

Abstract
The cell envelope of Gram-negative bacteria is an essential organelle that is important for cell shape and protection from toxic compounds. Proteins involved in envelope biogenesis are therefore attractive targets for the design of new antibacterial agents. In a search for new envelope assembly factors, we screened a collection of Escherichia coli deletion mutants for sensitivity to detergents and hydrophobic antibiotics, a phenotype indicative of defects in the cell envelope. Strains lacking yciM were among the most sensitive strains of the mutant collection. Further characterization of yciM mutants revealed that they display a thermosensitive growth defect on low-osmolarity medium and that they have a significantly altered cell morphology. At elevated temperatures, yciM mutants form bulges containing cytoplasmic material and subsequently lyse. We also discovered that yciM genetically interacts with envC, a gene encoding a regulator of the activity of peptidoglycan amidases. Altogether, these results indicate that YciM is required for envelope integrity. Biochemical characterization of the protein showed that YciM is anchored to the inner membrane via its N terminus, the rest of the protein being exposed to the cytoplasm. Two CXXC motifs are present at the C terminus of YciM and serve to coordinate a redox-sensitive iron center of the rubredoxin type. Both the N-terminal membrane anchor and the C-terminal iron center of YciM are important for function.
INTRODUCTION
Antibiotic resistance has become a worldwide problem that threatens the effectiveness of many medicines used today to treat bacterial infections. A particularly serious threat is the emergence of Gram-negative pathogens, including Escherichia coli and Pseudomonas aeruginosa, which are resistant to all of the available antibacterial agents (1). It is therefore urgent to develop new antibiotics active against Gram-negative bacteria, which requires a deep understanding of the biology of these organisms. Proteins playing a role in the assembly of the cell envelope are attractive targets for antibiotic development because this structure is essential for viability and protection against toxic compounds such as antibiotics.
The envelope of Gram-negative bacteria is characterized by the presence of two concentric membranes, the inner membrane (IM) and outer membrane (OM), which are separated by the periplasm, a compartment that represents between 10 and 20% of the total cell volume and contains the peptidoglycan layer, or sacculus (2–4). The peptidoglycan sacculus is a heteropolymeric macromolecule composed of relatively short glycan strands and peptide cross-links, which serve to maintain cell morphology and give protection from turgor pressure. Although several proteins that play a key role in the biogenesis of the cell envelope have been identified recently (5–7), we have a poor understanding of how this complex architecture is assembled and of how envelope biogenesis is coordinated with cell growth and division. For instance, we do not know how the phospholipids that are present in the inner leaflet of the OM are transported across the periplasm and inserted in the membrane, and we have a partial understanding of the mechanisms that ensure the transport and insertion of β-barrel proteins in the OM (8).
A straightforward method to search for proteins involved in a particular process is to screen for mutants that exhibit defects that could result from an impairment of this process. As mutants with a decreased integrity of the envelope typically display an increased sensitivity to hydrophobic antibiotics and detergents (9), we screened a systematic collection of E. coli deletion mutants (the Keio collection) (10) for sensitivity to rifampin, a hydrophobic antibiotic, and to SDS. In this screen, we found that the mutant lacking yciM, a gene coding for a heat shock protein of unknown function (11), was one of the most sensitive strains of the Keio collection, suggesting severe defects in the cell envelope.
Further characterization of yciM mutants revealed that their growth is severely affected both by the osmolarity of the medium and by the temperature. Moreover, deletion of yciM leads to changes in cell morphology and to the formation of bulges that contain cytoplasmic material and cause cell lysis. We also uncovered a genetic interaction between yciM and envC, a gene coding for a protein involved in septation by controlling the activity of peptidoglycan amidases (12). Altogether, these results indicate that the presence of YciM is required for envelope integrity. The biochemical characterization of YciM revealed that the protein is anchored to the IM via a highly conserved N-terminal segment and that two C-terminal CXXC motifs serve to coordinate a redox-sensitive iron center of the rubredoxin type. Both the N-terminal transmembrane segment and the C-terminal iron center are important for YciM function.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains are listed in Table S1. Strains CB23, VN26, NR1681, and VN250 were constructed by transferring the ΔyciM::kan, ΔpyrF::kan, ΔyciS::kan, and ΔenvC::kan alleles, respectively, from the Keio collection (10) into the MC1000 wild-type strain using P1 transduction standard procedures (13). The kanamycin resistance cassette of strain CB23 was excised using pCP20 (14) to generate NR1647. Double mutants VN64, VN90, VN94, VN110, VN255, VN262, VN264, VN266, and VN268 were generated in the NR1647 strain by P1 transduction using alleles from the Keio collection. Strain VN15 is an araD+ revertant of MC1000 selected on MacConkey agar supplemented with l-arabinose. The VN50 strain was constructed by transferring the ΔyciM::kan allele from the Keio collection into VN15 by P1 transduction. Unless otherwise noted, cultures were grown in LB medium (containing 1% NaCl) at 37°C. LB medium without NaCl (≤0.2%) was used for some experiments and is annotated as salt-free LB. When necessary, medium was supplemented with ampicillin (200 μg/ml), kanamycin (50 μg/ml), l-arabinose (0.2%), or d-glucose (0.2%). Growth was monitored by the optical density at 600 nm. We were unable to generate a ΔyciM ΔenvC::kan double mutant by introducing into NR1647 the ΔenvC::kan allele via P1 transduction and directly selecting for kanamycin-resistant transductants. However, we were able to construct the ΔyciM ΔenvC::kan double mutant by cotransduction of the ΔenvC::kan allele with a zib563::Tn10 allele (∼35% linkage) when we selected for tetracycline-resistant transductants.
Plasmid constructions.
The plasmids and primers used in the present study are listed in Tables S1 and S2, respectively, in the supplemental material. Plasmid constructions are described in the supplemental material.
Sensitivity to antibiotics, detergents, bile salts, and osmotic stress conditions.
To characterize the sensitivity of strains MC1000, CB23, and NR1681 to antibiotics, bacteria were grown in LB medium at 37°C. After 16 h, 100 μl of culture inoculum was mixed with 3 ml of molten LB top agar and poured over LB agar plates. BBL Sensi-Discs antimicrobial susceptibility test discs (6 mm) containing either rifampin (25 μg) or novobiocin (5 μg) were placed over the LB top agar, and the plates were incubated overnight at 37°C. The diameter of the zone of inhibition of growth around each disc was recorded in millimeters. The sensitivity to bile salts and osmotic stress was assayed by streaking isolated colonies of wild-type, yciM, yciS, and pyrF strains on MacConkey agar (which contains 0.15% bile salts) or on LB agar either lacking or containing 750 mM NaCl.
Complementation assays.
VN15 and VN50 carrying plasmids pVN2 (YciM), pVN182 encoding a truncated YciM protein lacking the first 22 amino acids (YciM*), or pVN173 encoding YciM in which the four cysteines of the CXXC motif are replaced by serines (YciMCC/SSSS) were streaked on MacConkey agar or on salt-free LB agar and incubated at 22°C and 42°C, respectively.
Expression and purification.
BL21(DE3) harboring the expression plasmid pHE72 encoding a C-terminally His-tagged version of YciM* (YciM*His), pVN55 encoding a C-terminally Strep-tagged version of YciM* (YciM*Strep), pVN74 (YciM*), pVN147 encoding a His-tagged mutant of YciM* in which the two cysteine residues outside the CXXC motif (Cys184 and Cys258) were replaced by serines (YciM*SS/CCCC−His), or pVN67 (YciM*SS/CCCC−Strep) was grown aerobically in LB medium at 37°C to an A600 of 0.5. Expression of the proteins was induced by adding 0.2% l-arabinose for YciM*His and YciM*SS/CCCC−His or 200 μg/liter of anhydrotetracycline for YciM*Strep, YciM*, and YciM*SS/CCCC−Strep. Shaking was resumed for 3 h. Cultures were then centrifuged for 20 min (8,000 × g at 4°C). From this point on, all steps were performed at 4°C. Bacteria expressing YciM*His and YciM*SS/CCCC−His were resuspended in 25 ml of buffer A (50 mM NaPi, pH 8.0, 300 mM NaCl). Bacteria expressing YciM*Strep, YciM*, or YciM*SS/CCCC−Strep were resuspended in 25 ml of buffer B (100 mM Tris, pH 8.0, 150 mM NaCl). Bacteria expressing YciM* were resuspended in 25 ml of buffer C (25 mM Tris, pH 8.0). Cells were then disrupted by two passages through a French press cell at 1,200 lb/in2, and the cell lysate was centrifuged for 40 min (20,000 × g at 4°C). The resulting supernatant was then loaded on a 1-ml HisTrap HP column (GE Health Care) for YciM*His and YciM*SS/CCCC−His, on a 5-ml Strep-Tactin column (IBA Tools) for YciM*Strep and YciM*SS/CCCC−Strep, or on a Q-Sepharose column (GE Health Care) for YciM*. The columns were washed with buffer A, B, and C, respectively. YciM*His and YciM*SS/CCCC−His were eluted with a 0 mM to 300 mM imidazole gradient in buffer A, YciM*Strep and YciM*SS/CCCC−Strep were eluted with 2.5 mM desthiobiotin in buffer B, and YciM* was eluted with a 0 mM to 400 mM NaCl gradient in buffer C. The fractions containing the proteins were concentrated using a Vivaspin-15 device and desalted on a PD10 column (GE Health Care) equilibrated with buffer C. The fractions containing YciM*His, YciM*Strep, YciM*SS/CCCC−His, or YciM*SS/CCCC−Strep were then loaded onto a Q-Sepharose column (GE Health Care) equilibrated with buffer C. The column was washed with buffer C, and the proteins were eluted with a 0 mM to 400 mM NaCl gradient in buffer C. The fractions containing the proteins were pooled and concentrated by ultrafiltration in an Amicon cell. The five different solutions containing each protein were then loaded on a Superdex 200 gel filtration column equilibrated with buffer D (25 mM HEPES, pH 7.1, 100 mM NaCl) at a flow rate of 0.5 ml/min. The fractions containing the proteins were concentrated, and the pure proteins were stored in buffer D. Optical spectra were recorded with a Varian Cary 50 Bio UV spectrophotometer.
Anaerobic purification.
YciM*Strep was expressed aerobically as described in the previous section. After centrifugation of the cultures for 20 min at 8,000 × g and 4°C, cells were broken anaerobically by two passages through a French press cell at 1,200 lb/in2. The protein was then purified as explained above in an anaerobic tent containing 95% N2 and 5% H2. Optical spectra were recorded anaerobically with a Varian Cary 50 Bio UV spectrophotometer.
Subcellular localization of YciM.
To determine the subcellular localization of YciM, BL21(DE3) cells harboring either the pVN2 (YciM), pVN182 (YciM*), or the pVN173 (YciMCC/SSSS) plasmid were grown in LB medium at 37°C to an A600 of 0.5. Expression of the proteins was induced by adding 0.2% l-arabinose. After 90 min, cultures were centrifuged for 20 min (8,000 × g at 4°C), and cell lysates were prepared as described above. The cell lysates were then ultracentrifuged 1 h at 100,000 × g at 4°C. The resulting supernatant containing the soluble proteins was stored. The pellet was resuspended in 15 ml of buffer A supplemented with 1% Triton X-100 and incubated at room temperature for 90 min. The solubilized proteins were then separated from the insoluble material by ultracentrifugation for 1 h at 100,000 × g at 4°C. The resulting supernatant containing the solubilized membrane proteins was kept for analysis.
Topology of YciM.
To determine the orientation of the C-terminal part of YciM, BL21(DE3) cells harboring the pHE43 (YciMHis) expression plasmid were grown aerobically in LB medium at 37°C until an A600 of 0.4 was reached. Expression of the protein was induced by adding 0.2% l-arabinose. After 1 h, cells were centrifuged for 5 min at 8,000 × g, and the pellet was resuspended in 500 μl of 50 mM Tris buffer, pH 8.0. Spheroplasts were prepared by adding 1 volume of SP buffer (50 mM Tris, pH 8.0, 1 M sucrose, 2 mM EDTA, 0.5 mg/ml lysozyme). The mixture was then incubated at room temperature. After 15 min, MgCl2 was added to a final concentration of 20 mM. Spheroplasts were collected by centrifugation at 13,000 × g for 10 min and resuspended in 500 μl of 50 mM Tris, pH 8.0, with or without Triton X-100 (0.2%). Proteinase K (50 μg/ml) was added to selected samples and incubated overnight at 37°C. Spheroplasts were then precipitated with 10% trichloroacetic acid (TCA) and left for 1 h on ice. After a 10-min centrifugation at 16,000 × g, the precipitated material was washed with 5% TCA and finally resuspended in Laemmli buffer (15). Protein degradation was then analyzed by Western blotting using an anti-YciM antibody produced from a rabbit immunized with the purified protein (Eurogentec, Liège, Belgium).
Metal analysis.
For the determination of the metal content of YciM, different preparations of YciM (50 μM each) were separated from exogenous metals by gel filtration using a PD10 column equilibrated with metal-free buffer (50 mM NaPi, pH 8.0) prepared by incubation with Chelex 100 resin. The samples were concentrated 2-fold by ultrafiltration using a Centricon (Millipore) and analyzed using an inductively coupled plasma mass spectrometer (Agilent 7500cx), located at the Laboratory of Toxicology of the Université Catholique de Louvain in Brussels, Belgium. The metal content of the protein-containing retentate was compared with that of the filtrate. Zinc, nickel, iron, cobalt, arsenic, aluminum, selenium, cadmium, barium, lead, chrome, and manganese concentrations were measured. As additional controls, buffer samples were analyzed using the same procedure.
PAR-PMPS assay.
Zinc concentration was determined using the zinc-complexing reagent PAR (4-(2-pyridylazo)resorcinol). PAR binds zinc to form a Zn(PAR)2 complex, which absorbs strongly at 500 nm (ε = 66,000 M−1 cm−1 in HEPES, pH 7.0). To determine the amount of zinc bound to the protein, p-hydroxymercuriphenylsulfonic acid (PMPS) was added to release zinc from the zinc center and allow the formation of the Zn(PAR)2 complex. PMPS forms mercaptide bonds with thiols using the following reaction mechanism: R-HgOH + P-SH → R-Hg-S-P + H2O, where the R stands for the phenylsulfonic acid group of PMPS and the P stands for a protein. This reaction mechanism proceeds as a substitution reaction whereby the hydroxyl anion is displaced by the thiol-containing protein to form a mercuric sulfide derivative of the protein (R-Hg-S-P). If the thiols on the protein are involved in coordinating a metal such as zinc, this covalent modification leads to the release of the zinc. To determine the number of cysteines involved in zinc binding, defined amounts of PMPS are added, and the changes in A500 are recorded (16, 17).
Sulfur content analysis.
The sulfur content of YciM*Strep was determined using a colorimetric assay described by Beinert et al. (18). Standard curves were established using a sodium sulfur solution (Na2S).
Microscopy.
For cell length analysis using phase-contrast microscopy, wild-type and yciM strains were grown at 37°C to exponential phase, placed on a 2% LB agarose pad, and mounted on a Nikon Ti-Eclipse inverted microscope equipped with a TI-CT-E motorized condenser and a CoolSnap HQ2 FireWire charge-coupled-device (CCD) camera. The cell length analysis was performed using the free, open-source software MicrobeTracker (19). This software package was specifically designed to detect and outline bacterial cells in microscopy images. To perform time-lapse microscopy, wild-type and yciM cells were grown at 37°C to an A600 of 0.5 and placed on a microscope slide layered with a pad of 1% agarose containing LB medium with different salt concentrations. These slides were placed on a microscope stage at different temperatures. Samples were observed using differential interference contrast (DIC) or phase-contrast imaging on a Nikon Eclipse E1000 microscope with a 100× objective and monitored with a Hamamatsu ORCA-ER LCD camera. Images were taken every 3 min and then processed with NIS Elements AR, version 3.0, software (Nikon). To generate images for Fig. S1 in the supplemental material, strains were grown on LB plates at 37°C overnight. Colonies were resuspended in LB medium, and cells were layered on an 1% agarose pad containing phosphate-buffered saline (PBS), pH 7.4 (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.46 mM KH2PO4), and imaged using phase-contrast with a 100× oil immersion objective lens and a Nikon Eclipse Ti-E microscope equipped with a Nikon DS-QI1 cooled digital camera.
Fluorescence microscopy.
To determine whether the bulges contain cytoplasmic material, HE89 cells were grown in LB medium at 37°C until an A600 of 0.5 was reached. Then, time-lapse microscopy was performed by placing the strains on a microscope slide that was layered with a pad of 1% agarose-containing LB medium. After the temperature was shifted to 42°C, DIC and fluorescence images were taken every 3 min. Fluorescence was observed with a BrightLine fluorescein isothiocyanate (FITC) filter set. To determine the localization of YciM and YciM* with a C-terminal fusion of the fluorescent protein mCherry (YciMmCherry and YciM*mCherry, respectively), HE64 and VN237 cells were grown in LB medium at 37°C to an A600 of 0.5. Production of YciMmCherry or YciM*mCherry was induced by adding l-arabinose to a final concentration of 0.2%. After 30 min, the bacteria were placed on a microscope slide layered with a pad of 1% agarose. Fluorescence was observed with a BrightLine TXRED-4040B filter set.
RESULTS
Deletion of yciM severely impairs the integrity of the cell envelope.
To identify new proteins involved in maintaining the integrity of the cell envelope, we screened the Keio collection for sensitivity to detergents and hydrophobic antibiotics, a phenotype indicative of envelope defects (9). The Keio collection is a systematic collection of E. coli deletion mutants that contain disruptions in 3,985 nonessential genes, including several hundreds of uncharacterized open reading frames (10). We streaked the mutants on LB plates containing either rifampin, a hydrophobic antibiotic which does not cross the OM easily (9), or a mixture of SDS and EDTA. EDTA complexes the cations inserted between the lipopolysaccharide (LPS) molecules and therefore destabilizes the OM, rendering the bacteria more sensitive to the detergent. Only 42 strains were sensitive to both SDS-EDTA and rifampin (see Table S3 in the supplemental material). Most of the sensitive mutants corresponded to genes already known to be involved in envelope assembly, such as mutants defective in LPS biosynthesis (rfaC, rfaD, rfaE, rfaF, rfaH, rfaI, and rfaP mutants) or in β-barrel insertion at the OM (surA, bamB, and bamE mutants), which validated our experimental approach (20, 21). Four mutants lacked proteins of unknown function (yciM, yfgC, yraP, and ybiX mutants) although recently YfgC has been implicated in the degradation of misfolded β-barrel proteins at the OM (22). We tested the sensitivity of these four mutants to other hydrophobic antibiotics and found that strains lacking yciM were the most sensitive (see Table S4), which prompted us to investigate the properties of the yciM mutant and to characterize the corresponding protein.
Characterization of the yciM mutant.
For our studies, we introduced the ΔyciM::kan allele from the Keio collection into MC4100 and MC1000, two well-characterized E. coli strains (23, 24). When we transferred the ΔyciM::kan allele in strain MC4100, we observed that the resulting mutant exhibited growth defects and accumulated suppressors rapidly. This strain was therefore not retained for further studies. In contrast, the ΔyciM::kan allele could be stably transduced into MC1000 (23). As shown in Table 1, and similar to the yciM strain from the Keio collection, the MC1000 yciM mutant exhibits increased sensitivity to rifampin and novobiocin, two hydrophobic antibiotics (Table 1), and to a mixture of SDS and EDTA.
TABLE 1.
Sensitivity of the MC1000 yciM and yciS mutants to antibiotics, SDS-EDTA, and osmotic stress conditions
| Strain | Inhibition zone (mm) with: |
Growth on LB plates with:a |
||
|---|---|---|---|---|
| Rifampin (25 μg) | Novobiocin (5 μg) | 0.05% SDS–1.25 mM EDTA | 750 mM NaCl | |
| Wild type | 12 ± 0.5 | < 6 | +++ | +++ |
| yciM strain | 16 ± 1.2 | 19 ± 3.1 | − | − |
| yciS strain | 13 ± 1.2 | 7.6 ± 1.5 | + | +++ |
Strains were streaked on plates containing the additives shown. Growth is reported from normal growth (+++) to no growth (−) after overnight incubation at 37°C.
We then investigated the growth properties of the MC1000 yciM mutant under various temperature and culture conditions and found that deletion of yciM causes a pleiotropic phenotype in E. coli. First, yciM mutants are unable to grow at low temperatures on MacConkey agar, a medium containing bile salts (Fig. 1A). Second, yciM mutants are sensitive to high salt concentrations, being unable to grow on LB medium containing 750 mM NaCl at 37°C (Table 1). Third, yciM mutants display a thermosensitive growth defect in low-osmolarity medium, being unable to grow at 42°C in low-salt LB liquid culture (Fig. 1B) and on salt-free LB agar (Fig. 1C). These mutants also exhibit a heat-sensitive phenotype at 45°C in LB liquid cultures (data not shown). These phenotypes are not caused by polar effects since mutants lacking pyrF, the gene located immediately downstream of yciM on the chromosome, are not sensitive to antibiotics, are able to grow on MacConkey plate at 22°C (Fig. 1A), and do not exhibit a thermosensitive growth defect on salt-free LB agar (Fig. 1C). Furthermore, expressing the wild-type YciM protein from a plasmid rescues the inability of the yciM mutant to grow on MacConkey agar at 22°C (Fig. 1A) and on salt-free LB agar at 42°C (Table 2). Thus, the aforementioned phenotypes are the result of the loss of YciM.
FIG 1.
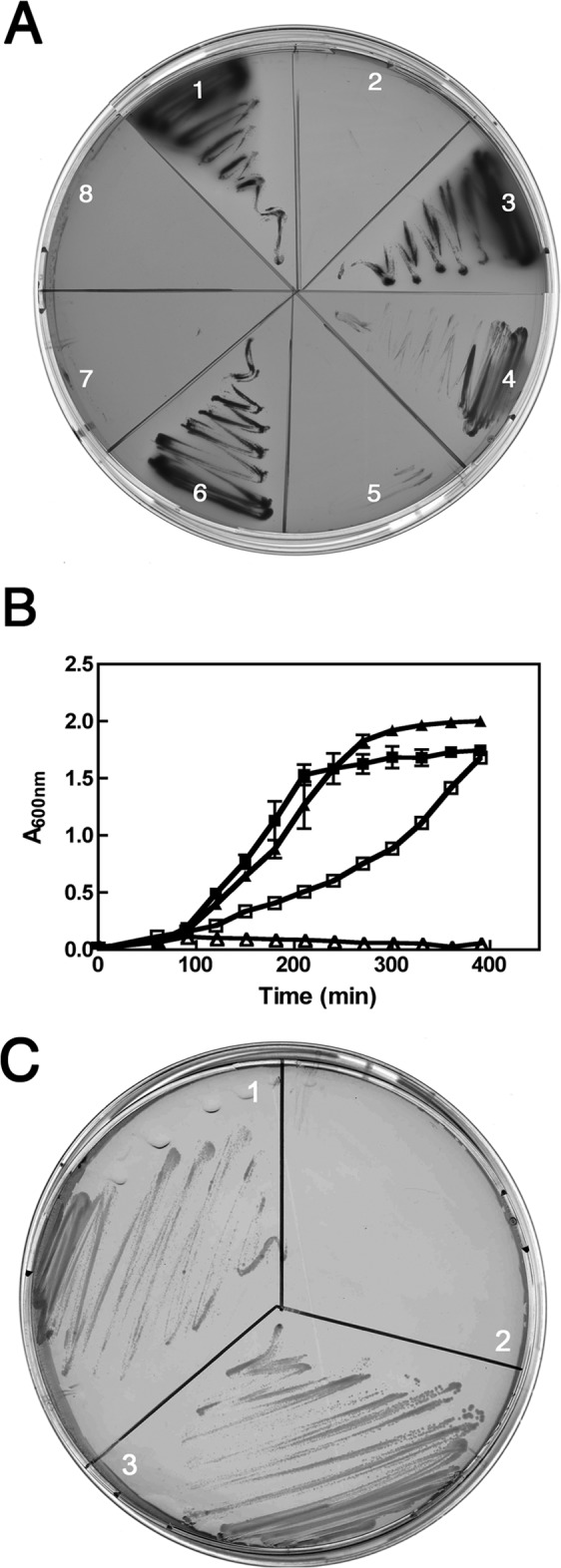
Characterization of the yciM mutant. (A) The growth of wild-type (1), yciM (2), and pyrF strains (3) on MacConkey agar was evaluated at 22°C. The ability of the various YciM constructs to complement the growth defect of yciM mutants on MacConkey agar at 22°C was tested by expressing YciM (4), YciM* (7), or YciMCC/SSSS (8). Wild-type (6) and yciM mutant (5) strains harboring the control pBAD-HisB plasmid were also analyzed. (B) Growth curves of wild-type (■) and yciM (▲) strains at 42°C in LB medium (filled symbols) or in salt-free LB medium (open symbols). (C) The growth of wild-type (1), yciM (2), and pyrF (3) strains was evaluated at 42°C on salt-free LB plates.
TABLE 2.
Complementation assays on salt-free LB medium
| Strain | Growth on salt-free LB plates at 42°Ca |
|---|---|
| Wild type (pBAD-HisB) | +++ |
| yciM (pBAD-HisB) strain | − |
| yciM (pBAD-HisB-YciM) strain | +++ |
| yciM (pBAD-HisB-YciM*) strain | − |
| yciM (pBAD-HisB-YciMCC/SSSS) strain | − |
The ability of the various YciM constructs to complement the growth defect of yciM mutants on salt-free LB medium at 42°C was tested. Wild-type and yciM mutant strains harboring the control pBAD-HisB plasmid were also analyzed. +++, normal growth; −, no growth.
As yciM has been reported to form a heat-inducible operon with yciS (11, 25), a gene encoding a small-membrane protein of unknown function (26), we investigated the phenotypes of the yciS mutant. Note that yciS is located upstream of yciM on the chromosome. Although yciS mutants are moderately sensitive to antibiotics and detergents (Table 1), their growth is not affected by modulating the salt concentration of the medium (Table 1) or the temperature (data not shown), indicating that YciS is not required for the full function of YciM. Thus, yciS was not considered further in the present study.
yciM mutants have an increased cell length and form bulges at elevated temperatures.
We then explored the morphology of the yciM mutants microscopically. First, we observed that there is a significant heterogeneity in the cell length of yciM mutants compared to the wild type (Fig. 2A) and that the average length of yciM mutant cells is ∼1.7-fold increased (n = 300) compared to the length of wild-type cells (from 4.45 ± 0.84 to 7.57 ± 3.98 μm; n = 300). The increase in length does not seem to result from failures in the division process as yciM mutant cells divide normally (see Movie S1 in the supplemental material). Moreover, using time-lapse microscopy, we observed that bacteria lacking yciM form bulges that are mostly localized at the division site and whose formation leads to cell lysis. Whereas only a small number of the cells (∼3%) were found to form bulges at 37°C, the number of bulging cells increases about 3-fold at 42°C (Fig. 2B). To check if the bulges contain cytoplasmic materials, green fluorescent protein (GFP) was constitutively expressed in the cytoplasm. The fluorescent protein was clearly visible within the bulges and then scattered in the environment upon lysis (Fig. 2D). This result strongly suggests that bulge formation involves both the IM and the OM and indicates that the bulges contain cytoplasmic materials.
FIG 2.

Loss of YciM causes morphological defects. (A) Wild-type and yciM strains grown at 37°C to exponential phase were placed on a 2% LB agarose pad. Phase-contrast images of 300 random cells were recorded, and the MicrobeTracker open-source software (19) was used to determine the length of individual cells. (B) yciM mutants were placed on a microscopy slide layered with a 1% agar pad containing LB medium. Bacteria were grown for 3 h at 42°C, and pictures were taken every 3 min using DIC. Bulges appear after about 60 min and are indicated by the black arrows. A enlarged image is shown in the lower part of the panel. (C) Phase-contrast images of wild-type (lower row) and yciM strains (upper row) grown on salt-free LB-agar medium at 42°C. yciM mutant cells have an altered morphology and lyse after a few divisions, whereas growth of the wild-type strain is only slightly affected. (D) DIC and fluorescence image (GFP) of strain HE89 (yciM mutant constitutively expressing cytoplasmic GFP from pFPV25) grown at 42°C. The displayed sequential images were taken 51 min after the shift from 37°C to 42°C. The presence of GFP in the bulges indicates that they involve both the IM and the OM and contain cytoplasmic materials. The cells numbered 1 and 2 lyse following bulge formation.
We also explored the effects of various salt concentrations on the growth and morphology of the yciM mutant at the single-cell level. On salt-free LB agar at 42°C (Fig. 2C), we observed that yciM mutant cells have irregular shapes and lyse after a few cell divisions (Fig. 2C). When grown on LB with high salt concentrations at 37°C, cells lacking yciM also rapidly lyse, sometimes following bulge formation (not shown).
yciM envC null alleles exhibit a synthetic phenotype.
The phenotypes of the yciM mutant described above are consistent with defects in the envelope, suggesting that loss of YciM alters envelope integrity. To obtain more information on the envelope assembly process specifically impacted by yciM deletion, we investigated whether yciM exhibits genetic synthetic interactions with null alleles of genes involved in envelope biogenesis. We focused on genes involved in LPS biosynthesis (rfaQ, rfaP, asmA, and rfaI) and peptidoglycan assembly (prc, envC, mrcA, ponB, pbpC, and nlpD) because the increased sensitivity to antibiotics and lysis reported above could result from defects in either pathway. Out of the 10 alleles tested, we found that only the inactivation of envC in the yciM mutant results in a synthetic phenotype. Initially, we were unable to introduce the ΔenvC::kan allele using P1 transduction by directly selecting for kanamycin resistance, suggesting that these alleles might be a synthetic lethal pair. However, we could generate the ΔyciM ΔenvC::kan double mutant by cotransducing at the expected ∼35% frequency the ΔenvC::kan allele with a nearby zib563::Tn10 allele when we selected for tetracycline-resistant transductants. This indicates that yciM and envC are not synthetic lethal. However, as shown in Fig. S1 in the supplemental material, yciM envC mutant cells exhibit aberrant morphology and often lyse, suggesting that they have severe cell envelope defects.
As the function of EnvC is to activate AmiA and AmiB (12), two peptidoglycan amidases that cooperate in septal peptidoglycan splitting, we tested whether yciM also genetically interacts with the genes coding for these two hydrolytic enzymes. We could construct the yciM amiA and yciM amiB double mutants. However, we were unable to construct the yciM amiA amiB triple mutant by selecting for kanamycin-resistant ΔamiA::kan transductants using a yciM amiB strain as recipient. Because yciM and envC are not synthetic lethal, the inability to construct this yciM amiA amiB triple mutant strain is likely caused by the same unknown reason for which we cannot select for kanamycin-resistant ΔenvC::kan transductants in a yciM mutant. Together, these phenotypes suggest that YciM is connected to peptidoglycan homeostasis (see Discussion).
YciM localizes to the IM.
YciM is a 44.5-kDa protein (Fig. 3A) which is annotated in the databases as a protein secreted to the periplasm. YciM presents, indeed, a highly hydrophobic N-terminal domain predicted to be a signal sequence by several computational methods such as Psort and SignalP (27). However, most periplasmic targeting sequences consist of a short, positively charged amino-terminal segment, followed by a longer hydrophobic domain (28). It is the hydrophobic character of this domain that is usually conserved, rather than the amino acid sequence. The sequence of the N-terminal domain of YciM does not fit well with that description: no positively charged residue is present among the first 20 amino acids. Moreover, although the sequence of the predicted export signal is hydrophobic, it is highly conserved among YciM homologues (Fig. 3B). These observations suggest that, instead of corresponding to a signal sequence that is released from the protein after export, the N-terminal segment of YciM could serve as a transmembrane α-helix anchoring the protein in the IM. We expressed both the full-length protein and a truncated version lacking the first 22 amino acids (YciM*) and studied their subcellular localizations. The wild-type protein was present in the membrane fraction, whereas YciM* was found in the soluble fraction (Fig. 4A). In a parallel approach, we fused the C terminus of YciM to the fluorescent protein mCherry. As shown in Fig. 4B, YciMmCherry localizes in the membrane while YciM*mCherry localizes in the cytoplasm. Thus, YciM is a membrane protein that is anchored in the IM by its N-terminal sequence. Interestingly, expression of YciM* is unable to rescue growth of the yciM mutant on MacConkey agar at 22°C (Fig. 1A), indicating that the N-terminal domain that anchors YciM to the membrane is required for its activity.
FIG 3.

Structural features of the YciM sequence. (A) YciM is a multidomain protein: the N-terminal part (transmembrane, TM) anchors the protein in the IM, the central part of the protein presents several TPR motifs that most likely mediate protein-protein interactions, and the C terminus presents two CXXC motifs that bind iron (Fe). (B) Multiple sequence alignment of E. coli YciM with homologous proteins from other Gram-negative bacteria from the Alpha-, Beta-, Gamma-, and Deltaproteobacteria. The N-terminal domain (underlined with the dashed line) is hydrophobic and highly conserved. All YciM proteins contain seven TPR motifs (marked from 1 to 7). The conserved iron-binding CXXC motifs are present at the C terminus (cysteine residues are marked with arrows). The sequences of Escherichia coli (UniProtKB accession no. P0AB58), Salmonella enterica (B5PPH9), Haemophilus influenzae (Q4QJV0), Colwellia psychrerythraea (Q482G0), Ralstonia eutropha (Q0KDH3), Cupriavidus taiwanensis (B3R3B7), Plesiocystis pacifica (A6G5X1), and Ehrlichia canis (Q3YT24) were aligned using Clustal W2.
FIG 4.
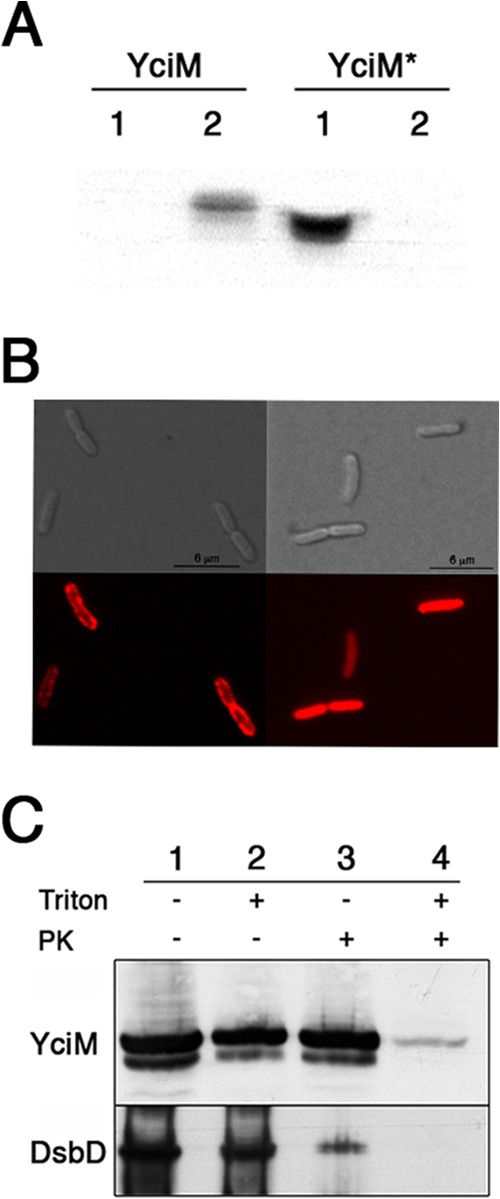
YciM is an inner membrane protein. (A)E. coli cells expressing YciM (VN241) or YciM* (VN198) were fractionated. YciM and YciM* were detected using an anti-YciM antibody (lane 1, soluble fraction; lane 2, membrane fraction). (B) DIC images (upper panel) and the corresponding fluorescence images (lower panel) of HE64 (expressing YciMmCherry) and VN237 (expressing YciM*mCherry) strains. YciMmCherry localizes in the membrane (left panel), whereas YciM*mCherry is present in the cytoplasm (right panel). Images were taken 30 min after the induction of the fluorescent proteins by addition of 0.2% l-arabinose. (C) Spheroplasts prepared from bacteria expressing YciM (VN241) were solubilized or not with Triton X-100 (1%) and then incubated with proteinase K (PK) as indicated. YciM was detected by Western blotting using an anti-YciM antibody. The fact that YciM is not degraded by proteinase K in the absence of Triton X-100 indicates that YciM is oriented toward the cytoplasm. DsbD, an inner membrane protein, was used as a control. DsbD was detected using an anti-DsbD antibody.
The presence of two arginine residues (Arg21 and Arg22) located immediately after the transmembrane α-helix suggests that the protein is oriented toward the cytoplasm (28). To confirm this topological orientation of YciM, spheroplasts were prepared from cells expressing YciM and incubated with proteinase K. As shown in Fig. 4C, YciM becomes accessible to the protease only when the membrane is solubilized with Triton X-100, thus confirming the cytoplasmic orientation of the protein.
YciM is a metal-binding protein.
There are two CXXC motifs at the C terminus of YciM (Fig. 3B). BLAST searches using the carboxy-terminal part of YciM revealed that this region is conserved in all YciM homologues (Fig. 3B) and aligns with the zinc-binding cysteine residues of the DNA repair protein RadA (not shown) (29). To test whether the CXXC motifs of YciM are also involved in zinc binding, we prepared a C-terminally His-tagged version of YciM* (YciM*His) to purify the protein by affinity chromatography. The fractions containing YciM*His were then successively loaded on a Q-Sepharose anion-exchange chromatography column and a Superdex S200 gel filtration column. After these steps, the protein was pure to near homogeneity (one minor contaminant, identified as the chaperone DnaK, was also present in the YciM-containing fractions) (see Fig. S2 in the supplemental material). The purified protein was then used to study the metal content of YciM. YciM*His was separated from free metals by gel filtration against metal-free buffer, and the metal content was analyzed by inductively coupled plasma-high-resolution mass spectrometry (ICP-HRMS). We found that zinc is present in the protein in a 1:1 molar stoichiometry (1.1 zinc/protein). No other metals were detected in the protein preparation.
Cysteines and histidines are frequently involved in zinc coordination. YciM contains six cysteine residues, including four in the carboxy-terminal region, and eight histidine residues. To determine if and how many cysteine residues are involved in zinc binding, we used the PAR/PMPS assay (16, 17). PAR is a dye that chelates zinc to form a red Zn(PAR)2 complex. This complex absorbs at 500 nm, and its formation can therefore be easily monitored. However, because the affinity of PAR for zinc (affinity constant [Ka], 2 × 1012 M−1) is lower than the affinity of most protein zinc centers (usually >1014 M−1), tightly bound zinc cannot be extracted by PAR, and thiol-modifying reagents such as PMPS are used.
When freshly purified YciM was incubated with PAR, we did not observe a significant increase in the A500, indicating that PAR was unable to extract the zinc from the protein. Stepwise additions of PMPS led to a progressive increase in the A500 (see Fig. S3 in the supplemental material), and zinc was fully released when four cysteine residues were titrated. This indicates that zinc is coordinated to YciM via four cysteine residues.
To determine whether the zinc-binding cysteine residues correspond to the four cysteines of the CXXC motifs, we purified a mutant version of YciM* in which the two cysteine residues outside the CXXC motifs (Cys184 and Cys258) were replaced by serines (YciM*SS/CCCC−His). ICP-HRMS analysis showed that YciM*SS/CCCC−His contains zinc in a 1:1 ratio, indicating that zinc is bound to YciM via the cysteines of the two CXXC motifs. This is consistent with the fact that Cys184 and Cys258 are not highly conserved (Fig. 3B), in contrast to the cysteine residues of the CXXC motifs.
YciM contains an iron cluster.
In the course of the project, we serendipitously purified YciM on a Strep-Tactin column by taking advantage of a Strep tag fused at the C terminus of the protein instead of a His tag. Surprisingly, the protein that eluted from the Strep-Tactin column, which was pure to near homogeneity, appeared bright pink, suggesting that it might bind iron (see Fig. S4 in the supplemental material). Whereas no peaks were observed in the visible range for YciM*His, the absorbance spectrum of purified YciM*Strep (Fig. 5A) shows a large peak around 500 nm and a smaller, narrow peak around 410 nm. We determined the metal content of YciM*Strep and found that both iron and zinc were present in 0.11 and 0.44 molar ratios, respectively. We tested whether Cys184 and Cys258 are involved in iron binding by expressing YciM*SS/CCCC fused to a Strep tag (YciM*SS/CCCC−Strep). However, replacement of Cys184 and Cys258 by serine residues does not alter the pink appearance of the protein and has no impact on the metal content, indicating that iron is also bound to YciM via the two C-terminal CXXC motifs. We confirmed the results obtained on YciM*Strep by purifying a nontagged version of the protein (YciM*). After purification of YciM* to homogeneity using ion exchange and size exclusion chromatography, the protein was found to contain iron and zinc in a ratio similar to that of YciM*Strep. Thus, the CXXC motifs of YciM are able to bind iron and/or zinc. In the Discussion, we explain why we think that iron, not zinc, is the metal which is coordinated by YciM in vivo. From now on, we will refer to YciM as an iron-binding protein. Because iron-binding centers are usually sensitive to oxygen, we also purified YciM*Strep anaerobically to see whether purification of the protein in the absence of oxygen would increase the iron-to-zinc ratio, which was not observed. Interestingly, while YciM is a slightly yellow protein under anaerobic conditions, exposure to oxygen turns it into a pink protein displaying the typical absorbance peak around 500 nm (Fig. 5B). This indicates that the iron center of YciM is redox sensitive.
FIG 5.
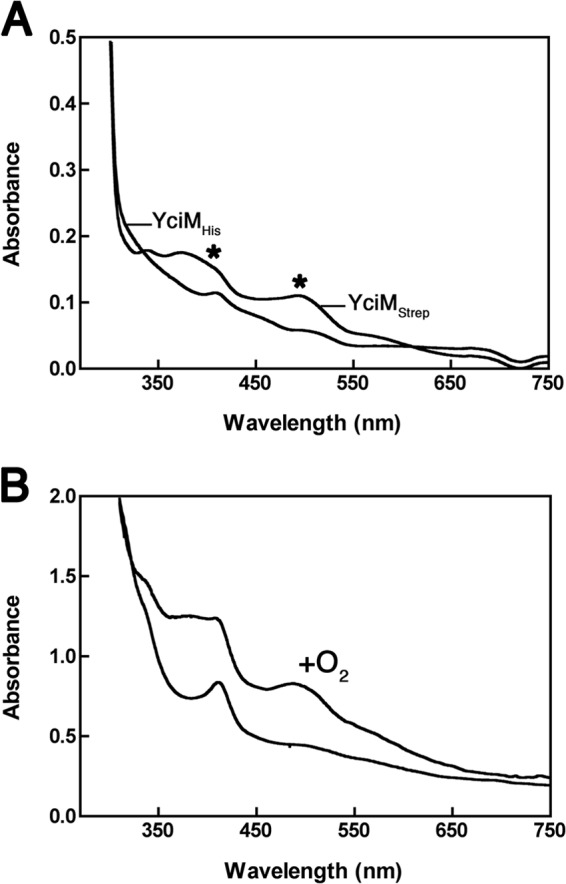
YciM is a metal binding protein. (A) UV-visible light absorption spectra of purified YciM*Strep and YciM*His (150 μM). The absorbance was measured between 250 nm and 750 nm. The spectrum of YciM*Strep shows a large peak around 500 nm and a smaller one around 410 nm (marked with asterisks). (B) YciM*Strep was purified on a Strep-Tactin column under anaerobic conditions. The protein that eluted from the column was yellow, and its absorbance spectrum presented a peak at 410 nm (lower curve). Exposure of the protein to oxygen turned it pink, presenting an absorbance peak around 500 nm (upper curve, + O2). The protein concentration was about 250 μM.
YciM has an iron center of the rubredoxin type which is required for protein stability.
CXXC motifs have been shown to be involved both in the coordination of iron-sulfur clusters and in the coordination of iron centers of the rubredoxin type (30). Analysis of the sulfur content of YciM by a colorimetric assay revealed that YciM does not contain any inorganic sulfur (data not shown), indicating that YciM binds an iron center of the rubredoxin type. This is further supported by the fact that the absorbance spectrum of YciM*Strep resembles that of other rubredoxin proteins (31, 32). We produced a mutant version of YciM in which the four cysteines of the CXXC motifs are replaced by serines (YciMCC/SSSS). This mutant mostly accumulates in inclusion bodies (data not shown), indicating that the metal center is required for the stability of the protein. However, as a significant portion of YciMCC/SSSS still localizes to the membrane, we tested whether it can rescue the growth defect of the yciM mutant on either MacConkey agar at 22°C or salt-free LB at 42°C. As shown in Fig. 1A and Table 2, we observed that the fraction of YciMCC/SSSS present in the membrane is unable to complement the yciM mutant, suggesting that the redox-sensitive iron center of YciM is important for YciM function.
DISCUSSION
In this study, we have demonstrated that YciM, a protein required for maintenance of cell envelope integrity in E. coli, contains two domains essential for function: an N-terminal transmembrane domain that anchors YciM to the IM and a C-terminal cytoplasmic domain that contains seven tetratricopeptide repeat (TPR) motifs and an iron cluster required for function. We have also described phenotypes associated with the loss of YciM that reveal both OM and peptidoglycan defects. Furthermore, its cellular localization and structural features suggest that YciM is likely to function in envelope biogenesis in association with other proteins that are yet to be identified.
YciM is anchored to the IM by a hydrophobic N-terminal domain that contains residues highly conserved among bacterial species from the Alpha-, Beta-, Gamma-, and Deltaproteobacteria (Fig. 3B). This high degree of conservation and the fact that this domain is required for YciM function suggest that it might mediate interactions between YciM and other polypeptides present in the IM. YciM also contains seven TPR motifs localized to the cytoplasm (Fig. 3B). TPR motifs serve in a variety of protein-protein interactions (33), which further suggests that YciM is involved in a multiprotein complex. Future studies targeting the identification of putative interacting partners may reveal the function that YciM plays in envelope biogenesis.
Another structural feature of the C-terminal portion of YciM is a metal center coordinated by four cysteine residues. Although we first identified zinc as the metal coordinated by the two CXXC motifs present in YciM, the protein also binds iron when the His tag is removed. Our results suggest that the His tag, which is located immediately downstream of the metal-chelating cysteines, interferes with the stability of the iron center in YciM, leading to the replacement of iron by zinc. The low iron content of YciM*Strep and YciM* very likely results from the notoriously low stability of iron centers, which often leads to the replacement of iron by zinc during protein overexpression and purification (34–36). Importantly, we have demonstrated that this iron center is essential for YciM function although whether it plays a structural or redox-related role remains unknown.
The sensitivity of yciM mutants to hydrophobic antibiotics and detergents results from an increased permeability of the OM, suggesting a defect in OM biogenesis (9). However, we have also uncovered phenotypes indicating that yciM mutants have defects in peptidoglycan biogenesis. First, the fact that yciM mutants have irregular shapes and lyse following bulge formation supports the presence of defects in the peptidoglycan layer (37). It has indeed been shown that bulge formation results from a defective peptidoglycan assembly leading to the formation of a weakened area in the peptidoglycan layer. For instance, treatment of E. coli with antibiotics that inhibit peptidoglycan synthesis, such as β-lactams, as well as overexpression of inactive variants of enzymes involved in peptidoglycan assembly causes bulge-induced lysis (38–40). Second, the sensitivity of the yciM deletion mutants to low-osmolarity medium is also consistent with defects in the peptidoglycan as a similar phenotype has been reported for mutants impaired in peptidoglycan assembly and/or cell division (41, 42). For instance, cells lacking FtsEX, an IM complex that recruits EnvC to the septum, also display a division defect in LB medium of low osmotic strength (43). Third, the facts that the simultaneous deletion of yciM and envC, a gene coding for a regulator of peptidoglycan hydrolysis (44, 45), leads to severe morphological defects and that yciM mutants are more sensitive to β-lactams (46) also support a role for YciM in peptidoglycan assembly. Nevertheless, although these phenotypes indicate that the loss of YciM leads to defects in peptidoglycan, we have also observed the aforementioned permeability phenotypes that are consistent with OM defects. In Gram-negative bacteria, the OM and peptidoglycan layer are intimately associated. For example, the Lpp OM lipoprotein covalently anchors the OM to the peptidoglycan layer (47, 48), while the OM lipoproteins LpoA and LpoB are needed for the proper synthesis of the peptidoglycan layer (49, 50). Consequently, defects in the biogenesis of one can affect the biogenesis of the other. Because the present studies cannot discriminate which of the reported defects are the primary consequence of the loss of YciM, additional work is required to understand the role that YciM plays in envelope biogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Asma Boujtat and Geneviève Connerotte for their excellent technical assistance, William Cenens (Katholieke Universiteit Leuven, Leuven, Belgium), Michael Deghelt (University of Namur, Namur, Belgium), and Emily K. Butler (The Ohio State University, USA) for the microscopy experiments, and Sander Govers (Katholieke Universiteit Leuven, Leuven, Belgium) for the cell length analysis. We also thank P. Rolf Thauer and Seigo Shima (Max-Planck Institute for Terrestrial Microbiology, Marburg, Germany) for their help in the anaerobic purification of YciM as well as Tricia Kiley, Benjamin Ezraty, and Frederic Barras for insightful discussions regarding YciM.
J.F.C. is Maître de Recherche of the FRS-FNRS. V.N. and M.D. are research fellows of the FRIA. P.L. is Chargée de Recherche of the FRS-FNRS. This work was supported by grants from the FRS-FNRS to J.-F.C. and the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM100951 (to N.R.), and start-up funds from The Ohio State University (to N.R.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00921-13.
REFERENCES
- 1.Gould IM. 2008. The epidemiology of antibiotic resistance. Int. J. Antimicrob. Agents 32(Suppl 1):S2–S9. 10.1016/j.ijantimicag.2008.06.016 [DOI] [PubMed] [Google Scholar]
- 2.Ruiz N, Kahne D, Silhavy TJ. 2006. Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4:57–66. 10.1038/nrmicro1322 [DOI] [PubMed] [Google Scholar]
- 3.Leverrier P, Vertommen D, Collet JF. 2010. Contribution of proteomics toward solving the fascinating mysteries of the biogenesis of the envelope of Escherichia coli. Proteomics 10:771–784. 10.1002/pmic.200900461 [DOI] [PubMed] [Google Scholar]
- 4.Van Wielink JE, Duine JA. 1990. How big is the periplasmic space? Trends Biochem. Sci. 15:136–137. 10.1016/0968-0004(90)90208-S [DOI] [PubMed] [Google Scholar]
- 5.Knowles TJ, Scott-Tucker A, Overduin M, Henderson IR. 2009. Membrane protein architects: the role of the BAM complex in outer membrane protein assembly. Nat. Rev. Microbiol. 7:206–214. 10.1038/nrmicro2069 [DOI] [PubMed] [Google Scholar]
- 6.Ricci DP, Silhavy TJ. 2012. The Bam machine: a molecular cooper. Biochim. Biophys. Acta 1818:1067–1084. 10.1016/j.bbamem.2011.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz N, Kahne D, Silhavy TJ. 2009. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 7:677–683. 10.1038/nrmicro2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rigel NW, Silhavy TJ. 2012. Making a beta-barrel: assembly of outer membrane proteins in Gram-negative bacteria. Curr. Opin. Microbiol. 15:189–193. 10.1016/j.mib.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317. 10.1016/j.cell.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 10.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA. 2006. Regulon and promoter analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes Dev. 20:1776–1789. 10.1101/gad.1428206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uehara T, Parzych KR, Dinh T, Bernhardt TG. 2010. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 29:1412–1422. 10.1038/emboj.2010.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bremer E, Silhavy TJ, Weisemann JM, Weinstock GM. 1984. Lambda placMu: a transposable derivative of bacteriophage lambda for creating lacZ protein fusions in a single step. J. Bacteriol. 158:1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 15.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 16.Collet JF, D'Souza JC, Jakob U, Bardwell JC. 2003. Thioredoxin 2, an oxidative stress-induced protein, contains a high affinity zinc binding site. J. Biol. Chem. 278:45325–45332. 10.1074/jbc.M307818200 [DOI] [PubMed] [Google Scholar]
- 17.Jakob U, Eser M, Bardwell JC. 2000. Redox switch of hsp33 has a novel zinc-binding motif. J. Biol. Chem. 275:38302–38310. 10.1074/jbc.M005957200 [DOI] [PubMed] [Google Scholar]
- 18.Beinert H. 1983. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131:373–378. 10.1016/0003-2697(83)90186-0 [DOI] [PubMed] [Google Scholar]
- 19.Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80:612–627. 10.1111/j.1365-2958.2011.07579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700. 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagan CL, Silhavy TJ, Kahne D. 2011. beta-Barrel membrane protein assembly by the Bam complex. Annu. Rev. Biochem. 80:189–210. 10.1146/annurev-biochem-061408-144611 [DOI] [PubMed] [Google Scholar]
- 22.Narita S, Masui C, Suzuki T, Dohmae N, Akiyama Y. 2013. Protease homolog BepA (YfgC) promotes assembly and degradation of beta-barrel membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 110:E3612–3621. 10.1073/pnas.1312012110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179–207. 10.1016/0022-2836(80)90283-1 [DOI] [PubMed] [Google Scholar]
- 24.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541–555. 10.1016/0022-2836(76)90119-4 [DOI] [PubMed] [Google Scholar]
- 25.Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BO. 2009. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 27:1043–1049. 10.1038/nbt.1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. 2005. Global topology analysis of the Escherichia coli inner membrane proteome. Science 308:1321–1323. 10.1126/science.1109730 [DOI] [PubMed] [Google Scholar]
- 27.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8:581–599. 10.1142/S0129065797000537 [DOI] [PubMed] [Google Scholar]
- 28.von Heijne G, Gavel Y. 1988. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 174:671–678. 10.1111/j.1432-1033.1988.tb14150.x [DOI] [PubMed] [Google Scholar]
- 29.Beam CE, Saveson CJ, Lovett ST. 2002. Role for radA/sms in recombination intermediate processing in Escherichia coli. J. Bacteriol. 184:6836–6844. 10.1128/JB.184.24.6836-6844.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lovenberg W, Sobel BE. 1965. Rubredoxin: a new electron transfer protein from Clostridium pasteurianum. Proc. Natl. Acad. Sci. U. S. A. 54:193–199. 10.1073/pnas.54.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee HJ, Lian LY, Scrutton NS. 1997. Recombinant two-iron rubredoxin of Pseudomonas oleovorans: overexpression, purification and characterization by optical, CD and 113Cd NMR spectroscopies. Biochem. J. 328:131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng P, Takayama SJ, Mauk AG, Li H. 2012. Hydrogen bond strength modulates the mechanical strength of ferric-thiolate bonds in rubredoxin. J. Am. Chem. Soc. 134:4124–4131. 10.1021/ja2078812 [DOI] [PubMed] [Google Scholar]
- 33.Blatch GL, Lassle M. 1999. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21:932–939. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Oganesyan N, Shin DH, Jancarik J, Yokota H, Kim R, Kim SH. 2005. Structural characterization of an iron-sulfur cluster assembly protein IscU in a zinc-bound form. Proteins 59:875–881. 10.1002/prot.20421 [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Kasciukovic T, White MF. 2012. The CRISPR associated protein Cas4 is a 5′ to 3′ DNA exonuclease with an iron-sulfur cluster. PLoS One 7:e47232. 10.1371/journal.pone.0047232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramelot TA, Cort JR, Goldsmith-Fischman S, Kornhaber GJ, Xiao R, Shastry R, Acton TB, Honig B, Montelione GT, Kennedy MA. 2004. Solution NMR structure of the iron-sulfur cluster assembly protein U (IscU) with zinc bound at the active site. J. Mol. Biol. 344:567–583. 10.1016/j.jmb.2004.08.038 [DOI] [PubMed] [Google Scholar]
- 37.Huang KC, Mukhopadhyay R, Wen B, Gitai Z, Wingreen NS. 2008. Cell shape and cell-wall organization in Gram-negative bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:19282–19287. 10.1073/pnas.0805309105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burdett ID, Murray RG. 1974. Septum formation in Escherichia coli: characterization of septal structure and the effects of antibiotics on cell division. J. Bacteriol. 119:303–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meisel U, Holtje JV, Vollmer W. 2003. Overproduction of inactive variants of the murein synthase PBP1B causes lysis in Escherichia coli. J. Bacteriol. 185:5342–5348. 10.1128/JB.185.18.5342-5348.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Z, Kahne D, Kishony R. 2012. Distinct single-cell morphological dynamics under beta-lactam antibiotics. Mol. Cell 48:705–712. 10.1016/j.molcel.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hara H, Yamamoto Y, Higashitani A, Suzuki H, Nishimura Y. 1991. Cloning, mapping, and characterization of the Escherichia coli prc gene, which is involved in C-terminal processing of penicillin-binding protein 3. J. Bacteriol. 173:4799–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohara M, Wu HC, Sankaran K, Rick PD. 1999. Identification and characterization of a new lipoprotein, NlpI, in Escherichia coli K-12. J. Bacteriol. 181:4318–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt KL, Peterson ND, Kustusch RJ, Wissel MC, Graham B, Phillips GJ, Weiss DS. 2004. A predicted ABC transporter, FtsEX, is needed for cell division in Escherichia coli. J. Bacteriol. 186:785–793. 10.1128/JB.186.3.785-793.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernhardt TG, de Boer PA. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 52:1255–1269. 10.1111/j.1365-2958.2004.04063.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters NT, Morlot C, Yang DC, Uehara T, Vernet T, Bernhardt TG. 2013. Structure-function analysis of the LytM domain of EnvC, an activator of cell wall remodelling at the Escherichia coli division site. Mol. Microbiol. 89:690–701. 10.1111/mmi.12304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamae C, Liu A, Kim K, Sitz D, Hong J, Becket E, Bui A, Solaimani P, Tran KP, Yang H, Miller JH. 2008. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol. 190:5981–5988. 10.1128/JB.01982-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braun V, Sieglin U. 1970. The covalent murein-lipoprotein structure of the Escherichia coli cell wall. The attachment site of the lipoprotein on the murein. Eur. J. Biochem. 13:336–346 [DOI] [PubMed] [Google Scholar]
- 48.Braun V, Wolff H. 1970. The murein-lipoprotein linkage in the cell wall of Escherichia coli. Eur. J. Biochem. 14:387–391. 10.1111/j.1432-1033.1970.tb00301.x [DOI] [PubMed] [Google Scholar]
- 49.Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. 2010. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143:1110–1120. 10.1016/j.cell.2010.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M, Breukink E, den Blaauwen T, Gross CA, Vollmer W. 2010. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143:1097–1109. 10.1016/j.cell.2010.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.