

Abstract
We have previously reported that interleukin-1 (IL-1) receptor-associated kinase (IRAK1) is essential for Epstein-Barr virus (EBV) latent infection membrane protein 1 (LMP1)-induced p65/RelA serine 536 phosphorylation and NF-κB activation but not for IκB kinase α (IKKα) or IKKβ activation (Y. J. Song, K. Y. Jen, V. Soni, E. Kieff, and E. Cahir-McFarland, Proc. Natl. Acad. Sci. U. S. A. 103:2689–2694, 2006, doi:10.1073/pnas.0511096103). Since the kinase activity of IRAK1 is not required for LMP1-induced NF-κB activation, IRAK1 is proposed to function as a scaffold protein to recruit a p65/RelA serine 536 kinase(s) to enhance NF-κB-dependent transcriptional activity. We now report that Ca2+/calmodulin-dependent protein kinase II (CaMKII) interacts with IRAK1 and is critical for LMP1-induced p65/RelA serine 536 phosphorylation and NF-κB activation. CaMKII bound the death domain of IRAK1 and directly phosphorylated p65/RelA at serine 536 in vitro. Downregulation of CaMKII activity or expression significantly reduced LMP1-induced p65/RelA serine 536 phosphorylation and NF-κB activation. Furthermore, LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation were significantly reduced in IRAK1 knockout (KO) mouse embryonic fibroblasts (MEFs). Thus, IRAK1 may recruit and activate CaMKII, which phosphorylates p65/RelA serine 536 to enhance the transactivation potential of NF-κB in LMP1-induced NF-κB activation pathway.
INTRODUCTION
The Epstein-Barr virus (EBV) latent infection membrane protein 1 (LMP1) is an integral membrane protein essential for EBV-infected primary B lymphocyte transformation into proliferating lymphoblastoid cell lines (LCLs) (reviewed in reference 2). LMP1 has an N-terminal cytoplasmic domain (amino acids [aa] 1 to 24), six transmembrane domains (aa 25 to 186), and a C-terminal cytoplasmic signaling domain (aa 187 to 386). Using the transmembrane domains, LMP1 self-aggregates in plasma membrane lipid rafts and barges and constitutively activates NF-κB, p38 mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK) through two C-terminal cytoplasm signaling domains referred to as C-terminal activation region 1 (CTAR1) and CTAR2 (3–10). Among signal transduction pathways activated by LMP1, NF-κB is critical for EBV-transformed LCL survival (11, 12).
NF-κB is a family of transcription factors, including RelA (p65), RelB, c-Rel, p105/p50 (NF-κB1), and p100/p52 (NF-κB2), that form homo- or heterodimers to regulate the expression of genes involved in cell proliferation, differentiation, and apoptosis (reviewed in references 13 and 14). The key regulator of NF-κB activation is the IκB kinase (IKK) complex, which is composed of the catalytic (IKKα and IKKβ) and regulatory (IKKγ) subunits. NF-κB is activated by two distinct signal transduction pathways called the canonical and noncanonical (alternative) pathways (15). A canonical pathway for NF-κB activation involves the p65/p50 complexes that are retained in the cytoplasm by inhibitor of κB (IκB) proteins. Upon activation, IκB proteins are phosphorylated by IKKβ and degraded by the ubiquitin-proteasome pathway, allowing the nuclear translocation of the p65/p50 complexes. A noncanonical pathway for NF-κB activation involves NF-κB-inducing kinase (NIK)- and IKKα-mediated proteolytic processing of p100 into p52 and translocation of the RelB/p52 or p65/p52 complexes into the nucleus (13, 14).
LMP1 activates both the noncanonical and the canonical NF-κB pathways, through CTAR1 and CTAR2, respectively. CTAR1 recruits tumor necrosis factor receptor (TNFR)-associated factor 1 (TRAF1), TRAF3, TRAF2, and TRAF5, and CTAR2 recruits TNFR-associated death domain proteins TRADD, RIP1, and interferon regulatory factor 7 (IRF7) (5–10, 16–19). In association with cellular adaptor proteins, CTAR1 and CTAR2 constitutively activate the noncanonical and canonical NF-κB pathways, respectively.
Interleukin-1 (IL-1) receptor-associated kinase 1 (IRAK1) is a serine/threonine kinase involved in Toll-like receptor (TLR)/interleukin-1 receptor (TIR)-mediated NF-κB activation (20–30). In TIR signaling pathways, IRAK1 functions as a scaffold protein to recruit TRAF6 to MyD88 and to induce subsequent TRAF6 activation, which is critical for IKKβ activation (reviewed in references 20 and 21). However, the role of IRAK1 in TIR-mediated NF-κB activation is debated and still unclear. In IRAK1 knockout (KO) mice, lipopolysaccharide (LPS)-induced IKKβ activation is attenuated but still intact, although DNA binding activity of NF-κB is inhibited (30). In addition, IRAK1 is not essential for IL-1β-induced IKKβ activation but is critical for NF-κB-dependent promoter activation in IRAK1 knockdown HEK293 (I1A-293) cells (31). In addition to the cytoplasmic function of IRAK1, IRAK1 translocates into the nucleus and enhances transcriptional activity of NF-κB or signal transducers and activators of transcription 3 (STAT3) in response to IL-1β or LPS (31, 32).
IRAK1 is also critical for LMP1-induced NF-κB activation (1, 33). In I1A-293 cells, CTAR1- or CTAR2-induced NF-κB-dependent promoter activation is significantly downregulated (33). Reconstitution of I1A-293 cells with kinase-inactive IRAK1 (K239S) restores LMP1-induced NF-κB activation, indicating that the kinase activity of IRAK1 is not required (1). Interestingly, IRAK1 is not critical for IKKα or β activation but is essential for p65/RelA serine 536 phosphorylation by LMP1 (1). Since the kinase activity of IRAK1 is not required for LMP1-induced p65/RelA serine 536 phosphorylation and NF-κB activation, IRAK1 may function as a scaffold protein to recruit and activate a p65/RelA serine 536 kinase(s). Therefore, the present study was undertaken to investigate a cellular p65/RelA serine 536 kinase(s) that interacts with IRAK1 in an LMP1-induced NF-κB activation pathway.
MATERIALS AND METHODS
Cells.
IRAK1 wild-type (WT) and KO mouse embryonic fibroblasts (MEFs) were kindly provided by James Thomas (University of Texas Southwestern Medical Center). Burkitt's lymphoma BL41 cells and their counterparts in which LMP1 expression can be negatively controlled by doxycycline (Dox) were described previously (11). IRAK1 null I1A 293 cells (I1A IRAK1−/Y) were kindly provided by Xiaoxia Li (Cleveland Clinic Foundation). Maintenance and propagation of HEK293 cells, the EBV-transformed lymphoblastoid cell line IB4, and MEFs were previously described (19, 34).
Antibodies, reagents, transfections, and reporter gene assays.
Antibodies to p65/RelA, phospho-IκBα, phospho-p65/RelA at serine 536, phospho-CaMKII at threonine 286, phospho-IKKα (serine 176)/IKKβ (serine 177), poly(ADP-ribose) polymerase (PARP), IκBα, and CaMKII were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to IRAK1, IKKγ, CaMKIIγ, hemagglutinin (HA), and Myc were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to FLAG and p100/p52 were purchased from Agilent Technologies (Santa Clara, CA) and EMB Millipore (Billerica, MA), respectively. An antitubulin antibody was purchased from Sigma-Aldrich (St. Louis, MO). Enhanced chemiluminescence detection reagents (Pierce, Rockford, IL) and secondary peroxidase-labeled anti-mouse or anti-rabbit immunoglobulin G antibodies (Amersham Biosciences, Piscataway, NJ) were used according to the manufacturer's directions. To avoid the hindrance caused by immunoprecipitating immunoglobulin heavy and light chains, TrueBlot secondary peroxidase-labeled anti-mouse or anti-rabbit immunoglobulin G antibodies were used (Rockland Immunochemicals, Gilbetsville, PA). Recombinant human IL-1β was purchased from R&D Systems (Minneapolis, MN). CaMKII-specific inhibitor KN-93 and its inactive analogue, KN-92, were purchased from EMB Millipore. Effectene for transient transfection was used according to the manufacturer's directions (Qiagen, Valencia, CA). Luciferase assays were performed as described previously (35).
Plasmid constructs.
Plasmids pSG5-FLAG-LMP1 WT, aa 1 to 231 (CTAR1), and Δ187–351(CTAR2) were previously described (19). Wild-type and kinase-dead (K239S) IRAK1 expression vectors driven by the thymidine kinase promoter were provided by Xiaoxia Li (The Cleveland Clinic Foundation). The construct expressing a Myc-tagged, constitutively active CaMKIIγ (pCMV-myc-CaMKIIγ1–290) was kindly provided by Ramnik Xavier (Massachusetts General Hospital). Catalytically inactive CaMKIIγ mutants (K43M) were generated by using the QuikChange site-directed mutagenesis kit (Agilent Technologies) with the following primers: CaMKIIγ K43M, 5′-GAGTACGCAGCAATGATCATCAATACC-3′ and 5′-GGTATTGATGATCATTGCTGCGTACTC-3′. To generate a construct encoding HA-tagged IRAK1 WT or death domain (DD, aa 1 to 103), a cDNA fragment was amplified by PCR using an IRAK1 expression vector driven by the thymidine kinase promoter. The following primers were used for PCR: IRAK1 WT, 5′-CGGCTAGCGCCACCATGTACCCATACGATGTTCCAGATTACGCTATGGCCGGGGGGCCG-3′ and 5′-GGCTCGAGGCCGCCTCAGCTCTGAAATTCATCACTTTCTTCGGGCCCCTG-3′; IRAK1 DD, 5′-CGGCTAGCGCCACCATGTACCCATACGATGTTCCAGATTACGCTATGGCCGGGGGGCCG-3′ and 5′-GGCTCGAGGCCGCCTCAGTGCCAGGCTGTGATGATGT-3′.The PCR products were digested with NheI-XhoI (New England BioLabs, Beverly, MA) and ligated into the pCEP4 vector (Life Technologies, Carlsbad, CA). To generate a construct encoding GST-tagged IκBα (aa 1 to 54) or p65/RelA (aa 365 to 551), a cDNA fragment was amplified by PCR using the following primers: IκBα (1–54), 5′-GGGGGATCCATGTTCCAGGCGGC-3′ and 5′-AGCCTCGAGTTAGAGGCGGATCT-3′; p65/RelA (365–551), 5′-GGGGGATCCATGACCATGGTGTT-3′ and 5′-AGCCTCGAGTTAGGAGCTGATCT-3′. The PCR products were digested with BamHI and XhoI (New England BioLabs) and ligated into the pGEX-4T-1 vector (GE Healthcare, Pittsburgh, PA).
Tandem affinity purification and mass spectrometry.
One billion IB4 cells stably expressing pcDNA3-HA-FLAG-IRAK1 were lysed in buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.5 mM EDTA, 1% NP-40, phosphatase inhibitor cocktail (EMB Millipore), and protease inhibitor cocktail (Roche, Indianapolis, IN). Lysates were precleared with protein A/G-agarose beads (Santa Cruz) and incubated at 4°C overnight with anti-HA antibody-conjugated agarose beads (Santa Cruz). After washing three times with lysis buffer, protein complexes were eluted with HA peptides (Covance, Princeton, NJ). The eluates were then incubated at 4°C for 2 h with anti-FLAG antibody-conjugated agarose beads (Sigma-Aldrich), and protein complexes were eluted with FLAG peptides (Sigma-Aldrich) after washing three times with lysis buffer. Protein complexes were analyzed by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (SDS-PAGE) using NuPAGE SDS-PAGE gels (Life Technologies). After SYPRO ruby (Life Technologies) staining, protein bands were cut out and analyzed by nanospray liquid chromatography-mass spectrometry at the Partners Center for Genetics and Genomics at Harvard Medical School as previously described (36).
siRNA transfections and qRT-PCR.
Accell nontargeting control pool small interfering RNA (siRNA) and Accell SMARTpool Human CaMKIIγ-specific siRNA were used according to the manufacturer's directions (Thermo Scientific, Pittsburgh, PA). Total RNA was isolated using the RNeasy kit and reverse transcribed into cDNA using the QuantiTect reverse transcription kit according to the manufacturer's directions (Qiagen) for quantitative reverse transcription-PCR (qRT-PCR). cDNAs were amplified and quantified in MxPro3000P QPCR System (Agilent Technologies) using Hot FirePol EvaGreen qPCR Mix Plus (Solis BioDyne, Tartu, Estonia) and the following primers: CaMKIIγ, 5′-ATGGCCACCACCGCCA-3′ and 5′-ACTGTCATGGAGGCGCACGA-3′; β-actin, 5′-ATCATGTTTGAGACCTTCAAC-3′ and 5′-CAGGAAGGAAGGCTGGAAGAG-3′.
Subcellular fractionation.
A method to fractionate nuclear and cytoplasmic proteins was used as previously described (1).
Immunoprecipitation and in vitro kinase assay.
Ten million cells were lysed in buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.5 mM EDTA, 1% NP-40, phosphatase inhibitor cocktail (EMB Millipore), and protease inhibitor cocktail (Roche). Lysates were precleared with protein A/G-agarose beads (Santa Cruz) and incubated at 4°C overnight with anti-HA antibody-conjugated agarose beads (Santa Cruz). After washing three times with lysis buffer, protein complexes were eluted with HA (Covance) peptides and subjected to Western blot analysis with antibody to Myc or HA. For in vitro kinase assay, precleared cell lysates were incubated at 4°C for 2 h with either anti-IKKγ antibody (for IκBα phosphorylation assay) or anti-Myc antibody (for p65/RelA serine 536 phosphorylation assay) plus protein A/G-agarose beads (Santa Cruz). Immunoprecipitates were washed three times with the lysis buffer and twice with 1× kinase buffer (Cell Signaling Technology). Kinase assays were at 30°C for 30 min in the kinase buffer containing 2 μg of glutathione S-transferase (GST) (for a negative control), GST-IκBα1–54 (for IκBα phosphorylation assay), or GST-p65/RelA365–551 (for p65/RelA serine 536 phosphorylation assay) and 0.2 mM ATP. Reactions were stopped by addition of an equal volume of 2× SDS-PAGE loading buffer (100 mM Tris-HCl [pH 6.8], containing 4% SDS, 0.02% bromophenol blue, and 2% 2-mercaptoethanol) and subjected to Western blot analysis with antibody to phospho-IκBα, phospho-p65/RelA at serine 536, IKKγ, or Myc. The intensity of each band was measured by using the Quantitative Molecular Imaging system (GE Healthcare).
RESULTS
IRAK1 interacts with CaMKII through the death domain.
To determine an unknown p65/RelA serine 536 kinase(s) that interacts with IRAK1, tandem affinity purification combined with mass spectrometry (TAP-MS) was employed by using EBV-transformed LCLs stably expressing IRAK1 fused to an HA-FLAG-tag. The list of IRAK1-interacting proteins was narrowed down by searching for serine/threonine protein kinases involved in NF-κB activation, and the γ and δ isoforms of CaMKII (CaMKIIγ and CaMKIIδ) were identified (data not shown). Indeed, CaMKIIγ interacts with IRAK1 at the endogenous level in LCLs (Fig. 1A, lane 2).
FIG 1.
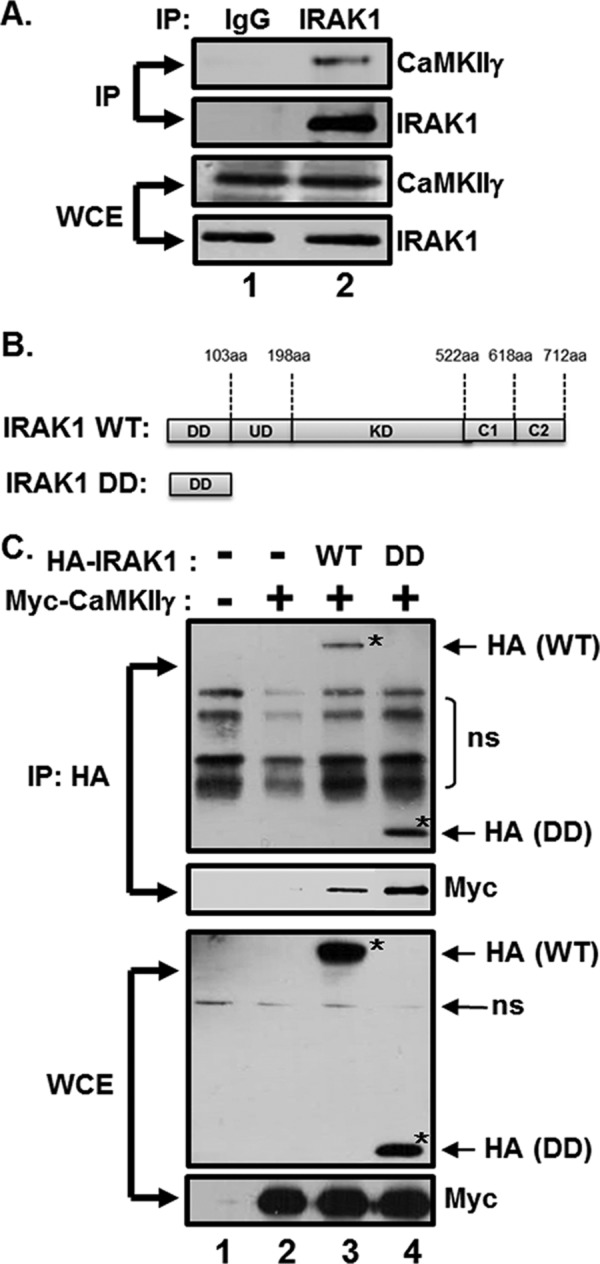
The death domain of IRAK1 interacts with CaMKII. (A) LCL lysates were immunoprecipitated with either normal IgG or anti-IRAK1 antibodies and analyzed by CaMKIIγ and IRAK1 Western blotting. (B) Schematic representation of IRAK1 WT and IRAK1 DD mutant. DD, death domain; UD, undetermined domain; KD, kinase domain; C1, C-terminal domain 1; C2, C-terminal domain 2. (C) HEK293 cells were cotransfected with pCEP4 (lanes 1 and 2), pCEP4-HA-IRAK1 WT (lane 3), or pCEP4-HA-IRAK1 DD (lane 4) plus pCMV-myc-CaMKIIγ1–290. HA-IRAK1 immunoprecipitates (IP) and whole-cell extracts (WCE) were analyzed by HA and Myc Western blotting. *, HA-IRAK1 WT or DD. Additional nonspecific (ns) bands were detected, possibly due to a nonspecific binding of antibodies generated by insufficient blocking and/or washing of the membrane.
To further analyze the interaction between IRAK1 and CaMKIIγ or CaMKIIδ, HEK293 cells were cotransfected with expression vectors for HA epitope-tagged IRAK1 WT or DD (Fig. 1B) and Myc-tagged constitutively active CaMKIIγ1–290 or CaMKIIδ1–290. The N-terminal residues 1 to 290 of CaMKII are highly conserved within the isoforms (37). Cell lysates were immunoprecipitated with anti-HA–agarose beads, and CaMKIIγ or CaMKIIδ binding was assessed by Western blotting (Fig. 1C and data not shown). IRAK1 WT immunoprecipitated with CaMKIIγ and, at low levels, with CaMKIIδ (Fig. 1C, lane 3, and data not shown). Since the death domain of IRAK1 is critical for interaction with signaling proteins, whether IRAK1 binds to CaMKIIγ and CaMKIIδ through the death domain was further examined. Interestingly, the death domain of IRAK immunoprecipitated at high levels with CaMKIIγ and CaMKIIδ (Fig. 1C, lane 4, and data not shown). In addition, the interaction between IRAK1 and CaMKIIγ, not CaMKIIδ, was slightly increased by LMP1 expression (data not shown). These data suggest that IRAK1 interacts with CaMKIIγ or CaMKIIδ through the death domain.
CaMKII activity is critical for LMP1-induced p65/RelA serine 536 phosphorylation.
To assess the role of CaMKII in LMP1-induced IKK activation and p65/RelA serine 536 phosphorylation, parental BL41 cells or their LMP1-expressing counterparts were treated with either KN93, a specific inhibitor of CaMKII, or KN-92, an inactive KN-93 analogue, and IKK activation and p65/RelA serine 536 phosphorylation were tested by in vitro kinase assay and Western blot analysis (Fig. 2). In cells treated with KN-92, LMP1 expression significantly induced CaMKII phosphorylation at threonine 286, which activates the catalytic domain of CaMKII, approximately 3-fold (Fig. 2A, compare lane 2 with lane 1). In addition, in cells treated with KN-92, LMP1 expression induced p65/RelA serine 536 phosphorylation 2-fold (Fig. 2A, compare lane 2 with lane 1), while LMP1-induced p65/RelA serine 536 phosphorylation was significantly reduced by 90% in cells treated with KN-93 (Fig. 2A, compare lane 4 with lane 2). Surprisingly, KN-93 treatment did not affect LMP1-induced phosphorylation of IKKα and IKKβ at serines 176 and 177, respectively (Fig. 2A, compare lane 4 with lane 2). Furthermore, KN-93 had no effect on LMP1-induced IKKα or IKKβ activation (Fig. 2B and C, compare lane 4 with lane 2). Similar to KN-92, dimethyl sulfoxide (DMSO) had no adverse effect on LMP1-induced IKK activation and p65/RelA serine 536 phosphorylation (data not shown). Consistent with the IRAK1 data, CaMKII is not required for LMP1-induced IKKα or IKKβ activation but is essential for p65/RelA serine 536 phosphorylation.
FIG 2.
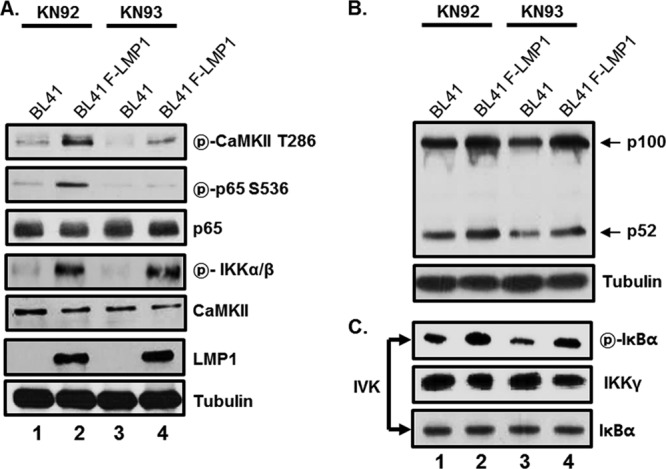
Effect of CaMKII-specific inhibitor KN93 on LMP1-induced IKK activation and p65/RelA serine 536 phosphorylation. BL41 cells and their FLAG-tagged LMP1-expressing counterparts (BL41-F-LMP1) were treated with either KN-93, a specific inhibitor of CaMKII (lanes 3 and 4), or KN-92, an inactive KN-93 analogue (lanes 1 and 2), at 10 μM for 18 h. (A and B) Equal amounts of cell extracts were subjected to Western blot analysis with antibody to phospho-CaMKII threonine 286, phospho-p65/RelA serine 536, p65/RelA, phospho-IKKα/β, CaMKII, LMP1, tubulin, or p100/p52. (C) Equal amounts of cell extracts were immunoprecipitated with anti-IKKγ antibody, and the in vitro IKKβ assay was performed as described in Materials and Methods. The reaction mixtures were then subjected to Western blot analysis with antibody to phospho-IκBα, IKKγ, or IκBα. IVK, in vitro kinase assay.
Both LMP1 CTAR1 and CTAR2 induce CaMKII activation and p65/RelA serine 536 phosphorylation.
Since LMP1 activates CaMKII in BL41 cells, the roles of the two LMP1 C-terminal signaling domains (CTAR1 and CTAR2) in CaMKII activation and p65/RelA serine 536 phosphorylation were assessed by using LMP1 mutants with CTAR1 or CTAR2 deletion (Fig. 3A). Both LMP1 CTAR1 and CTAR2 strongly induced CaMKII activation and p65/RelA serine 536 phosphorylation in mouse embryonic fibroblasts (MEFs) (Fig. 3B, compare lanes 2 to 4 with lane 1). CTAR1- or CTAR2-induced CaMKII activation and p65/RelA serine 536 phosphorylation were significantly downregulated by KN-93 treatment without affecting the protein levels of CaMKII, p65/RelA, or tubulin (Fig. 3B, compare lanes 6 to 8 with lanes 2 to 4). These data suggest that both CTAR1 and CTAR2 induce CaMKII activation and p65/RelA serine 536 phosphorylation.
FIG 3.
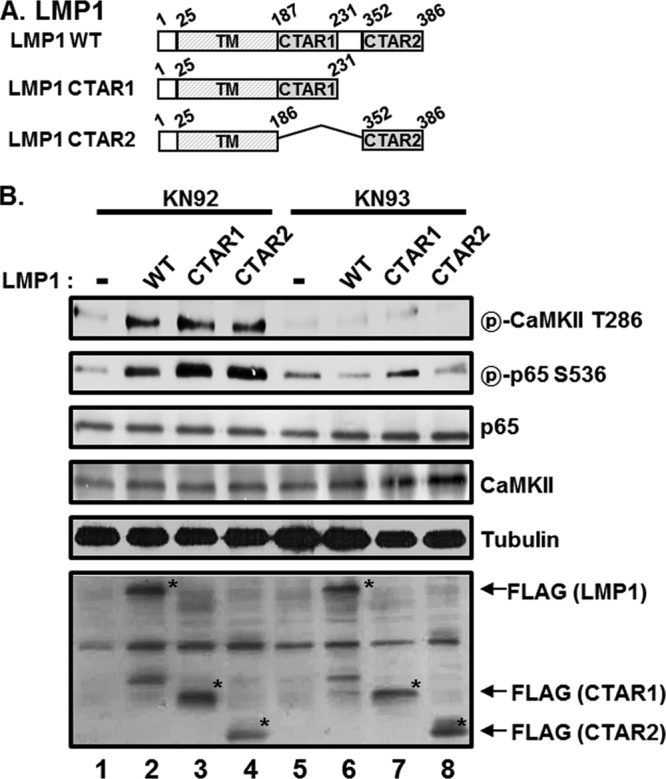
Both LMP1 CTAR1 and CTAR2 induce CaMKII activation and p65/RelA serine 536 phosphorylation. (A) Schematic representation of LMP1 WT, LMP1 1–231 (CTAR1), and LMP1 Δ187–351 (CTAR2). TM, transmembrane domain. (B) MEFs were transfected with pSG5 (lanes 1 and 5), pSG5-FLAG-LMP1 WT (lanes 2 and 6), pSG5-FLAG-LMP1 1–231 (lanes 3 and 7), or pSG5-FLAG-LMP1 Δ187–351 (lanes 4 and 8). After 12 h, cells were treated with either KN-92 (lanes 1 to 4) or KN-93 (lanes 5 to 8) at 10 μM for 18 h, and equal amounts of cell extracts were subjected to Western blot analysis with antibody to phospho-CaMKII threonine 286, phospho-p65/RelA serine 536, p65/RelA, CaMKII, tubulin, or FLAG. *, FLAG-LMP1 WT, CTAR1, or CTAR2. Additional nonspecific bands were detected, possibly due to a nonspecific binding of antibodies generated by insufficient blocking and/or washing of the membrane.
CaMKII phosphorylates p65/RelA at serine 536 in vitro.
Since CaMKII interacts with IRAK1 and is critical for LMP1-induced p65/RelA serine 536 phosphorylation, the possibility that CaMKII directly phosphorylates p65/RelA at serine 536 was assessed by using an in vitro kinase assay. HEK293 cells were transfected with either Myc-tagged constitutively active CaMKIIγ1–290 (WT) or its catalytically inactive counterpart (K43M), and cell lysates were immunoprecipitated with anti-myc antibody. The kinase reaction was performed on immunoprecipitated Myc-CaMKIIγ1–290 WT or K43M using GST-tagged p65/RelA365–551 as described in Materials and Methods. Phosphorylation of p65/RelA at serine 536 was determined by using Western blot analysis with antibody to phospho-p65/RelA serine 536 (Fig. 4). Interestingly, CaMKIIγ WT, but not K43M mutants, phosphorylated p65/RelA at serine 536 in vitro (Fig. 4, compare lanes 2 with lane 3). In addition to CaMKIIγ, CaMKIIδ also phosphorylated p65/RelA at serine 536 in vitro (data not shown). Thus, CaMKIIγ directly phosphorylates p65/RelA at serine 536 and may play important roles in LMP1-induced NF-κB activation.
FIG 4.
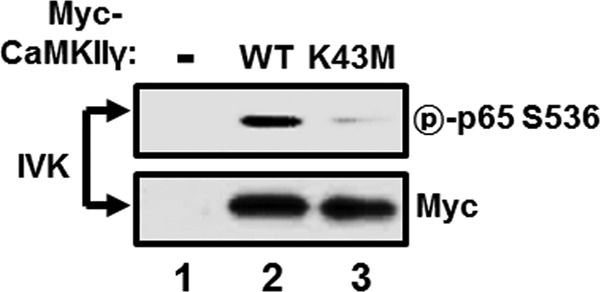
CaMKIIγ directly phosphorylates p65/RelA at serine 536 in vitro. HEK293 cells were transfected with pCMV (lane 1), pCMV-myc-CaMKIIγ1–290 (lane 2), or pCMV-myc-CaMKIIγ1–290 K43M (lane 3). After 24 h, equal amounts of cell extracts were immunoprecipitated with anti-myc antibody, and the in vitro p65/RelA serine 536 phosphorylation assay was performed as described in Materials and Methods. The reaction mixtures were then subjected to Western blot analysis with antibody to phospho-p65/RelA serine 536 or Myc.
CaMKII is critical for LMP1-induced NF-κB activation.
To determine the role of CaMKII in LMP1-induced NF-κB activation, HEK293 cells were cotransfected with the expression vector for LMP1 WT, CTAR1, or CTAR2 plus an NF-κB-dependent luciferase reporter and then treated with either KN-92 or KN-93. At 18 h of treatment, NF-κB-dependent luciferase activities were measured (Fig. 5A). KN-93 treatment significantly reduced LMP1 WT-, CTAR1-, or CTAR2-induced NF-κB activation by 49%, 41%, or 53%, respectively (Fig. 5A, lanes 2 to 4). Interestingly, KN-93 reduced both CTAR-1- and CTAR-2-induced NF-κB activation (Fig. 5A, lanes 3 and 4). CaMKII may regulate LMP1 CTAR1-induced NF-κB activation by affecting noncanonical p65/p52 complexes. Interestingly, in the absence of CTAR1, CTAR2 was more potent than LMP1 WT in inducing NF-κB activation (Fig. 5A and B, compare lane 4 with lane 2). In IKKα KO MEFs, LMP1-induced NF-κB activation is elevated (33). Thus, the CTAR1-induced noncanonical pathway for NF-κB activation may attenuate the CTAR2-induced canonical pathway for NF-κB activation.
FIG 5.

CaMKIIγ is critical for LMP1-induced NF-κB activation. (A) HEK293 cells were cotransfected with pSG5 (bars 1), pSG5-FLAG-LMP1 WT (bars 2), pSG5-FLAG-LMP1 1–231 (bars 3), or pSG5-FLAG-LMP1 Δ187–351 (bars 4) plus NF-κB-dependent firefly luciferase and control Renilla luciferase plasmids. Cells were then treated with either KN-92 or KN-93 at 10 μM for 18 h, and luciferase activity was measured using a dual-luciferase assay system. NF-κB-dependent luciferase activity was expressed in relative luciferase units (RLU) by normalizing firefly luciferase activity with constitutive Renilla luciferase activity. To calculate relative luciferase activity, LMP1-induced luciferase activity in the presence of KN-92 was set at 100%. Luciferase data shown here represent three independent experiments. (B) HEK293 cells were pretreated with either nonsilencing control siRNA or siRNA against CaMKIIγ and then cotransfected with pSG5 (bars 1), pSG5-FLAG-LMP1 WT (bars 2), pSG5-FLAG-LMP1 1–231 (bars 3), or pSG5-FLAG-LMP1 Δ187–351 (bars 4) plus NF-κB-dependent firefly luciferase and control Renilla luciferase plasmids. After 72 h, luciferase activity was measured as described above. (C) The mRNA levels for CaMKIIγ in cells treated with nonsilencing control siRNA or siRNA against CaMKIIγ were analyzed by qRT-PCR analysis. Significant differences between samples were determined by the P value of a two-sample t test (P < 0.05).
To further determine the role of the γ isoform of CaMKII in LMP1-induced NF-κB activation, HEK293 cells were pretreated with CaMKIIγ-specific siRNAs and then cotransfected with the expression vector for LMP1 WT, CTAR1, or CTAR2 plus an NF-κB dependent luciferase reporter. At 72 h after the siRNA treatment, NF-κB-dependent luciferase activities were measured (Fig. 5B). The treatment of CaMKIIγ-specific siRNAs significantly reduced the expression of CaMKIIγ by 54.27% (Fig. 5C, lane 2) and downregulated LMP1 WT-, CTAR1-, or CTAR2-induced NF-κB activation by 46%, 48%, or 54%, respectively (Fig. 5B, compare lanes 2 to 4). Residual NF-κB activities in cells treated with KN-93 or CaMKIIγ-specific siRNAs were possibly due to insufficient knockdown of CaMKII activity or expression, respectively (Fig. 5C). In addition, the IRAK1-independent (possibly also CaMKII-independent) LMP1-induced NF-κB activation pathway may contribute to the residual NF-κB activities (1). Taken together, these data indicate that CaMKII is critical for both LMP1 CTAR1- and CTAR2-induced NF-κB activation.
IRAK1 is essential for LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation.
Since CaMKII interacts with IRAK1 and is required for LMP1-induced NF-κB activation and p65/RelA serine 536 phosphorylation, the role of IRAK1 in LMP1-induced CaMKII activation was determined using IRAK1 KO MEFs. IRAK1 WT or KO MEFs were transfected with the expression vector for LMP1 WT, CTAR1, or CTAR2, and CaMKII activation and p65/RelA serine 536 phosphorylation were determined (Fig. 6). In IRAK1 WT MEFs, LMP1 WT, CTAR1, or CTAR2 induced CaMKII activation and p65/RelA serine 536 phosphorylation. However, LMP1 WT, CTAR1, or CTAR2 failed to induce CaMKII activation and p65/RelA serine 536 phosphorylation in IRAK1 KO MEFs. Although LMP1-induced p65/RelA serine 536 phosphorylation was significantly reduced in IRAK1 KO MEFs, LMP1 still induced p65/RelA serine 536 phosphorylation at low levels (Fig. 6, compare lanes 6 to 8 with lane 5). Another p65/RelA serine 536 kinase(s) may play a minor role in LMP1-induced p65/RelA serine 536 phosphorylation. Nonetheless, these data clearly indicate that IRAK1 is required for LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation.
FIG 6.
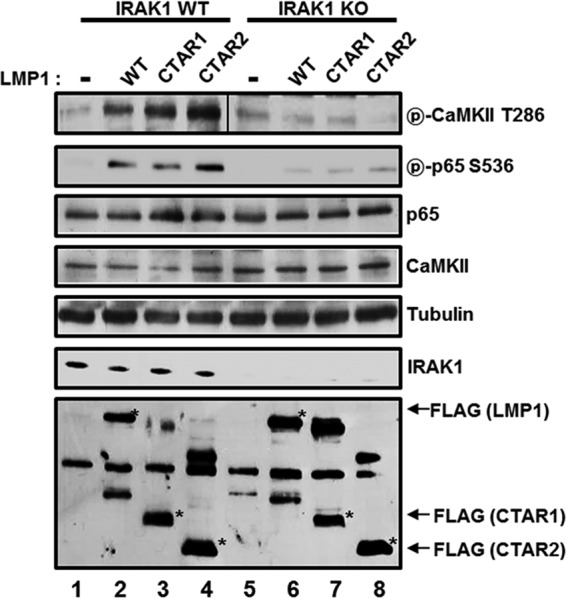
IRAK1 is required for LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation. IRAK1 WT (lanes 1 to 4) or KO (lanes 5 and 6) MEFs were transfected with pSG5 (lanes 1 and 5), pSG5-FLAG-LMP1 WT (lanes 2 and 6), pSG5-FLAG-LMP1 1–231 (lanes 3 and 7), or pSG5-FLAG-LMP1 Δ187–351 (lanes 4 and 8). After 48 h, equal amounts of cell extracts were subjected to Western blot analysis with antibody to phospho-CaMKII threonine 286, phospho-p65/RelA serine 536, p65/RelA, CaMKII, tubulin, IRAK1, or FLAG. *, FLAG-LMP1 WT, CTAR1, or CTAR2. Additional nonspecific bands were detected, possibly due to a nonspecific binding of antibodies generated by insufficient blocking and/or washing of the membrane.
To further determine whether IRAK1 kinase activity is required for LMP1-induced CaMKII activation, WT, IRAK1-null (I1A IRAK1−/Y), or I1A IRAK1−/Y 293 cells transfected with kinase-dead IRAK1 expression vectors (I1A IRAK1−/Y K239S) were transfected with the expression vector for LMP1, and LMP1-induced CaMKII phosphorylation at threonine 286 was assessed by Western blotting (Fig. 7). Consistent with IRAK1 KO MEFs, LMP1-induced CaMKII activation was strongly reduced in I1A IRAK1−/Y cells (Fig. 7, compare lane 2 with lane 6). By reconstituting I1A IRAK1−/Y cells with the kinase-dead IRAK1 (I1A IRAK1−/Y K239S), both the basal and LMP1-induced CaMKII activations were restored (Fig. 7, compare lanes 3 and 4 with lanes 5 and 6). Increased basal CaMKII activities in I1A IRAK1−/Y K239S cells were possibly due to overexpression of IRAK1 K239S (Fig. 7, lane 3). These data indicate that IRAK1 kinase activity is not required for LMP1-induced CaMKII activation.
FIG 7.
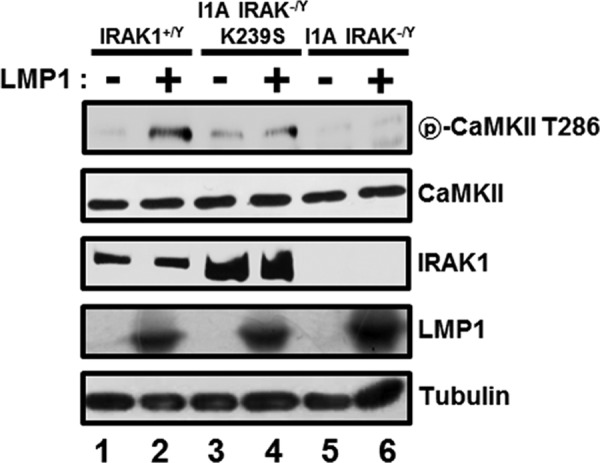
IRAK1 kinase activity is not required for LMP1-induced CaMKII activation. WT cells (lanes 1 and 2), IRAK1 null cells (I1A IRAK1−/Y) (lanes 5 and 6), or I1A IRAK1−/Y 293 cells transfected with kinase-dead IRAK1 expression vectors (I1A IRAK1−/Y K239S) (lanes 3 and 4) were transfected with the expression vector for LMP1. After 36 h, equal amounts of cell extracts were subjected to Western blot analysis with antibody to phospho-CaMKII threonine 286, CaMKII, IRAK1, LMP1, or tubulin.
LMP1 induced p65/RelA serine 536 phosphorylation in the cytoplasm.
To further determine whether p65/RelA serine 536 phosphorylation occurs in the cytoplasm or the nucleus, subcellular fractionation of parental BL41 cells or their LMP1-expressing counterparts was performed (Fig. 8). In LMP1-expressing BL41 cells, p65/RelA serine 536 phosphorylation occurred in the cytoplasm, and phosphorylated p65/RelA translocated into the nucleus (Fig. 8). Although IRAK1 was localized in both the cytoplasm and the nucleus (31, 32), the nuclear localization of IRAK1 was not induced by LMP1 expression in BL41 cells (Fig. 8, compare lanes 3 and 4 with lanes 1 and 2). T286-phosphorylated CaMKII was localized only in the cytoplasm as previously reported (Fig. 8, compare lanes 3 and 4 with lanes 1 and 2) (38). Interestingly, in BL41 cells, CaMKII proteins were localized more in the nucleus than in the cytoplasm (Fig. 8, compare lanes 3 and 4 with lanes 1 and 2). Taken together, both LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation occur in the cytoplasm. After phosphorylation at serine 536, p65/RelA translocates into the nucleus to induce NF-κB-dependent gene expression.
FIG 8.
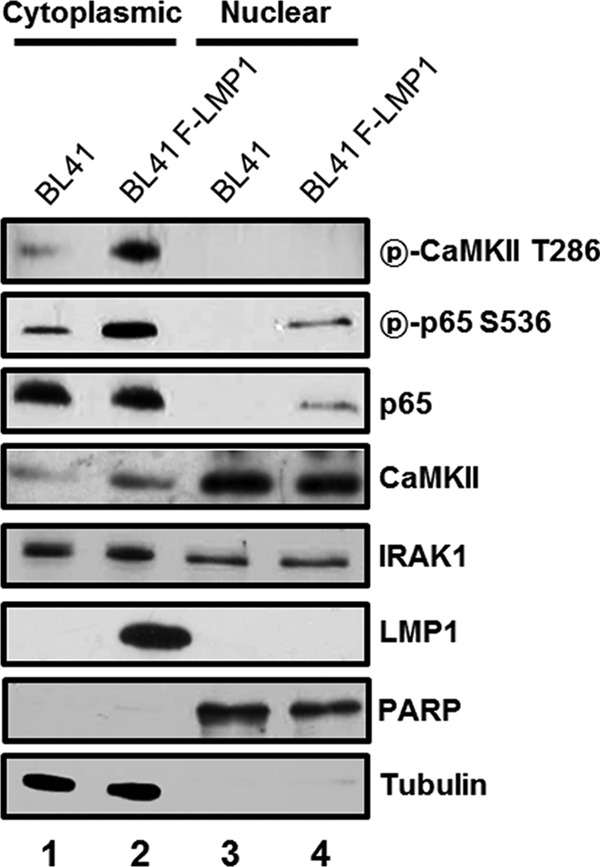
LMP1 induces p65/RelA serine 536 phosphorylation in the cytoplasm. Cytoplasmic (lanes 1 and 2) or nuclear (lanes 3 and 4) extracts of BL41 (lanes 1 and 3) or BL41-F-LMP1 (lanes 2 and 4) cells were subjected to Western blot analysis with antibody to phospho-CaMKII threonine 286, phospho-p65/RelA serine 536, p65/RelA, CaMKII, IRAK1, LMP1, PARP, or tubulin.
Both IRAK1 and CaMKII are not essential for IL-1β-induced IKKβ activation but are critical for p65/RelA serine 536 phosphorylation.
Although IRAK1 is required for IL-1β-induced NF-κB activation, it is dispensable for IKKβ activation in IRAK1 knockdown I1A 293 cells (31). To assess the role of IRAK1 in IL-1β-induced IKKβ activation and p65/RelA serine 536 phosphorylation, IRAK1 WT or KO MEFs were treated with IL-1β, and cell extracts were subjected to Western blot analyses for IκBα and phospho-p65/RelA serine 536 (Fig. 9A). In both IRAK1 WT and KO MEFs, IL-1β induced the degradation of IκBα, indicating that IRAK1 is not required for IL-1β-induced IKKβ activation (Fig. 9A, compare lanes 6 to 10 with lanes 1 to 5). However, IL-1β-induced p65/RelA serine 536 phosphorylation was significantly impaired in IRAK1 KO MEFs (Fig. 9A, compare lanes 6 to 10 with lanes 1 to 5). To further determine whether CaMKII is required for IL-1β-induced IKKβ activation and p65/RelA serine 536 phosphorylation, IRAK1 WT MEFs were pretreated with KN-93, and cell extracts were harvested for Western blot analysis after IL-1β stimulation (Fig. 9B). While KN-93 treatment had no effect on IL-1β-induced IKKβ activation, p65/RelA serine 536 phosphorylation was significantly attenuated in KN-93-treated MEFs (Fig. 9B, compare lanes 6 to 10 with lanes 1 to 5). Thus, both IRAK1 and CaMKII are not essential for IL-1β-induced IKKβ activation but are critical for p65/RelA serine 536 phosphorylation.
FIG 9.
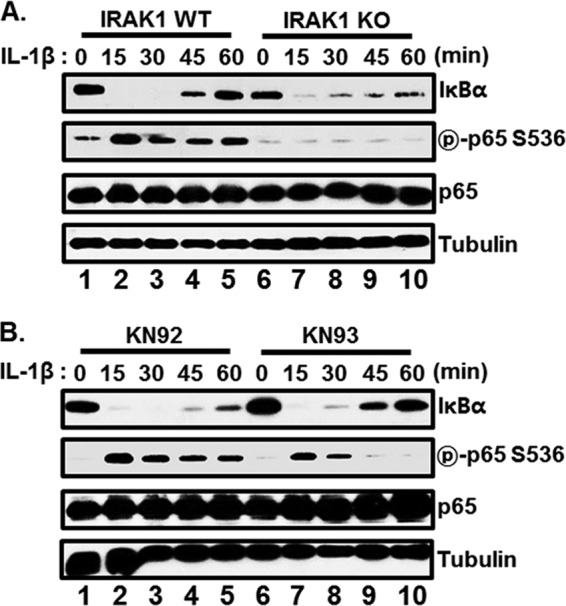
Both CaMKII and IRAK1 are critical for IL-1β-induced p65/RelA serine 536 phosphorylation. (A) IRAK1 WT (lanes 1 to 5) or KO (lanes 6 to 10) MEFs were treated with IL-1β, and cell extracts were harvested at 0, 15, 30, 45, and 60 min after IL-1β treatment. Equal amounts of cell extracts were subjected to Western blot analysis with antibody to IκBα, phospho-p65/RelA serine 536, p65/RelA, or tubulin. (B) MEFs were pretreated with either KN-92 (lanes 1 to 5) or KN-93 (lanes 6 to 10) at 10 μM for 3 h and stimulated with IL-1β. At 0, 15, 30, 45, and 60 min after IL-1β treatment, cell extracts were harvested and subjected to Western blot analysis with antibody to IκBα, phospho-p65/RelA serine 536, p65/RelA, or tubulin.
DISCUSSION
Phosphorylation of p65/RelA plays important roles in regulating NF-κB-dependent gene expression (reviewed in references 39, 40, and 41). p65/RelA contains multiple phosphorylation sites within and adjacent to the N-terminal Rel homology domain (RHD) and the C-terminal transactivation domain (TAD) (reviewed in references 39, 40, and 41). Among them, phosphorylation of serine 536 in the TAD of p65/RelA is critical for enhancing the transcriptional activity of NF-κB by recruitment of histone acetyltransferases and/or TATA-binding protein-associated factor II31 (42–46). In addition, p65/RelA phosphorylation at serine 536 specifies the NF-κB transcriptional response by inducing selective target gene expression (47).
We have previously reported that IRAK1 is essential for LMP1-induced p65/RelA serine 536 phosphorylation and NF-κB-dependent promoter activation (19, 33). Since the kinase activity of IRAK1 is not required for LMP1-induced NF-κB activation, IRAK1 may function as a scaffold protein to recruit p65/RelA serine 536 kinase(s) in the LMP1-induced NF-κB activation pathway (19). Thus, this study was initiated to determine a cellular serine/threonine protein kinase(s) that interacts with IRAK1 and phosphorylates p65/RelA at serine 536.
Using the TAP-MS analysis, CaMKIIγ and CaMKIIδ were identified as serine/threonine protein kinases that interact with IRAK1. CaMKII consists of four isoforms, α, β, γ, and δ, with numerous alternatively spliced variants (48). The α and β isoforms are predominantly expressed in neural tissues, and the γ and δ isoforms are expressed in most tissues. The CaMKII isoforms can homo- or hetero-oligomerize via their C-terminal domains to form a holoenzyme (48). Indeed, CaMKII is involved in various NF-κB activation pathways. Upon T-cell receptor activation, CaMKII regulates NF-κB activation by inducing CARMA1 or Bcl10 phosphorylation (49, 50). In addition, CaMKII is critical for Toll-like receptor 3 (TLR3)-, TLR4-, or TLR9-induced NF-κB and MAPK activation (51). Consistent with the TAP-MS data, IRAK1 interacts with CaMKIIγ or CaMKIIδ through the death domain in HEK293 cells.
Interestingly, LMP1 expression activates CaMKII (Fig. 2A and 3B). Dellis et al. reported that LMP1 increases calcium influx in B lymphocytes (52). Since KN-93, which blocks calcium-dependent calmodulin binding to CaMKII, diminished LMP1-induced CaMKII activation (Fig. 2A and 3B), LMP1 may activate CaMKII by modulating calcium influx.
In IRAK1 knockdown I1A 293 cells, both CTAR1- and CTAR2-induced NF-κB-dependent promoter activation is significantly reduced (33). In LMP1-induced NF-κB activation, IRAK1 is not required for IKKα or IKKβ activation but is essential for p65/RelA serine 536 phosphorylation (1). Consistent with these results, both CTAR1 and CTAR2 induced p65/RelA serine 536 phosphorylation and activated CaMKII (Fig. 3B). CaMKII directly phosphorylated p65/RelA at serine 536 (Fig. 4), and its activity was required for LMP1-induced p65/RelA serine 536 phosphorylation and NF-κB activation without affecting IKKα or IKKβ activity (Fig. 2, 3B, and 5). Furthermore, LMP1-induced CaMKII activation and p65/RelA serine 536 phosphorylation were significantly downregulated in IRAK1 KO MEFs (Fig. 6). Consistent with p65/RelA serine 536 phosphorylation, IRAK1 kinase activity is not required for LMP1-induced CaMKII activation (Fig. 7) (1). LMP1 may induce p65/RelA serine 536 phosphorylation in the cytoplasm because phosphorylated p65 was detected in the cytoplasm as well as in the nucleus (Fig. 8). Although CaMKII is reported to shuttle between the cytoplasm and the nucleus (53–57), more CaMKII was detected in the nucleus than in the cytoplasm in BL41 cells (Fig. 8).
In IRAK1 knockdown I1A-293 cells, IL-1β-induced IKKβ activation is attenuated but still intact, although NF-κB-dependent gene expression is significantly reduced (31). Consistent with these results, IL-1β-induced IKKβ activation was not inhibited in IRAK1 KO MEFs (Fig. 9). Interestingly, IL-1β-induced p65/RelA serine 536 phosphorylation was significantly reduced in IRAK1 KO MEFs (Fig. 9). In addition, CaMKII inhibition significantly inhibited IL-1β-induced p65/RelA serine 536 phosphorylation without affecting IKKβ activation (Fig. 9). Thus, IRAK1 and CaMKII interaction may also be involved in IL-1β-induced p65/RelA serine 536 phosphorylation and NF-κB activation.
Using p65/RelA KO MEFs stably expressing p65/RelA WT or S536A mutant, Moreno et al. reported that p65/RelA serine 536 phosphorylation is critical for the expression of Saa3, Mmp3, and Mmp13 genes in response to TNF-α (47). However, LMP1 had almost no effect on the expression of Saa3, Mmp3, and Mmp13 genes in MEFs (data not shown). Thus, LMP1 may induce different sets of genes that are dependent on p65/RelA serine 536 phosphorylation. Future studies are warranted to determine p65/RelA serine 536 phosphorylation-dependent genes induced by LMP1 and the role of CaMKII in regulating the expression of these genes. Since IRAK1 is not required for TNF-α but is required for LMP1 and IL-1β, CaMKII may be differently utilized by various signaling pathways.
CaMKII is also involved in type I interferon (IFN) production by directly binding and phosphorylating transforming growth factor β-activated kinase 1 (TAK1) and interferon regulatory factor 3 (IRF3) (51). Since IRAK1 is involved in TLR7- or TLR9-mediated induction of type I IFN (58), the interaction between IRAK1 and CaMKII may play an important role(s) in type I IFN signaling pathway.
This study suggests that, in the LMP1-induced NF-κB activation pathway, IRAK1interacts with CaMKII and mediates CaMKII activation. Activated CaMKII phosphorylates p65/RelA at serine 536 in the cytoplasm, and CTAR1-mediated noncanonical p65/p52 complexes or CTAR2-mediated canonical p65/p50 complexes, in turn, translocate into the nucleus to activate NF-κB-dependent promoters (Fig. 10). How LMP1 utilizes IRAK1 to activate CaMKII is unclear and is the subject of future studies.
FIG 10.
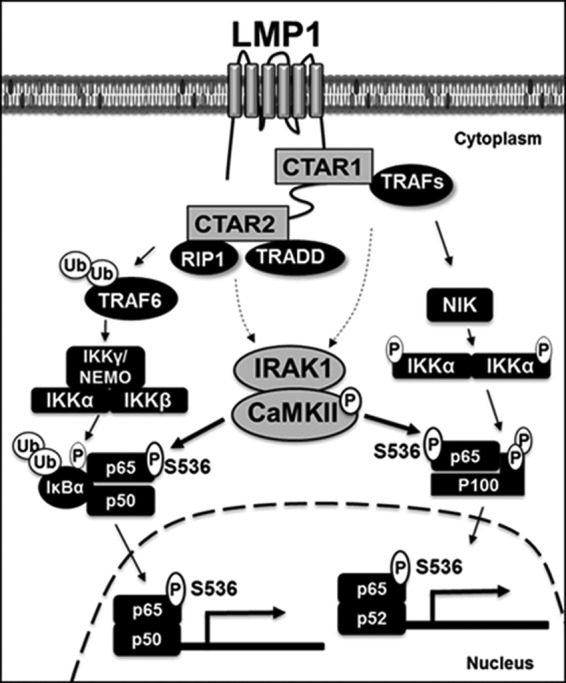
Model for the functional interaction between IRAK1 and CaMKII in LMP1-induced NF-κB activation. LMP1 CTAR1 activates NIK- and IKKα-mediated proteolytic processing of p100 to produce p52 and nuclear translocation of the p65/p52 complexes. LMP1 CTAR2 activates IKKβ-mediated phosphorylation and degradation of IκBα and nuclear localization of the p65/p50 complexes. IRAK1 is not required for LMP1-induced IKK activation. However, IRAK1 interacts with CaMKII and is critical for LMP1-induced CaMKII activation, p65/RelA serine 536 phosphorylation, and subsequent p65/p50- or p65/p52-mediated transactivation. CaMKII directly phosphorylates p65/RelA at serine 536 in the cytoplasm and is required for LMP1-induced NF-κB activation. Ub, ubiquitin.
ACKNOWLEDGMENTS
We thank members of the laboratory for helpful discussion and technical support.
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) (no. 2010-0003301). This work was also supported by a National Institutes of Health Public Health Service grant (CA085180).
Footnotes
Published ahead of print 18 November 2013
REFERENCES
- 1.Song YJ, Jen KY, Soni V, Kieff E, Cahir-McFarland E. 2006. IL-1 receptor-associated kinase 1 is critical for latent membrane protein 1-induced p65/RelA serine 536 phosphorylation and NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 103:2689–2694. 10.1073/pnas.0511096103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kieff ED, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2655 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams, and Wilkins, Philadelphia, PA [Google Scholar]
- 3.Mitchell T, Sugden B. 1995. Stimulation of NF-kappa B-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J. Virol. 69:2968–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huen DS, Henderson SA, Croom-Carter D, Rowe M. 1995. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10:549–560 [PubMed] [Google Scholar]
- 5.Devergne O, Cahir McFarland ED, Mosialos G, Izumi KM, Ware CF, Kieff E. 1998. Role of the TRAF binding site and NF-kappaB activation in Epstein-Barr virus latent membrane protein 1-induced cell gene expression. J. Virol. 72:7900–7908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, Kieff E, Mosialos G. 1996. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-kappaB activation. Mol. Cell. Biol. 16:7098–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. 1995. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80:389–399. 10.1016/0092-8674(95)90489-1 [DOI] [PubMed] [Google Scholar]
- 8.Kaye KM, Devergne O, Harada JN, Izumi KM, Yalamanchili R, Kieff E, Mosialos G. 1996. Tumor necrosis factor receptor associated factor 2 is a mediator of NF-kappa B activation by latent infection membrane protein 1, the Epstein-Barr virus transforming protein. Proc. Natl. Acad. Sci. U. S. A. 93:11085–11090. 10.1073/pnas.93.20.11085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaye KM, Izumi KM, Mosialos G, Kieff E. 1995. The Epstein-Barr virus LMP1 cytoplasmic carboxy terminus is essential for B-lymphocyte transformation; fibroblast cocultivation complements a critical function within the terminal 155 residues. J. Virol. 69:675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izumi KM, Cahir McFarland ED, Riley EA, Rizzo D, Chen Y, Kieff E. 1999. The residues between the two transformation effector sites of Epstein-Barr virus latent membrane protein 1 are not critical for B-lymphocyte growth transformation. J. Virol. 73:9908–9916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. 2004. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J. Virol. 78:4108–4119. 10.1128/JVI.78.8.4108-4119.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. 2000. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. U. S. A. 97:6055–6060. 10.1073/pnas.100119497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S, Karin M. 2002. Missing pieces in the NF-kappaB puzzle. Cell 109(Suppl):S81–S96. 10.1016/S0092-8674(02)00703-1 [DOI] [PubMed] [Google Scholar]
- 14.Hayden MS, Ghosh S. 2004. Signaling to NF-kappaB. Genes Dev. 18:2195–2224. 10.1101/gad.1228704 [DOI] [PubMed] [Google Scholar]
- 15.Bonizzi G, Karin M. 2004. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25:280–288. 10.1016/j.it.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 16.Izumi KM, Cahir McFarland ED, Ting AT, Riley EA, Seed B, Kieff ED. 1999. The Epstein-Barr virus oncoprotein latent membrane protein 1 engages the tumor necrosis factor receptor-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require RIP for NF-kappaB activation. Mol. Cell. Biol. 19:5759–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izumi KM, Kaye KM, Kieff ED. 1997. The Epstein-Barr virus LMP1 amino acid sequence that engages tumor necrosis factor receptor associated factors is critical for primary B lymphocyte growth transformation. Proc. Natl. Acad. Sci. U. S. A. 94:1447–1452. 10.1073/pnas.94.4.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izumi KM, Kieff ED. 1997. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. U. S. A. 94:12592–12597. 10.1073/pnas.94.23.12592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song YJ, Izumi KM, Shinners NP, Gewurz BE, Kieff E. 2008. IRF7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc. Natl. Acad. Sci. U. S. A. 105:18448–18453. 10.1073/pnas.0809933105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottipati S, Rao NL, Fung-Leung WP. 2008. IRAK1: a critical signaling mediator of innate immunity. Cell. Signal. 20:269–276. 10.1016/j.cellsig.2007.08.009 [DOI] [PubMed] [Google Scholar]
- 21.Janssens S, Beyaert R. 2003. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell 11:293–302. 10.1016/S1097-2765(03)00053-4 [DOI] [PubMed] [Google Scholar]
- 22.Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. 2002. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol. Cell. Biol. 22:7158–7167. 10.1128/MCB.22.20.7158-7167.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Handoyo H, Stafford MJ, McManus E, Baltzis D, Peggie M, Cohen P. 2009. IRAK1-independent pathways required for the interleukin-1-stimulated activation of the Tpl2 catalytic subunit and its dissociation from ABIN2. Biochem. J. 424:109–118. 10.1042/BJ20091271 [DOI] [PubMed] [Google Scholar]
- 24.Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. 2008. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9:684–691. 10.1038/ni.1606 [DOI] [PubMed] [Google Scholar]
- 25.Kim TW, Yu M, Zhou H, Cui W, Wang J, DiCorleto P, Fox P, Xiao H, Li X. 2012. Pellino 2 is critical for Toll-like receptor/interleukin-1 receptor (TLR/IL-1R)-mediated post-transcriptional control. J. Biol. Chem. 287:25686–25695. 10.1074/jbc.M112.352625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim YI, Park JE, Martinez-Hernandez A, Yi AK. 2008. CpG DNA prevents liver injury and shock-mediated death by modulating expression of interleukin-1 receptor-associated kinases. J. Biol. Chem. 283:15258–15270. 10.1074/jbc.M709549200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas JA, Haudek SB, Koroglu T, Tsen MF, Bryant DD, White DJ, Kusewitt DF, Horton JW, Giroir BP. 2003. IRAK1 deletion disrupts cardiac Toll/IL-1 signaling and protects against contractile dysfunction. Am. J. Physiol. Heart Circ. Physiol. 285:H597–H606. 10.1152/ajpheart.0655.2001 [DOI] [PubMed] [Google Scholar]
- 28.Windheim M, Stafford M, Peggie M, Cohen P. 2008. Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IkappaBalpha kinase. Mol. Cell. Biol. 28:1783–1791. 10.1128/MCB.02380-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. 2008. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol. Cell. Biol. 28:3538–3547. 10.1128/MCB.02098-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swantek JL, Tsen MF, Cobb MH, Thomas JA. 2000. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164:4301–4306 http://www.jimmunol.org/content/164/8/4301 [DOI] [PubMed] [Google Scholar]
- 31.Liu G, Park YJ, Abraham E. 2008. Interleukin-1 receptor-associated kinase (IRAK)-1-mediated NF-kappaB activation requires cytosolic and nuclear activity. FASEB J. 22:2285–2296. 10.1096/fj.07-101816 [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Li T, Sane DC, Li L. 2004. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J. Biol. Chem. 279:51697–51703. 10.1074/jbc.M410369200 [DOI] [PubMed] [Google Scholar]
- 33.Luftig M, Prinarakis E, Yasui T, Tsichritzis T, Cahir-McFarland E, Inoue J, Nakano H, Mak TW, Yeh WC, Li X, Akira S, Suzuki N, Suzuki S, Mosialos G, Kieff E. 2003. Epstein-Barr virus latent membrane protein 1 activation of NF-kappaB through IRAK1 and TRAF6. Proc. Natl. Acad. Sci. U. S. A. 100:15595–15600. 10.1073/pnas.2136756100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JE, Jun S, Song M, Kim JH, Song YJ. 2012. The extract of Chrysanthemum indicum Linne inhibits EBV LMP1-induced NF-kappaB activation and the viability of EBV-transformed lymphoblastoid cell lines. Food Chem. Toxicol. 50:1524–1528. 10.1016/j.fct.2012.02.034 [DOI] [PubMed] [Google Scholar]
- 35.Bari W, Song YJ, Yoon SS. 2011. Suppressed induction of proinflammatory cytokines by a unique metabolite produced by Vibrio cholerae O1 El Tor biotype in cultured host cells. Infect. Immun. 79:3149–3158. 10.1128/IAI.01237-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, Shi Y, Gill G. 2006. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc. Natl. Acad. Sci. U. S. A. 103:5308–5313. 10.1073/pnas.0601066103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tombes RM, Faison MO, Turbeville JM. 2003. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene 322:17–31. 10.1016/j.gene.2003.08.023 [DOI] [PubMed] [Google Scholar]
- 38.Heist EK, Srinivasan M, Schulman H. 1998. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J. Biol. Chem. 273:19763–19771. 10.1074/jbc.273.31.19763 [DOI] [PubMed] [Google Scholar]
- 39.Chen LF, Greene WC. 2004. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5:392–401. 10.1038/nrm1368 [DOI] [PubMed] [Google Scholar]
- 40.Smale ST. 2011. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 12:689–694. 10.1038/ni.2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Viatour P, Merville MP, Bours V, Chariot A. 2005. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 30:43–52. 10.1016/j.tibs.2004.11.009 [DOI] [PubMed] [Google Scholar]
- 42.Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. 2004. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 279:55633–55643. 10.1074/jbc.M409825200 [DOI] [PubMed] [Google Scholar]
- 43.Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. 2003. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 23:7780–7793. 10.1128/MCB.23.21.7780-7793.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr 2001. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 276:18934–18940. 10.1074/jbc.M101103200 [DOI] [PubMed] [Google Scholar]
- 45.Sizemore N, Leung S, Stark GR. 1999. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol. Cell. Biol. 19:4798–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. 2003. The NF-kappa B activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J. Biol. Chem. 278:919–926. 10.1074/jbc.M208696200 [DOI] [PubMed] [Google Scholar]
- 47.Moreno R, Sobotzik JM, Schultz C, Schmitz ML. 2010. Specification of the NF-kappaB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res. 38:6029–6044. 10.1093/nar/gkq439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braun AP, Schulman H. 1995. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 57:417–445. 10.1146/annurev.ph.57.030195.002221 [DOI] [PubMed] [Google Scholar]
- 49.Ishiguro K, Ando T, Goto H, Xavier R. 2007. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. Mol. Immunol. 44:2095–2100. 10.1016/j.molimm.2006.09.012 [DOI] [PubMed] [Google Scholar]
- 50.Ishiguro K, Green T, Rapley J, Wachtel H, Giallourakis C, Landry A, Cao Z, Lu N, Takafumi A, Goto H, Daly MJ, Xavier RJ. 2006. Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-kappaB activation. Mol. Cell. Biol. 26:5497–5508. 10.1128/MCB.02469-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X. 2008. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112:4961–4970. 10.1182/blood-2008-03-144022 [DOI] [PubMed] [Google Scholar]
- 52.Dellis O, Arbabian A, Papp B, Rowe M, Joab I, Chomienne C. 2011. Epstein-Barr virus latent membrane protein 1 increases calcium influx through store-operated channels in B lymphoid cells. J. Biol. Chem. 286:18583–18592. 10.1074/jbc.M111.222257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caran N, Johnson LD, Jenkins KJ, Tombes RM. 2001. Cytosolic targeting domains of gamma and delta calmodulin-dependent protein kinase II. J. Biol. Chem. 276:42514–42519. 10.1074/jbc.M103013200 [DOI] [PubMed] [Google Scholar]
- 54.Donai H, Murakami T, Amano T, Sogawa Y, Yamauchi T. 2000. Induction and alternative splicing of delta isoform of Ca(2+)/calmodulin-dependent protein kinase II during neural differentiation of P19 embryonal carcinoma cells and during brain development. Brain Res. Mol. Brain Res. 85:189–199. 10.1016/S0169-328X(00)00221-7 [DOI] [PubMed] [Google Scholar]
- 55.Donai H, Nakamura M, Sogawa Y, Wang JK, Urushihara M, Yamauchi T. 2000. Involvement of Ca2+/calmodulin-dependent protein kinase II in neurite outgrowth induced by cAMP treatment and serum deprivation in a central nervous system cell line, CAD derived from rat brain. Neurosci. Lett. 293:111–114. 10.1016/S0304-3940(00)01500-7 [DOI] [PubMed] [Google Scholar]
- 56.Johnson LD, Willoughby CA, Burke SH, Paik DS, Jenkins KJ, Tombes RM. 2000. delta Ca(2+)/calmodulin-dependent protein kinase II isozyme-specific induction of neurite outgrowth in P19 embryonal carcinoma cells. J. Neurochem. 75:2380–2391. 10.1046/j.1471-4159.2000.0752380.x [DOI] [PubMed] [Google Scholar]
- 57.Kutcher LW, Beauman SR, Gruenstein EI, Kaetzel MA, Dedman JR. 2003. Nuclear CaMKII inhibits neuronal differentiation of PC12 cells without affecting MAPK or CREB activation. Am. J. Physiol. Cell Physiol. 284:C1334–C1345. 10.1152/ajpcell.00510.2002 [DOI] [PubMed] [Google Scholar]
- 58.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. 2005. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 201:915–923. 10.1084/jem.20042372 [DOI] [PMC free article] [PubMed] [Google Scholar]