

Abstract
In budding yeast (Saccharomyces cerevisiae), the cell cycle-dependent telomere elongation by telomerase is controlled by the cyclin-dependent kinase 1 (Cdk1). The telomere length homeostasis is balanced between telomerase-unextendable and telomerase-extendable states that both require Cdc13. The recruitment of telomerase complex by Cdc13 promotes telomere elongation, while the formation of Cdc13-Stn1-Ten1 (CST) complex at the telomere blocks telomere elongation by telomerase. However, the cellular signaling that regulates the timing of the telomerase-extendable and telomerase-unextendable states is largely unknown. Phosphorylation of Cdc13 by Cdk1 promotes the interaction between Cdc13 and Est1 and hence telomere elongation. Here, we show that Cdk1 also phosphorylates Stn1 at threonine 223 and serine 250 both in vitro and in vivo, and these phosphorylation events are essential for the stability of the CST complexes at the telomeres. By controlling the timing of Cdc13 and Stn1 phosphorylations during cell cycle progression, Cdk1 regulates the temporal recruitment of telomerase complexes and CST complexes to the telomeres to facilitate telomere maintenance.
INTRODUCTION
Telomeres are specialized nucleoprotein structures found at the chromosome ends that are essential for maintaining chromosomal stability and genome integrity (1). In most eukaryotes, the telomeres are synthesized by the enzyme telomerase, a specialized ribonucleoprotein complex. Telomere elongation by telomerase counteracts the loss of telomeric DNA repeats due to the incomplete replication by DNA polymerase as well as nucleolytic degradation. The balance between telomere elongation and telomere attrition defines the telomere length homeostasis in different species. Loss of telomerase activity can lead to progressive shortening of telomeric DNA repeats and telomere dysfunction, which eventually inhibits the proliferation capacity of the cells and induces replicative senescence (2, 3). In humans, telomerase deficiencies due to mutations in DKC1, hTER, and hTERT as well as other telomere maintenance genes have been identified in patients suffering from conditions such as dyskeratosis congenita, aplastic anemia, myelodysplastic syndrome, and idiopathic fibrosis (4). These disease manifestations are invariably associated with a decreased capacity in cell and tissue renewal. Recent studies by genome-wide association (GWAS) also identify hCTC1 and hSTN1/OBFC1 (subunit of human hCTC1-hSTN1-hTEN1 complex) genomic loci that are associated with the mean human leukocyte telomere length (5–9). Similarly, the Saccharomyces cerevisiae Cdc13 and Stn1 (the homologue of hCTC1 and hSTN1) are essential for yeast telomere maintenance.
In budding yeast, the telomeric DNA is composed of 250 to 350 bp of double-stranded C1-3A/TG1-3 repeats with a short single-stranded TG1-3 3′ overhang (10, 11). To date, more than 150 genes have been identified which code for various proteins that contribute to telomere length maintenance in yeast (12, 13). One major protein involved in the maintenance of telomere length homeostasis is Cdc13, a DNA-binding protein that binds to single-stranded telomeric DNA. A loss of Cdc13 function (such as in a cdc13-1 mutant) results in the uncapping of the telomeres, excessive telomeric C-strand loss, and activation of the DNA damage checkpoint (14, 15). Exo1 nuclease and Rad9 and Rad24 checkpoint proteins each influence the end resection process at such uncapped telomeres that is also regulated by cyclin-dependent kinase 1 (Cdk1) (16–19). These data highlight the essential role that Cdc13 plays in protecting the chromosome ends. The telomere end protection function of Cdc13 requires at least two additional proteins, Stn1 and Ten1. Similarly to Cdc13, a loss of Stn1 or Ten1 function also results in telomere uncapping, generation of excessive G-rich single-stranded telomere overhangs, and activation of the DNA damage response (20, 21). In particular, Stn1 contains binding domains for both Cdc13 and Ten1, which are essential for the formation of the heterotrimeric Cdc13-Stn1-Ten1 (CST) complex at the chromosome ends. In the absence of Stn1, the interaction between Cdc13 and Ten1 is unstable (22). Recent bioinformatic analysis and protein structure modeling have indicated that Stn1 and Ten1 share several structural similarities with Rpa2 and Rpa3, the subunits of the replication protein A (RPA) complex (5, 23, 24). The heterotrimeric RPA complex binds nonspecifically to the single-stranded DNA and mediates diverse functions in eukaryotic DNA enzymology. These results have led to the proposal that Cdc13, Stn1, and Ten1 proteins form an RPA-like complex that protects telomeric ends specifically, a function dominated by the conventional RPA complex elsewhere in the genome. Such an RPA-like heterotrimeric CST complex is well conserved in different species, including yeast, plants, and mammals (5, 6, 25), highlighting the functional significance of the CST complex in telomere maintenance during evolution.
In addition to telomere end protection, Cdc13 is also essential for the recruitment of telomerase complex, which contains the protein catalytic subunit Est2 and the integral RNA template TLC1, as well as Est1 and Est3 in budding yeast (26, 27). Recruitment of the telomerase complex by Cdc13 relies on the direct interaction between Cdc13 and the Est1 subunit of telomerase (28). Disruption of this interaction or deletion of any of the telomerase components will result in telomere shortening and eventually senescence (29).
The telomere elongation by telomerase is cell cycle dependent and restricted to the late S/G2 phase of the cell cycle in vivo (30, 31). This is consistent with the notion that telomere elongation is coupled to the DNA replication machinery that is necessary for the synthesis of the opposite C1-3A strand of telomere DNA. Previous results from chromatin immunoprecipitation studies have demonstrated the interactions between protein factors involved in telomere elongation (including Est1, Est2, and Cdc13) and telomeres in the late S/G2 phase (32, 33). These results indicate that the regulation of telomere elongation by telomerase occurs at the recruitment and assembly of functional telomerase complexes on the telomeres. In budding yeast, the cell cycle-dependent telomere elongation by telomerase is controlled by a single cyclin-dependent kinase, Cdk1 (Cdc28). Inhibiting Cdk1 activity prevents the de novo addition of telomere repeats by telomerase (18). In addition, the generation of extended telomeric single-strand overhang, which is usually 12 to 14 nucleotides and becomes longer (>30 nucleotides) in length in late S/G2 phase (29, 34, 35), is also dependent on the Cdk1 kinase activity (18, 19). In budding yeast, the MRX complex coordinates with Sae2 to generate short 3′-terminal overhangs. More extensive end resection is then mediated by several pathways dependent on Exo1 or Sgs1/Dna2 (36, 37). Similar results are shown in mammals, as the 3′-overhang formation in mouse embryonic fibroblasts is controlled by shelterin complex in a cell cycle-dependent manner (38). The direct involvement of Cdk1 in telomere length homeostasis is further confirmed by the identification of a Cdk1 phosphorylation site (T308) in budding yeast Cdc13 (39–41). A defect in Cdc13 T308 phosphorylation results in the reduced recruitment of telomerase to the telomere and an ∼75-bp shortening of yeast telomere length (39). The Cdc13 T308 phosphorylation mutant affects telomerase-dependent telomere elongation but not the telomere end protection.
Previous results have shown that the recruitment of telomerase complex and the formation of CST complex counteract each other in terms of telomere length regulation (27, 39, 42–44). In fact, the telomeres bound by the yeast CST complexes are unextendable by telomerase in vitro (45). Recent results further demonstrate that the human CST complex inhibits telomere elongation by telomerase directly through primer sequestration and physical interaction with POT1-TPP1 (46). Similarly to telomerase, the CST complex is also recruited to the telomeres during the late S/G2 phase of the cell cycle in budding yeast (39, 44). These results indicate that the timing for the recruitment of telomerase complex by Cdc13 and the timing for the formation of CST complex at the telomere overlap during the cell cycle progression. How the timing of Cdc13-Stn1-Ten1-unextendable and Cdc13-telomerase-extendable states (47) is regulated during the cell cycle progression is largely unknown. Here, we present data showing that Stn1 threonine 223 and serine 250 are phosphorylated by Cdk1 in vivo and that the phosphorylations of Stn1 are essential for maintaining the stability of the CST complexes at the telomeres. We also show that the Cdk1-dependent phosphorylation of Cdc13 precedes the phosphorylations of Stn1. Therefore, by controlling the timing at which Cdc13 and Stn1 are phosphorylated during the cell cycle, Cdk1 plays an essential role in regulating the temporal recruitment of the telomerase complexes and the CST complexes. This is crucial for the telomere length homeostasis in budding yeast since there are only a limited number of telomeres that are elongated by telomerase during a single cell cycle (47). The remaining telomeres that are not bound by the telomerase complex would require the binding of CST complex to prevent uncontrolled end processing by nucleases such as Exo1.
MATERIALS AND METHODS
Yeast strains and plasmids.
For more details of yeast strains and plasmids, please see the supplemental material.
Immunoprecipitation and Western blot analysis.
For asynchronous culture, yeast strains were grown at 30°C in yeast extract-peptone-dextrose (YPD) until the optical density at 600 nm (OD600) reached 0.6 to 0.8. Typically, yeast pellets from a 50-ml cell culture were lysed in 1 ml of lysis buffer (50 mM HEPES, pH 7.0, 10% glycerol, 150 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1% Tween 20, 50 mM NaF, 50 mM β-glycerol phosphate, 1 mM Na3VO4 with Complete EDTA-free protease inhibitor tablet) by bead beating. The total proteins were normalized according to protein concentration determined by OD280 (Nanodrop ND-2000 spectrophotometer). The lysates were incubated with appropriate antibodies at 4°C for an hour. Protein G-agarose beads (Sigma) were then added and incubated with end-over-end rotation at 4°C overnight. The beads were then washed three times with yeast lysis buffer and resolved on 7.5% SDS-PAGE gels before Western blotting. For immunoprecipitation and Western blot analysis, anti-Myc 9E10 (Covance), anti-Flag M2 (Sigma), and affinity-purified rabbit anti-Stn1 serine 250 phosphopeptide-specific antibodies (Anaspec) were used.
For 4-amino-1-tert-butyl-3-(1′-naphthylmethyl)pyrazolo[3,4-d]pyrimidine (1-NM-PP1) treatments, 10 μM 1-NM-PP1 (final concentration) was added to asynchronous yeast culture at an OD600 of 0.6 for 15 min before harvest for immunoprecipitation and Western blot analysis.
Yeast synchronization.
For studies using synchronous cultures, overnight yeast cultures were diluted to an OD600 of 0.05 in fresh YPD medium. The cultures were then arrested in late G1 phase by the addition of 0.01 μg/ml α-factor at an OD of 0.3 and grown for an additional 2 h at 30°C. The α-factor was then removed, and cells were diluted at a 1:2 ratio into fresh YPD medium to allow the yeast culture to progress synchronously through the cell cycle at room temperature. Samples were collected at 15-min interval for subsequent analysis by fluorescence-activated cell sorting (FACS), immunoprecipitation, and chromatin immunoprecipitation (ChIP) analysis.
Phosphatase treatment of yeast lysates.
For phosphatase treatment, yeast lysates were divided into three aliquots. For treatment with lambda protein phosphatase (Upstate), MnCl2 (final concentration, 2 mM), dithiothreitol (DTT) (final concentration, 1 mM), and lambda protein phosphatase (final concentration, 1,000 U/ml) were added. For lambda protein phosphatase treatment with phosphatase inhibitor, NaF (final concentration, 50 mM), β-glycerol phosphate (final concentration, 50 mM), and Na3VO4 (final concentration, 1 mM) were added. The lysates were incubated at 37°C for an hour followed by immunoprecipitation and Western blot analysis as described above.
In vitro kinase assays.
Active Cdk1-as1-TAP/cyclin complexes were purified from sic1Δ pGAL-Cdc28-as1-TAP yeast cells as previously described (48), except that yeast lysates were prepared by bead beating. His-tagged Cdc13, Ten1, Est1, and Est3 recombinant proteins were expressed and batch purified from Tuner (DE3) Codon Plus bacteria (Novagen) using nickel-nitrilotriacetic acid (Ni-NTA) beads. For in vitro kinase assays, 5 to 10 μg of recombinant protein on Ni-NTA beads was incubated with 7 nM Cdc13-as1-TAP/cyclin complex, 1 mM creatine kinase-based ATP regeneration system, 1× kinase reaction buffer (25 mM HEPES-NaOH, pH 7.4, 10 mM NaCl, and 2 mM MgCl2), 1× phosphatase inhibitors (50 mM NaF, 80 mM β-glycerophosphate, and 1 mM Na3VO4), and 5 to 10 μCi N6-(benzyl)-[γ-32P]ATP in a final 100-μl volume at room temperature for 30 min as previously described (49). The His-tagged recombinant proteins were resolved by 10% SDS-PAGE, stained with Coomassie brilliant blue R-250, and exposed in a PhosphoImager cassette for 48 h. The protein phosphorylation levels were measured using ImageQuant (Molecular Dynamics). The protein phosphorylation levels were normalized to protein loading on Coomassie blue stain. The relative phosphorylation efficiency compared to the wild-type 6×His-Stn1(M) was shown.
Southern blot analysis and telomere length measurement.
Yeast genomic DNAs were digested with XhoI, separated by 0.8% agarose gel electrophoresis, and transferred to Hybond-XL membrane (GE). The blot was probed for telomeric C1–3A sequence using the 32P-end-labeled oligonucleotide (5′-TGTGGTGTGTGGGTGTGGTGT-3′). The in-gel native hybridization was performed as previously described (34) using 32P-end-labeled oligonucleotide (5′-CCCACCACACACACCCACACCC-3′). The quantification of the relative intensity of single-stranded telomeric DNA was done by ImageQuantTL (GE). Given that the telomere in the STN1 mutant is longer, the relative intensity of single-stranded telomere DNA is calculated as follows: total signal intensity of single-stranded X′ and Y′ telomere on nondenatured gel/X′ telomere signal intensity on denatured gel, normalized to the average signal from 0 min in STN1. The data are derived from three independent experiments.
Coimmunoprecipitation of TLC1 RNA.
The coimmunoprecipitation of TLC1 and Cdc13 was performed as previously described (39). Briefly, the lysates were incubated with 1 μl anti-Myc 9E10 ascites (Covance) at 4°C for 1 h. Dynabeads protein G magnetic beads (Invitrogen) were then added and incubated for an additional 4 h at 4°C. The beads were then washed 3 times with yeast lysis buffer. The total RNA on the beads was purified using the RNeasy minikit (Qiagen) followed by on-column DNase digestion as described in the RNA purification protocol. The amounts of TLC1 and actin mRNA copurified were then quantified using real-time quantitative reverse transcription-PCR (qRT-PCR) (Bio-Rad).
ChIP.
ChIP analysis was performed as previously described (33, 39), except that the cross-linking time was increased to 30 min. Cross-linked chromatin complexes were sheared by sonication using Qsonica sonicator Q700-Output 20%, with 1 s on and 1 s off for 6 × 12 pulses. Quantification of immunoprecipitated DNA was performed via real-time quantitative PCR (qPCR) analysis using the Bio-Rad CFX96 real-time PCR system. The PCR primers used were specific in amplifying a 114-bp sequence of the XII-L Y′ subtelomeric sequence and a 372-bp sequence of ARO1. The enrichment of telomere sequence was expressed as the fold change of Y′ subtelomeric DNA immunoprecipitated per reaction mixture from the tagged strain relative to that from the nontagged strain after normalization to the total amount of ARO1 enriched and the DNA input.
Yeast two-hybrid assay.
The two-hybrid interaction of Pol12 and Stn1 was done in the DY5736 reporter strain as previously described (43). The two-hybrid interaction of Stn1 with Cdc13 and Ten1 was done in the pJ694a reporter strain. The β-galactosidase activity was measured by liquid assay (50).
RESULTS
Stn1 is phosphorylated by Cdk1 in vivo.
When endogenously expressed Stn1-13×Myc protein was immunoprecipitated and resolved on SDS-PAGE gels, a distinct upward band shift was observed (Fig. 1A, lane 1), indicative of posttranslational modification of the protein. When the immunoprecipitated Stn1-13×Myc was treated with lambda protein phosphatase, no band shift was observed (Fig. 1A, lane 2). The loss of mobility shift can be prevented when the immunoprecipitated Stn1-13×Myc was treated with lambda protein phosphatase in the presence of a phosphatase inhibitor cocktail (Fig. 1A, lane 3). These results suggest that endogenously expressed Stn1 is phosphorylated in vivo. In order to identify the kinase that is responsible for the phosphorylation of Stn1 in vivo, we used a yeast strain expressing the Cdk1-as1 allele. The kinase activity of Cdk1-as1 can be inactivated by 5 to 25 μM 1-NM-PP1 (a small molecule that specifically inhibits the analogue-sensitive Cdk1-as1) within 10 min (51). The Stn1-13×Myc proteins immunoprecipitated from a yeast strain expressing the cdk1-as1 allele show an obvious mobility shift on SDS-PAGE, similar to Stn1-13×Myc proteins immunoprecipitated from a yeast strain harboring the CDK1 allele (Fig. 1A, compare lanes 4 and 5). Upon the addition of 10 μM 1-NM-PP1, the mobility shift of Stn1-13×Myc proteins in a yeast strain expressing the cdk1-as1 allele disappeared within 15 min of treatment (Fig. 1A, compare lane 6 to lane 5). These results suggest that Stn1 phosphorylation is dependent on Cdk1 in vivo. As a control, we also checked whether the gel mobility shift induced by the phosphorylation of Stn1-13×Myc is dependent on Tel1 and Mec1 (homologue of human ATM and ATR). As shown in Fig. 1A (lane 7), deletion of TEL1 and MEC1 simultaneously does not affect the mobility shift of Stn1-13×Myc. This implies that the mobility shift induced by the phosphorylation of Stn1-13×Myc is not dependent on Tel1 and Mec1. However, we cannot completely rule out the possibility that Tel1 and Mec1 also phosphorylate Stn1, which may not result in an obvious shift of Stn1-13×Myc protein mobility on SDS-PAGE gels.
FIG 1.

Phosphorylation of Stn1 by Cdk1. Endogenously expressed Stn1 is tagged at its C terminus with a 13×Myc tag. (A) Phosphorylation of Stn1 in vivo is dependent on Cdk1 but not Tel1 and Mec1. (B) Stn1 threonine 223 and serine 250 are phosphorylated in vivo. The expression of phosphoglycerate kinase (PGK) was used as a protein loading control. (C) Change in protein mobility due to S250A mutation is more obvious when the protein is resolved further by SDS-PAGE. (D) The Stn1 phosphoserine 250-specific antibody detects wild-type Stn1 and the Stn1-T223A mutant but not the Stn1-S250A and Stn1-T223A,S250A mutant proteins immunoprecipitated from asynchronous yeast cell lysates. (E) In yeast harboring the Cdk1-as1 allele, Stn1 phosphoserine 250-specific antibody detects Stn1-13×Myc immunoprecipitated from asynchronous yeast cell lysate that is abolished upon 1-NM-PP1 treatment for 15 min. (F) Phosphorylation of Stn1 threonine 223 and serine 250 by Cdk1-as1 in vitro. The top panel shows a Coomassie blue-stained gel showing the input of recombinant wild-type and mutant 6×His-Stn1(M) proteins that contain mutations at individual putative Cdk1 phosphorylation sites as indicated. Asterisks mark 6×His-Stn1(M) proteins. WT, wild type.
Threonine 223 and serine 250 of Stn1 are phosphorylated in vivo.
To identify the putative phosphorylation sites in Stn1 that are phosphorylated by Cdk1 in vivo, we engineered knock-in mutations that replace a threonine or serine residue with an alanine residue at each of the four potential Cdk1 phosphorylation sites in Stn1 (as indicated in Fig. S1A in the supplemental material). Mutation of these potential Cdk1 phosphorylation sites in Stn1 does not result in any obvious cell growth defect (data not shown). As shown in Fig. 1B, mutation of threonine 183 and threonine 203 to alanine does not affect the gel mobility of Stn1-13×Myc proteins immunoprecipitated from asynchronous A364a yeast cell lysates (Fig. 1B, compare lanes 2 and 3 to lane 1). In contrast, mutation of Stn1 threonine 223 to alanine results in the complete loss of the Stn1-13×Myc protein's gel mobility shift (Fig. 1B, compare lane 4 to lane 1). Mutation of Stn1 serine 250 to alanine also results in a slight change in the Stn1-13×Myc protein's gel mobility shift (Fig. 1B, compare lane 5 to lane 6). The effect of serine 250 mutation to alanine is more obvious when the protein is resolved further on SDS-PAGE gels (Fig. 1C, compare lane 2 to lane 1). These data suggest that the phosphorylation of threonine 223 in Stn1 is dependent on Cdk1 and responsible for the obvious mobility shift of Stn1-13×Myc on SDS-PAGE gels.
Since the serine-250-to-alanine mutation did not result in an obvious mobility change of Stn1 on SDS-PAGE gels, we raised a serine 250 phosphopeptide-specific antibody in the rabbit to further confirm whether serine 250 is also phosphorylated in vivo. Affinity-purified Stn1 serine 250 phosphopeptide-specific antibody detects both wild-type Stn1 and Stn1-T223A mutant proteins but not Stn1 mutant proteins with serine 250 mutated to alanine (Stn1-S250A and Stn1-T223A,S250A) immunoprecipitated from asynchronous yeast cell lysates (Fig. 1D, both panels; compare lanes 1 and 2 to lanes 3 and 4). The Stn1 S250 phosphopeptide-specific antibody detects only wild-type Stn1 recombinant protein that is phosphorylated by Cdk1/cyclin complexes in vitro, not the Stn1-S250A mutant protein (see Fig. S2 in the supplemental material). These findings validate the specificity of affinity-purified Stn1 serine 250 phosphopeptide-specific antibody and confirm the phosphorylation of serine 250 in vivo. These results also indicate that the faster-migrating form of Stn1-13×Myc contains only the serine 250-phosphorylated protein subpopulation while the slower-migrating form of Stn1-13×Myc contains a protein subpopulation that is phosphorylated at both threonine 223 and serine 250. The phosphorylation of T223 likely stimulates the phosphorylation of S250 in vivo as the phosphorylation of S250 is reduced in T223A mutant yeast (Fig. 1D, top panel, compare lane 2 to lane 1). In yeast harboring the Cdk1-as1 allele, the affinity-purified serine 250 phosphopeptide-specific antibody was able to detect wild-type Stn1-13×Myc protein immunoprecipitated from asynchronous yeast cell lysate that is abolished upon 1-NM-PP1 treatment (Fig. 1E, compare lane 1 to lane 2). These results confirm that the phosphorylation of Stn1 serine 250 is also dependent on Cdk1 in vivo. Both threonine 223 and serine 250 are located in the linker region of Stn1 away from its Ten1 and Cdc13 binding domains (see Fig. S1B in the supplemental material), and they are well conserved between different species of Saccharomyces (see Fig. S1C). Introduction of 13×Myc to the C terminus of endogenous STN1 does not affect yeast growth and results only in a slight telomere shortening (see Fig. S3A in the supplemental material). Loss-of-function mutations in Stn1 usually result in telomere elongation.
Cdk1 phosphorylates Stn1 threonine 223 and serine 250 directly in vitro.
To further validate whether Cdk1 can phosphorylate Stn1 threonine 223 and serine 250 directly, we incubated bacterially expressed recombinant 6×His-Stn1(M) proteins with Cdk1-as1/cyclin complexes purified from asynchronous yeast culture for in vitro kinase assay as previously described (39). As shown in Fig. 1F, wild-type 6×His-Stn1(M) protein can be phosphorylated by Cdk1-as1/cyclin complex efficiently in vitro. Similar results were observed for 6×His-Stn1(M) proteins with an alanine substitution at threonine 183 or threonine 203. However, when the threonine 223 or serine 250 is mutated to alanine, the phosphorylations of 6×His-Stn1(M) proteins by Cdk1-as1/cyclin complex are dramatically reduced. Mutations of both threonine 223 and serine 250 to alanines result in a complete abolishment of phosphorylation by Cdk1-as1/cyclin complex in vitro. Altogether, these results indicate that Cdk1 phosphorylates Stn1 at threonine 223 and serine 250 directly in vitro.
Cell cycle-dependent phosphorylation of Cdc13 and Stn1.
To identify the specific cell cycle stage in which Stn1 is phosphorylated and to compare the phosphorylation timings of Stn1 and Cdc13 during cell cycle progression, α-factor-synchronized yeast cultures (coexpressing Cdc13-3×Flag and Stn1-13×Myc) were analyzed in a series of time points every 15 min. As shown in Fig. 2A, phosphorylation of Cdc13 was readily detected in the time span from 30 to 60 min after α-factor synchronization release. Fluorescence-activated cell sorting (FACS) analysis of the cellular DNA content reveals that the phosphorylation of Cdc13 occurs in S to early G2 phases of the cell cycle (Fig. 2B), which coincides with telomere elongation by telomerase in vivo as shown previously (30–33). Interestingly, the phosphorylations of Stn1-T223 (as indicated by the lower-mobility shift of Stn1-13×Myc proteins on SDS-PAGE gels) and Stn1-S250 (as indicated by the serine 250 phosphopeptide-specific antibody) were detected at about the same time and peaked around 45 to 90 min after α-factor synchronization release. FACS analysis of the cellular DNA content showed that the phosphorylation of Stn1 occurred at the late S phase and peaked at the G2 phase. These results suggest that the Cdk1-dependent phosphorylations of Cdc13 and Stn1 occur sequentially and overlap during cell cycle progression.
FIG 2.
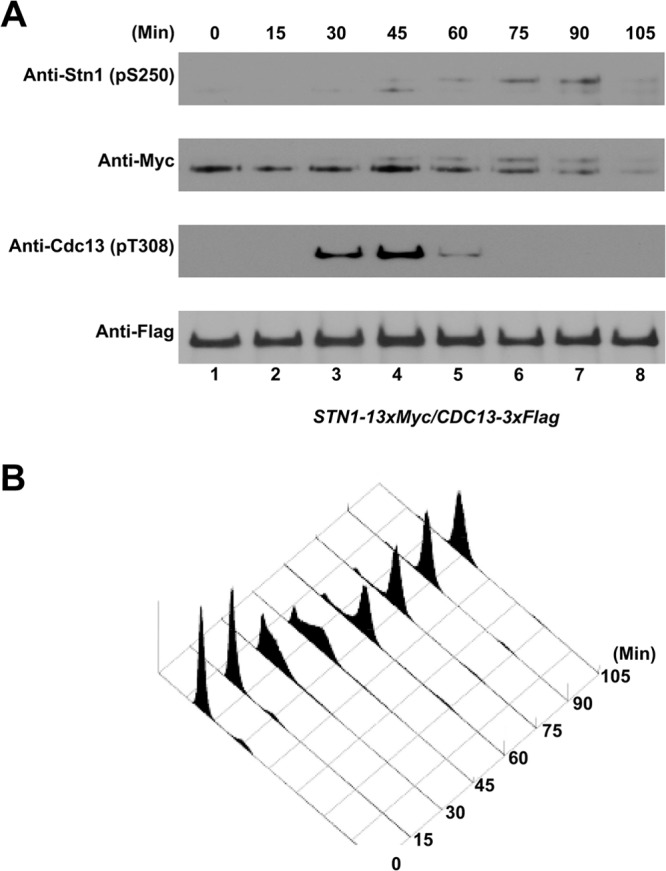
Cell cycle-dependent phosphorylation of Cdc13-T308 and Stn1-T223,S250 in vivo. A yeast strain coexpressing Cdc13-3×Flag and Stn1-13×Myc (under the control of endogenous promoters) was synchronized using α-factor. The yeast culture was then released into the cell cycle progression, and lysates were collected and analyzed at 15-min intervals. (A) Phosphorylation of Cdc13-T308 was detected 30 to 60 min after α-factor synchronization release. The phosphorylations of Stn1-T223 and Stn1-S250 were detected 45 to 90 min after α-factor synchronization release. (B) FACS analysis of the cell cycle progression after α-factor synchronization release.
Mutations in the Stn1 phosphorylation sites result in telomere lengthening.
Loss of Cdk1-dependent phosphorylation of Cdc13 results in telomere shortening (39, 41). To further understand the functional significance of cell cycle-dependent phosphorylation of Stn1, we first asked whether eliminating the phosphorylation of Stn1 would affect the telomere length in vivo. A diploid yeast strain (A364a background) heterozygous for the threonine 183 (T183A)-, threonine 203 (T203A)-, threonine 223 (T223A)-, or serine 250 (S250A)-to-alanine mutation was sporulated and dissected. The telomere length of each individual yeast colony derived from the dissected spores was measured after ∼100 cell divisions on YPD plates at 30°C. Southern blot hybridization using a telomere probe (teloblot) showed that the stn1-T183A and stn1-T203A mutations did not affect telomere length maintenance in vivo (Fig. 3A, compare lanes 1 and 2 to lanes 3 and 4 and lanes 5 and 6 to lanes 7 and 8). In contrast, the stn1-T223A and stn1-S250A mutations resulted in significant telomere lengthening (Fig. 3A, compare lanes 9 and 10 to lanes 11 and 12 and lanes 13 and 14 to lanes 15 and 16) and suggest that the Cdk1-dependent phosphorylations of Stn1 are important for telomere maintenance. Simultaneous mutation of threonine 223 and serine 250 to alanine (stn1-T223A,S250A) results in further telomere lengthening compared to the stn1-T223A or stn1-S250A mutation alone (Fig. 3A, compare lanes 17 and 18 to lanes 19 and 20). Progressive telomere lengthening can be observed in stn1-T223A,S250A mutant yeast strains at early passages and stabilized thereafter (see Fig. S3B in the supplemental material). To further investigate the functional interaction of Cdk1-dependent phosphorylations of Cdc13 and Stn1, we also probed the telomere length phenotype in yeast strains harboring both cdc13-T308A and stn1-T223A,S250A mutations. As shown in Fig. 3B, mutation of CDC13 threonine 308 to alanine resulted in telomere shortening (Fig. 3B, compare lane 3 to lane 1) while mutations of STN1 threonine 223 and serine 250 to alanine resulted in telomere elongation (Fig. 3B, compare lane 2 to lane 1). Interestingly, the yeast cells containing both CDC13 and STN1 phosphorylation site mutations have an intermediate telomere length phenotype (Fig. 3B, lane 4), suggesting additive interaction between Cdk1-dependent phosphorylation of Cdc13 and Stn1. Replacement of STN1 threonine 223 or serine 250 with aspartic acid or glutamic acid, which simulates the negative charge produced by phosphorylation, failed to rescue the telomere phenotype of alanine mutant (see Fig. S4, lanes 1 to 16, in the supplemental material), suggesting that the phosphorylation moiety rather than the negative charge associated with phosphorylation is necessary for the function of Stn1 in vivo. In addition, mutations of proline 224 (stn1-P224A) or proline 251 (stn1-P251A) (on the Cdk1 phosphorylation consensus motif) to alanine, which prevent Cdk1-dependent phosphorylation, also resulted in a telomere-lengthening phenotype similar to that of the stn1-T223A or stn1-S250A mutation, respectively (see Fig. S4, lanes 17 to 24). Therefore, these results further validate the functional significance of the Cdk1-dependent phosphorylation of Stn1 in telomere length maintenance in vivo.
FIG 3.

Mutations in STN1 phosphorylation sites cause telomere elongation in vivo. (A) Only mutation of STN1 threonine 223 or serine 250 to alanine results in telomere elongation. Double mutations of both threonine 223 and serine 250 to alanines result in further telomere lengthening. (B) Yeast cells that contain both the CDC13 and STN1 phosphorylation site mutations exhibit an intermediate telomere length phenotype compared to CDC13 or STN1 mutant alone. (C) Enhanced efficiency of telomerase recruitment by Cdc13 in STN1 phosphorylation mutants, as shown by an increased association between Cdc13 and TLC1 in coimmunoprecipitation experiments followed by real-time qRT-PCR (TLC1, RT-PCR primers for specific amplification of TLC1 RNA; actin, RT-PCR primers for specific amplification of actin mRNA as negative control). The data are derived from three independent experiments. (D) Straight Western blotting shows that the expression of Cdc13-13×Myc is not affected by stn1 phosphorylation mutations in vivo.
Telomere lengthening in the STN1 phosphorylation mutants is telomerase dependent.
To test whether the telomere-lengthening phenotype observed in STN1 phosphorylation mutants relies on telomerase-dependent telomere maintenance pathways, we compared the telomere length in the stn1-T223A,A250A single mutant with those in other single mutants of known telomerase-dependent telomere maintenance factors (yku70Δ, yku80Δ, est2Δ, tel1Δ, and mec1Δ/sml1Δ) as well as double mutants containing the stn1-T223A,A250A mutation in combination with yku70Δ, yku80Δ, est2Δ, tel1Δ, or mec1Δ/sml1Δ mutation. As shown in Fig. S5 in the supplemental material, telomere lengthening induced by the stn1-T223A,A250A single mutant is telomerase dependent, as compromised telomerase-dependent telomere lengthening inhibits the telomere-lengthening phenotype of the stn1-T223A,A250A mutant. Consistent with these data, deletion of EST2 completely abolished the telomere elongation induced by stn1-T223A,A250A mutation. These results suggest that the telomere-lengthening phenotype observed in STN1 phosphorylation mutants relies on telomerase-dependent telomere elongation.
To address whether the telomere lengthening observed in the STN1 phosphorylation mutant is due to increased recruitment of telomerase complex to the telomere, we proceeded to quantify the amount of TLC1 that was coimmunoprecipitated with Cdc13 by qRT-PCR. Increased TLC1 that coimmunoprecipitated with Cdc13 has been shown to be correlated with increased recruitment of telomerase complex to the telomere and telomere elongation (39). As shown in Fig. 3C, mutations of the Cdk1 phosphorylation sites in STN1 to alanine (stn1-T223A, stn1-S250, and stn1-T223A,S250A) resulted in an increased amount of TLC1 that was coimmunoprecipitated with Cdc13 but not the control actin mRNA (Fig. 3C). The expression of TLC1 is not affected by the loss of Stn1 phosphorylation (data not shown). Similarly, the expression of Cdc13 was not significantly affected by the loss of Stn1 phosphorylation (Fig. 3D). These data indicated that the loss of Cdk1-dependent phosphorylation of Stn1 resulted in an increased recruitment of the telomerase complex to the telomeres. These results are consistent with telomere elongation observed in STN1 phosphorylation site mutants. These data also imply that the Cdk1-dependent phosphorylation of Stn1 inhibits telomere elongation by telomerase.
STN1 phosphorylation site mutations resulted in a cell cycle-dependent increase in single-stranded telomere G-rich overhangs.
Formation of the Cdc13-Stn1-Ten1 complex could compete with the recruitment of the telomerase complex by Cdc13 and thereby inhibit telomerase-dependent telomere addition (27, 28, 45, 46). Therefore, it is possible that the telomere-lengthening phenotype observed in STN1 phosphorylation mutants is due to reduced binding of the CST complexes at the telomeres, which in turn will affect the processing of single-stranded telomere overhangs (15, 20, 21). Therefore, we performed Southern blot analysis to determine if there was a change in the amount of single-stranded telomere overhangs during cell cycle progression. The single-stranded telomere overhangs were visualized by in-gel native hybridization using an end-labeled C-rich oligonucleotide under nondenaturing conditions. As shown in Fig. 4A, the single-stranded TG1–3 DNA appeared in a cell cycle-dependent manner, which peaked between 30 and 75 min after α-factor synchronization release in both wild-type and stn1-T223A,A250A mutant yeast strains. The cells are in S/G2 phase between 30 and 75 min after α-factor synchronization release as shown by FACS in Fig. 4C and D. An increase in signal from single-stranded telomere G-rich overhangs can be detected in a yeast strain harboring mutations in the STN1 phosphorylation sites (Fig. 4A and E, compare STN1 to stn1-T223A,S250A). Since the amounts of telomeric DNA for both STN1 and stn1-T223A,S250A were comparable (Fig. 4B, in-gel hybridization under denaturing conditions), these data suggest that the loss of Cdk1-dependent phosphorylation of Stn1 results in an increase in single-stranded telomere overhangs. Similar results were observed in stn11-T223A and stn1-S250A single mutants to a lesser extent (see Fig. S6 in the supplemental material).
FIG 4.

Mutations in STN1 phosphorylation sites result in a cell cycle-dependent increase in single-stranded telomere G-rich overhangs. (A) In-gel hybridization under nondenaturing conditions showed that increased signal from the single-stranded telomere G-rich overhangs can be detected in a yeast strain harboring the Cdk1 phosphorylation site mutations (stn1-T223A,S250A). (B) The same gel as in panel A, probed with a telomere-specific probe under denaturing conditions. (C and D) FACS analysis showing the DNA content of wild-type and STN1 phosphorylation mutant yeast strains from α-factor synchronization release that were used for the in-gel hybridization. (E) Quantification of the relative intensity of cell cycle-dependent generation of single-stranded telomere DNA in STN1 and stn1-T223A,S250A yeast strains. The data are derived from three independent experiments.
This increase in single-stranded telomere overhangs can be caused by either an increase in telomere elongation by telomerase or an increase in telomere end processing. In order to distinguish between these two possibilities, we first asked whether deletion of EST2 can suppress the increase in single-stranded telomere overhangs observed in the stn1-T223A,A250A mutant. As shown in Fig. S7 in the supplemental material, an increased signal from single-stranded telomere G-rich overhang was detected in the est2Δ/stn1-T223A,S250A yeast strain compared to the est2Δ yeast strain. These results suggest that telomere elongation by telomerase does not contribute significantly to the marked increase in single-stranded overhangs as we observed in the stn1-T223A,S250A yeast strain. Exo1 has been shown to be a key nuclease involved in the end processing of telomere and generation of single-stranded G-rich overhangs (17). Therefore, we investigated next whether the increase in single-stranded telomere overhangs observed in the stn1-T223A,A250A mutant can be suppressed by deleting EXO1. As shown in Fig. 5A to E, in-gel hybridization under nondenaturing conditions indicated that the increased single-stranded telomere G-rich overhangs in the stn1-T223A,S250A mutant yeast strain were suppressed after the deletion of EXO1. These results further validate that the increase in single-stranded telomere overhangs is a result of an increase in telomere end processing, a process that can be suppressed by recruitment of the CST complexes to the telomeres. The increase in single-stranded telomere G-rich overhangs in the stn1-T223A,S250A mutant yeast strain during S/G2 phase does not trigger a DNA damage response, as indicated by the lack of Rad53 phosphorylation (see Fig. S8 in the supplemental material).
FIG 5.

The increase in single-stranded telomere G-rich overhangs in the STN1 phosphorylation mutant is due to an increase in telomere end processing. (A) In-gel hybridization under nondenaturing conditions showing that the increased single-stranded telomere G-rich overhangs in the stn1-T223A,S250A phosphorylation mutant can be suppressed by deletion of EXO1 (stn1-T223A,S250A/exo1Δ). (B) Same gel as in panel A, probed with telomere-specific probe under denaturing conditions. (C and D) FACS analysis showing the DNA content of stn1-T223A,S250A and stn1-T223A,S250A/exo1Δ mutant yeast strains that were used for the in-gel hybridization after α-factor synchronization release. (E) Quantification of the relative intensity of cell cycle-dependent generation of single-stranded telomere DNA in stn1-T223A,S250A and stn1-T223A,S250A/exo1Δ mutant yeast strains. The data are derived from three independent experiments.
Cdk1-dependent phosphorylation of Stn1 is essential for the recruitment of the CST complex to the telomere.
Since the loss of Cdk1-dependent phosphorylation of Stn1 results in telomere elongation and a significant increase in single-stranded telomere overhangs, we hypothesized that there could be a defect in telomere end protection by the CST complex. As threonine 223 and serine 250 are located in the linker region connecting the Cdc13 and Ten1 binding domain on Stn1 (see Fig. S1B in the supplemental material), we proceeded to investigate if the loss of Cdk1-dependent phosphorylation of Stn1 would affect its interaction with Cdc13 or Ten1. As a control, we first tested whether the loss of the Cdk1-dependent phosphorylation of Stn1 affects its interaction with Pol12 (Fig. 6A). The yeast two-hybrid results shown in Fig. 6A indicate that the Cdk1-dependent phosphorylation of Stn1 is not necessary for the efficient interaction between Stn1 and Pol12. As shown in Fig. 6B and C, loss of Cdk1-dependent phosphorylation in Stn1 does not affect the interaction of Stn1 with Cdc13 or Ten1 in a yeast two-hybrid assay. Consistent with these results, the interaction of endogenous Ten1 and Stn1 is not affected by the Stn1 phosphorylation site mutations either (see Fig. S9 in the supplemental material).
FIG 6.

Cdk1-dependent phosphorylations of Stn1 are essential for the recruitment of the CST complex to the telomere. (A) Stn1 phosphorylations are not necessary for the interaction between Stn1 and Pol12, as shown by the yeast two-hybrid assay. (B) Stn1 phosphorylations are not necessary for the interaction between Stn1 and Cdc13, as shown by the yeast two-hybrid assay. (C) Stn1 phosphorylations are not necessary for the interaction between Stn1 and Ten1, as shown by the yeast two-hybrid assay. LexA-DBD, LexA DNA-binding domain alone; AD, Gal4 activation domain alone; POL12(N), POL12 N-terminal 381 amino acids fused to the LexA DNA-binding domain; Gal-DBD, Gal4 DNA-binding domain alone; CDC13, CDC13 with deletion in the DNA-binding domain fused to Gal4 DNA-binding domain; TEN1, full-length wild-type TEN1 fused to Gal4 DNA-binding domain; STN1, full-length wild-type STN1 fused to Gal4 activation domain; stn1, full-length stn1-T223A,S250A mutant fused to Gal4 activation domain. (D) Coimmunoprecipitation (Co-IP) of Cdc13-13×Myc and Ten1-3×Flag is dramatically reduced in the stn1-T223A,S250A mutant yeast strain compared to the STN1 yeast strain. The asterisks denote nonspecific bands in the background. The Cdc13-13×Myc and Ten1-3×Flag proteins are marked by the arrows. (E) Coimmunoprecipitation of Ten1-13×Myc and Cdc13-3×Flag is dramatically reduced in the stn1-T223A,S250A mutant yeast strain compared to the STN1 yeast strain. The Ten1-13×Myc and Cdc13-3×Flag proteins are marked by the arrowheads. (F) ChIP assays in synchronous yeast cultures show that the cell cycle-dependent recruitment of Cdc13 to the telomere is not affected in the stn1-T223A,S250A mutant. (G) ChIP assays in synchronous yeast cultures show that the cell cycle-dependent recruitment of Ten1 to telomere is dramatically reduced in the stn1-T223A,S250A mutant. (H) ChIP assays in synchronous yeast cultures show that the cell cycle-dependent recruitment of Stn1 to the telomere is dramatically reduced in the stn1-T223A,S250A mutant. (I) In contrast, ChIP assays in synchronous yeast cultures show the prolonged association of Est1 and the telomere in the stn1-T223A,S250A mutant.
These results indicate that the Cdk1-dependent phosphorylations of Stn1 are not essential for the interaction of Stn1 with Cdc13 or Ten1, respectively. Since the end protection function of the CST complex relies on the stable formation of the trimeric CST complex on the telomere and the interaction between Cdc13 and Ten1 is unstable without Stn1 (22), we asked whether the Cdk1-dependent phosphorylation of Stn1 would affect the stability of the CST complex by probing the association of Cdc13 and Ten1. As shown in Fig. 6D (compare lane 2 to lane 3), immunoprecipitation of Ten1-3×Flag results in efficient coimmunoprecipitation of Cdc13-13×Myc in the STN1 yeast strain but is dramatically reduced in the stn1-T223A,A250A mutant yeast strain. Similar results are observed for the reciprocal coimmunoprecipitation of Cdc13-3×Flag and Ten1-13×Myc (Fig. 6E). To further validate our observation, we used a chromatin immunoprecipitation (ChIP) assay to investigate the recruitment of the CST complex and telomerase complex to the telomere during the cell cycle in synchronous yeast cultures (see Fig. S10 in the supplemental material). As shown in Fig. 6F, ChIP assays in synchronous yeast cultures show that the cell cycle-dependent recruitment of Cdc13 to the telomere is not affected in the stn1-T223A,S250A mutant. However, Cdk1-dependent Stn1 phosphorylation is necessary for the efficient recruitment of Stn1 and Ten1 to the telomere, as dramatic reduction of the recruitment of Ten1 (Fig. 6G; see also Fig. S11 in the supplemental material) and Stn1 (Fig. 6H) to the telomere was observed in a yeast strain harboring the stn1-T223A,S250A mutant allele. Similar results were also observed in stn1-T223A and stn1-S250A single mutants to a lesser extent (see Fig. S12 and S13 in the supplemental material). Consistent with previous studies showing the cell cycle-dependent recruitment of both Cdc13 and Stn1 to the telomere (39, 44), our results also show that the recruitment of Ten1 to telomere is cell cycle dependent. Interestingly, in a yeast strain harboring the stn1-T223A, stn1-S250A, stn1-T223A,S250A mutant allele, we observed prolonged association of Est1 and the telomere late in G2/M phase (Fig. 6I; see also Fig. S12D). These results indicate that the Cdk1-depdendent phosphorylations of Stn1 at threonine 223 and serine 250 are essential for the stable formation of CST complexes at the telomeres. Loss of such Cdk1-dependent phosphorylations results in the destabilization of the CST complexes at the telomeres, which in turn leads to the increased telomere end processing as well as prolonged telomere elongation by telomerase.
DISCUSSION
The regulation of telomere length homeostasis in vivo is balanced between Stn1-Ten1-unextendable and telomerase-extendable states (47) that both require Cdc13. Our data indicate that the Cdk1-dependent phosphorylations of Stn1 threonine 223 and serine 250 are essential for the stable formation of CST heterotrimeric complex on the telomeres. Although the phosphorylation of Stn1 is not necessary for the interaction of Stn1 with Cdc13 or Ten1 individually, it is necessary for Stn1 to interact with Cdc13 and Ten1 simultaneously to form a functional heterotrimeric complex. As the phosphorylation sites for Stn1 threonine 223 and serine 250 are located in the linker region connecting the binding domains for Cdc13 and Ten1, it is likely that the phosphorylations of these sites induces a conformational change of the protein, which allows it to bind to both Cdc13 and Ten1 simultaneously. In the absence of Cdk1-dependent phosphorylation, Stn1 may adopt a more constrained conformation that prevents it from interacting with both Cdc13 and Ten1 at the same time, which disrupts the stability of CST complex. This is consistent with recent results showing that the interaction between Cdc13 and Ten1 is unstable in the absence of Stn1 (22).
Mutation of Stn1 threonine 223 and serine 250 to alanines results in telomere elongation. However, the phosphomimetic mutations do not result in telomere shortening. These data indicate that the phosphorylation moiety rather than the negative charges is essential for the function of Stn1. The increased single-stranded telomere G tail in Stn1 phosphorylation mutants is restricted to S/G2 phase and does not trigger the DNA damage checkpoint. This is unlike typical capping-deficient strains with structure mutations in CDC13 and STN1, which have long single-stranded telomere G tail persistent through the cell cycle and activation of the cellular DNA damage checkpoint (19).
Cdk1-dependent phosphorylations of Cdc13 and Stn1 occur at specific stages during the cell cycle progression. Our results indicate that the phosphorylation of Cdc13 precedes the phosphorylation of Stn1. Briefly, the phosphorylation of Cdc13 occurs in the S phase to the early G2 phase of the cell cycle, while the phosphorylation of Stn1 occurs close to the late S phase and peaks at the G2 phase of the cell cycle. How Cdk1 controls the timing of Cdc13 and Stn1 phosphorylations remains to be addressed. Based on our findings, we would like to propose the following mechanism for the Cdk1-dependent phosphorylation of Cdc13 and Stn1 (Fig. 7): when cells enter the S phase, the telomeres are replicated by DNA polymerase. Subsequent telomere end processing by the MRX complex as well as Exo1 and Sgs1/Dna2 will produce single-stranded telomere overhangs for the binding of Cdc13. The phosphorylation of Cdc13 threonine 308 stimulates the recruitment of telomerase complexes to telomeres through the interaction between Est1 and phosphorylated Cdc13. Since there are a limited number of telomerase complexes available in any given yeast cell, and only ∼7% of telomeres are elongated by telomerase during a single cell cycle (47), the remaining telomeres that are not bound by the telomerase complex will require the binding of CST complexes to prevent uncontrolled end processing by nucleases, such as Exo1 and Sgs1/Dna2. The subsequent phosphorylations of Stn1 threonine 223 and serine 250 result in the stabilization of the Stn1 interaction with both Cdc13 and Ten1, thus facilitating the formation of functional CST complexes at the telomeres. By controlling the timing at which phosphorylation of Cdc13 and Stn1 occurs, Cdk1 is able to regulate the recruitment of telomerase complexes to telomeres for telomere elongation and subsequently the formation of CST complexes on telomeres for telomere protection as well as telomerase inhibition during cell cycle progression in yeast.
FIG 7.

Cdk1 regulates the recruitment of telomerase complex and CST complex to the telomere during cell cycle progression. During the S phase, only a small fraction of telomeres are elongated. Phosphorylation of Cdc13 by Cdk1 results in the preferential recruitment of telomerase complexes to the telomeres. The subsequent phosphorylation of Stn1 by Cdk1 results in the stable formation of Cdc13-Stn1-Ten1 (CST) complex. On the one hand, the binding of CST complex is necessary to protect the telomeres that are not elongated by telomerase complexes from being further processed by the nucleases; on the other hand, the CST complex is necessary to inhibit the telomere elongation by telomerase. Thus, the cell cycle-dependent phosphorylations of Cdc13 and Stn1 by Cdk1 regulate the recruitment of telomerase complexes and CST complexes to the telomeres.
Recent studies have revealed that the Cdc13-Stn1-Ten1 (CST) complex is the functional homologue of the heterotrimeric replication protein A (RPA) complex, the major single-stranded DNA-binding activity in eukaryotic cells (23, 24, 52). Subunits of the trimeric RPA complex, such as RPA1 and RPA2, are phosphorylated in vivo (53–55). These phosphorylation events serve to regulate the activity of RPA in vivo (56–58). The regulation of RPA function by phosphorylations also resembles the regulation of telomere elongation and telomere protection through Cdk1-dependent phosphorylations of Cdc13 and Stn1. The telomeres bound by the yeast CST complexes are unextendable by telomerase in vitro (45). The human CST complex has been identified and plays an important role in telomere maintenance (5, 6, 59–62). Recent results have shown that the human CST complex inhibits telomere elongation by telomerase directly through primer sequestration and physical interaction with POT1-TPP1 (46). Consistent with this finding, inhibition of Cdc13-dependent recruitment of Stn1-Ten1 to the telomere results in significant telomere elongation, while overexpression of Stn1 results in telomere shortening (27, 39, 42–44). Such Stn1-Ten1-Cdc13-unextendable and telomerase-Cdc13-extendable states of the telomere must be well coordinated during cell cycle progression in order to maintain telomere length homeostasis. Our results provide a clue of how Cdk1 regulates telomere elongation by controlling the recruitment of telomerase complex and the competing Stn1-Ten1 complex by Cdc13 to the telomeres during different stages of the cell cycle. Recent identification of a mammalian CST complex that plays multiple roles in telomere maintenance and nontelomeric functions (5, 6, 63–65) provides further evidence that yeast and humans share similar regulatory mechanisms in telomere length homeostasis. Genome-wide association also identified single nucleotide polymorphisms (SNPs) in OBFC1 (human homologue of yeast Stn1) with shortened human leukocyte telomere length (8). One of the two SNPs (rs2487999) that result in nonsynonymous alterations resides in a region that is aligned with the linker region of budding yeast Stn1 that contains the two Cdk1 phosphorylation sites. As such, it is important to illustrate how the CST complex is regulated during telomere elongation and telomere DNA replication in order to gain insights into the pathology caused by the mutations in these factors.
In conclusion, the data that we present here provide compelling evidence for the roles of Cdk1 in controlling the telomere elongation and telomere protection by directly phosphorylating both Cdc13 and Stn1 in vivo. Given that Rpa2 (the middle subunit of an RPA trimeric complex, like Stn1) is a target for multiple kinases, it would be interesting to determine whether Stn1 is phosphorylated by other kinases as well and whether these phosphorylations have any effect on the regulation of telomere elongation by Stn1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Victoria Lundblad for providing the yeast two-hybrid plasmids pVL705 and pVL859, as well as the pJ694a yeast strain. We thank Constance I. Nugent for providing the yeast two-hybrid plasmids pCN124 (pAS1-TEN1) and pCN181 (pACT2-STN1). We thank Michel Charbonneau for providing the yeast two-hybrid plasmids pAS2-TEN1, pACT2-TEN1, pAS2-STN1, pACT2-STN1, pAS2-CDC13, and pACT2-CDC13. We thank XiaoLan Zhao for providing the yeast two-hybrid plasmids pOBD-CDC13 and pOAD-STN1. We thank Elizabeth H. Blackburn and Uttam Surana for critical reading of the manuscript.
This work was supported by grants from MOE tier 2 funding to S.L.
Footnotes
Published ahead of print 28 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01235-13.
REFERENCES
- 1.Blackburn EH. 2000. Telomere states and cell fates. Nature 408:53–56. 10.1038/35040500 [DOI] [PubMed] [Google Scholar]
- 2.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, II, Greider CW, DePinho RA. 1998. Essential role of mouse telomerase in highly proliferative organs. Nature 392:569–574. 10.1038/33345 [DOI] [PubMed] [Google Scholar]
- 3.Lundblad V, Szostak JW. 1989. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57:633–643. 10.1016/0092-8674(89)90132-3 [DOI] [PubMed] [Google Scholar]
- 4.Lansdorp PM. 2009. Telomeres and disease. EMBO J. 28:2532–2540. 10.1038/emboj.2009.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F. 2009. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell 36:193–206. 10.1016/j.molcel.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 6.Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE. 2009. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol. Cell 36:207–218. 10.1016/j.molcel.2009.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Codd V, Mangino M, van der Harst P, Braund PS, Kaiser M, Beveridge AJ, Rafelt S, Moore J, Nelson C, Soranzo N, Zhai G, Valdes AM, Blackburn H, Mateo Leach I, de Boer RA, Kimura M, Aviv A, Goodall AH, Ouwehand W, van Veldhuisen DJ, van Gilst WH, Navis G, Burton PR, Tobin MD, Hall AS, Thompson JR, Spector T, Samani NJ. 2010. Common variants near TERC are associated with mean telomere length. Nat. Genet. 42:197–199. 10.1038/ng.532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy D, Neuhausen SL, Hunt SC, Kimura M, Hwang SJ, Chen W, Bis JC, Fitzpatrick AL, Smith E, Johnson AD, Gardner JP, Srinivasan SR, Schork N, Rotter JI, Herbig U, Psaty BM, Sastrasinh M, Murray SS, Vasan RS, Province MA, Glazer NL, Lu X, Cao X, Kronmal R, Mangino M, Soranzo N, Spector TD, Berenson GS, Aviv A. 2010. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl. Acad. Sci. U. S. A. 107:9293–9298. 10.1073/pnas.0911494107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangino M, Hwang SJ, Spector TD, Hunt SC, Kimura M, Fitzpatrick AL, Christiansen L, Petersen I, Elbers CC, Harris T, Chen W, Srinivasan SR, Kark JD, Benetos A, El Shamieh S, Visvikis-Siest S, Christensen K, Berenson GS, Valdes AM, Vinuela A, Garcia M, Arnett DK, Broeckel U, Province MA, Pankow JS, Kammerer C, Liu Y, Nalls M, Tishkoff S, Thomas F, Ziv E, Psaty BM, Bis JC, Rotter JI, Taylor KD, Smith E, Schork NJ, Levy D, Aviv A. 2012. Genome-wide meta-analysis points to CTC1 and ZNF676 as genes regulating telomere homeostasis in humans. Hum. Mol. Genet. 21:5385–5394. 10.1093/hmg/dds382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellinger RJ, Wolf AJ, Zakian VA. 1993. Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell 72:51–60. 10.1016/0092-8674(93)90049-V [DOI] [PubMed] [Google Scholar]
- 11.Zakian VA. 1996. Structure, function, and replication of Saccharomyces cerevisiae telomeres. Annu. Rev. Genet. 30:141–172. 10.1146/annurev.genet.30.1.141 [DOI] [PubMed] [Google Scholar]
- 12.Askree SH, Yehuda T, Smolikov S, Gurevich R, Hawk J, Coker C, Krauskopf A, Kupiec M, McEachern MJ. 2004. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc. Natl. Acad. Sci. U. S. A. 101:8658–8663. 10.1073/pnas.0401263101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatbonton T, Imbesi M, Nelson M, Akey JM, Ruderfer DM, Kruglyak L, Simon JA, Bedalov A. 2006. Telomere length as a quantitative trait: genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet. 2:e35. 10.1371/journal.pgen.0020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinert TA, Hartwell LH. 1993. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics 134:63–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garvik B, Carson M, Hartwell L. 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 15:6128–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zubko MK, Guillard S, Lydall D. 2004. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics 168:103–115. 10.1534/genetics.104.027904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonetti D, Martina M, Clerici M, Lucchini G, Longhese MP. 2009. Multiple pathways regulate 3′ overhang generation at S. cerevisiae telomeres. Mol. Cell 35:70–81. 10.1016/j.molcel.2009.05.015 [DOI] [PubMed] [Google Scholar]
- 18.Frank CJ, Hyde M, Greider CW. 2006. Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Mol. Cell 24:423–432. 10.1016/j.molcel.2006.10.020 [DOI] [PubMed] [Google Scholar]
- 19.Vodenicharov MD, Wellinger RJ. 2006. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol. Cell 24:127–137. 10.1016/j.molcel.2006.07.035 [DOI] [PubMed] [Google Scholar]
- 20.Grandin N, Damon C, Charbonneau M. 2001. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J. 20:1173–1183. 10.1093/emboj/20.5.1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grandin N, Reed SI, Charbonneau M. 1997. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev. 11:512–527. 10.1101/gad.11.4.512 [DOI] [PubMed] [Google Scholar]
- 22.Qian W, Wang J, Jin NN, Fu XH, Lin YC, Lin JJ, Zhou JQ. 2009. Ten1p promotes the telomeric DNA-binding activity of Cdc13p: implication for its function in telomere length regulation. Cell Res. 19:849–863. 10.1038/cr.2009.67 [DOI] [PubMed] [Google Scholar]
- 23.Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V. 2007. RPA-like proteins mediate yeast telomere function. Nat. Struct. Mol. Biol. 14:208–214. 10.1038/nsmb1205 [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Yu EY, Yang Y, Confer LA, Sun SH, Wan K, Lue NF, Lei M. 2009. Stn1-Ten1 is an Rpa2-Rpa3-like complex at telomeres. Genes Dev. 23:2900–2914. 10.1101/gad.1851909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin V, Du LL, Rozenzhak S, Russell P. 2007. Protection of telomeres by a conserved Stn1-Ten1 complex. Proc. Natl. Acad. Sci. U. S. A. 104:14038–14043. 10.1073/pnas.0705497104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lendvay TS, Morris DK, Sah J, Balasubramanian B, Lundblad V. 1996. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 144:1399–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandra A, Hughes TR, Nugent CI, Lundblad V. 2001. Cdc13 both positively and negatively regulates telomere replication. Genes Dev. 15:404–414. 10.1101/gad.861001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pennock E, Buckley K, Lundblad V. 2001. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 104:387–396. 10.1016/S0092-8674(01)00226-4 [DOI] [PubMed] [Google Scholar]
- 29.Nugent CI, Hughes TR, Lue NF, Lundblad V. 1996. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 274:249–252. 10.1126/science.274.5285.249 [DOI] [PubMed] [Google Scholar]
- 30.Diede SJ, Gottschling DE. 1999. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell 99:723–733. 10.1016/S0092-8674(00)81670-0 [DOI] [PubMed] [Google Scholar]
- 31.Marcand S, Brevet V, Mann C, Gilson E. 2000. Cell cycle restriction of telomere elongation. Curr. Biol. 10:487–490. 10.1016/S0960-9822(00)00450-4 [DOI] [PubMed] [Google Scholar]
- 32.Schramke V, Luciano P, Brevet V, Guillot S, Corda Y, Longhese MP, Gilson E, Geli V. 2004. RPA regulates telomerase action by providing Est1p access to chromosome ends. Nat. Genet. 36:46–54. 10.1038/ng1284 [DOI] [PubMed] [Google Scholar]
- 33.Taggart AK, Teng SC, Zakian VA. 2002. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science 297:1023–1026. 10.1126/science.1074968 [DOI] [PubMed] [Google Scholar]
- 34.Dionne I, Wellinger RJ. 1996. Cell cycle-regulated generation of single-stranded G-rich DNA in the absence of telomerase. Proc. Natl. Acad. Sci. U. S. A. 93:13902–13907. 10.1073/pnas.93.24.13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wellinger RJ, Wolf AJ, Zakian VA. 1993. Origin activation and formation of single-strand TG1-3 tails occur sequentially in late S phase on a yeast linear plasmid. Mol. Cell. Biol. 13:4057–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994. 10.1016/j.cell.2008.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774. 10.1038/nature07312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu P, Takai H, de Lange T. 2012. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150:39–52. 10.1016/j.cell.2012.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Makovets S, Matsuguchi T, Blethrow JD, Shokat KM, Blackburn EH. 2009. Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 136:50–61. 10.1016/j.cell.2008.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smolka MB, Albuquerque CP, Chen SH, Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. U. S. A. 104:10364–10369. 10.1073/pnas.0701622104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng SF, Shen ZJ, Tsai HJ, Lin YH, Teng SC. 2009. Rapid Cdc13 turnover and telomere length homeostasis are controlled by Cdk1-mediated phosphorylation of Cdc13. Nucleic Acids Res. 37:3602–3611. 10.1093/nar/gkp235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dahlseid JN, Lew-Smith J, Lelivelt MJ, Enomoto S, Ford A, Desruisseaux M, McClellan M, Lue N, Culbertson MR, Berman J. 2003. mRNAs encoding telomerase components and regulators are controlled by UPF genes in Saccharomyces cerevisiae. Eukaryot. Cell 2:134–142. 10.1128/EC.2.1.134-142.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grossi S, Puglisi A, Dmitriev PV, Lopes M, Shore D. 2004. Pol12, the B subunit of DNA polymerase alpha, functions in both telomere capping and length regulation. Genes Dev. 18:992–1006. 10.1101/gad.300004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puglisi A, Bianchi A, Lemmens L, Damay P, Shore D. 2008. Distinct roles for yeast Stn1 in telomere capping and telomerase inhibition. EMBO J. 27:2328–2339. 10.1038/emboj.2008.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeZwaan DC, Toogun OA, Echtenkamp FJ, Freeman BC. 2009. The Hsp82 molecular chaperone promotes a switch between unextendable and extendable telomere states. Nat. Struct. Mol. Biol. 16:711–716. 10.1038/nsmb.1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen LY, Redon S, Lingner J. 2012. The human CST complex is a terminator of telomerase activity. Nature 488:540–544. 10.1038/nature11269 [DOI] [PubMed] [Google Scholar]
- 47.Teixeira MT, Arneric M, Sperisen P, Lingner J. 2004. Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell 117:323–335. 10.1016/S0092-8674(04)00334-4 [DOI] [PubMed] [Google Scholar]
- 48.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229. 10.1006/meth.2001.1183 [DOI] [PubMed] [Google Scholar]
- 49.Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. 2003. Targets of the cyclin-dependent kinase Cdk1. Nature 425:859–864. 10.1038/nature02062 [DOI] [PubMed] [Google Scholar]
- 50.Moretti P, Freeman K, Coodly L, Shore D. 1994. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 8:2257–2269. 10.1101/gad.8.19.2257 [DOI] [PubMed] [Google Scholar]
- 51.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. 2000. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407:395–401. 10.1038/35030148 [DOI] [PubMed] [Google Scholar]
- 52.Gelinas AD, Paschini M, Reyes FE, Heroux A, Batey RT, Lundblad V, Wuttke DS. 2009. Telomere capping proteins are structurally related to RPA with an additional telomere-specific domain. Proc. Natl. Acad. Sci. U. S. A. 106:19298–19303. 10.1073/pnas.0909203106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binz SK, Sheehan AM, Wold MS. 2004. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst.) 3:1015–1024. 10.1016/j.dnarep.2004.03.028 [DOI] [PubMed] [Google Scholar]
- 54.Nuss JE, Patrick SM, Oakley GG, Alter GM, Robison JG, Dixon K, Turchi JJ. 2005. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry 44:8428–8437. 10.1021/bi0480584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anantha RW, Vassin VM, Borowiec JA. 2007. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 282:35910–35923. 10.1074/jbc.M704645200 [DOI] [PubMed] [Google Scholar]
- 56.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. 2006. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem. 281:39517–39533. 10.1074/jbc.M605121200 [DOI] [PubMed] [Google Scholar]
- 57.Vassin VM, Wold MS, Borowiec JA. 2004. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol. 24:1930–1943. 10.1128/MCB.24.5.1930-1943.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. 1999. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 18:1397–1406. 10.1093/emboj/18.5.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu P, Min JN, Wang Y, Huang C, Peng T, Chai W, Chang S. 2012. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 31:2309–2321. 10.1038/emboj.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang F, Stewart JA, Kasbek C, Zhao Y, Wright WE, Price CM. 2012. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2:1096–1103. 10.1016/j.celrep.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD, II, Wright WE, Price CM. 2012. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 31:3537–3549. 10.1038/emboj.2012.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang C, Dai X, Chai W. 2012. Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res. 22:1681–1695. 10.1038/cr.2012.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafe L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenco CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ. 2012. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 44:338–342. 10.1038/ng.1084 [DOI] [PubMed] [Google Scholar]
- 64.Keller RB, Gagne KE, Usmani GN, Asdourian GK, Williams DA, Hofmann I, Agarwal S. 2012. CTC1 mutations in a patient with dyskeratosis congenita. Pediatr. Blood Cancer 59:311–314. 10.1002/pbc.24193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Polvi A, Linnankivi T, Kivela T, Herva R, Keating JP, Makitie O, Pareyson D, Vainionpaa L, Lahtinen J, Hovatta I, Pihko H, Lehesjoki AE. 2012. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am. J. Hum. Genet. 90:540–549. 10.1016/j.ajhg.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.