

Abstract
Differentiation programs require strict spatial and temporal control of gene transcription. Genes expressed during meiotic development in Saccharomyces cerevisiae display transient induction and repression. Early meiotic gene (EMG) repression during mitosis is achieved by recruiting both histone deacetylase and chromatin remodeling complexes to their promoters by the zinc cluster DNA binding protein Ume6p. Ume6p repression is relieved by ubiquitin-mediated destruction that is stimulated by Gcn5p-induced acetylation. In this report, we demonstrate that Gcn5p acetylation of separate lysines within the zinc cluster domain negatively impacts Ume6p DNA binding. Mimicking lysine acetylation using glutamine substitution mutations decreased Ume6p binding efficiency and resulted in partial derepression of Ume6p-regulated genes. Consistent with this result, molecular modeling predicted that these lysine side chains are adjacent to the DNA phosphate backbone, suggesting that acetylation inhibits Ume6p binding by electrostatic repulsion. Preventing acetylation did not impact final EMG induction levels during meiosis. However, a delay in EMG induction was observed, which became more severe in later expression classes, ultimately resulting in delayed and reduced execution of the meiotic nuclear divisions. These results indicate that Ume6p acetylation ensures the proper timing of the transient transcription program during meiotic development.
INTRODUCTION
Meiosis is the process in which a diploid cell produces haploid gametes capable of sexual reproduction. In Saccharomyces cerevisiae, many genes required for meiotic landmark events are transcriptionally repressed during mitotic cell division and then induced at particular stages in meiosis (1, 2). Posttranslational modifications of histones allow the coordinated execution of these gene transcription programs (3, 4). For example, the acetylation and deacetylation of histone lysine residues are associated with transcriptional activation and repression, respectively (5). Histone acetyltransferases (HATs) are directed to promoters through interaction with methylated histones or chromatin binding proteins (6). Conversely, transcriptional repression is mediated by histone deacetylases (HDACs) that are recruited to promoters by sequence-specific DNA binding proteins (7).
In budding yeast, the SAGA complex containing the Gcn5p acetyltransferase promotes the transcription of several genes, including those involved in the stress response (8) and meiosis (9). Gcn5p is not required for mitotic cell division, although mutants lacking this factor grow slower (10). However, gcn5Δ mutants are defective for meiosis, arresting before the first meiotic division (9). Similarly, the HDAC Rpd3p and its adaptor Sin3p display relatively minor growth defects but also fail to undergo any meiotic nuclear divisions or make spores (11, 12). The findings that Rpd3p and Sin3p are required for meiosis are even more remarkable, as both activities are required for full early meiotic gene (EMG) repression (12, 13). These results indicate that posttranslational histone modifications are critical for execution of a differentiation pathway, while their loss is more tolerated in mitotically dividing cells.
In addition to histones, there is a rapidly growing list of nonhistone proteins whose acetylation also stimulates transcription (7, 14–17). In these cases, gene expression is enhanced by upregulation of the modified factor's activity. We recently identified a new mechanism for acetylation-dependent gene induction through targeted destruction of the yeast transcriptional repressor Ume6p (18). Ume6p destruction is initiated by Gcn5p-dependent acetylation at a cluster of three lysines (15). Ume6p is a Zn2Cys6 zinc cluster protein that binds EMG promoters through the URS1 DNA element (19). Ume6p represses EMG transcription by recruiting the Sin3p-Ume1p-Rpd3p deacetylase and Isw2p chromatin remodeling complexes (20–23). Here, we show that Gcn5p acetylates Ume6p at a second set of lysines located adjacent to the Zn2Cys6 zinc cluster domain. Rather than regulating protein stability, acetylation at this additional site helps release Ume6p from early meiotic gene promoters, allowing timely initiation of the meiotic transient transcription program.
MATERIALS AND METHODS
Yeast strains, plasmids, and culture conditions.
Strains and plasmids used in this study are described in Tables 1 and 2, respectively. UME6 mutations were generated by site-directed mutagenesis and then introduced into the chromosome by using a two-step transplacement strategy (24). All genomic mutations were verified by sequencing of PCR products derived from genomic DNA. Gene deletions were performed by using homologous recombination as described previously (25). Meiotic cultures were generated and analyzed as described previously (26).
TABLE 1.
Yeast strains used in this study
| Strain | Genotypea | Reference |
|---|---|---|
| RSY335 | MATa/α cyh2R-z ho::LYS2 leu2::hisG lys2 trp1::hisG ura3 | 18 |
| RSY1079 | T7-UME6 | 18 |
| RSY1091 | gcn5::TRP1 | This study |
| RSY1152 | ume6::leu2::KanMX | 18 |
| RSY1404 | gcn5::TRP1 ume6::leu2::KanMX | This study |
| RSY1407 | T7-UME6K3R | This study |
| RSY1408 | T7-UME6K3Q gcn5::TRP1 | This study |
| RSY1420 | T7-UME6K3Q | This study |
| RSY1500 | ADA2-TAP-his3MX6::LEU2 | This study |
| RSY1563 | ADA2-TAP-his3MX6::LEU2 gcn5::TRP1 | This study |
| RSY1632 | rpd3::TRP1 | This study |
| RSY1717 | MATα cyh2R-z ho::LYS2 leu2::hisG lys2 trp1::hisG ura3 T7-UME6K1R K3R | This study |
| RSY1718 | MATα cyh2R-z ho::LYS2 leu2::hisG lys2 trp1::hisG ura3 T7-UME6K1Q K3Q | This study |
| RSY1732 | MATα cyh2R-z ho::LYS2 leu2::hisG lys2 trp1::hisG ura3 T7-UME6K1R K3Q | This study |
| RSY1733 | MATα cyh2R-z ho::LYS2 leu2::hisG lys2 trp1::hisG ura3 T7-UME6K1Q K3R | This study |
| RSY1743 | T7-UME6K3R rpd3::TRP1 | This study |
| RSY1835 | MATa/α lys2 ura3 LYS2::hoΔ ume6::KANMX | This study |
All strains are isogenic to RSY335 except as noted. All alleles are homozygous except as noted.
TABLE 2.
Plasmids used in this study
| Plasmid | Genotype | Reference |
|---|---|---|
| pRS306 | YIp URA3 | 66 |
| pMM285 | T7-UME6 in pRS315 CEN6 LEU2 | 18 |
| pML2 | T7-UME6K812R,K813R,K814R in pMM285 | This study |
| pML3 | T7-UME6K812Q,K813Q,K814Q in pMM285 | This study |
| pML11 | T7-UME6K813R,K814R in pMM285 | This study |
| pML16 | T7-UME6K813Q,K814Q in pMM285 | This study |
| pML23 | T7-UME6K813R,K814R in pRS306 | This study |
| pML24 | T7-UME6K813Q,K814Q in pRS306 | This study |
| pMM288 | HIS6-UME6726–836 | This study |
| pML38 | HIS6-UME6K736R,K737R,K745R | This study |
| pMM293 | HIS6-UME6K812R,K813R,K814R | This study |
| pML40 | HIS6-UME6K736Q,K737Q,K745Q | This study |
| pML41 | HIS6-UME6K736R,K737R,K745R,K812R,K183R,K814R | This study |
| pML53 | HIS6-UME6K812Q,K813Q,K814Q | This study |
| pML63 | T7-Ume6p in pRS316; CEN6 URA3 | This study |
| pML64 | T7-Ume6pK3R in pRS316; CEN6 URA3 | This study |
| pML65 | T7-Ume6pK3Q in pRS316; CEN6 URA3 | This study |
In vitro acetyltransferase assays.
In vitro acetyltransferase assays were conducted as described previously (27), with the following modifications. Whole-cell lysates were prepared from mid-log-phase cultures of RSY1500 (TAP-ADA2) or RSY1563 (TAP-ADA2 gcn5Δ) as described previously (28), except that the ultracentrifugation step was replaced with centrifugation at 20,000 × g for 30 min. Since standard tandem affinity purification (TAP) methodology resulted in purification of inactive acetyltransferase complexes, it was necessary to perform a modified purification protocol. Total acetyltransferase complexes were enriched by using Ni2+ affinity chromatography followed by a modified TAP procedure (29, 30). HAT complexes were isolated directly following tobacco etch virus (TEV) endopeptidase cleavage without further purification. We assayed the ability of these purified HAT complexes to incorporate [3H]acetyl coenzyme A ([3H]acetyl-CoA) into recombinant hexahistidine (His6)-tagged proteins containing the carboxyl-terminal 110 amino acids (aa) of Ume6p harboring either the wild-type (wt) sequence, a truncation removing lysine clusters 4 and 5 (Δ4,5) (carboxyl-terminal 15 amino acids), or lysine-to-arginine mutations in lysine cluster 1 (K736R, K737R, and K745R) and lysine cluster 3 (K812R, K813R, and K814R) in isolation or in tandem. Histone H3.3 and histone H4 were used as positive controls. Approximately 2.5 μg of each substrate was incubated in HAT assay buffer (27) containing [3H]acetyl coenzyme A for 30 min at 30°C and resolved by using SDS-PAGE. The resulting gels were Coomassie stained prior to fluorography.
Protein identification by nanoLC-ESI-MS/MS analyses.
SDS-PAGE-separated samples for both control and test samples (∼500 ng) were subjected to in-gel digestion by chymotrypsin and subsequent extraction as reported previously (31). The chymotryptic digest was subjected to nanoscale liquid chromatography-electrospray ionization-tandem mass spectrometry (nanoLC-ESI-MS/MS) analysis using an LTQ-Orbitrap Velos (Thermo-Fisher Scientific, San Jose, CA) mass spectrometer equipped with a Plug and Play nanoscale ion source device (CorSolutions LLC, Ithaca, NY). Dynamic exclusion parameters were set at repeat count 1 with a 20-s repeat duration, an exclusion list size of 500, a 30-s exclusion duration, and a ±10-ppm exclusion mass width. All data were acquired by using Xcalibur 2.1 operation software (Thermo-Fisher Scientific) and processed by using Proteome Discoverer 1.1 (PD1.1; Thermo) against a yeast protein database. All MS/MS spectra for identified acetylated peptides were manually inspected and validated by using PD1.1 and Xcalibur 2.1 software.
In vitro binding analysis.
Surface plasmon resonance (SPR) binding experiments were performed on a Biacore 3000 instrument (Biacore Inc., Piscataway, NJ). URS1SPO13 and mutant URS1ΔGC DNA probes (Table 2) were chemically synthesized carrying a 5′ biotin tag on one strand of DNA and then annealed to their complement to allow immobilization of the double-stranded DNA (dsDNA) onto streptavidin-coated sensor chips (SA chips; Biacore Inc.). The DNA probes were diluted in running buffer [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 μM Zn(O2CCH3)2, 5% glycerol, 125 μg/ml salmon sperm DNA, 62.5 μg/ml acetylated bovine serum albumin, 1 mM dithiothreitol, 0.05% surfactant P20] and injected over the sensor chip surface at 10 μl/min at 20°C. His6 fusion proteins containing the 110-amino-acid carboxyl-terminal DNA binding domain of Ume6p containing either the wild type, lysine-to-glutamine (His6-3QQQ), or lysine-to-arginine (His6-3RRR) mutations in lysine cluster 3 sequences were purified from Escherichia coli and quantitated by using SDS-PAGE followed by Coomassie staining. Recombinant proteins were serially diluted in 3-fold steps in running buffer with a maximum concentration of 350 nM and injected in triplicate at 20°C at a flow rate of 50 μl min−1 for 2 min. Disruption of any complex that remained bound after a 5-min dissociation was achieved by using a 1-min injection of 1 M NaCl at 20 μl min−1. Data were processed and analyzed by using Scrubber2 (BioLogic Software, Australia). Electrophoretic mobility shift assays (EMSAs) were performed as previously described (32). Unlabeled competitors were added at 75- and 150-fold molar excesses prior to incubation with the protein extracts.
Molecular modeling.
Side chain conformations for the model were built from the known backbone coordinates from the Gal4p DNA binding domain/DNA cocrystal structure (33), and sequence alignments were performed by using SCWRL (34) and a backbone-dependent rotamer library (35, 36). Loops with gaps in the sequence alignment were built with the program MODELLER (37). SCWRL was then used to rebuild the side chains on the structure built by MODELLER.
Western blot and chromatin immunoprecipitation assays.
Western blot analysis was performed as described previously (18). Chromatin immunoprecipitation (ChIP) assays were performed essentially as described previously (38), with the following modifications. Twenty milliliters of meiotic cultures or 50 ml of mid-log-phase dextrose cultures was cross-linked with 1% formaldehyde (15 min at room temperature), followed by quenching of cross-linked protein-DNA complexes with 140 mM glycine for 5 min. The anti-acetylated lysine antibodies do not cross-react with acetylated Ume6p (15). The Ume6p antibody was specific for Ume6p in ChIPs (see Fig. S3 in the supplemental material). Immune complexes were collected, washed, and eluted prior to reversing cross-links. DNA was precipitated, treated with proteinase K, and subjected to quantitative PCR (qPCR). Relative ChIP signals were calculated by using the formula 2−IP(CT target −CT control)/input(CT target − CT control). The NUP85 coding region was used as an internal control for nonspecific anti-Ume6p or anti-acetylated lysine immunoprecipitations.
RT-qPCR.
Total nucleic acids were prepared from either 5 ml of a sporulation culture (meiotic analyses) or 10 ml of mid-log-phase cultures (steady-state measurements). Total nucleic acid preparations were then treated with DNase I (New England BioLabs) and incubated with Moloney murine leukemia virus (Mu-MLV) reverse transcriptase (RT) (New England BioLabs) in oligo(dT)-primed reaction mixtures to allow reverse transcription of poly(A) mRNA. Subsequent qPCRs were prepared by using Power SYBR Master mix (Applied Biosystems) containing primers (Table 3). For meiotic experiments, all threshold cycle (CT) values were normalized to NUP85 values to allow relative mRNA abundance estimates (ΔCT). For steady-state experiments, CT values were first normalized to NUP85 values, followed by normalization to wild-type values (ΔΔCT). Values reported are the averages of three or more independent biological replicates; error bars represent the standard errors of the means.
TABLE 3.
qPCR primers used in this study
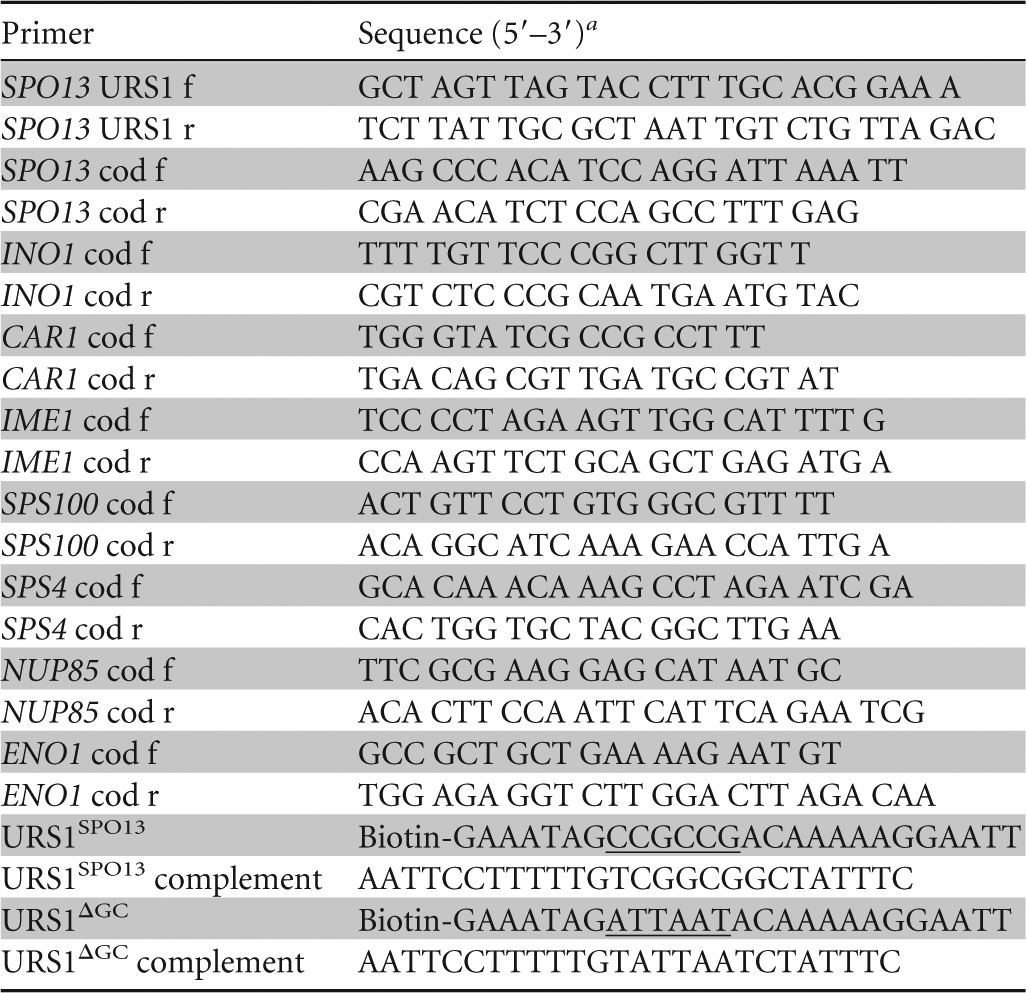
Underlining indicates the location of the core URS1 consensus element within the oligonucleotide.
RESULTS
Ume6p contains a second lysine cluster acetylated by SAGA.
Previously, we identified three lysines in the carboxyl-terminal 110 amino acids of Ume6p (aa 726 to 836) (Ume6p726–836) that are a substrate for SAGA (lysine cluster 1) (Fig. 1A) (15). However, mutation of all three sites to arginine did not abolish in vitro acetylation of His6-Ume6p726–836 (Fig. 1B, compare lanes 3 and 5), suggesting that additional modified residues are present. This region of Ume6p contains four additional lysine cluster motifs that are potential SAGA targets (Fig. 1A). Although deletion of lysine clusters 4 and 5 did not reduce acetylation (Fig. 1C), Lys-to-Arg substitution mutations in lysine cluster 3 (KKK→RRR) produced a significant reduction in acetylation (Fig. 1B, compare lanes 3 and 4). This activity was dependent on GCN5, as SAGA purified from gcn5Δ mutants failed to modify Ume6p (Fig. 1B, lanes 9 to 12). Mutation of clusters 1 and 3 reduced acetylation more than did either single mutant, suggesting that both clusters contribute to Ume6p acetylation. The lysine cluster 1 and 3 double mutant did not completely abolish all acetylation, indicating that additional modified residues exist in this region of the protein (Fig. 1B, lane 6 [long exposure]). Mass spectroscopic analysis confirmed that lysine cluster 3 is acetylated by SAGA in vitro (Fig. 1D). Although this analysis indicated that one of the three lysines was modified, it could not be determined whether acetylation was restricted to one of the triplet residues. In addition, mass spectroscopic analysis of purified Ume6p from yeast extracts revealed the presence of a signal consistent with a peptide that was both acetylated and phosphorylated (see Fig. S1 in the supplemental material). However, no peak was observed for the unmodified or only acetylated species (data not shown), suggesting that phosphorylation and acetylation occur coincidently at high efficiency within the lysine triplet 3 region. Taken together, these results identified a new Ume6p modification target for SAGA.
FIG 1.

Ume6p repressor function is reduced by SAGA acetylation. (A) Schematic diagram displaying full-length Ume6p and the amino acid sequence (aa 726 to 836) of the His6 peptides used for in vitro acetylation assays. The five lysine clusters (boxed) and six cysteines in the zinc cluster domain (underlined) are indicated. Acetylated lysine residues in cluster 1 are denoted with asterisks. The ⌉ symbols delimit the chymotryptic peptide generated during mass spectroscopic analysis shown in panel D. (B) Ume6p peptides containing the indicated lysine cluster 1 and 3 configurations were incubated with SAGA purified from a wild-type GCN5 strain (left) or a gcn5Δ mutant (right), in the presence of [3H]acetyl-CoA. Long- and short-exposure autoradiographs of reaction mixtures separated by SDS-PAGE are shown. The same gel was Coomassie stained to control for substrate loading. Purified recombinant histones H3 and H4 served as positive controls. (C) In vitro HAT assays for a Ume6p derivative truncated for lysine clusters 3 and 4. (D) Representative spectra from mass spectrometry showing lysine cluster 3 acetylation. His6-Ume6p726–836 was in vitro acetylated by SAGA using Ada2p-TAP-purified extracts. Either unmodified (top) or acetylated (bottom) Ume6p was digested with chymotrypsin and evaluated via mass spectroscopy (see Materials and Methods for details). Peaks corresponding to mass increases indicative of acetylation are indicated along with the peptide sequence. Although modification the first lysine of triplet 3 is indicated, mass spectroscopy is unable to determine which lysine in the triplet was actually acetylated.
Glutamine substitution at lysine cluster 3 derepresses Ume6p-regulated genes.
The mass spectroscopic data described above revealed that at least one amino acid in the third lysine triplet is acetylated. To help ascertain which of these residues is modified, we took advantage of our previous finding that Ume6p acetylation at lysine cluster 1 allowed EMG derepression (15). Therefore, we reasoned that modification of an individual lysine in cluster 3 may have a similar impact. To test this possibility, we substituted glutamine, an acetylation mimic, for each individual lysine in triplet 3 and tested their impact on the ability of Ume6p to repress an EMG reporter gene (spo13-lacZ) during vegetative growth. Previous studies have established that substituting lysine with glutamine can mimic acetylation, due to the similarity of chemical structure and charge (39–42). To avoid complications from acetylation of the first lysine cluster, these experiments were conducted in dextrose medium, which limits the modification of these residues (15). Substituting glutamine for the second or third lysine located in lysine cluster 3, but not the first, derepressed spo13-lacZ (Fig. 2A). The double-glutamine substitution of the second and third lysines did not significantly reduce Ume6p repressor function relative to that of each individual mutation, suggesting that modification of either residue is sufficient to adversely affect repressor function. The conservative lysine-to-arginine substitution (KRR) mutant did not alter Ume6p repressor function (Fig. 2A). Therefore, we utilized the KQQ or KRR substitution mutation in the third lysine triplet (termed Ume6pK3Q and Ume6pK3R, respectively) to represent the acetylated or unmodified Ume6p species, respectively.
FIG 2.
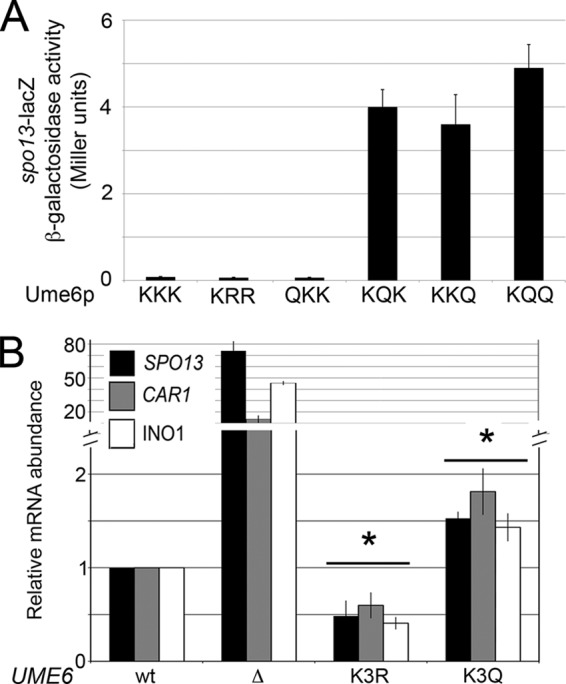
The second and third lysines in cluster 3 regulate Ume6p repressor function. (A) A ume6Δ strain harboring plasmids expressing wild-type UME6 or the mutant derivatives, as indicated, was transformed with a spo13-lacZ reporter plasmid, and β-galactosidase activity was determined as described previously (65). Mean values from three independent biological replicates are presented (±standard errors of the means). (B) SPO13, INO1, and CAR1 mRNA levels were monitored by RT-qPCR from mid-logarithmic-phase UME6-, UME6K3R-, or UME6K3Q-expressing cultures. Mean values from three independent biological replicates are presented (±standard errors of the means) relative to ENO1 mRNA levels. Asterisks indicate statistically significantly differences from wild-type values as determined by the Student t test (P < 0.05).
We next addressed whether the Ume6pK3R or Ume6pK3Q substitution mutation affected the regulation of other Ume6p target genes. The wild-type UME6 allele was replaced with either UME6K3R or UME6K3Q, and the mRNA levels of SPO13 (23), CAR1 (19, 43), and INO1 (44) were determined by RT-qPCR. The UME6K3R-expressing mutant displayed a small, but statistically significant, enhanced repressor activity over the wild type in all three loci tested (Fig. 2B). Conversely, UME6K3Q cultures exhibited a similar loss of repression. These results indicate that acetylation disrupts Ume6p repressor function at several loci. However, the derepression observed in Ume6pK3Q-expressing cultures was still ∼35-fold lower than that observed in cells lacking UME6, suggesting that cluster 3 acetylation only partially antagonizes Ume6p repressor function.
Glutamine substitutions in cluster 3 diminish Ume6p DNA binding.
We next sought a mechanism to explain why cluster 3 acetylation reduced Ume6p repressor function. We previously reported that cluster 1 acetylation promoted ubiquitin-mediated proteolysis (15). However, Western blot analysis revealed only minor differences in steady-state levels of Ume6p, Ume6pK3R, or Ume6pK3Q (Fig. 3A), suggesting that protein stability was unlikely to be responsible for this phenotype. The proximity of these lysines to the Zn2Cys6 zinc cluster domain (Fig. 1A) suggested that acetylation may impact Ume6p binding to the URS1SPO13 control element. To test this possibility, electrophoretic mobility shift assays (EMSAs) were performed by using crude extracts containing Ume6p, Ume6pK3R, or Ume6pK3Q incubated with radiolabeled URS1SPO13. Normally, six complexes (C1 to C6) are formed, with C2 being specific for Ume6p (32). In these experiments, C1, C4, C5, and C6 were underexposed to allow straightforward inspection of C2 intensity. In the absence of a competitor, the C2 signal intensity was reduced in Ume6pK3Q extracts compared to the wild type or Ume6pK3R (Fig. 3B, compare lanes 1, 4, and 7). The addition of unlabeled competitor resulted in the rapid loss of C2 in the Ume6pK3Q extracts compared to the wild type or Ume6pK3R. These data suggest that mimicking acetylation at lysine cluster 3 reduces Ume6p-URS1SPO13 complex stability.
FIG 3.

Mimicking acetylation at lysine cluster 3 but not cluster 1 diminishes Ume6p DNA binding in vitro. (A) Cells containing genomically integrated K3R and K3Q mutations at lysine cluster 3 were grown to mid-log phase in dextrose medium. Total protein extracts (50 μg) were subjected to Western blot analysis to probe for the T7 epitope. The blots were stripped and reprobed for Tub1p as a loading control. Phosphorimaging of the Western blot images was performed, and the ratio of Ume6p to Tub1p was set to 1. The ratios of the lysine cluster 3 mutants to Tub1p are given relative to the wild type. (B) EMSAs were performed with a 32P-labeled URS1SPO13 probe and crude extracts expressing the indicated UME6 allele. The reaction mixtures contained either no (Ø) or a 75- or 150-fold excess of unlabeled URS1SPO13 probe as a competitor. Six complexes are formed in the EMSA (C1 to C6), with C2 (arrow) being Ume6p specific (32). (C) EMSA was performed as in described above for panel B except that the crude extract was replaced with recombinant His6-Ume6p726–836 containing the wild type or KKK→QQQ substitution mutations in either lysine cluster 3 or cluster 1. A Coomassie-stained gel indicating the amounts of different proteins that were added to the reaction mixture is shown (inset). (D) EMSA was performed as described above for panel C, using increasing amounts of the KKK→QQQ mutation in lysine cluster 3. Protein loading for each reaction is shown by Coomassie staining of a separate gel. (E) Surface plasmon resonance was performed by using peptides containing the last 110 amino acids of Ume6p with either the wild-type (KKK), triple-arginine (RRR), or triple-glutamine (QQQ) substitutions at lysine triplet 3. Proteins were serially diluted in 3-fold steps, starting at a 1 μM concentration. Following loading of each protein onto a conjugated chip, buffer was introduced, and the dissociation of the complex was monitored. (F) Table showing dissociation rates (kd) and half-life (t1/2) calculations generated from data obtained in Fig. 2E.
To ensure that the results described above are due to glutamine mutations in lysine cluster 3 specifically and not a general phenomenon of mutagenesis in the DNA binding region, we next assessed DNA binding of Ume6p containing lysine-to-glutamine mutations in lysine cluster 1. As discussed above, the Ume6pK1Q mutation results in rapid turnover of Ume6p, making EMSA using crude yeast extracts not feasible (15). To avoid this complication, His6 fusion proteins containing the 110-amino-acid carboxyl-terminal DNA binding domain containing either the wild-type or glutamine substitutions in either lysine cluster 1 or 3 (1KKK→1QQQ and 3KKK→3QQQ, respectively) were purified from E. coli. EMSAs using these recombinant proteins showed that while both His6-wt and His6-1QQQ proteins interact with URS1SPO13 with similar affinities, His6-3QQQ failed to form a stable protein-DNA complex (Fig. 3C). These data demonstrate that mutagenesis of lysine cluster 3 but not cluster 1 negatively impacts URS1SPO13 DNA binding.
To further quantitate the impact of cluster 3 glutamine substitutions on the Ume6p-URS1 interaction, we employed two strategies. To estimate the difference in equilibrium binding between His6-wt and His6-3QQQ proteins, we employed EMSA to compare the DNA binding abilities of increasing concentrations of His6-3QQQ to His6-wt (Fig. 3D). These experiments showed an ∼5-fold decrease in equilibrium binding between His6-wt and His6-3QQQ. To understand the nature of this decrease in affinity, we assayed the binding of recombinant His6-wt, His6-3QQQ, and His6-3RRR using surface plasmon resonance (SPR). Wild-type URS1SPO13 (CGGCGG) or URS1ΔGC (ATTAAT) oligonucleotides were used as DNA targets. URS1ΔGC served as a negative control for nonspecific binding, as it lacks the Ume6p core recognition sequence (32). As expected, the URS1ΔGC DNA target failed to form stable protein-DNA complexes with Ume6p or either derivative (see Fig. S2A in the supplemental material). With URS1SPO13, all three peptides exhibited a dissociation pattern that was best fit by incorporating two curves (Fig. 3E; see also Fig. S2B in the supplemental material). Quantification of these data revealed that the wild-type and 3RRR mutant proteins exhibited similar dissociation rates (Fig. 3F). Consistent with our EMSA results, the 3QQQ mutant displayed dissociation curves with an off rate that was ∼3-fold higher than that of the wild type. These results support the model that lysine cluster 3 acetylation enhances Ume6p dissociation from URS1SPO13.
Glutamine mutagenesis in cluster 3 reduces Ume6p-URS1SPO13 binding in vivo.
To determine whether mimicking acetylation affected Ume6p DNA binding in vivo, we employed chromatin immunoprecipitation (ChIP) studies. Using an antibody directed toward endogenous Ume6p (15), ChIP analyses were performed to examine Ume6p occupancy at URS1SPO13. Preliminary studies utilizing this antibody in ChIP experiments validated its specificity for Ume6p bound to URS1SPO13 (see Fig. S3 in the supplemental material). In Ume6pK3R-expressing cultures, no significant difference in DNA binding relative to the wild type was observed (Fig. 4A). Conversely, Ume6pK3Q exhibited a statistically significant ∼5-fold loss in SPO13 promoter binding. These results suggest that lysine cluster 3 acetylation reduces Ume6p repressor binding in vivo.
FIG 4.
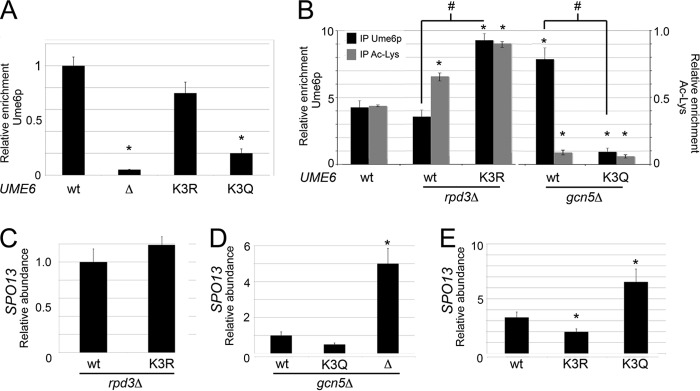
Gcn5p- and Rpd3p-dependent regulation of lysine triplet 3 acetylation controls Ume6p DNA binding and repression activity. (A) Ume6p occupancy at the SPO13 promoter was monitored by ChIP in mid-log-phase cultures expressing Ume6p, Ume6pK3R, or Ume6pK3Q. (B) Same as panel A except in host strains with the indicated genotypes. The anti-acetylated Lys (Ac-Lys) antibodies detect acetylated proteins associated with the SPO13 promoter but not acetylated Ume6p (15). Mean values from three independent biological replicates are presented (±standard errors of the means) relative to the values for the NUP85 internal control. (C and D) SPO13 mRNA levels from mid-log-phase cultures expressing the indicated UME6 allele in either the rpd3Δ (C) or the gcn5Δ (D) background were monitored by RT-qPCR. The different UME6 alleles examined are given below the graph. (E) SPO13 mRNA abundance measured as described above for panel C except that the cultures expressing the indicated UME6 alleles were grown to mid-log phase in acetate medium. Mean values from three independent biological replicates are presented (±standard errors of the means) relative to the values for the NUP85 internal control. In all panels, the asterisks indicate a P value of <0.05 (Student's t test) for the difference from the wild type. In panel B, # indicates that the two values indicated by the line are significantly different (P < 0.05).
We have previously demonstrated that Ume6p is acetylated and deacetylated by the opposing activities of Gcn5p and Rpd3p, respectively (15). If our model that cluster 3 acetylation diminishes Ume6p-DNA interactions is correct, we would predict that preventing Ume6p acetylation should enhance binding. First, we examined Ume6p occupancy of URS1SPO13 by ChIP in wild-type or gcn5Δ mutant strains. The loss of Gcn5p activity should lead to reduced Ume6p acetylation and elevated levels of binding. As shown in Fig. 4B, Ume6p displayed a 2-fold increase in URS1SPO13 occupancy when GCN5 was deleted. This stimulation of Ume6p binding in the gcn5Δ strain was lost when glutamines were substituted for lysine triplet 3. Similarly, we would predict that Ume6p binding is reduced in the rpd3Δ mutant due to elevated acetylation of triplet 3 and that this reduction would be suppressed in the K3R mutant. Although the reduction of Ume6p binding in the rpd3Δ strain was minimal compared to that in the wild type, the level of Ume6pK3R binding was significantly elevated (Fig. 4B). This elevation in the level of Ume6pK3R binding was not anticipated and may be explained by the mutation of Ume6p affecting other posttranslational modifications or by RPD3 removal affecting the local histone environment (see Discussion). These results indicate that the Ume6p-URS1 interaction is controlled by the opposing activities of Gcn5p and Rpd3p at lysine cluster 3. In addition, the relative binding of Ume6p was independent of the acetylation state of the surrounding chromatin. For example, the level of Ume6p or Ume6pK3R binding was elevated under conditions of hypoacetylated (gcn5Δ) or hyperacetylated (rpd3Δ) histones, respectively.
Preventing or mimicking Ume6p acetylation does not replace the requirements of Rpd3p or Gcn5p for SPO13 transcriptional regulation.
We next investigated the relative contributions of the acetylation/deacetylation status of Ume6p and promoter histones to regulation of SPO13 transcription. First, we determined whether the requirement of Rpd3p for SPO13 repression could be partially suppressed if lysine triplet 3 was no longer a substrate for Gcn5p. Using the same strategy as that described above, we assayed SPO13 mRNA levels in an rpd3Δ mutant expressing either wild-type Ume6p or Ume6pK3R using RT-qPCR. These studies revealed that full Ume6p repressor function requires Rpd3p regardless of the enhanced binding ability of Ume6pK3R (Fig. 4C). This result agrees well with previous reports that established this relationship (20, 22). Similarly, although Ume6pK3Q binding was reduced in gcn5Δ mutants, SPO13 derepression still required this HAT (Fig. 4D). Taken together, these results indicate that histone modification is epistatic to Ume6p acetylation.
Glutamine substitution at lysine cluster 3 suppresses cluster 1-dependent Ume6p turnover.
Previously, we found that lysine cluster 1 acetylation enhanced Ume6p degradation, resulting in SPO13 mRNA derepression (15). Therefore, we next determined the functional relationship of mutating lysine clusters 1 and 3 in tandem. First, we constructed yeast strains harboring all possible combinations of lysine cluster 1 and 3 mutations (i.e., K1R K3R, K1R K3Q, K1Q K3Q, and K1Q K3R). These mutant yeast strains were grown to mid-log phase in dextrose medium and then analyzed for their ability to repress three Ume6p targets, SPO13, CAR1, and INO1, using RT-qPCR. Interestingly, the introduction of glutamine into lysine cluster 3 resulted in modest derepression of SPO13 and CAR1 (P < 0.05) regardless of cluster 1 status (Fig. 5A, top and middle). This derepression was observed in the UME6K1Q K3Q strain only for INO1, and this trend did not reach statistical significance (Fig. 5A, bottom). On the other hand, the K3R mutations did not affect Ume6p repressor function regardless of lysine cluster 1 status. These results were not due to changes in steady-state protein levels as determined by Western blotting (Fig. 5B), indicating that mimicking acetylation of lysine cluster 3 suppresses the protein turnover phenotype of cluster 1 (15). In addition, our results suggest that not all promoter contexts respond to cluster 3 acetylation equally.
FIG 5.
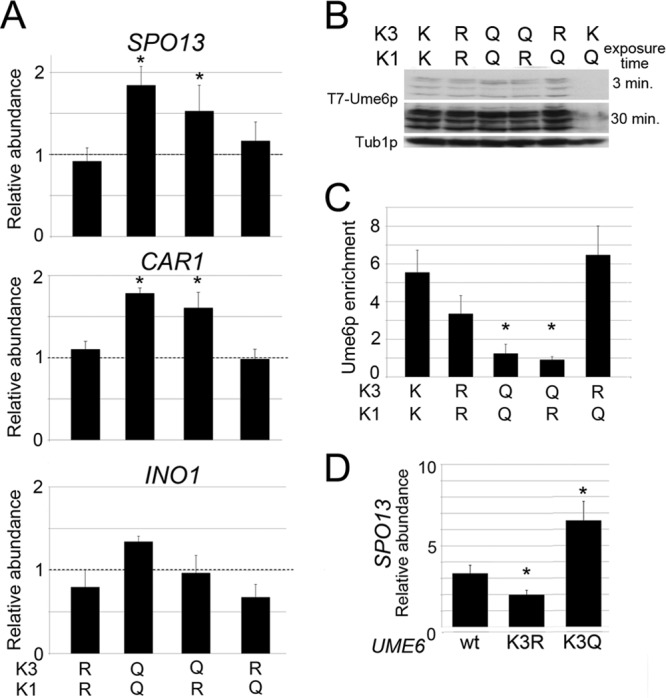
Lysine cluster 3 acts upstream of lysine cluster 1 to repress URS1-containing transcripts. (A) SPO13, CAR1, and INO1 mRNA levels were monitored by RT-qPCR in mid-log-phase dextrose cultures expressing Ume6p, Ume6pK1R K3R, Ume6pK1Q K3Q, Ume6pK1R K3Q, or Ume6pK1Q K3R. Wild-type levels of each RNA target were set equal to 1, as indicated by the dashed line. Mean values from three independent biological replicates are presented (±standard errors of the means) relative to the values for the NUP85 internal control. (B and C) Steady-state protein levels (B) and URS1SPO13 occupancy (C) for the Ume6p derivatives described above for panel A were determined by Western blotting and ChIP, respectively. Ume6pK1Q mutants served as a control in panel B. (D) SPO13 mRNA levels were monitored by RT-qPCR in mid-log-phase acetate cultures expressing Ume6p, Ume6pK3R, or Ume6pK3Q. SPO13 mRNA levels in wild-type dextrose cultures were set equal to 1. Mean values from three independent biological replicates are presented (±standard errors of the means); asterisks indicate significant differences relative to the wild type (P < 0.05).
Finally, to determine if the SPO13 derepression observed in K1Q K3Q and K1R K3Q double mutants is due to a DNA binding defect, ChIP was employed by using antibodies specific for Ume6p and primers directed at the SPO13 promoter region. These experiments show that Ume6p DNA binding is reduced in the presence of the K3Q mutation regardless of lysine cluster 1 status (Fig. 5C). These data indicate that acetylation-induced Ume6p turnover requires the identity of lysine cluster 3 to be maintained (see Discussion).
Next, we examined the relationship between lysine cluster 1 acetylation and cluster 3 mutagenesis by assaying Ume6p, Ume6pK3R, and Ume6pK3Q repressor functions in acetate cultures. Our previous study revealed that Gcn5p-dependent acetylation of lysine cluster 1 occurs only in medium containing a nonfermentable carbon source (e.g., acetate), resulting in partial degradation of Ume6p and an ∼3-fold derepression of SPO13 mRNA (15). Therefore, cells growing in acetate should phenocopy the glutamine substitution mutation in lysine cluster 1. Compared to wild-type cells, UME6K3R acetate cultures displayed a modest but statistically significant reduction in SPO13 mRNA levels (Fig. 5D), consistent with the strong URS1 occupancy observed for the Ume6pK1Q K3R mutant. Conversely, the presence of Ume6pK3Q elevated SPO13 mRNA levels about 2-fold above those normally observed in acetate medium alone. Taken together, these results show that Ume6p DNA binding controls acetate-dependent SPO13 derepression, indicating that a complex relationship exists between cluster 1 and cluster 3 acetylation marks.
Lysine cluster 3 integrity is required for normal meiotic gene induction and nuclear divisions.
Ume6p proteolysis is required for EMG induction and meiotic progression (18). Failure to acetylate lysine cluster 1 delays Ume6p destruction, resulting in defects in both the timing and efficiency of meiotic nuclear divisions (15). To determine whether lysine cluster 3 acetylation is required for normal meiotic progression, samples were taken from cultures expressing UME6, UME6K3R, or UME6K3Q before and following transfer into sporulation medium (SPM). Western blot analysis revealed identical destruction kinetics for the wild type and Ume6pK3Q (Fig. 6A). However, Ume6pK3R displayed a more complex degradation profile. The initial reduction in Ume6pK3R levels following transfer to sporulation medium was delayed 3 h. When meiotic destruction did occur, it was incomplete, as Ume6pK3R was still detected throughout the time course. These findings suggest that lysine cluster 3 modification is required for meiotic Ume6p degradation.
FIG 6.

Cluster 3 acetylation is required for timely meiotic gene induction and meiotic progression. (A) Ume6p, Ume6pK3R, or Ume6pK3Q levels were determined by Western blotting of samples taken before and following the shift to SPM (hours). Tub1p served as a loading control. (B and C) URS1SPO13 occupancy by either Ume6p (B) or Ume6pK3R (C) was determined by ChIP. The presence of acetylated lysine proteins at the SPO13 promoter was determined by ChIP using pan-anti-acetylated Lys antibodies. Mean values from three independent biological replicates are presented (±standard errors of the means) relative to the values for the NUP85 internal control. (D to G) Meiotic SPO13 (D), SPS4 (E), SPS100 (F), and IME1 (G) mRNA levels were determined by RT-qPCR in Ume6p- or Ume6pK3R-expressing cultures, as indicated. Mean values from three independent biological replicates are presented (±standard errors of the means) relative to the values for the NUP85 internal control. Asterisks indicate statistical differences at a given time point using the Student t test (P < 0.05). (H) The percentages of Ume6p-, Ume6pK3R-, or Ume6pK3Q-expressing cultures exhibiting bi- and tetranucleated cells were determined by DAPI analysis. Mean values from three independent biological replicates are given (±standard errors of the means). Asterisks indicate statistically significant differences relative to the wild type (P < 0.05).
The delay in Ume6pK3R destruction raised the question of whether it was still bound to URS1SPO13 and able to recruit the Rpd3p HDAC. ChIP analysis revealed that Ume6p was lost from URS1SPO13 at approximately 7.5 h (Fig. 6B). The loss of Ume6p binding correlated with enhanced protein (i.e., histone) acetylation at the SPO13 locus. To repeat these experiments with the Ume6pK3R-expressing strain, an extended time course was conducted to account for the delay in destruction of this mutant. ChIP analysis indicated that Ume6pK3R remained associated with URS1SPO13 until about 15 h following transfer to SPM. Again, the retention of Ume6pK3R corresponded well with the reduced acetylation of proteins associated with the SPO13 promoter (Fig. 6C). These results indicate that lysine cluster 3 acetylation is important for efficient Ume6p removal from URS1SPO13. In addition, the loss of Ume6p or Ume6pK3R binding correlated well with increased overall protein acetylation at URS1SPO13. These findings suggest that Rpd3p removal from the EMG promoter coincides with loss of Ume6p binding.
The presence of Ume6pK3R at the SPO13 promoter coupled with continued protein deacetylation suggested that EMG induction would also be delayed. RT-qPCR analysis revealed that in the wild type, peak SPO13 mRNA accumulation occurred 9 h after transfer into SPM (Fig. 6D). This time frame is consistent with the loss of Ume6p binding and elevated levels of acetylation at URS1SPO13. The Ume6pK3R-expressing strain displayed a delay in peak SPO13 mRNA accumulation that mirrored the retention of Ume6pK3R at URS1SPO13 (Fig. 6D). These results indicate that the delay in removing Ume6pK3R resulted in a similar delay in EMG induction. A more dramatic effect was observed for the expression profile of SPS4, a member of the “middle” class of meiotic genes (45, 46). RT-qPCR analysis indicated a >6-h delay in SPS4 peak expression (Fig. 6E). The delay appeared even greater for the late-expressing gene SPS100 (Fig. 6F). Notably, these delays were not due to a failure to enter meiosis, as both Ume6p- and Ume6pK3R-expressing strains displayed similar induction kinetics of IME1 (Fig. 6G). Ime1p expression initiates meiosis, and its transcriptional control is independent of Ume6p (13). Taken together, these results indicate that the relatively small delay in removing Ume6pK3R from EMG promoters results in a similar retardation in EMG induction. However, the transcriptional delay becomes exaggerated as meiosis progresses.
Since the middle and late genes are important for executing the meiotic nuclear divisions and spore morphogenesis, respectively, we next assessed the impact of delayed Ume6pK3R destruction on meiotic progression. The percentages of the population undergoing meiosis I and/or meiosis II were compared in Ume6p-, Ume6pK3Q-, and Ume6pK3R-expressing strains. 4′,6-Diamidino-2-phenylindole (DAPI) analysis revealed that the appearance of bi- and tetranucleated cells was similar in wild-type and Ume6pK3Q-expressing strains (Fig. 6H). However, strains expressing Ume6pK3R demonstrated a statistically significant delay in binucleated cell accumulation and overall sporulation efficiency. Taken together, these data demonstrate that Ume6p acetylation at lysine cluster 3 is required for normal execution of a cellular differentiation pathway.
DISCUSSION
Histone acetylation is an established component of the gene induction program. Here, we demonstrate a new mechanism by which acetylation activates gene expression. Rather than opening chromatin or enhancing the function of a transcriptional activator, we find that Gcn5p-dependent acetylation stimulates early meiotic gene (EMG) transcription by reducing the DNA binding ability of the transcriptional repressor Ume6p. Both in vitro and in vivo studies revealed that substituting glutamine for the lysines in cluster 3 to mimic acetylation reduced Ume6p binding to its target element, resulting in partial EMG derepression. Conversely, preventing acetylation slowed removal of Ume6p from EMG promoters following meiotic entry, resulting in delayed induction of all meiotic gene expression classes. Importantly, a defect was observed in both the timing and efficiency of the meiotic nuclear divisions. These results indicate that Ume6p acetylation plays an important role in establishing the timing of the transient transcription cascade and, ultimately, meiotic progression.
Our data demonstrate a modest derepression of three target genes when mutations are introduced into Ume6p to mimic its acetylated state at lysine cluster 3 (K3Q). The finding that the derepression of these genes is >20-fold greater in ume6Δ strains suggests that the impact of Ume6p acetylation in vegetative cells represents a minor component of the overall mitotic repression system. Differences in gene expression of this magnitude are not uncommon in mutants defective for histone modification in mitotic cells. For example, loss of NuA4 or Gcn5p function results in less than a 2-fold reduction in GAL1-dependent transcription (47). Similarly, sin3Δ or rpd3Δ mutants generally derepress target genes only 2- to 4-fold (11, 12). Therefore, it may not be surprising that the impact of Ume6p acetylation on EMG repression is not dramatic. However, just like the chromatin-modifying proteins, a more robust phenotype is observed during meiotic development when Ume6p acetylation is prevented. We find that SPO13 expression reaches similar meiotic levels in both Ume6p- and Ume6pK3R-expressing strains. However, peak expression is delayed 3 h when Ume6p acetylation is prevented, which expands to twice that value for middle genes and even longer for late-expressing loci. These findings indicate that Ume6p acetylation represents an additional cue designed to fine-tune the timing, but not the absolute levels, of the meiotic gene transcriptional cascade.
Our observation that Ume6p DNA binding is enhanced in yeast cells harboring RPD3 deletions while expressing Ume6pK3R was surprising. We have no explanation for this result at present. One possibility is that enhancement in global chromatin due to the loss of Rpd3p function (reviewed in references 3, 5, 48, and 49) alters overall ChIP efficiencies independent of lysine 3 status. Alternatively, lysine cluster 3 acetylation may induce a series of posttranslational modifications in Ume6p that ultimately leads to Ume6p release from DNA. For example, methylation of E2F-1 inhibits both acetylation and phosphorylation at nonproximal sites contained within the full-length protein, which ultimately leads to protein ubiquitylation and degradation (50). Similarly, p53 methylation is induced during the DNA damage response and is required for subsequent p53 acetylation, which acts to stimulate p53 activity and transcriptional induction (51). Perhaps, substituting arginines for lysines in cluster 3 prevents not only acetylation but also the modification of Ume6p by other enzymes, which may change Ume6p behavior.
Approximately 50 proteins in budding yeast contain a Zn2Cys6 motif (reviewed in reference 52), and the structures of several of these proteins have been solved (33, 53–55). In all examples, the region occupied by lysine triplet 3 contains the homodimerization domain that stabilizes the protein-DNA complex (56). However, Ume6p is an exception in that it binds URS1 as a monomer (57). Therefore, our results suggest that the additional DNA binding affinity afforded by lysine cluster 3 functions in place of dimerization to increase Ume6p binding affinity. Modeling of Ume6p on the crystal structure of Gal4p bound to DNA revealed a close juxtaposition of the third lysine cluster to the DNA phosphate backbone (Fig. 7A, arrows). Therefore, lysine cluster 3 may mediate an electrostatic interaction with the DNA phosphate backbone, contributing to protein-DNA complex stability. In support of this possibility, DNase I footprinting revealed that Ume6p protected this region flanking the URS1 core sequences (57). These results suggest that the additional interaction of lysine cluster 3 with DNA is an adaptive response to the loss of a dimerization domain.
FIG 7.

Model for acetylation regulating Ume6p transcriptional repression. (A) The space-filling model of Ume6p interacting with URS1SPO13 was modeled on the Gal4p DNA binding domain-DNA crystal structure (33). The Zn2Cys6 DNA binding domain and lysine cluster 3 (arrows) are indicated. The first lysine in triplet 3 is highlighted in magenta. (White, carbon; blue, amines; red, oxygen; yellow, sulfur; blue, plus strand; green, minus strand; orange/red, phosphate backbone.) (B) Proposed mechanism for lysine acetylation regulating Ume6p target gene transcription. In response to environmental signals, Ume6p is acetylated at lysine cluster 3, reducing its DNA binding ability. In the absence of additional meiosis-inducing signals, this reaction is reversible via Rpd3p-dependent deacetylation. Meiotic induction induces Ume6p acetylation at both the first and third lysine clusters, leading to a loss in DNA binding followed by its subsequent destruction. (Blue stars, acetylation; black partial circle, anaphase-promoting complex/cyclosomeCdc20; red shapes, degraded Ume6p.)
Previous studies found many roles for nonhistone acetylation in controlling transcription. Similar to Ume6p, the acetylation of the FOXO transcription factor reduces its DNA binding ability (58, 59). Although structural studies were unable to model the acetylated lysines of FOXO proposed to interact with DNA (60), the DNA binding loss when these residues were modified to glutamine is similar to what we observed for Ume6p. However, one significant difference between Ume6p and FOXO acetylation is the result. While acetylation of FOXO reduced gene transcription, Ume6p acetylation had the opposite effect on target genes. Therefore, the readout for acetyltransferase activity can change depending on the function of the substrate. Another example is Gcn5p-dependent acetylation of the chromatin remodeling proteins Rsc4 and Snf2 (14, 61). In these examples, the DNA binding ability of these factors was not affected. Rather, the acetylation marks appear to be autoregulatory, affecting the association of these complexes with chromatin. In the case of Ume6p, lysine cluster 3 acetylation clearly functions to reduce its DNA binding ability. Therefore, similar to phosphorylation, acetylation marks can have a positive or negative impact on gene function depending on the specific nature of the process being regulated.
Previously, we described additional Ume6p acetylation sites that lie just upstream of the DNA binding region (15). Acetylation of these lysines (lysine cluster 1) accelerates Ume6p destruction as cells enter meiosis. Does lysine cluster 3 acetylation enhance lysine cluster 1 acetylation and destruction, or are these two marks independent? During meiosis, Ume6p degradation is accelerated in UME6K1Q cultures (15) but is not affected in UME6K3Q cultures (Fig. 6B), suggesting that lysine cluster 3 acetylation does not accelerate modification of lysine cluster 1. Furthermore, the meiotic stabilization of Ume6p in cultures containing UME6K3R shows that without acetylation of cluster 3, one may not induce acetylation-dependent Ume6p degradation. Why would the cell want Ume6p downregulation via two independent mechanisms? Ume6p regulates several gene classes, including those involved in arginine metabolism, phospholipid biosynthesis, and meiosis (19, 43, 62–64). To regulate these genes, Ume6p must respond to different internal and external cues. Ume6p acetylation by SAGA at lysine triplet 3 would result in promoter dissociation and subsequent transcriptional activation (Fig. 7B). Since this acetylation is reversible via Rpd3p-mediated deacetylation (15), Ume6p could return to the promoter in its full repressive state. However, as cells enter meiosis, complete Ume6p destruction occurs. Therefore, combining these two acetylation marks may direct the cell toward a commitment point for EMG induction and meiotic S phase. Taken together, the combination of chromatin and transcription factor acetylation provides a multitiered regulatory platform to allow flexibility prior to meiotic commitment and complete implementation of the program once the decision to proceed has been made.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Cooper, M. Primig, A. Andrews, P. Katsamba, and M. Henry for helpful discussions and critical reading of the manuscript. We also thank S. Zhang and the Proteomics and Mass Spectrometry Core Facility at Cornell University for analysis of acetylated Ume6p.
This work was supported by Public Health Service grants GM082013 (M.J.L.) and GM086788 (R.S.) from the General Medical Institute.
We have no financial conflicts with this study.
Footnotes
Published ahead of print 2 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00256-13.
REFERENCES
- 1.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. 1998. The transcriptional program of sporulation in budding yeast. Science 282:699–705. 10.1126/science.282.5389.699 [DOI] [PubMed] [Google Scholar]
- 2.Primig M, Williams RM, Winzeler EA, Tevzadze GG, Conway AR, Hwang SY, Davis RW, Esposito RE. 2000. The core meiotic transcriptome in budding yeasts. Nat. Genet. 26:415–423. 10.1038/82539 [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- 4.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. 2007. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14:1025–1040. 10.1038/nsmb1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger SL. 2007. The complex language of chromatin regulation during transcription. Nature 447:407–412. 10.1038/nature05915 [DOI] [PubMed] [Google Scholar]
- 6.Couture JF, Trievel RC. 2006. Histone-modifying enzymes: encrypting an enigmatic epigenetic code. Curr. Opin. Struct. Biol. 16:753–760. 10.1016/j.sbi.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 7.Kaluarachchi Duffy S, Friesen H, Baryshnikova A, Lambert JP, Chong YT, Figeys D, Andrews B. 2012. Exploring the yeast acetylome using functional genomics. Cell 149:936–948. 10.1016/j.cell.2012.02.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huisinga KL, Pugh BF. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13:573–585. 10.1016/S1097-2765(04)00087-5 [DOI] [PubMed] [Google Scholar]
- 9.Burgess SM, Ajimura M, Kleckner N. 1999. GCN5-dependent histone H3 acetylation and RPD3-dependent histone H4 deacetylation have distinct, opposing effects on IME2 transcription, during meiosis and during vegetative growth, in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 96:6835–6840. 10.1073/pnas.96.12.6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshikawa K, Tanaka T, Ida Y, Furusawa C, Hirasawa T, Shimizu H. 2011. Comprehensive phenotypic analysis of single-gene deletion and overexpression strains of Saccharomyces cerevisiae. Yeast 28:349–361. 10.1002/yea.1843 [DOI] [PubMed] [Google Scholar]
- 11.Vidal M, Gaber RF. 1991. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol. 11:6317–6327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vidal M, Strich R, Esposito RE, Gaber RF. 1991. RPD1 (SIN3/UME4) is required for maximal activation and repression of diverse yeast genes. Mol. Cell. Biol. 11:6306–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strich R, Slater MR, Esposito RE. 1989. Identification of negative regulatory genes that govern the expression of early meiotic genes in yeast. Proc. Natl. Acad. Sci. U. S. A. 86:10018–10022. 10.1073/pnas.86.24.10018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanDemark AP, Kasten MM, Ferris E, Heroux A, Hill CP, Cairns BR. 2007. Autoregulation of the rsc4 tandem bromodomain by gcn5 acetylation. Mol. Cell 27:817–828. 10.1016/j.molcel.2007.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallory MJ, Law MJ, Sterner DE, Berger SL, Strich R. 2012. Gcn5p-dependent acetylation induces degradation of the meiotic transcriptional repressor Ume6p. Mol. Biol. Cell 23:1609–1617. 10.1091/mbc.E11-06-0536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi JK, Grimes DE, Rowe KM, Howe LJ. 2008. Acetylation of Rsc4p by Gcn5p is essential in the absence of histone H3 acetylation. Mol. Cell. Biol. 28:6967–6972. 10.1128/MCB.00570-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Q, Lu J, Duan J, Su D, Hou X, Li F, Wang X, Huang B. 2008. Gcn5- and Elp3-induced histone H3 acetylation regulates hsp70 gene transcription in yeast. Biochem. J. 409:779–788. 10.1042/BJ20070578 [DOI] [PubMed] [Google Scholar]
- 18.Mallory MJ, Cooper KF, Strich R. 2007. Meiosis-specific destruction of the Ume6p repressor by the Cdc20-directed APC/C. Mol. Cell 27:951–961. 10.1016/j.molcel.2007.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strich R, Surosky RT, Steber C, Dubois E, Messenguy F, Esposito RE. 1994. UME6 is a key regulator of nitrogen repression and meiotic development. Genes Dev. 8:796–810 [DOI] [PubMed] [Google Scholar]
- 20.Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T. 2000. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103:423–433. 10.1016/S0092-8674(00)00134-3 [DOI] [PubMed] [Google Scholar]
- 21.Kurdistani SK, Robyr D, Tavazoie S, Grunstein M. 2002. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat. Genet. 31:248–254. 10.1038/ng907 [DOI] [PubMed] [Google Scholar]
- 22.Kadosh D, Struhl K. 1997. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89:365–371. 10.1016/S0092-8674(00)80217-2 [DOI] [PubMed] [Google Scholar]
- 23.Mallory MJ, Strich R. 2003. Ume1p represses meiotic gene transcription in S. cerevisiae through interaction with the histone deacetylase Rpd3p. J. Biol. Chem. 278:44727–44734. 10.1074/jbc.M308632200 [DOI] [PubMed] [Google Scholar]
- 24.Scherer S, Davis RW. 1979. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl. Acad. Sci. U. S. A. 76:4951–4955. 10.1073/pnas.76.10.4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.3.CO;2-U [DOI] [PubMed] [Google Scholar]
- 26.Cooper KF, Strich R. 2002. Saccharomyces cerevisiae C-type cyclin Ume3p/Srb11p is required for efficient induction and execution of meiotic development. Eukaryot. Cell 1:66–74. 10.1128/EC.01.1.66-74.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eberharter A, John S, Grant PA, Utley RT, Workman JL. 1998. Identification and analysis of yeast nucleosomal histone acetyltransferase complexes. Methods 15:315–321. 10.1006/meth.1998.0635 [DOI] [PubMed] [Google Scholar]
- 28.Arcangioli B, Lescure B. 1985. Identification of proteins involved in the regulation of yeast iso-1-cytochrome C expression by oxygen. EMBO. 4:2627–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229. 10.1006/meth.2001.1183 [DOI] [PubMed] [Google Scholar]
- 30.Rigaut G, Shevchenko A, Rurz B, Wilm M, Mann M, Seraphin B. 1999. A genetic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032. 10.1038/13732 [DOI] [PubMed] [Google Scholar]
- 31.Dominy JE, Jr, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. 2008. Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J. Biol. Chem. 283:12188–12201. 10.1074/jbc.M800044200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mallory M, Law M, Buckingham L, Strich R. 2010. The Sin3p PAH domains provide separate functions repressing meiotic gene transcription in yeast. Eukaryot. Cell 9:1835–1844. 10.1128/EC.00143-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marmorstein R, Carey M, Ptashne M, Harrison SC. 1992. DNA recognition by GAL4: structure of a protein-DNA complex. Nature 356:408–414. 10.1038/356408a0 [DOI] [PubMed] [Google Scholar]
- 34.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 12:2001–2014. 10.1110/ps.03154503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunbrack RL., Jr 2002. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 12:431–440. 10.1016/S0959-440X(02)00344-5 [DOI] [PubMed] [Google Scholar]
- 36.Dunbrack RL, Jr., Cohen FE. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6:1661–1681. 10.1002/pro.5560060807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 38.Meluh PB, Koshland D. 1997. Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev. 11:3401–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M, Luo J, Brooks CL, Gu W. 2002. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277:50607–50611. 10.1074/jbc.C200578200 [DOI] [PubMed] [Google Scholar]
- 40.De Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. 2004. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature 427:370–374. 10.1038/nature02258 [DOI] [PubMed] [Google Scholar]
- 41.Sabet N, Volo S, Yu C, Madigan JP, Morse RH. 2004. Genome-wide analysis of the relationship between transcriptional regulation by Rpd3p and the histone H3 and H4 amino termini in budding yeast. Mol. Cell. Biol. 24:8823–8833. 10.1128/MCB.24.20.8823-8833.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Hayes JJ. 2008. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol. Cell. Biol. 28:227–236. 10.1128/MCB.01245-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park H-D, Luche RM, Cooper TG. 1992. The yeast UME6 gene product is required for transcriptional repression mediated by the CAR1 URS1 repressor binding site. Nucleic Acids Res. 20:1909–1915. 10.1093/nar/20.8.1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson JC, Lopes JM. 1996. The yeast UME6 gene is required for both negative and positive transcriptional regulation of phospholipid biosynthetic gene expression. Nucleic Acids Res. 24:1322–1329. 10.1093/nar/24.7.1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Law DTS, Segall J. 1988. The SPS100 gene of Saccharomyces cerevisiae is activated late in the sporulation process and contributes to spore wall maturation. Mol. Cell. Biol. 8:912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garber AT, Segall J. 1986. The SPS4 gene of Saccharomyces cerevisiae encodes a major sporulation-specific mRNA. Mol. Cell. Biol. 6:4478–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ginsburg DS, Govind CK, Hinnebusch AG. 2009. NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5. Mol. Cell. Biol. 29:6473–6487. 10.1128/MCB.01033-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8:284–295. 10.1038/nrm2145 [DOI] [PubMed] [Google Scholar]
- 49.Zheng C, Hayes JJ. 2003. Structures and interactions of the core histone tail domains. Biopolymers 68:539–546. 10.1002/bip.10303 [DOI] [PubMed] [Google Scholar]
- 50.Kontaki H, Talianidis I. 2010. Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39:152–160. 10.1016/j.molcel.2010.06.006 [DOI] [PubMed] [Google Scholar]
- 51.Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, Herrera-Medina EM, Rauscher F, III, Reinberg D, Barlev NA. 2007. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol. 27:6756–6769. 10.1128/MCB.00460-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol. Mol. Biol. Rev. 70:583–604. 10.1128/MMBR.00015-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marmorstein R, Harrison SC. 1994. Crystal structure of a PPR1-DNA complex: DNA recognition by proteins containing a Zn2Cys6 binuclear cluster. Genes Dev. 8:2504–2512 [DOI] [PubMed] [Google Scholar]
- 54.Swaminathan K, Flynn P, Reece RJ, Marmorstein R. 1997. Crystal structure of a PUT3-DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat. Struct. Biol. 4:751–759. 10.1038/nsb0997-751 [DOI] [PubMed] [Google Scholar]
- 55.King DA, Zhang L, Guarente L, Marmorstein R. 1999. Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat. Struct. Biol. 6:64–71. 10.1038/4940 [DOI] [PubMed] [Google Scholar]
- 56.Baleja JD, Marmorstein R, Harrison SC, Wagner G. 1992. Solution structure of the DNA-binding domain of Cd2-GAL4 from S. cerevisiae. Nature 356:450–453. 10.1038/356450a0 [DOI] [PubMed] [Google Scholar]
- 57.Anderson SF, Steber CM, Esposito RE, Coleman JE. 1995. UME6, a negative regulator of meiosis in Saccharomyces cerevisiae, contains a C-terminal Zn2Cys6 binuclear cluster that binds the URS1 DNA sequence in a zinc-dependent manner. Protein Sci. 4:1832–1843. 10.1002/pro.5560040918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brent MM, Anand R, Marmorstein R. 2008. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 16:1407–1416. 10.1016/j.str.2008.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. 2005. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 102:11278–11283. 10.1073/pnas.0502738102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015. 10.1126/science.1094637 [DOI] [PubMed] [Google Scholar]
- 61.Kim JH, Saraf A, Florens L, Washburn M, Workman JL. 2010. Gcn5 regulates the dissociation of SWI/SNF from chromatin by acetylation of Swi2/Snf2. Genes Dev. 24:2766–2771. 10.1038/nrm2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jani NM, Lopes JM. 2008. Transcription regulation of the Saccharomyces cerevisiae PIS1 gene by inositol and the pleiotropic regulator, Ume6p. Mol. Microbiol. 70:1529–1539. 10.1111/j.1365-2958.2008.06506.x [DOI] [PubMed] [Google Scholar]
- 63.Messenguy F, Vierendeels F, Scherens B, Dubois E. 2000. In Saccharomyces cerevisiae, expression of arginine catabolic genes CAR1 and CAR2 in response to exogenous nitrogen availability is mediated by the Ume6 (CargRI)-Sin3 (CargRII)-Rpd3 (CargRIII) complex. J. Bacteriol. 182:3158–3164. 10.1128/JB.182.11.3158-3164.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams RM, Primig M, Washburn BK, Winzeler EA, Bellis M, Sarrauste de Menthiere C, Davis RW, Esposito RE. 2002. The Ume6 regulon coordinates metabolic and meiotic gene expression in yeast. Proc. Natl. Acad. Sci. U. S. A. 99:13431–13436. 10.1073/pnas.202495299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 66.Sikorski R, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.