

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is etiologically associated with Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL). KS lesions are characterized by endothelial cells with multiple copies of the latent KSHV episomal genome, lytic replication in a low percentage of infiltrating monocytes, and inflammatory cytokines plus growth factors. We demonstrated that KSHV utilizes inflammatory cyclooxygenase 2/prostaglandin E2 to establish and maintain latency (Sharma-Walia, N., A. G. Paul, V. Bottero, S. Sadagopan, M. V. Veettil, N. Kerur, and B. Chandran, PLoS Pathog 6:e1000777, 2010 [doi:10.1371/journal.ppat.1000777]). Here, we evaluated the role of 5-lipoxygenase (5LO) and its chemotactic metabolite leukotriene B4 (LTB4) in KSHV biology. Abundant staining of 5LO was detected in human KS tissue sections. We observed elevated levels of 5LO and high levels of secretion of LTB4 during primary KSHV infection of endothelial cells and in PEL B cells (BCBL-1 and BC-3 cells). Blocking the 5LO/LTB4 cascade inhibited viral latent ORF73, immunomodulatory K5, viral macrophage inflammatory protein 1 (MIP-1), and viral MIP-2 gene expression, without much effect on lytic switch ORF50, immediate early lytic K8, and viral interferon-regulatory factor 2 gene expression. 5LO inhibition significantly downregulated latent viral Cyclin and latency-associated nuclear antigen 2 levels in PEL cells. 5LO/LTB4 inhibition downregulated TH2-related cytokine secretion, elevated TH1-related cytokine secretion, and reduced human monocyte recruitment, adhesion, and transendothelial migration. 5LO/LTB4 inhibition reduced fatty acid synthase (FASN) promoter activity and its expression. Since FASN, a key enzyme required in lipogenesis, is important in KSHV latency, these findings collectively suggest that 5LO/LTB4 play important roles in KSHV biology and that effective inhibition of the 5LO/LTB4 pathway could potentially be used in treatment to control KS/PEL.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) is etiologically associated with KS, primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). KS is a highly disseminated enigmatic angiogenic tumor of proliferative endothelial cells (ECs) and resembles chronic inflammation (1–5). KS is responsible for significant morbidity and mortality in HIV-infected patients in the developing world (1, 2, 4). KS lesions are histologically complex and are characterized by proliferating spindle-shaped ECs, neovascular structures, leukocyte infiltrate (monocytes, lymphocytes, and mast cells), and an abundance of inflammatory cytokines (ICs), growth factors, angiogenic factors, and invasive factors. KSHV-associated PEL is an aggressive form of non-Hodgkin's B cell lymphoma (NHL) that accounts for 4% of all AIDS-associated NHLs, and patients with PEL have a poor prognosis and a median survival of approximately 6 months (6, 7). Critical components of the pathogenesis of KS, PEL, and MCD are a persistent KSHV genome, deregulated secretion of autocrine/paracrine cytokines and chemokines, an aggressive neoangiogenic inflammatory network, and a subverted host immune response.
During latency, KSHV expresses a battery of genes, such as ORF73 (latency-associated nuclear antigen 1 [LANA-1]), ORF72 (viral Cyclin [vCyclin]), ORF71 (K13/vFLIP), and ORFK12 (kaposins A, B and C), as well as 12 distinct microRNAs, to facilitate the establishment of lifelong latency in its host and survival against the host intrinsic, innate, and adaptive immune surveillance mechanisms (8–10). KSHV encodes >86 open reading frames (ORFs), of which at least 22 are potentially immunomodulatory (K3 [modulator of immune recognition 1 {MIR-1}], K5 [MIR-2], K4 [viral macrophage-inflammatory protein II], K6 [viral macrophage inflammatory protein 1 {vMIP-1}], K9 [viral interferon-regulatory factor {vIRF}], K11.1 [vIRF2]) and antiapoptotic (K7, viral Bcl-2) (11, 12), regulate cytokine secretion levels, antagonize host interferon (IFN)-mediated antiviral responses, and regulate immune evasion. Host immune responses against KSHV control viral replication and viral spread and exert selective pressure on the virus to establish a latent state which allows the virus to evade the subsequent wave of adaptive host immune responses following an effective innate immune response.
KSHV has been shown to hijack cellular signaling pathways, transcription factors, and cytokines and secrete the arachidonic acid (AA) pathway's lipid metabolite prostaglandin E2 (PGE2) for its own advantage, especially to remain latent in the host cell (13–20). Here, we demonstrate that, apart from induction of cyclooxygenase 2 (COX-2)/PGE2 of the AA pathway, KSHV infection also induces components of the lipoxygenase pathway, such as 5-lipoxygenase (5LO; arachidonate:oxygen 5-oxidoreductase [EC 1.13.11.34]) and leukotriene (LT) A4 hydrolase (LTA4H), and infected cells secrete a highly potent chemotactic lipid mediator of the 5LO pathway called leukotriene B4 (LTB4).
LTB4, the first LT discovered, is produced by enzymatically catalyzed serial reactions. Therefore, LTB4 activity is much faster and more potent than that of the peptide chemokines, which require transcription and translation. LTB4 is a pivotal mediator of host defenses that brings the novel paradigm of lipid-cytokine-chemokine cascades in orchestrating the recruitment of immune cells responding at the initial stage of infection. The initial responding immune cells guided by LTB4 produce cytokines locally, which in turn induce the local release of chemokines, which then markedly amplify subsequent waves of leukocyte recruitment.
LTB4 mediates its actions by binding to two heterotrimeric G-protein-coupled seven-transmembrane receptors (GPCRs), LTB4R1 (BLT1R; a high-affinity receptor) and LTB4R2 (BLT2R; a low-affinity receptor) (21–35). LTB4 is a potent chemotactic mediator for granulocytes and T lymphocytes, plays critical functions in the immune system as a stimulator of monocytes and T and B lymphocytes, and allows ECs to promote transmigration, activation, and/or effector functions. LTB4 can also induce B cell activation and proliferation (36–43). LTB4 has been shown to impact the immune response and spread of viral infections, including those caused by HIV, respiratory syncytial virus, and Epstein-Barr virus (EBV) (44–49). 5LO, LTA4H, and LTB4 are found at high levels in most of the inflammation and oxidative stress-associated cancers, such as breast, lung, prostate, pancreatic, colon, bladder, esophageal, and testicular cancers; glioma; chronic myelogenous leukemia; and Mantle cell lymphoma (MCL) non-Hodgkin's lymphomas (41, 42, 50–54). 5LO pathway inhibitors have been tested as chemoprevention agents in many cancers (55, 56).
Since LTB4 is linked to inflammation and immune modulation and KS, PEL, and MCD are chronic inflammation-associated malignancies, we hypothesized that regulation of the 5LO/LTB4 cascade is one of the virus's triggered host factors which plays key roles in KSHV pathogenesis. Our studies show that KSHV infection induces the 5LO/LTB4 cascade to aid in its latent infection and in the induction of the inflammatory milieu and that targeting of 5LO/LTB4 provides a new avenue of treatment against KSHV-associated malignancies.
MATERIALS AND METHODS
Cells and reagents.
Low-passage-number human microvascular dermal endothelial cells (HMVEC-ds; Lonza, Walkersville, MD), human umbilical vein endothelial cells (HUVEC; Lonza), and telomerase-immortalized human umbilical vein endothelial (TIVE) cells and long-term-infected TIVE (TIVE-LTC) cells (a gift from Rolf Renne, University of Florida) were cultured in lipopolysaccharide-free endothelial basal medium 2 with growth factors, as described before (19). PELs (KSHV positive [KSHV+]/EBV negative [EBV−]; BCBL-1 and BC-3 cells) and noninfected human Burkitt lymphoma (BL) cells (KSHV negative [KSHV−]/EBV−; BJAB cells) were cultured in RPMI 1640 medium (Gibco BRL, Grand Island, NY) with 10% heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT), 2 mM l-glutamine (Gibco BRL), and penicillin-streptomycin (Gibco BRL). BC-3 and BJAB cells were purchased from the American Type Culture Collection (ATCC), Manassas, VA. The BCBL-1 cell line was a gift from M. McGrath (University of California, San Francisco). KSHV-BJAB cells (57) were a gift from Blossom Damania, University of North Carolina. KSHV-BJAB (KSHV+/EBV−) cells were cultured in PEL cell growth medium supplemented with 0.2 mg/ml hygromycin B (Sigma, St. Louis, MO) (57). All cell lines were tested for mycoplasma contamination using a MycoAlert Plus mycoplasma detection kit (Lonza) per the manufacturer's instructions.
Reagents.
The antibodies used here were monoclonal antibodies, including anti-α tubulin clone DM (50 kDa; catalog no. T9026; Sigma), anti-β-actin clone AC-15 (42 kDa; catalog no. A5441; Sigma), anti-TATA binding protein (TBP; catalog no. ab818; 38 kDa; Abcam, Cambridge, MA), anti-KSHV–LANA-2 (amino acids 143 to 310; 70 kDa; LifeSpan BioSciences, Seattle, WA), and anti-KSHV-vCyclin (catalog no. ab12208; 30 kDa; Abcam), and polyclonal antibodies, including anti-lamin B1-nuclear envelope marker (66 kDa; catalog no. ab16048; Abcam,), anti-5LO-activating protein (anti-FLAP; band of approximately 48 kDa; predicted molecular mass, 19 kDa; catalog no. ab39535; Abcam), anti-5-lipoxygenase (78 kDa; catalog no. 160402; Cayman Chemical, Ann Arbor, MI), anti-leukotriene A4 hydrolase (69 kDa; catalog no. 160250, Cayman Chemical), anti-cyclooxygenase 1 (catalog no. 4842; Cell Signaling Technology), and LANA-1 antibody (from Bala Chandran, Rosalind Franklin University of Medicine and Science [RFUMS]). The reagents used in this study were the COX-2-specific inhibitor NS-398 [N-(2-cyclohexyloxy-4-nitrophenyl)-methanesulfonamide; Calbiochem, La Jolla, CA], the 5LO activation inhibitor MK866 {1-[(4-chlorophenyl)methyl]-3-[(1,1-dimethylethyl)thio]-α,α-dimethyl-5-(1-methylethyl)-1H-indole-2-propanoic acid, sodium salt; catalog no. 10133; Cayman Chemical}, and the 5LO inhibitor zileuton {N-(1-benzo[b]thien-2-ylethyl)-N-hydroxyurea; catalog no. 3308; Tocris Bioscience, Minneapolis, MN}. These were reconstituted in dimethyl sulfoxide (DMSO), and DMSO was used as the solvent control for all experiments involving treatments with inhibitors.
Virus.
Induction of the KSHV lytic cycle in BCBL-1 cells, supernatant collection, and virus purification procedures were described previously (20). Viral DNA was extracted, and the copy numbers were quantitated by real-time DNA PCR using primers amplifying the KSHV ORF73 gene as described previously (20). All stock preparations of purified KSHV were monitored for endotoxin contamination by standard Limulus assay as described previously (20). Replication-defective virus (UV-inactivated KSHV) was generated by inactivating KSHV with UV light (365 nm) for 20 min at a 10-cm distance (58, 59). KSHV DNA was extracted from live KSHV and UV-inactivated KSHV, and viral copy numbers were quantitated by real-time DNA PCR (19, 20, 58, 59).
Plasmid.
A plasmid encoding firefly luciferase under the control of the wild-type (WT) FASN promoter was obtained from Qiang Liu (Western College of Veterinary Medicine, University of Saskatchewan, Saskatoon, SK, Canada) and has been described previously (60).
Cytotoxicity assay.
HMVEC-d, HUVEC, and PEL cells were incubated with Dulbecco modified Eagle medium (Lonza) containing different concentrations of various inhibitors for 4 h or 4 days. At different time points, supernatants were collected and assessed for cellular toxicity using a cytotoxicity assay kit (Promega, Madison, WI) as described previously (58, 59).
Luciferase reporter assays.
KSHV infection's effect on the FASN full-length promoter (WT FASN) (60) was measured using a dual-luciferase kit according to the manufacturer's protocol (Promega). 293 cells were transfected using methods described before (20). The relative FASN promoter activity or number of relative luciferase units (RLU) was normalized to Renilla luciferase protein levels.
Viral gene expression profiling by real-time RT-PCR.
Thirty copies of KSHV DNA per cell were used for infection of HMVEC-d and HUVEC, and the cells were observed until 48 h postinfection (p.i.). KSHV ORF73 (forward primer 5′-CGCGAATACCGCTATGTACTCA-3′, reverse primer 5′-GGAACGCGCCTCATACGA-3′, TaqMan probe 6FAM-ACATCACCACCCCACAGACCTGGAG-TAMRA, where 6FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine), ORF50 (forward primer 5′-CGCAATGCGTTACGTTGTTG-3′, reverse primer 5′-GCCCGGACTGTTGAATCG-3′, TaqMan probe 6FAM-ACCTGTGCCCCCTCTTCGACACC-TAMRA), K5 (forward primer 5′-GAGCGTCCAGGTGCACAAC-3′, reverse primer 5′-TTGAACTGTTTCTGCTGATGTCTG-3′, TaqMan probe 6FAM-ACGCCGACAAGCCCAGCCAC-TAMRA), K8 (forward primer 5′-CCTGGACGCTCTCTCACACA-3′, reverse primer 5′-GGATCTGCGAGTTGGAAGCT-3′, TaqMan probe 6FAM-CCAAGAGGACCACACATTTCGCA-TAMRA), and vIRF2 (forward primer 5′-CATTTTTGGAGGAGCGACGTA-3′, reverse primer 5′-AGCCCAGGCCAGTCTCAGT-3′, TaqMan probe 6FAM-CGGGCTGCCAGAAATCCCGG-TAMRA) gene expression was assessed by real-time reverse transcription-PCR (RT-PCR) using amplification cycles as described before (20). KSHV vMIP-1 (forward primer 5′-ATGCTGCGTTAGCGTACTGCT, reverse primer 5′-GAACCCGTAGCAGCAGCTAT-3′) and vMIP-2 (forward primer 5′-TTGTCCGGTCTATGCCAGG-3′, reverse primer 5′-CTGCCTTGCTTTGTTTGCAA-3′) primer sequences were used to quantitate vMIP-1 and vMIP-2 gene expression. Prior to reverse transcription, the RNA samples were subjected to DNase I (amplification grade; Invitrogen, Carlsbad, CA) treatment to remove contaminating DNA, as described previously (20). The relative copy numbers of the transcripts were calculated from a standard graph plotted using the threshold cycle values for different dilutions of in vitro-transcribed transcripts. Maximum care was taken to keep the slope of the standard curve close to 3.3. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control.
TaqMan gene expression analysis for 5LO, LTA4H, and FLAP.
Thirty copies of KSHV DNA per cell were used for infection of HMVEC-d and HUVEC for different times. Gene expression of 5LO, LTA4H, and FLAP was analyzed using TaqMan gene expression assays (5LO, Hs00167536; LTA4H, Hs00168505; FLAP, Hs00970921; GAPDH, Hs99999905; Applied Biosystems, Foster City, CA) by the methods described in the manufacturer's protocol. PCR amplifications without cDNA were performed as negative controls.
Immunohistochemistry (IHC).
Sections from lymph nodes, skin biopsy samples of healthy subjects, and KS patients, sections of lung from healthy individuals, and lesions of lung from PEL patients were obtained from the AIDS and Cancer Specimen Resource (ACSR). Sections were deparaffinized with HistoChoice clearing reagent and hydrated with water before microwave treatment in 1 mmol/liter EDTA (pH 8.0) for 15 min for antigen retrieval and then blocked with blocking solution (2% donkey serum, 0.3% Triton X-100 in phosphate-buffered saline). Sections were incubated with the primary antibodies against 5LO or LTA4H overnight at 4°C. These sections were incubated with rabbit-polymer-horseradish peroxidase (Biocare Medical) for 15 min, washed, and developed using the 3,3′-diaminobenzidine reagent (Dako). Counterstaining with hematoxylin was done as described previously (20).
Immunofluorescence assay (IFA).
BCBL-1 cells were stained with anti-5LO and anti-FLAP primary antibodies, developed, and visualized with Alexa 594- and Alexa 488-coupled secondary antibody staining by the methods described before (19). Stained BCBL-1 cells were washed and viewed with appropriate filters under a fluorescence microscope with a Nikon Metamorph digital imaging system.
Preparation of nuclear extracts.
HMVEC-d or HUVEC were serum starved for 10 h and then infected with 30 DNA copies of KSHV per cell for various times at 37°C, and nuclear extracts were prepared using a nuclear extract kit (Active Motif Corp., Carlsbad, CA), in accordance with the manufacturer's instructions. After measurement of protein concentrations with bicinchoninic acid protein assay reagent (Pierce Biotechnology, Rockford, IL), extracts were aliquoted and stored at −80°C until use. The purity of the nuclear extracts was assessed by immunoblotting using anti-lamin B antibodies, and cytoskeletal contamination was checked using an anti-α-tubulin antibody.
Isolation of lymphocytes and mononuclear cells.
These studies were approved by the RFUMS Institutional Review Board. After informed consent was obtained in accordance with the Declaration of Helsinki, peripheral blood was collected in sodium heparin Vacutainer tubes (Becton, Dickinson, Franklin Lakes, NJ). The peripheral blood mononuclear cell (PBMC) fraction was collected after differential density centrifugation over Ficoll-Paque Plus (GE Healthcare, Piscataway, NJ). After washing the cells twice in Hanks' balanced salt solution (Lonza), monocytes were purified from the PBMCs via magnetic separation using beads coated with anti-CD14 antibodies, in accordance with the manufacturer's instructions (Miltenyi Biotec, San Diego, CA). Purified CD14+ monocytes were cultured in RPMI 1640 medium (Life Technologies) supplemented with fetal bovine/calf serum (HyClone, Rockford, IL) and penicillin-streptomycin (Life Technologies) at 37°C with 5% CO2 in 96-well polypropylene plates until assayed. The CD14− fraction was retained and termed lymphocytes.
ELISA for LTB4 and PGE2.
LTB4 and PGE2 levels in the supernatants of uninfected or KSHV-infected HMVEC-d or HUVEC, untreated or LO inhibitor-treated BCBL, BC-3, KSHV-BJAB, BJAB, TIVE, and TIVE-LTC cells were measured by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN) as described previously (16, 17, 19, 20). Data are expressed as the amount of LTB4 or PGE2 produced (pg/ml) per 105 cells.
Western blot and immunoprecipitation (IP) analysis.
Cell extracts (20 μg/lane) were separated on SDS-polyacrylamide gels and electrotransferred to 0.45-μm-pore-size nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin (BSA), probed with anti-5LO, anti-FLAP, anti-LTA4H, anti-β-actin, and anti-α-tubulin antibodies, and visualized using an enhance chemiluminescence detection system (20). Lysates prepared in low-salt buffer were clarified by centrifugation and immunoprecipitated for 4 h at 4°C with specific antibodies. Immune complexes were collected on protein A-Sepharose, separated by SDS-PAGE, and transferred onto nitrocellulose. Immunoblots were incubated in 5% BSA, 10 mM Tris HCl, pH 7.5, 1 mM EDTA, and 0.1% Tween 20 for 1 h at room temperature and probed first with specific antibodies and then with secondary antibodies.
TH1/TH2 cytokine analysis.
Analysis of TH1/TH2 cytokines in the supernatants obtained from untreated or LO inhibitor-treated HMVEC-d was done using a BD cytometric bead array (CBA) human TH1/TH2 cytokine kit per the manufacturer's instructions (BD Biosciences, San Diego, CA). The BD CBA human TH1/TH2 cytokine kit is used to quantitatively measure interleukin-2 (IL-2), interleukin-4 (IL-4), interleukin-5 (IL-5), interleukin-10 (IL-10), tumor necrosis factor (TNF), and gamma interferon (IFN-γ) protein levels in a single sample. The BD CBA human TH1/TH2 cytokine kit uses the bead array technology to simultaneously detect multiple cytokine proteins in research samples. Six bead populations with distinct fluorescence intensities were coated with capture antibodies specific for IL-2, IL-4, IL-5, IL-10, tumor necrosis factor alpha (TNF-α), and IFN-γ proteins. The six bead populations were mixed together to form the bead array, which was resolved in the red channel of a flow cytometer. During the assay procedure, we mixed the cytokine capture beads with recombinant standards or supernatants collected from various treatments and incubated them with phycoerythrin (PE)-conjugated detection antibodies to form sandwich complexes. The intensity of PE fluorescence of each sandwich complex revealed the concentration of that cytokine. After acquiring samples on a flow cytometer, FCAP Array software was used to generate results.
Monocyte adhesion assay.
Endothelial cells (ECs) were allowed to form a monolayer. Monocytes were purified and labeled with LeukoTracker solution (2 μl of 500× LeukoTracker solution was added to 1.0 ml of a leukocyte cell suspension at 1.0 × 106 cells/ml). An equal number of labeled monocytes was resuspended in various supernatants obtained from uninfected or infected cells and 5LO inhibitor-treated or 5LO-silenced and then infected cells. These monocytes were allowed to adhere to the EC monolayer for 60 min, and adhesion was quantitated by a CytoSelect leukocyte-endothelium adhesion assay (Cell Biolabs Inc., San Diego, CA). Adherent leukocytes were lysed, and fluorescence measurement was done using a fluorescence plate reader at 480 nm/520 nm.
Monocyte recruitment and transendothelial migration assay.
The in vitro effect of LO inhibitors (MK866 and zileuton) and 5LO silencing on the uninfected and infected cell supernatants in monocyte recruitment was determined by a CytoSelect 24-well cell migration assay (3 μm, fluorometric format) per the manufacturer's instructions (Cell Biolabs Inc.).
RESULTS
5LO and LTA4H enzymes are expressed in human KS lesion tissue sections.
KS lesions show a histopathology characterized by spindle-shaped ECs with latent KSHV infection expressing EC markers (CD31, CD34, CD36, and endothelium antigen clone EN4), extensive neoangiogenesis, and inflammatory infiltration (1, 7, 61, 62). To study the role of 5LO and LTA4H in KS, we analyzed the skin tissue sections of healthy subjects and KS patients for the presence of LO pathway enzymes 5LO and LTA4H by IHC. Healthy control skin tissue sections showed negligible expression of 5LO (Fig. 1A, panels a and c) and LTA4H (Fig. 1B, panels a and c). In contrast, abundant 5LO (Fig. 1A, panels b and d) and LTA4H (Fig. 1B, panels b and d) expression was detected in KS skin tissue. Specificity was confirmed by the nonreactivity of the isotype control for 5LO in human KS skin tissue specimen (Fig. 1A, panel f) and human healthy skin tissue specimens (Fig. 1A, panel e). Similarly, specificity was confirmed by the nonreactivity of the isotype control for LTA4H in human KS skin tissue specimens (Fig. 1B, panel f) and human healthy skin tissue specimens (Fig. 1B, panel e). These results demonstrate that 5LO and LTA4H are abundant in KS skin lesions and suggest that the LO pathway might be playing a role in KSHV pathogenesis.
FIG 1.

Lipoxygenase expression in human tissues and KSHV-infected cells. (A and B) Human KS and healthy skin tissue sections were analyzed by immunohistochemical staining for 5LO (A) and LTA4H (B) and counterstained with hematoxylin. (Aa, c, and e and Ba, c, and e) Healthy human skin tissue sections; (Ab, d, and f and Bb, d, and f) human KS skin tissue sections. Arrows (yellow) in all panels, 5LO or LTA4H staining. Magnifications: ×20 (Aa, b, e, and f, Ba, b, e, and f) and ×40 (Ac and d and Bc and d). (C) Immunofluorescence analysis of LANA-1 and 5LO in long-term KSHV latently infected ECs. TIVE and TIVE-LTC cells were grown to 80 to 90% confluence, fixed, permeabilized, and examined with LANA-1-specific (red) and 5LO-specific (green) antibodies. Nuclei were counterstained with DAPI (blue). Red arrows in panel Cb, nuclear staining for KSHV latency protein LANA-1; green arrows in panels Cc and i, 5LO staining; yellow arrows in panels Cd and j, LANA-1 and 5LO colocalization in nuclei; white arrows in panel Ce, k, f, and l, LANA-1 and 5LO colocalization with the nuclear stain DAPI. Magnifications: ×40 (Ca to k) and further enlargement by an additional ×40 (Cf and l).
5LO is expressed in KSHV latently infected ECs.
Since we observed a strong staining of 5LO in KS tissues, we next wanted to confirm this with in vitro cultures of KSHV latently infected ECs, which are an in vitro model for KS. We stained TIVE-LTC cells for 5LO and LANA-1, a marker for KSHV latent infection. TIVE-LTC cells show tight latent KSHV gene expression similar to the viral gene expression seen in the majority of KS lesion spindle cells and support long-term episomal maintenance (63). Punctate nuclear staining of KSHV latent LANA-1 protein was observed in many TIVE-LTC cells (Fig. 1C, panel b). TIVE-LTC cells were positive for 5LO, with diffuse cytoplasmic and dense nuclear 5LO staining seen in a majority of the TIVE-LTC cells (Fig. 1C, panel c). TIVE cells showed very faint nuclear staining and appreciable levels of cytoplasmic staining for 5LO (Fig. 1C, panel i) and no staining for LANA-1 (Fig. 1C, panel h). Nuclear localization of 5LO in TIVE-LTC cells was evident in the merged images (white color) stained with the nuclear stain DAPI (4′,6-diamidino-2-phenylindole; blue) and for LANA-1 (red) and host protein 5LO (green). Positioning of 5LO within the nucleus of resting cells is a powerful determinant of the capacity to generate LTB4 upon subsequent activation; therefore, these results suggest that KSHV latently infected cells (TIVE-LTC cells) might have an active 5LO pathway and a higher capacity to synthesize LTB4 than uninfected cells (TIVE cells) and 5LO might be playing a role in KSHV pathogenesis.
De novo KSHV infection of ECs induces 5LO pathway enzymes.
To study 5LO expression during primary infection, HMVEC-d and HUVEC were infected for different times. KSHV ORF73 gene expression, as assessed by real-time RT-PCR with ORF73 gene-specific primers and TaqMan probes (data not shown), confirmed the successful infection of HUVEC and HMVEC-d. To evaluate the expression of 5LO pathway genes during in vitro KSHV infection of target HMVEC-d and HUVEC, we examined the time kinetics of 5LO, LTA4H, and FLAP gene expression (Fig. 2A to F). KSHV infection induced higher levels of 5LO (Fig. 2A), FLAP (Fig. 2C), and LTA4H (Fig. 2E) in HMVEC-d than uninfected cells. Similarly, we observed the induction of the 5LO (Fig. 2B), FLAP (Fig. 2D), and LTA4H (Fig. 2F) genes in KSHV-infected HUVEC. Higher levels of LO pathway enzyme gene expression were observed at later time points (6 h onwards) of infection (Fig. 2A to F). LTA4H gene expression showed a bimodal induction pattern during de novo KSHV infection of HMVEC-d (Fig. 2E) and HUVEC (Fig. 2F). These results demonstrating upregulation of the 5LO pathway strongly support its role in KSHV pathogenesis.
FIG 2.

Detection of 5LO pathway enzymes during de novo KSHV infection in latently infected ECs and in PEL cells. (A to F) Gene expression of 5LO pathway enzymes during primary infection of HMVEC-d and HUVEC. Cells grown to 80 to 90% confluence were serum starved for 8 h and infected with 30 DNA copies/cell of KSHV for different time points. At different times p.i. (up to 48 h), total RNA was isolated, DNase I treated, and then subjected to qRT-PCR using 5LO (A, B), FLAP (C, D), and LTA4H (E, F) gene-specific TaqMan primers. (G) Expression of LTA4H, FLAP, and 5LO in TIVE and TIVE-LTC cells. Cells were serum starved for 24 h and then used to prepare total RNA, and the expression of LTA4H, FLAP, and 5LO genes was analyzed. (H) Expression of LTA4H, FLAP, and 5LO genes in BCBL-1 BC-3, BJAB-KSHV, and BJAB cells. B cells grown to about 106 cells per ml were serum starved (0.2% FBS) for 8 h. Total RNA was isolated, DNase I treated, and then subjected to qRT-PCR using LTA4H, FLAP, and 5LO gene-specific TaqMan primers. Fold induction in panels A to F was calculated by considering induction in uninfected HMVEC-d (A, C, and E), HUVEC (B, D, and F), TIVE cells (G), and BJAB cells (H) to be 1-fold. (I) Expression of the gene for the 5LO enzyme during primary infection of HMVEC-d with 30 DNA copies/cell of live KSHV or UV-inactivated KSHV (UV-KSHV) for 24 h or 48 h. At different time points postinfection (24 h or 48 h), total RNA was isolated, DNase I treated, and then subjected to qRT-PCR using 5LO-specific TaqMan primers and ABI expression assays. Each bar represents the average ± SD of three independent experiments.
5LO pathway enzymes are expressed in long-term KSHV-infected ECs.
Since we observed strong staining of 5LO in TIVE-LTC cells, we analyzed the expression of all three LO pathway enzymes required for LTB4 secretion in these cells by gene expression analysis (Fig. 2G). Compared to TIVE cells, TIVE-LTC cells showed increased expression of the 5LO (8.5-fold), FLAP (5.2-fold), and LTA4H (17.8-fold) genes (Fig. 2G). Among the entire set of LO pathway enzymes tested, LTA4H showed the highest level of expression, which was found in TIVE-LTC cells but not in TIVE cells (Fig. 2G).
5LO pathway enzymes are expressed in KSHV+ PEL cells.
We examined 5LO pathway gene expression in BC-3 and BCBL-1 cells as well as BJAB-KSHV cells (Fig. 2H). BCBL-1 cells expressed higher levels of 5LO (7-fold), FLAP (3-fold), and LTA4H (14.5-fold) than BJAB cells (Fig. 2H), while BC-3 cells expressed higher levels of 5LO (8.3-fold), FLAP (3-fold), and LTA4H (18-fold) than BJAB cells (Fig. 2H). BJAB-KSHV cells expressed higher levels of 5LO (6.6-fold), FLAP (3-fold), and LTA4H (15.4-fold) than BJAB cells (Fig. 2H). The level of LTA4H expression was the highest among all of the LO pathway genes tested (Fig. 2H). Higher 5LO pathway gene expression in latently infected ECs and B cells suggests a potential connection between viral latent gene expression and host 5LO.
5LO induction depends predominantly on viral transcription.
To determine whether KSHV gene expression is essential for 5LO upregulation, replication-incompetent KSHV was prepared by UV irradiation (58). UV-inactivated KSHV did not induce any KSHV gene expression, whereas an equal number of live virus DNA copies per cell induced a quantitative increase in latency-associated ORF73 gene expression as well as lytic cycle-associated ORF50, K8, and K5 gene expression, as shown previously (58). We observed a dramatic reduction in 5LO gene expression in UV-inactivated KSHV-infected cells compared to that in live KSHV-infected cells (Fig. 2I), suggesting that 5LO induction is dependent primarily on KSHV gene expression. Since infection with UV-radiated KSHV virions still resulted in a 2- to 4-fold induction of 5LO gene expression, the possibility of involvement of virion components during the binding, entry, and nuclear entry stages of infection in 5LO induction cannot be ruled out.
KSHV infection increases protein levels of 5LO pathway enzymes.
Since we observed the induction of 5LO pathway enzyme gene expression, we evaluated the levels of 5LO pathway proteins upon KSHV infection. De novo KSHV infection (up to 48 h) induced increased expression of LTA4H (Fig. 3A) and 5LO (Fig. 3A) in HMVEC-d. A similar induction was observed in HUVEC (data not shown). Compared to the levels of expression in TIVE cells, TIVE-LTC cells showed increased expression of 5LO (2.6-fold) and LTA4H (4.2-fold) (Fig. 3B). BCBL-1 cells expressed higher levels of 5LO (3.2-fold) and LTA4H (4.6-fold) than BJAB cells (Fig. 3C). Similarly, BC-3 cells expressed higher levels of 5LO (2.9-fold) and LTA4H (4.4-fold) (Fig. 3C). KSHV-BJAB cells expressed higher levels of 5LO (4.6-fold) and LTA4H (5.4-fold) than BJAB cells (Fig. 3D). Higher 5LO pathway enzyme protein levels in the whole-cell lysates of latently infected ECs and B cells suggest a probable connection of viral latent gene expression and induction of the 5LO pathway.
FIG 3.

Protein levels of 5LO/LTA4H and secretion of LTB4 during de novo KSHV infection and latency. (A to D) Lysates from serum-starved (8 h) uninfected, and KSHV (30 DNA copies/cell)-infected HMVEC-d for the indicated times (0 h, 30 min, 1 h, 2 h, 4 h, 8 h, 24 h, and 48 h) and TIVE, TIVE-LTC cells, BCBL-1, BC-3, KSHV-BJAB, and BJAB cells were Western blotted for 5LO or LTA4H, stripped, and immunoblotted for β-actin. A representative blot from three independent experiments is shown. (E to I) Secretion of LTB4 in de novo-infected HMVEC-d (E), HUVEC (F), TIVE and TIVE-LTC cells (G), BCBL-1, BC-3, and BJAB cells (H), and KSHV-BJAB and BJAB cells (I). HUVEC and HMVEC-d were serum starved for 8 h and infected with 30 DNA copies/cell of KSHV for 8 h, 24 h, and 48 h, and supernatants were collected for LTB4 immunoassay. TIVE and TIVE-LTC cells were serum starved for 8 h in medium containing low levels (0.2%) of FBS, and supernatants were collected for LTB4 quantification. BCBL-1, BC-3, KSHV-BJAB, and BJAB cells were serum starved for 8 h in medium containing low levels (0.2%) of FBS and collected for LTB4 assay. Values for LTB4 secretion (in pg/ml) are indicated in the histograms. Each bar represents the average ± SD of three independent experiments. Statistical significance (t test) for panels E and F was calculated with respect to the results for uninfected cells. Statistical significance (t test) for panel G was calculated with respect to the results for TIVE cells. Statistical significance (t test) for panels H and I was calculated with respect to the results for BJAB cells. The statistical analysis was carried out using a two-tailed Student's test. *, P < 0.05; **, P < 0.01.
KSHV infection induces LTB4 secretion.
5LO activation leads to the synthesis and secretion of the chemotactic bioactive lipid metabolite LTB4. To evaluate the consequences of 5LO activation, we quantitated the release of LTB4 upon KSHV infection. KSHV infection in HMVEC-d and HUVEC induced significant levels of LTB4 secretion (Fig. 3E and F) which increased over the time course of infection. Compared to TIVE cells, TIVE-LTC cells showed significantly increased secretion of LTB4 under unstarved as well as serum-starved conditions (Fig. 3G). Compared to BJAB, BCBL-1, and BC-3 cells (Fig. 3H) as well as KSHV-BJAB cells (Fig. 3I), significantly higher levels of LTB4 were secreted under unstarved than under serum-starved conditions. Taken together, the results in Fig. 1 to 3 demonstrate that there is an active involvement of the 5LO pathway during KSHV infection.
KSHV-infected cells express higher levels of 5LO pathway components in both the cytoplasmic and nuclear fractions.
LTB4 is synthesized from membrane-bound arachidonic acid via the 5LO pathway enzymes (64). Activation of 5LO is connected with its translocation from the cytosol to the nuclear membrane (64) (Fig. 4A). Nuclear localization of 5LO is also a determinant of the LTB4 synthetic capacity of the cell. 5LO activation leads to the formation of unstable LTA4, which can be metabolized to either LTB4 (a potent lipid mediator of inflammation synthesized predominantly by leukocytes, such as monocytes/macrophages and neutrophils) or cysteinyl (Cys) LTs/peptide leukotrienes (LTC4, LTD4, and LTE4).
FIG 4.

Nuclear levels of 5LO expression during KSHV infection and dependency on NF-κB activation. (A) Schematic showing the 5LO and FLAP interactions that generate LTB4. (B to G) Western blots of nuclear and cytoplasmic fractions in de novo KSHV-uninfected (30 DNA copies/cell) (B) or KSHV-infected (C) HMVEC-d, TIVE and TIVE-LTC cells (D), BCBL-1 and BJAB cells (E), BC-3 and BJAB cells (F), and BJAB and KSHV-BJAB cells (G). These fractions were collected and immunoblotted for host proteins (5LO, LTA4H, FLAP, α-tubulin, and lamin B) and KSHV protein LANA-1. (H) Western blots of nuclear fractions of untreated, NF-κB inhibitor solvent-treated, or NF-κB inhibitor (5 μM Bay11-7082)-treated BCBL-1 and BC-3 cells. Unt., untreated; Sol., solvent treated; Bay, Bay11-7082. Representative blots from three independent experiments are shown in panels B to H.
5LO is localized in the nucleus as well as the cytosol, but on cellular activation, 5LO undergoes Ca2+- or NF-κB-dependent translocation to the nuclear envelope (65–68). The mechanism of nuclear translocation involves the necessary association of 5LO with a novel 18-kDa membrane protein known as 5LO-activating protein, or FLAP (69). Cytoplasmic and nuclear fractions were prepared from uninfected (8 h, 24 h, and 48 h) (Fig. 4B) and KSHV-infected (8 h, 24 h, and 48 h) HMVEC-d (Fig. 4C). Cytoplasmic and nuclear fractions were checked for purity by α-tubulin and lamin B staining, respectively (Fig. 4B and C). The purity of the nuclear extracts was verified by the absence of α-tubulin staining. Higher levels of 5LO and LTA4H proteins were detected in the nuclear fractions of KSHV-infected (8 h, 24 h, and 48 h) HMVEC-d than uninfected HMVEC-d (Fig. 4B and C). Appreciable levels of FLAP were observed in the nuclear fractions of uninfected and infected (8 h, 24 h, and 48 h) HMVEC-d (Fig. 4B and C).
Similar to the higher nuclear levels of 5LO in de novo KSHV-infected ECs (Fig. 4B and C), latently infected TIVE-LTC cells (Fig. 4D), latently infected BCBL-1 cells (Fig. 4E), and BC-3 cells (Fig. 4F) expressed high levels of 5LO in the nuclear fractions. KSHV-BJAB cells expressed higher nuclear levels of 5LO than BJAB cells (Fig. 4G). Surprisingly, FLAP levels appeared to be slightly lower in KSHV-BJAB cells than uninfected BJAB cells. FLAP levels in KSHV-BJAB cells seemed to be abundant enough to form complexes with 5LO to generate LTB4. Higher 5LO protein levels in the nuclear fraction, along with abundant levels of its activating protein (FLAP) and LTA4H, indicate the possibility of their interaction, which can lead to the efficient synthesis and secretion of LTB4. These results also indicate that KSHV de novo infection of ECs tightly controls the synthesis of inflammatory LTB4 via regulating the reorganization of the LT biosynthetic enzymes. This tight regulation might be allowing virus to establish latency in the host cells and would prevent the untimely onset of proinflammatory LTB4 at the very early stages of infection.
High nuclear fraction levels or high levels of activated components of the 5LO pathway have been reported to be a consequence of the high nuclear translocation of 5LO, which is in turn dependent upon NF-κB activation and Ca2+ induction. Since KSHV infection has been shown to induce NF-κB, we checked the levels of 5LO in the nuclear fractions obtained from untreated, Bay11-7082 solvent-treated, or 5 μM Bay11-7082-treated BCBL-1 and BC-3 cells. We observed a drastic decline in the nuclear levels of 5LO and LTA4H (Fig. 4H). This reduction was related to the reduction in the level of active NF-κB (the p65 nuclear fraction of NF-κB) (Fig. 4H). Since KSHV infection also induces Ca2+, we checked the effect of pharmacological inhibitors of Ca2+ [1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) and 8-(N,N-diethylamino)octyl-3,4,5-trimethoxybenzoate (TMB-8)] (55). Treatment with Ca2+ inhibitors did not change the nuclear levels of 5LO in the KSHV-infected cells (data not shown). These results suggest that KSHV infection-induced NF-κB signaling is critical for the activation of 5LO pathway enzymes.
KSHV infection induces 5LO and FLAP association.
5LO is a 78-kDa protein that catalyzes the conversion of arachidonic acid to LTs. FLAP is an 18-kDa membrane-bound protein that is essential for LTB4 synthesis in cells. In LTB4-generating cells, FLAP and 5LO interact and form a 5LO/FLAP complex (∼100 kDa). By IP and reverse IP experiments with 5LO and FLAP antibodies, we detected the possible 5LO and FLAP association in uninfected endothelial (TIVE) cells or KSHV−/EBV− BJAB cells and KSHV-infected TIVE-LTC or BCBL-1 cells (Fig. 5A). Briefly, lysates were immunoprecipitated with anti-5LO and Western blotted with anti-5LO antibody. We detected higher levels of a 78-kDa band in KSHV latently infected BCBL-1 and TIVE-LTC cells than uninfected BJAB and TIVE cells (Fig. 5A, panel a). Besides the 78-kDa band, we also detected a 100-kDa band (Fig. 5A, panel a). At this point, we do not know the exact identity of the 100-kDa band but will ascertain it by mass spectrometric analysis in our future studies. We speculate it to be some posttranslationally modified form of 5LO, since 5LO is known to get posttranslationally modified by phosphorylation, glycosylation, nitrosylation, and peroxidation. We also performed a reverse IP with anti-5LO antibody and Western blotting with anti-FLAP and detected the 18-kDa band. The 5LO and FLAP bands in IP and reverse IP experiments were stronger in TIVE-LTC or BCBL-1 cells than TIVE or BJAB cells (Fig. 5A, panel b). The increased intensity of the FLAP band in the 5LO immunoprecipitate may be caused by higher levels of 5LO (definitely) and FLAP (probably) in TIVE-LTC cells. These results also suggest that the KSHV infection-induced overall high levels of 5LO and FLAP probably initiate some interactions between 5LO and FLAP components of the 5LO pathway, which probably result in the increased synthesis and secretion of LTB4 in latently infected cells (Fig. 5A). 5LO predominantly associates with FLAP in KSHV-infected cells but not in uninfected cells. The results in Fig. 5 (FLAP and 5LO possible association) were also confirmed by IFA for 5LO and FLAP in BCBL-1 cells (Fig. 5B). We observed FLAP staining (Fig. 5B, panel 2) colocalizing with nuclear membrane staining of lamin B (Fig. 5B, panel 1) in Fig. 5B, panel 3. We also observed intense colocalization of 5LO staining (panel 4) with FLAP (panel 5) in Fig. 5B, panel 6. Lamin B/FLAP and 5LO/FLAP colocalization is clear in panels in Fig. 5B with enlarged pictures. These results clearly suggest that KSHV infection induces activation of the 5LO pathway, which leads to the production of LTB4 secretion in the infected cell microenvironment.
FIG 5.
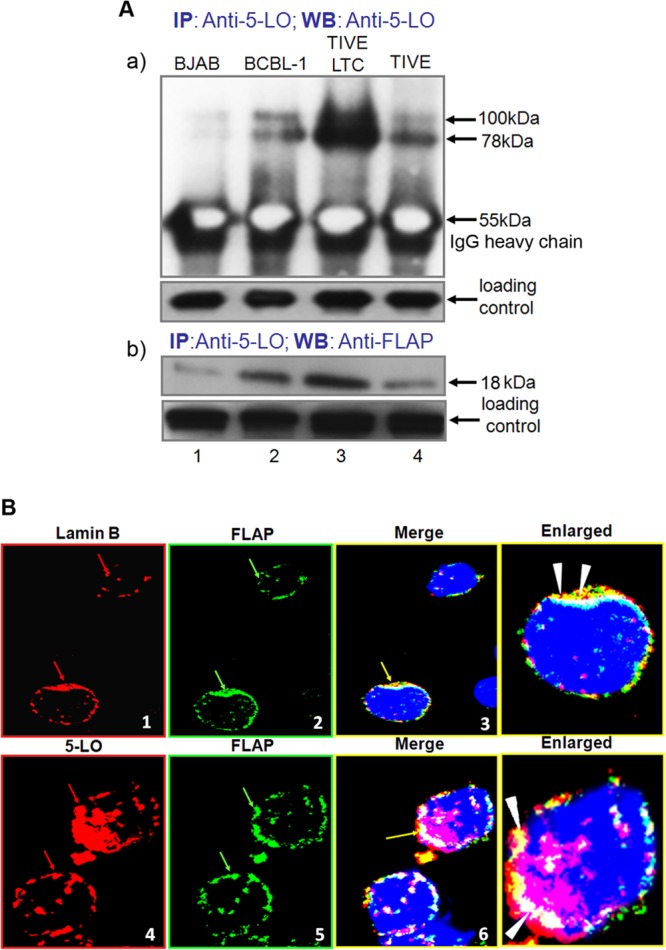
5LO and FLAP interaction during KSHV infection. (A, panel a) Uninfected endothelial (TIVE) cells, BJAB cells, KSHV-infected endothelial (TIVE-LTC) cells, or BCBL-1 cells were lysed and immunoprecipitated with anti-5LO and Western blotted (WB) with anti-5LO antibody. (A, panel b) TIVE, BJAB, TIVE-LTC, or BCBL-1 cells were lysed and immunoprecipitated with anti-5LO and Western blotted with anti-FLAP antibody. (B) BCBL-1 cells were immunostained with anti-lamin B (red) and anti-FLAP (green) or with 5LO (red) and FLAP (green) antibody and analyzed by confocal microscopy. Red arrows, nuclear membrane staining in panel 1 and nuclear localization of 5LO in panel 4; green arrows in panels 2 and 5, nuclear membrane localization of FLAP; yellow arrows, colocalization of FLAP with the nuclear membrane in panel 3 and colocalization of 5LO and FLAP in panel 6; white arrowheads in enlarged panels, colocalization of lamin B and FLAP and 5LO and FLAP.
5LO silencing reduces 5LO gene expression and LTB4 secretion.
To study the downstream consequences of 5LO pathway blocking, we opted for the chemical inhibition and 5LO gene silencing strategies. To determine the specificity of 5LO involvement in KSHV pathogenesis and to avoid the 5LO-independent effects of chemical inhibitors, we standardized the 5LO silencing method. The effect of 5LO silencing in HMVEC-d was determined by infecting serum-starved (8 h) control (construct encoding a scrambled sequence that will not lead to the specific degradation of any known cellular mRNA) short hairpin RNA lentiviral particle (sh-C)- or 5-lipoxygenase short hairpin RNA (h) lentiviral particle (5LO)-transduced cells for 4 h, 8 h, 24 h, and 48 h. Silencing of 5LO reduced the level of KSHV infection-induced 5LO gene expression and LTB4 secretion in HMVEC-d (Fig. 6A and B). KSHV-infected sh-5LO-induced HMVEC-d showed significantly reduced 5LO expression compared to sh-C-transduced KSHV-infected HMVEC-d (Fig. 6A).
FIG 6.

5LO silencing or 5LO chemical inhibition reduces 5LO gene expression and LTB4 secretion. HMVEC-d were transduced with sh-C or sh-5LO at a multiplicity of infection of 10, as described previously (19). sh-C- or sh-5LO-transduced HMVEC-d were serum starved for 8 h and infected with KSHV for 4 h, 8 h, 24 h, and 48 h. RNA from these cells was analyzed by qRT-PCR for 5LO expression (A), and the supernatants were used to quantify LTB4 levels by ELISA (B). (A) Percent gene (5LO) expression by considering the expression in sh-C-transduced cells at the respective time points to be 100%. Similarly, 5LO gene expression was examined in sh-C- or sh-5LO-transduced TIVE-LTC, BCBL-1, and BC-3 cells. (C and D) Supernatants from sh-C or sh-5LO transduced TIVE-LTC cells (C) and BCBL-1 and BC-3 cells (D) were used to detect LTB4 secretion. (E) Lysates prepared from sh-C- or sh-5LO-transduced TIVE-LTC, BC-3, and BCBL-1 cells were tested for the protein levels of 5LO and COX-2. These blots were reprobed with anti-β-actin antibody to confirm equal loading. 5LO chemical inhibition reduces LTB4 secretion. HMVEC-d were pretreated with solvent for MK866 (MK), MK866, solvent for zileuton (Zil.), or zileuton for 1 h and then infected with 30 DNA copies KSHV/cell for different times by the methods described previously (19). (F) Supernatants were used to quantify LTB4 levels by ELISA. (G and H) Supernatants from solvent- or inhibitor-treated TIVE-LTC cells (G) and BCBL-1 and BC-3 cells (H) were used to detect LTB4. LTB4 levels in quadruplicate samples were measured, and values are presented in pg/ml. Each point represents the average ± SD from three independent experiments. The statistical analysis was carried out using a two-tailed Student's t test. *, P < 0.05; **, P < 0.01. Red arrows facing down, percent inhibition.
We next assessed the functional consequences of 5LO silencing by quantifying the secreted LTB4 levels in the supernatant of sh-C or sh-5LO lentivirus-transduced and then KSHV-infected HMVEC-d (Fig. 6B). KSHV-infected sh-5LO-transduced HMVEC-d showed a significantly reduced level of LTB4 secretion compared to that of sh-C-transduced and KSHV-infected HMVEC-d (Fig. 6B). Our results suggested that 5LO silencing could effectively abrogate KSHV infection-induced LTB4 secretion in ECs. A similar reduction in 5LO gene expression and LTB4 secretion was observed in TIVE-LTC, BC-3, and BCBL-1 cells (Fig. 6C and D). 5LO silencing did not change COX-2 protein levels, further validating the specificity of the silencing procedure (Fig. 6E). 5LO silencing did not interfere with PGE2 (a metabolite of the COX-2 pathway) secretion (data not shown).
5LO chemical inhibition reduces LTB4 secretion.
For chemical blockage of the 5LO pathway, we used two specific inhibitors: MK866 and zileuton. MK866 blocks binding of 5LO to the membrane by specifically interacting with the membrane-bound activating protein FLAP, which is necessary for cellular LT biosynthesis. MK866 is an orally active anticancer drug blocking the synthesis of the highly chemotactic and procarcinogenic leukotriene LTB4 (70). Zileuton is an orally active inhibitor of 5LO and thus inhibits LTB4 formation. Zileuton is rapidly absorbed, with a mean time to the peak blood serum concentration of 1.7 h, and is primarily excreted in the urine (∼95%). Zileuton is FDA approved and is used to prevent difficulty in breathing, chest tightness, wheezing, and coughing due to asthma and has also been tested for its antineoplastic properties in colon, lung, and prostate cancers (21, 31). It is in clinical trials (clinicalTrials.gov) for the treatment of cancers, including chronic myeloid leukemia (CML), and of sickle cell disease (SCD), smoking-related lung disease, and chronic obstructive pulmonary disease (COPD).
The effect of MK866 or zileuton treatment in HMVEC-d was determined by infecting serum-starved (8 h) HMVEC-d for 4 h, 8 h, 24 h, and 48 h. We assessed the functional consequences of MK866 or zileuton treatment by quantifying the secreted LTB4 levels in the supernatants of KSHV-infected HMVEC-d and MK866-treated, zileuton-treated,journal MK866 solvent-treated, or zileuton solvent-treated and then KSHV-infected HMVEC-d (Fig. 6F). KSHV-infected and MK866- or zileuton-treated HMVEC-d showed significantly reduced LTB4 secretion (Fig. 6F) compared to solvent-pretreated (for MK866 or for zileuton) and then infected HMVEC-d, thus suggesting that 5LO inhibition could effectively abrogate KSHV infection-induced LTB4 secretion in ECs. A similar reduction in LTB4 secretion was observed in TIVE-LTC cells and KSHV+ PEL cells (Fig. 6G and H). Results presented in Fig. 6 demonstrated that gene silencing as well as chemical inhibition can effectively downregulate 5LO and reduce LTB4 secretion and could be used to understand the role of the LO pathway during KSHV infection and viral latency.
Inhibition of 5LO affects KSHV latent and lytic gene expression.
To analyze the potential role of 5LO pathway activation in KSHV gene expression, HMVEC-d were preincubated with inhibitory/noncytotoxic concentrations of either MK866 or zileuton or their respective solvent controls at 37°C for 1 h and then infected with KSHV for 2 h, and viral messages, collected at different times after KSHV infection (8 h, 24 h, and 48 h), were quantitated by real-time RT-PCR. Since significant levels of LTB4 secretion were observed in the later hours of infection, we chose to analyze the 5LO inhibition effects at 8 h, 24 h, and 48 h. Similarly, sh-C- or sh-5LO-transduced HMVEC-d were infected with KSHV for 2 h, and viral messages, collected at different time points after KSHV infection, were quantitated by real-time RT-PCR. De novo KSHV-infected HMVEC-d displayed the sustained expression of latency-associated ORF73, ORF72, and K13 and transient expression of a limited number of lytic genes, including the ORF50 (regulator of transcription activation [RTA]), vIRF2, K8, and K5 genes, as reported before (58, 71). 5LO inhibition reduced ORF73 gene expression (Fig. 7A) by 30 to 48%. An approximately 20% to 32% induction in the ORF50 gene was observed upon 5LO inhibition (Fig. 7B). These results demonstrate that the 5LO induced by KSHV late during target cell infection plays a role in the viral life cycle. 5LO inhibition could not reduce LANA-1 protein levels, as indicated in Fig. 7H.
FIG 7.

Effect of 5LO inhibition on KSHV gene expression in HMVEC-d. (A to G) HMVEC-d were treated with 5LO chemical inhibitors (1 μM MK866 and 1 μM zileuton) or solvent, or HMVEC-d were transduced with sh-C or sh-5LO and then infected with KSHV (30 DNA copies/ml) for different times. RNA was isolated, and viral transcripts were quantitated. Percent inhibition was calculated by considering viral gene expression in solvent- or sh-C-transduced cells to be 100%. Each point represents the average ± SD from three independent experiments. The statistical analysis was carried out using a two-tailed Student's t test. *, P < 0.05; **, P < 0.01. Red arrows facing down, percent inhibition; green arrows facing up, percent induction. (H) Effect of 5LO inhibition on viral protein LANA-1. Cell lysates prepared from HMVEC-d treated with 5LO chemical inhibitors (1 μM MK866 and 1 μM zileuton) or solvent and then infected with KSHV (30 DNA copies/ml) for different time points were Western blotted for LANA-1, stripped, and immunoblotted for α-tubulin. Inf., infection. A representative blot from three independent experiments is shown.
Inhibition of 5LO effectively downregulates KSHV immediate early lytic K5 gene expression but not K8 or vIRF2 gene expression in de novo-infected HMVEC-d.
Our previous studies have shown that, in addition to the latent ORF73 and lytic ORF50 genes, KSHV also expresses a limited number of lytic cycle genes early during infection (71). Unlike its effect on K8 (Fig. 7D) and vIRF2 (Fig. 7E) gene expression, 5LO inhibition had a maximal impact on K5 gene expression in HMVEC-d, with >86% inhibition (Fig. 7C). K5, which is detected for up to 5 days in infected HMVEC-d, is involved in the downregulation of major histocompatibility complex class I, intercellular adhesion molecule 1 (ICAM-1), and B7.2 molecules and thus probably blocks the elimination of infected cells by cytotoxic T and NK cells (72, 73). K5 downregulation by 5LO inhibition suggests an important role for LTs in enabling virus to evade the host immune system. The differential effects over KSHV gene expression by 5LO inhibition also suggested the specificity of the observed reduction in viral RNA copy numbers.
Inhibition of 5LO effectively downregulates the expression of KSHV immediate early lytic K4 (vMIP-2) and K6 (vMIP-1) gene expression in de novo-infected HMVEC-d.
KSHV also expresses a limited number of several viral macrophage inflammatory proteins (vMIPs), KSHV ORFs K4 (vMIP-2) and K6 (vMIP-1), early during de novo infection (71). These viral genes encode chemokines showing homology to cellular CC chemokines, such as MIP-1α and RANTES. vMIP-1 and vMIP-2 bind to CCR8 and are highly angiogenic. vMIP-2 has also been shown to be a potent chemoattractant, induce signal transduction and chemotaxis in monocytic cells, and contribute to KSHV pathogenesis (74). We did not observe high levels of vMIP-1 and vMIP-2 gene expression at 24 h and 48 h of infection. Therefore, we studied the role of the 5LO pathway at very early times (2 h, 4 h, and 8 h) of infection in HMVEC-d. 5LO inhibition had a huge impact on vMIP-1 and vMIP-2 gene expression in HMVEC-d at 8 h of viral infection, with ∼60 to 80% inhibition (Fig. 7F and G). vMIP-1 and vMIP-2 downregulation by 5LO inhibition suggests an important role for LTs in viral pathogenesis.
Inhibition of 5LO downregulates KSHV latent vCyclin and LANA-2 gene expression and protein levels in PEL cells.
We next examined the effect of blocking 5LO on viral gene expression in BC-3 and BCBL-1 cells (Fig. 8A to H). 5LO inhibition significantly downregulated latent vCyclin gene expression in BCBL-1 cells (Fig. 8A) and BC-3 cells (Fig. 8B). We also observed a significant reduction in LANA-2 gene expression upon 5LO inhibition in BCBL-1 cells (Fig. 8C) and BC-3 cells (Fig. 8D). 5LO blocking downregulated (20 to 30%) LANA-1 gene expression in BCBL-1 and BC-3 cells (Fig. 8E and F, respectively), with no effect on v-FLIP expression in BCBL-1 and BC-3 cells (Fig. 8G and H, respectively). All these results support the role of 5LO in the KSHV life cycle. 5LO inhibition effectively reduced vCyclin and LANA-2 protein levels, as indicated in BCBL-1 cells (Fig. 8I) and BC-3 cells (Fig. 8J). 5LO inhibition had much less of an effect on LANA-1 protein levels in BCBL-1 and BC-3 cells (Fig. 8I and J, respectively).
FIG 8.

Effect of 5LO inhibition on KSHV gene expression and protein levels in PEL cells. (A to H) BCBL-1 or BC-3 cells were treated with 5LO chemical inhibitors (1 μM MK866 and 1 μM zileuton) or their respective solvents or transduced with sh-C or sh-5LO, and RNA was prepared. Viral transcripts (vCyclin, LANA-2, LANA-1, and vFLIP) were quantitated by real time-RT PCR as described before (19). Percent inhibition was calculated by considering viral gene expression in solvent-treated or sh-C-transduced cells to be 100%. Each point represents the average ± SD from three independent experiments. The statistical analysis was carried out using a two-tailed Student's t test. *, P < 0.05; **, P < 0.01. Red arrows facing down, inhibition. (I and J) Effect of 5LO inhibition on viral proteins LANA-1, vCyclin, and LANA-2. Cell lysates prepared from BCBL-1 or BC-3 cells treated with 5LO chemical inhibitors (1 μM MK866 and 1 μM zileuton) or their respective solvents or transduced with sh-C or sh-5LO were Western blotted for LANA-1, vCyclin, and LANA-2, stripped, and immunoblotted for α-tubulin. Representative blots from three independent experiments are shown.
KSHV-induced 5LO regulates the secretion of TH1/TH2-related cytokines.
KS lesions are noted for their prominent leukocyte infiltrate consisting of monocytes, lymphocytes, and mast cells (75, 76). KSHV infection has been shown to shift the immune response from a TH1 to a TH2 microenvironment (77). Endothelial cells and circulating endothelial progenitor cells have been reported to secrete TH1/TH2/TH17-related cytokines (78, 79). To determine the role of KSHV-induced 5LO in TH1/TH2-related cytokine secretion during de novo infection of HMVEC-d, we used 5LO inhibitors in conjunction with 5LO silencing methods and examined cytokine secretion. We collected supernatants from serum-starved (8 h) uninfected HMVEC-d, infected HMVEC-d, and cells pretreated with inhibitor (MK866 or zileuton) or solvent and then infected with KSHV. Similarly, supernatants were collected from serum-starved HMVEC-d (8 h) transduced with sh-C or sh-5LO and then left uninfected or infected with KSHV. 5LO inhibition enhanced the secretion of the TH1-related cytokines IFN-γ (Fig. 9A) and IL-2 (Fig. 9B) at 8 h, 24 h, and 48 h postinfection, suggesting that 5LO keeps TH1-related cytokines in a low profile and helps KSHV evade the host immune response. 5LO inhibition marginally reduced the TH1-related cytokine TNF-α (Fig. 9C). Uninfected cells also secreted low levels of TH1-related cytokines, and their fold levels of induction at various time points are shown in Table 1.
FIG 9.
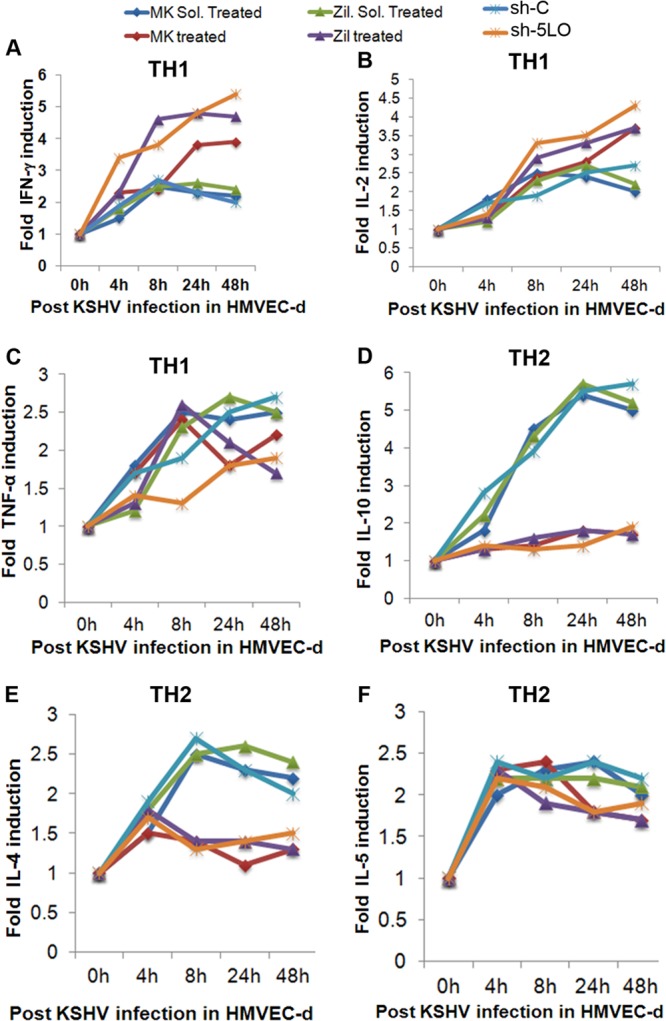
Effect of 5LO inhibition on KSHV infection-induced TH1/TH2-related cytokines in HMVEC-d. Supernatants collected from serum-starved (8 h) cells either uninfected or infected (4 h, 8 h, 24 h, and 48 h) or pretreated with either MK866, solvent for MK866, zileuton, or solvent for zileuton and then infected with KSHV for 4 h, 8 h, 24 h, and 48 h were used to analyze the levels of TH1- and TH2-related cytokines. Similarly, supernatants collected from serum-starved (8 h) sh-C- or sh-5LO-transduced HMVEC-d either uninfected or infected (4 h, 8 h, 24 h, and 48 h) were used to analyze the levels of TH1- and TH2-related cytokines. Fold induction was calculated by considering the secretion at 0 h infection to be 1-fold. Cytokine levels with respect to those of 0 h of infection are represented in a line graph format. (A) IFN-γ; (B) IL-2; (C) TNF-α; (D) IL-10; (E) IL-4; (F) IL-5.
TABLE 1.
Effect of 5LO pathway inhibition on TH1 cytokine secretion in uninfected and KSHV-infected cells at different time points
| TH1 cytokine, time, and infection statusa | Fold inductionb |
|||||
|---|---|---|---|---|---|---|
| MK sol. treat. | MK treat. | Zil. sol. treat. | Zil. treat. | sh-C | sh-5LO | |
| IFN-γ | ||||||
| 4 h, uninf. | 1.2 | 0.9 | 1.2 | 1.1 | 1.3 | 1.1 |
| 4 h, inf. | 1.5 | 2.3 | 1.8 | 2.3 | 1.9 | 3.4 |
| 8 h, uninf. | 1.4 | 1.2 | 1.3 | 0.9 | 1.5 | 1.1 |
| 8 h, inf. | 2.5 | 2.4 | 2.5 | 4.6 | 2.7 | 3.8 |
| 24 h, uninf. | 1.5 | 1.1 | 1.4 | 1.1 | 1.4 | 0.8 |
| 24 h, Inf. | 2.3 | 3.8 | 2.6 | 4.8 | 2.3 | 4.8 |
| 48 h, uninf. | 1.6 | 1.1 | 1.5 | 1.1 | 1.1 | 0.7 |
| 48 h, inf. | 2.2 | 3.9 | 2.4 | 4.7 | 2.0 | 5.4 |
| IL-2 | ||||||
| 4 h, uninf. | 1.2 | 1.1 | 1.1 | 1.1 | 1.1 | 1.2 |
| 4 h, inf. | 1.8 | 1.3 | 1.2 | 1.3 | 1.7 | 1.7 |
| 8 h, uninf. | 1.2 | 1.1 | 1.2 | 1.1 | 1.2 | 1.2 |
| 8 h, inf. | 2.5 | 2.4 | 2.3 | 2.9 | 1.9 | 3.3 |
| 24 h, uninf. | 1.2 | 1.1 | 1.3 | 1.2 | 1.1 | 1.1 |
| 24 h, inf. | 2.4 | 2.8 | 2.7 | 3.3 | 2.5 | 3.5 |
| 48 h, uninf. | 1.2 | 1.1 | 1.3 | 0.8 | 1.1 | 0.7 |
| 48 h, inf. | 2.0 | 3.7 | 2.2 | 3.7 | 2.7 | 4.3 |
| TNF-α | ||||||
| 4 h, uninf. | 1.4 | 1.2 | 1.3 | 1.1 | 1.5 | 0.9 |
| 4 h, inf. | 1.8 | 1.7 | 1.2 | 1.3 | 1.7 | 1.4 |
| 8 h, uninf. | 1.3 | 1.1 | 1.3 | 1.1 | 1.4 | 1.1 |
| 8 h, inf. | 2.5 | 2.4 | 2.3 | 2.6 | 1.9 | 1.3 |
| 24 h, uninf. | 1.4 | 1.1 | 1.5 | 1.1 | 1.6 | 1.1 |
| 24 h, inf. | 2.4 | 1.8 | 2.7 | 2.1 | 2.5 | 1.8 |
| 48 h, uninf. | 1.1 | 1.1 | 1.4 | 1.1 | 1.5 | 1.1 |
| 48 h, inf. | 2.5 | 2.2 | 2.5 | 1.7 | 2.7 | 1.9 |
uninf., uninfected; inf., infected.
The fold induction at different time points was calculated by considering the secretion at 0 h to be 1-fold. Boldface data indicate ∼2- or more than 2-fold induction. MK sol. treat., MK866 solvent treatment; MK treat., MK866 treatment; Zil. sol. treat., zileuton solvent treatment; Zil. treat., zileuton treatment.
5LO inhibition drastically reduced the secretion of TH2-related anti-inflammatory cytokines IL-10 (Fig. 9D) and IL-4 (Fig. 9E), suggesting that 5LO augments the release of TH2-related cytokines. 5LO inhibition had almost no effect on the release of TH2-related anti-inflammatory cytokine IL-5 (Fig. 9F). We also observed a decrease in the secretion of TH2 cytokines upon 5LO inhibition in the uninfected cells (Table 2). Since uninfected cells had minimal changes in the level of 5LO and subsequent induction of cytokines involved in the TH2 response, the percent inhibition observed was also very low (Table 2). Collectively, our results suggest that KSHV infection induces 5LO to help maintain the TH2-related cytokine microenvironment to evade host immune responses and to maintain viral latency.
TABLE 2.
Effect of 5LO pathway inhibition on TH2 cytokine secretion in uninfected and KSHV-infected cells at different time points
| TH2 cytokine, time, and infection statusa | Fold induction (% inhibition)b |
|||||
|---|---|---|---|---|---|---|
| MK sol. treat. | MK treat. | Zil. sol. treat. | Zil. treat. | sh-C | sh-5LO | |
| IL-10 | ||||||
| 4 h, uninf. | 1.2 | 1.1 (9) | 1.4 | 1.1 (22) | 1.4 | 0.9 (36) |
| 4 h, inf. | 1.8 | 1.3 (28) | 2.2 | 1.3 (51) | 2.8 | 1.4 (50) |
| 8 h, uninf. | 1.3 | 1.1 (16) | 1.3 | 1.2 (8) | 1.2 | 0.8 (34) |
| 8 h, inf. | 4.5 | 1.4 (60) | 4.3 | 1.6 (63) | 3.9 | 1.3 (66) |
| 24 h, uninf. | 1.6 | 0.9 (44) | 1.8 | 1.1 (39) | 1.9 | 1.1 (43) |
| 24 h, inf. | 5.4 | 1.8 (67) | 5.7 | 1.8 (69) | 5.5 | 1.4 (75) |
| 48 h, uninf. | 1.7 | 1.2 (30) | 1.7 | 1.1 (36) | 1.4 | 0.7 (50) |
| 48 h, inf. | 5.0 | 1.7 (66) | 5.2 | 1.7 (68) | 5.7 | 1.9 (66) |
| IL-4 | ||||||
| 4 h, uninf. | 1.2 | 1.1 (9) | 1.2 | 1.1 (9) | 1.3 | 0.8 (39) |
| 4 h, inf. | 1.5 | 1.5 | 1.8 | 1.8 | 1.9 | 1.7 (11) |
| 8 h, uninf. | 1.4 | 0.8 (43) | 1.3 | 1.1 (16) | 1.7 | 1.2 (30) |
| 8 h, inf. | 2.5 | 1.4 (44) | 2.5 | 1.4 (54) | 2.7 | 1.3 (52) |
| 24 h, uninf. | 1.4 | 1.2 (15) | 1.5 | 1.1 (17) | 1.5 | 1.1 (27) |
| 24 h, inf. | 2.3 | 1.1 (53) | 2.6 | 1.4 (46) | 2.3 | 1.4 (40) |
| 48 h, uninf. | 1.1 | 0.8 (28) | 1.2 | 0.8 (34) | 1.3 | 0.7 (47) |
| 48 h, inf. | 2.2 | 1.3 (41) | 2.4 | 1.3 (46) | 2.0 | 1.5 (25) |
| IL-5 | ||||||
| 4 h, uninf. | 1.2 | 0.8 (34) | 1.3 | 0.9 (31) | 1.3 | 0.9 (31) |
| 4 h, inf. | 2.0 | 2.3 | 2.2 | 2.3 | 2.4 | 2.2 (9) |
| 8 h, uninf. | 1.2 | 0.9 (25) | 1.3 | 1.1 (9) | 1.4 | 0.8 (43) |
| 8 h, inf. | 2.3 | 2.4 | 2.2 | 1.9 (14) | 2.2 | 2.1 (5) |
| 24 h, uninf. | 1.2 | 1.1 (9) | 1.5 | 0.9 (40) | 1.2 | 0.8 (34) |
| 24 h, inf. | 2.4 | 1.8 (25) | 2.2 | 1.8 (19) | 2.4 | 1.8 (25) |
| 48 h, uninf. | 1.3 | 1.1 (16) | 1.2 | 0.9 (25) | 1.2 | 0.8 (34) |
| 48 h, inf. | 2.0 | 1.7 (15) | 2.1 | 1.7 (20) | 2.2 | 1.9 (14) |
uninf., uninfected; inf., infected.
The fold induction at different time points was calculated by considering the secretion at 0 h to be 1-fold. Percent inhibition of TH2-related cytokine secretion was calculated by considering the levels or the percent induction in solvent-treated or sh-C-treated cells at the respective time points to be 100%. Boldface data indicate ∼2- or more than 2-fold induction. MK sol. treat., MK866 solvent treatment; MK treat., MK866 treatment; Zil. sol. treat., zileuton solvent treatment; Zil. treat., zileuton treatment.
5LO inhibition blocks the monocyte adhesion induced by KSHV-infected cell supernatant.
LTs are found at high levels in most inflammatory lesions and contribute to the physiological changes characteristic of the inflammatory process (41, 42, 50–54). LTs are lipid-signaling molecules which initiate and amplify innate and adaptive immunity (41, 42, 50–54). LTB4 acts not only on neutrophils and eosinophils but also on monocytes, macrophages, T cells, and ECs to promote transmigration, activation, and/or effector functions and participates actively in tissue inflammation (36–43). LTB4 can also induce B cell activation and proliferation (36–43). LTs are lipid-signaling molecules derived from arachidonic acid that initiate and amplify innate and adaptive immune responses by orchestrating the recruitment and activation of leukocytes in inflamed tissues.
To address the role of LTB4 in the infected cell supernatants in monocyte adhesion, we collected supernatants from uninfected or KSHV-infected HMVEC-d at the 8-h, 24-h, and 48-h time points. We purified monocytes (Fig. 10B) from the blood of healthy volunteers and labeled those with LeukoTracker solution. An equal number of labeled monocytes was resuspended in various supernatants obtained from uninfected or infected cells (8 h, 24 h, and 48 h). These monocytes were allowed to adhere to the EC monolayer for 60 min, and adhesion was quantitated as described in the Materials and Methods section and shown in the schematic in Fig. 10C. The difference in the level of monocytes that adhered in the presence of supernatants obtained from uninfected cells or infected cells was low (24 h) to negligible (8 h). In contrast, we observed a drastic increase in monocyte adhesion in the presence of supernatants obtained from KSHV-infected HMVEC-d at 48 h (Fig. 10D).
FIG 10.

Effect of 5LO pathway on monocyte adhesion, recruitment, and transendothelial migration. (A) Schematic of monocyte recruitment, rolling, adhesion, and transendothelial migration during inflammation. (B) Purification of monocytes as described in Materials and Methods. FITC, fluorescein isothiocyanate. (C) Schematic of monocyte adhesion assay. (D) Supernatants from HMVEC-d uninfected or KSHV infected (30 DNA copies/ml) for 8 h, 24 h, and 48 h were assessed for their ability to stimulate monocyte adhesion, as described in Materials and Methods. (E) The effect of 5LO inhibition on monocyte adhesion was examined by using supernatants from chemical inhibitor-treated or 5LO-silenced cells, as described in Materials and Methods. (F) Schematic of monocyte recruitment assay. (G) Supernatants from HMVEC-d uninfected or KSHV infected (30 DNA copies/ml) for 8 h, 24 h, and 48 h were assessed for their chemotactic potential to recruit monocytes, as described in Materials and Methods. (H) The effect of 5LO inhibition on the chemotactic potential of infected cell supernatants to recruit monocytes was evaluated by using supernatants from chemical inhibitor-treated or 5LO-silenced cells, as described in Materials and Methods. (I) Schematic of monocyte transendothelial migration assay. (J) Supernatants from HMVEC-d uninfected or KSHV infected (30 DNA copies/ml) for 8 h, 24 h, and 48 h were assessed for their potential to mediate transendothelial migration of monocytes, as described in Materials and Methods. (K) The effect of 5LO inhibition on monocyte transendothelial migration was evaluated by using supernatants from chemical inhibitor-treated or 5LO-silenced cells, as described in Materials and Methods. (D, E, G, H, J, and K) The fluorescence associated with the adhered, recruited, or transendothelial migrated monocytes is shown, and the values correspond to the mean ± SD of three independent experiments. supnts., supernatants; Fluoresc., fluorescent. The statistical analysis was carried out using a two-tailed Student's t test. *, P < 0.05; **, P < 0.01.
To ensure that monocyte adhesion was largely due to the presence of LTB4 levels in the infected cell supernatants, we performed the monocyte adhesion assay in the presence of supernatants obtained from solvent (MK866 or zileuton)-treated, inhibitor (MK866 or zileuton)-treated, or sh-C- or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. We observed reduced monocyte adhesion in the presence of supernatants obtained from inhibitor (MK866 or zileuton)-pretreated or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. A drastic decrease in monocyte adhesion was observed in the presence of supernatants obtained from cells pretreated with inhibitor (MK866 or zileuton) or transduced with sh-5LO and then infected with KSHV for 48 h. All these results suggest that the LTB4 generated later during KSHV infection favors monocyte adhesion to the ECs (Fig. 10E).
5LO inhibition blocks monocyte recruitment induced by KSHV-infected cell supernatant.
Next, to evaluate the role of LTB4 in the infected cell supernatants in monocyte recruitment, we collected supernatants from uninfected HMVEC-d or HMVEC-d infected with KSHV for 8 h, 24 h, and 48 h, purified the monocytes, and labeled the monocytes with LeukoTracker. Equal numbers of labeled monocytes resuspended in serum-free medium were allowed to migrate to the lower chamber in the presence of supernatants (used as the chemoattractant) obtained from the uninfected or KSHV-infected HMVEC-d at the 8-h, 24-h, and 48-h time points. Increased migration of monocytes from the upper chamber to the lower chamber containing various supernatants is an index of increased chemoattractant levels in the medium. Monocyte recruitment was quantitated as described in Materials and Methods and in the schematic in Fig. 10F. The difference in the level of monocytes recruited in the presence of supernatants obtained from uninfected cells or infected cells was low (24 h) to negligible (8 h). In contrast, we observed an increase in monocyte recruitment in the presence of supernatants obtained from HMVEC-d infected with KSHV for 48 h (Fig. 10G).
To evaluate the role of the LTB4 present in the infected cell supernatants in monocyte recruitment, we performed the monocyte chemotaxis assay in the presence of supernatants obtained from solvent (MK866 or zileuton)-treated, inhibitor (MK866 or zileuton)-treated, or sh-C- or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. We observed reduced monocyte migration in the presence of supernatants obtained from inhibitor (MK866 or zileuton)-pretreated or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. A substantial decrease in monocyte migration was observed in the presence of supernatants obtained from cells pretreated with inhibitor (MK866 or zileuton) or transduced with sh-5LO and then infected with KSHV for 48 h. All these results suggest that LTB4 enhances monocyte recruitment to the ECs by 48 h of primary infection (Fig. 10H).
5LO inhibition blocks the monocyte transendothelial migration induced by KSHV-infected cell supernatant.
ECs play a pivotal role in the regulation of leukocyte recruitment. Under conditions of inflammation, ECs express receptors, which initiate the capture and rolling adhesion of migrated leukocytes, and also synthesize and present chemokines and other chemotactic agents that can induce stable adhesion between leukocyte integrins and their corresponding EC receptors, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), promoting subsequent migration into tissue. To examine the role of LTB4 in the infected cell supernatants in monocyte transendothelial migration, we collected supernatants from uninfected or KSHV-infected HMVEC-d. We purified monocytes and labeled those with LeukoTracker. Equal numbers of labeled monocytes resuspended in serum-free medium were allowed to migrate to the lower chamber through the EC monolayer in the presence of supernatants (used as the chemoattractant) obtained from uninfected or KSHV-infected HMVEC-d for 8 h, 24 h, and 48 h. Transendothelial monocyte migration was quantitated as described in Materials and Methods and in the schematic in Fig. 10I. The difference in the level of monocytes that migrated through the EC monolayer in the presence of supernatants obtained from uninfected cells or infected cells was low (24 h) or negligible (8 h). In contrast, we observed an increase in transendothelial monocyte migration in the presence of supernatants obtained from HMVEC-d infected with KSHV for 48 h (Fig. 10J).
To evaluate the role of the LTB4 present in the infected cell supernatants in monocyte recruitment, we performed the monocyte chemotaxis assay in the presence of supernatants obtained from solvent (MK866 or zileuton)-treated, inhibitor (MK866 or zileuton)-treated, or sh-C- or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. A significant reduction in monocyte migration was observed in the presence of supernatants obtained from inhibitor (MK866 or zileuton)-pretreated or sh-5LO-transduced cells infected with KSHV for 8 h, 24 h, and 48 h. A drastic decrease in monocyte migration was observed in the presence of supernatants obtained from cells pretreated with inhibitor (MK866 or zileuton) or transduced with sh-5LO and then infected with KSHV for 8 h, 24 h, and 48 h. All these results suggest that LTB4 enhances transendothelial monocyte migration to the ECs by 48 h of primary infection (Fig. 10K).
Inhibition of 5LO blocks lipogenesis at later time points of KSHV infection.
It has been reported that fatty acid synthesis is required for the survival of KSHV latently infected ECs and that inhibition of key enzymes in this pathway leads to the apoptosis of infected cells (80). Addition of palmitic acid to latently infected cells treated with a fatty acid synthesis inhibitor protected the cells from death, indicating that the products of this pathway are essential (80). KSHV infection has been shown to lead to elevated levels of over half of the detectable metabolite products of de novo fatty acid synthesis (lipogenesis) and was concurrent with increased lipid synthesis in latently infected ECs. KSHV gene expression has been shown to regulate lipogenesis and support latency (80). FASN, a complex multifunctional enzyme, is a 250- to 270-kDa multifunctional, homodimeric enzyme responsible for energy storage by converting excess carbohydrate to fatty acids that are then sterified to store triacylglycerols (60). 5LO is also known as a lipid-metabolizing enzyme (60, 81). Its LTB4 metabolite is established to induce transcription factors, such as STAT1, STAT3, NF-κB, peroxisome proliferator-activated receptors (PPARs), and early growth response protein 1 (EGR-1), which play significant roles in lipid metabolism and cancer. LTB4, the final metabolite in the 5LO pathway, is able to enhance proliferation, increase survival, and suppress the apoptosis of human cells. Blockade of the LTB4-signaling pathway induced apoptosis in colon cancer cells (82).
To evaluate the status of FASN gene expression during de novo KSHV infection, we infected HMVEC-d for 2 h, 4 h, 8 h, 24 h, and 48 h. KSHV ORF73 gene expression, as assessed by quantitative RT-PCR (qRT-PCR) as well as by real-time RT-PCR with ORF73 gene-specific primers and TaqMan probes (data not shown), confirmed the successful infection of ECs (HMVEC-d). Compared to uninfected HMVEC-d, KSHV infection induced expression of FASN (Fig. 11A). Higher expression of FASN was observed at later time points (8 h and onwards) of infection (Fig. 11A). Similarly, compared to TIVE cells, TIVE-LTC cells showed increased expression of FASN (Fig. 11B), further confirming the high FASN expression during KSHV latency.
FIG 11.

Effect of 5LO and LTB4 on fatty acid synthase gene expression. (A) FASN gene expression during primary infection of HMVEC-d. Cells grown to 80 to 90% confluence were serum starved for 8 h and infected with KSHV (30 DNA copies/cell) for different times. At different time points postinfection (up to 48 h), total RNA was isolated, DNase I treated, and then subjected to qRT-PCR using FASN-specific primers. (B) Expression of FASN in TIVE and TIVE-LTC cells. TIVE and TIVE-LTC cells were used to prepare total RNA, and the expression of FASN was analyzed using FASN-specific primers. (C, D) FASN expression in 5LO inhibitor-treated or 5LO-silenced and then KSHV-infected HMVEC-d (C) or 5LO inhibitor-treated or 5LO-silenced TIVE-LTC cells (D) was measured by the real-time PCR methods described in the text. Each bar represents the average ± SD of three independent experiments. (E) Lysates from sh-C- or Sh-5LO-transduced TIVE-LTC cells (a), BC-3 cells (b), or BCBL-1 cells (c) were Western blotted for 5LO and FASN, stripped, and immunoblotted for β-actin, and representative blots from three independent experiments are shown. (F) Effect of KSHV gene expression on FASN promoter activities. 293 cells were transfected with the control basic vector pGL3 or WT FASN promoter luciferase constructs. After 24 h, cells were serum starved, infected for different times, lysed, and assayed for luciferase activity as described previously. Data are expressed as the mean numbers of RLU after normalization with the cotransfected Renilla luciferase activity. Each reaction was done in triplicate, and each point represents the average ± SD of three independent experiments. (G) Effects of various concentrations of exogenous LTB4 on the FASN promoter. 293 cells transfected with WT FASN promoter luciferase constructs were serum starved (24 h) and stimulated with various concentrations of LTB4 for different times, lysed, and assayed for luciferase activity, as described in the text. Data are expressed as the mean numbers of RLU after normalization by cotransfected Renilla luciferase activity. Each reaction was done in triplicate, and each point represents the average ± SD of three independent experiments. (H) Effect of LTB4 in KSHV-infected culture supernatants on FASN promoter activity. Supernatants obtained from HMVEC-d treated with 5LO inhibitor or silenced for 5LO prior to KSHV infection were used to assess FASN promoter activation. 293 cells were transfected with control PGL3 basic and WT FASN luciferase constructs. After 24 h, cells were serum starved (10 h) and incubated with various supernatants obtained from uninfected or infected cells (as described above). Data are expressed as the mean numbers of RLU after normalization by the cotransfected Renilla luciferase activity. Each reaction was done in triplicate, and each point represents the average ± SD of three independent experiments. Percent inhibition was calculated by taking the level of FASN promoter activity in the presence of supernatant obtained from either untreated, solvent-treated, or sh-C-transduced cells to be 100%. The statistical analysis was carried out using a two-tailed Student's t test. *, P < 0.05; **, P < 0.01.
To analyze the potential role of the 5LO pathway in regulating KSHV infection-induced FASN gene expression, HMVEC-d were preincubated with inhibitory/noncytotoxic concentrations of either MK866, zileuton, or their respective solvent controls at 37°C for 1 h, infected with KSHV for different times (8 h, 24 h, and 48 h), and then quantitated by real-time RT-PCR. Similarly sh-C- or sh-5LO-transduced HMVEC-d were infected with KSHV for different times (8 h, 24 h, and 48 h). Inhibition of 5LO by chemical inhibitor (MK866 and zileuton) treatment or by 5LO gene silencing effectively reduced FASN gene expression (Fig. 11C). We evaluated the effect of 5LO inhibition (by chemical inhibitor treatment or 5LO silencing) on FASN gene expression in TIVE-LTC cells. MK866 and zileuton treatment or 5LO gene silencing markedly reduced FASN gene expression in TIVE-LTC cells (Fig. 11D). 5LO silencing also reduced the protein levels of FASN in TIVE-LTC, BC-3, and BCBL-1 cells (Fig. 11E). All these results suggest a strong connection between the 5LO pathway status, viral latent gene expression, and fatty acid synthesis.
To evaluate the transcriptional regulation of FASN during KSHV infection, we studied FASN promoter activation in transiently transfected 293 cells. Compared to the results obtained with the promoterless basic vector (pGL3), the FASN full-length promoter construct containing the human fatty acid synthase gene (WT FASN) was significantly activated during KSHV infection at the indicated time points compared to the levels in the respective uninfected cells (Fig. 11F). Exogenous LTB4 supplementation has been shown to stimulate FASN promoter activity (60) in many cancers. Therefore, we hypothesized that LTB4 secreted in the supernatant of KSHV-infected cells might be contributing to the induction of FASN transcriptional regulation. To investigate the direct role of exogenous LTB4 on the FASN promoter, we stimulated 293 cells transfected with the WT FASN promoter with various concentrations (50 to 200 pg/ml) of LTB4 for different times (Fig. 11G). We observed that LTB4 in amounts even as low as 50 pg/ml was sufficient to induce FASN promoter activity (Fig. 11G). These results also indicate the specificity of LTB4-mediated FASN transcriptional regulation.
To evaluate the role of LTB4 present in the KSHV-infected culture supernatant in FASN transcriptional regulation, we used supernatants collected from HMVEC-d (solvent control pretreated, 5LO inhibitor pretreated, and 5LO silenced) and then KSHV infected (Fig. 11H) by the methods described before (19). Effective reduction was observed in the presence of supernatants obtained from cells pretreated with 5LO inhibitors or silenced for 5LO (Fig. 11H). Though inhibition of 5LO primarily affects LTB4 levels, the possibility that it affects the secretion of cytokines cannot be ruled out. KSHV-infected culture supernatants had no effect on the activity of the promoterless PGL3 vector (data not shown).
DISCUSSION
KS, a chronic inflammation-associated tumor, and its progression are believed to be profoundly driven by the autocrine and paracrine loops of inflammatory cytokines and growth factors present in the lesion microenvironment (4). To establish a successful lifelong infection in an immunocompetent host, KSHV has to evade innate and adaptive immune responses and control host immune surveillance. Our novel findings in this study provide substantial evidence that KSHV utilizes the 5LO/LTB4 cascade as one of the key immunomodulatory mechanisms to evade the host immune system. Our study underscores the importance of the 5LO/LTB4 axis in KS/PEL pathogenesis and suggests that clinically approved 5LO inhibitors have tremendous therapeutic potential in controlling KSHV-linked malignancies.
Status of LO/LTB4 cascade components during KSHV infection.
The abundant expression of 5LO and LTA4H in human KS lesions and latently infected ECs and the significant level of induction of 5LO and LTA4H expression during de novo infection of ECs (Fig. 1 and 2) with subsequent secretion of LTB4 (Fig. 3) emphasize that 5LO/LTB4 cascade components are actively involved in KS/PEL pathogenesis and strengthen the role of 5LO/LTB4 in KSHV biology. To our knowledge, this is the first report of 5LO expression in TIVE-LTC cells, PEL cells, and de novo-infected ECs (Fig. 4A to F). Strikingly, we observed that PEL cells secrete higher levels of LTB4 than latently infected ECs (when the number of cells is considered to be constant). This might be due to either cell type differences or KSHV latent infection in all PEL cells.
The underlying role of 5LO in the growth of several tumor types, including pancreatic, colorectal, prostate, and breast cancers (41, 42, 50–54), has been reported. Numerous studies demonstrated overexpression of 5LO, especially increased nuclear levels in tissue samples of primary tumor cells as well as in established cancer cell lines (41, 42, 50–54). 5LO is localized in the nucleus and cytosol of resting cells, but on cellular activation, the enzyme translocates to the nuclear envelope, which initiates its association with a novel 18-kDa membrane protein called FLAP. Surprisingly, we observed higher nuclear expression of 5LO in TIVE-LTC cells, PEL cells, and de novo-infected ECs (Fig. 4A to F). Interestingly, TIVE-LTC and PEL cells showed some indication of 5LO and FLAP interaction, suggesting an active 5LO pathway (Fig. 5) which might lead to high levels of LTB4 secretion (Fig. 3). 5LO nuclear translocation and subsequent LTB4 production have been shown to be regulated by Src, ATP, reactive oxygen species (ROS), NF-κB, and Ca2+. Previous reports have shown the sustained induction of NF-κB not only during the early stages of KSHV infection but also during the establishment and subsequent maintenance of latency (18), which might be upstream of 5LO translocation and LTB4 secretion. Here we demonstrate that NF-κB induction is upstream of 5LO nuclear localization (Fig. 4H). Though we show evidence for NF-κB involvement in 5LO nuclear localization in PEL cells (Fig. 4H), the possibility that other KSHV infection-induced factors, such as Src and ROS, play a role cannot be ruled out (13, 83, 84).
Role of KSHV-induced 5LO/LTB4 cascade in the viral life cycle.
KS is an enigma among the known human tumors, due to its multifocal nature and the variety of cell types, such as spindle-shaped cells and inflammatory cells, in the lesions. KSHV ORF73, ORF72, K13, and K12 were detected in vascular endothelial and spindle cells of KS lesions, and the lytic cycle proteins were detected in about 1 to 10% of infiltrating monocytic cells of KS lesions (10, 85–90). Hence, to effectively control and eliminate KS, it is important to understand the relationship between the timeline of viral gene expression and the consequent secretion of the inflammatory molecules required for the progression of KS lesions. Our studies show that LO inhibition downregulated KSHV latent ORF73 gene expression and upregulated major lytic switch ORF50 gene expression during de novo KSHV infection (Fig. 7). Interestingly, 5LO inhibition drastically downregulated KSHV immunomodulatory genes (K5, vMIP-1, and vMIP-2) (Fig. 7), which strongly suggests the induction of the 5LO/LTB4 cascade as KSHV's strategy to create an environment conducive to its host immune evasion (71, 91). ORF50/RTA activates the KSHV immediate early (K8, K5, K2, K12, Nut1, and ORFs 6, 57, and 74), early (K9, ORF59, ORF65, and K3), and late (K1, K8.1A, and ORF21) lytic genes, either by itself or in synergy with other viral genes (71). Here, we observed that blocking 5LO-induced ORF50 expression resulted in a massive reduction in K5 gene expression which could be explained by the ORF50-independent expression of K5 (71). The dramatic reduction in K5, vMIP-1, and vMIP-2 gene expression upon 5LO blocking is very exciting, and further studies are ongoing to decipher the mechanism of regulation of these genes via 5LO. These studies would provide new targets and better insight to understand an important avenue used by infected cells to escape immune surveillance.
LTB4 is known to stimulate several signaling cascades by extracellular signal-regulated kinases 1 and 2, Jun N-terminal protein kinases 1 and 2, and phosphoinositide 3-kinase/Akt) (92). LTB4 stabilizes NADPH oxidase-derived ROS (93), enhances the secretion of superoxide anions (94), and modulates the activation of transcription factors (NF-κB, STAT1, STAT3, EGR-1, c-Fos, c-Jun) (92, 95). It is interesting to note that some of the LTB4-activated transcription factors (NF-κB, STAT1, STAT3, EGR-1, c-Fos, and c-Jun) are established for their role(s) in the modulation of viral latency (ORF73) and lytic promoter activity (ORF50) (13, 58). Therefore, LTB4 in the infected cell microenvironment could be responsible for the transcriptional regulation of viral genes.
Our results indicate a strong interrelation of 5LO and KSHV latency in PEL cells. We observed a significant reduction in vCyclin and LANA-2 gene expression (Fig. 8A to D) and protein levels (Fig. 8I and J) in BCBL-1 and BC-3 PEL cell lines. This decrease was not accompanied by cell death, but treatment with the drugs for 3 or 5 days induced cell death (data not shown). Since vCyclin is a positive cell cycle regulator that favors G1 progression and is critical for the survival of infected cells, we are currently studying the mechanism of 5LO/LTB4-mediated downregulation of vCyclin and its effect on viral latency. Nucleophosmin (NPM), a multifunctional nuclear phosphoprotein and a histone chaperone implicated in chromatin organization and transcription control, has been reported to be a novel link between LANA/vCyclin and KSHV latency. NPM phosphorylation by the vCyclin–cyclin-dependent kinase 6 complex supports its interaction with LANA and enables the transcriptional silencing of the KSHV lytic genes needed for latency (96). Interestingly, NPM phosphorylation has been shown to be regulated by lipoxygenases (97). Further studies are ongoing to decipher the role of lipoxygenases in manipulating NPM to regulate viral latency.
Besides overcoming host intrinsic, innate, and adaptive immune responses, survival of latently infected cells requires the constant blockage of apoptosis. 5LO inhibition significantly downregulated LANA-2 gene expression and its protein levels in PEL (BCBL-1 and BC-3) cells. LANA-2 inhibits apoptosis induced by p53 (and is also called a p53 antagonist) and is absolutely required for the viability and proliferation of PEL cells. LANA-2 inhibits the transcriptional repression of the survivin promoter mediated by PML (tumor suppressor), disrupts promyelocytic leukemia (PML) nuclear bodies, and contributes to cellular transformation and carcinogenesis. Interestingly, 5LO has been shown to selectively block p53-induced apoptotic pathways by reducing p53 relocalization within PML nuclear bodies and inhibit the physical interaction between p53 and PML in many cancers. Therefore, it would be important to evaluate whether KSHV utilizes 5LO induction (i) to block p53-induced apoptotic pathways; (ii) to inhibit the interaction between P53 and PML nuclear bodies; and (iii) to manipulate LANA-2 to disrupt PML nuclear bodies, block their antiviral defense functions, and induce transformation and tumorigenesis.
Role of the KSHV-induced 5LO/LTB4 cascade on the balance of TH1/TH2 cytokines.
Escape from antigen presentation is a central strategy of evasion from the adaptive immune response used by KSHV (98). KSHV immunomodulatory genes and inflammatory cytokine (IC)-rich infected cell microenvironments orchestrate host cell immune evasion. The KS lesion microenvironment is enriched with proangiogenic factors, ICs (IL-1β, IL-6, IFN-γ, TNF, granulocyte-macrophage colony-stimulating factor), and chemokines (monocyte chemoattractant protein 1, IL-8), which play key roles in the growth, survival, and spread of infected cells (7, 86). Cytokines play important roles in viral immune evasion and lytic replication. ICs like IL-1β, IL-6, and TNF-α have been shown to inhibit KSHV lytic gene transcription in ECs (99), and KSHV de novo infection of human ECs provides a good in vitro model for studying the viral and host factors involved in the establishment and maintenance of latent infection.
Our comprehensive investigation of TH1/TH2 cytokines illustrates that cytokine secretion upon KSHV infection is not a random event but follows tightly regulated kinetics at different time points of KSHV infection (Fig. 9; Tables 1 and 2). It also indicates that regulation of cytokines via 5LO is highly selective (Fig. 9; Tables 1 and 2). For example, the levels of TH1 cytokines IFN-γ and IL-2 were induced by 5LO blockade, whereas TH2 cytokines such as IL-10 and IL-4 were significantly inhibited by 5LO blockade. We did not observe any significant change in the level of TNF-α (TH1) or IL-5 (TH2) secretion upon 5LO inhibition. A pathogenic role of IFN-γ in KSHV reactivation and KS progression has been suggested (100), and IFN-γ has been shown to induce ECs to acquire the phenotypic and functional characteristics of KS spindle cells and to induce angiogenic lesions (101, 102). Since we observed that IFN-γ levels were tightly regulated by LTB4, it is possible that 5LO/LTB4 plays important roles in KSHV reactivation and lytic replication. 5LO inhibition could not impair cytokine secretion by 100%, which strongly suggests the role of viral and other host factors in controlling the TH1/TH2-related cytokine balance.
Role of KSHV-induced 5LO/LTB4 in guiding monocyte recruitment and extravasation.
KS, a highly disseminated tumor, is often involved with visceral organs, causing KS edema in patients at advanced stages, and is associated with a poor prognosis (103–105). KS, the most common AIDS-associated malignancy, is a multifocal tumor characterized by deregulated angiogenesis, proliferation of spindle cells, and extravasation of inflammatory cells (monocytes) and red blood cells (103–105). In KS lesion cells, KSHV is in a latent form with about 10 to 20 copies of the viral episome per cell, and lytic replication is observed in a low percentage of infiltrating monocytes. The recruitment of immune cells (monocytes and neutrophils) into peripheral tissues is choreographed by chemoattractants, a chemically diverse group of molecular guidance signals, including lipid mediator LTB4. LTB4 is known to stimulate the synthesis of interleukins (IL-6, IL-8) and monocyte chemoattractant protein 1 (106). To date, the effect of KSHV infection on the ability of ECs to recruit monocytes has not been much studied. There is some evidence that KSHV augments inflammatory cell recruitment by inducing EC expression of various ICs and chemokines (107, 108). There are some reports focusing on the existence of KSHV-mediated immune evasion strategies likely to inhibit leukocyte recruitment, for example, lysosomal degradation of EC ICAM-1 and CD31 through the action of the KSHV-encoded E3 ubiquitin ligases K5 and K3 (73, 109–111). We hypothesized that the LT-rich microenvironment might be contributing to the extravasation of inflammatory cells and LTB4 augments monocyte recruitment, adhesion, and transendothelial migration. Our results showing the role of KSHV-induced LTB4 in promoting monocyte adhesion, recruitment, and transendothelial migration at later stages of infection (24 h and 48 h) (Fig. 10C to K) fit well in context with previous reports. The lower levels of monocyte adhesion, recruitment, and transendothelial migration early during infection suggest that LTB4 secretion is tightly regulated. KSHV strategically controls the LTB4 storm at early times of infection to avoid the bulk recruitment of immune cells, which thus provides enough time for the virus to establish latency.
KSHV establishes infection within monocytes in the peripheral circulation (112) as well as monocytes and their mature macrophage counterparts in KSHV-associated tumors (4, 113, 114). We speculate that the LTB4 in the infected cell microenvironment recruits monocytes to the site of infection. These monocytes can potentially get infected by KSHV, which could add to the severity of viral pathogenesis by spreading the infection. Wu et al. (2006) (115) reported the detection of CD14+/green fluorescent protein-positive cells in the bone marrow and spleens of NOD/SCID-hu mice inoculated with recombinant KSHV.219-infected hematopoietic progenitor cells. Their study suggested that monocytes/macrophages may participate in viral maintenance and dissemination in vivo and could serve as cellular reservoirs for KSHV. It is feasible that infected monocytes transport KSHV to tissues such as the skin, spread viral infection to neighboring cells, and differentiate into the latently infected spindle-like endothelial macrophages (a special subset of macrophages with endothelial features) found in KS lesions and may contribute to lymphomagenesis.
Role of KSHV-induced 5LO/LTB4 cascade on lipogenesis: an important event in latency.
Novel and very interesting studies from Bhatt et al. (2012) (116) and Delgado et al. (2012) (80) showed that the induction of fatty acid synthesis and lipogenesis is critical for the survival of KSHV latently infected cells. 5LO is a lipid-metabolizing enzyme (60, 81), and LTB4 activates transcription factor NF-κB and PPARs, which play significant roles in lipid metabolism and cancer (117). 5LO is an emerging target in obesity, insulin resistance, obesity-related fatty liver disease, metabolic dysfunction (118, 119), and nonalcoholic fatty liver and nonalcoholic steatohepatitis (120). Since 5LO/LTB4 has been shown to induce lipogenesis in human sebocytes (121), breast cancer cells (60), and hepatitis B virus X protein-mediated development of hepatocellular carcinoma (122) and since 5LO inhibitors are known to directly downregulate lipogenesis in sebocytes (121) and hepatic steatosis (119), we hypothesized that KSHV infection induces the 5LO/LTB4 cascade to enhance lipogenesis (123, 124) and favor latency. Our results show that LTB4 promotes FASN transcription and gene expression and silencing of 5LO effectively reduces the protein levels of FASN (Fig. 11), suggesting that 5LO/LTB4 induction is another strategy used to maintain KSHV latency.
Collectively, the current study unravels the complexity of the KSHV-host interactions governing KS progression/pathogenesis in conjunction with the pleiotropic effects of host factor 5LO and its chemotactic metabolite LTB4 (Fig. 12). Here, by using FDA-approved clinical inhibitors of LO/LTB4 (zileuton and MK866) or by 5LO silencing, we uncovered the biological significance of 5LO pathway induction and subsequent LTB4 secretion in the viral life cycle (latent and lytic gene expression during de novo KSHV infection in ECs and latently infected PEL cells) and pathogenesis (TH1/TH2 response, monocyte recruitment, adhesion, and transendothelial migration). We demonstrate that 5LO induction during de novo KSHV infection specifically regulates the expression of KSHV's immediate early immunomodulatory genes K5, vMIP-1, and vMIP-2. Interestingly, we deciphered the role of LTB4 in regulating KSHV-induced lipogenesis, an important event contributing to viral latency in the host cells. Current treatment strategies against KSHV-associated cancers are limited by low efficacy, and thus, there is a critical need to design therapies that can target tumor formation, eradicate the KSHV load, and control disease progression. 5LO inhibition-based therapy to target latent herpesviral persistence and pathogenesis might provide an effective way to treat the angioproliferative KS lesions and lymphoproliferative PEL malignancy. Since we have extensively shown the activation of the COX-2/PGE2 pathway during KSHV infection (13, 16, 17, 19, 20, 84), it will be important to investigate whether COX-2 and 5LO coregulate or act independently in controlling KSHV latency and pathogenesis and whether dual inhibition of COX-2 and 5LO has improved antiviral/anticancer effects in the KS and PEL tumors.
FIG 12.

Schematic diagram depicting the diverse outcomes of KSHV-induced 5LO and LTB4 in ECs and the consequences and roles of KSHV in viral pathogenesis. In vitro KSHV infection of HMVEC-d involves binding of the virus to cell surface heparan sulfate (HS) molecules via its envelope glycoproteins, followed by interaction with integrins and cystine transporter (xCT) molecules (125). Virus interaction with target cell triggers preexisting signal cascades facilitating virus entry and delivery of the viral genome into the target cell nucleus and reprograms the host gene expression required for various growth, angiogenic, and invasive factors (15). Data presented here show 5LO being one of the host factors upregulated during later time points of de novo KSHV infection of ECs. We also observed high 5LO and LTA4H gene expression in TIVE-LTC cells, PEL cells, human KS lesions, and PEL tissue sections. We show that in infected cells, 5LO, along with FLAP, catalyzes the synthesis of LTB4 from LTA4 utilizing the LTA4H enzyme. LTB4 is released to the infected cell supernatant, where it can mediate its downstream effects through either an autocrine or a paracrine mechanism on a neighboring infected or uninfected cell via interaction through the family of highly conserved seven-transmembrane G-protein-coupled rhodopsin-type LTB4R (LTB4R1 [BLT1] and LTB4R2 [BLT2]) receptors. The KSHV-induced 5LO/LTB4 cascade regulates multiple events involved in KS pathogenesis, such as the balance of TH1/TH2 cytokines, monocyte recruitment, adhesion, and transmigration. Interestingly, our study demonstrated that the LTB4 released into the infected cell microenvironment induces the FASN promoter and its gene expression to enhance lipogenesis, another pathway to promote viral latency, in the infected cells. In summary, together with the downregulation of viral K5, vMIP-1, and vMIP-2 gene expression upon 5LO/LTB4 inhibition, our study suggests that KSHV hijacks the host cellular machinery and manipulates the 5-lipoxygenase/LTB4 pathway to its advantage to aggravate pathogenesis and lipogenesis and its persistence in the target cell. Green arrows facing up, induction; PI3K, phosphoinositide 3-kinase; ERK1/2, extracellular signal-regulated kinases 1 and 2.
ACKNOWLEDGMENTS
We thank Bala Chandran and Keith Philibert for critically reading the manuscript. We gratefully acknowledge Qiang Liu from the University of Saskatchewan, Saskatoon, SK, Canada, for providing us with the wild-type FASN promoter. We gratefully acknowledge Rolf Renne (University of Florida) for providing the TIVE-LTC and TIVE cells and Blossom Damania for providing KSHV-BJAB cells. We thank the NIH AIDS Research and Reference Reagent Program for normal, KS, and solid PEL specimens. We thank Bala Chandran and his lab members for their valuable input. We thank Robert Dickinson from the RFUMS Cell Sorting Core Facility for his kind help in fluorescence-activated cell-sorting analyses. We gratefully acknowledge Sudeshna Goswami for assistance in developing a few Western blots during manuscript revision.
Footnotes
Published ahead of print 11 December 2013
REFERENCES
- 1.Boshoff C, Gao SJ, Healy LE, Matthews S, Thomas AJ, Coignet L, Warnke RA, Strauchen JA, Matutes E, Kamel OW, Moore PS, Weiss RA, Chang Y. 1998. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood 91:1671–1679 [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss RA. 1998. Kaposi's sarcoma-associated herpesvirus. Adv. Cancer Res. 75:57–86. 10.1016/S0065-230X(08)60739-3 [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. 2000. Angiogenesis in cancer and other diseases. Nature 407:249–257. 10.1038/35025220 [DOI] [PubMed] [Google Scholar]
- 4.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551. 10.1073/pnas.96.8.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fidler IJ, Singh RK, Yoneda J, Kumar R, Xu L, Dong Z, Bielenberg DR, McCarty M, Ellis LM. 2000. Critical determinants of neoplastic angiogenesis. Cancer J. 6(Suppl 3):S225–S236 [PubMed] [Google Scholar]
- 6.Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF. 2009. HIV-associated lymphomas and gamma-herpesviruses. Blood 113:1213–1224. 10.1182/blood-2008-09-180315 [DOI] [PubMed] [Google Scholar]
- 7.Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273–296. 10.1146/annurev.pathol.1.110304.100133 [DOI] [PubMed] [Google Scholar]
- 8.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McClure LV, Sullivan CS. 2008. Kaposi's sarcoma herpes virus taps into a host microRNA regulatory network. Cell Host Microbe 3:1–3. 10.1016/j.chom.2007.12.002 [DOI] [PubMed] [Google Scholar]
- 10.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, Friedman-Kien AE, Fleckenstein B. 1997. Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 71:839–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867. 10.1073/pnas.93.25.14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George Paul A, Sharma-Walia N, Kerur N, White C, Chandran B. 2010. Piracy of prostaglandin E2/EP receptor-mediated signaling by Kaposi's sarcoma-associated herpes virus (HHV-8) for latency gene expression: strategy of a successful pathogen. Cancer Res. 70:3697–3708. 10.1158/0008-5472.CAN-09-3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524–1539. 10.1128/JVI.77.2.1524-1539.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84. 10.1158/0008-5472.CAN-03-2767 [DOI] [PubMed] [Google Scholar]
- 16.Paul AG, Chandran B, Sharma-Walia N. 2013. Concurrent targeting of EP1/EP4 receptors and COX-2 induces synergistic apoptosis in KSHV and EBV associated non-Hodgkin lymphoma cell lines. Transl. Res. 161:447–468. 10.1016/j.trsl.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paul AG, Sharma-Walia N, Chandran B. 2011. Targeting KSHV/HHV-8 latency with COX-2 selective inhibitor nimesulide: a potential chemotherapeutic modality for primary effusion lymphoma. PLoS One 6:e24379. 10.1371/journal.pone.0024379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968. 10.1128/JVI.02333-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 6:e1000777. 10.1371/journal.ppat.1000777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma-Walia N, Raghu H, Sadagopan S, Sivakumar R, Veettil MV, Naranatt PP, Smith MM, Chandran B. 2006. Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J. Virol. 80:6534–6552. 10.1128/JVI.00231-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Wang S, Wu N, Sood S, Wang P, Jin Z, Beer DG, Giordano TJ, Lin Y, Shih WC, Lubet RA, Yang CS. 2004. Overexpression of 5-lipoxygenase in rat and human esophageal adenocarcinoma and inhibitory effects of zileuton and celecoxib on carcinogenesis. Clin. Cancer Res. 10:6703–6709. 10.1158/1078-0432.CCR-04-0838 [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Li D, Li S. 2009. The Alox5 gene is a novel therapeutic target in cancer stem cells of chronic myeloid leukemia. Cell Cycle 8:3488–3492. 10.4161/cc.8.21.9852 [DOI] [PubMed] [Google Scholar]
- 23.Ding XZ, Hennig R, Adrian TE. 2003. Lipoxygenase and cyclooxygenase metabolism: new insights in treatment and chemoprevention of pancreatic cancer. Mol. Cancer 2:10. 10.1186/1476-4598-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding XZ, Iversen P, Cluck MW, Knezetic JA, Adrian TE. 1999. Lipoxygenase inhibitors abolish proliferation of human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 261:218–223. 10.1006/bbrc.1999.1012 [DOI] [PubMed] [Google Scholar]
- 25.Ding XZ, Kuszynski CA, El-Metwally TH, Adrian TE. 1999. Lipoxygenase inhibition induced apoptosis, morphological changes, and carbonic anhydrase expression in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 266:392–399. 10.1006/bbrc.1999.1824 [DOI] [PubMed] [Google Scholar]
- 26.Ding XZ, Talamonti MS, Bell RH, Jr, Adrian TE. 2005. A novel anti-pancreatic cancer agent, LY293111. Anticancer Drugs 16:467–473. 10.1097/00001813-200506000-00001 [DOI] [PubMed] [Google Scholar]
- 27.Ding XZ, Tong WG, Adrian TE. 2001. 12-Lipoxygenase metabolite 12(S)-HETE stimulates human pancreatic cancer cell proliferation via protein tyrosine phosphorylation and ERK activation. Int. J. Cancer 94:630–636. 10.1002/ijc.1527 [DOI] [PubMed] [Google Scholar]
- 28.Ding XZ, Tong WG, Adrian TE. 2001. Cyclooxygenases and lipoxygenases as potential targets for treatment of pancreatic cancer. Pancreatology 1:291–299. 10.1159/000055827 [DOI] [PubMed] [Google Scholar]
- 29.Ding XZ, Tong WG, Adrian TE. 2003. Multiple signal pathways are involved in the mitogenic effect of 5(S)-HETE in human pancreatic cancer. Oncology 65:285–294. 10.1159/000074640 [DOI] [PubMed] [Google Scholar]
- 30.Gunning WT, Kramer PM, Steele VE, Pereira MA. 2002. Chemoprevention by lipoxygenase and leukotriene pathway inhibitors of vinyl carbamate-induced lung tumors in mice. Cancer Res. 62:4199–4201 [PubMed] [Google Scholar]
- 31.Li N, Sood S, Wang S, Fang M, Wang P, Sun Z, Yang CS, Chen X. 2005. Overexpression of 5-lipoxygenase and cyclooxygenase 2 in hamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clin. Cancer Res. 11:2089–2096. 10.1158/1078-0432.CCR-04-1684 [DOI] [PubMed] [Google Scholar]
- 32.Sun Z, Sood S, Li N, Ramji D, Yang P, Newman RA, Yang CS, Chen X. 2006. Involvement of the 5-lipoxygenase/leukotriene A4 hydrolase pathway in 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamster cheek pouch, and inhibition of carcinogenesis by its inhibitors. Carcinogenesis 27:1902–1908. 10.1093/carcin/bgl039 [DOI] [PubMed] [Google Scholar]
- 33.Tong WG, Ding XZ, Adrian TE. 2002. The mechanisms of lipoxygenase inhibitor-induced apoptosis in human breast cancer cells. Biochem. Biophys. Res. Commun. 296:942–948. 10.1016/S0006-291X(02)02014-4 [DOI] [PubMed] [Google Scholar]
- 34.Tong WG, Ding XZ, Talamonti MS, Bell RH, Adrian TE. 2005. LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun. 335:949–956. 10.1016/j.bbrc.2005.07.166 [DOI] [PubMed] [Google Scholar]
- 35.Tong WG, Ding XZ, Witt RC, Adrian TE. 2002. Lipoxygenase inhibitors attenuate growth of human pancreatic cancer xenografts and induce apoptosis through the mitochondrial pathway. Mol. Cancer Ther. 1:929–935 [PubMed] [Google Scholar]
- 36.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. 1980. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 286:264-265. 10.1038/286264a0 [DOI] [PubMed] [Google Scholar]
- 37.Ford-Hutchinson AW, Bray MA, Smith MJ. 1979. The aggregation of rat neutrophils by arachidonic acid: a possible bioassay for lipoxygenase activity. J. Pharm. Pharmacol. 31:868–869. 10.1111/j.2042-7158.1979.tb13687.x [DOI] [PubMed] [Google Scholar]
- 38.Ford-Hutchinson AW, Bray MA, Smith MJ. 1980. Lipoxygenase products and the polymorphonuclear leucocyte. Agents Actions 10:548–550. 10.1007/BF02024162 [DOI] [PubMed] [Google Scholar]
- 39.Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. 2003. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 4:965–973. 10.1038/ni972 [DOI] [PubMed] [Google Scholar]
- 40.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. 2003. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat. Immunol. 4:974–981. 10.1038/ni971 [DOI] [PubMed] [Google Scholar]
- 41.Tager AM, Dufour JH, Goodarzi K, Bercury SD, von Andrian UH, Luster AD. 2000. BLTR mediates leukotriene B(4)-induced chemotaxis and adhesion and plays a dominant role in eosinophil accumulation in a murine model of peritonitis. J. Exp. Med. 192:439–446. 10.1084/jem.192.3.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tager AM, Luster AD. 2003. BLT1 and BLT2: the leukotriene B(4) receptors. Prostaglandins Leukot. Essent. Fatty Acids 69:123–134. 10.1016/S0952-3278(03)00073-5 [DOI] [PubMed] [Google Scholar]
- 43.Taube C, Miyahara N, Ott V, Swanson B, Takeda K, Loader J, Shultz LD, Tager AM, Luster AD, Dakhama A, Gelfand EW. 2006. The leukotriene B4 receptor (BLT1) is required for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J. Immunol. 176:3157–3164 http://www.jimmunol.org/content/176/5/3157 [DOI] [PubMed] [Google Scholar]
- 44.Gosselin J, Borgeat P. 1997. Epstein-Barr virus modulates 5-lipoxygenase product synthesis in human peripheral blood mononuclear cells. Blood 89:2122–2130 [PubMed] [Google Scholar]
- 45.Lipschik GY, Doerfler ME, Kovacs JA, Travis WD, Andrawis VA, Lawrence MG, Dichter JR, Ognibene FP, Shelhamer JH. 1993. Leukotriene B4 and interleukin-8 in human immunodeficiency virus-related pulmonary disease. Chest 104:763–769. 10.1378/chest.104.3.763 [DOI] [PubMed] [Google Scholar]
- 46.Medina JF, Odlander B, Funk CD, Fu JY, Claesson HE, Radmark O. 1989. B-lymphocytic cell line Raji expresses the leukotriene A4 hydrolase gene but not the 5-lipoxygenase gene. Biochem. Biophys. Res. Commun. 161:740–745. 10.1016/0006-291X(89)92662-4 [DOI] [PubMed] [Google Scholar]
- 47.Volovitz B, Faden H, Ogra PL. 1988. Release of leukotriene C4 in respiratory tract during acute viral infection. J. Pediatr. 112:218–222. 10.1016/S0022-3476(88)80058-1 [DOI] [PubMed] [Google Scholar]
- 48.Volovitz B, Welliver RC, De Castro G, Krystofik DA, Ogra PL. 1988. The release of leukotrienes in the respiratory tract during infection with respiratory syncytial virus: role in obstructive airway disease. Pediatr. Res. 24:504–507. 10.1203/00006450-198810000-00018 [DOI] [PubMed] [Google Scholar]
- 49.Wahl LM, Corcoran ML, Pyle SW, Arthur LO, Harel-Bellan A, Farrar WL. 1989. Human immunodeficiency virus glycoprotein (gp120) induction of monocyte arachidonic acid metabolites and interleukin 1. Proc. Natl. Acad. Sci. U. S. A. 86:621–625. 10.1073/pnas.86.2.621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claesson HE. 2009. On the biosynthesis and biological role of eoxins and 15-lipoxygenase-1 in airway inflammation and Hodgkin lymphoma. Prostaglandins Other Lipid Mediat. 89:120–125. 10.1016/j.prostaglandins.2008.12.003 [DOI] [PubMed] [Google Scholar]
- 51.Claesson HE, Griffiths WJ, Brunnstrom A, Schain F, Andersson E, Feltenmark S, Johnson HA, Porwit A, Sjoberg J, Bjorkholm M. 2008. Hodgkin Reed-Sternberg cells express 15-lipoxygenase-1 and are putative producers of eoxins in vivo: novel insight into the inflammatory features of classical Hodgkin lymphoma. FEBS J. 275:4222–4234. 10.1111/j.1742-4658.2008.06570.x [DOI] [PubMed] [Google Scholar]
- 52.Claesson HE, Odlander B, Jakobsson PJ. 1992. Leukotriene B4 in the immune system. Int. J. Immunopharmacol. 14:441–449. 10.1016/0192-0561(92)90174-J [DOI] [PubMed] [Google Scholar]
- 53.Claesson S, Morrison A, Wertheimer AI, Berger ML. 1999. Compliance with prescribed drugs: challenges for the elderly population. Pharm. World Sci. 21:256–259. 10.1023/A:1008786004974 [DOI] [PubMed] [Google Scholar]
- 54.Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Jr, Ford-Hutchinson AW, Caskey CT, Evans JF. 1999. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399:789–793. 10.1038/21658 [DOI] [PubMed] [Google Scholar]
- 55.Goossens L, Pommery N, Henichart JP. 2007. COX-2/5-LOX dual acting anti-inflammatory drugs in cancer chemotherapy. Curr. Top. Med. Chem. 7:283–296. 10.2174/156802607779941369 [DOI] [PubMed] [Google Scholar]
- 56.Rioux N, Castonguay A. 1998. Inhibitors of lipoxygenase: a new class of cancer chemopreventive agents. Carcinogenesis 19:1393–1400. 10.1093/carcin/19.8.1393 [DOI] [PubMed] [Google Scholar]
- 57.Nun TK, Kroll DJ, Oberlies NH, Soejarto DD, Case RJ, Piskaut P, Matainaho T, Hilscher C, Wang L, Dittmer DP, Gao SJ, Damania B. 2007. Development of a fluorescence-based assay to screen antiviral drugs against Kaposi's sarcoma associated herpesvirus. Mol. Cancer Ther. 6:2360–2370. 10.1158/1535-7163.MCT-07-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308–10329. 10.1128/JVI.79.16.10308-10329.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207–4223. 10.1128/JVI.78.8.4207-4223.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu N, Li Y, Zhao Y, Wang Q, You JC, Zhang XD, Ye LH. 2011. A novel positive feedback loop involving FASN/p-ERK1/2/5-LOX/LTB4/FASN sustains high growth of breast cancer cells. Acta Pharmacol. Sin. 32:921–929. 10.1038/aps.2011.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 62.Cornelissen M, van der Kuyl AC, van den Burg R, Zorgdrager F, van Noesel CJ, Goudsmit J. 2003. Gene expression profile of AIDS-related Kaposi's sarcoma. BMC Cancer 3:7. 10.1186/1471-2407-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. 2006. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi's sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol. 80:4833–4846. 10.1128/JVI.80.10.4833-4846.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo M, Jones SM, Peters-Golden M, Brock TG. 2003. Nuclear localization of 5-lipoxygenase as a determinant of leukotriene B4 synthetic capacity. Proc. Natl. Acad. Sci. U. S. A. 100:12165–12170. 10.1073/pnas.2133253100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balcarek JM, Theisen TW, Cook MN, Varrichio A, Hwang SM, Strohsacker MW, Crooke ST. 1988. Isolation and characterization of a cDNA clone encoding rat 5-lipoxygenase. J. Biol. Chem. 263:13937–13941 [PubMed] [Google Scholar]
- 66.Dixon RA, Jones RE, Diehl RE, Bennett CD, Kargman S, Rouzer CA. 1988. Cloning of the cDNA for human 5-lipoxygenase. Proc. Natl. Acad. Sci. U. S. A. 85:416–420. 10.1073/pnas.85.2.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Funk CD. 2001. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875. 10.1126/science.294.5548.1871 [DOI] [PubMed] [Google Scholar]
- 68.Peters-Golden M, Brock TG. 2001. Intracellular compartmentalization of leukotriene synthesis: unexpected nuclear secrets. FEBS Lett. 487:323–326. 10.1016/S0014-5793(00)02374-7 [DOI] [PubMed] [Google Scholar]
- 69.Miller DK, Gillard JW, Vickers PJ, Sadowski S, Leveille C, Mancini JA, Charleson P, Dixon RA, Ford-Hutchinson AW, Fortin R, Gauthier JY, Rodkey J, Rosen R, Rouzer C, Sigal IS, Strader CD, Evans JF. 1990. Identification and isolation of a membrane protein necessary for leukotriene production. Nature 343:278–281. 10.1038/343278a0 [DOI] [PubMed] [Google Scholar]
- 70.Rouzer CA, Ford-Hutchinson AW, Morton HE, Gillard JW. 1990. MK886, a potent and specific leukotriene biosynthesis inhibitor blocks and reverses the membrane association of 5-lipoxygenase in ionophore-challenged leukocytes. J. Biol. Chem. 265:1436–1442 [PubMed] [Google Scholar]
- 71.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620. 10.1128/JVI.78.7.3601-3620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Means RE, Ishido S, Alvarez X, Jung JU. 2002. Multiple endocytic trafficking pathways of MHC class I molecules induced by a herpesvirus protein. EMBO J. 21:1638–1649. 10.1093/emboj/21.7.1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tomescu C, Law WK, Kedes DH. 2003. Surface downregulation of major histocompatibility complex class I, PE-CAM, and ICAM-1 following de novo infection of endothelial cells with Kaposi's sarcoma-associated herpesvirus. J. Virol. 77:9669–9684. 10.1128/JVI.77.17.9669-9684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Damania B. 2004. Modulation of cell signaling pathways by Kaposi's sarcoma-associated herpesvirus (KSHVHHV-8). Cell Biochem. Biophys. 40:305–322. 10.1385/CBB:40:3:305 [DOI] [PubMed] [Google Scholar]
- 75.Ensoli B, Sirianni MC. 1998. Kaposi's sarcoma pathogenesis: a link between immunology and tumor biology. Crit. Rev. Oncog. 9:107–124. 10.1615/CritRevOncog.v9.i2.20 [DOI] [PubMed] [Google Scholar]
- 76.Ensoli B, Sturzl M. 1998. Kaposi's sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 9:63–83. 10.1016/S1359-6101(97)00037-3 [DOI] [PubMed] [Google Scholar]
- 77.Stine JT, Wood C, Hill M, Epp A, Raport CJ, Schweickart VL, Endo Y, Sasaki T, Simmons G, Boshoff C, Clapham P, Chang Y, Moore P, Gray PW, Chantry D. 2000. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 95:1151–1157 [PubMed] [Google Scholar]
- 78.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. 1998. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91:3527–3561 [PubMed] [Google Scholar]
- 79.Magen E, Feldman A, Cohen Z, Alon DB, Minz E, Chernyavsky A, Linov L, Mishal J, Schlezinger M, Sthoeger Z. 2010. Circulating endothelial progenitor cells, Th1/Th2/Th17-related cytokines, and endothelial dysfunction in resistant hypertension. Am. J. Med. Sci. 339:117–122. 10.1097/MAJ.0b013e3181c6a968 [DOI] [PubMed] [Google Scholar]
- 80.Delgado T, Sanchez EL, Camarda R, Lagunoff M. 2012. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog. 8:e1002866. 10.1371/journal.ppat.1002866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y, Wang W, Wang Q, Zhang X, Ye L. 2012. Lipid metabolism enzyme 5-LOX and its metabolite LTB4 are capable of activating transcription factor NF-kappaB in hepatoma cells. Biochem. Biophys. Res. Commun. 418:647–651. 10.1016/j.bbrc.2012.01.068 [DOI] [PubMed] [Google Scholar]
- 82.Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. 2007. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J. Pharmacol. Sci. 103:24–32. 10.1254/jphs.FP0060651 [DOI] [PubMed] [Google Scholar]
- 83.Bottero V, Chakraborty S, Chandran B. 2013. Reactive oxygen species are induced by Kaposi's sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J. Virol. 87:1733–1749. 10.1128/JVI.02958-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharma-Walia N, George Paul A, Patel K, Chandran K, Ahmad W, Chandran B. 2010. NFAT and CREB regulate Kaposi's sarcoma-associated herpesvirus-induced cyclooxygenase 2 (COX-2). J. Virol. 84:12733–12753. 10.1128/JVI.01065-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175–212. 10.1128/MMBR.67.2.175-212.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ganem D. 1997. KSHV and Kaposi's sarcoma: the end of the beginning? Cell 91:157–160. 10.1016/S0092-8674(00)80398-0 [DOI] [PubMed] [Google Scholar]
- 87.Moore PS, Kingsley LA, Holmberg SD, Spira T, Gupta P, Hoover DR, Parry JP, Conley LJ, Jaffe HW, Chang Y. 1996. Kaposi's sarcoma-associated herpesvirus infection prior to onset of Kaposi's sarcoma. AIDS 10:175–180. 10.1097/00002030-199602000-00007 [DOI] [PubMed] [Google Scholar]
- 88.Schulz TF. 1998. Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8). J. Gen. Virol. 79:1573–1591 [DOI] [PubMed] [Google Scholar]
- 89.Schulz TF, Sheldon J, Greensill J. 2002. Kaposi's sarcoma associated herpesvirus (KSHV) or human herpesvirus 8 (HHV8). Virus Res. 82:115–126 [DOI] [PubMed] [Google Scholar]
- 90.Zhong W, Wang H, Herndier B, Ganem D. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U. S. A. 93:6641–6646. 10.1073/pnas.93.13.6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liang C, Lee JS, Jung JU. 2008. Immune evasion in Kaposi's sarcoma-associated herpes virus associated oncogenesis. Semin. Cancer Biol. 18:423–436. 10.1016/j.semcancer.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanchez-Galan E, Gomez-Hernandez A, Vidal C, Martin-Ventura JL, Blanco-Colio LM, Munoz-Garcia B, Ortega L, Egido J, Tunon J. 2009. Leukotriene B4 enhances the activity of nuclear factor-kappaB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovasc. Res. 81:216–225. 10.1093/cvr/cvn277 [DOI] [PubMed] [Google Scholar]
- 93.Barcellos-de-Souza P, Canetti C, Barja-Fidalgo C, Arruda MA. 2012. Leukotriene B(4) inhibits neutrophil apoptosis via NADPH oxidase activity: redox control of NF-kappaB pathway and mitochondrial stability. Biochim. Biophys. Acta 1823:1990–1997. 10.1016/j.bbamcr.2012.07.012 [DOI] [PubMed] [Google Scholar]
- 94.Steiner DR, Gonzalez NC, Wood JG. 2001. Leukotriene B(4) promotes reactive oxidant generation and leukocyte adherence during acute hypoxia. J. Appl. Physiol. 91:1160–1167 http://jap.physiology.org/content/91/3/1160.full [DOI] [PubMed] [Google Scholar]
- 95.Stankova J, Rola-Pleszczynski M. 1992. Leukotriene B4 stimulates c-fos and c-jun gene transcription and AP-1 binding activity in human monocytes. Biochem. J. 282:625–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. 2010. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 6:e1000818. 10.1371/journal.ppat.1000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thornber K, Colomba A, Ceccato L, Delsol G, Payrastre B, Gaits-Iacovoni F. 2009. Reactive oxygen species and lipoxygenases regulate the oncogenicity of NPM-ALK-positive anaplastic large cell lymphomas. Oncogene 28:2690–2696. 10.1038/onc.2009.125 [DOI] [PubMed] [Google Scholar]
- 98.Areste C, Blackbourn DJ. 2009. Modulation of the immune system by Kaposi's sarcoma-associated herpesvirus. Trends Microbiol. 17:119–129. 10.1016/j.tim.2008.12.001 [DOI] [PubMed] [Google Scholar]
- 99.Milligan S, Robinson M, O'Donnell E, Blackbourn DJ. 2004. Inflammatory cytokines inhibit Kaposi's sarcoma-associated herpesvirus lytic gene transcription in in vitro-infected endothelial cells. J. Virol. 78:2591–2596. 10.1128/JVI.78.5.2591-2596.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chang J, Renne R, Dittmer D, Ganem D. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17–25. 10.1006/viro.1999.0077 [DOI] [PubMed] [Google Scholar]
- 101.Fiorelli V, Gendelman R, Sirianni MC, Chang HK, Colombini S, Markham PD, Monini P, Sonnabend J, Pintus A, Gallo RC, Ensoli B. 1998. Gamma-interferon produced by CD8+ T cells infiltrating Kaposi's sarcoma induces spindle cells with angiogenic phenotype and synergy with human immunodeficiency virus-1 Tat protein: an immune response to human herpesvirus-8 infection? Blood 91:956–967 [PubMed] [Google Scholar]
- 102.Mesri EA. 1999. Inflammatory reactivation and angiogenicity of Kaposi's sarcoma-associated herpesvirus/HHV8: a missing link in the pathogenesis of acquired immunodeficiency syndrome-associated Kaposi's sarcoma. Blood 93:4031–4033 [PubMed] [Google Scholar]
- 103.Bower M, Nelson M, Young AM, Thirlwell C, Newsom-Davis T, Mandalia S, Dhillon T, Holmes P, Gazzard BG, Stebbing J. 2005. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J. Clin. Oncol. 23:5224–5228. 10.1200/JCO.2005.14.597 [DOI] [PubMed] [Google Scholar]
- 104.Feller L, Masipa J, Wood N, Raubenheimer E, Lemmer J. 2008. The prognostic significance of facial lymphoedema in HIV-seropositive subjects with Kaposi sarcoma. AIDS Res. Ther. 5:2. 10.1186/1742-6405-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Flore O, Rafii S, Ely S, O'Leary JJ, Hyjek EM, Cesarman E. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588–592. 10.1038/29093 [DOI] [PubMed] [Google Scholar]
- 106.Huang L, Zhao A, Wong F, Ayala JM, Struthers M, Ujjainwalla F, Wright SD, Springer MS, Evans J, Cui J. 2004. Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes. Arterioscler. Thromb. Vasc. Biol. 24:1783–1788. 10.1161/01.ATV.0000140063.06341.09 [DOI] [PubMed] [Google Scholar]
- 107.Ensoli B, Sgadari C, Barillari G, Sirianni MC, Sturzl M, Monini P. 2001. Biology of Kaposi's sarcoma. Eur. J. Cancer 37:1251–1269. 10.1016/S0959-8049(01)00121-6 [DOI] [PubMed] [Google Scholar]
- 108.Sun Q, Matta H, Lu G, Chaudhary PM. 2006. Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene 25:2717–2726. 10.1038/sj.onc.1209298 [DOI] [PubMed] [Google Scholar]
- 109.Coscoy L, Ganem D. 2001. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 107:1599–1606. 10.1172/JCI12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ishido S, Choi JK, Lee BS, Wang C, DeMaria M, Johnson RP, Cohen GB, Jung JU. 2000. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13:365–374. 10.1016/S1074-7613(00)00036-4 [DOI] [PubMed] [Google Scholar]
- 111.Manes TD, Hoer S, Muller WA, Lehner PJ, Pober JS. 2010. Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. J. Immunol. 184:5186–5192. 10.4049/jimmunol.0902938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sirianni MC, Vincenzi L, Fiorelli V, Topino S, Scala E, Uccini S, Angeloni A, Faggioni A, Cerimele D, Cottoni F, Aiuti F, Ensoli B. 1998. Gamma-interferon production in peripheral blood mononuclear cells and tumor infiltrating lymphocytes from Kaposi's sarcoma patients: correlation with the presence of human herpesvirus-8 in peripheral blood mononuclear cells and lesional macrophages. Blood 91:968–976 [PubMed] [Google Scholar]
- 113.Blasig C, Zietz C, Haar B, Neipel F, Esser S, Brockmeyer NH, Tschachler E, Colombini S, Ensoli B, Sturzl M. 1997. Monocytes in Kaposi's sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71:7963–7968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Valmary S, Richard P, Brousset P. 2005. Frequent detection of Kaposi's sarcoma herpesvirus in germinal centre macrophages from AIDS-related multicentric Castleman's disease. AIDS 19:1229–1231. 10.1097/01.aids.0000176225.56108.b2 [DOI] [PubMed] [Google Scholar]
- 115.Wu W, Vieira J, Fiore N, Banerjee P, Sieburg M, Rochford R, Harrington W, Jr, Feuer G. 2006. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood 108:141–151. 10.1182/blood-2005-04-1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, Damania B. 2012. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. U. S. A. 109:11818–11823. 10.1073/pnas.1205995109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Narala VR, Adapala RK, Suresh MV, Brock TG, Peters-Golden M, Reddy RC. 2010. Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-alpha agonist. J. Biol. Chem. 285:22067–22074. 10.1074/jbc.M109.085118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. 2010. Inflammatory lipid mediators in adipocyte function and obesity. Nat. Rev. Endocrinol. 6:71–82. 10.1038/nrendo.2009.264 [DOI] [PubMed] [Google Scholar]
- 119.Lopez-Parra M, Titos E, Horrillo R, Ferre N, Gonzalez-Periz A, Martinez-Clemente M, Planaguma A, Masferrer J, Arroyo V, Claria J. 2008. Regulatory effects of arachidonate 5-lipoxygenase on hepatic microsomal TG transfer protein activity and VLDL-triglyceride and apoB secretion in obese mice. J. Lipid Res. 49:2513–2523. 10.1194/jlr.M800101-JLR200 [DOI] [PubMed] [Google Scholar]
- 120.Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H, Watkins SM, Sanyal AJ. 2009. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 50:1827–1838. 10.1002/hep.23229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zouboulis CC, Seltmann H, Alestas T. 2010. Zileuton prevents the activation of the leukotriene pathway and reduces sebaceous lipogenesis. Exp. Dermatol. 19:148–150. 10.1111/j.1600-0625.2009.00929.x [DOI] [PubMed] [Google Scholar]
- 122.You X, Liu F, Zhang T, Li Y, Ye L, Zhang X. 2013. Hepatitis B virus X protein up-regulates oncogene Rab18 to result in the dysregulation of lipogenesis and proliferation of hepatoma cells. Carcinogenesis 34:1644–1652. 10.1093/carcin/bgt089 [DOI] [PubMed] [Google Scholar]
- 123.Wang Q, Zhang W, Liu Q, Zhang X, Lv N, Ye L, Zhang X. 2010. A mutant of hepatitis B virus X protein (HBxDelta127) promotes cell growth through a positive feedback loop involving 5-lipoxygenase and fatty acid synthase. Neoplasia 12:103–115 http://www.neoplasia.com/pdf/manuscript/v12i02/neo091298.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Q, Zhang WY, Ye LH, Zhang XD. 2010. A mutant of HBx (HBxDelta127) promotes hepatoma cell growth via sterol regulatory element binding protein 1c involving 5-lipoxygenase. Acta Pharmacol. Sin. 31:367–374. 10.1038/aps.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J. Virol. 84:2188–2199. 10.1128/JVI.01334-09 [DOI] [PMC free article] [PubMed] [Google Scholar]