts? J. Biol. Chem. 276:42761–42766. 10.1074/jbc.M106690200 [DOI] [PubMed] [Google Scholar]
- 22.Frobel J, Rose P, Muller M. 2012. Twin-arginine-dependent translocation of folded proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1029–1046. 10.1098/rstb.2011.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10:483–496. 10.1038/nrmicro2814 [DOI] [PubMed] [Google Scholar]
- 24.Berks BC. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22:393–404. 10.1046/j.1365-2958.1996.00114.x [DOI] [PubMed] [Google Scholar]
- 25.Stanley NR, Palmer T, Berks BC. 2000. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275:11591–11596. 10.1074/jbc.275.16.11591 [DOI] [PubMed] [Google Scholar]
- 26.Kol S, Majczak W, Heerlien R, van der Berg JP, Nouwen N, Driessen AJ. 2009. Subunit a of the F(1)F(0) ATP synthase requires YidC and SecYEG for membrane insertion. J. Mol. Biol. 390:893–901. 10.1016/j.jmb.2009.05.074 [DOI] [PubMed] [Google Scholar]
- 27.Stanley NR, Sargent F, Buchanan G, Shi J, Stewart V, Palmer T, Berks BC. 2002. Behaviour of topological marker proteins targeted to the Tat protein transport pathway. Mol. Microbiol. 43:1005–1021. 10.1046/j.1365-2958.2002.02797.x [DOI] [PubMed] [Google Scholar]
- 28.Tullman-Ercek D, DeLisa MP, Kawarasaki Y, Iranpour P, Ribnicky B, Palmer T, Georgiou G. 2007. Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 282:8309–8316. 10.1074/jbc.M610507200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu L, Wasey A, White SH, Dalbey RE. 2013. Charge composition features of model single-span membrane proteins that determine selection of YidC and SecYEG translocase pathways in Escherichia coli. J. Biol. Chem. 288:7704–7716. 10.1074/jbc.M112.429431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 31.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8 [DOI] [PubMed] [Google Scholar]
- 32.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 33.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. 10.1038/417141a [DOI] [PubMed] [Google Scholar]
- 34.Fyans JK, Bignell D, Loria R, Toth I, Palmer T. 2013. The ESX/type VII secretion system modulates development, but not virulence, of the plant pathogen Streptomyces scabies. Mol. Plant Pathol. 14:119–130. 10.1111/j.1364-3703.2012.00835.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546. 10.1073/pnas.0337542100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng L, Baumann U, Reymond JL. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32:e115. 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margoliash E, Walasek OF. 1967. Cytochrome c from vertebrate and invertebrate sources. Methods Enzymol. 10:339–348. 10.1016/0076-6879(67)10064-5 [DOI] [Google Scholar]
- 38.Zoltner M, Fyfe PK, Palmer T, Hunter WN. 2013. Characterization of Staphylococcus aureus EssB, an integral membrane component of the type VII secretion system: atomic resolution crystal structure of the cytoplasmic segment. Biochem. J. 449:469–477. 10.1042/BJ20121209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 40.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76:4350–4354. 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joshi MV, Mann SG, Antelmann H, Widdick DA, Fyans JK, Chandra G, Hutchings MI, Toth I, Hecker M, Loria R, Palmer T. 2010. The twin arginine protein transport pathway exports multiple virulence proteins in the plant pathogen Streptomyces scabies. Mol. Microbiol. 77:252–271. 10.1111/j.1365-2958.2010.07206.x [DOI] [PubMed] [Google Scholar]
- 42.Schaerlaekens K, Van Mellaert L, Lammertyn E, Geukens N, Anne J. 2004. The importance of the Tat-dependent protein secretion pathway in Streptomyces as revealed by phenotypic changes in tat deletion mutants and genome analysis. Microbiology 150:21–31. 10.1099/mic.0.26684-0 [DOI] [PubMed] [Google Scholar]
- 43.Widdick DA, Dilks K, Chandra G, Bottrill A, Naldrett M, Pohlschroder M, Palmer T. 2006. The twin-arginine translocation pathway is a major route of protein export in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U. S. A. 103:17927–17932. 10.1073/pnas.0607025103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicholls DG, Ferguson SJ. 2002. Bioenergetics 3 Academic Press, London, United Kingdom [Google Scholar]
- 45.Megehee JA, Hosler JP, Lundrigan MD. 2006. Evidence for a cytochrome bc1-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology 152:823–829. 10.1099/mic.0.28723-0 [DOI] [PubMed] [Google Scholar]
- 46.Nicholls P, Petersen LC, Miller M, Hansen FB. 1976. Ligand-induced spectral changes in cytochrome c oxidase and their possible significance. Biochim. Biophys. Acta 449:188–196. 10.1016/0005-2728(76)90132-8 [DOI] [PubMed] [Google Scholar]
- 47.Ahuja U, Kjelgaard P, Schulz BL, Thony-Meyer L, Hederstedt L. 2009. Haem-delivery proteins in cytochrome c maturation System II. Mol. Microbiol. 73:1058–1071. 10.1111/j.1365-2958.2009.06833.x [DOI] [PubMed] [Google Scholar]
- 48.Aigle B, Wietzorrek A, Takano E, Bibb MJ. 2000. A single amino acid substitution in region 1.2 of the principal sigma factor of Streptomyces coelicolor A3(2) results in pleiotropic loss of antibiotic production. Mol. Microbiol. 37:995–1004. 10.1046/j.1365-2958.2000.02022.x [DOI] [PubMed] [Google Scholar]
- 49.Palmer T, Hutchings MI. 2010. Protein secretion in Streptomyces, p 87–104 In Dyson PJ. (ed), Streptomyces molecular biology and biotechnology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 50.Saint-Joanis B, Demangel C, Jackson M, Brodin P, Marsollier L, Boshoff H, Cole ST. 2006. Inactivation of Rv2525c, a substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases beta-lactam susceptibility and virulence. J. Bacteriol. 188:6669–6679. 10.1128/JB.00631-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84. 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 52.Casadaban MJ, Cohen SN. 1979. Lactose genes fused to exogenous promoters in one step using a Mu-lac bacteriophage: in vivo probe for transcriptional control sequences. Proc. Natl. Acad. Sci. U. S. A. 76:4530–4533. 10.1073/pnas.76.9.4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61–68. 10.1016/0378-1119(92)90603-M [DOI] [PubMed] [Google Scholar]
- 55.Paget MS, Chamberlin L, Atrih A, Foster SJ, Buttner MJ. 1999. Evidence that the extracytoplasmic function sigma factor sigmaE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J. Bacteriol. 181:204–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gust B, Chandra G, Jakimowicz D, Yuqing T, Bruton CJ, Chater KF. 2004. Lambda red-mediated genetic manipulation of antibiotic-producing Streptomyces. Adv. Appl. Microbiol. 54:107–128. 10.1016/S0065-2164(04)54004-2 [DOI] [PubMed] [Google Scholar]
- 57.Bierman M, Logan R, O'Brien K, Seno ET, Rao RN, Schoner BE. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49. 10.1016/0378-1119(92)90627-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.