

Abstract
In response to suboptimal activation, T cells become hyporesponsive, with a severely reduced capacity to proliferate and produce cytokines upon reencounter with antigen. Chromatin analysis of T cells made tolerant by use of different in vitro and in vivo approaches reveals that the expression of gamma interferon (IFN-γ) is epigenetically silenced in anergic effector TH1 cells. In those T cells, calcium signaling triggers the expression of Tle4, a member of the Groucho family of corepressors, which is then recruited to a distal regulatory element in the Ifng locus and causes the establishment of repressive epigenetic marks at the Ifng gene regulatory elements. Consequently, impaired Tle4 activity results in a markedly reduced capacity to inhibit IFN-γ production in tolerized T cells. We propose that Blimp1-dependent recruitment of Tle4 to the Ifng locus causes epigenetic silencing of the expression of the Ifng gene in anergic TH1 cells. These results define a novel function of Groucho family corepressors in peripheral T cells and demonstrate that specific mechanisms are activated in tolerant T helper cells to directly repress expression of effector cytokines, supporting the hypothesis that stable epigenetic imprinting contributes to the maintenance of the tolerance-associated hyporesponsive phenotype in T cells.
INTRODUCTION
T cells that escape negative selection in the thymus while still bearing T cell receptors (TCRs) with potential to respond against self-antigens pose a threat and can cause autoimmune disease. Several mechanisms of peripheral tolerance are in place to neutralize or prevent the activation of self-reactive T cells, including, among others, peripheral deletion, suppression mediated by regulatory T cells, and T cell anergy (1). Anergy is a cell-intrinsic program that is engaged in T cells to induce functional unresponsiveness (2) and occurs in T cells in response to suboptimal stimulation. For instance, clonal anergy is established following encounter with cognate antigen in the absence of a costimulatory signal, most frequently transmitted by CD28 (3, 4), or in the presence of inhibitory signals that can block costimulation (5–7).
In T cells, anergizing stimuli in the form of TCR engagement without costimulatory signals lead to a sustained increase in the levels of intracellular calcium, which in turn activate the calmodulin-dependent phosphatase calcineurin. Activated calcineurin dephosphorylates nuclear factor of activated T cells (NFAT) proteins, which then translocate into the nucleus (8, 9). In contrast to activated T cells, where NFAT can partner with activator protein 1 (AP-1) proteins to induce activation-induced genes, anergizing stimuli induce the activation of NFAT in the presence of suboptimal AP-1 activity. This triggers the expression of anergy-specific genes in an NFAT-dependent manner (2, 10). These genes encode a series of proteins that are responsible for TCR-signaling blockade and inhibition of interleukin-2 (IL-2) expression in anergic cells (11).
Epigenetic regulation of gene expression forms an integral part of the mechanisms that govern numerous programs of T cell differentiation. The ability to synthesize IL-2 following antigen reencounter is severely restricted in anergic CD4+ T cells (4). This is a consequence of two different mechanisms: a blockade that prevents efficient transduction of signaling downstream of the TCR (12) and a direct epigenetic regulation of the expression of the Il2 gene (13). In anergic T cells, the transcription factor Ikaros is a critical regulator of the expression of the Il2 gene through the induction of suppressive chromatin modifications at the Il2 promoter (14, 15). The regulation of expression of effector cytokines in anergic T cells has, however, remained poorly understood. Gamma interferon (IFN-γ) is one of the defining cytokines responsible for T helper 1 (TH1) differentiation and function (16–18). This TH1 cell signature cytokine is rapidly produced in response to antigen encounter and regulates, among other processes, macrophage activation, expression of major histocompatibility complex (MHC) molecules, and antitumor immune responses. We and others have shown that IFN-γ expression is also downregulated in anergic TH1 cells, but the mechanisms that inhibit Ifng expression in anergic cells remain unknown (2, 19–22).
Transducin-like enhancer of split 4 (Tle4), a member of the Groucho family of transcriptional corepressors, is one of the proteins expressed in T cells in response to anergizing stimuli (2). Tle proteins have been shown to oligomerize, to associate with amino-terminal domains of histone-modifying proteins, and to form higher-order structures as parts of repressive complexes (23). Tle4 does not possess DNA binding activity but can be recruited to a target site by different proteins, such as Runt domain proteins, high-mobility-group box proteins, and B lymphocyte-induced maturation protein (Blimp), to induce transcriptional repression of target genes (24–26). Because Blimp1 has been shown to repress IFN-γ expression in TH2 cells (27), we intended to investigate whether Tle4 could induce epigenetic and chromatin-modifying changes that could regulate IFN-γ expression in anergic T cells.
In this study, we show that calcium signaling during anergy induction causes epigenetic silencing of both the Ifng promoter and a conserved noncoding sequence (CNS) 21 kb upstream of the proximal Ifng promoter (−21kb CNS) (27). We also show that this effect is mediated by the calcium-induced expression of the corepressor protein Tle4, which, under anergizing conditions, is recruited to the −21kb CNS of the Ifng locus, likely through partnership with Blimp1, to induce repressive chromatin modifications. This transcriptional checkpoint at a key regulatory locus may be a critical part of the complex pathways through which anergic effector T cells preserve their unresponsiveness by controlling effector cytokine production.
MATERIALS AND METHODS
Mice.
Four- to 6-week-old female C57BL/6J and B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased from Jackson Laboratories and maintained under pathogen-free conditions. All animal experiments were carried out in accordance with the guidelines set by the Institutional Animal Care Committee at the Albert Einstein College of Medicine.
Cell culture.
Primary CD4+ T cells from mouse lymph nodes and spleens were isolated using CD4-coated Dynal magnetic beads (Invitrogen). To generate TH1 cultures, CD4+ T cells were activated with plate-bound anti-CD3ε and anti-CD28 antibodies (BD Biosciences) and differentiated for 6 days in the presence of 10 ng/ml of mouse IL-12 (eBioscience), 10 μg/ml of anti-mouse IL-4 antibody, and recombinant human IL-2 (Biological Resources Branch of the National Cancer Institute). To generate TH2 cells, CD4+ T cells were activated with plate-bound anti-CD3ε and anti-CD28 antibodies (BD Biosciences) and differentiated for 6 days in the presence of mouse IL-4 (10 ng/ml) and anti-IFN-γ (10 μg/ml) (BD Biosciences). All T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, nonessential amino acids with essential vitamins (Cambrex), and 50 μM 2-mercaptoethanol. The B16-OVA cell line was a kind gift from E. M. Lord (University of Rochester Medical Center) and was cultured, maintained, and used to induce tumors as described elsewhere (28). Jurkat and Phoenix ecotropic cells (kindly provided by G. P. Nolan, Stanford University) were cultured in DMEM supplemented with 10% FBS, 2 mM l-glutamine, and 50 μM 2-mercaptoethanol.
Anergy induction.
TH1 cells, differentiated and expanded over 6 days to the TH1 phenotype, were treated with 1 μM ionomycin (Calbiochem) for 16 h for induction of anergy. Alternatively, cells were stimulated for 20 h with 2.5 μg/ml plate-bound anti-CD3. Following anergizing treatments, T cells were detached from wells, washed, and rested for 4 and 48 h, respectively, before being restimulated. In some experiments, the global histone deacetylase (HDAC) inhibitor trichostatin A (TSA; Upstate Biotechnology) was added to a final concentration of 10 nM 1 h before starting the anergizing treatment and allowed to stay in the culture medium during anergy induction.
ELISA.
Culture supernatants were harvested from 2.5 × 104 to 5 × 104 T cells and then left to rest or stimulated with either anti-CD3ε/anti-CD28 antibodies or T cell-depleted, OVA323–339-loaded splenocytes at a 1:5 T cell/splenocyte ratio. Typically, supernatants were collected 12 to 18 h after stimulation, and IFN-γ levels were measured by a sandwich enzyme-linked immunosorbent assay (ELISA) (BD Biosciences).
RT-qPCR.
Total RNA was isolated from cells by use of TRIzol (Invitrogen), and cDNA was synthesized using a Superscript III first-strand synthesis system (Invitrogen). Real-time quantitative PCR (RT-qPCR) was performed using PowerSYBR (Applied Biosystems) on a StepOnePlus real-time PCR system (Applied Biosystems). Expression of all the genes studied was normalized to that of beta-actin. The following primer pairs were used for amplifications: Ifng forward primer, TCAAGTGGCATAGATGTGGAAGAA; Ifng reverse primer, TGGCTCTGCAGGATTTTCATG; Tle4 forward primer, TCACTCAAGTTTGCCCACTG; Tle4 reverse primer, CACAGCTAAGCACCGATGAG; Prdm1 forward primer, GACGGGGGTACTTCTGTTCA; Prdm1 reverse primer, GGCATTCTTGGGAACTGTGT; Tbx21 forward primer, GGTGTCTGGGAAGCTGAGAG; Tbx21 reverse primer, CCACATCCACAAACATCCTG; actin forward primer, GGCTGTATTCCCCTCCATCG; and actin reverse primer, CCAGTTGGTAACAATGCCATGT.
Immunoblotting.
Total protein lysates were prepared by lysing cells in 1× RIPA buffer containing protease inhibitors. Heat-denatured samples from total cell lysates or immunoprecipitates were run in SDS-polyacrylamide gels, transferred onto nitrocellulose membranes, and probed with anti-Tle4 (Santa Cruz Biotechnology), anti-Blimp1 (eBioscience), or anti-Myc (Cell Signaling) antibodies. Mouse anti-beta-actin was used as a loading control.
Coimmunoprecipitation.
HEK293 cells were transfected with plasmids expressing green fluorescent protein (GFP), Blimp1, or Myc-tagged Tle4 and lysed in lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 0.25% Nonidet P-40, protease inhibitor cocktail [Roche]). Lysates were precleared with protein G Dynabeads (Invitrogen) and incubated overnight at 4°C with an anti-Myc monoclonal antibody (Cell Signaling). Immunoprecipitates were recovered using protein G Dynabeads, washed, and analyzed by immunoblotting.
ChIP.
Nuclear lysates from 106 to 107 paraformaldehyde-fixed T cells were incubated overnight with the relevant chromatin immunoprecipitation (ChIP)-grade antibodies for acetylated histone H3 or H4 (Upstate-Millipore), trimethylated H3-K9 (Abcam), HDAC1 or HDAC2 (Abcam), Suv39H1 (Upstate-Millipore), Tle4 (Abcam), and Blimp1 (eBioscience). For HDAC1, HDAC2, and Suv39H1 ChIPs, cells were treated with 10 mM dimethyl adipimidate dihydrochloride (Aldrich) for 10 min at room temperature before being fixed with freshly prepared paraformaldehyde at a final concentration of 1%. Immune complexes were collected by use of protein G Dynabeads (Invitrogen), and the recovered DNA fragments were subjected to real-time PCR. Specific primer pairs were designed to amplify regions of the Ifng promoter (forward, TCAGCTGATCCTTTGGACCC; and reverse, CTCAGAGCTAGGCCGCAGG), the −21kb CNS (forward, CACCTGGGGTGAAAAGAAAT; and reverse, GTGAATCCCCAGAGAAGCAG), the Il2 promoter (forward, GCCACCTAAGTGTGGGCTAA; and reverse, ATATGGGGGTGTCACGATGT), and the Cd3e promoter (forward, TTCCTGCCTCCGCTGGAGGG; and reverse, GGCAGAAGCCTCCGCCTTGG). Specific enrichments were calculated and expressed as percent recovery of inputs or ratios after subtracting background recovery obtained with nonspecific isotype-matched antibodies.
Transfections and reporter assays.
Primary TH1 cells were transfected using an Amaxa nucleofection system (Lonza) following the manufacturer's protocol. Cells were electroporated either with pMax-GFP plasmid (Lonza) or with Tle4 or Tle5 expression vectors together with pMax-GFP. Thirty-six hours after transfection, GFP-expressing cells were sorted using a FACSAria cell sorter (BD) for subsequent analysis. For reporter assay experiments, Jurkat cells were transfected by electroporation with the reporter plasmid pGL3-IFNγ-Luc, containing the murine Ifng proximal promoter (bps +15 to −845) and a 300-bp region of the −21kb CNS, harboring a Blimp1 binding site, subcloned immediately upstream of the promoter. The reporter plasmid was cotransfected with pCDNA3.1-Blimp1, pCDNA3.1-Blimp1 with the 6th exon truncated, pCDNA3.1-Blimp1 together with pCDNA3.1-Tle4, or the truncated Blimp1 vector along with pCDNA3.1-Tle4. Twenty-four hours after transfection, cells were stimulated with 500 nM ionomycin (Calbiochem) and 20 nM phorbol myristate acetate (PMA; Calbiochem) or plate-bound anti-human CD3 and CD28 (BD) at 0.5 μg/ml. Six to 8 h after stimulation, cells were lysed, and luciferase activity was assayed using a dual-luciferase reporter assay system (Promega). In all experiments, cotransfection with a Renilla luciferase plasmid was included for normalization.
Retroviral transduction of primary TH1 cells.
The retroviral vector RV-IRES-GFP has been described before (2). RV-Tle4-IRES-GFP was constructed by inserting the mouse Tle4 cDNA into the RV-IRES-GFP vector. Phoenix ecotropic retroviral packaging cells were transfected with the retroviral vector, and viral supernatants were harvested 48 h after transfection. The supernatants were filtered, supplemented with Polybrene (6 μg/ml), and used to transduce T cells 24 and 48 h after activation with anti-CD3 and anti-CD28 antibodies. Positively infected cells were sorted for GFP expression and used for subsequent analyses.
RESULTS
IFN-γ expression is impaired in anergic TH1 cells.
Anergic T cells show a profound blockade of their response to subsequent encounter with antigen. Even when costimulation is present, recall responses in anergic cells are characterized by marked inhibition of activation-induced cell proliferation and IL-2 production (4). In order to investigate whether the expression of an effector cytokine could also be inhibited in TH cells, we analyzed the production of IFN-γ in anergic TH1 cells. We had previously shown that inhibition of proliferation and IL-2 expression was induced in T cells through the calcium/calcineurin/NFAT-mediated activation of the expression of anergy-associated genes (2, 14). To determine whether this calcium-dependent mechanism would also be responsible for the inhibition of IFN-γ expression, we employed a well-established method of anergy induction used to isolate calcium/NFAT signaling by treating cells with the calcium ionophore ionomycin (2). Confirming our hypothesis, anergic TH1 cells showed a markedly reduced capacity to produce IFN-γ following restimulation with engagement of the TCR and CD28 compared to control nonanergic cells (Fig. 1A), which reflected inhibition of Ifng gene transcription (Fig. 1B).
FIG 1.
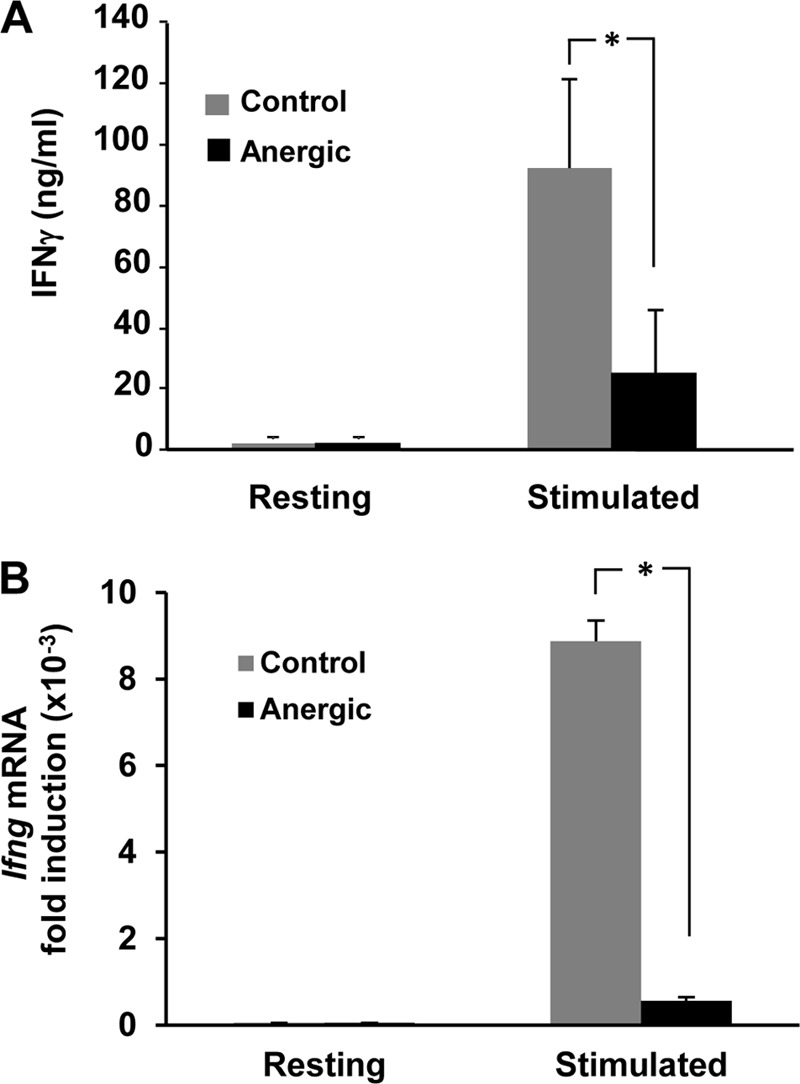
IFN-γ expression is downregulated in anergic TH1 cells. (A) TH1 cells were anergized with 1 μM ionomycin for 16 h and then left resting or stimulated with anti-CD3 and anti-CD28 antibodies for 12 h. IFN-γ production was measured by ELISA. (B) TH1 cells were anergized as for panel A and then left resting or stimulated with anti-CD3 and anti-CD28 antibodies for 6 h. RNA was obtained and Ifng mRNA expression measured by RT-qPCR. Results show fold induction of Ifng mRNA expression compared to that of resting cells. Both graphs show means and standard errors of the means (SEM) for 3 different experiments. *, P < 0.05.
Repressive chromatin marks are established in the Ifng promoter in anergic T cells.
Chromatin marks associated with silencing, including histone H3 and H4 deacetylation, have been described to be associated with inhibition of expression of the Il2 gene in anergic T cells (13–15). In order to determine the possibility that those modifications also occur at the Ifng promoter and could be responsible for the inhibition of expression of this cytokine gene in anergic TH1 cells, we measured the levels of H3 and H4 acetylation at this locus by ChIP using anti-acetylated H3 or H4 antibodies. These assays revealed that the Ifng promoter was clearly acetylated at H3 and H4 in resting TH1 cells and, as expected, that histone acetylation was markedly increased upon stimulation with anti-CD3 and anti-CD28, a condition under which these cells produce high levels of IFN-γ (Fig. 1 and Fig. 2A to C). However, when cells received anergizing stimuli, either through ionomycin treatment or by partial stimulation with anti-CD3 in the absence of CD28 engagement, T cells exhibited significant decreases in H3 and H4 acetylation (Fig. 2A to C), suggesting that epigenetic silencing of the Ifng promoter may contribute to the inhibition of IFN-γ expression in anergic TH1 cells. Similar results were obtained (2.5-fold reduction in H4 acetylation when anergic cells were compared with control resting cells) when TH1 cells differentiated from CD4+ T cells isolated from DO11.10 mice (bearing a transgenic TCR that recognizes the ovalbumin peptide OVA323–339 on MHC I-Ad) were stimulated with CHO cells expressing MHC class II I-Ad molecules but no B7 loaded with OVA323–339 to induce anergy (4.5% H4-acetylated chromatin recovery in resting cells, compared to 1.8% in anergic cells). In support of the presence of an active mechanism of histone deacetylation at the Ifng locus in anergic cells, anergizing stimulus-induced H4 deacetylation at the Ifng promoter was prevented when the global HDAC inhibitor TSA was added to the culture 45 min before administration of the anergizing stimulus (Fig. 2D). To further elucidate the mechanisms responsible for histone deacetylation at the Ifng locus in anergic TH1 cells, we determined whether HDACs would be recruited to the Ifng promoter during anergy induction. Class I HDACs have been shown to participate in the remodeling and repression of the Il2 gene in anergic T cells (13–15, 29), and to determine if they could also be responsible for the deacetylation of core histone tails at the Ifng promoter, we performed ChIP assays using anti-HDAC1 antibodies. These assays detected a strong recruitment of HDAC1 to the Ifng promoter in anergic TH1 cells, in contrast to minimal occupation in resting control cells (Fig. 2E). Recruitment of HDAC1 appeared to be specific for the Ifng promoter, as we could not detect significant HDAC1 binding when control primers specific for the promoter of the Cd3e gene, which does not undergo silencing in anergic cells, were used on the same samples (Fig. 2E).
FIG 2.

Anergizing stimuli induce histone deacetylation and H3K9 trimethylation at the Ifng promoter. (A to C) TH1 cells were anergized either with ionomycin (A) or through stimulation with anti-CD3 antibody in the absence of costimulation (B and C) for 16 h. Cells were then collected and analyzed for H4 (A and B) or H3 (C) acetylation at the Ifng promoter by ChIP using an anti-acetyl H4 (AcH4) or H3 (AcH3) antibody. Controls included resting cells and cells activated through stimulation with anti-CD3 and anti-CD28 antibodies. Results show means and SEM for 3, 4, and 5 independent experiments for panels A, B, and C, respectively. *, P < 0.05; **, P < 0.01. (D) ChIP assays were carried out with AcH4 antibody on resting (Rest) and anergized (Anerg) TH1 cells in the presence or absence of the histone deacetylase inhibitor TSA. Results show means and SEM for 4 independent experiments. (E) ChIP assays were performed on resting and anergized TH1 cells with an anti-HDAC1 antibody to assess HDAC1 occupancy of the Ifng promoter. The Cd3e promoter was used as a control. Results show means and SEM for 3 independent experiments. *, P < 0.05. (F and G) TH1 cells were treated as described for panels B and D, respectively, and H3K9 trimethylation (Me3-H3K9) at the Ifng promoter was assessed by ChIP. Results show means and SEM for 4 independent experiments. *, P < 0.05. (H) Control and anergized (Anerg) TH1 cells were either kept resting (Rest) or restimulated with anti-CD3 and anti-CD28 antibodies (Stim). The Ifng promoter was probed for the Me3-H3K9 histone modification mark by ChIP. Results are means and SEM for 3 independent experiments. (I) Resting, activated (Activ), and anergized (Anerg) TH1 cells were analyzed by ChIP using an antibody specific for the histone methyltransferase Suv39H1 to assess occupancy of the Ifng promoter by Suv39H1. Results show means and SEM for 3 independent experiments. *, P < 0.05.
Our previous studies on the Il2 gene had shown that silencing of this gene in anergic T cells was associated with additional silencing epigenetic marks, including trimethylation of H3 at lysine 9, that stabilize silencing of the expression of that gene in anergic cells (13). To determine whether those additional silencing epigenetic modifications were in place to stabilize suppression of Ifng gene expression in anergic TH1 cells, we analyzed the Ifng promoter for possible targeting by the histone methylation machinery. Anergy was induced in TH1 cells, and the Ifng promoter was analyzed by ChIP to assess methylation at lysine 9 of H3, using an anti-trimethylated H3K9 (Me3-H3K9) antibody. We found that H3 at the Ifng promoter was clearly trimethylated at K9 in anergic T cells compared to nonanergic control cells (Fig. 2F). This effect lent additional support to the possibility that the Ifng promoter assumes a more complex pattern of repressive chromatin modifications not restricted to hypoacetylation alone in response to anergizing stimuli. Interestingly, in separate experiments, addition of TSA to cells subjected to anergizing treatments also prevented the establishment of the Me3-H3K9 mark on the Ifng promoter, as the level of H3K9 methylation in cells pretreated with TSA was found to be comparable to that in control resting cells (Fig. 2G). This observation suggested that H3K9 tri-methylation at the Ifng promoter was dependent on previously acquired histone deacetylation of the locus.
Our results indicated that anergic TH1 cells failed to produce IFN-γ in response to full stimulation (Fig. 1). Continued retention of repressive epigenetic marks might explain such an outcome. This prompted us to investigate if Me3-H3K9 was still retained at the Ifng promoter in anergic cells following restimulation. Supporting the stable nature of this epigenetic modification, we observed that the Ifng promoter strongly retained the repressive Me3-H3K9 mark in anergic cells that were restimulated with anti-CD3 and anti-CD28 (Fig. 2H). To identify the mechanism that could cause increased Me3-H3K9 at the Ifng promoter in anergic T cells, we looked for the involvement of histone methyltransferases (HMTs) that could account for the trimethylation of H3K9 in T cells. Among several such candidates present in mammalian cells, Suv39H1 has been well characterized to be responsible for the Me3-H3K9 mark, and its involvement in gene silencing in T cells was reported earlier (13, 30, 31). Thus, we determined if Suv39H1 recruitment to the Ifng promoter could be detected in anergic T cells. Indeed, ChIP experiments showed an increased presence of Suv39H1 at the proximal promoter of the Ifng gene in anergic TH1 cells compared to control resting or stimulated cells, which showed negligible binding of this HMT (Fig. 2I). Overall, these results indicate that repressive epigenetic marks such as Me3-H3K9 are actively established at the Ifng promoter in anergic TH1 cells.
Tle4 suppresses IFN-γ expression in anergic T cells.
We previously reported that a specific subset of genes is upregulated in anergic T cells, in a calcium/calcineurin/NFAT-dependent manner. Among those genes, we identified genes encoding proteins with transcriptional repressor activity, including Tle4 (2). To support a possible role of Tle4 in T cell anergy, we first confirmed that Tle4 mRNA was upregulated in anergic TH1 cells in response to sustained calcium signaling (Fig. 3A), which led to increased levels of Tle4 protein (Fig. 3B). We then set out to determine whether Tle4 could inhibit IFN-γ production. For this purpose, we used bicistronic retroviral vectors that expressed Tle4 and GFP to transduce primary TH1 cells. Infected GFP+ T cells were sorted, and IFN-γ expression was measured following activation with anti-CD3 and anti-CD28 antibodies. We could not detect any appreciable changes in IL-2 expression (data not shown), but IFN-γ production was significantly reduced in cells transduced with Tle4-expressing retroviruses (Fig. 3C). To further characterize the role that Tle4 could have in the regulation of IFN-γ expression in anergic cells, we transduced TH1 cells with retroviruses expressing GFP and Tle5, a shorter, 197-amino-acid, naturally occurring form of Tle4 that acts as a dominant negative protein by preventing Tle4 from undergoing multimerization, which is essential for its ability to recruit HDACs in a repressive complex (24). Confirming the involvement of Tle4 in the regulation of IFN-γ expression in anergic cells, T cells that expressed Tle5 showed reduced downregulation of IFN-γ expression after receiving an anergizing stimulus and produced 2.5-fold more IFN-γ than anergized control cells expressing only GFP (Fig. 3D). It is important to note that these results reflected the partial recovery of IFN-γ expression that could be expected of Tle5-mediated prevention of Tle4-induced silencing of the Ifng gene expression, without affecting the interference of TCR signaling that is characteristic of anergic T cells.
FIG 3.

Ifng is suppressed by Tle4 in anergic T cells. (A and B) Tle4 mRNA (A) and protein (B) in resting and anergic TH1 cells were quantified by RT-qPCR and immunoblotting, respectively. Data in panel A show means and SEM for 3 independent experiments. (C) IFN-γ expression in TH1 cells transduced with retroviruses expressing either GFP alone or GFP and Tle4 (Tle4). GFP+ cells were sorted and stimulated with anti-CD3 and anti-CD28 antibodies. IFN-γ production was measured by ELISA. Bars represent means and SEM for 3 independent experiments. *, P < 0.05. (D) IFN-γ production was measured in anergic control cells and cells expressing the Tle4 dominant negative protein, Tle5. TH1 cells were transduced with retroviruses expressing either GFP alone or GFP and Tle5 and were sorted for GFP expression. Cells were either left untreated or anergized and then stimulated (Stim) with anti-CD3 and anti-CD28 antibodies for 24 h. The anergy index value (ratio of IFN-γ produced by control cells and anergized cells) is also shown. Bars represent means and SEM for 3 independent experiments. *, P < 0.05. (E and F) Tle4 occupancy of the Ifng −21kb CNS was analyzed by ChIP using an anti-Tle4 antibody on resting TH1 cells, cells anergized with either ionomycin (E) or anti-CD3 treatment (E and F), and activated cells (F). Graphs show means and SEM for 6 and 2 independent experiments, respectively. *, P < 0.05.
This suppressive effect on IFN-γ expression prompted us to question whether Tle4 could directly repress Ifng transcription. In order to identify where the recruitment sites for Tle4 in the Ifng locus might be located, we first carried out ChIP experiments using anti-Tle4 antibodies to assess binding to the Ifng promoter. However, we could not detect any significant binding of Tle4 to that region of the Ifng locus. A CNS located 21 kb upstream of the proximal Ifng promoter (−21kb CNS) has been shown to have a critical regulatory role in the regulation of IFN-γ expression in T cells (27, 32, 33). We therefore explored the possibility that Tle4 targets this regulatory CNS. Indeed, anergizing treatments induced a strong recruitment of Tle4 to the −21kb CNS region compared to that in control resting cells (Fig. 3E). Moreover, Tle4 occupancy of this CNS in activated, IFN-γ-producing TH1 cells was found to be even lower than that in resting cells (Fig. 3F). Our data thus support the hypothesis that recruitment of Tle4 to the −21kb CNS of the Ifng gene may result in inhibition of IFN-γ expression.
The Ifng −21kb CNS undergoes repressive histone modifications in anergic T cells.
The experiments described above indicated that the Ifng −21kb CNS was targeted by Tle4 in anergic cells. Since Tle4-induced transcriptional repression has been known to be mediated by the recruitment of the histone deacetylation machinery, we sought to evaluate if this regulatory region underwent histone deacetylation in anergic T cells. ChIP experiments using anti-acetylated H4 antibodies revealed that the −21kb CNS became hypoacetylated under anergizing conditions, in contrast to fully activated cells, which displayed marked hyperacetylation (Fig. 4A). Groucho family corepressors interact with the N-terminal tails of core histones and recruit complexes containing class I HDACs to silenced loci (24, 34). Supporting the involvement of HDACs in a process of active deacetylation of the −21kb CNS, hypoacetylation was prevented when T cells receiving anergizing stimuli were cultured in the presence of TSA (Fig. 4B), while ChIP analyses revealed HDAC1 and HDAC2 binding to the −21kb CNS in anergic T cells (Fig. 4C). Similar to what we had detected in the Ifng promoter, the −21kb CNS also acquired strong H3K9 trimethylation under anergic conditions (Fig. 4D), which was maintained even when anergic cells were restimulated with anti-CD3 and anti-CD28 antibodies (Fig. 4E). Methylation of H3K9 appeared to be mediated by Suv39H1, as we could detect markedly increased recruitment of this HMT to the −21kb CNS in anergic T cells (Fig. 4F).
FIG 4.

Anergic TH1 cells show repressive histone modifications at the −21kb CNS. (A) H4 acetylation at the Ifng −21kb CNS was assessed by ChIP assay of resting, anergic, and activated TH1 cells by use of anti-acetyl H4 antibodies. Data are means and SEM for 4 independent experiments. *, P < 0.05. (B) H4 acetylation at the Ifng −21kb CNS was measured by ChIP assay of resting cells (Rest) and cells anergized (Anerg) in the presence or absence of the histone deacetylase inhibitor TSA (10 nM). Data are means and SEM for 2 independent experiments performed in triplicate. (C) HDAC1 and HDAC2 binding to the Ifng −21kb CNS in resting and anergic TH1 cells was determined by ChIP using an anti-HDAC1 or anti-HDAC2 antibody. Results are means and SEM for 3 and 2 independent experiments, respectively. *, P < 0.05. (D) Me3-H3K9 repressive chromatin modification at the Ifng −21kb CNS in resting, anergic (anti-CD3), and activated (anti-CD3 plus anti-CD28) TH1 cells was assessed by ChIP using an anti-Me3-H3K9 antibody. Results are means and SEM for 4 independent experiments. *, P < 0.05. (E) Stability of the Me3-H3K9 modification in anergic cells. Me3-H3K9 repressive chromatin modification at the Ifng −21kb CNS was determined by ChIP assay of resting, anergic TH1 cells and anergic cells restimulated for 24 h with anti-CD3 and anti-CD28 antibodies. Data are means and SEM for 3 independent experiments. (F) Suv39H1 occupancy of the Ifng −21kb CNS was analyzed by ChIP using an anti-Suv39H1 antibody on resting, anergic, and stimulated cells. Bars represent means and SEM for 4 independent experiments. *, P < 0.05.
Tle4 induces histone deacetylation at the Ifng regulatory regions in anergic T cells.
On the basis of our previous observations that overexpression of Tle4 led to diminished production of IFN-γ, we set out to determine whether increased Tle4 expression could be responsible for the establishment of a hypoacetylated state in the Ifng locus in anergic T cells. To address this question, we first analyzed the consequences of overexpressing Tle4 in T cells and sought to identify repressive epigenetic marks at the Ifng regulatory regions. TH1 cells were transfected with a GFP-expressing vector, with or without cotransfection of a Tle4-expressing plasmid. ChIP analysis of sorted GFP+ cells revealed marked decreases in histone H3 and H4 acetylation of the −21kb CNS and the Ifng promoter in cells that overexpressed Tle4 (Fig. 5A to D). These results supported the observation that the elevated levels of Tle4 in anergic T cells could induce suppressive chromatin modifications at these critical regulatory regions to contribute to the functional unresponsiveness observed in anergy in terms of IFN-γ output. To strengthen support for this observation, we again utilized the properties of the dominant negative Tle5 protein and transfected T cells with an expression plasmid for that protein. Following an anergizing treatment, Tle5-expressing TH1 cells showed approximately 2-fold higher levels of histone acetylation at the −21kb CNS than similarly treated control cells (Fig. 5E), further supporting the hypothesis that upregulation of Tle4 in response to anergizing stimuli was responsible for the deacetylation of the Ifng gene regulatory regions in anergic T cells. Notably, an examination of the Il2 promoter, which is also actively deacetylated in anergic cells, did not reveal any reversal in deacetylation in the Tle5-positive cells, with the promoter remaining hypoacetylated, in marked contrast to the Ifng promoter (Fig. 5F), indicating that the effect of Tle4 was specific for IFN-γ and did not extend to the regulation of IL-2 expression.
FIG 5.

Histone deacetylation at two Ifng regulatory regions is regulated by Tle4. (A to D) TH1 cells were transfected by nucleofection with a GFP-expressing vector alone or cotransfected with GFP and murine Tle4 expression plasmids. Chromatin samples from sorted GFP+ cells were subjected to ChIP assays with anti-AcH4 and anti-AcH3 antibodies to analyze the H4 and H3 acetylation status at the Ifng −21kb CNS (A and C) and promoter (B and D). Graphs show means and SEM for 4 to 6 independent experiments. *, P < 0.05. (E and F) TH1 cells were transfected with a GFP-expressing vector alone or cotransfected with GFP and Tle5 expression plasmids. GFP+ cells were sorted and subjected to anergizing treatment followed by ChIP analysis using an anti-AcH4 antibody. Data show comparisons between anergized cells and Tle5-expressing anergized cells in terms of relative % acetylation of the Ifng −21kb CNS (E) or the Ifng and Il2 promoters (F). Graphs show means and SEM for 4 independent experiments. *, P < 0.05.
Blimp1 is a potential recruiter of Tle4 to the −21kb CNS.
Tle4 is a corepressor that does not possess a DNA binding domain (35). Therefore, it needs to be recruited to its target loci by a transcriptional repressor complex with core component proteins with DNA binding capacity. To further elucidate how Tle4 would be recruited to the −21kb CNS, we focused our studies on Blimp1, which had previously been reported to be a DNA-binding transcriptional repressor that could partner with Tle4 (26). Importantly, Blimp1 interacts with HDAC1 and HDCA2 and has been shown to inhibit TH1 differentiation by repressing the Ifng and Tbx1 gene expression targeting multiple regulatory sites in CD4+ T cells (27, 36). Highly conserved consensus Blimp1 binding sites have been identified in the −21kb CNS and have been described to play critical roles in the regulation of Ifng transcription in T cells (27, 32). We monitored Blimp1 expression in TH1 cells under both resting and anergizing conditions and detected that Prdm1 (encoding Blimp1) mRNA expression was upregulated 5-fold in anergic TH1 cells (Fig. 6A). These data were supported by a concomitant increase in Blimp1 protein (Fig. 6B). These results prompted us to investigate if Blimp1 could bind to the −21kb CNS, which we had earlier found to be a target of Tle4 (Fig. 3E). ChIP assays revealed that the −21kb CNS showed significantly increased binding of Blimp1 in anergic cells (Fig. 6C). The Il2 promoter was used as a control region, as it was unaffected by overexpression of Tle5 (Fig. 5D and 6C). These results indicated that Tle4 could be recruited to this Ifng regulatory region in a Blimp1-dependent manner. In support of this possibility, protein-protein interaction between Blimp-1 and Tle4 could readily be detected in HEK293 cells that were transfected with plasmids expressing these proteins (Fig. 6D). We next explored the functional cooperation of these two transcription factors in a luciferase-based reporter assay system. We transfected Jurkat cells with a Blimp1 and/or Tle4 expression vector along with a pGL3-luciferase reporter plasmid containing the murine Ifng promoter as well as a 300-bp fragment of the −21kb CNS containing the Blimp1 binding site. Transcription of the Ifng promoter-CNS reporter was modestly inhibited by Blimp1 expression alone, whereas cotransfection with both Blimp1- and Tle4-expressing plasmids caused reporter activity to be reduced approximately 45% (Fig. 6D). The proline-rich domain of Blimp1 has been implicated in its interaction with the Groucho family of transcriptional corepressors (26). In contrast to the results obtained when wild-type Blimp1 was expressed, a Blimp1 mutant with the proline-rich domain deleted failed to engage in functional cooperation with Tle4 and could not significantly repress reporter activity (Fig. 6D). These results further support the prediction that cooperation between Tle4 and Blimp1 is necessary to repress IFN-γ expression in anergic cells.
FIG 6.

Blimp1 can recruit Tle4 to the Ifng −21kb CNS. (A and B) Resting and anergic TH1 cells were analyzed for Prdm1 mRNA expression by RT-qPCR (A) and for Blimp1 protein levels by immunoblotting (B). Data in panel A are expressed as means and SEM for the fold induction of Blimp1 expression in anergic T cells compared to resting cells for 5 independent experiments. (C) ChIP assays using an anti-Blimp1 antibody were carried out to compare relative Blimp1 binding to the Ifng promoter (first bar) and the Ifng −21kb region (second bar). The Il2 promoter was used as a control. Data (means and SEM for 5 independent experiments) are presented as fold enrichment of Blimp1 binding in anergic cells compared to resting cells. *, P < 0.05. (D) HEK293 cells were transfected with plasmids expressing GFP, Myc-tagged Tle4, or Blimp1, as indicated. Twenty-four hours later, cell lysates were prepared and immunoprecipitated (IP) using an anti-Myc antibody. Input and immunoprecipitates were analyzed by immunoblotting (WB) using anti-Myc or anti-Blimp1 antibody. A control immunoprecipitation using beads with no antibody was also included. Representative blots from one of two independent experiments are shown. (E) Jurkat cells were transfected by electroporation with either wild-type or PR region-deleted/Tle4 interaction-deficient (Blimp1*) Blimp1 and/or Tle4 expression plasmids together with a pGL3-luciferase reporter vector containing the mouse Ifng promoter as well as a 300-bp fragment from the −21kb CNS region. A Renilla luciferase expression vector was included in every transfection mix to normalize relative light units (RLU) as a measurement of the Ifng reporter activity. Data show means and SEM for 4 independent experiments. *, P < 0.05; ns, not significant. (F and G) Naive CD4+ T cells were differentiated in vitro into TH1 or TH2 cells for 1 week. Total RNA was prepared and expression of Tle4 and Prdm1 measured by RT-qPCR. Data (2−ΔΔCT, using actin as a housekeeping control) show means and SEM for 6 different experiments. **, P < 0.01. (H to J) TH2 cells were transfected with plasmids expressing GFP alone or GFP and the Tle4 dominant negative protein, Tle5. Twenty-four hours after transfection, GFP+ cells were sorted and stimulated with anti-CD3 and anti-CD28 for 12 h. IFN-γ (H) and IL-4 (J) production was measured by ELISA, and expression of Tbx21 (I) was quantified by RT-qPCR. Graphs show means and SEM for data obtained from 5 independent experiments. ns, not significant.
Tle4 mediates Ifng repression in TH2 cells.
Blimp1 has been reported to inhibit TH1 differentiation through transcriptional repression of the Ifng and Tbx21 genes (27). Since our results indicated that Tle4 was a corepressor for Blimp1 to silence Ifng expression in anergic cells, we explored the possibility that the function of Tle4 extends beyond anergy to the maintenance of other T cell phenotypes that required repression of Ifng gene expression, including that of TH2 cells. RT-PCR analyses confirmed that TH2 cells expressed Tle4, though at levels similar to those found in TH1 cells (Fig. 6F). However, the expression of Blimp1 was much higher in TH2 than in TH1 cells (Fig. 6G). These results suggested that in TH2 cells, the level of Blimp1 might be the limiting factor to achieve Ifng repression, though they did not rule out the possibility that Blimp1 still complexes with Tle4 to inhibit the expression of the Ifng gene. To address this possibility, we transfected TH2 cells with a plasmid expressing the dominant negative Tle5 protein. Confirming the role of Tle4 as a repressor of the expression of Ifng in TH2 cells, expression of Tle5 allowed TH2 cells to regain the ability to produce IFN-γ (Fig. 6H). This effect appeared to be specific for IFN-γ, as Tle5 had no effect on Tbx21 expression (Fig. 6I) and did not affect the production of the TH2 cytokine IL-4 (Fig. 6J).
Tumor-specific CD4+ T cells show elevated Tle4 and Blimp1 levels and reduced IFN-γ production.
An effector immune response against tumor cells is often neutralized by a tumor microenvironment that is immunosuppressive in nature. Among the mechanisms that govern the induction of tumor antigen-specific tolerance in T cells, it has been shown that tumor-specific CD4+ T cells frequently become anergic (28, 37, 38). To corroborate that Ifng gene silencing also occurred in a model of tumor-induced anergy in vivo, we used a B16 melanoma line that stably expresses ovalbumin (B16-OVA) as a surrogate tumor antigen. Tumors were induced in OT-II mice, whose T cells express an MHC class II-restricted TCR that recognizes the OVA323–339 peptide. B16-OVA cells were injected into the lumbar flanks of OT-II mice, and tumors were allowed to grow to a size of 1 cm3. Mice were then sacrificed and CD4+ T cells harvested from the tumor draining inguinal lymph nodes (DLN) and also from nondraining contralateral distal cervical lymph nodes (NDLN). CD4+ T cells isolated from the DLN were found to produce significantly smaller amounts of IFN-γ than cells from NDLN (Fig. 7A). Accordingly, T cells from DLN were also found to express higher levels of both Tle4 and Prdm1 mRNAs (Fig. 7B). Based on these observations, we tested if the upregulation of these two genes would result in chromatin modulatory changes in the Ifng regulatory regions. In order to maximize cell recovery, we injected tumors into both flanks of the mice and therefore used cells from non-tumor-bearing mice as controls. ChIP experiments showed that both the −21kb CNS and the Ifng promoter were clearly hypoacetylated in T cells isolated from tumor-bearing mice compared to cells obtained from tumor-free animals (Fig. 7C), further supporting our ex vivo findings indicating that Tle4 and Blimp1 cooperate to suppress the expression of the effector cytokine IFN-γ and maintain TH1 cell tolerance.
FIG 7.
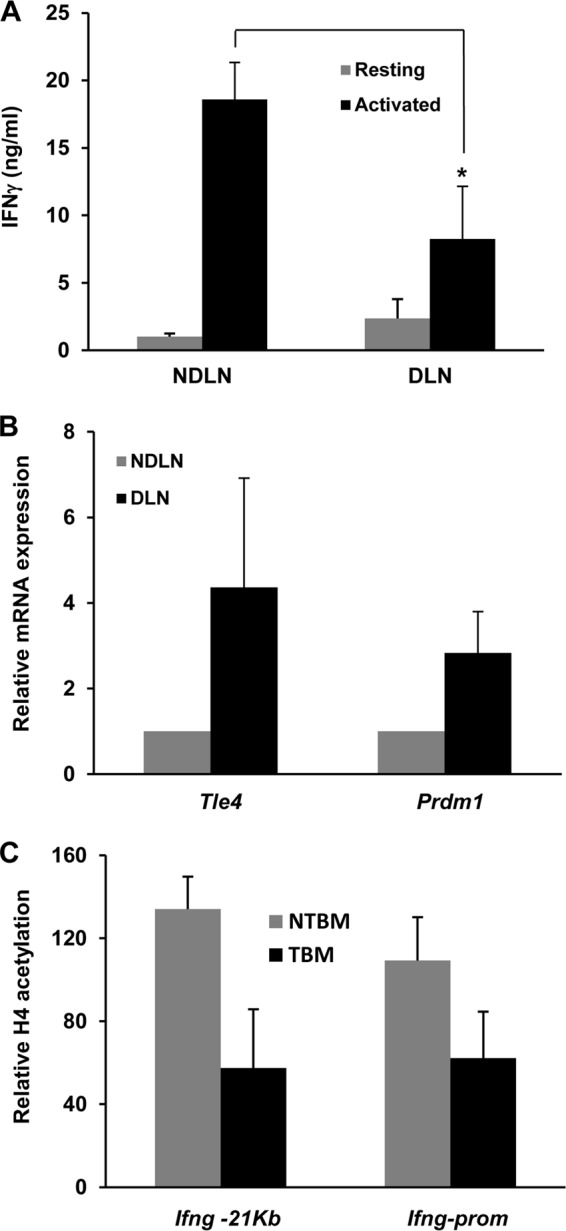
Tumor-specific CD4+ T cells exhibit increased Tle4 and Blimp1 expression as well as reduced IFN-γ production. (A) IFN-γ expression in T cells isolated from draining (DLN) and nondraining (NDLN) lymph nodes from B16-OVA melanoma-bearing OT-II mice and stimulated for 18 h with T cell-depleted splenocytes loaded with OVA323–339 peptide, as measured by ELISA. Data are means and SEM for 5 different experiments. *, P < 0.05. (B) Similarly isolated CD4+ T cells from tumor-bearing mice were used to prepare total mRNA. Relative expression levels of mRNAs for Tle4 and Prdm1 were determined by qPCR. Bars represent relative expression in T cells isolated from DLN compared to cells from NDLN, expressed as means and SEM for 6 different experiments. (C) ChIP experiments were carried out to assess H4 acetylation at the Ifng −21kb CNS and promoter in CD4+ T cells isolated from DLN of tumor-bearing mice and from tumor-free mice. Bars show relative H4 acetylation in T cells isolated from DLN compared to cells from NDLN in tumor-bearing (TBM) and non-tumor-bearing (NTBM) mice, expressed as means and SEM for 3 different experiments.
DISCUSSION
Among the mechanisms that regulate self-tolerance, anergy represents an intrinsic process of inactivation that occurs in T cells in response to suboptimal stimulation that renders them unable to respond to subsequent encounters with antigen. Anergic T cells fail to proliferate and produce IL-2 when restimulated, even in the presence of costimulation (39, 40). In CD4+ T cells, anergy is established as a consequence of the expression of a specific set of anergy-related genes that are transcribed in a calcium/NFAT- and Egr2-dependent manner in response to tolerizing stimuli (2, 41–43). Initial characterizations of anergic T cells identified a blockade in the Ras/mitogen-activated protein (MAP) kinase signaling pathway (12, 44). Proteins encoded by specific anergy genes, including several ubiquitin ligases, such as Grail, Itch, and Cbl-b, caspase 3, diacylglycerol kinase alpha, and Sirtuin 1, inhibit activation of the MAP kinase and other signaling cascades downstream of the TCR when anergic T cells reencounter antigen (45–52).
Recent studies have demonstrated that signaling blockade is not the only process that is engaged to prevent activation-induced responses in anergic T cells. Epigenetic silencing of cytokine gene expression has been shown to occur at the Il2 locus, adding a second checkpoint at the transcriptional level to inhibit IL-2 expression. In anergic T cells, Ikaros binds to the Il2 promoter and recruits histone-modifying complexes that imprint silencing epigenetic marks, including H3 and H4 deacetylation and H3K9 trimethylation (14, 15). These changes allow binding of the heterochromatin binding protein HP-1 and cause segregation of the Il2 locus to heterochromatin-rich regions in the nucleus (13). Whereas decreased proliferation and IL-2 production have classically been identified as the defining properties of anergic T cells, several reports have shown data to support the prediction that effector cytokine production is also reduced in anergic cells (2, 19–22). Corroborating prior in vitro and in vivo data, our results show that anergic TH1 cells also present a defect in their ability to produce IFN-γ when restimulated. Increased DNA methylation and decreased histone acetylation were previously reported to occur at the Ifng locus in T cells isolated from mice that had been anergized using a viral superantigen (21). These results supported the hypothesis that epigenetic control might extend beyond the regulation of IL-2 expression to also modulate the transcription of the TH1 effector cytokine IFN-γ. During T helper cell differentiation, Ifng gene transcription is silenced in TH2 cells through a process that involves chromatin remodeling to close the Ifng locus and make it transcriptionally inactive (53, 54). Anergy can also be considered a stable state of T cell differentiation, and it was recently shown that in CD8+ T cells, a tolerance-maintaining program may be epigenetically imprinted (55). Anergic CD8+ T cells in which the tolerant state has been transiently reversed by adoptive transfer into lymphopenic hosts eventually regain their unresponsive status, even in the absence of any new tolerogenic stimulus (55). In fact, our data confirm that once established, histone deacetylation and silencing H3K9 trimethylation at the Ifng locus are not reversed even by strong stimulation in the presence of CD28 engagement.
Whereas Ikaros is a transcriptional repressor that triggers chromatin remodeling at the Il2 locus (14, 15), the mechanisms responsible for the regulation of Ifng gene expression have so far remained unknown. We previously reported that the expression of Tle4, a member of the Groucho family of corepressors, was upregulated in anergic T cells in a calcium/calcineurin/NFAT1-dependent manner (2). Our data now indicate that in anergic TH1 cells, Tle4 is recruited to a region located approximately 21 kb upstream of the Ifng proximal promoter, likely through interactions with Blimp1. Tle proteins are corepressors that lack intrinsic DNA binding activity but interact with other transcription factors to form repressor complexes (35). For instance, Tle4 binding to Pax2 has been shown to displace a Pax2 coactivator complex and instead induce recruitment of histone methyltransferases and polycomb proteins, turning Pax2 from an activator into a repressor (56). This result supports a crucial role for Tle4 in the determination of the specific function of a transcription factor and suggests that the availability of Tle4 could be a determining factor in that process. Anergic TH1 cells also upregulate Tle4 expression. This increased Tle4 availability could therefore change the cell's transcriptome by turning otherwise silent or activating transcriptional complexes into repressors and thus contributing to the establishment of an anergy-defining epigenetic imprinting.
Anergy in tumor antigen-specific T cells is a mechanism of immune evasion that contributes to the overall inefficiency of antitumor T cell responses (38). In fact, when anergy is prevented from occurring, tumor-specific T cells maintain strong antitumor responses, including IFN-γ production, and exert a better control of tumor growth (28, 37). Here we show that tumor-induced anergic T cells also present increased expression of Tle4, which lowers levels of histone acetylation of the Ifng locus and decreases production of IFN-γ, supporting the observation that, as we have seen in our in vitro models, in vivo-induced anergy is also accompanied by epigenetic silencing of Ifng gene expression and increased Tle4 and Blimp1 expression. Interestingly, increased Blimp1 expression has also been detected in exhausted T cells in the context of a chronic infection (57). Since exhausted cells show decreased production of IFN-γ when stimulated, it might be interesting to determine if Tle4 is also used by Blimp1 as a corepressor in that situation, where a stable repression of Ifng expression is also established, not due to suboptimal activation but as a consequence of chronic antigen stimulation.
Our data support the prediction that Blimp1 can recruit Tle4 to the Ifng −21kb CNS in anergic TH1 cells to silence this gene's expression. Blimp1 exerts a similar repressive role in TH2 cells, by inhibiting the expression of several TH1 lineage-specific genes, including Ifng (27). Whether Tle4 or other Tle family proteins are involved in that process was unknown. Our data support the hypothesis that Tle4 is also involved in the suppression of expression of the Ifng gene in TH2 cells. When Tle4 activity was inhibited using a dominant negative Tle protein, TH2 cells became able to produce IFN-γ. We have to note that the level of IFN-γ expressed by these cells was still much lower (20- to 40-fold) than production of this cytokine in TH1 cells. This could be attributed to the fact that inhibiting Tle4 may not affect other targets of Blimp1 in TH2 cells. Accordingly, the expression of Tbx21, which is inhibited by Blimp1 in TH2 cells (27), was not increased by the dominant negative Tle5 protein. These results point to a specific role of Tle4 in T cells as a repressor of Ifng expression and imply the existence of Blimp1-dependent Tle4-independent mechanisms that regulate the expression of other genes involved in T helper differentiation. A similar situation would occur in anergic cells, where Tle4 would control Ifng expression but would not affect Il2 transcription, which would be regulated instead by Ikaros (14, 15). Blimp1-deficient mice have been characterized, their T cells present a hyperactive state with increased IFN-γ production, and these mice develop a severe colitis with an early onset (58). Based on our results, it is possible that this phenotype is a consequence not only of a TH1 bias but also of an inability of Blimp1-deficient T cells to recruit Tle4 to the Ifng locus and repress its expression in response to tolerizing stimuli, which would result in inefficient T cell tolerance and altered T cell homeostasis.
Tle4 is recruited to a region located approximately 21 kb upstream of the Ifng promoter. However, silencing epigenetic marks can also be detected at the Ifng promoter in anergic cells. Furthermore, Tle4 overexpression induces repressive marks at both loci, and expression of a dominant negative Tle5 protein prevents histone deacetylation at both loci as well. Though we cannot rule out the possibility that other transcriptional repressors directly or indirectly regulated or aided by Tle4 are responsible for those changes, it is also possible that changes initiated at the distal Tle4 recruitment site spread toward the promoter to ensure epigenetic silencing of Ifng gene expression. Bending of the chromatin at the Ifng locus could bring the corepressor complex in close proximity to the promoter region and allow chromatin remodeling of that region to inhibit Ifng gene expression. DNA bending at this locus has already been reported to account for enhancer-promoter interactions (59). A similar mechanism may be in place to allow repressor-promoter interactions in anergic T cells.
Epigenetic silencing of effector cytokine expression can account for the stable nature of the anergic status and contribute to an imprinting program that defines those cells and prevents them from becoming fully functional T cells. Nevertheless, it is important that signaling blockade also plays an important role in keeping anergic T cells unresponsive. Indeed, when we blocked Tle4-induced repression of Ifng transcription, we could only partially reverse the inhibition of IFN-γ production in anergic cells. This likely reflects the coexistence of several signaling blocks that, by preventing TCR- and/or CD28-mediated signal transduction, also determine the capacity of anergic T cells to produce cytokines (11). Nevertheless, the importance of the epigenetic control of IFN-γ expression is underlined by the fact that even in the presence of that signaling block, inhibition of Tle4 activity led to an almost 3-fold increase in the ability of anergic cells to produce IFN-γ upon restimulation.
In summary, our data show that epigenetic regulation of effector cytokine expression is a key component of the anergy program of TH1 cells and identify Tle4 as the transcriptional repressor responsible for the establishment of the epigenetic repressive marks at the Ifng locus that result in silencing of Ifng gene expression. Inhibition of Tle4 could therefore constitute a possible target to prevent downregulation of IFN-γ expression and the inhibition of effector functions of tumor antigen-specific T cells in order to improve antitumor immune responses.
ACKNOWLEDGMENT
This work was supported by NIH grant AI059738 (to F.M.).
Footnotes
Published ahead of print 4 November 2013
REFERENCES
- 1.Walker LS, Abbas AK. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2:11–19. 10.1038/nri701 [DOI] [PubMed] [Google Scholar]
- 2.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. 2002. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109:719–731. 10.1016/S0092-8674(02)00767-5 [DOI] [PubMed] [Google Scholar]
- 3.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. 1993. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc. Natl. Acad. Sci. U. S. A. 90:6586–6590. 10.1073/pnas.90.14.6586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. 1990. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J. Immunol. 144:16–22 [PubMed] [Google Scholar]
- 5.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. 2001. CTLA-4 regulates induction of anergy in vivo. Immunity 14:145–155. 10.1016/S1074-7613(01)00097-8 [DOI] [PubMed] [Google Scholar]
- 6.Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. 1997. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity 6:411–417. 10.1016/S1074-7613(00)80284-8 [DOI] [PubMed] [Google Scholar]
- 7.Wells AD, Walsh MC, Bluestone JA, Turka LA. 2001. Signaling through CD28 and CTLA-4 controls two distinct forms of T cell anergy. J. Clin. Invest. 108:895–903. 10.1172/JCI13220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. 1993. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365:352–355. 10.1038/365352a0 [DOI] [PubMed] [Google Scholar]
- 9.Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A. 1996. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J. Biol. Chem. 271:10884–10891. 10.1074/jbc.271.18.10884 [DOI] [PubMed] [Google Scholar]
- 10.Macian F, Garcia-Rodriguez C, Rao A. 2000. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 19:4783–4795. 10.1093/emboj/19.17.4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baine I, Abe BT, Macian F. 2009. Regulation of T-cell tolerance by calcium/NFAT signaling. Immunol. Rev. 231:225–240. 10.1111/j.1600-065X.2009.00817.x [DOI] [PubMed] [Google Scholar]
- 12.Fields PE, Gajewski TF, Fitch FW. 1996. Blocked Ras activation in anergic CD4+ T cells. Science 271:1276–1278. 10.1126/science.271.5253.1276 [DOI] [PubMed] [Google Scholar]
- 13.Bandyopadhyay S, Montagna C, Macian F. 2012. Silencing of the Il2 gene transcription is regulated by epigenetic changes in anergic T cells. Eur. J. Immunol. 42:2471–2483. 10.1002/eji.201142307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, Macian F. 2007. Interleukin 2 gene transcription is regulated by Ikaros-induced changes in histone acetylation in anergic T cells. Blood 109:2878–2886. 10.1182/blood-2006-07-037754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas RM, Chunder N, Chen C, Umetsu SE, Winandy S, Wells AD. 2007. Ikaros enforces the costimulatory requirement for IL2 gene expression and is required for anergy induction in CD4+ T lymphocytes. J. Immunol. 179:7305–7315 [DOI] [PubMed] [Google Scholar]
- 16.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 292:1907–1910. 10.1126/science.1059835 [DOI] [PubMed] [Google Scholar]
- 17.Murphy KM, Reiner SL. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2:933–944. 10.1038/nri954 [DOI] [PubMed] [Google Scholar]
- 18.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655–669. 10.1016/S0092-8674(00)80702-3 [DOI] [PubMed] [Google Scholar]
- 19.Knoechel B, Lohr J, Zhu S, Wong L, Hu D, Ausubel L, Abbas AK. 2006. Functional and molecular comparison of anergic and regulatory T lymphocytes. J. Immunol. 176:6473–6483 [DOI] [PubMed] [Google Scholar]
- 20.Powell JD, Lerner CG, Schwartz RH. 1999. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J. Immunol. 162:2775–2784 [PubMed] [Google Scholar]
- 21.Thomas RM, Saouaf SJ, Wells AD. 2007. Superantigen-induced CD4+ T cell tolerance is associated with DNA methylation and histone hypo-acetylation at cytokine gene loci. Genes Immun. 8:613–618. 10.1038/sj.gene.6364415 [DOI] [PubMed] [Google Scholar]
- 22.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. 2008. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111:251–259. 10.1182/blood-2007-03-081646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palaparti A, Baratz A, Stifani S. 1997. The Groucho/transducin-like enhancer of split transcriptional repressors interact with the genetically defined amino-terminal silencing domain of histone H3. J. Biol. Chem. 272:26604–26610. 10.1074/jbc.272.42.26604 [DOI] [PubMed] [Google Scholar]
- 24.Brantjes H, Roose J, van De Wetering M, Clevers H. 2001. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res. 29:1410–1419. 10.1093/nar/29.7.1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Javed A, Guo B, Hiebert S, Choi JY, Green J, Zhao SC, Osborne MA, Stifani S, Stein JL, Lian JB, van Wijnen AJ, Stein GS. 2000. Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J. Cell Sci. 113:2221–2231 [DOI] [PubMed] [Google Scholar]
- 26.Ren B, Chee KJ, Kim TH, Maniatis T. 1999. PRDI-BF1/Blimp-1 repression is mediated by corepressors of the Groucho family of proteins. Genes Dev. 13:125–137. 10.1101/gad.13.1.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cimmino L, Martins GA, Liao J, Magnusdottir E, Grunig G, Perez RK, Calame KL. 2008. Blimp-1 attenuates Th1 differentiation by repression of ifng, tbx21, and bcl6 gene expression. J. Immunol. 181:2338–2347 [DOI] [PubMed] [Google Scholar]
- 28.Abe BT, Shin DS, Mocholi E, Macian F. 2012. NFAT1 supports tumor-induced anergy of CD4(+) T cells. Cancer Res. 72:4642–4651. 10.1158/0008-5472.CAN-11-3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kametani Y, Wang L, Koduka K, Sato T, Katano I, Habu S. 2008. Rapid histone deacetylation and transient HDAC association in the IL-2 promoter region of TSST-1-stimulated T cells. Immunol. Lett. 119:97–102. 10.1016/j.imlet.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 30.du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. 2007. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 26:424–435. 10.1038/sj.emboj.7601517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reed-Inderbitzin E, Moreno-Miralles I, Vanden-Eynden SK, Xie J, Lutterbach B, Durst-Goodwin KL, Luce KS, Irvin BJ, Cleary ML, Brandt SJ, Hiebert SW. 2006. RUNX1 associates with histone deacetylases and SUV39H1 to repress transcription. Oncogene 25:5777–5786. 10.1038/sj.onc.1209591 [DOI] [PubMed] [Google Scholar]
- 32.Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, Weaver CT. 2006. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25:717–729. 10.1016/j.immuni.2006.09.007 [DOI] [PubMed] [Google Scholar]
- 33.Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. 2007. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 8:732–742. 10.1038/ni1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flores-Saaib RD, Courey AJ. 2000. Analysis of Groucho-histone interactions suggests mechanistic similarities between Groucho- and Tup1-mediated repression. Nucleic Acids Res. 28:4189–4196. 10.1093/nar/28.21.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jennings BH, Ish-Horowicz D. 2008. The Groucho/TLE/Grg family of transcriptional co-repressors. Genome Biol. 9:205. 10.1186/gb-2008-9-1-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. 2000. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell. Biol. 20:2592–2603. 10.1128/MCB.20.7.2592-2603.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. 2003. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 198:569–580. 10.1084/jem.20030590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staveley-O'Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. 1998. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl. Acad. Sci. U. S. A. 95:1178–1183. 10.1073/pnas.95.3.1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandyopadhyay S, Soto-Nieves N, Macian F. 2007. Transcriptional regulation of T cell tolerance. Semin. Immunol. 19:180–187. 10.1016/j.smim.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz RH. 2003. T cell anergy. Annu. Rev. Immunol. 21:305–334. 10.1146/annurev.immunol.21.120601.141110 [DOI] [PubMed] [Google Scholar]
- 41.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, Powell JD. 2005. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 6:472–480. 10.1038/ni1193 [DOI] [PubMed] [Google Scholar]
- 42.Zheng Y, Zha Y, Driessens G, Locke F, Gajewski TF. 2012. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med. 209:2157–2163. 10.1084/jem.20120342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, Macian F. 2009. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J. Exp. Med. 206:867–876. 10.1084/jem.20082731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Whaley CD, Mondino A, Mueller DL. 1996. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science 271:1272–1276. 10.1126/science.271.5253.1272 [DOI] [PubMed] [Google Scholar]
- 45.Anandasabapathy N, Ford GS, Bloom D, Holness C, Paragas V, Seroogy C, Skrenta H, Hollenhorst M, Fathman CG, Soares L. 2003. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 18:535–547. 10.1016/S1074-7613(03)00084-0 [DOI] [PubMed] [Google Scholar]
- 46.Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. 2004. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 5:255–265. 10.1038/ni1047 [DOI] [PubMed] [Google Scholar]
- 47.Kriegel MA, Rathinam C, Flavell RA. 2009. E3 ubiquitin ligase GRAIL controls primary T cell activation and oral tolerance. Proc. Natl. Acad. Sci. U. S. A. 106:16770–16775. 10.1073/pnas.0908957106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nurieva RI, Zheng S, Jin W, Chung Y, Zhang Y, Martinez GJ, Reynolds JM, Wang SL, Lin X, Sun SC, Lozano G, Dong C. 2010. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity 32:670–680. 10.1016/j.immuni.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. 2006. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 7:1174–1181. 10.1038/ni1400 [DOI] [PubMed] [Google Scholar]
- 50.Puga I, Rao A, Macian F. 2008. Targeted cleavage of signaling proteins by caspase 3 inhibits T cell receptor signaling in anergic T cells. Immunity 29:193–204. 10.1016/j.immuni.2008.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, Praveen K, Stang S, Stone JC, Gajewski TF. 2006. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat. Immunol. 7:1166–1173. 10.1038/ni1394 [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, Divekar R, McBurney MW, Braley-Mullen H, Zaghouani H, Fang D. 2009. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Invest. 119:3048–3058. 10.1172/JCI38902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agarwal S, Rao A. 1998. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9:765–775. 10.1016/S1074-7613(00)80642-1 [DOI] [PubMed] [Google Scholar]
- 54.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. 2002. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 3:643–651. 10.1038/nrg904 [DOI] [PubMed] [Google Scholar]
- 55.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. 2012. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335:723–727. 10.1126/science.1214277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel SR, Bhumbra SS, Paknikar RS, Dressler GR. 2012. Epigenetic mechanisms of Groucho/Grg/TLE mediated transcriptional repression. Mol. Cell 45:185–195. 10.1016/j.molcel.2011.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, Wherry EJ. 2009. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31:309–320. 10.1016/j.immuni.2009.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martins GA, Cimmino L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, Calame K. 2006. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 7:457–465. 10.1038/ni1320 [DOI] [PubMed] [Google Scholar]
- 59.Eivazova ER, Aune TM. 2004. Dynamic alterations in the conformation of the Ifng gene region during T helper cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 101:251–256. 10.1073/pnas.0303919101 [DOI] [PMC free article] [PubMed] [Google Scholar]