

Abstract
Avian leukosis virus (ALV) subgroups A, B, and J are very common in poultry flocks and have caused serious economic losses in recent years. A multiplex PCR (mPCR) method for the detection of these three subgroups was developed and optimized in this study. We first designed a common forward primer, PF, and three downstream primers, AR, BR, and JR, which can amplify 715 bp for subgroup A, 515 bp for subgroup B, and 422 bp for subgroup J simultaneously in one reaction. The mPCR method produced neither cross-reactions with other subgroups of ALVs nor nonspecific reactions with other common avian viruses. The detection limit of the mPCR was as low as 1 × 103 viral DNA copies of each of the three subgroups. In animal experiments, the mPCR detected ALVs 2 to 4 days earlier than did virus isolation from whole-blood samples and cloaca swabs. Furthermore, a total of 346 clinical samples (including 127 tissue samples, 86 cloaca swabs, 59 albumen samples, and 74 whole-blood samples) from poultry flocks with suspected ALV infection were examined by mPCR, routine PCR, and virus isolation. The positive sample/total sample ratios for ALV-A, ALV-B, and ALV-J were 48% (166/346) as detected by mPCR and 48% (166/346) as detected by routine PCR. However, the positive sample/total sample ratio detected by virus isolation was 40% (138/346). The results of the mPCR and routine PCR were confirmed by sequencing the specific fragments. These results indicate that the mPCR method is rapid, specific, sensitive, and convenient for use in epidemiological studies of ALV, clinical detection of ALV, and ALV eradication programs.
INTRODUCTION
Avian leukosis viruses (ALVs) are type C retroviruses associated with a variety of neoplasms, including lymphoid and myeloid leukosis infections (1). In commercial poultry flocks worldwide, ALVs are prevalent in several breeding flocks, causing serious economic losses from tumor mortality, carcass condemnation, and loss of pedigree birds (1, 2). ALVs have been divided into 10 different viral subgroups (designated A to J) based on their host range, viral envelope interference, and cross-neutralization patterns (3). The 10 different viral subgroups are also classified as being either exogenous or endogenous ALVs. Subgroups A to D and J are exogenous viruses, while members of the ALV subgroup E are endogenous viruses (4).
In commercial poultry, subgroups A, B, and J are the most common ALVs. The classical subgroup A avian leukosis virus (ALV-A) primarily induces lymphocytoma and several other types of cellular tumors, such as hemangioma. ALV-A has also been associated with subcutaneous tumors in young layer chickens (5). The subgroup B avian leukosis virus (ALV-B) mainly presents as lymphocytic leukosis and sarcomas (6). Subgroups C and D have rarely been reported. Subgroup E is the ubiquitous endogenous leukosis virus of low pathogenicity (7). Subgroup J avian leukosis virus (ALV-J) was first isolated from meat-type chickens in 1988, and it was designated an exogenous virus, primarily causing myeloid leukosis in meat-type chickens (8). No field cases of ALV-J infection or tumors in layer chickens were observed worldwide until 2004 (9–11). However, parent and commercial layer flocks in China have experienced outbreaks of this virus in recent years, causing serious economic losses (10). Thus, ALV-A, ALV-B, and ALV-J are not only the most common but also the most dangerous viruses to the poultry industry. In addition, ALV-A and ALV-J can infect the same chicken, and ALV-A and ALV-B have also been detected in the same commercial laying hens (12, 13). These types of coinfection provide a potential opportunity for recombination between different ALV subgroups. Thus, it is essential to develop a rapid and convenient method of detecting the three subgroups of avian leukosis viruses (ALV-A, ALV-B, and ALV-J).
The existing detection methods for ALVs include enzyme-linked immunosorbent assay (ELISA), real-time PCR, immunofluorescence assay (IFA), virus isolation, and routine PCR. Antigen capture ELISA (AC-ELISA) has been used widely and has played an important role in the eradication of ALVs. However, ELISA can detect only group-specific antigen p27 and cannot differentiate endogenous viruses (14). Real-time PCR and immunofluorescence assays have been developed for antigen detection and the differentiation of endogenous and exogenous ALVs. Nevertheless, both of these techniques require sophisticated instrumentation (such as quantitative fluorescence PCR machines and fluorescence microscopes) and cannot be used widely in the field (15, 16). Virus isolation in cell culture is often used as the gold standard. However, this method is time-consuming because a minimum of 7 days is required to obtain the results, and this method also cannot differentiate between virus subgroups (17). Routine PCR using primer pairs such as H5-H7 can detect only ALV-J, while the primer pair H5-AD1, which can detect all ALVs except ALV-J, cannot distinguish different subgroups (18). Multiplex PCR (mPCR) is a useful technique for the rapid differential diagnosis of avian viruses and the detection of multiple infections of avian viruses under field conditions (19). In the present study, a sensitive and specific mPCR method for the detection of ALV-A, ALV-B, and ALV-J has been developed. The mPCR DNA products consisted of three specific fragments of 715 bp (ALV-A), 515 bp (ALV-B), and 422 bp (ALV-J) that can be visualized by gel electrophoresis. This novel method allows for three very common subgroups of exogenous ALVs (ALV-A, ALV-B, and ALV-J) to be detected and differentiated in one reaction.
MATERIALS AND METHODS
Viruses.
ALV-A strain RAV-1, ALV-B strain RAV-2, ALV-C strain RAV-49, ALV-D strain RAV-50, ALV-E strain RAV-0, and ALV-J strain HLJ09SH01, chicken infectious anemia virus (CAV), avian reticuloendotheliosis virus (REV), infectious bursa disease virus (IBDV), and avian reovirus (ARV) were maintained in our laboratory. Avian infectious bronchitis virus (IBV), Marek's disease virus (MDV), avian infectious laryngotracheitis virus (ILTV), fowlpox virus (FPV), and Newcastle disease virus (NDV) were previously maintained in the State Key Laboratory of Veterinary Biotechnology at the Harbin Veterinary Research Institute.
Virus isolation.
All virus isolations were performed in DF-1 cells, which are known to be susceptible to exogenous ALVs only (20). The procedures for the isolation and identification of ALVs in cell culture were performed as previously described (21). Briefly, 200 μl of the filtered supernatant of a freshly abraded specimen was inoculated into DF-1 cells, which were cultured in 48-well cell culture plates with Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, CA) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. Two hours postinoculation, the cells were overlaid with DMEM supplemented with 2% FBS and incubated at 37°C in a 5% CO2 incubator with daily monitoring for 7 days. The infected DF-1 cells were harvested and tested for the ALV group-specific antigen (p27) by AC-ELISA with an avian leukosis virus antigen test kit (Idexx Laboratories, Inc., MA).
Nucleic acid extraction.
The proviral DNA of viruses, cloaca swabs, albumen, whole blood, cell cultures, and tissue samples were extracted using an established method (10). Briefly, the samples were lysed in tissue lysis buffer (4 M guanidine hydrochloride, 25 mM sodium citrate, and 1% Triton X-100) and extracted twice with phenol-chloroform-isoamyl alcohol (25:24:1). The DNA was precipitated with absolute isopropanol, washed with 70% ethanol, and dried at room temperature. Subsequently, the DNA was resuspended in nuclease-free water. The RNA extraction was performed according to the TRIzol LS manufacturer's protocol (22). The RNA was reverse transcribed to cDNA as described by the Thermo Scientific Maxima H Minus reverse transcriptase kit (catalog no. EP0759). The concentrations of DNA were determined by spectrophotometry using the Spectronic BioMate 5 (Thermo Spectronic, Rochester, NY) and stored at −20°C.
Primer design.
The complete sequences of the ALV-A, ALV-B, and ALV-J strains were retrieved from the GenBank database (GenBank accession no. DQ365814, DQ412726, DQ412727, EU070900, EU070901, EU352877, HM452341, L10923, M19113, M14902, JX848322, JQ935966, JF932004, and Z46390) and aligned using DNAStar (DNAStar, Inc., Madison, WI). On the basis of the results of the sequence comparison and the sequence characteristics of the three subgroups, we first designed a common forward primer, PF, based on the 3′ region of the pol gene, which was conserved across ALV-A, ALV-B, and ALV-J. The downstream primers (AR, BR, and JR) were chosen from the env gene, which allows for discrimination of the three subgroups. The primer PF amplifies a 715-bp fragment with primer AR to detect ALV-A, amplifies a 515-bp fragment with primer BR to detect ALV-B, and amplifies a 422-bp fragment with primer JR to detect ALV-J. The optimal primers (Table 1) were synthesized by the Huada Gene Company (Beijing, China).
TABLE 1.
Sequences of oligonucleotide primers, targets, and expected PCR product sizes
| Primer name | Sequence (5′ to 3′) | Product size with PF (bp) | Amplification target |
|---|---|---|---|
| PF | CGGAGAAGACACCCTTGCT | ||
| AR | GCATTGCCACAGCGGTACTG | 715 | ALV-A |
| BR | GTAGACACCAGCCGGACTATC | 515 | ALV-B |
| JR | CGAACCAAAGGTAACACACG | 422 | ALV-J |
Multiplex PCR method.
A proviral DNA mixture of equal concentrations of ALV-A, ALV-B, and ALV-J was utilized as a template to amplify the target fragment and optimize the protocol for annealing temperatures, primer concentrations, extension time, and cycle quantity. Finally, the reaction was performed in a 25-μl volume containing 2 μl template proviral DNA, 12.5 μl Premix Taq (TaKaRa, China), 2 μl forward primer PF, 0.8 μl downstream primer AR, 1 μl downstream primer BR, 1 μl downstream primer JR, and the appropriate volume of double-distilled water (ddH2O). All primers were diluted to 10 pM/μl.
The mPCR procedure consisted of an initial denaturation at 94°C for 5 min and then 30 cycles that each consisted of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, and extension at 72°C for 1 min. The sample was then heated at 72°C for 5 min for a final extension. A negative control was run with each test; the negative control did not contain template cDNA and consisted of Premix Taq (TaKaRa, China), all four primers, and deionized water. The mPCR products were evaluated by 1.0% agarose gel electrophoresis.
Routine PCR.
The primer set H5, 5′-GGATGAGGTGACTAAGAAAG-3′, and H7, 5′-CGAACCAAAGGTAACACACG-3′, was used to amplify ALV-J (18). A 545-bp band was amplified by this primer pair. The primer set H5 and AD1, 5′-GGGAGGTGGCTGACTGTGT-3′, was used for the detection of subgroup A to E ALVs, which generate a 295- to 326-bp PCR product (18). The reaction was performed in a 25-μl mixture containing 1 μl of proviral DNA, 2.5 μl of 10 × Ex Taq buffer, 2 μl of deoxynucleoside triphosphate (dNTP) (2.5 mM), 1 μl of H5 primer (10 pM/μl), 1 μl of H7 or AD1 primer (10 pM/μl), 1 U of Ex Taq HS (TaKaRa, China), and the appropriate volume of ddH2O. The PCR procedure was as follows: 95°C for 5 min, 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 10 min. The PCR products were evaluated by 1.0% agarose gel electrophoresis.
Standard plasmid preparation.
To obtain well-characterized positive controls and determine the sensitivity of the mPCR method, a 715-bp fragment of ALV-A, a 515-bp fragment of ALV-B, and a 422-bp fragment of ALV-J were amplified with the mPCR method and cloned into the pMD-18T vector (TaKaRa, China) to obtain the recombinant plasmids pMD-A (ALV-A), pMD-B (ALV-B), and pMD-J (ALV-J).
Specificity of the mPCR method.
Proviral DNA was extracted from all viruses, including ALV-A, ALV-B, ALV-C, ALV-D, ALV-E, ALV-J, and REV, and then DNA was extracted from MDV, CAV, ILTV, and FPV. The proviral DNA and the DNA were used as templates to determine the specificity of the mPCR method. The RNA extracted from IBDV, ARV, NDV, and IBV was reverse transcribed into cDNA (23) and was also used to measure the specificity of the mPCR method.
Sensitivity of the mPCR method.
The concentration of the constructed plasmids was determined by UV spectrophotometry, and the plasmid copy number was calculated using the following formula: copy number (copies/μl) = NA (copies/mol) × concentration (g/μl)/MW (g/mol), where NA is Avogadro's number and MW is the base number times 340 (24). Each of the three plasmids (pMD-A, pMD-B, and pMD-J) and the three mixed plasmids (pMD-A/pMD-B/pMD-J) was diluted from 1 × 1010 to 1 × 101 copies/μl and was used to detect the sensitivity of the mPCR method. Furthermore, DNA extracted from the liver of a specific-pathogen-free (SPF) chicken was added to the mPCR reaction mixtures to determine the sensitivity of mPCR under “real conditions.” In order to determine if a low concentration of one virus subtype in a mixed infection affected the sensitivity of the mPCR, we reduced the copies of the plasmid pMD-A (ALV-A) from 3 × 1010 to 3 × 105 copies/μl and mixed it with the other two plasmids, pMD-B (ALV-B) (3 × 1010 copies/μl) and pMD-J (ALV-J) (3 × 1010 copies/μl), in the same volume. The mixed plasmids were serially diluted from 1 × 1010 copies/μl to 1 × 101 copies/μl and tested using the mPCR.
Experimental infections.
To examine the practicality of the mPCR method, 60 1-day-old SPF chickens were randomly divided into four groups. Each group had 15 chickens. Three groups were challenged intra-abdominally with 0.2 ml per chicken of the 105/ml 50% tissue culture infective dose (TCID50) of the RAV-1 (ALV-A), RAV-2 (ALV-B), or HLJ09SH01 (ALV-J) strain of ALV; these infected groups were, respectively, designated group A (inoculated with ALV-A), group B (inoculated with ALV-B), and group J (inoculated with ALV-J). One group of chickens was inoculated with DMEM and used as the control group. The four groups were kept in separate rooms. Cloaca swabs and whole-blood samples were collected from all chickens from each of the four groups every other day for a total of 15 sampling times (from 2 days postinoculation [dpi] to 30 dpi). The cloaca swabs and whole-blood samples were analyzed by mPCR and the results were compared with the results of the virus isolation method. All animal studies were approved by the Institutional Review Board of the Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. All animal procedures were performed according to international standards for animal welfare.
Clinical specimens.
A total of 346 clinical samples (including 127 tissue samples, 86 cloaca swabs, 59 albumen samples, and 34 whole-blood samples) were collected from several different poultry flocks from 2010 to 2013 in China and stored at −80°C in an ultra-low-temperature freezer. All chickens from which the samples were taken were suspected of having avian leukosis disease based on clinical symptoms, including hemorrhages in the skin of the phalanges and feather follicles. Some birds had gray-white nodules in the liver, spleen, or kidneys, and the liver and spleen were enlarged to several times their normal sizes. Tissue samples were homogenized in phosphate-buffered saline (PBS) containing 1,000 IU/ml penicillin and streptomycin and were subsequently centrifuged at 6,000 × g for 5 min at 4°C. Before virus isolation was carried out, the cloaca swabs and whole-blood samples were centrifuged, and the albumen was double diluted. An aliquot of the supernatant of all samples was used to extract proviral DNA, which was utilized as a template for mPCR and routine PCR detection. The remaining supernatant was passed through 0.22-μm filters to carry out virus isolation (16).
DNA sequencing.
To further confirm the results, all specific fragments amplified from the clinical samples by mPCR and routine PCR were excised from a 1.0% agarose gel, purified using an AxyPrep DNA gel extraction kit (Axygen Scientific, Inc., CA), and sequenced by the Beijing Genomics Institute (Beijing, China).
RESULTS
Establishment of the mPCR method.
Throughout this research, manipulations were made to optimize the parameters of the assay, such as the annealing temperatures, primer concentrations, extension times, and cycle quantity. We determined the most optimal reaction conditions and developed a multiplex PCR. The target fragments were amplified with the combination of DNA from the three different subgroups of ALVs (ALV-A, ALV-B, and ALV-J), and each product was visualized by electrophoresis on a 1.0% agarose gel followed by ethidium bromide staining and UV transillumination (Fig. 1).
FIG 1.
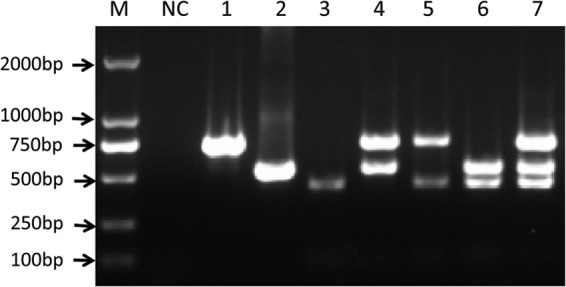
Agarose gel electrophoresis (1.0%) of specific fragments amplified by mPCR from purified proviral DNAs of known ALV-A, ALV-B, and ALV-J. Lane M, DL2000 marker (TaKaRa, China); lane NC, negative control; lane 1, ALV-A (715-bp fragment); lane 2, ALV-B (515-bp fragment); lane 3, ALV-J (422-bp fragment); lane 4, ALV-A/B; lane 5, ALV-A/J; lane 6, ALV-B/J; lane 7, ALV-A/B/J.
Specificity of the mPCR method.
We determined the specificity of the mPCR by examining the ability of the method to detect and differentiate ALV-A, ALV-B, and ALV-J. First, an assessment of the mPCR specificity was carried out by testing the other subgroup ALVs (ALV-C, ALV-D, and ALV-E). Furthermore, the mPCR was tested using other common avian viruses (including MDV, REV, CAV, ILTV, IBV, FPV, IBDV, ARV, and NDV). There were neither cross-reactions with other subgroups of ALVs nor nonspecific reactions with other common avian viruses. Only the positive control (the mixture of the proviral DNA of ALV-A, ALV-B, and ALV-J at the same concentrations) produced three specific fragments consisting of 715 bp for ALV-A, 515 bp for ALV-B, and 422 bp for ALV-J (Fig. 2).
FIG 2.

Specificity of the mPCR method. Results of 1.0% agarose gel electrophoresis of products amplified by mPCR from purified proviral DNAs of known avian viruses. Lane 1, DL2000 marker (TaKaRa, China); lane 2 (+), positive control (mixture of the same concentrations of proviral cDNA from ALV-A, ALV-B, and ALV-J); lane 3, Marek's disease virus (MDV); lane 4, avian reticuloendotheliosis virus (REV); lane 5, chicken infectious anemia virus (CAV); lane 6, avian infectious laryngotracheitis virus (ILTV); lane 7, avian infectious bronchitis virus (IBV); lane 8, fowlpox virus (FPV); lane 9, infectious bursa disease virus (IBDV); lane 10, avian reovirus (ARV); lane 11, Newcastle disease virus (NDV); lane 12, DF-1 cells; lane 13, ALV-C; lane 14, ALV-D; lane 15, ALV-E.
Sensitivity of the mPCR method.
Three constructed plasmids (pMD-A, pMD-B, and pMD-J) and the mixed plasmids (pMD-A/pMD-B/pMD-J) were serially diluted from 1 × 1010 copies/μl to 1 × 101 copies/μl. All single plasmids were detected to 1 × 102 copies/μl, and the mixed plasmids were detected to 1 × 103 copies/μl using the mPCR method (Fig. 3). The sensitivity of the mPCR was also determined with the mixture of plasmids at different concentrations (plasmid pMD-A at 3 × 105 copies/μl and plasmids pMD-B and pMD-J at 3 × 1010 copies/μl). The detection limit of the mPCR was as low as 1 × 103 viral DNA copies of each of the three subgroups in mixed infections (data not shown). The sensitivity of the mPCR was determined under “real conditions” with target DNA, and the host DNA was also extracted from the liver of the SPF chicken. The results show that the detection limit of the mPCR was 1 × 103 viral DNA copies of each of the three subgroups (data not shown). These data indicate that the low concentration of one virus subtype in mixed infections and the host DNA did not affect the sensitivity of the mPCR method.
FIG 3.

Sensitivity of the mPCR method. Three constructed plasmids (pMD-A, pMD-B, and pMD-J) and the three mixed plasmids (pMD-A/pMD-B/pMD-J) were diluted from 1010 to 101 DNA copies/μl to evaluate the sensitivity of the mPCR method. Shown are the sensitivity of pMD-A for ALV-A (A), the sensitivity of pMD-B for ALV-B (B), the sensitivity of pMD-J for ALV-J (C), and the sensitivity of pMD-A/pMD-B/pMD-J for ALV-A/B/J (D). M, DL2000 marker (TaKaRa, China).
Evaluation of the mPCR method using infectious experimental samples.
Cloaca swabs and whole-blood samples collected from the SPF chickens that were artificially inoculated with the three subgroup ALVs (ALV-A, ALV-B, and ALV-J) were analyzed by the mPCR method and virus isolation. The mPCR detected ALVs as early as 8 dpi for ALV-A and ALV-B and 6 dpi for ALV-J from the whole-blood samples. However, virus isolation detected ALVs 2 to 4 days later than the mPCR method (Fig. 4). Furthermore, the positive ratio of viremia in group A reached 93.3% on the 16th day postinoculation. It lasted 5 days at this high level, and then the positive ratio of viremia declined significantly and remained at a low level (<30%). The positive ratio of viremia in group B achieved 60% on the 14th day postinoculation and never increased in subsequent samplings. However, the positive ratio of viremia in group J reached 100% on the 16th day postinoculation and remained at a high level (>80%) (Fig. 4). The positive ratio detected by the mPCR was almost the same as that detected by virus isolation on every day tested.
FIG 4.

Ratios of samples positive for viremia from the three infected groups as detected by the mPCR method and virus isolation. Shown are the results of group A inoculated with ALV-A (A), group B inoculated with ALV-B (B), and group J inoculated with ALV-J (C).
At the same time, the mPCR method detected ALVs 2 days earlier than virus isolation from cloaca swabs of all three infected groups. Another phenomenon of concern was that the ratio of ALV-positive cloaca swabs from group J remained at a high level (>66.7%) from the 18th day postinoculation. Few ALV-positive chickens were detected from the cloaca swabs of both group A and group B at the end of the testing period (Table 2). In the control group, no viruses were detected from the cloaca swabs or the whole-blood samples.
TABLE 2.
Results for cloaca swabs from the three infected groups as detected by mPCR and virus isolation
| No. of dpi | No. of ALV-positive chickensa by virus subgroup and detection method: |
|||||
|---|---|---|---|---|---|---|
| ALV-A |
ALV-B |
ALV-J |
||||
| Virus isolation | mPCR | Virus isolation | mPCR | Virus isolation | mPCR | |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | 0 | 0 | 0 | 0 | 0 | 2 |
| 12 | 0 | 0 | 0 | 0 | 1 | 2 |
| 14 | 0 | 0 | 0 | 0 | 1 | 3 |
| 16 | 0 | 0 | 0 | 0 | 4 | 4 |
| 18 | 0 | 0 | 0 | 0 | 8 | 8 |
| 20 | 0 | 0 | 0 | 0 | 9 | 10 |
| 22 | 0 | 1 | 0 | 1 | 11 | 11 |
| 24 | 1 | 1 | 1 | 1 | 11 | 11 |
| 26 | 1 | 2 | 2 | 2 | 10 | 11 |
| 28 | 1 | 2 | 1 | 1 | 9 | 10 |
| 30 | 2 | 2 | 3 | 4 | 10 | 10 |
The total number of chickens examined per subgroup was 15.
Evaluation of the mPCR method using clinical samples.
A total of 346 clinical samples (including 127 tissue samples, 86 cloaca swabs, 59 albumen samples, and 34 whole-blood samples) collected from several different poultry flocks from 2010 to 2013 were examined by the mPCR, routine PCR, and virus isolation methods. The mPCR method detected 166 positive samples (17 of ALV-A, 16 of ALV-B, and 133 of ALV-J). The routine PCR with the primer pair H5-H7 detected 133 samples positive for ALV-J. The routine PCR with the primer pair H5-AD1 detected 278 positive samples in clinical samples. Among the 278 positive samples, 17 samples positive for ALV-A and 16 samples positive for ALV-B were confirmed by sequencing. There were only 138 positive samples detected by virus isolation. The positive sample/total sample ratios for ALV-A, ALV-B, and ALV-J were 48% (166/346) as detected by mPCR and 48% (166/346) as detected by routine PCR. However, the positive sample/total sample ratio detected by virus isolation was 40% (138/346) (Table 3). Furthermore, among the 166 positive samples detected by mPCR, there was 1 sample positive for coinfection with ALV-A and ALV-B and 2 samples positive for coinfections with ALV-A and ALV-J.
TABLE 3.
Results for clinical samples examined by multiplex PCR, virus isolation, and routine PCR
| Specimen source | Multiplex PCR resultsa by subgroup: |
Virus isolation resultsa | Routine PCR resultsa by primer pair: |
|||
|---|---|---|---|---|---|---|
| A | B | J | H5-H7b | H5-AD1c | ||
| Tissue | 7a/127b | 10/127 | 67/127 | 79/127 | 67/127 | 17/127 |
| Cloaca swabs | 3/86 | 2/86 | 27/86 | 21/86 | 27/86 | 5/86 |
| Albumen | 2/59 | 0/59 | 16/59 | 10/59 | 16/59 | 2/59 |
| Whole blood | 5/74 | 4/74 | 23/74 | 28/74 | 23/74 | 9/74 |
| Total | 17/346 | 16/346 | 133/346 | 138/346 | 133/346 | 33/346 |
| Positive-sample rate (%) | 166/346 (48) | 138/346 (40) | 166/246 (48) | |||
Number of positive samples/number of samples examined.
The number of positive samples here detected by the primer pair H5-H7 was for ALV-J.
The number of positive samples here detected by the primer pair H5-AD1 was for ALV-A and ALV-B only (18).
Sequencing results.
To further confirm the result, the products of all clinical positive samples amplified by the mPCR and routine PCR (primer pairs H5-H7 and H5-AD1) (18) were sequenced. The sequencing results showed that 17 products belonged to ALV-A, 16 products belonged to ALV-B, and 133 products belonged to ALV-J (data not shown). The sequencing results agreed with the results of the mPCR method. The coincidence ratio between the mPCR and routine PCR was 100%.
DISCUSSION
Avian leukosis virus (ALV) is the most common naturally occurring avian retrovirus associated with neoplastic diseases and other production problems in chickens (25). ALV-A, ALV-B, and ALV-J are the three most prevalent and dangerous subgroups of ALVs. No effective vaccine or medication against ALVs is currently available. The control of ALV infection is dependent upon the early detection and removal of virus-shedding birds to reduce the spread of congenital and contact infections to other birds (26). Therefore, achieving rapid detection of infection is imperative in effective control of the spread of ALVs (27). Antigen detection is used routinely for the detection of ALVs (28). In this study, a multiplex PCR with high specificity and sensitivity was developed and optimized to detect ALV-A, ALV-B, and ALV-J simultaneously in an ordinary 25-μl reaction mixture system.
Primer design plays a very important role in developing a successful multiplex PCR method. ALVs belong to the genus Alpharetrovirus of the Retroviridae family and contain the overall structure of a typical slow-transforming replication-competent ALV: 5′-LTR-leader-gag/pol-env-LTR-3′ (29, 30). ALVs are divided into exogenous viruses (ALV-A, -B, -C, -D, and -J) and endogenous viruses (ALV-E) according to sequence differences in their long terminal repeats (LTRs) (23). The gag genes encode Gag (group-specific antigen), the pol genes encode reverse transcriptase and integrase, and the env gene encodes the envelope glycoproteins (2, 31). The pol genes show >96% sequence identity among the exogenous ALVs (32). The classification of ALV subgroups is based mainly on the gene sequence of the ALV env gene (gp85) (33). In this study, after aligning a number of ALV-A, ALV-B, and ALV-J strain sequences published in GenBank, we used only a highly conserved region of all three subgroups to design the common forward primer PF. Three downstream primers (AR, BR, and JR) were chosen from the most discrepant region that was conservative for each subgroup.
The specificity of the mPCR method was evaluated with different subgroup ALVs (ALV-A to ALV-E and ALV-J) and other related avian viruses. The mPCR method produced neither cross-reactions with other subgroups of ALVs nor nonspecific reactions with other common avian viruses (MDV, REV, CAV, ILTV, IBV, FPV, IBDV, ARV, and NDV). Moreover, during testing of the 346 clinical samples, the number of samples positive for ALV-J as detected by mPCR was equal to the number detected by the primer pair H5-H7, and the number of samples positive for ALV-A and ALV-B as detected by mPCR was equal to the number of ALV-A- and ALV-B-positive samples detected by the primer pair H5-AD1 (18). In total, 166 samples positive for ALV-A, ALV-B, and ALV-J were detected by both methods, and the coincidence ratio was 100%. Furthermore, all sequences of the specific fragments of the positive samples of the three subgroups had >90% homology with the ALV-A, ALV-B, and ALV-J reference strains. Thus, the mPCR method was shown to have a high specificity for detecting all ALVs subgroups, common avian viruses, and different types of clinical samples.
Three recombinant plasmids (pMD-A, pMD-B, and pMD-J) were used to determine the sensitivity of the mPCR method. The detection limit of this method was as low as 1 × 102 viral DNA copies for each of the three plasmids. The mPCR method was compared with the virus isolation method because virus isolation is considered to be the gold standard for viral detection (10). In animal experiments, the mPCR detected ALVs 2 to 4 days earlier than virus isolation from whole-blood samples and 2 days earlier than virus isolation from cloaca swabs. The results of testing using whole-blood samples and cloaca swabs from the infected groups demonstrated that the sensitivity of mPCR was slightly higher than that of virus isolation. Additionally, the positive-sample ratios of 346 clinical samples were 48% (166/346) by the mPCR and 40% (138/346) by virus isolation. All data confirmed that the mPCR method has a higher sensitivity. One limitation of virus isolation might be that it can detect only samples containing a certain amount of live virus, which may be the reason for its low sensitivity (28). Another reason for the lower positive ratio for samples examined by virus isolation for clinical detection was that long-distance transportation and long-term preservation may affect the viability of the virus in clinical samples.
In animal experiments, the viremia in group A was temporary and the viremia in group B was always at a low level (<60%). The chickens of groups A and B showed little evidence of virus shedding in the 30 days postinoculation. These results are perhaps an important reason why infection with ALV-A or ALV-B has always been at a low level and why ALV-A and ASV-B do not appear to cause large-scale outbreaks in poultry flocks. In contrast, the viremia in group J stayed at a high level (>80%) from the 16th day postinoculation, and the ratio of virus shedding for group J remained at >60% from the 18th day postinoculation. Persistent viremia and a high proportion of virus shedding from chickens infected with ALV-J might be related to the outbreak of ALV-J in recent years in China (9, 11, 34). Persistent viremia is closely related to the generation of tumors (27), and virus shedding has an important link with the horizontal transmission of virus, which played an important role in the epidemic and outbreaks of ALVs (35).
The coinfection status of the different subgroups of avian leukosis viruses was previously found in poultry flocks. Gingerich et al. (36) and Lupiani et al. (37) found the recombinant ALVs ALV-B and ALV-J in commercial white leghorn egg layer flocks. Both ALV-A and ALV-J were isolated from sarcomas of 817 broiler hybrids (38). Liu et al. (39) detected ALV-B in an egg-type chicken that was infected with ALV-J. One positive sample with coinfection with ALV-A and ALV-B and 2 positive samples with coinfections with ALV-A and ALV-J were found in clinical samples in the present study. Coinfection, which may have a more powerful pathogenicity and which brings about more serious economic losses, has become widespread among flocks in China (40). Furthermore, coinfection with different subgroups provides the opportunity for the recombination of ALVs and makes it possible for the emergence of new subgroup viruses, such as ALV-J, with stronger pathogenicity and epidemicity (8, 31). Therefore, achieving rapid clinical detection of infection is imperative for the effective control of the spread of ALVs (41). The mPCR method developed in this study is an easy and effective method for the detection of coinfection of ALV-A, ALV-B, and/or ALV-J.
In conclusion, the use of the mPCR method was found to be rapid, specific, sensitive, and cost-effective for the detection of ALV-A, ALV-B, and ALV-J. Testing of experimental and clinical samples (including cloaca swabs and albumen, whole-blood, and tissue samples) demonstrated that the mPCR method is practical in laboratory and clinical diagnoses and will be useful in epidemiological studies and eradication programs.
ACKNOWLEDGMENTS
The study was supported by the Special Fund for Agro-scientific Research in the Public Interest (grant no. 201203056), the Natural Science Foundation of China (grant no. 31072146), the Earmarked Fund for the Modern Agro-industry Technology Research System (grant no. nycytx-42-G3-01), and the Harbin Programs for Application Technology Research and Development (grant no. 2013FB6CJ092).
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.Pham TD, Spencer JL, Johnson ES. 1999. Detection of avian leukosis virus in albumen of chicken eggs using reverse transcription polymerase chain reaction. J. Virol. Methods 78:1–11. 10.1016/S0166-0934(98)00157-8 [DOI] [PubMed] [Google Scholar]
- 2.Payne L, Nair V. 2012. The long view: 40 years of avian leukosis research. Avian. Pathol. 41:11–19. 10.1080/03079457.2011.646237 [DOI] [PubMed] [Google Scholar]
- 3.Payne LN. 1991. Developments in avian leukosis research. Leukemia 6:150S–152S [PubMed] [Google Scholar]
- 4.McNally MM, Wahlin KJ, Canto-Soler MV. 2010. Endogenous expression of ASLV viral proteins in specific pathogen free chicken embryos: relevance for the developmental biology research field. BMC Dev. Biol. 10:106. 10.1186/1471-213X-10-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono M, Tsukamoto K, Tanimura N, Haritani M, Kimura KM, Suzuki G, Okuda Y, Sato S. 2004. An epizootic of subcutaneous tumors associated with subgroup A avian leukosis/sarcoma virus in young layer chickens. Avian. Dis. 48:940–946. 10.1637/7162-020204R [DOI] [PubMed] [Google Scholar]
- 6.Venugopal K. 1999. Avian leukosis virus subgroup J: a rapidly evolving group of oncogenic retroviruses. Res. Vet. Sci. 67:113–119. 10.1053/rvsc.1998.0283 [DOI] [PubMed] [Google Scholar]
- 7.Adkins HB, Blacklow SC, Young JA. 2001. Two functionally distinct forms of a retroviral receptor explain the nonreciprocal receptor interference among subgroups B, D, and E avian leukosis viruses. J. Virol. 75:3520–3526. 10.1128/JVI.75.8.3520-3526.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Payne L. 1998. HPRS-103: a retro virus strikes back. The emergence of subgroup J avian leukosis virus. Avian. Pathol. 27:36–45 [Google Scholar]
- 9.Gao Y, Qin L, Pan W, Wang Y, Qi X, Gao H, Wang X. 2010. Avian leukosis virus subgroup J in layer chickens, China. Emerg. Infect. Dis. 10:1639. 10.3201/eid1610.100780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y, Yun B, Qin L, Pan W, Qu Y, Liu Z, Wang Y, Qi X, Gao H, Wang X. 2012. Molecular epidemiology of avian leukosis virus subgroup J in layer flocks in China. J. Clin. Microbiol. 50:953–960. 10.1128/JCM.06179-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu B, Dong W, Yu C, He Z, Lv Y, Sun Y, Feng X, Li N, Lee LF, Li M. 2004. Occurrence of avian leukosis virus subgroup J in commercial layer flocks in China. Avian. Pathol. 33:13–17. 10.1080/03079450310001636237a [DOI] [PubMed] [Google Scholar]
- 12.Fenton SP, Reddy MR, Bagust TJ. 2005. Single and concurrent avian leukosis virus infections with avian leukosis virus-J and avian leucosis virus-A in Australian meat-type chickens. Avian. Pathol. 34:48–54. 10.1080/03079450400025356 [DOI] [PubMed] [Google Scholar]
- 13.Spencer JL, Benkel B, Chan M, Nadin-Davis S. 2003. Evidence for virus closely related to avian myeloblastosis-associated virus type 1 in a commercial stock of chickens. Avian. Pathol. 32:383–390. 10.1080/0307945031000121130 [DOI] [PubMed] [Google Scholar]
- 14.Yun B, Li D, Zhu H, Liu W, Qin L, Liu Z, Wu G, Wang Y, Qi X, Gao H, Wang X, Gao Y. 2012. Development of an antigen-capture ELISA for the detection of avian leucosis virus p27 antigen. J. Virol. Methods 187:278–283. 10.1016/j.jviromet.2012.11.027 [DOI] [PubMed] [Google Scholar]
- 15.Qin A, Liu Y, Zhou Z, Huang W. 2001. Detection of subgroup J avian leukosis virus by immunofluorescence assay. Chin. J. Prev. Vet. Med. 23:214–216 (In Chinese.) 10.3969/j.issn.1008-0589.2001.03.016 [DOI] [Google Scholar]
- 16.Qin L, Gao Y, Ni W, Sun M, Wang Y, Yin C, Qi X, Gao H, Wang X. 2013. Development and application of real-time PCR for detection of subgroup J avian leukosis virus. J. Clin. Microbiol. 51:149–154. 10.1128/JCM.02030-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.García M, El-Attrache J, Riblet SM, Lunge VR, Fonseca AS, Villegas P, Ikuta N. 2003. Development and application of reverse transcriptase nested polymerase chain reaction test for the detection of exogenous avian leukosis virus. Avian. Dis. 47:41–53. 10.1637/0005-2086(2003)047[0041:DAAORT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 18.Smith LM, Brown SR, Howes K, McLeod S, Arshad SS, Barron GS, Venugopal K, McKay JC, Payne LN. 1998. Development and application of polymerase chain reaction (PCR) tests for the detection of subgroup J avian leukosis virus. Virus Res. 54:87–98. 10.1016/S0168-1702(98)00022-7 [DOI] [PubMed] [Google Scholar]
- 19.Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. 2000. Multiplex PCR: optimization and application in diagnostic virology. Clin. Microbiol. Rev. 13:559–570. 10.1128/CMR.13.4.559-570.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagust TJ, Fenton SP, Reddy MR. 2004. Detection of subgroup J avian leukosis virus infection in Australian meat-type chickens. Aust. Vet. J. 82:701–706. 10.1111/j.1751-0813.2004.tb12163.x [DOI] [PubMed] [Google Scholar]
- 21.Maas R, van Zoelen D, Oei H, Claassen I. 2006. Replacement of primary chicken embryonic fibroblasts (CEF) by the DF-1 cell line for detection of avian leucosis viruses. Biologicals 34:177–181. 10.1016/j.biologicals.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 22.Xie Z, Fadl AA, Girshick T, Khan MI. 1997. Amplification of avian reovirus RNA using the reverse transcriptase-polymerase chain reaction. Avian. Dis. 41:654–660. 10.2307/1592157 [DOI] [PubMed] [Google Scholar]
- 23.Reddy R, Henning D, Das G, Harless M, Wright D. 1987. The capped U6 small nuclear RNA is transcribed by RNA polymerase III. J. Biol. Chem. 262:75–81 [PubMed] [Google Scholar]
- 24.Zhou G, Cai W, Liu X, Niu C, Gao C, Si C, Zhang W, Qu L, Han L. 2011. A duplex real-time reverse transcription polymerase chain reaction for the detection and quantitation of avian leucosis virus subgroups A and B. J. Virol. Methods 173:275–279. 10.1016/j.jviromet.2011.02.017 [DOI] [PubMed] [Google Scholar]
- 25.Mohammadi KA, Masoudian M. 2008. Detection of avian leucosis virus (ALV) in albumen of Shiraz commercial and local layer flocks using ELISA and RT-PCR. Iran. J. Vet. Res. 9:24 http://ijvr.shirazu.ac.ir/?_action=articleInfo&article=572 [Google Scholar]
- 26.Davidson I, Borovskaya A, Perl S, Malkinson M. 1995. Use of the polymerase chain reaction for the diagnosis of natural infection of chickens and turkeys with Marek's disease virus and reticuloendotheliosis virus. Avian. Pathol. 24:69–94. 10.1080/03079459508419050 [DOI] [PubMed] [Google Scholar]
- 27.Fadly AM, Smith EJ. 1999. Isolation and some characteristics of a subgroup J-like avian leukosis virus associated with myeloid leukosis in meat-type chickens in the United States. Avian. Dis. 17:391–400 [PubMed] [Google Scholar]
- 28.Zhang X, Liao M, Jiao P, Luo K, Zhang H, Ren T, Zhang G, Xu C, Xin C, Cao W. 2010. Development of a loop-mediated isothermal amplification assay for rapid detection of subgroup J avian leukosis virus. J. Clin. Microbiol. 48:2116–2121. 10.1128/JCM.02530-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai J, Howes K, Payne LN, Skinner MA. 1995. Sequence of host-range determinants in the env gene of a full-length, infectious proviral clone of exogenous avian leukosis virus HPRS-103 confirms that it represents a new subgroup (designated J). J. Gen. Virol. 76:181–187. 10.1099/0022-1317-76-1-181 [DOI] [PubMed] [Google Scholar]
- 30.Sacco MA, Flannery DM, Howes K, Venugopal K. 2000. Avian endogenous retrovirus EAV-HP shares regions of identity with avian leucosis virus subgroup J and the avian retrotransposon ART-CH. J. Virol. 74:1296–1306. 10.1128/JVI.74.3.1296-1306.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Gao Y, Wang Y, Qin L, Qi X, Qu Y, Gao H, Wang X. 2012. A 205-nucleotide deletion in the 3′ untranslated region of avian leukosis virus subgroup J, currently emergent in China, contributes to its pathogenicity. J. Virol. 86:12849–12860. 10.1128/JVI.01113-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chesters PM, Howes K, Petherbridge L, Evans S, Payne LN, Venugopal K. 2002. The viral envelope is a major determinant for the induction of lymphoid and myeloid tumours by avian leukosis virus subgroups A and J, respectively. J. Gen. Virol. 83:2553–2561 http://vir.sgmjournals.org/content/83/10/2553.long. [DOI] [PubMed] [Google Scholar]
- 33.Bai J, Payne LN, Skinner MA. 1995. HPRS-103 (exogenous avian leucosis virus, subgroup J) has an env gene related to those of endogenous elements EAV-0 and E51 and an E element found previously only in sarcoma viruses. J. Virol. 69:779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Z, Liu J, Cui Z, Zhang L. 2010. Tumors associated with avian leukosis virus subgroup J in layer hens during 2007 to 2009 in China. J. Vet. Med. Sci. 72:1027. 10.1292/jvms.09-0564 [DOI] [PubMed] [Google Scholar]
- 35.de Boer GF, Maas HJ, van Vloten J, Groenendal JE. 1981. Horizontal transmission of lymphoid leukosis virus. Influence of age, maternal antibodies and degree of contact exposure. Avian. Pathol. 10:343–358. 10.1080/03079458108418483 [DOI] [PubMed] [Google Scholar]
- 36.Gingerich E, Porter RE, Lupiani B, Fadly AM. 2002. Diagnosis of myeloid leukosis induced by a recombinant avian leukosis virus in commercial white leghorn egg laying flocks. Avian. Dis. 46:745–748. 10.1637/0005-2086(2002)046[0745:DOMLIB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 37.Lupiani B, Pandiri AR, Mays J, Hunt HD, Fadly AM. 2006. Molecular and biological characterization of a naturally occurring recombinant subgroup B avian leukosis virus with a subgroup J-like long terminal repeat. Avian. Dis. 50:572–578. 10.1637/7656-053006R.1. [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Wang B, Zhang Z, Wang J, Sun S, Cui Z. 2011. The separation of subgroup A and J ALV in soft tissue sarcomas of “817” broiler hybrids. Chin. J. Anim. Vet. Sci. 3:012 (In Chinese.) [Google Scholar]
- 39.Liu GZ, Zhang HH, Liu Q, Qiu B, Wang F, Wang XW, Chen HB, Cheng ZQ. 2009. Isolation and identification of avian leukosis virus-B from layer chickens infected with avian leukosis virus-J. Bing Du Xue Bao 25:445–451 (In Chinese.) [PubMed] [Google Scholar]
- 40.Cui Z, Sun S, Zhang Z, Meng S. 2009. Simultaneous endemic infections with subgroup J avian leukosis virus and reticuloendotheliosis virus in commercial and local breeds of chickens. Avian Pathol. 38:443–448. 10.1080/03079450903349188 [DOI] [PubMed] [Google Scholar]
- 41.Caterina KM, Frasca SJ, Jr, Girshick T, Khan MI. 2004. Development of a multiplex PCR for detection of avian adenovirus, avian reovirus, infectious bursal disease virus, and chicken anemia virus. Mol. Cell Probes 18:293–298. 10.1016/j.mcp.2004.04.003 [DOI] [PubMed] [Google Scholar]