

Abstract
The minichromosome maintenance (MCM) complex, which plays multiple important roles in DNA replication, is loaded onto chromatin following mitosis, remains on chromatin until the completion of DNA synthesis, and then is unloaded by a poorly defined mechanism that involves the MCM binding protein (MCM-BP). Here we show that MCM-BP directly interacts with the ubiquitin-specific protease USP7, that this interaction occurs predominantly on chromatin, and that MCM-BP can tether USP7 to MCM proteins. Detailed biochemical and structure analyses of the USP7–MCM-BP interaction showed that the 155PSTS158 MCM-BP sequence mediates critical interactions with the TRAF domain binding pocket of USP7. Analysis of the effects of USP7 knockout on DNA replication revealed that lack of USP7 results in slowed progression through late S phase without globally affecting the fork rate or origin usage. Lack of USP7 also resulted in increased levels of MCM proteins on chromatin, and investigation of the cause of this increase revealed a defect in the dissociation of MCM proteins from chromatin in mid- to late S phase. This role of USP7 mirrors the previously described role for MCM-BP in MCM complex unloading and suggests that USP7 works with MCM-BP to unload MCM complexes from chromatin at the end of S phase.
INTRODUCTION
The faithful replication of eukaryotic genomes relies on the complex interplay of multiple factors. Starting with the assembly of the prereplicative complex (pre-RC) at replication origins in G1, the coordinated loading of replication factors onto DNA and origin activation at the start of S phase result in the bidirectional progression of the replication machinery (replisome) to carry out DNA synthesis. The minichromosome maintenance (MCM) complex is a hexameric complex consisting of MCMs 2 to 7 (MCM2-7) and is an essential component of the pre-RC as well as the replisome (1). It is loaded onto DNA as double heterohexamers in G1 phase through the concerted actions of Cdc6, Cdt1, and ORC (together, the pre-RC) (1). In eukaryotes, the MCM complex is loaded in an ∼20-fold excess over the number of replication origins, with only a fraction being required for replication (2–7). At the start of S phase, phosphorylation events mediated by S-CDK (S-phase cyclin-dependent kinase) and DDK (Dbf4-dependent kinase) result in the association of the MCM complex with Cdc45 and the GINS complex (Psf1-3 and Sld5) forming an active “CMG” helicase (8–10). However, only ∼10% of loaded MCM double hexamers form active CMG helicases, and the remaining “inactive” MCM complexes are thought to license dormant origins (11). During replication, these “inactive” MCM complexes pose a barrier to replication and must be removed ahead of the replication fork. While Pif1 family helicases have been implicated in this process (12–14), a mechanism for MCM complex unloading from dormant origins and from terminating replisomes at the end of S phase is not well characterized.
Proteomics experiments performed with MCM proteins in human cells revealed a strong interaction with a previously uncharacterized protein, MCM-BP (MCM binding protein), which is conserved in most eukaryotes (with the exception of budding yeast and Caenorhabditis elegans) (15). In human cells, we have shown that MCM-BP depletion results in altered nuclear morphology and replication stress, resulting in G2 checkpoint activation (16). Similarly, inactivation or loss of the MCM-BP homologues in Schizosaccharomyces pombe (Mcb1) and Arabidopsis thaliana (ETG1) resulted in defects in DNA replication and G2 checkpoint activation (17–20). MCM-BP may contribute to DNA replication in multiple ways. For example, studies in S. pombe support a role for MCM-BP in pre-RC formation (21), and the interaction of human MCM-BP with Dbf4 also suggests a role for MCM-BP at the onset of S phase (22). In addition, a role for MCM-BP in unloading MCM complexes from chromatin at the end of S phase was identified using the Xenopus egg extract system, where MCM-BP depletion resulted in increased levels of MCM proteins on chromatin in mid- to late S phase and MCM-BP was shown to disrupt MCM complexes (23). Similar observations were subsequently made for MCM-BP in human cells (16, 22, 23). However, the mechanism by which MCM-BP regulates MCM complex unloading is unclear.
A putative interaction between MCM-BP (also called C10orf119) and the deubiquitylating enzyme USP7 was identified from a proteomic screen of ∼100 deubiquitylating enzymes in human cells (24). USP7 (also called HAUSP) was originally discovered as a binding partner of the ICP0 protein of herpes simplex virus and was subsequently shown to be targeted by several viral proteins from different DNA viruses (25–31). USP7 has multiple roles in the cell that impact oncogenesis in a variety of ways (32). For example, USP7 regulates the p53 pathway by binding either p53, MDM2, or MDMX through its N-terminal TRAF domain and cleaving polyubiquitin chains from them with its central catalytic domain (33–38). In addition to cleaving polyubiquitin chains, USP7 reverses the monoubiquitylation of FOXO4 and PTEN, thereby regulating their localization (39, 40). Similarly, USP7, in complex with GMP synthetase, removes monoubiquitin from histone H2B, impacting gene expression (41, 42). Finally, USP7 can also affect cellular processes independent of its catalytic activity, since its role in regulating the levels of promyelocytic leukemia (PML) proteins and nuclear bodies was shown to involve only its protein interaction domains (43).
In this work, we investigated the interaction between MCM-BP and USP7 and its functional significance. We showed that MCM-BP and USP7 interact directly, and we defined the mechanism of this interaction. We also demonstrated that USP7 functions in DNA replication, and we present evidence that USP7 acts in unloading MCM complexes from chromatin in late S phase in conjunction with MCM-BP.
MATERIALS AND METHODS
Cell lines.
HeLa cells were maintained in Dulbecco's minimal essential medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS). The colon carcinoma HCT116 cell line was obtained from Daniel Durocher and maintained in alpha minimal essential media (Gibco) supplemented with 10% fetal bovine serum. USP7-null HCT116 cells were obtained from Bert Vogelstein and cultured as for HCT116 cells.
RNAi and plasmids.
For RNA interference (RNAi) against USP7, we synthesized a previously characterized small interfering RNA (siRNA) targeting USP7 (5′CCCAAATTATTCCGCGGCAAA3′) using Sigma Genosys (44). AllStars negative-control siRNA was obtained from Qiagen. MCM-BP was depleted using short hairpin RNA (shRNA) (corresponding to shMCM-BP1 in the work of Jagannathan et al. [16]) expressed from pRS, which contains a puromycin resistance cassette. For experiments involving MCM-BP transient expression, we first generated pCMV3FC by excising the yellow fluorescent protein (YFP) tag from pEYFP-N1 (Clontech) with BamHI and NotI and replacing it with a triple FLAG tag. MCM-BP was PCR amplified from pMZS3F MCM-BP (16) and subcloned between the SalI and XbaI sites in pCMV3FC. To generate the MCM-BP S158A point mutant, PCR-mediated site-directed mutagenesis was performed on MCM-BP in pCMV3FC using the primer 5′ GTCAGTCCCTCAACAGCCTACACTCCTAGTC 3′.
Antibodies.
Antibodies used in this work targeted MCM2 (9839; Santa Cruz), MCM3 (9850; Santa Cruz), MCM4 (22779; Santa Cruz), MCM5 (165993; Santa Cruz), MCM6 (9843; Santa Cruz), MCM7 (22782; Santa Cruz), Flag M2 (Sigma), ECS/DDDDK (A190-102A; Bethyl), bromodeoxyuridine (BrdU)-fluorescein isothiocyanate (FITC) (556028; BD Pharmingen), actin (1616; Santa Cruz), USP7 (A300-033A; Bethyl), Psf2 (16247-1-AP; Proteintech), histone H3 (A300-823A; Bethyl), MEK2 (A302-141A; Bethyl), and MCM-BP (A303-477A; Bethyl). The antibody used to immunoprecipitate MCM-BP has been described previously (15), and FLAG-tagged proteins were immunoprecipitated using FLAG M2 resin (Sigma). All secondary antibodies were obtained from Santa Cruz Biotechnology.
Transfections.
Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For USP7 silencing, 0.25 × 106 HeLa cells were transfected with 100 pmol Allstars negative-control or USP7-targetting siRNA at time of plating and every 24 h subsequently for a total of 4 separate transfections. Cells were harvested by trypsinization at 144 h after initial transfection. For MCM-BP silencing, 0.5 × 106 to 1.0 × 106 HeLa cells were transfected with 6 μg pRS plasmid expressing shMCM-BP1 (or empty pRS as a negative control). Forty-eight hours later, puromycin was added to a final concentration of 2.5 μg/ml, and cells were grown under selection for 3 days prior to harvesting. For transient expression of MCM-BP and MCM-BP S158A, 1.0 × 106 HeLa cells were transfected with 6 μg of plasmid and harvested 48 h later.
Cell fractionation.
Transfected cells were fractionated into soluble and chromatin-bound fractions as described previously (15, 16). Briefly, 2 × 106 cells were resuspended in 150 μl hypotonic lysis buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, and 0.1% Triton X-100), and after 15 min on ice, the lysate was subjected to centrifugation at 1,300 × g for 5 min. The supernatant comprising the soluble protein fraction was removed, and the pellet was washed with the same buffer. Chromatin-associated proteins were released from the pellet using either 150 μl buffer with micrococcal nuclease (Sigma) (0.2 U/ml for 1 min at 37°C) or 150 μl protein sample buffer. Thirty micrograms of the soluble fraction from each sample was analyzed by Western blotting. For the chromatin-associated fraction of each sample, a volume equivalent to that used for the soluble fraction was analyzed by Western blotting. Total cell extracts were prepared by sonicating cells in 9 M urea in 50 mM Tris (pH 6.8) for 15 s at 50% amplitude, and 30 μg of each sample was analyzed by Western blotting.
Coimmunoprecipitation.
For immunoprecipitation (IP) from whole-cell extracts, HeLa cells were lysed with 6 volumes of 50 mM Tris, pH 8.0, 150 mM NaCl, 5% glycerol, 2 mM EDTA, and 0.1% Triton X-100 on ice for 30 to 60 min, sonicated at low amplitude for 10 s, and clarified by centrifugation at 15,000 rpm for 10 min. The lysate (1 mg) was incubated with specific antibody against MCM-BP (described in reference 16) overnight at 4°C. Thirty microliters protein A/G-agarose (Sc-2003; Santa Cruz Biotechnology Inc.) was then added for 2 h, and the resin was washed three times with lysis buffer, resuspended in SDS sample buffer, and analyzed by SDS-PAGE and Western blotting. For IP from fractionated HeLa cells, fractions were incubated with specific antibody against MCM-BP followed by protein A/G-agarose as described above. For FLAG IP, HeLa cells were transfected with pMZS3F-LacZ (16) or pCMV3FC expressing MCM-BP or the MCM-BP S158A mutant. Harvested cells were lysed 48 h later as described above, and the lysate was incubated with FLAG M2 resin (Sigma) overnight at 4°C. The resin was washed three times with lysis buffer, resuspended in SDS samples buffer, and analyzed by SDS-PAGE and Western blotting.
Cell synchronization.
HeLa cells were transfected with siRNA as described above. Twenty-four hours after the final transfection, thymidine (Sigma) was added to cells to a final concentration of 2 mM for 19 h, followed by two washes in phosphate-buffered saline (PBS) and release into complete DMEM for 10 h. Thymidine was added again to the cells to a final concentration of 2 mM for 17 h, and cells were either harvested directly or released into complete medium for the indicated times.
Flow cytometry.
For DNA content analysis, cells were fixed in 70% ethanol overnight at −20°C, washed with PBS with 0.5% bovine serum albumin (BSA), treated with 100 μg/ml RNase A for 1 h at 37°C, and stained with 50 μg/ml propidium iodide. All samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences), and data were collected using CellQuest software. Cell cycle analysis was performed using FlowJo software (Treestar Inc.). For BrdU pulse experiments, HCT116 wild-type (WT) and USP7-null cells were pulsed with 10 μg/ml BrdU for 30 min. The labeled cells were washed twice with PBS and either harvested and fixed or released into BrdU-free complete medium for the indicated times before harvesting and fixation in 70% ethanol overnight at −20°C. DNA was denatured using 2 N HCl at room temperature for 30 min. BrdU was detected using FITC-conjugated anti-BrdU antibody (BD Pharmingen), while DNA was stained with 50 μg/ml propidium iodide.
Expression of MCM-BP proteins in insect cells.
Baculoviruses expressing WT or S158A MCM-BP proteins fused to an N-terminal hexahistidine tag were generated using the pFastBacHT system. The MCM-BP proteins were expressed and purified from High Five insect cells as described previously (15).
Purification of His-tagged USP7 proteins for protein interactions.
Full-length USP7 was expressed in High Five insect cells and purified using the hexahistidine tag as previously described (45). USP7 fragments coding for amino acids 1 to 205, 1 to 540, and 202 to 540 were PCR amplified and cloned between the NdeI and BamHI sites of pET15b (Novagen). Proteins were expressed in BL21(DE3)pLysS cells after overnight induction with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 18°C. Cells were lysed by sonication in 50 mM Tris, pH 8.0, 500 mM NaCl, 20 mM imidazole, 5% glycerol containing 0.25% Triton X-100, 0.25% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), and protease inhibitor cocktail (8340; Sigma). The lysates were clarified by centrifugation for 30 min in an SS34 Sorvall rotor at 15,000 rpm. Clarified lysates were incubated with nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) for 1 h, and after extensive washing with loading buffer lacking detergent, the His-tagged proteins were eluted with 250 mM imidazole and dialyzed against 50 mM Tris, pH 8.0, 150 to 200 mM NaCl, 5% glycerol, 0.1 mM EDTA, and 0.1 mM dithiothreitol (DTT).
Expression and purification of GST-USP7 fragments.
USP7 fragments coding for amino acids 1 to 205 (UPS7 1–205), 1 to 540 (USP7 1–540), 1 to 560 (USP7 1–560), 560 to 1102 (UPS7 560–1102), 202 to 560 (USP7 202–560), and 1 to 560 with the D164A and W165A mutations (USP7 1–560 DW) were PCR amplified and cloned between the NdeI and BamHI sites of a modified pGEX-2TK vector (a gift from Yi Sheng). Proteins were expressed in BL21(DE3)pLysS cells after overnight induction with 0.5 mM IPTG at 18°C. Cells were lysed by sonication in PBS containing 1% Triton X-100 and protease inhibitor cocktail (Sigma). The lysates were clarified by centrifugation for 30 min in an SS34 Sorvall rotor at 15,000 rpm. Clarified lysates were incubated with glutathione-agarose (Pierce, Thermo Scientific) for 1 h, and after extensive washing with PBS lacking detergent, the glutathione S-transferase (GST)-tagged proteins were eluted with 20 mM glutathione and dialyzed against 50 mM Tris, pH 8.0, 150 to 200 mM NaCl, 5% glycerol, 0.1 mM EDTA, and 0.1 mM DTT. The USP7 1-560 DW mutant was subcloned by PCR amplification from full-length USP7 with D164A and W165A mutations in pCAN (a gift from Yi Sheng [46]).
Gel filtration analysis of MCM-BP–USP7 interactions.
Purified MCM-BP (70 μg) was incubated with equimolar amounts of purified his-tagged USP7 fragments for 30 min on ice in 500 μl of 50 mM Tris, pH 8.0, 150 mM NaCl, and 5% glycerol and then loaded on a 10/300 GL Superose 6 column (GE Healthcare) and developed in the same buffer. Forty-eight 500-μl fractions were collected, and 35 μl of each fraction was analyzed by SDS-PAGE and Coomassie staining.
Analysis of MCM-BP interactions with GST-USP7 fusion proteins.
Purified GST-USP7 fusion protein (1 nmol) was bound to 30 μl of glutathione-agarose in PBS, and then 1 nmol of purified MCM-BP (70 μg) in 500 μl of 50 mM Tris, pH 8.0, 150 mM NaCl, and 5% glycerol was incubated with the resin for 1 h at 4°C. Unbound proteins were removed with four 500-μl washes in loading buffer, and bound proteins were eluted with two 40-μl incubations in the same buffer containing 25 mM glutathione. One-tenth of the first wash (flowthrough) and 100% of both elutions were analyzed by SDS-PAGE and Coomassie staining.
Expression and purification of USP7 54–205 for crystallization.
USP7 fragment 54–205 was expressed from pET15b in Escherichia coli BL21(DE3) cells and purified by virtue of the hexahistidine tag as described previously (45). Briefly, the cells were lysed by sonication in 50 mM Tris (pH 7.5), 500 mM NaCl, 10 mM imidazole, 1 mM benzamidine, and 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and the clarified lysate was incubated with Ni-NTA beads (Qiagen). After extensive washing with 50 mM Tris (pH 7.5), 500 mM NaCl, and 30 mM imidazole, protein was eluted with 50 mM Tris (pH 7.5), 500 mM NaCl, and 250 mM imidazole. The His tag was cleaved using thrombin, and USP7 54–205 was further purified by size exclusion chromatography using a Sephacryl S200 16/60 column on a purifier 10 UPC system (GE Healthcare).
Crystallization and structure determination of USP7–MCM-BP complex.
USP7 54–205 (100 mg/ml) was cocrystallized with 5-fold-excess MCM-BP (152RVSPSTSYTP161) peptide (synthesized by CanPeptide Inc., with amino-terminal acetylation and carboxy-terminal amidation) using microseeding with USP7-HdmXAHSS crystals (38). The crystals appeared after several days at 4°C in the dark in 30% polyethylene glycol (PEG) 4000, 0.1 M Tris (pH 8.5), and 0.2 M lithium sulfate in crystallization tools (Qiagen). Diffraction data from a frozen USP7–MCM-BP complex crystal was collected at 100 K on a Rigaku MicroMax007 rotating anode diffractometer with a Saturn 944+ charge-coupled-device (CCD) detector. The space group was P41 with a = b = 70.0 Å and c = 45.7 Å unit cell parameters. All of the diffraction data were integrated and scaled using the HKL2000 software program (47). The data collection statistics are shown in Table 1. Molecular replacement was used to determine the structure using the CNS program (version 1.3) with USP7 54-205 (PDB identifier [ID] 1YY6) as the search model without any peptide (48). The molecular graphics program O was used for electron density visualization and model building (49). Rigid body, simulated annealing torsion angle, and individual B-factor refinement were performed using CNS 1.3. Several rounds of refinement were combined with model rebuilding in O after inspection of both 2Fo-Fc and Fo-Fc maps. Water molecules were picked using CNS 1.3 and manually verified. The refinement statistics are shown in Table 1. The figure was prepared using the software program Pymol.
TABLE 1.
X-ray data collection and refinement parametersa
| Parametera | Value for USP7–MCM-BPb |
|---|---|
| X-ray data | |
| Space group | P41 |
| Resolution (Å) | 50.0–1.70 |
| Unit cell axes (Å3) | 69.8 × 69.8 × 45.7 |
| Molecules/AU | 1 |
| No. of total observations | 267,608 |
| No. of unique reflections | 22,977 |
| Intensity (I/σ<I>) | 21.9 (1.6) |
| Completeness (%) | 94.3 (80.1) |
| Rsymc | 0.066 (0.509) |
| Refinement | |
| Rwork | 0.207 |
| Rfree | 0.233 |
| No. of protein atoms | 1,146 |
| No. of water molecules | 130 |
| RMSD, bonds (Å) | 0.006 |
| RMSD, angles (°) | 1.3 |
| RMSD, dihedrals (°) | 25.3 |
| RMSD, improper (°) | 0.87 |
| Thermal factors (Å2) | 19.6 |
| Ramachandran plot | |
| Most favored | 0.891 |
| Additionally allowed | 0.109 |
RMSD, root mean square deviation; AU, arbitrary units.
Numbers in parentheses refer to the highest-resolution shell, 1.73 Å to 1.70 Å.
Rsym = Σ|I − <I>|/ΣI, where “I” is the observed intensity and “<I>” is the average intensity from multiple observations of symmetry-related reflections.
Assay of USP7 interaction with MCM5 and MCM7.
High Five insect cells in a 15-cm plate were coinfected with baculovirus expressing WT or S158A MCM-BP proteins and FLAG-tagged MCM5 or MCM7 as described previously (22). Some of the infections also included baculovirus expressing full-length USP7. Cells were lysed 3 days postinfection in 1 ml of 50 mM Tris, pH 8.0, 150 mM NaCl, 5% glycerol, 2 mM EDTA, and 0.1% Triton X-100 on ice for 30 min. Lysates were clarified by centrifugation at 13,000 rpm for 10 min and then incubated with 25 μl FLAG-M2 Resin (Sigma) for 1 h at 4°C. The resin was washed three times with 1 ml of lysis buffer, and then proteins were eluted with sample buffer containing 2% SDS, analyzed by SDS-PAGE, and stained with Coomassie blue.
Assay of USP7 ubiquitin cleavage activity.
Purified USP7 (0.2 μg) was incubated with purified MCM-BP (0 to 4.0 μg) in 5 μl of the reaction buffer (40 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2 mM EDTA, 10 mM DTT) on ice for 30 min and then at 37°C for 10 min. At the same time, the 3 μg of K48-linked diubiquitin substrate (catalogue no. UC-200; Boston Biochem) in 5 μl of reaction buffer was incubated at 37°C for 10 min. The reaction was started by combining the USP7 and diubiquitin samples and quenched with sample buffer after a 10-min incubation. Products were analyzed by SDS-PAGE on NuPAGE Novex 4 to 12% Bis-Tris gels (Invitrogen), followed by Coomassie blue staining.
Molecular combing.
Molecular combing of DNA fibers was performed as previously described (50) with the following exceptions. Cells were cast into 1% low-melting-grade agarose plugs to a final concentration of 2.5 × 106. Agarose plugs were melted in two steps at 74°C. At least 70 interorigin distances and 130 fork rates were measured per experiment, and reported values are pooled distributions for 2 independent replicates.
Protein structure accession number.
Coordinates and structure factors for the USP7–MCM-BP structure have been deposited in the RCSB PDB under PDB identifier (ID) 4KG9.
RESULTS
MCM-BP binds directly to the USP7 TRAF domain.
A genome-wide study of protein interactions with human deubiquitylating enzymes detected an interaction between USP7 and MCM-BP (called C10orf119) in 293 cells (24). To determine if this interaction could be verified by other methods and in other cell lines, we immunoprecipitated endogenous MCM-BP from HeLa cells and BJ primary fibroblasts and examined recovery of USP7. Immunoprecipitated MCM-BP consistently recovered USP7 from both cell lines at levels above that seen with nonspecific IgG (Fig. 1A). As expected, MCM6 was also recovered with MCM-BP, serving as a positive control.
FIG 1.
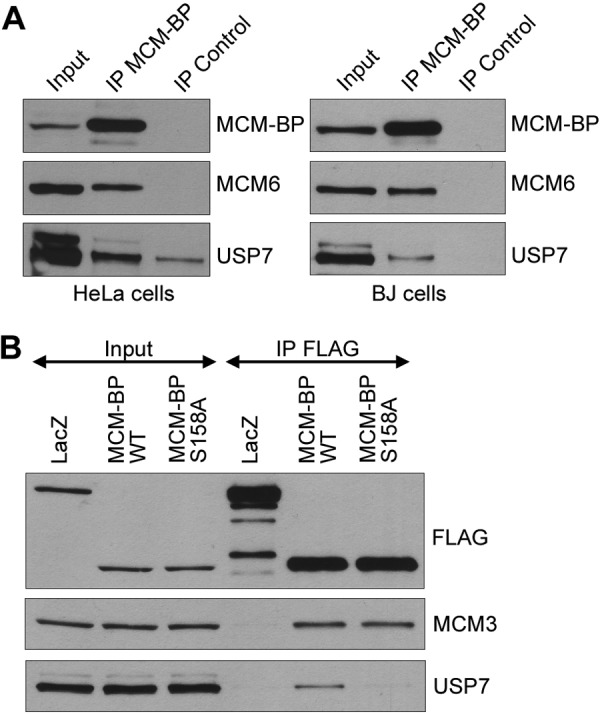
Coimmunoprecipitation of USP7 with MCM-BP. (A) Equal amounts of whole-cell extracts from HeLa cells or BJ cells were incubated with antibody against MCM-BP or nonspecific IgG (control). Immunoprecipitants were analyzed by Western blotting against the indicated proteins. Note that USP7 is known to run as a doublet due to the presence of two isoforms (69). (B) HeLa cells were transfected with FLAG-LacZ, FLAG–MCM-BP, and FLAG–MCM-BP S158A expression plasmids and 48 h later were subjected to anti-FLAG immunoprecipitation. Western blots for the indicated proteins are shown for the immunoprecipitates (IP FLAG) and the starting cell lysates (5% input).
Having verified the USP7–MCM-BP interaction, we next asked whether this interaction was direct. To this end, human MCM-BP and USP7 (shown in Fig. 2A) were expressed and purified from insect cells and analyzed by gel filtration individually and after combining them at equal molar concentrations. As expected, the migration of MCM-BP on its own was slower than that of USP7 due to its smaller size (Fig. 2B, top 2 panels). When the two proteins were combined, a significant proportion of both proteins comigrated at a faster-migrating position, consistent with the formation of a complex (Fig. 2B, bottom panel). This interaction also occurred when the same assay was performed using MCM-BP and the N-terminal half of USP7 (1–540) containing both the TRAF and catalytic domains (Fig. 2A), as evidenced by their size shift (relative to individual proteins) and comigration (Fig. 2C).
FIG 2.

Gel filtration analyses of USP7–MCM-BP complexes. (A) Schematic representation of the domain organization of USP7, showing amino acid numbers. (B to E) Purified MCM-BP was incubated with equimolar quantities of purified full-length USP7 (B), USP7 1–540 (C), USP7 1–205 (D), or USP7 202–540 (E). In panel F, purified MCM-BP S158A was incubated with an equimolar quantity of purified full-length USP7. All protein mixtures were run through a Suparose gel filtration column, and equal-volume fractions were collected. Equal volumes of each of the indicated fractions were then analyzed by SDS-PAGE and Coomassie staining. The positions of the gel filtration standards myoglobin (17 kDa), ovalbumin (44 kDa), aldolase (158 kDa), and thyroglobulin (669 kDa) on the same column are indicated at the top of each panel.
We further investigated the requirements for the USP7 TRAF and catalytic domains for MCM-BP binding, initially by repeating the gel filtration analyses with USP7 fragments containing either the N-terminal TRAF domain (1–205) or the catalytic domain (202–540). However, neither of these USP7 fragments caused an obvious shift in the migration of MCM-BP (Fig. 2D and E), suggesting that stable complex formation involved interactions with both USP7 domains. We then investigated whether these domains individually mediate transient interactions with MCM-BP using a GST pulldown assay, which can detect more transient interactions than gel filtration. In this assay, purified USP7 domains linked to GST were immobilized on glutathione resin and then incubated with purified full-length MCM-BP and eluted with glutathione (Fig. 3). Consistent with the gel filtration analysis, MCM-BP was retained on the resin by USP7 1–540 or UPS7 1–560 (Fig. 3E and F) but not by the USP7 catalytic domain (Fig. 3C) or GST alone (Fig. 3A). However, in this assay, some interaction of MCM-BP with USP7 1–205 was detected (Fig. 3B), suggesting that MCM-BP interacts weakly with the USP7 TRAF domain and that this interaction is stabilized by additional contacts with the catalytic domain. We also detected some weak binding to the USP7 C-terminal half (560 to 1102) (Fig. 3D). Other proteins that bind the USP7 TRAF domain do so through a binding pocket that involves contacts with D164 and W165 (28, 36, 38). To further examine the importance of TRAF domain contacts for MCM-BP binding to USP7 1–560, we repeated the GST pulldown assays with USP7 1–560 containing the D164A and W165A point mutations (Fig. 3G). These mutations abrogated the interaction with MCM-BP, confirming the importance of the TRAF binding pocket.
FIG 3.

GST pulldown assays of USP7–MCM-BP interactions. Purified MCM-BP was incubated with equimolar quantities of purified GST (A), GST-USP7 1–205 (B), GST-USP7 202–560 (C), GST-USP7 560–1102 (D), GST-USP7 1–540 (E), GST-USP7 1–560 (F), or GST-USP7 1–560 DW (G) that was immobilized on glutathione-agarose beads. In panel H, MCM-BP S158A was incubated with an equimolar quantity of purified GST USP7 1–560. Input and 1/10 flowthrough for GST-tagged proteins are indicated by L1 and F1, respectively. Input and 1/10 flowthrough for MCM-BP/MCM-BP S158A are indicated by L2 and F2, respectively. Bound proteins were eluted with two glutathione incubations (E1 and E2) and analyzed by SDS-PAGE and Coomassie staining.
We have previously identified a P/AxxS consensus sequence that mediates the interaction of p53, Mdm2, and MdmX with the USP7 TRAF binding pocket (28, 36, 38). There are five putative P/AxxS motifs in MCM-BP (135PGES138, 155PSTS158, 266PVLS269, 299PPAS302, and 397PRNS400), but 155PSTS158 was the only one that exactly matched known USP7 binding sequences (156PSTS159 and 397PSTS400 from Mdm2) and therefore seemed most likely to mediate the USP7 interaction. To test the importance of 155PSTS158 for USP7 binding, we generated MCM-BP with an S158A point mutation and tested its ability to bind USP7 in the gel filtration assay (Fig. 2F) and USP7 1–560 in the GST pulldown assay (Fig. 3H). In both cases, this mutation greatly decreased the interaction with MCM-BP relative to that seen with the WT protein, indicating that this PxxS motif is part of the USP7 binding sequence. This result further supports the importance of the TRAF domain contacts for the MCM-BP–USP7 interaction. Finally, we verified that S158 is important for MCM-BP binding to USP7 by expressing FLAG-tagged WT or S158A versions of MCM-BP in HeLa cells, recovering the tagged protein with anti-FLAG resin, and blotting for USP7. As shown in Fig. 1B, USP7 was recovered with the WT but not with the S158A version of MCM-BP. Importantly, both versions of MCM-BP recovered MCM3 equally, showing that the S158A mutation did not cause misfolding of MCM-BP.
Crystal structure of USP7 TRAF domain with the MCM-BP peptide.
The molecular mechanism of the interaction between USP7 and MCM-BP was further examined by cocrystallizing the USP7 TRAF domain with the MCM-BP 152RVSPSTSYTP161 peptide and determining the structure by molecular replacement (Fig. 4A). As previously reported, this USP7 domain is an eight-stranded antiparallel beta sandwich closely resembling TRAF domains in other proteins (28, 36). The MCM-BP peptide binds within a groove on the surface of the TRAF domain in proximity to strand β7. The final model of the complex was refined to an Rwork of 0.206 and an Rfree of 0.231 at 1.7-Å resolution with 130 water molecules. Residues 54 to 62 and 106 to 111 are disordered and were not built into the final model of the complex. The MCM-BP peptide forms several interactions with the USP7 β-strand 7 (Fig. 4B). MCM-BP S158 makes the most contacts with USP7 54–205 by forming H bonds with D164. MCM-BP residues V153, S154, and Y159 were also modeled but do not form any direct contacts with USP7 54–205. The electron density for the remainder of the peptide is disordered and cannot be modeled. Comparison of the MCM-BP peptide (154VPSTSY159) to the Mdm2 396QPSTSS401 peptide bound to the USP7 TRAF domain (38) indicated that the two peptides were superimposable over the six Cα atoms with a root mean square deviation of 0.1 Å2 (Fig. 4C). However, unlike the Mdm2 peptide, which has water-mediated contacts with USP7 outside the PSTS sequence, contacts of the MCM-BP peptide with the USP7 TRAF domain were limited to the PSTS sequence.
FIG 4.

Crystal structure of USP7 54–205 bound to MCM-BP peptide 152RVSPSTSYTP161. (A) Ribbon representation of USP7 54–205 (cyan) bound to the MCM-BP peptide, shown in stick representation (yellow). PDB ID 4KG9. (B) Detailed interactions of MCM-BP (yellow), with β-strand 7 of USP7 54–205 (cyan) shown in stick representation. The hydrogen bonds are shown with black dashed lines. (C) Overlay of MCM-BP peptide (yellow) with Mdm2 396QPSTSS401 peptide (green) from previously determined Mdm2-USP7 structure (38).
A role for USP7 in the S/G2 transition.
Since MCM-BP is known to function in DNA replication, the interaction of USP7 with MCM-BP suggests that USP7 might also affect DNA replication. We examined this possibility using the HCT116 USP7 knockout cells generated previously (33). These cells are known to have a longer G1 phase, presumably due to high p53 levels (33), but their progression through S phase has not been examined previously. We compared the S-phase progression of WT and USP7-null HCT116 cells by pulse labeling log-phase cells with BrdU and following the change in the cell cycle profile of BrdU-incorporated cells over time by fluorescence-activated cell sorting (FACS) (Fig. 5A). The two cell lines were found to progress through early and mid-S phase at similar rates, but differences became apparent at the 6- and 8-h post-BrdU pulse. At these time points, the cell cycle profile indicates that USP7-null cells have more cells in late S phase, as well as an accumulation of G2 or M cells that have not yet transitioned back to G1. The percentage of S-phase cells from the FACS analysis were quantified from three independent experiments (Fig. 5B), confirming that the USP7-null cells had significantly more cells in S phase at the 6- and 8-h time points than the WT cells.
FIG 5.

USP7 affects progression through late S phase. (A) WT and USP7-null cells were pulsed with BrdU and chased in the absence of BrdU for the indicated times. The DNA content of BrdU-positive cells was measured by flow cytometry at each time point. For each sample, the polygon in the top panel shows BrdU-incorporated cells, while the bottom panel shows cell phase distribution as determined by DNA content. (B) The percentages of S phase cells from three independent experiments performed as for panel A were determined using FlowJo software (Treestar Inc.) and graphed. Error bars represent standard deviations, and “∗∗” represents P < 0.01. (C) Distributions of interorigin distance (IOD) and replication fork progression were obtained by DNA combing in USP7-null (KO) and WT HCT116 cells. Median values for IOD are 102 kbp (WT) and 100 kbp (KO). Median values for the fork rate are 0.86 kbp/min (WT) and 0.96 kbp/min (KO). P values were determined by a two-tailed Mann-Whitney U test used to compare nonnormal distributions.
We further examined possible contributions of USP7 to DNA replication using molecular combing experiments to measure the replication fork rate and origin usage by examining single DNA molecules (51–53). To this end, log-phase USP7-null or WT HCT116 cells were sequentially pulse labeled with the thymidine analogues chlorodeoxyuridine (CldU) and iododeoxyuridine (IdU). DNA fibers from these cells were then stretched uniformly on glass slides, and incorporated thymidine analogues were detected by immunofluorescence microscopy using specific antibodies. From the length of fluorescent DNA tracks and the pattern of incorporation, both the rate of replication fork progression and interorigin distance were determined in two independent experiments (a schematic diagram explaining how the fork rate and interorigin distance were obtained can be found elsewhere (50)). As shown in Fig. 5C, a lack of USP7 did not have a significant effect on the rate of replication fork movement. Lack of USP7 also did not affect interorigin distance, suggesting that it does not significantly contribute to origin activation (Fig. 5C). In addition, we found no significant change in the rate of fork movement or interorigin distance after silencing USP7 in HeLa cells (data not shown). Therefore, the contributions of USP7 to DNA replication appear to be limited to late S phase.
USP7 affects MCM protein levels on chromatin.
MCM-BP depletion in human cells and in Xenopus egg extracts results in increased levels of MCM proteins on chromatin (16, 23). Since USP7 interacts with MCM-BP, we asked whether USP7 depletion also affected MCM protein levels on chromatin. We began by comparing MCM protein levels in whole-cell extracts and in soluble and chromatin-associated fractions from WT and USP7-null HCT116 cells (Fig. 6A). Total MCM protein levels were slightly decreased in the USP7-null cells relative to those in WT cells (left panel). However, the most striking difference was in the ratio of chromatin associated with soluble MCM proteins, where USP7-null cells had a higher fraction of each MCM protein on chromatin (relative to the soluble fraction) than wild-type cells (Fig. 6A, right panel). Blots for MEK2 (a soluble protein) and histone H3 (a chromatin-associated protein) verified that the extracts were fractionated appropriately and showed that their degree of chromatin association was unaffected by USP7. The ratio of chromatin associated with soluble proteins was determined for each MCM protein from three independent experiments, showing that each MCM protein was consistently increased on chromatin in the absence of USP7 (Fig. 6B).
FIG 6.

USP7 depletion increases the chromatin association of MCM proteins. (A) Whole-cell (WCE), soluble (Sol.), and chromatin-associated (Chr.) lysates from WT and USP7-null (KO) cells were analyzed by SDS-PAGE and Western blotting against the indicated MCM proteins and USP7. In the left panel, histone H3 and actin are used as loading controls. (B) MCM protein bands from the soluble and chromatin-associated lysate were quantified from three independent experiments by densitometry (GelEval software). The ratio of chromatin-associated to soluble protein was calculated for each MCM protein from the WT and USP7-null cells and graphed. Error bars represent standard deviations. (C) Whole-cell, soluble, and chromatin-associated lysates from HeLa cells transfected with control or USP7-targeted siRNA (siControl and siUSP7, respectively) were analyzed by SDS-PAGE and Western blotting for the indicated MCM proteins and USP7. Blots against H3 (chromatin associated) and MEK2 (soluble) are used to indicate appropriate fractionation and also serve as loading controls. Note that since only a small proportion of MCM-BP is chromatin associated, the amount of chromatin lysate loaded in this experiment is not enough to detect MCM-BP in this fraction. (D) MCM protein bands from the whole-cell, soluble, and chromatin-associated lysate were quantified from two independent experiments by densitometry. The ratio of siUSP7 band density to siControl band density was calculated for each MCM protein in the whole-cell, soluble, and chromatin-associated lysate and graphed. Error bars represent standard deviations. (E) DNA content of HeLa cells transfected with control or USP7-targeted siRNA was measured by flow cytometry in three independent experiments and graphed using FlowJo software (Treestar Inc.). The percentages of cells in G1, S, and G2/M were estimated using FlowJo software and are shown in the graph. Error bars represent standard deviations. (F) Soluble (Sol.) and chromatin-associated (Ch.) lysates from WT and USP7-null cells (KO) were analyzed by SDS-PAGE and Western blotting against the indicated MCM proteins, Psf2 and USP7. Histone H3 (chromatin-associated protein) and MEK2 (soluble protein) indicate appropriate fractionation and also serve as loading controls. (G) Chromatin-associated lysates from HeLa cells transfected with control plasmid or shMCM-BP1 were analyzed by SDS-PAGE and Western blotting for the indicated MCM proteins, Psf2, and histone H3 as a loading control (left panel). Since only a small proportion of MCM-BP is chromatin associated, MCM-BP silencing was assessed by comparing equal volumes of the soluble lysate, compared to MEK2 as a loading control (right panel).
To verify that the increased levels of MCMs on chromatin were a direct effect of USP7 depletion, as opposed to an indirect effect caused by G1 accumulation or long-term growth in the absence of USP7, we repeated the above-described experiments in HeLa cells transfected with siRNA targeting USP7 or negative-control siRNA (Fig. 6C and D). Unlike the results for USP7-null cells, USP7 silencing did not significantly change the cell cycle distribution in comparison to that of control cells (Fig. 6E). USP7 silencing also did not affect the total cellular levels of MCM proteins (compare lanes 1 and 2 in Fig. 6C). However, like the USP7 knockout, it did result in an increased level of each MCM protein on chromatin (compare lanes 5 and 6 in Fig. 6C). Quantification of the total, soluble, and chromatin-associated MCM protein levels from two independent experiments is shown in Fig. 6D and confirms that USP7 depletion results in increased chromatin association of MCM proteins.
Two populations of the MCM complex are associated with chromatin during S phase. The first population comprises active CMG complexes (containing Cdc45, GINS, and the MCM complex) at the replication fork, while the second population comprises inactive MCM complexes that license dormant origins and that are not associated with GINS or Cdc45. We tested whether USP7 affected the chromatin association of CMG by blotting for the Psf2 subunit of GINS (Fig. 6F). We observed a consistent increase in the amount of chromatin-associated Psf2 in USP7-null cells (2-fold on average) compared to that in WT HCT116 cells, suggesting that USP7 can affect the chromatin association of GINS. Since MCM-BP is important for MCM complex unloading and directly binds USP7, we also tested whether MCM-BP silencing resulted in the accumulation of Psf2 on chromatin (23). To this end, HeLa cells were transfected with plasmids expressing shRNA against MCM-BP or empty plasmid, and then chromatin fractions were prepared. As shown in Fig. 6G, MCM-BP silencing resulted in increased levels of MCM proteins and Psf2 on chromatin. Together, our data indicate that at least some of the MCM complexes that are regulated by USP7 and MCM-BP are associated with GINS and therefore are likely CMG complexes.
USP7 contributes to MCM protein unloading.
MCM proteins are loaded onto chromatin throughout G1 and unloaded over the course of S phase. Therefore, the increased levels of MCM proteins on chromatin resulting from USP7 depletion could either be due to increased loading of MCM proteins (resulting in higher MCM levels on chromatin in G1 or early S) or decreased unloading (resulting in higher MCM levels on chromatin in late S or G2). To differentiate between these possibilities, we synchronized control and USP7-depleted HeLa cells at the G1/S boundary using a double thymidine block and released cells into fresh medium for the indicated times. Chromatin-associated protein fractions were prepared for each time point and analyzed by Western blotting for MCM proteins (Fig. 7A). In the negative-control sample, the levels of chromatin-associated MCM proteins decreased in the 4- to 8-h samples, which corresponds to mid-S phase and late S phase, respectively (based on FACS analysis of the DNA content) (Fig. 7B). This is the expected profile for MCM protein unloading. Interestingly, USP7 levels on the chromatin slightly increased in the 4- to 8-h time points, corresponding to the unloading of the MCM proteins. In USP7-depleted cells, the levels of MCM proteins on chromatin were similar to those of the control cells at the 0- and 2-h time points, but there was no obvious decrease in MCM proteins on chromatin at 4 to 8 h. This effect was consistent in three independent experiments, and the quantification of MCM5 levels on chromatin from the 0-h (G1/S) and 8-h (late S) time points is shown in Fig. 7C. We conclude that USP7 depletion inhibits MCM protein dissociation from chromatin in mid- to late S phase and therefore, like MCM-BP, USP7 may contribute to MCM complex unloading during S phase.
FIG 7.

USP7 depletion affects unloading of the MCM complex at the end of S phase. (A) HeLa cells transfected with control or USP7-targeted siRNA were synchronized to the G1/S boundary using a double thymidine block and released for the indicated times (0 h to 8 h). The chromatin-associated extract for each time point was analyzed by SDS-PAGE and Western blotting for the indicated MCM proteins and USP7. PCNA was used as a loading control. (B) The DNA content of the above samples was measured using flow cytometry and graphed using FlowJo software (Treestar Inc.). (C) MCM5 protein bands from the chromatin-associated fraction at 0 and 8 h after release from a double thymidine block were quantified from three independent experiments by densitometry. Average values with standard deviations are shown relative to results for 0-h siControl samples (set to 100). ∗, P < 0.05.
USP7 interacts with MCM-BP on chromatin and forms a ternary complex with MCM-BP and MCMs.
The impaired unloading of MCM proteins from chromatin after USP7 depletion mimics the previously described effect of MCM-BP depletion (16, 23). This suggests that USP7 may be working together with MCM-BP to unload the MCM complex. If this were correct, we would expect USP7 to bind the chromatin-associated MCM-BP and to form a ternary complex with the MCM-BP and MCM proteins. To test whether USP7 interacts with chromatin-associated or soluble MCM-BP, we fractionated HeLa cells into soluble and chromatin-associated extracts, immunoprecipitated MCM-BP from each fraction, and blotted for USP7 (Fig. 8A). Although USP7 and MCM-BP were present in both the soluble and chromatin-associated extracts (see input lanes), USP7 was recovered only with MCM-BP (at levels above those of the nonspecific IgG control) from the chromatin-associated fraction. MCM6 was also recovered along with USP7 in this MCM-BP IP, which could reflect the formation of a USP7–MCM-BP–MCM6 ternary complex or individual dimeric complexes with MCM-BP.
FIG 8.
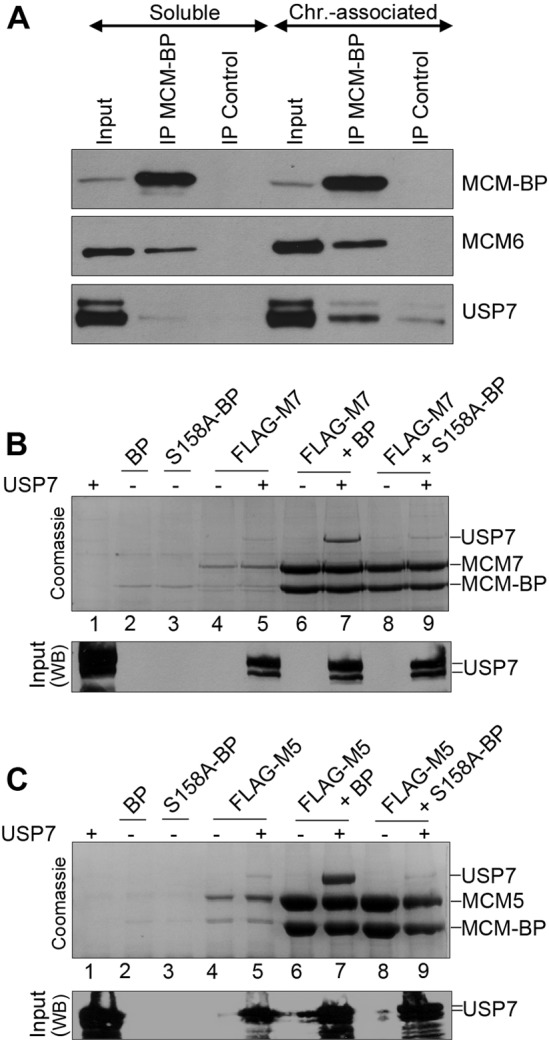
MCM-BP binds USP7 on chromatin and can mediate an interaction between the USP7 and MCM proteins. (A) Immunoprecipitations were performed with soluble and chromatin-associated lysates from HeLa cells using antibody against MCM-BP or negative-control IgG. Immunoprecipitants were analyzed by SDS-PAGE and Western blotting against the indicated proteins. (B and C) FLAG-tagged MCM7 (B) or FLAG-tagged MCM5 (C) was expressed individually or coexpressed with MCM-BP (BP) or MCM-BP S158A (S158A-BP) in insect cells. In each case, these proteins were expressed with and without USP7, as indicated. Insect cell lysates were then incubated with anti-FLAG resin to recover FLAG-MCM7 or FLAG-MCM5 with associated proteins. Bound proteins were eluted with protein sample buffer and analyzed by SDS-PAGE and Coomassie staining (top panel) or Western blotting for USP7 (bottom panel).
We then investigated whether USP7 could form a ternary complex with the MCM-BP and MCM proteins by coexpressing USP7 and MCM-BP with FLAG-tagged MCM5 or MCM7 in insect cells, isolating the FLAG-MCM protein on anti-FLAG resin, and assessing recovery of MCM-BP and USP7 by Coomassie blue staining (Fig. 8B and C). Both USP7 and MCM-BP were recovered with FLAG-MCM5 or FLAG-MCM7 (lane 7 in Fig. 8B and 8C). To determine whether USP7 bound directly to the MCM protein or was tethered to it through MCM-BP, we repeated the experiment by replacing MCM-BP with the S158A MCM-BP point mutant, which is unable to bind USP7. In this case, MCM-BP was still efficiently recovered with FLAG-MCM5 or FLAG-MCM7, but USP7 was not recovered (lane 9 in 8B and C). This indicates that USP7 is tethered to MCM proteins through MCM-BP. Note that this experiment cannot be performed in the absence of MCM-BP because MCM-BP dramatically increases the solubility and recovery of MCM proteins (compare MCM bands in lanes 4 and 5 to those in lanes 6 to 9). We conclude that USP7 binding to MCM-BP does not block MCM-BP binding to MCM proteins but rather that MCM-BP mediates the formation of a ternary complex with the USP7 and MCM proteins. Together, our results suggest that USP7 regulates MCM complex unloading at the end of S phase in an MCM-BP-dependent manner.
DISCUSSION
The MCM complex plays critical roles in DNA replication, both in the activation of origins of replication and in enabling the progression of replication forks, by virtue of its DNA helicase activity (54–56). Although there have been many studies of the proteins and events that enable the loading of MCM complexes on DNA during G1, their subsequent activation at the onset of S phase, and their role as a DNA helicase throughout S phase, little is known about how these complexes are unloaded from chromatin during the course of S phase and as replication forks terminate. Insight into MCM complex unloading recently came from studies of MCM-BP, a highly conserved protein that forms a variety of complexes with MCM proteins (15, 22). MCM-BP from both humans and Xenopus was shown to promote the dissociation of MCM complexes from chromatin in late S phase. The data presented here identify a second protein, USP7, as participating in this process and suggest that it does so through a direct interaction with MCM-BP.
An association of USP7 with MCM-BP and MCM proteins was suggested from a proteomics study of the protein interactions of deubiquitylating enzymes (24). We have verified this interaction in both primary and transformed human cells and have shown using purified proteins that USP7 binds directly to MCM-BP. Using a combination of GST pulldown assays, which can detect transient interactions, and gel filtration analysis, which detects stable complexes, we have shown that MCM-BP contacts the N-terminal TRAF domain of USP7 but requires additional contacts with the USP7 catalytic domain for stable binding. The interaction with both the TRAF and catalytic domains resembles the mechanism of interaction of the viral interferon regulatory factor 4 (vIRF4) protein of Kaposi sarcoma-associated herpesvirus (KSHV), which also contacts both of these USP7 domains (29). The contact of vIRF4 with the USP7 catalytic domain was shown to inhibit its ubiquitin cleavage activity. However, this is not the case for MCM-BP, since the addition of MCM-BP to in vitro assays of K48-linked diubiquitin cleavage by full-length USP7 (a substrate efficiently hydrolyzed by USP7 [57]) failed to have any effect on the ability of USP7 to cleave this substrate (Fig. 9). This suggests that unlike vIRF4, MCM-BP does not contact the catalytic site of the USP7 catalytic domain but rather contacts the catalytic domain in a way that preserves the enzymatic function of USP7.
FIG 9.
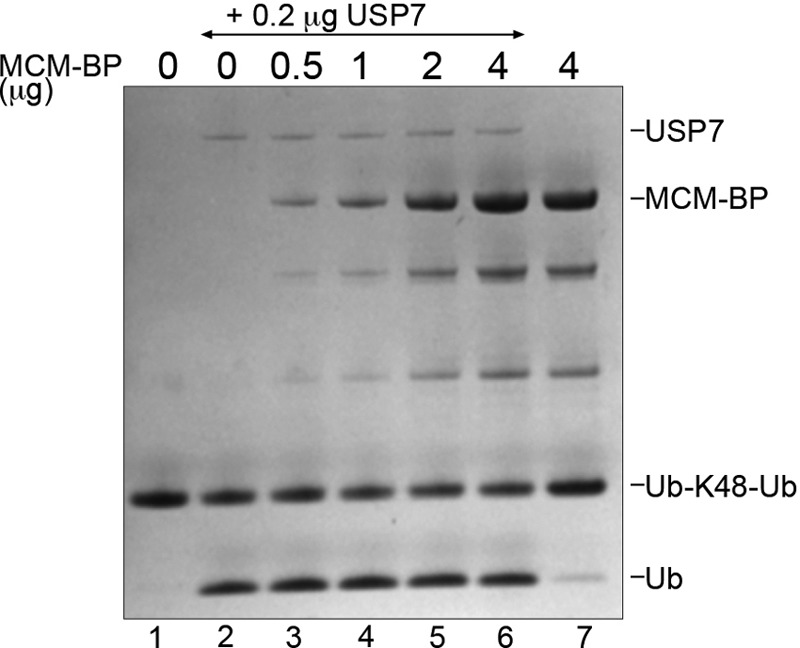
MCM-BP does not inhibit the catalytic activity of USP7. Purified USP7 (0.2 μg) was preincubated with increasing amounts of MCM-BP (as indicated) and then incubated with K48-linked diubiquitin (3 μg) for 10 min at 37°C. A Coomassie-stained SDS-PAGE gel is shown, where the positions of the diubiquitin substrate (Ub-K48-Ub) and monoubiquitin product (Ub) are indicated. Lanes 1 and 7 are controls lacking USP7.
Our data on the MCM-BP–USP7 interaction show that contacts of MCM-BP with the peptide binding pocket in the USP7 TRAF domain are critical for this interaction. Specifically, mutation of D164 and W165 in this binding pocket, which were previously shown to be critical for specific peptide recognition, abrogated the MCM-BP interaction. From our studies of other protein interactions with the USP7 TRAF domain, we previously defined an A/PxxS motif that mediates interactions with the TRAF binding pocket (28, 36, 38). Although MCM-BP contains five potential PxxS motifs, we identified one (155PSTS158) as being critical for the MCM-BP–USP7 interaction, since mutation of S158 to alanine greatly reduced binding of MCM-BP to USP7 both in vitro and in vivo. The crystal structure of the USP7 TRAF domain bound to the MCM-BP peptide 152RVSPSTSYTP161 confirmed the importance of S158 from MCM-BP and D164 from USP7 for this interaction. It also showed that this interaction is very similar to those reported previously for the p53, Mdm2, and MdmX peptides with the USP7 TRAF domain (36, 38, 58).
The interaction of USP7 with MCM-BP prompted an investigation of the possible role of USP7 in DNA replication. Comparison of the S-phase progression of USP7-null and WT HCT116 cells indicated that progression through early to mid-S phase was unaffected by loss of USP7; however, the progression through late S phase and G2 was noticeably affected. We also tested possible roles of USP7 in replication fork progression and replication origin activation using DNA combing experiments performed on USP7-null and WT HCT116 cells. No statistically significant differences were observed, indicating that USP7 does not globally affect replication the fork rate or origin function. This was an important possibility to consider because monoubiquitylation of histone H2B was recently shown to affect replication fork progression and replisome stability in S. cerevisiae (59), and USP7 is one of the enzymes that reverses this modification in higher eukaryotes (41, 42). Our data indicate that the contribution of USP7 to DNA replication is specific for late S phase/G2 progression.
The defect in late S phase/G2 progression caused by lack of USP7 could result from impaired replication fork termination, such as would be expected to occur if MCM complexes were not unloaded from chromatin. In keeping with this possibility, examination of the effect of USP7 on MCM proteins showed that the level of each MCM protein on chromatin increased as a result of USP7 silencing or knockout. Further analysis of MCM-chromatin interactions through S phase indicated that MCM proteins failed to dissociate from chromatin in late S phase in USP7-depleted cells. This effect on MCM-chromatin interactions mimics the effect of MCM-BP depletion, as observed in both human cells and Xenopus egg extracts (16, 23). The requirement for MCM-BP to dissociate MCM proteins from chromatin in late S phase, along with the ability of MCM-BP to disrupt MCM complexes, led to the conclusion that one of the functions of MCM-BP is to unload MCM complexes following DNA replication. Our data suggest that USP7 can function with MCM-BP to unload MCM complexes, although we cannot completely exclude abnormal scenarios, such as MCM reloading late in S phase in the absence of USP7.
There are two forms of MCM complexes on chromatin during S phase: (i) MCM complexes that interact with GINS and CDC45 to form the active CMG helicase at the replication fork and (ii) MCM complexes that bind dormant origins and are not associated with GINS or Cdc45 (11, 60, 61). We found that the lack of USP7 (in USP7-null cells) resulted in an increase in the Psf2 subunit of GINS on chromatin, suggesting that some of the MCM complexes that are unloaded by USP7 are part of the CMG complex or at least contain GINS. However, the MCM-chromatin association (typically increased 4- to 5-fold in USP7-null cells) was affected more than the Psf2-chromatin association (∼2-fold increase in USP7-null cells), suggesting that USP7 contributes to the unloading of both “active” CMG complexes and “inactive” MCM complexes at dormant origins. We also observed that MCM-BP silencing increased Psf2 levels on chromatin, consistent with a role for MCM-BP in unloading both MCM and GINS complexes. This also fits with MCM-BP's function in MCM unloading in the Xenopus egg system, in which there is an ∼10-fold increase in CMG complexes in comparison to findings for human cells (62–65).
Since USP7 stabilizes several of its interacting proteins by deubiquitylating them, we considered that USP7 might stabilize the MCM-BP and/or MCM protein. However, we did not find support for this possibility, since USP7 silencing did not result in lower levels of the MCM-BP or MCM protein. While USP7-null HCT116 cells did have somewhat lower levels of MCM proteins compared to WT cells, they also had lower levels of other S-phase-associated proteins, such as Sld5, Cdc45, and cyclin A (data not shown). This suggests that the lower levels of total MCM proteins in the USP7-null cells are more likely due to the slower cell cycle rather than specific stabilization by USP7.
An important feature of the MCM-BP–USP7 interaction, is that it occurs in such a way that MCM-BP retains the ability to bind MCM proteins. While the MCM-BP residues important for binding MCM proteins have not been determined, we found that the S158A mutation, which abrogates binding to USP7, had no effect on the ability of MCM-BP to bind MCM proteins (Fig. 1B). In addition, we showed that MCM-BP bridges an interaction between the USP7 and MCM proteins (Fig. 8). This bridging effect through MCM-BP may account for the recovery of some MCM proteins with USP7 previously observed in the Sowa et al. proteomic study (24). The ability of MCM-BP to bridge USP7-MCM interactions, combined with the observation that USP7 is preferentially associated with the fraction of MCM-BP that is on chromatin, suggests that MCM-BP recruits USP7 to MCM complexes on chromatin, where it could affect their function. Interestingly, the association of USP7 with chromatin was observed to increase at mid- to late S phase, a time when the MCM proteins are observed to dissociate from chromatin. This supports a scenario where the increased chromatin association of USP7 promotes the unloading of the MCM complex.
The role of USP7 in MCM unloading may or may not involve the catalytic activity of USP7, since some but not all of the cellular functions of USP7 involve its ability to cleave ubiquitin. While there is currently no evidence that MCM proteins are ever polyubiquitylated in human cells, MCM5 and MCM7 were identified as potentially monoubiquitylated proteins in a recent proteomic screen (66). Biochemical and structural studies have suggested that the MCM2-7 ring-shaped complex opens at the interface between MCM2 and MCM5, referred to as the MCM2/5 gate (67, 68). While it is not known what triggers this opening, it is possible that the opening and closing of the MCM2/5 gate is regulated by the ubiquitylation state of MCM5 or MCM7. In such a scenario, USP7 could either affect MCM unloading by cleaving ubiquitin from the MCM protein, or, since USP7 is known to interact with several E3 ubiquitin ligases (24), it might act as a scaffold to recruit the ubiquitin ligase that modifies the MCM protein.
In summary, we have shown that USP7 binds directly to MCM-BP and mirrors its function in enabling the dissociation of MCM complexes from chromatin at the end of S phase. Although USP7 has been reported to contribute to several cellular functions, this is the first time it has been shown to directly contribute to DNA replication. Our data support a model in which MCM-BP recruits USP7 to MCM complexes on chromatin to facilitate their unloading as replication terminates. Further studies will be necessary to determine how USP7 exerts this effect.
ACKNOWLEDGMENTS
We thank Dionne White for assistance with FACS analyses and Kathy Shire for excellent technical assistance. We also acknowledge Anthony La Delfa for early studies of MCM-BP peptide interactions with USP7 and Jay Yang for assistance with DNA combing experiments.
This work was supported by operating grants to L.F. from the Canadian Institutes of Health Research (CIHR) (grant number 84306) and from the Cancer Research Society. It was also supported by operating grants from the CIHR to V.S. (grant number 106583) and to G.W.B. (MOP-79368). L.F. is a tier 1 Canada Research Chair in Molecular Virology.
Footnotes
Published ahead of print 4 November 2013
REFERENCES
- 1.Boos D, Frigola J, Diffley JF. 2012. Activation of the replicative DNA helicase: breaking up is hard to do. Curr. Opin. Cell Biol. 24:423–430. 10.1016/j.ceb.2012.01.011 [DOI] [PubMed] [Google Scholar]
- 2.Burkhart R, Schulte D, Hu D, Musahl C, Gohring F, Knippers R. 1995. Interactions of human nuclear proteins P1Mcm3 and P1Cdc46. Eur. J. Biochem. 228:431–438. 10.1111/j.1432-1033.1995.tb20281.x [DOI] [PubMed] [Google Scholar]
- 3.Lei M, Kawasaki Y, Tye BK. 1996. Physical interactions among Mcm proteins and effects of Mcm dosage on DNA replication in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:5081–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowles A, Chong JP, Brown L, Howell M, Evan GI, Blow JJ. 1996. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell 87:287–296. 10.1016/S0092-8674(00)81346-X [DOI] [PubMed] [Google Scholar]
- 5.Mahbubani HM, Chong JP, Chevalier S, Thommes P, Blow JJ. 1997. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J. Cell Biol. 136:125–135. 10.1083/jcb.136.1.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donovan S, Harwood J, Drury LS, Diffley JF. 1997. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 94:5611–5616. 10.1073/pnas.94.11.5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. 2002. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 277:33049–33057. 10.1074/jbc.M204438200 [DOI] [PubMed] [Google Scholar]
- 8.Ilves I, Petojevic T, Pesavento JJ, Botchan MR. 2010. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 37:247–258. 10.1016/j.molcel.2009.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheu YJ, Stillman B. 2010. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463:113–117. 10.1038/nature08647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zegerman P, Diffley JF. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445:281–285. 10.1038/nature05432 [DOI] [PubMed] [Google Scholar]
- 11.Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. 2006. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 173:673–683. 10.1083/jcb.200602108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. 2003. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 12:1525–1536. 10.1016/S1097-2765(03)00456-8 [DOI] [PubMed] [Google Scholar]
- 13.Sabouri N, McDonald KR, Webb CJ, Cristea IM, Zakian VA. 2012. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 26:581–593. 10.1101/gad.184697.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinacher R, Osman F, Dalgaard JZ, Lorenz A, Whitby MC. 2012. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 26:594–602. 10.1101/gad.184663.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakwe AM, Nguyen T, Athanasopoulos V, Shire K, Frappier L. 2007. Identification and characterization of a novel component of the human minichromosome maintenance complex. Mol. Cell. Biol. 27:3044–3055. 10.1128/MCB.02384-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jagannathan M, Sakwe AM, Nguyen T, Frappier L. 2012. The MCM-associated protein MCM-BP is important for human nuclear morphology. J. Cell Sci. 125:133–143. 10.1242/jcs.089938 [DOI] [PubMed] [Google Scholar]
- 17.Ding L, Forsburg SL. 2011. Schizosaccharomyces pombe minichromosome maintenance-binding protein (MCM-BP) antagonizes MCM helicase. J. Biol. Chem. 286:32918–32930. 10.1074/jbc.M111.282541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JJ, Schnick J, Hayles J, MacNeill SA. 2011. Purification and functional inactivation of the fission yeast MCM(MCM-BP) complex. FEBS Lett. 585:3850–3855. 10.1016/j.febslet.2011.10.033 [DOI] [PubMed] [Google Scholar]
- 19.Takahashi N, Quimbaya M, Schubert V, Lammens T, Vandepoele K, Schubert I, Matsui M, Inze D, Berx G, De Veylder L. 2010. The MCM-binding protein ETG1 aids sister chromatid cohesion required for postreplicative homologous recombination repair. PLoS Genet. 6:e1000817. 10.1371/journal.pgen.1000817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi N, Lammens T, Boudolf V, Maes S, Yoshizumi T, De Jaeger G, Witters E, Inze D, De Veylder L. 2008. The DNA replication checkpoint aids survival of plants deficient in the novel replisome factor ETG1. EMBO J. 27:1840–1851. 10.1038/emboj.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santosa V, Martha S, Hirose N, Tanaka K. 2013. The fission yeast minichromosome maintenance (MCM)-binding protein (MCM-BP), Mcb1, regulates MCM function during prereplicative complex formation in DNA replication. J. Biol. Chem. 288:6864–6880. 10.1074/jbc.M112.432393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen T, Jagannathan M, Shire K, Frappier L. 2012. Interactions of the human MCM-BP protein with MCM complex components and Dbf4. PLoS One 7:e35931. 10.1371/journal.pone.0035931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishiyama A, Frappier L, Mechali M. 2011. MCM-BP regulates unloading of the MCM2-7 helicase in late S phase. Genes Dev. 25:165–175. 10.1101/gad.614411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:1519–1530. 10.1093/emboj/16.7.1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J, Frappier L. 2003. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 278:29987–29994. 10.1074/jbc.M303977200 [DOI] [PubMed] [Google Scholar]
- 27.Salsman J, Jagannathan M, Paladino P, Chan PK, Dellaire G, Raught B, Frappier L. 2012. Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J. Virol. 86:806–820. 10.1128/JVI.05442-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, Zhang RG, Liao J, Lee W, Edwards AM, Arrowsmith CH, Frappier L. 2005. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 18:25–36. 10.1016/j.molcel.2005.02.029 [DOI] [PubMed] [Google Scholar]
- 29.Lee HR, Choi WC, Lee S, Hwang J, Hwang E, Guchhait K, Haas J, Toth Z, Jeon YH, Oh TK, Kim MH, Jung JU. 2011. Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat. Struct. Mol. Biol. 18:1336–1344. 10.1038/nsmb.2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ching W, Koyuncu E, Singh S, Arbelo-Roman C, Hartl B, Kremmer E, Speiseder T, Meier C, Dobner T. 2013. A ubiquitin-specific protease possesses a decisive role for adenovirus replication and oncogene-mediated transformation. PLoS Pathog. 9:e1003273. 10.1371/journal.ppat.1003273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jager W, Santag S, Weidner-Glunde M, Gellermann E, Kati S, Pietrek M, Viejo-Borbolla A, Schulz TF. 2012. The ubiquitin-specific protease USP7 modulates the replication of Kaposi's sarcoma-associated herpesvirus latent episomal DNA. J. Virol. 86:6745–6757. 10.1128/JVI.06840-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholson B, Suresh Kumar KG. 2011. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem. Biophys. 60:61–68. 10.1007/s12013-011-9185-5 [DOI] [PubMed] [Google Scholar]
- 33.Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. 2004. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428:486–487. 10.1038/nature02501 [DOI] [PubMed] [Google Scholar]
- 34.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. 2002. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416:648–653. 10.1038/nature737 [DOI] [PubMed] [Google Scholar]
- 35.Li M, Brooks CL, Kon N, Gu W. 2004. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13:879–886. 10.1016/S1097-2765(04)00157-1 [DOI] [PubMed] [Google Scholar]
- 36.Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, Frappier L. 2006. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 13:285–291. 10.1038/nsmb1067 [DOI] [PubMed] [Google Scholar]
- 37.Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. 2005. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 18:565–576. 10.1016/j.molcel.2005.04.024 [DOI] [PubMed] [Google Scholar]
- 38.Sarkari F, La Delfa A, Arrowsmith CH, Frappier L, Sheng Y, Saridakis V. 2010. Further insight into substrate recognition by USP7: structural and biochemical analysis of the HdmX and Hdm2 interactions with USP7. J. Mol. Biol. 402:825–837. 10.1016/j.jmb.2010.08.017 [DOI] [PubMed] [Google Scholar]
- 39.Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. 2007. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128:141–156. 10.1016/j.cell.2006.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM. 2006. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 8:1064–1073. 10.1038/ncb1469 [DOI] [PubMed] [Google Scholar]
- 41.van der Knaap JA, Kumar BR, Moshkin YM, Langenberg K, Krijgsveld J, Heck AJ, Karch F, Verrijzer CP. 2005. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 17:695–707. 10.1016/j.molcel.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 42.Sarkari F, Sanchez-Alcaraz T, Wang S, Holowaty MN, Sheng Y, Frappier L. 2009. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5:e1000624. 10.1371/journal.ppat.1000624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkari F, Wang X, Nguyen T, Frappier L. 2011. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One 6:e16598. 10.1371/journal.pone.0016598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, El-Deiry WS, Yang X. 2006. Critical role for Daxx in regulating Mdm2. Nat. Cell Biol. 8:855–862. 10.1038/ncb1442 [DOI] [PubMed] [Google Scholar]
- 45.Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L. 2003. Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J. Biol. Chem. 278:47753–47761. 10.1074/jbc.M307200200 [DOI] [PubMed] [Google Scholar]
- 46.Sarkari F, Wheaton K, La Delfa A, Mohamed M, Shaikh F, Khatun R, Arrowsmith CH, Frappier L, Saridakis V, Sheng Y. 2013. USP7/HAUSP is a regulator of UbE2E1/UbcH6. J. Biol. Chem.. 10.1074/jbc.M113.469262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT. 2007. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2:2728–2733. 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 49.Jones TA, Kjeldgaard M. 1997. Electron-density map interpretation. Methods Enzymol. 277:173–208. 10.1016/S0076-6879(97)77012-5 [DOI] [PubMed] [Google Scholar]
- 50.Yang J, O'Donnell L, Durocher D, Brown GW. 2012. RMI1 promotes DNA replication fork progression and recovery from replication fork stress. Mol. Cell Biol. 32:3054–3064. 10.1128/MCB.00255-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, Bensimon A. 2007. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 18:3059–3067. 10.1091/mbc.E06-08-0689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conti C, Herrick J, Bensimon A. 2007. Unscheduled DNA replication origin activation at inserted HPV 18 sequences in a HPV-18/MYC amplicon. Genes Chromosomes Cancer 46:724–734. 10.1002/gcc.20448 [DOI] [PubMed] [Google Scholar]
- 53.Tuduri S, Tourriere H, Pasero P. 2010. Defining replication origin efficiency using DNA fiber assays. Chromosome Res. 18:91–102. 10.1007/s10577-009-9098-y [DOI] [PubMed] [Google Scholar]
- 54.Ishimi Y. 1997. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 272:24508–24513. 10.1074/jbc.272.39.24508 [DOI] [PubMed] [Google Scholar]
- 55.Bochman ML, Schwacha A. 2009. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 73:652–683. 10.1128/MMBR.00019-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costa A, Onesti S. 2008. The MCM complex: (just) a replicative helicase? Biochem. Soc. Trans. 36:136–140. 10.1042/BST0360136 [DOI] [PubMed] [Google Scholar]
- 57.Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. 2011. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 44:147–159. 10.1016/j.molcel.2011.06.034 [DOI] [PubMed] [Google Scholar]
- 58.Hu M, Gu L, Li M, Jeffrey PD, Gu W, Shi Y. 2006. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 4:e27. 10.1371/journal.pbio.0040027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trujillo KM, Osley MA. 2012. A role for H2B ubiquitylation in DNA replication. Mol. Cell 48:734–746. 10.1016/j.molcel.2012.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ge XQ, Jackson DA, Blow JJ. 2007. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 21:3331–3341. 10.1101/gad.457807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ibarra A, Schwob E, Mendez J. 2008. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U. S. A. 105:8956–8961. 10.1073/pnas.0803978105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dimitrova DS, Gilbert DM. 1998. Regulation of mammalian replication origin usage in Xenopus egg extract. J. Cell Sci. 111(Part 19):2989–2998 [DOI] [PubMed] [Google Scholar]
- 63.Walter J, Newport JW. 1997. Regulation of replicon size in Xenopus egg extracts. Science 275:993–995. 10.1126/science.275.5302.993 [DOI] [PubMed] [Google Scholar]
- 64.Arias EE, Walter JC. 2007. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 21:497–518. 10.1101/gad.1508907 [DOI] [PubMed] [Google Scholar]
- 65.Blow JJ. 2001. Control of chromosomal DNA replication in the early Xenopus embryo. EMBO J. 20:3293–3297. 10.1093/emboj/20.13.3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. 2011. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44:325–340. 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bochman ML, Schwacha A. 2010. The Saccharomyces cerevisiae Mcm6/2 and Mcm5/3 ATPase active sites contribute to the function of the putative Mcm2-7 ‘gate.’ Nucleic Acids Res. 38:6078–6088. 10.1093/nar/gkq422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. 2011. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 18:471–477. 10.1038/nsmb.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antrobus R, Boutell C. 2008. Identification of a novel higher molecular weight isoform of USP7/HAUSP that interacts with the Herpes simplex virus type-1 immediate early protein ICP0. Virus Res. 137:64–71. 10.1016/j.virusres.2008.05.017 [DOI] [PubMed] [Google Scholar]