

Abstract
Pathogen transmission cycles require many steps: initial colonization, growth and persistence, shedding, and transmission to new hosts. Alterations in the membrane components of the bacteria, including lipid A, the membrane anchor of lipopolysaccharide, could affect any of these steps via its structural role protecting bacteria from host innate immune defenses, including antimicrobial peptides and signaling through Toll-like receptor 4 (TLR4). To date, lipid A has been shown to affect only the within-host dynamics of infection, not the between-host dynamics of transmission. Here, we investigate the effects of lipid A modification in a mouse infection and transmission model. Disruption of the Bordetella bronchiseptica locus (BB4268) revealed that ArnT is required for addition of glucosamine (GlcN) to B. bronchiseptica lipid A. ArnT modification of lipid A did not change its TLR4 agonist activity in J774 cells, but deleting arnT decreased resistance to killing by cationic antimicrobial peptides, such as polymyxin B and β-defensins. In the standard infection model, mutation of arnT did not affect B. bronchiseptica colonization, growth, persistence throughout the respiratory tract, recruitment of neutrophils to the nasal cavity, or shedding of the pathogen. However, the number of bacteria necessary to colonize a host (50% infective dose [ID50]) was 5-fold higher for the arnT mutant. Furthermore, the arnT mutant was defective in transmission between hosts. These results reveal novel functions of the ArnT lipid A modification and highlight the sensitivity of low-dose infections and transmission experiments for illuminating aspects of infectious diseases between hosts. Factors such as ArnT can have important effects on the burden of disease and are potential targets for interventions that can interrupt transmission.
INTRODUCTION
Lipopolysaccharide (LPS), the major component of the outer membrane of Gram-negative bacteria is known to affect interactions with the host in a variety of ways that have been illuminated using host infection models. Upon initial contact with the host mucosa, LPS can protect pathogens from innate host defenses, such as complement and cationic antimicrobial peptides (CAMPs) (1). When the lipid A portion of LPS binds to host membrane complexes, including MD2 and CD14, a signaling cascade is initiated through Toll-like receptor 4 (TLR4) (2). This signaling pathway mobilizes the transcription factor NF-κB and induces the expression of proinflammatory cytokines and chemokines in cells of the innate immune system (2). TLR4 signaling also facilitates the recruitment of adaptive immune responses, particularly through the activation of dendritic cells (DC), which are induced by LPS to migrate to regional lymph nodes and present antigens to T cells (3). Lipid A-TLR4 interactions are therefore central to host-pathogen dynamics during infections by Gram-negative bacteria. Consequently, it is not surprising that pathogens regulate their lipid A structures through a number of covalent modifications, which can affect interactions with host immunity (1, 4).
Bordetella bronchiseptica is a Gram-negative coccobacillus, closely related to Bordetella pertussis and Bordetella parapertussis, the causative agents of whooping cough in humans. B. bronchiseptica is highly infectious in mice, providing a model system in which the role of specific Bordetella virulence factors during infection can be probed in the context of a natural host infection (5). Adhesins, toxins, and other factors that enable B. bronchiseptica to thrive within the host are chiefly controlled by the two-component regulatory system, BvgAS (6, 7). These virulence-associated genes are expressed maximally in the Bvg+ phase and transcriptionally repressed in the Bvg− phase (8). Modifications of the lipid A of B. bronchiseptica are regulated by BvgAS (9–11). B. bronchiseptica lipid A consists of a glucosamine disaccharide backbone anchored to the bacterial outer membrane by a series of acyl groups (9) (Fig. 1C, D, and E, showing the structure of the lipid A). Normally, B. bronchiseptica lipid A is penta-acylated with 3-OH C14 acyl groups at the 2 and 2′ positions and a 3-OH C10 at the 3′ position. The 3 position is “empty” due to the deacylase activity of the outer membrane enzyme PagL (9). Secondary, or piggyback, acylations at the 2 position are either a 2OH-C12 or C12, with the presence of 2OH-C12 dependent on the lipid A dioxygenase, LpxO (9). PagP is a Bvg-regulated lipid A palmitoyl transferase that adds palmitate as a secondary acylation at the 3′ position, generating a hexa-acylated structure (12, 13). Finally, the major lipid A species contains a single phosphate at the C-4′ position although some molecules are also phosphorylated at the C-1 position (10). In monophosphorylated lipid A species, the C-4′ phosphate is decorated with a GlcN molecule; however, in lipid A molecules that possess two phosphate groups, only one GlcN modification is observed at either the C-1 or the C-4′ position. Orthologues of the lipid A modification enzyme, ArnT, which decorates the lipid A phosphates of Salmonella enterica serovar Typhimurium with aminoarabinose are conserved among Bordetella species (14). The modification of phosphate groups with aminoarabinose decreases the net negative charge on LPS and renders S. Typhimurium resistant to the antimicrobial cationic peptide, polymyxin (15). In B. pertussis, the ArnT activity of the homologue LgmB is induced in the Bvg+ phase and mediates the addition of GlcN to both terminal phosphate groups of the lipid A, which is associated with increased stimulation of TLR4 activity in an HEK-Blue assay and upon infection of human macrophages (10, 11, 16). Deletion of arnT from B. pertussis did not affect resistance to killing by polymyxin B (10). ArnT-mediated addition of GlcN has also been reported for B. parapertussis (17) and B. bronchiseptica strain 4650 (11).
FIG 1.

Ions corresponding to glucosamine additions in RB50 are not seen in RB50 ΔarnT. (A) Mass spectra of B. bronchiseptica RB50 ions corresponding to glucosamine additions are identified by mass-to-charge ratios (m/z) of 1,651, 1,667, and 1,731 (*). (B) RB50 ΔarnT mutant strain with no glucosamine peaks. Lipid A structures corresponding to m/z 1,570 (C), 1,808 (D), and 1,651 (E) are shown.
In this work, we characterized the function of the B. bronchiseptica arnT homologue, BB4268, by the construction and analysis of an arnT mutant in B. bronchiseptica strain RB50, henceforth referred to as RB50 ΔarnT. We found that, similar to its function in B. pertussis, B. bronchiseptica arnT was required for Bvg+ phase-dependent addition of GlcN to the lipid A. No change in the TLR4 agonist activity of the mutant strain was observed; however, loss of resistance against polymyxin B and β-defensin (BD)-mediated killing was detected. Loss of arnT had no effect on bacterial growth or persistence when bacteria were seeded throughout the respiratory tract by standard high-dose inoculation; however, the arnT mutant was not transmitted between mice even though the mutant was shed from index cases at the same level as the wild type. Furthermore, RB50 ΔarnT required approximately a 5-fold increase in mean inoculation dose compared to the wild type to initiate infections. Together, these results show that deleting arnT had no observable effects in standard virulence and pathogenesis assays but did affect LPS modification, which had a major impact on shedding and transmission of B. bronchiseptica.
MATERIALS AND METHODS
Bacterial strains and growth.
Bordetella bronchiseptica strains RB50 (8), RB50 Δwbm (18), and RB50 ΔarnT were maintained on Bordet-Gengou (BG) agar (Difco) supplemented with 10% defibrinated sheep blood (Hema Resources) and 200 μg/ml streptomycin (Sigma-Aldrich) and cultured in Stainer-Scholte broth (19) at 37°C until grown to mid-log phase, approximately an optical density at 600 nm (OD600) of 0.5. For a bacterial killing assay using murine β-defensin 3 (mBD3), Escherichia coli K-12 bacteria were grown in Luria Bertani broth at 37°C until mid-log phase.
Mutation of BB4268.
Genomic DNA template was made by resuspending several colonies of plate-grown bacteria in 0.5 ml of water, boiling the samples in a water bath for 5 min, spinning them at top speed in a benchtop microcentrifuge for 2 min, and then taking a 0.2-ml sample of the supernatant. One microliter of supernatant was used per PCR. Each PCR mixture comprised genomic DNA template, buffer as directed by the manufacturer, deoxynucleoside triphosphate (dNTPs; 25 mM each), 20 ng of each primer, 5% (vol/vol) dimethyl sulfoxide (DMSO), 5 mM MgCl2, and 2.5 units of Taq DNA polymerase (Promega). Primers used to amplify an approximately 1-kb section of B. bronchiseptica 4268 were 5′-ATGTAGCCGACCAGCTTG-3′ and 5′-ATCCATGCAACCCCATGC-3′, corresponding to bases 4544225.0.4544242 and 4545223.0.4545206, respectively, of the B. bronchiseptica RB50 genome sequence, GenBank accession number BX470250 (20). PCRs were incubated at 94°C for 5 min, followed by 30 cycles of 94°C for 75 s, 60°C for 75 s, and 72°C for 90 s, with a final step of 72°C for 7 min.
The PCR product was cloned into pGEM-T Easy (Promega) according to the manufacturer's instructions. A nonpolar kanamycin resistance cassette was ligated into a unique StuI site residing in the middle of the cloned BB4268 region. The resulting BB4268-kan region was subcloned into pEX100T (21), and the resulting construct was moved into the conjugation donor strain SM10 (22) by transformation. Bacterial conjugations were performed as described previously (18). The expected chromosomal rearrangements in conjugants were confirmed by Southern hybridization analyses.
B. bronchiseptica arnT was amplified by PCR using primers incorporating NdeI and HindIII restriction endonuclease recognition sites at the 5′ and 3′ ends of the amplicon, respectively. Following digestion of the PCR product with NdeI and HindIII, this fragment was cloned behind the B. bronchiseptica pagP promoter into pBBRkanpagP (12), replacing the pagP coding sequence (CDS) in this construct. The arnT-containing construct was moved into wild-type B. bronchiseptica and the B. bronchiseptica arnT mutant by conjugation as described previously (12).
Lipid A purification.
LPS was purified from 1 liter of overnight B. bronchiseptica cultures as described previously (23). Further treatment of LPS with RNase A, DNase I, and proteinase K ensured removal of contaminating nucleic acids and proteins (24). Hydrolysis of LPS to isolate lipid A was accomplished with 1% sodium dodecyl sulfate (SDS) at pH 4.5 as described previously (19).
Confirmation of lipid A structures.
The lipid A structures were confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry. LPS was isolated using a rapid small-scale isolation method (25). Cell culture pellets (1 to 10 ml of an overnight culture) were resuspended in a 1.0-ml aliquot of TriReagent (Molecular Research Center, Cincinnati, OH, USA) and incubated at room temperature for 15 min. Chloroform (200 ml) was added, and the samples were vortexed and incubated at room temperature for 15 min. Samples were centrifuged for 10 min at 13 400 × g, and the aqueous layers were collected. Water (500 ml) was added to the lower layer and vortexed. After 30 min, the samples were centrifuged as above, and the aqueous layers were pooled. Two more aliquots of water were added to each sample, for a total of four extractions. The combined aqueous layers were frozen and lyophilized. The LPS was then hydrolyzed to lipid A. Lyophilized LPS was resuspended in 0.5 ml of 1% sodium dodecyl sulfate (SDS) in 10 mM sodium acetate buffer, pH 4.5 (19). Samples were incubated at 100°C for 1 h, frozen, and lyophilized. The dried pellets were washed in 0.1 ml of water and 1 ml of acidified ethanol (EtOH) (100 ml of 4 N HCl in 20 ml of 95% EtOH). Samples were centrifuged at 2,300 × g for 5 min, and the supernatant was discarded. The lipid A pellet was further washed (twice for a total of three washes) in 1 ml of 95% EtOH. The entire series of washes was repeated twice. A final wash step was carried out in 100% ethanol. Lipid A was extracted in a mixture of chloroform, methanol, and water (3:1:0.25, vol/vol/vol). One microliter of this extract was then spotted onto a MALDI target plate followed by 1 μl of Norharmane matrix and air dried. Samples were analyzed on a Bruker AutoFlex Speed (Bruker Daltonics, Billerica, MA) mass spectrometer, which was calibrated using Agilent Tuning Mix (Agilent Technologies, Foster City, CA).
In vitro macrophage stimulation and TNF-α detection.
J774 murine macrophages were cultured in Dulbecco's modified Eagle's medium (DMEM; Difco) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% nonessential amino acids, and 1% sodium pyruvate (wt/vol). The cells were grown to approximately 85% confluence in a 96-well plate. In order to compare tumor necrosis factor alpha (TNF-α) secretion, J774 cells were inoculated with RB50 or RB50 ΔarnT at a multiplicity of infection (MOI) of 0.001, 0.01, 0.1, or 1. Medium was harvested 2 h postinoculation, and an enzyme-linked immunosorbent assay (ELISA) to detect TNF-α concentrations was performed according to the manufacturer's instructions (R&D Systems). These assays were performed two times in quadruplicate.
Adherence assay.
Rat epithelial (L2) cells were grown to 80% confluence in 96-well plates using DMEM (Difco)–F-12 medium supplemented with 10% fetal bovine serum. These cells were inoculated with 104 CFU of RB50 or RB50 ΔarnT (MOI of 0.2). Plates were then centrifuged for 5 min at 250 × g, followed by incubation at 37°C with 5% CO2 for 40 min. Wells were then washed four times with 1 ml of the growth medium to remove nonadherent bacteria. L2 cells were then treated with 0.5 ml of 0.125% trypsin (Sigma-Aldrich), followed by incubation for 10 min at 37°C. The total volume of each well was brought up to 1 ml with growth medium and homogenized by pipetting. Dilutions were plated on BG plates containing 40 μg of streptomycin/ml to determine CFU counts, which were then used to calculate the proportion of adherent bacteria, expressed as a percentage of the original inoculum.
Serum killing assay.
Approximately, 103 CFU of RB50, RB50 Δwbm (O-antigen mutant), or RB50 ΔarnT in 50 μl of phosphate-buffered saline (PBS) was incubated with serum from mice naive to Bordetella at concentrations ranging from 0 to 15% solution by volume. After 1 h of incubation at 37°C followed by a 5-min incubation on ice, the entire 50-μl sample was plated onto Bordet-Gengou blood agar containing streptomycin (20 μg/ml). Colonies were enumerated after 2 days of incubation at 37°C. This assay was performed two times in quadruplicate.
β-Defensin killing assay.
A total of 106 CFU of RB50 and RB50 ΔarnT was incubated with 0, 5, and 10 μg/ml of synthetic porcine β-defensin 1 (pBD1) (26) and mBD3 (R&D Systems) in 100 μl of PBS at 37°C for 2 h, after which the reaction mixture was plated on BG agar and incubated at 37°C for 2 days to calculate the number of CFU. For the mBD3 assay, E. coli K-12 bacteria were used as a positive control.
Polymyxin B susceptibility assay.
Cultures of RB50 or RB50 ΔarnT were diluted to 106 CFU/ml into a final 1-ml volume of PBS, PBS containing 10 mg/ml polymyxin B, or PBS containing 100 mg/ml of polymyxin B. Suspensions were incubated for 2 h at 37°C, following which the number of organisms remaining in each sample was determined by quantitative culture on BG agar plates.
Colonization studies.
C57BL/6 (wild type) and C3H/HEJ (TLR4 deficient) mice were obtained from Jackson Laboratories and bred in our Bordetella-free, specific-pathogen-free facilities at The Pennsylvania State University. Bacteria grown overnight to an optical density at 600 nm of approximately 0.3 in liquid culture were diluted in PBS to approximately 2 × 106 CFU/ml. For a high-dose/high-volume inoculation, 50 μl of the inoculum (104 CFU) was pipetted on to the external nares of 4- to 6-week-old mice that had been lightly sedated with 5% isoflurane in oxygen. For low-dose/low-volume inoculations, bacterial cultures were further diluted in PBS to concentrations of 103 CFU/ml, and for high-dose/low-volume inoculations, they were diluted to 4 × 104 CFU/ml. Mice were inoculated with doses of 5 CFU and 200 CFU in 5 μl using the previously described procedure. Groups of three or four animals were sacrificed on days 3, 7, 14, and 28 postinoculation or as indicated (see Fig. 5A), and the nasal cavity, trachea, and lungs were excised. Bacterial numbers in the respiratory tract were quantified by homogenization of each tissue in PBS, followed by plating onto Bordet-Gengou blood agar containing streptomycin (20 μg/ml). Colonies were enumerated after 2 days of growth at 37°C. All protocols were reviewed and approved by The Pennsylvania State University Institutional Animal Care and Use Committee (IACUC), and all animals were handled in accordance with institutional guidelines.
FIG 5.

ArnT decreases the mean infectious dose of B. bronchiseptica. (A) C57BL/6 mice were inoculated with 5 CFU in 5 μl of either RB50 or RB50 ΔarnT. Dots represent the number of CFU recovered from the nasal cavity of individual mice dissected 7 days after inoculation. The whiskers span the 1st and 4th quartiles. The limit of detection is marked by a dashed line. The proportion of infected mice in each group is listed above the box-and-whisker plot. (B) Percentage of C57BL/6 mice infected after inoculation with either 5 CFU (10 mice/group), 50 CFU (12 mice/group), or 200 CFU (16 mice/group) of RB50 or RB50 ΔarnT.
Shedding analysis.
Shedding was assessed by lightly swabbing the external nares for 10 s using a Dacron-polyester tipped swab. Swab tips were cut off and placed into 1 ml of PBS. Samples were vortexed vigorously and cultured on Bordet-Gengou agar (Himedia).
Analysis of leukocyte recruitment.
Prior to dissection, 10 to 20 ml of PBS was perfused through the left ventricles of the mice while venous runoff was collected from the orbits. Nasal bones were dissected and placed in 1 ml of DMEM containing 5% FBS and 1 mg/ml collagenase D. Samples were incubated for 45 min at 37°C and subsequently disaggregated into a single-cell suspension by mechanical disruption over a 70-μm mesh screen. A total of 2 × 106 cells per well were then added to 96-well plates. Samples were resuspended in FC blocking buffer (200:1, anti-CD16/32 [BD Biosciences] in PBS–2% FBS) and incubated on ice for 20 min. Following a washing step, cell surface markers were labeled with the following antibodies in PBS–2% FBS: anti-CD45 allophycocyanin (APC)-Cy7 (400:1; BD Biosciences), anti-CD11b Horizon V450 (BD Biosciences), and anti-Ly6G APC (E Bioscience).
Statistical analysis.
Data analysis between groups was performed using a two-tailed Student's t test to evaluate statistical significance.
RESULTS
ArnT is required for modification of lipid A with glucosamine.
To investigate the role of ArnT enzymatic activity in GlcN modification of lipid A, the B. bronchiseptica RB50 arnT homologue, previously designated BB4268, was mutated to generate strain RB50 ΔarnT. Purified lipid A isolated from RB50 or RB50 ΔarnT was analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry in the negative-ion mode. Bordetella bronchiseptica lipid A had previously been characterized by Preston et al. (12). Peaks indicating a glucosamine addition were present at m/z 1,651 and 1,667, representing the monophosphorylated penta-acylated lipid A with a two acyl-oxo-acyl C12 and 2OH-C12, respectively, and m/z 1,731, corresponding to the 1,651 species with diphosphate additions (Fig. 1A) as previously described (12). These peaks were entirely absent in the RB50 ΔarnT spectrum (Fig. 1B). Complementation with the arnT gene restored the phenotype (data not shown), demonstrating that ArnT is required for addition of glucosamine to Bordetella bronchiseptica lipid A. All other characteristic peaks were observed for both the wild type and RB50 ΔarnT.
TLR4 stimulation in murine macrophages, serum resistance, and adhesion are not affected by arnT mutation.
ArnT-mediated modification of lipid A has been shown to enhance stimulation of TLR4 by B. pertussis LPS (10). To determine if the increased TLR4 agonist activity of B. bronchiseptica LPS was dependent on GlnN addition, cultures of murine macrophages (J774) were inoculated with either wild-type strain RB50 or the arnT mutant. Release of the proinflammatory cytokine TNF-α into the medium was used as a surrogate measurement of TLR4 receptor activity. Cultures were inoculated at MOIs ranging from 0.001 to 1, and the concentration of TNF-α in the medium was determined 2 h after inoculation by quantitative ELISA (Fig. 2A). The amount of TNF-α detected in the supernatant of cells exposed to B. bronchiseptica increased in a dose-dependent manner; however, at each MOI, the amounts of TNF-α released by macrophages were similar between RB50- and RB50 ΔarnT-inoculated cell cultures. These data suggest that agonist activity of lipid A for TLR4 is not affected by ArnT-mediated addition of glucosamine.
FIG 2.

ArnT is not required for induction of TNF-α in murine macrophages, complement resistance, or adherence to the lung epithelial cells. (A) J774 macrophages were incubated for 2 h in the presence of RB50 or RB50 ΔarnT at an MOI of 0.001, 0.01, 0.1, or 1.0. Gray bars represent cultures treated with medium only. (B) A total of 103 CFU of either B. bronchiseptica strain RB50, RB50 ΔarnT (black diamonds), or RB50 Δwbm (black circles) was incubated for 1 h in PBS only or PBS with 5, 10, or 15% naive mouse serum. Symbols represent the mean log10 CFU ± standard error of four individual samples recovered after incubation. The limit of detection is marked by a dashed line. (C) Increasing MOIs of RB50 or RB50 ΔarnT were inoculated onto cultured L2 rat lung epithelial cells. The quantity of CFU of RB50 or RB50 ΔarnT adhering to epithelial cells following incubation was determined by culture. Symbols represent the mean CFU counts ± standard error of four individual samples.
B. bronchiseptica is protected from the antimicrobial activities of serum complement by its LPS; strains lacking either O-antigen, such as RB50 Δwbm (18), or the outer core oligosaccharide and O-antigen portions of LPS are highly susceptible to killing by serum complement. The B. bronchiseptica RB50 mutant with a deletion of wbm, a locus necessary for the assembly of the O antigen, lacks this structure and has the aforementioned defect in resistance to complement-mediated killing (18). To determine whether mutation of arnT affects serum resistance, approximately 103 CFU of RB50, RB50 Δwbm, or RB50 ΔarnT was incubated with concentrations of serum ranging from 0 to 15% (by volume). Whereas RB50 Δwbm was killed by 5% serum, serum concentrations of up to 15% had no effect on either wild-type or arnT mutant bacteria, demonstrating that mutation of arnT does not affect serum resistance (Fig. 2B).
Loss of the GlcN substitution might alter the structure of the outer membrane and destabilize interactions that facilitate bacterial adherence to respiratory epithelial cells. To test this, L2 rat lung epithelial cells were inoculated with between 5 and 1,000 CFU of the wild-type strain or RB50 ΔarnT. No significant difference was observed in the number of wild-type or mutant bacteria recovered from the trypsin-treated epithelial cells (Fig. 2C). This result suggests that binding of RB50 to the respiratory epithelium is not affected by deletion of arnT.
Glucosamine additions to lipid A contribute to resistance against cationic antimicrobial peptides.
Host epithelial cells and leukocytes produce small cationic peptides, defensins, and cathelicidins that exhibit antimicrobial activity against numerous bacterial species (27–29). Defensins are amphipathic molecules proposed to intercalate into the bacterial cell membrane and use like-charge repulsion to disrupt membrane integrity (29). Disruption of the arnT locus rendered S. Typhimurium susceptible to killing by cationic peptides. To test whether arnT provided a similar adaptation for B. bronchiseptica, we compared the ability of RB50 and RB50 ΔarnT to survive in the presence of various concentrations of antimicrobial peptides. RB50 was resistant to killing by polymyxin B at concentrations up to 100 μg/ml. However, greater than 90% of RB50 ΔarnT bacteria were killed by 10 μg/ml, and 99.99% of the mutant bacteria were killed by 100 μg/ml of polymyxin B (Fig. 3A). In addition, wild-type RB50 survived a concentration of 10 μg/ml of the porcine β-defensin 1 (pBD1), whereas more than 99% of RB50 ΔarnT was killed by 5 μg/ml of pBD1. Surprisingly, RB50 and the arnT mutant showed no significant difference in susceptibilities to killing by mouse β-defensin 3, which shares homology with pBD1 (Fig. 3C). Our results suggest that modification of lipid A by GlcN renders RB50 more resistant to killing by some, but not all, antimicrobial cationic peptides.
FIG 3.

ArnT is required for resistance to killing by cationic antimicrobial peptides. Bacteria (106 to 108 CFU) were incubated for 2 h in PBS (white bars) or PBS with increasing concentrations, as indicated, of polymyxin B (A), pBD1 (B), or synthetic mBD3 (C). B. bronchiseptica strains RB50 and RB50 ΔarnT were incubated with mBD3, or E. coli K-12, whose susceptibility to mBD3 was previously shown, was used as a control to determine whether mBD3 was active. Bars represent the mean log10 CFU ± standard error of four individual samples in each group. *, P < 0.05; **, P < 0.01. The limit of detection is marked by a dashed line.
B. bronchiseptica arnT is not necessary for infection of the mouse respiratory tract.
To determine whether decreased resistance to CAMPs would result in reduced fitness within a host, we compared the ability of the RB50 and the arnT mutant to grow and persist during experimental infections of mice. Following inoculation of C57BL/6 mice with 104 CFU of either RB50 or RB50 ΔarnT in 50 μl of PBS, mice were dissected after 3, 7, 14, and 28 days. At each time point the number of CFU recovered from the respiratory tract of mice inoculated with RB50 ΔarnT was similar to that recovered from RB50-inoculated mice (Fig. 4). This result suggests that addition of GlcN to the lipid A is not required for growth and persistence of B. bronchiseptica in the murine respiratory tract when it is introduced in a high-dose inoculum.
FIG 4.
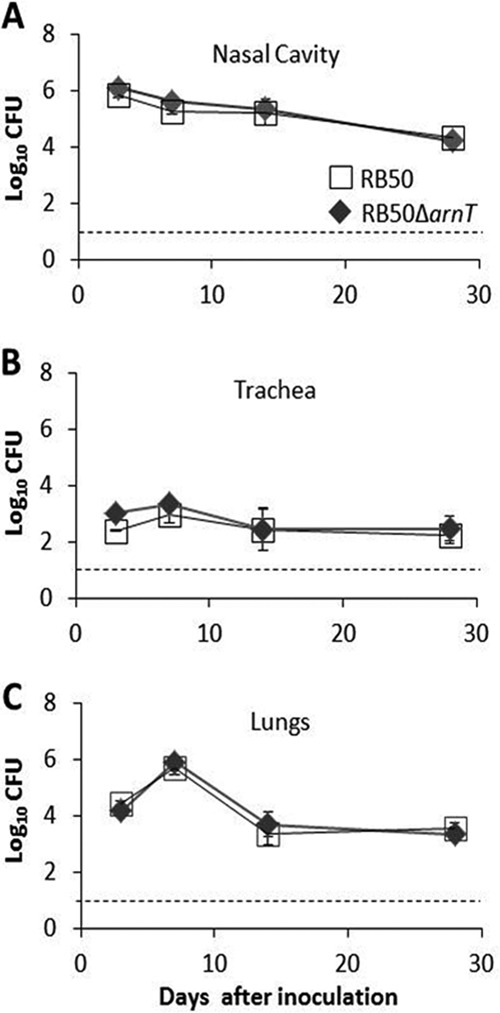
ArnT does not contribute to growth or resistance in the respiratory tract in a high-dose model of respiratory infection. Groups of four mice were inoculated with 5 × 104 CFU of RB50 (white squares) or RB50 ΔarnT (black diamonds) in 50 μl of PBS. Symbols represent the mean log10 CFU count ± standard error recovered from either the nasal cavity (A), trachea (B), or lungs (C) at 3, 7, 14, and 28 days following inoculation. The limit of detection is marked by a dashed line.
RB50 ΔarnT is unable to colonize mice when inoculated in a low dose.
Although RB50 ΔarnT grew and persisted similarly to the wild-type strain following high-dose inoculation challenge, we hypothesized that high-dose infections may not accurately reproduce the interactions taking place when B. bronchiseptica initially seeds the respiratory mucosa. For example, CAMPs may not be sufficiently potent to control the initial influx of large quantities of infectious organisms, or the epithelial cells that produce CAMPs may be rapidly damaged by bacterial toxins. A functional defect resulting from decreased CAMP resistance could therefore be more effectively determined using low-dose inoculations. To approximate a mean infectious dose, 5, 50, or 200 CFU of either RB50 or RB50 ΔarnT was inoculated in a 5-μl droplet onto the external nares of C57BL/6 mice. Seven days postinoculation, the presence of B. bronchiseptica in the nasal cavity was determined (Fig. 5A). Five CFU of wild-type RB50 was sufficient to infect 5 of 10 mice, whereas 50 CFU resulted in stable colonization of 11/12 mice. Additionally, 12/12 mice became infected following inoculation with 200 CFU of RB50. By comparison, 5 CFU of RB50 ΔarnT infected only 1/16 mice, 50 CFU infected 4/12 mice, and 200 CFU infected 12/12 mice (Fig. 5B). Using the Reed-Muench calculation (30), we determined that the 50% infectious dose (ID50) of wild-type RB50 was approximately 6.2 CFU, whereas the ID50 of RB50 ΔarnT was 30 CFU.
RB50 ΔarnT fails to transmit between mice.
Although the presence of ArnT did not affect the infectious burden following high-dose inoculation, subtle functional deficiency resulting from mutation of arnT would be more likely resolved by analysis of transmission efficiency. We have previously demonstrated that RB50 transmits efficiently between C3H/HeJ mice (32). To evaluate whether the increased experimental ID50 of RB50 ΔarnT corresponded to changes in transmission efficiency between animals, mice were inoculated with 500 CFU of RB50 or RB50 ΔarnT. Inoculated individuals (index mice) were placed in a cage with two to three naive mice (secondary mice). After mice were housed together for 21 days, mice were dissected, and bacterial numbers in the nasal cavity were determined (Fig. 6). Five out of six secondary mice exposed to RB50-inoculated index mice became colonized, whereas none of the secondary mice exposed to RB50 ΔarnT-infected index mice became colonized (Fig. 6).
FIG 6.

ArnT is required for transmission of B. bronchiseptica between mice. Two index C3H/HeJ mice per group were inoculated with 500 CFU of either RB50 or RB50 ΔarnT in 5 μl of PBS. Index mice were housed with two to three secondary mice for 3 weeks following inoculation. The quantity of CFU of RB50 or of RB50 ΔarnT recovered from the nasal cavity of individual secondary mice is represented by dots. The box spans the interquartile range, and the median value is represented by a bar. The whiskers span the 1st and 4th quartiles. The proportion of infected mice in each group is listed above the box-and-whisker plot. The limit of detection is marked by a dashed line.
RB50 ΔarnT is not defective at being shed by the host.
Although the inability of RB50 ΔarnT to be transmitted corresponded with an increased ID50, this did not exclude the possibility that RB50 ΔarnT might also be shed at a reduced rate. We have demonstrated that the shedding intensity of index mice correlates with the probability of transmission to other hosts (32). To determine whether RB50 ΔarnT is shed at a lower rate than wild-type RB50, C57BL/6 mice were inoculated with 500 CFU of either RB50 or RB50 ΔarnT. Shedding was monitored by swabbing the external nares at multiple time points over the course of 14 days. Neutrophil recruitment to the nasal cavity, previously shown to correlate with shedding intensity (31), was also determined at 7 and 14 days after inoculation. Shedding intensity of RB50 ΔarnT was not different from that of RB50 at any time point over a 14-day course (Fig. 7B). Furthermore, neutrophil numbers in the nasal cavity of mice inoculated with RB50 ΔarnT were similar to those obtained from RB50-inoculated mice at day 7 and day 14 postinoculation (Fig. 7A). These results demonstrate that ArnT is not required to induce neutrophil recruitment or enhance bacterial shedding from the murine nasal cavity.
FIG 7.

ArnT does not affect neutrophil recruitment or shedding from the host. C57BL/6 mice were infected with 500 CFU in 5 μl of PBS of either RB50 or RB50 ΔarnT. (A) Mice were sacrificed at either 7 or 14 days after inoculation, and the mean ± standard error neutrophil counts from nasal cavities of mice infected with RB50 (white bars) or RB50 ΔarnT (black bars) were obtained by flow cytometric detection of CD45+ CD11b+ Ly6G+ cells. (B) Shedding was detected by culture of bacteria from a 10-s swab of the external nares. Symbols represent the mean log10 CFU ± standard error shed from four mice/group infected with RB50 or RB50 ΔarnT. The limit of detection is marked by a dashed line.
DISCUSSION
Phenotypic assessment of an arnT-deficient derivative of B. bronchiseptica strain RB50 showed that this gene is required for the modification of lipid A with GlcN (9). Deletion of arnT was associated with decreased resistance to killing mediated by cationic antimicrobial peptides (CAMPs). ArnT did not affect the growth or persistence within the host but was required for transmission. The failure of mice to transmit RB50 ΔarnT corresponded with a 5-fold increase in the mean infectious dose. Together, these results suggest that ArnT-mediated addition of glucosamine to lipid A confers resistance to certain CAMPs and increases the frequency with which pathogens that seed the nasal mucosa are able to successfully colonize and initiate infections.
The protein encoded by arnT shares homology with Salmonella ArnT, a periplasmic enzyme that catalyzes the transfer of aminoarabinose from an undecaprenyl donor to the phosphate groups of lipid A and results in resistance to CAMPs (14). This protein is 100% identical at the amino acid level to the B. pertussis ArnT homologue, encoded by the gene designated BP0398. While the B. pertussis ArnT homologue mediates substitution of GlcN for lipid A(11), this activity was not associated with decreased resistance to CAMP-mediated attack but, rather, with increased TLR4 agonist potency (15). Deletion of the B. bronchiseptica ortholog did not alter the TLR4 stimulatory activity of the bacteria, suggesting that ArnT activity is not required for maximal stimulation of TLR4 by B. bronchiseptica. The differences between the observed effects of ArnT in B. bronchiseptica and B. pertussis may relate to their relative TLR4 stimulatory activities, which differ by an order of magnitude. The other structural differences between the LPSs (actually lipooligosaccharide [LOS] for B. pertussis) of these two organisms and the basis for these differences remain to be elucidated.
B. bronchiseptica is strongly resistant to the bactericidal activity of porcine β-defensin 1 (26). In the absence of arnT, B. bronchiseptica is more susceptible to the bactericidal effects of polymyxin B and pBD1. These results are analogous to findings reported for deletion mutants of arnT in S. Typhimurium. As with 4-amino l-arabinose, replacement of the phosphate group with GlcN could decrease the anionic character of the outer membrane. The charge-neutralizing effect of GlcN may therefore represent an adaptation that destabilizes CAMP binding within the membrane and thus mitigates its activity. It is unclear why RB50 ΔarnT is more susceptible to killing by pBD1 but is unaffected in its sensitivity to mBD3. Although CAMPs are hypothesized to have a common mechanism of action based on conserved structural motifs, the multitude of genes encoding CAMPs in eukaryotic genomes and the divergence of sequences from one host species to another suggests that individual CAMPs may have some specificity regarding the microbes they target and/or their mechanisms of action (29). Alternatively, it is possible that mBD3 is not highly active on its own but acts in cooperation with other antimicrobial mechanisms in vivo.
Since ArnT activity is increased in the Bvg+ phase, which is thought to contribute to growth during infection, one would expect this gene to contribute to fitness within the host. When RB50 and RB50 ΔarnT are compared in mouse infections, differences between the two strains were observed upon titration of the infectious dose toward the lower limits. When mice were inoculated with RB50 ΔarnT at a volume and dose sufficient to seed the entire respiratory tract with high bacterial numbers, no difference between the wild-type parental strain and the arnT mutant was detected. When the fitness levels of the wild type and the mutant introduced in very low numbers (50 CFU or less) were compared, the mean infectious dose required to stably infect 50% of mice (ID50) with RB50 ΔarnT was approximately 5-fold greater than the ID50 of the wild-type strain. These data suggest that the likelihood of B. bronchiseptica successfully colonizing the respiratory tract of a new host is greatly enhanced by ArnT. The lipid A modification mediated by ArnT is likely an adaptation to host antimicrobial defenses, which, while effective against small numbers of pathogens, may be overwhelmed by increased doses of bacteria. When pathogens seed mucosal surfaces and begin the process of invasion and colonization, CAMP molecules, in particular β-defensins, are among the first elements of host resistance encountered. Resistance to CAMP killing may play a role in the survival of bacteria during the initial colonization process. However, due to the number of genes encoding CAMPs, knockout systems may not be a practical way of demonstrating that ArnT activity enables B. bronchiseptica to colonize mice by conferring resistance to CAMP-mediated attack.
Our results indicate that ArnT mediates GlcN modification of lipid A and contributes significantly to the infectiousness and transmissibility of B. bronchiseptica. We also demonstrate that directly quantifying transmission and infectivity using low-dose infection models can be a more sensitive approach for probing interactions that are important for the initial colonization of new hosts, aspects not clearly observed when higher-dose inocula are delivered. An approach focusing on lower, more natural, doses and on transmission between animals revealed phenotypes dependent on ArnT that are likely to have important implications to the spread of infection and, therefore, the burden of this infectious disease at the population scale.
ACKNOWLEDGMENTS
We thank Ashutosh Pathak and Daryl Nowacki for performing preliminary experiments to characterize this strain. We also thank Alexia Karanikas, Sara Hester, Heather Feaga, and Laura Goodfield for invaluable editorial input.
This work was supported by National Institutes of Health grants GM083113 and T32AI074551.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1.Raetz CR, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76:295–329. 10.1146/annurev.biochem.76.010307.145803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 3.Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. 2006. Modulation of dendritic cell trafficking to and from the airways. J. Immunol. 176:3578–3584 http://www.jimmunol.org/content/176/6/3578.abstract?ijkey=3c1ef3e82db0173c9e2e7e9db48ad1d89be5f477&keytype2=tf_ipsecsha [DOI] [PubMed] [Google Scholar]
- 4.Miller SI, Ernst RK, Bader MW. 2005. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3:36–46. 10.1038/nrmicro1068 [DOI] [PubMed] [Google Scholar]
- 5.Mattoo S, Cherry JD. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 18:326–382. 10.1128/CMR.18.2.326-382.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arico B, Scarlato V, Monack DM, Falkow S, Rappuoli R. 1991. Structural and genetic analysis of the bvg locus in Bordetella species. Mol. Microbiol. 5:2481–2491. 10.1111/j.1365-2958.1991.tb02093.x [DOI] [PubMed] [Google Scholar]
- 7.Merkel TJ, Stibitz S, Keith JM, Leef M, Shahin R. 1998. Contribution of regulation by the bvg locus to respiratory infection of mice by Bordetella pertussis. Infect. Immun. 66:4367–4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter PA, Miller JF. 1994. BvgAS-mediated signal transduction: analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect. Immun. 62:3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacArthur I, Jones JW, Goodlett DR, Ernst RK, Preston A. 2011. Role of pagL and lpxO in Bordetella bronchiseptica lipid A biosynthesis. J. Bacteriol. 193:4726–4735. 10.1128/JB.01502-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marr N, Hajjar AM, Shah NR, Novikov A, Yam CS, Caroff M, Fernandez RC. 2010. Substitution of the Bordetella pertussis lipid A phosphate groups with glucosamine is required for robust NF-κB activation and release of proinflammatory cytokines in cells expressing human but not murine Toll-like receptor 4-MD-2-CD14. Infect. Immun. 78:2060–2069. 10.1128/IAI.01346-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marr N, Tirsoaga A, Blanot D, Fernandez R, Caroff M. 2008. Glucosamine found as a substituent of both phosphate groups in Bordetella lipid A backbones: role of a BvgAS-activated ArnT ortholog. J. Bacteriol. 190:4281–4290. 10.1128/JB.01875-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preston A, Maxim E, Toland E, Pishko EJ, Harvill ET, Caroff M, Maskell DJ. 2003. Bordetella bronchiseptica PagP is a Bvg-regulated lipid A palmitoyl transferase that is required for persistent colonization of the mouse respiratory tract. Mol. Microbiol. 48:725–736. 10.1046/j.1365-2958.2003.03484.x [DOI] [PubMed] [Google Scholar]
- 13.Pilione MR, Pishko EJ, Preston A, Maskell DJ, Harvill ET. 2004. pagP is required for resistance to antibody-mediated complement lysis during Bordetella bronchiseptica respiratory infection. Infect. Immun. 72:2837–2842. 10.1128/IAI.72.5.2837-2842.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. 2001. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-l-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 276:43122–43131. 10.1074/jbc.M106961200 [DOI] [PubMed] [Google Scholar]
- 15.Herrera CM, Hankins JV, Trent MS. 2010. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol. Microbiol. 76:1444–1460. 10.1111/j.1365-2958.2010.07150.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah NR, Albitar-Nehme S, Kim E, Marr N, Novikov A, Caroff M, Fernandez RC. 2013. Minor modifications to the phosphate groups and the C3′ acyl chain length of lipid A in two Bordetella pertussis strains, BP338 and 18–323, independently affect Toll-like receptor 4 protein activation. J. Biol. Chem. 288:11751–11760. 10.1074/jbc.M112.434365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geurtsen J, Dzieciatkowska M, Steeghs L, Hamstra HJ, Boleij J, Broen K, Akkerman G, El Hassan H, Li J, Richards JC, Tommassen J, van der Ley P. 2009. Identification of a novel lipopolysaccharide core biosynthesis gene cluster in Bordetella pertussis, and influence of core structure and lipid A glucosamine substitution on endotoxic activity. Infect. Immun. 77:2602–2611. 10.1128/IAI.00033-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns VC, Pishko EJ, Preston A, Maskell DJ, Harvill ET. 2003. Role of Bordetella O antigen in respiratory tract infection. Infect. Immun. 71:86–94. 10.1128/IAI.71.1.86-94.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caroff M, Tacken A, Szabo L. 1988. Detergent-accelerated hydrolysis of bacterial endotoxins and determination of the anomeric configuration of the glycosyl phosphate present in the “isolated lipid A” fragment of the Bordetella pertussis endotoxin. Carbohydr. Res. 175:273–282. 10.1016/0008-6215(88)84149-1 [DOI] [PubMed] [Google Scholar]
- 20.Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O'Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35:32–40. 10.1038/ng1227 [DOI] [PubMed] [Google Scholar]
- 21.Schweizer HP, Hoang TT. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22. 10.1016/0378-1119(95)00055-B [DOI] [PubMed] [Google Scholar]
- 22.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 23.Westphal O, Jann JK. 1965. Bacterial lipopolysaccharides: extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 5:83–91 [Google Scholar]
- 24.Fischer W, Koch HU, Haas R. 1983. Improved preparation of lipoteichoic acids. Eur. J. Biochem. 133:523–530. 10.1111/j.1432-1033.1983.tb07495.x [DOI] [PubMed] [Google Scholar]
- 25.Yi EC, Hackett M. 2000. Rapid isolation method for lipopolysaccharide and lipid A from gram-negative bacteria. Analyst 125:651–656. 10.1039/b000368i [DOI] [PubMed] [Google Scholar]
- 26.Elahi S, Buchanan RM, Attah-Poku S, Townsend HG, Babiuk LA, Gerdts V. 2006. The host defense peptide beta-defensin 1 confers protection against Bordetella pertussis in newborn piglets. Infect. Immun. 74:2338–2352. 10.1128/IAI.74.4.2338-2352.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doss M, White MR, Tecle T, Hartshorn KL. 2010. Human defensins and LL-37 in mucosal immunity. J. Leukoc. Biol. 87:79–92. 10.1189/jlb.0609382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song J, Bishop BL, Li G, Duncan MJ, Abraham SN. 2007. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe 1:287–298. 10.1016/j.chom.2007.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganz T. 2004. Defensins: antimicrobial peptides of vertebrates. C R Biol. 327:539–549. 10.1016/j.crvi.2003.12.007 [DOI] [PubMed] [Google Scholar]
- 30.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 31.Gopinath S, Hotson A, Johns J, Nolan G, Monack D. 2013. The systemic immune state of super-shedder mice is characterized by a unique neutrophil-dependent blunting of TH1 responses. PLoS Pathog. 9:e1003408. 10.1371/journal.ppat.1003408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rolin O, Smallridge W, Henry M, Goodfield L, Place D, Harvill E. Toll-like receptor 4 limits transmission of Bordetella bronchiseptica. PLoS One, in press [DOI] [PMC free article] [PubMed] [Google Scholar]