

Abstract
As an alternative to targeting human immunodeficiency virus (HIV), we have developed vaccines targeting CCR5, a self-protein critically involved in HIV replication and pathogenesis. By displaying peptides derived from CCR5 at high density on the surface of virus-like particles, we can efficiently induce high-titer IgG antibodies against this self-molecule. Here, we investigated whether prophylactic immunization of rhesus macaques with a particle-based vaccine targeting two regions of macaque CCR5 could prevent or suppress vaginal infection with highly virulent SIVmac251. Twelve macaques were vaccinated with a bacteriophage Qß-based vaccine targeting macaque CCR5 (Qß.CCR5). Six control animals were immunized with the Qß platform alone. All animals immunized with Qß.CCR5 developed high-titer anti-CCR5 antibody responses. Macaques were vaginally challenged with a high dose of SIVmac251. The mean peak viral RNA levels in the vaccinated groups were 30-fold lower than in the control group (106.8 versus 108.3 copies/ml plasma). Three of the 12 vaccinated macaques dramatically suppressed simian immunodeficiency virus (SIV) replication: peak viral loads were low (103 to 104 RNA copies/ml), and SIV RNA became undetectable from 6 weeks onward. No viral RNA or DNA could be detected in colon and lymph node biopsy specimens collected 13 months after challenge. In vivo depletion of CD8+ cells failed to induce a viral rebound. However, once anti-CCR5 antibody responses had waned, the 3 animals became infected after intravaginal and/or intravenous rechallenge. In conclusion, vaccination against CCR5 was associated with dramatic suppression of virus replication in a subset (25%) of macaques. These data support further research of vaccination against CCR5 to combat HIV infection.
INTRODUCTION
Human immunodeficiency virus (HIV) sequence diversity and antigenic variation are major and perhaps insurmountable barriers that hinder the development of vaccines against the virus. As an alternative strategy to conventional HIV vaccines, we have developed a vaccine that targets CCR5, a self-molecule that is not subject to antigenic variation and is critically involved in HIV acquisition.
During infection, HIV uses chemokine receptors as coreceptors in addition to its primary receptor, CD4, to gain entry into cells (1–3). Although HIV can potentially utilize several coreceptors, the viruses isolated from infected individuals early after infection are predominantly CCR5-tropic, indicating a selective advantage for these viruses during transmission and/or during the early stages of infection (4, 5). Individuals harboring a homozygous genetic mutation of the CCR5 allele (termed Δ32) are resistant to HIV infection, and infected heterozygous individuals (who express lower levels of CCR5) progress more slowly to AIDS (6, 7). In addition, HIV-1 entry inhibitors targeting CCR5 became an important component in the arsenal of antiretroviral drugs when, in 2008, the first small-molecule CCR5 inhibitor, maraviroc (Pfizer), was approved for clinical use. Maraviroc binds to CCR5 and induces a conformational change to prevent recognition by the coreceptor binding sites present on the HIV envelope glycoprotein, gp120. HIV-infected patients receiving maraviroc monotherapy have viral loads that are dramatically decreased, often to undetectable levels (8–10). These data, in addition to the effects of the Δ32 mutation on HIV infection, indicate that a reduction in the availability of functional CCR5 on target cells profoundly diminishes both virus acquisition and viral pathogenesis, without adverse effects on the host.
Unlike viral antigens, in which variants are rapidly selected in response to host immune pressures, CCR5 is a cellular protein and is, therefore, genetically stable. We hypothesized that a vaccine that induced antibodies (Ab) against CCR5—either by inducing internalization and downregulating its expression on the cell surface or by blocking virus-receptor interactions—could prevent viral transmission and block viral replication. In support of this concept, a monoclonal Ab (MAb) against CCR5 has shown some efficacy in early-stage therapeutic clinical trials (11, 12). However, because CCR5 is a self-protein, the ability to initiate an antibody response to the molecule is seemingly limited by the mechanisms of B cell tolerance, which normally prevent the induction of antibody responses to self-molecules. In spite of this, we have shown that by arraying self-molecules at high density on the surface of virus-like particles (VLPs), we can completely abrogate these tolerance mechanisms and induce high titer IgG antibodies against diverse self-antigens (13–18).
Our laboratory has taken advantage of these findings to develop several VLP-based vaccines that elicit anti-CCR5 antibodies, and other laboratories have used other tolerance-breaking strategies to target CCR5 (19–23). For example, we developed a papillomavirus (PV) VLP-based vaccine targeting the N-terminal extracellular domain of macaque CCR5 that, in macaques, induces antibodies that bind to native macaque CCR5 and block virus infection in vitro (13). Although the antibody responses to immunization with this vaccine were somewhat variable, prophylactic vaccination of macaques with the CCR5 vaccine reduced viral loads and time to clearance in pig-tailed macaques infected intravenously with a weakly pathogenic CCR5-tropic simian-human immunodeficiency virus (SHIV), and SHIV clearance was correlated with anti-CCR5 antibody titer and avidity (13). Thus, our data, and similar results from Misumi and colleagues (19), suggest that prophylactic vaccination against CCR5 may play a role in controlling viral replication in a SHIV-macaque model. In contrast to these encouraging reports in the SHIV-macaque model, a DNA vaccine expressing human CCR5 fused to tetanus toxoid failed to protect macaques from SIVsm challenge (20).
More recently, we have developed bacteriophage particle-based vaccines that target two extracellular domains of CCR5 that are involved in HIV binding. These vaccines induce high-titer anti-CCR5 antibodies in rodents that bind to native macaque CCR5 and can inhibit simian immunodeficiency virus (SIV) infection in vitro (24). A bivalent vaccine targeting the N-terminal domain and the second extracellular loop of CCR5 was more effective at inducing SIV-inhibitory antibodies than each vaccine separately (24).
In the current study, we investigated whether vaccination against CCR5 could protect female macaques against intravaginal (VAG) infection with highly virulent SIVmac251. SIVmac251 was selected because it uses CCR5 as its main coreceptor, as also demonstrated in vivo by its inhibition by maraviroc (25, 26). We demonstrated that the vaccine was immunogenic and safe and was able to drastically reduce virus replication in a subset of the animals. These results support further research on the development of CCR5-targeting vaccines to reduce HIV transmission.
MATERIALS AND METHODS
Vaccine construct preparation.
Qß bacteriophage was prepared by infecting a 500-ml culture of Escherichia coli strain A/λ at A600 = 0.25 with Qß at a multiplicity of infection of 0.5. After lysis, unlysed cells and insoluble cellular debris were removed by centrifugation. Phages were precipitated from the supernatant at 4°C overnight after addition of NaCl at 0.5 M and polyethylene glycol (PEG) 8000 at 10%. Precipitate was collected by centrifugation and then dissolved in TNME (0.1 M NaCl, 10 mM Tris-HCl, 0.1 mM MgCl2, 0.01 mM EDTA). CsCl was added to achieve a density of 1.40 g/ml, and the solution was centrifuged to equilibrium at 40,000 rpm using an SW50.1 rotor. The phage band was recovered and dialyzed against phosphate-buffered saline (PBS; pH 7.4).
A 21-amino-acid peptide (designated EC1) representing the N-terminal 21 amino acids (MDYQVSSPTYDIDYYTSEPC; sulfated at Y10 and Y14) of macaque CCR5 (mCCR5) was synthesized by American Peptide (Sunnyvale, CA). A second peptide representing the second extracellular loop (ECL2) of mCCR5 was synthesized by Celtek Peptides (Nashville, TN). The ECL2 peptide (DRSQREGLHYTG) is a cyclic peptide spanning amino acids 168 to 177 of pig-tailed macaque CCR5 in which the Arg and Thr residues are linked through an Asp-Gly dipeptide spacer (Fig. 1). The EC1 and ECL2 peptides were directly linked to Qß bacteriophage using a bifunctional cross-linker (SMPH; Pierce Endogen, IL) as described previously (24), which allowed us to link the C-terminal cysteine on the peptides to exposed surface lysine residues on the coat protein of Qß. Conjugation efficiency was monitored by SDS-PAGE analysis. Each Qß.EC1 particle displayed an average of 90 EC1 peptides, and each Qß.ECL2 particle displayed an average of 270 ECL2 peptides (data not shown and reference 24). Equal amounts of the 2 constructs (Qß.EC1 and Qß.ECL2) were mixed to formulate the Qß.CCR5 vaccine. Wild-type Qß was used as a placebo vaccine construct.
FIG 1.

Preparation of two CCR5 vaccine constructs. A linear peptide representing the N terminus (EC1) and a cyclic peptide representing the undecapeptidyl arch of the second extracellular loop (ECL2) of macaque CCR5 (A), representing two different regions that interact with HIV gp120 during infection, were synthesized and then conjugated at high density to Qß bacteriophage using the bifunctional cross-linker SMPH to obtain Qß.EC1 and Qß.ECL2 particles, respectively (B). The image of Qß bacteriophage was generated using RasMol and a structure of Qß from the Brookhaven protein database.
Animals.
All 18 animals were primiparous (n = 1) or multiparous (n = 17) adult female rhesus macaques (Macaca mulatta), with an age range of 10 to 14 years, and were housed at the California National Primate Research Center (CNPRC) in accordance with American Association for Accreditation of Laboratory Animal Care standards. We strictly adhered to the Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Resource Council (27). The study was approved by the Institutional Animal Care and Use Committee of the University of California Davis. Of the 18 animals, 15 were Indian-origin rhesus macaques. The remaining 3 animals (29741, 31380, and 32214) were one-fourth Chinese origin and three-fourth Indian origin and were assigned as one animal per study group.
Animal immunizations.
The CCR5 vaccine constructs were administered by the intramuscular (IM) or intravaginal (VAG) route. For the intramuscular immunizations, 0.5 ml of Qß.CCR5 vaccine (consisting of 25 μg each of Qß.EC1 and Qß.ECL2 constructs in PBS–0.25 ml of incomplete Freund adjuvant [Sigma-Aldrich, St. Louis, MO]) was injected into the right and left quadriceps muscles in an alternating manner. The intravaginal immunizations consisted of 2 steps: 1 ml of 3% CMC (carboxymethylcellulose; an inert gel) plus 4% nonoxynol-9 (N-9) (Spectrum, Gardena, CA) was slowly administered to the vaginal lumen, followed 6 h later with the actual immunization of 50 μg each of the Qß.EC1 and Qß.ECL2 constructs in 100 μl via the use of an aerosolizing high-pressure syringe (model FMJ-250; Penn-Century Inc., Wyndmoor, PA). A control group of six macaques received four intramuscular inoculations of 50 μg of wild-type Qß (with 0.25 ml incomplete Freund adjuvant for a total volume of 0.5 ml per immunization).
SIV inoculations.
To test the efficacy of the vaccine regimens, we used a previously established high-dose intravaginal inoculation regimen, designed to ensure a nearly 100% infection rate among unimmunized control animals (28, 29). Twice in a single day (with the two occasions separated by a 4-h interval), 1 ml of undiluted SIVmac251 was administered atraumatically and slowly via a 1-ml needleless syringe into the vagina. The SIVmac251 stock (reference number 6/04), which was propagated on rhesus peripheral blood mononuclear cells [PBMC]), contained approximately 109 SIV RNA particles per ml, or 105 50% tissue culture infective doses (TCID50) per ml (30). No Depo-Provera treatment was used, and all animals were inoculated on the same day without regard for the stage of the menstrual cycle.
Collection and processing of blood and tissue specimens.
When necessary, animals were immobilized with ketamine HCl (Parke-Davis, Morris Plains, New Jersey) injected intramuscularly at 10 mg/kg of body weight. Blood samples were collected regularly for monitoring viral and immunologic parameters as described previously (31). Complete blood counts (CBC) were performed on EDTA-anticoagulated blood samples. Samples were analyzed using a Pentra 60C+ analyzer (ABX Diagnostics); differential cell counts were determined manually.
To collect cervical secretions, a speculum was inserted into the vagina and two Weck-Cel sponges (Eye Spears; Beaver-Visitic, Waltham, MA) were placed sequentially in the endocervical canal for 1 min each and then carefully removed and placed in tubes and frozen.
Lymphoid tissues collected at euthanasia were processed to obtain cell suspensions by dissecting them with scalpels in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gemini BioProducts, Calabasas, CA) (complete RPMI) and passing the cell homogenate through a cell strainer (Fisher, Pittsburgh, PA). Mononuclear cells were isolated from the splenic cell suspensions and the blood by density gradient centrifugation with Lymphocyte Separation Medium from MP Biomedicals (Aurora, OH), followed by two washes with RPMI 1640.
For some animals, biopsy specimens of axillary lymph node and traverse colon were collected and processed to obtain mononuclear cell suspensions according to previously described methods, including collagenase digestion of the colon samples (32, 33).
Determination of host genetics.
Major histocompatibility complex (MHC) typing for 9 class I alleles (Mamu-A*01, Mamu-A*02, Mamu-A*08, Mamu-A*11, Mamu-B*01, Mamu-B*03, Mamu-B*04, Mamu-B*08, and Mamu-B*17) was performed using methods previously described (34, 35). TRIM5α genotyping was not performed, because a previous study demonstrated that mucosal SIVmac251 infection is not susceptible to TRIM5α restriction (36).
Quantitation of SIV RNA and DNA.
SIV RNA levels in plasma samples were tested by a real-time reverse transcription-PCR (RT-PCR) assay for SIV gag, as previously described in detail (37); this assay has a cutoff value of 30 copies for a 0.5-ml plasma sample. To determine cell-associated viral RNA and DNA levels, cell pellets of approximately 2 million PBMC or lymphoid cells isolated from lymphoid tissues (axillary lymph node or colon biopsy specimens) were snap frozen and stored at −70°C. The cell pellets were subsequently tested for SIV gag RNA and DNA and CCR5 DNA (as a reference for cell equivalents) according to methods described previously (38). Assays were routinely run in duplicate. For these assays, variability on replicate samples run on separate days was on the order of a 25% to 30% coefficient of variation (CV). Based on samples from known uninfected and unexposed animals, the false-positive rate was extremely low (<0.2%) and was always resolved by duplicate testing.
The plasma viral RNA set point was defined as the mean viral RNA level from week 6 after SIV infection to week 20 or at the time of euthanasia (if earlier).
Viral envelope sequence analysis using single-genome amplification.
Plasma was obtained from each animal at 7 to 21 days postchallenge, and viral RNA was extracted using a QIAamp Viral RNA Minikit (Qiagen). RNA was eluted and immediately subjected to cDNA synthesis. Reverse transcription of RNA to single-stranded cDNA was performed using SuperScript III reverse transcription according to the manufacturer's recommendations (Invitrogen). In brief, a cDNA reaction mixture consisting of 1× RT buffer, 0.5 mM each deoxynucleoside triphosphate, 5 mM dithiothreitol, 2 U/ml RNaseOUT (RNase inhibitor), 10 U/ml of Superscript III reverse transcriptase, and 0.25 mM antisense primer SIVmacV4R1 (5′-TTT ACW ATT TGY CTW ATT CT-3′ [where Y is C or T and W is A or T]) was incubated at 50°C for 60 min and at 55°C for 60 min and was then heat inactivated at 70°C for 15 min followed by treatment with 1 U of RNase H at 37°C for 20 min. The newly synthesized cDNA was used immediately.
The V1-to-V4 loop of the env gene was amplified and sequenced using a limiting dilution PCR where only a single genome is amplified (SGA) per reaction. PCR was performed with 1× PCR buffer, 2 mM MgCl2, 0.2 mM each deoxynucleoside triphosphate, 0.2 μM each primer, and 0.025 U/μl Platinum Taq polymerase (Invitrogen) in a 20-μl reaction mixture. First-round PCR was performed with sense primer SIVmacV1F1 (5′-TGT GCA ACC AAG AAT AGG GAT ACT TG-3′) and antisense primer SIVmacV4R1 under the following conditions: 1 cycle of 94°C for 2 min and 35 cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 4 min, followed by a final extension of 72°C for 10 min. Next, 1 μl from the first-round PCR product was added to a second-round PCR that included the sense primer SIVEnvF2 (5′-CAG TCA CAG AAC AGG CAA TAG AGG-3′) and antisense primer SIVEnvR2 (5′-TAR AAR AAT TCT CCT CCA CA-3′) performed under the same conditions used for first-round PCR but with a total of 45 cycles. Correctly sized amplicons were identified by agarose gel electrophoresis and directly sequenced with second-round PCR primers using BigDye Terminator technology. To confirm PCR amplification from a single template, chromatograms were manually examined for multiple peaks, indicative of the presence of amplicons resulting from PCR-generated recombination events, Taq polymerase errors, or multiple-variant templates. All 92 sequences were deposited in GenBank under accession numbers KF542561 to KF542652).
Virus isolation.
PBMC or lymph node mononuclear cells were cocultured with CEMx174 cells in T25 flasks, and subsequent p27 core antigen measurement was performed according to methods previously described (39).
In vivo CD8+ cell depletion.
CD8+ cells were depleted in three macaques through a single administration of anti-CD8 monoclonal antibody M-T807R1, given at a dose of 50 mg/kg via slow intravenous infusion according to the manufacturer's instructions (http://www.nhpreagents.org/NHP/default.aspx).
Phenotyping of lymphocyte populations.
Multiparameter flow cytometric analysis was performed to characterize lymphocyte populations in PBMC and tissue cell suspensions. All antibodies were from BD Biosciences (San Jose, CA) unless otherwise stated. Cell suspensions were stained with peridinin chlorophyll protein (PerCP)-conjugated anti-human CD8 (clone SK1; Becton, Dickinson Immunocytometry Inc., San Jose, CA), fluorescein isothiocyanate (FITC)-conjugated anti-human CD3 (clone SP34; Pharmingen), phycoerythrin (PE)-conjugated anti-human CD4 (clone M-T477; Pharmingen), and allophycocyanin (APC)-conjugated anti-human CD20 (clone L27; Becton, Dickinson) for 4-color flow cytometry and analyzed on a FACSCalibur flow cytometer, as described previously (31). CD4+ T lymphocytes, CD8+ T lymphocytes, NK cells, and B cells were defined as CD4+ CD3+, CD8+ CD3+, CD8+ CD3−, and CD20+ CD3− lymphocytes, respectively. During the in vivo CD8+ cell depletion experiment, as suggested elsewhere (http://www.nhpreagents.org/NHP/default.aspx), the anti-CD8 antibody was replaced by the DK25 clone (Dako, Carpinteria, CA) conjugated to PE (and combined with anti-CD3-PerCP, anti-CD4-FITC, and anti-CD20-APC).
A separate aliquot of blood was stained with anti-CD3-PerCP (clone SP34-2), anti-CD4-APC (clone L200), anti-CD195 (CCR5)-PE (clone 3A9), and anti-CD95-FITC (clone DX2). The number of anti-CCR5-PE antibodies bound per CD4+ CD3+ T cell was measured via the use of QuantiBRITE PE beads according to the manufacturer's instructions.
In separate tubes, T cell subpopulations of CD3+ (clone SP34-2) and CD4+ (L200) or CD8+ (SK1) T cells were further defined by markers for memory [CD45RA (5H9) and CCR7 (3D12)], activation [Ki-67 (B56) and CD69 (FN-50)], and apoptosis [PD-1 (J105; eBioscience), Bcl-2 (Bcl-2/100), and caspase 3 (C92-605)]. In addition, blood or tissue single-cell suspensions were added to round-bottom tubes and treated with 0.5 μg purified anti-CD28 and anti-CD49d (BD Biosciences) in 1 ml supplemented RPMI medium plus (ii) no stimulant, (ii) 10 μg SIVmac239 p27 Gag peptide pool (peptides 5243 to 5299; AIDS Research and Reference Reagent Program no. 6204), (iii) 10 μg SIVmac239 Env peptide pool (peptides 6708 to 6719; AIDS Research and Reference Reagent Program no. 6883), or (iv) phorbol myristate acetate (PMA; 5 ng/ml)-ionomycin (500 ng/ml) (Sigma, St. Louis, MO) as a positive control. Stimulated cells were incubated at 37°C and 5% CO2 for 1 h and then treated with 1× brefeldin A (eBioscience) and incubated for an additional 5 h. Cells were then washed and stained using a fixable Live/Dead discriminator (Invitrogen) and antibodies against intracellular cytokines interleukin-2 (IL-2) (MQ1-17H12), tumor necrosis factor alpha (TNF-α) (MAb11), gamma interferon (IFN-γ) (B27), IL-17A (64CAP17; eBioscience), and granzyme B (B11). Cells were fixed in 1% paraformaldehyde and stored in the dark at 4°C until analysis. Samples were acquired on a LSR II instrument and analyzed using FlowJo software with Boolean gating (TreeStar, Ashland, OR). Staining panels were evaluated using fluorescence-minus-one (FMO) controls. Antigen-specific T cell responses were represented as the percentage of positive cells within the CD4+ or CD8+ T cell populations after subtraction of background responses in unstimulated cultures.
Detection of anti-CCR5 antibody responses.
Sera were tested for antibodies specific for the CCR5-EC1 and ECL2 peptides and Qβ bacteriophage VLPs by enzyme-linked immunosorbent assay (ELISA). Briefly, Immulon II ELISA plates (Dynex Technologies, Chantilly, VA) were coated overnight at 4°C with 0.5 μg of either EC1 or ECL2 peptide (conjugated to streptavidin using the cross-linker SMPH; Thermo Scientific, Rockford, IL) or 0.5 μg Qβ VLPs per well. Wells were then blocked with 50 μl of PBS–0.5% milk (wt/vol) per well for 2 h at room temperature. An initial 1:40 dilution of serum was serially diluted 4-fold and applied to wells for 2.5 h at room temperature. All dilutions were done in 0.5% milk (wt/vol)–PBS unless otherwise noted. Reactivity to target peptides was determined by using horseradish peroxidase (HRP)-labeled goat anti-monkey IgG (Fitzgerald Industries, Acton, MA) at a dilution of 1:4,000, and the reaction mixtures were incubated for 1 h at room temperature. Upon development, the optical density at 405 nm (OD405) was determined using a Thermo Max microplate reader (ThermoLab Systems; Fisher Scientific, Pittsburgh, PA). Absorbance values greater than twice the background were considered positive.
Detection of SIV-specific antibody responses.
SIV-specific immunoglobulin G (IgG) in plasma samples was detected by ELISA as described previously (40). Briefly, Costar enzyme immunoassay-radioimmunoassay (EIA/RIA) plates (Fisher Scientific, Santa Clara, CA) were coated with purified whole SIVmac251, and serial 4-fold dilutions of plasma or serum, starting from a 1:100 dilution, were tested. Titers were determined as the highest dilution with an OD above the cutoff value.
Viral inhibition assay.
The viral inhibition assay was performed via a modification of a neutralization assay that measures a reduction in luciferase reporter gene expression after a single round of infection in TZM-bl cells (which express human CCR5) as described previously (41, 42). TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program, as contributed by John Kappes and Xiaoyun Wu. Briefly, freshly trypsinized cells (10,000 cells in 100 μl of growth medium containing 75 μg/ml DEAE dextran) were preincubated for 1 h at 37°C in 96-well flat-bottom culture plates with serial 3-fold dilutions of plasma prior to addition of virus (∼150,000 relative luminescence units [RLU] equivalents) in duplicate, in a total volume of 250 μl. Samples were tested against the very neutralization-sensitive T cell line-adapted (TCLA)-SIVmac251 stock (grown in H9 cells) and two neutralization-resistant virus stocks, SIVmac251CS (grown in human PBMC) and SIVmac239CS (grown in 293 T cells). One set of control wells received cells plus virus (virus control), another set received cells only (background control), and a third set received murine leukemia virus (MLV)-pseudotyped virus simian virus amphotropic MLV (SVA-MLV) (grown in 293T cells) as a control for nonspecific viral inhibition. After a 48-h incubation, cells were lysed using a Britelite luminescence reporter gene assay system (PerkinElmer Life Sciences) and three-fourths of the lysate was transferred to 96-well black solid plates (Costar) for measurements of luminescence. Neutralization titers were determined as the dilution at which RLU were reduced by 50% compared to virus control wells after subtraction of background RLU.
Criteria for euthanasia and necropsy.
Euthanasia of animals with simian AIDS was determined by established criteria of one or more of the following clinical observations indicative of a severe life-threatening situation: weight loss of >15% in 2 weeks or >25% over any time course; chronic diarrhea or another opportunistic infection(s) unresponsive to treatment; inability to maintain body heat or fluids without supplementation; obtundation; neurologic deficits; and persistent, marked hematologic abnormalities, including anemia (<20%), thrombocytopenia with petechiae or ecchymosis, and hypoproteinemia with edema. A complete necropsy with collection of formalin-fixed tissues was performed.
Statistical analysis.
Statistical analyses were performed with Prism 5 for Mac and Instat 3 (GraphPad Software Inc., San Diego, CA). A value of P of <0.05 was considered statistically significant.
Nucleotide sequence accession numbers.
All 92 sequences determined in this work were deposited in GenBank under accession numbers KF542561 to KF542652).
RESULTS
Overview of experimental design.
Eighteen adult female macaques were divided into three groups of 6 animals each. Two of these groups were immunized with Qß.CCR5, a vaccine targeting two extracellular domains of the CCR5 receptor that were selected because of their role in HIV binding to CCR5 (43, 44) (Fig. 1). One of these targets was a 21-amino-acid peptide that corresponds to the amino terminus of macaque CCR5. The tyrosines at positions 10 and 14 of the peptide were sulfated to reflect the fact that sulfation of these residues in native CCR5 is thought to be important in HIV binding (45). The second antigen was a cyclic peptide spanning amino acids 168 to 177 of the second extracellular loop of macaque CCR5 in which the Arg and Thr residues are linked through an Asp-Gly dipeptide spacer. This peptide was originally identified as an immunogen capable of inducing anti-CCR5 antibodies by Misumi and colleagues presumably because it mimics the native conformation of the epitope (46). Both peptides are highly conserved between macaques and humans; the N-terminal sequence contains only two amino acid differences (at residues 9 and 13), and the cyclic peptide contains one (at residue 171). Peptides were individually conjugated to Qß VLPs and then combined to formulate the Qß-CCR5 vaccine.
Group 1 (IM) received Qß.CCR5 as 4 intramuscular immunizations at weeks −24, −20, −16, and −2 (relative to SIV inoculation), and the 2nd group (IM/VAG) first received an intramuscular prime immunization with Qß-CCR5 at week −24, followed by three vaginal booster immunizations at weeks −20, −16, and −2 (Fig. 2). As outlined in Materials and Methods, each vaginal immunization was performed using a high-pressure microsprayer syringe at 6 h after chemical disruption of vaginal epithelia with nonoxynol-9. The last group (“placebo”) received four intramuscular inoculations with unmodified Qß. At time zero, all animals received a high-dose intravaginal challenge of SIVmac251 (two doses of 105 TCID administered 4 h apart), after which the animals were monitored regularly for infection status and disease progression.
FIG 2.
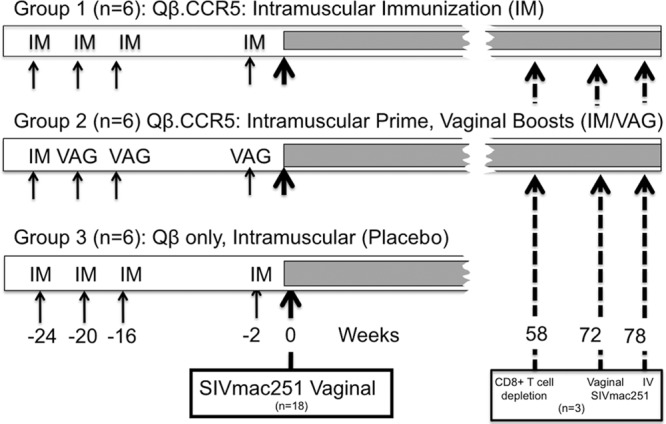
Experimental design of animal studies. Eighteen animals were divided in groups of 6 animals each. Group 1 (IM) received 4 intramuscular immunizations with Qß-CCR5 at weeks −24, −20, −16, and −2. Group 2 (IM/VAG) received an intramuscular prime immunization with Qß-CCR5 at week −24 followed by vaginal boosts with vaginal booster immunizations at weeks −20, −16, and −2. Group 3 (placebo) received 4 intramuscular immunizations with the control vector Qß. At time zero, all animals received a high-dose intravaginal challenge of SIVmac251 (2× 105 TCID). Three animals (1 of group 1 and two of group 2) which had transient viremia were depleted of CD8+ cells at 58 weeks of infection and rechallenged intravaginally at week 72 and, if still not viremic, intravenously at week 78.
Observations during the immunization period prior to SIV challenge.
The Qß.CCR5 vaccine was immunogenic, as high-titer IgG responses to both domains of CCR5 were detected in plasma of all 12 of the macaques that received Qß.CCR5 immunizations. Compared with that of the responses to Qß.CCR5 immunization in inbred mice (24), the magnitude of the antibody responses was somewhat more variable in the outbred macaques. Titers of antibody against the ECL2 peptide were slightly higher than those against the EC1 peptide (Fig. 3), likely reflecting the fact that the ECL2 peptide was displayed on Qß particles at a higher density. Intravaginal boosting successfully increased antibody levels in the serum; between weeks −20 and −10, anti-EC1 and anti-ECL2 antibody levels increased 6.5-fold and 17-fold, respectively (P < 0.05), which is similar to the boosting we previously observed upon intravaginal vaccination of mice (47). However, macaques that received four intramuscular injections developed higher antibody titers than those seen with the group that received intravaginal boosts (Fig. 3). Nevertheless, all 12 macaques had peak anti-CCR5 endpoint dilution titers of greater than 103 (EC1) and 104 (ECL2). These antibody titers remained relatively stable throughout the immunization period. At the time of SIV challenge (2 weeks after the last immunization), antibody titers in individual macaques were no lower than 1 4-fold dilution from each animal's respective peak antibody titer. Although we tried to collect cervical secretions using Weck-Cel sponges, the protein yield in the samples was too low for further antibody analysis.
FIG 3.

Anti-CCR5 responses in macaques immunized with Qβ.CCR5. EC1 and ECL2 peptide-specific antibody titers (expressed as endpoint dilution titers) were measured by ELISA in plasma from macaques immunized by the routes indicated. Macaques were immunized at weeks −24, −20, −16, and −2 and were challenged at week 0 (vertical line). Asterisks indicate the macaques that controlled SIV infection.
There were no detectable adverse effects of the anti-CCR5 immunizations, based on clinical observations, CBC data, and values (percentages and cell counts per μl) of the major lymphocyte subsets in peripheral blood (CD4+ and CD8+ T lymphocytes, NK cells, and B lymphocytes; data not shown). Flow cytometry techniques were used to determine if the development of plasma anti-CCR5 antibodies reduced CCR5 expression on CD4+ T lymphocytes. There was no difference between the animal groups in the numbers of CCR5 molecules on the surface of CD4+ T lymphocytes; the estimated average numbers of anti-CCR5-PE antibodies bound per cell ranged from 738 to 4,883 (Fig. 4A), which is similar to results previously described for human CD4+ T cells (48). In addition, during the immunization period prior to SIV inoculation, there was no difference between the immunized groups and the placebo group in percentages or absolute numbers of CCR5+ CD4+ T lymphocytes and CD4+ T lymphocytes or in the CD4+/CD8+ T cell ratio in peripheral blood (Fig. 4B to D).
FIG 4.

Immunological markers in macaques immunized with Qβ.CCR5 and subsequently challenged with virulent SIVmac251. Animals received 4 immunizations (dotted arrows) with placebo or Qß-CCR5 and were then inoculated intravaginally with SIVmac251 (solid arrows). Flow cytometry techniques were used on whole blood to measure the density of CCR5 molecules on the surface of CD4+ T cells (A), the number of CCR5+ CD4+ CD3+ lymphocytes (B), the number of CD4+ CD3+ lymphocytes (C), and the CD4+/CD8+ T cell ratio (D) in peripheral blood.
Plasma collected at time zero (i.e., the time of virus inoculation) was tested in an in vitro viral inhibition assay using TZM-bl cells. No antiviral activity against the control virus SVA-MLV or against SIVmac239CS was detected (all titers < 1:20). Sporadic but very low inhibitory activity (titers < 70) was detected against SIVmac251/CS and SIVmac251-TCLA; however, some samples from control animals also had low inhibitory activity, and there was no correlation with vaccine groups and outcome after SIV challenge (data not shown).
As expected, because the Qß.CCR5 and Qß vaccine constructs did not contain any SIV sequences, no SIV-specific antibody responses were detected in any of the animals (all titers < 1:100 by ELISA; data not shown) prior to the SIV challenge.
Virologic and clinical outcome after vaginal SIVmac251 challenge.
Following the high-dose SIVmac251 challenge, all animals became infected based on detection of viral RNA in plasma during the first 4 weeks after inoculation. Overall, the mean peak viral RNA levels in the 12 vaccinated animals were approximately 30-fold lower than in the control animals (106.8 versus 108.3 copies per ml plasma; P = 0.08, one-tailed t test). However, two distinct patterns emerged: 15 animals (including all 6 controls) demonstrated a typical course of SIVmac251 infection with persistent viremia, while 3 immunized animals had an atypical course of infection characterized by transient viremia (Fig. 5).
FIG 5.

Plasma SIV RNA levels after vaginal SIVmac251 inoculation. Panel A shows the means (after log transformation) of viral RNA levels in plasma of the 3 experimental groups. Panels B through D show the results for the individual animals within each group. At time zero, all animals were inoculated intravaginally with SIVmac251. Three animals (32787, 30285, and 32214) received another intravaginal inoculation at 72 weeks, and the 2 protected animals (32787 and 32214) then received an intravenous inoculation at 78 weeks. In panels C and D, the anti-CCR5 IgG titers in plasma at the time of SIV challenge are shown. Survival is indicated by the time when animals were euthanized because they met the established clinical criteria; > indicates that the animals were euthanized at the indicated time point without meeting these clinical criteria. The time points of CD8+ depletion and SIV rechallenges on 3 animals that controlled initial infection are indicated.
All 6 control animals, 5 of the 6 animals of the CCR5 IM group, and 4 of the 6 animals of the CCR5 IM-IVAG group developed high peak viremia (≥107 SIV RNA copies per ml plasma) within the first 3 weeks after inoculation, followed by persistent viremia (Fig. 5). While the viral RNA set points for the control animals were all ≥106 RNA copies/ml, the CCR5 IM group had one animal with a viral set point of ∼104 copies/ml (animal 31380; Fig. 5C). Viral RNA levels were predictive of disease-free survival. Of these 15 persistently viremic animals, the 7 animals with the highest level of viremia (>107 RNA copies/ml) showed a rapid decline in CCR5+ and CD4+ T lymphocyte numbers and CD4+/CD8+ T cell ratio (Fig. 4B to D) and met the clinical criteria for euthanasia within 18 weeks of infection. These 7 rapid progressors mounted low anti-SIV antibody responses (peak titers ≤ 1,600 by ELISA), which is consistent with previous observations of rapid progressors in this animal model (49, 50). The remaining 8 persistently viremic animals had generally lower virus levels (Fig. 5), associated with a slower depletion of CD4+ T lymphocytes (Fig. 4) and higher SIV-specific antibody responses (peak titers ≥ 25,600; data not shown). Five of these 8 animals met the criteria for euthanasia at between 25 and 50 weeks of infection, while the remaining 3 animals (29741, 32571, and 31380) were clinically stable when euthanized at the experimental endpoint of 1 year after SIV infection (Fig. 5).
The remaining 3 animals (32787, 30285, and 32214), which all belonged to the two CCR5-immunized groups and whose the anti-CCR5 antibody titers were in the upper range within their respective groups, had a remarkably different outcome of infection. All 3 animals had detectable plasma viral RNA at 4 consecutive time points (weeks 1, 2, 3, and 4 postinoculation), with peak levels of 740 to 21,000 RNA copies per ml (Fig. 5). However, from week 6 onward, all plasma samples tested negative for SIV RNA (i.e., the results were below the detection limit of 10 to 30 copies per ml, depending on specimen volume). For these 3 animals, no infectious virus could be isolated from 5 million PBMC at the time point of peak viral RNA levels. Despite this transient detection of RNA in plasma, these 3 animals did not seroconvert, as no SIV-specific IgG could be detected in any plasma samples, including all early time points (all titers < 100 by ELISA; data not shown). These 3 animals maintained normal CD4+ and CD8+ T cell counts, including CCR5+ CD4+ T cell counts (Fig. 4).
In an attempt to explain the different disease courses, we evaluated viral and host genetics. Sequence analysis of the V1-to-V4 loop of the env gene was performed on plasma collected at week 2 (for the persistently viremic animals) or at week 1 or 3 (i.e., at the respective times of peak viremia) for the animals with transient viremia. Very little total diversity was detected, and the sequence data were consistent with those reported previously for this particular SIVmac251 stock (51). There were no obvious differences between the three experimental groups in early viral envelope sequences and no differences between the 7 rapid progressors, 8 normal progressors, and 3 animals with transient viremia (data not shown; GenBank accession numbers KF542561 to KF542652).
Animals were typed for MHC class I alleles that in some studies correlated with lower viremia (Mamu-A*01, Mamu-B*08, and Mamu-B*17) or higher viremia (Mamu-B*01) of some, but not all, SIV isolates, particularly SIVmac239 (35, 52–54). In this study, similar to previous studies conducted with our SIVmac251 stocks (31, 32), no correlation was observed between these MHC class I alleles and disease progression; in particular, the control group had the highest frequency of so-called “protective” alleles, while two of the three animals that had transient viremia had no protective alleles (Table 1).
TABLE 1.
Typing of MHC class I alleles
| Group | Animal no. | Presence or absence of allele or % CNPRC frequencya |
MHC scoreb | Survival (wks)c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A01 | A02 | A08 | A11 | B01 | B03 | B04 | B08 | B17 | ||||
| 1 (IM) | 29469 | − | − | − | − | − | − | − | − | − | 0 | 26 |
| 30640 | + | − | − | − | + | − | − | − | − | 0 | 17 | |
| 30784 | − | − | − | − | − | − | − | − | + | +1 | 25 | |
| 31380 | − | − | − | − | − | − | − | − | − | 0 | >52 | |
| 31393 | − | + | − | − | − | − | − | − | − | 0 | 50 | |
| 32787* | − | − | − | − | − | − | − | − | − | 0 | >86 | |
| 2 (IM/VAG) | 29709 | − | − | − | + | − | − | − | − | − | 0 | 18 |
| 30285* | − | + | − | − | − | − | − | − | − | 0 | >75 | |
| 30793 | − | − | + | − | − | − | − | − | − | 0 | 26 | |
| 31512 | − | + | − | − | + | − | − | − | − | −1 | 14 | |
| 32060 | − | − | − | − | + | − | − | − | − | −1 | 11 | |
| 32214* | − | − | − | − | − | − | − | − | + | +1 | >86 | |
| 3 (control) | 29741 | − | + | − | − | − | − | − | + | − | +1 | >52 |
| 29806 | − | − | − | − | − | − | − | − | − | 0 | 34 | |
| 31336 | − | − | − | − | + | − | − | − | − | −1 | 10 | |
| 31621 | − | + | + | − | − | − | − | − | − | 0 | 16 | |
| 32499 | + | − | − | − | − | − | − | − | + | +2 | 14 | |
| 32571 | + | − | − | − | − | − | − | − | − | +1 | >52 | |
| CNPRC frequency | 18.8 | 13.8 | 13.0 | 9.1 | 29.9 | 1.2 | 1.5 | 7.1 | 11.2 | |||
+, present; −, absent. Numerical data represent frequencies of alleles in the general rhesus macaque colony of the California National Primate Research Center.
The MHC scores were calculated by adding one point for each protective allele (Mamu-A*01, Mamu-B*08, and Mamu-B*17) and subtracting a point for Mamu-B*01, which has been associated with higher viral levels; other alleles were given a zero score (35, 52–54). There were no statistically significant differences in the frequencies of the individual alleles or MHC scores between the groups (P values > 0.5). The mean MHC scores for groups 1 (IM), 2 (IM/VAG), and 3 (control) were +0.17, −0.17, and +0.5, respectively.
Survival is indicated by the time (in weeks) when animals met established clinical criteria for euthanasia; the > sign indicates animals subjected to euthanasia at this time prior to meeting the clinical criteria.
In the absence of Qβ.CCR5 booster immunizations, antibody titers in plasma against the EC1 and ECL2 peptides declined after SIV challenge. At 55 weeks after SIV challenge, for the 3 animals with transient viremia, the antibody titers in animals 30285 and 32214 had declined to below the limit of detection (<40), while anti-EC1 and anti-ECL2 titers in animal 32787 were 2,560 and 640, respectively (data not shown).
Biopsy collection, CD8+ cell depletion, and rechallenge of the 3 animals with transient viremia.
To look for viral reservoirs in the 3 animals with transient plasma viremia, PBMC and mononuclear cells isolated from axillary lymph node and colon biopsy specimens collected at week 56 were tested by PCR for cell-associated SIV RNA and DNA. All samples were negative (results were below the detection limit of 1 to 2 copies of SIV RNA or DNA per 100,000 cellular equivalents). In addition, for all 3 animals, no virus could be isolated from 4 to 10 million PBMC and axillary lymph node mononuclear cells using coculture experiments. Thus, despite the initial viremia, there was no detectable evidence of infection afterward based on the viral and immunological markers assessed in this study.
To further substantiate that conclusion, and to determine if the atypical course of infection in the 3 animals with transient viremia was due to strong cell-mediated antiviral immune responses, an in vivo CD8+ cell depletion experiment was performed. At 58 weeks after SIV inoculation, the three animals were administered an anti-CD8 antibody that in previous studies induced a transient increase in viremia in SIV-infected animals (31, 55, 56). Following a single infusion of anti-CD8 antibody M-T807R1, the CD3+ CD8+ T cell and CD3− CD8+ NK cell counts in peripheral blood were undetectable or very low (≤5 per μl) for 3, 8, and 9 weeks in animals 32787, 30285, and 32214, respectively (Fig. 6). Throughout the close observation period of 14 weeks after CD8+ cell depletion, 14 blood samples were collected from each animal at frequent intervals (i.e., twice a week for the first 2 weeks, then weekly for the next 6 weeks, and then biweekly for the following 6 weeks). None of the plasma samples had detectable viral RNA levels (all values were below the detection limit of 10 RNA copies/ml). In addition, no SIV-specific antibodies could be detected (all titers < 1:100). In conclusion, the CD8+ cell depletion experiment failed to induce viremia and seroconversion.
FIG 6.
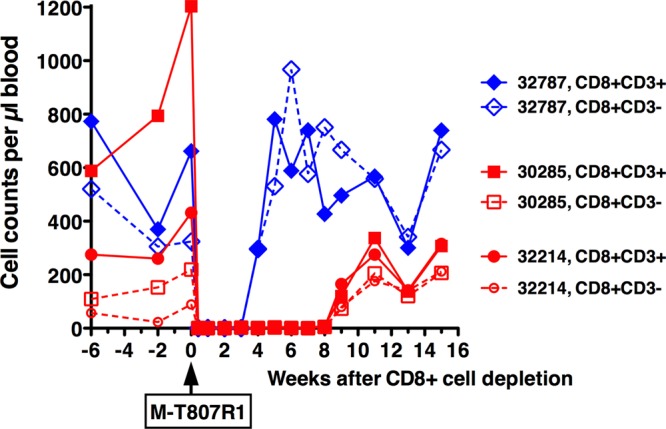
CD8+ cell depletion of three aviremic animals. At 58 weeks after the initial intravaginal SIV inoculation, the three aviremic animals were given a single infusion of anti-CD8 antibody M-T807R1. The absolute cell counts of CD3+ CD8+ T cells and CD3− CD8+ NK cells in peripheral blood are presented.
At 14 weeks after the CD8+ cell depletion experiment (i.e., at 72 weeks after the first vaginal SIV inoculations), all three animals were rechallenged using exactly the same regimen, namely, two intravaginal administrations (administered 4 h apart in a single day) of 1 ml containing 105 TCID50 of SIVmac251. This time, animal 30285 became highly viremic; although we monitored this animal for only 6 weeks after rechallenge, the available evidence indicated features of a rapid progressor (persistently high viremia > 107 copies/ml [Fig. 5D]; low SIV-specific IgG titer of 1:100 at week 6). This animal had also low SIV-specific cell-mediated immune responses in PBMC (1.2% of CD4+ cells and CD8+ cells expressed at least one cytokine after in vitro stimulation with SIV Gag or Env peptides; data not shown). In contrast, the other 2 animals (32214 and 32787) maintained undetectable plasma viral RNA levels (<10 copies/ml), with the exception that the plasma collected from animal 32214 2 weeks after intravaginal rechallenge had 14 SIV RNA copies per ml. Neither of these animals developed detectable SIV-specific antibodies or SIV-specific cell-mediated immune responses (as measured by flow cytometry with intracellular cytokine staining; data not shown).
Because the resistance of these animals to the repeated mucosal challenges was remarkable, we investigated whether these 2 animals had any inherent genetically defined resistance to infection by exposing them to the most stringent route of exposure, namely, intravenous inoculation. Therefore, at 6 weeks after the vaginal SIV rechallenge (i.e., at 78 weeks after the initial vaginal challenge), the same high dose of SIVmac251 (1 ml of 105 TCID50) was administered to both animals by the intravenous route. Both animals became persistently infected (peak viremia > 107 RNA copies/ml plasma) and seroconverted by week 4 (SIV-specific IgG titer of 1:1,600). At week 3, no SIV-specific cell-mediated immune responses were detectable in PBMC of either animal. However, animal 32214, which experienced an approximately 2-log reduction in plasma SIV RNA levels following peak viremia (Fig. 5D), had high levels of SIV-specific cell-mediated immune responses in both CD4+ and CD8+ lymphocyte populations at the time of euthanasia (i.e., 7 weeks after intravenous challenge), when ≥25% of CD4+ T cells and CD8+ T cells secreted at least one cytokine upon in vitro stimulation with SIV Gag or Env peptides (data not shown). In contrast, animal 32787, which maintained higher plasma viremia, had very low SIV-specific immune responses by the time of euthanasia at week 7 of infection (<0.12% of CD4+ cells and ≤0.2% of CD8+ cells expressing at least one cytokine; data not shown).
DISCUSSION
The current pilot study was aimed at exploring—in a preclinical animal model with highly virulent challenge virus—the feasibility of an anti-HIV vaccine that, instead of targeting viral proteins, would exert antiviral activity through the induction of antibodies to the CCR5 coreceptor.
Considering that any vaccine that induces an antibody response to a self-protein engenders safety concerns that generate substantial regulatory and public perception hurdles, it was encouraging that immunization with Qß.CCR5 elicited high-titer antibody responses in macaques without overt adverse effects. Although the primary goal of the current nonhuman primate study was the evaluation of immunogenicity and efficacy, the available data indicate that the CCR5 vaccine was well tolerated, as no safety concerns were raised during the 24-week immunization period. Although the safety assessment was limited in duration because of the subsequent SIV challenge which induced immunopathology in animals with persistent viremia, the 3 animals with transient viremia continued to have lymphocyte phenotypic values and clinical observations that were indistinguishable from those of uninfected animals for an additional year of observation. The existence of the CCR5-Δ32 allele in human population gives some clues as to the potential side effects of anti-CCR5 therapy. To date, the only suggestion that the CCR5-Δ32 mutation may be associated with enhanced susceptibility to disease is in individuals infected with two flaviviruses, West Nile Virus and tickborne encephalitis virus (57, 58). Nevertheless, the consequences of functional inactivation of CCR5 in normal individuals cannot necessarily be predicted by examination of knockout individuals. Of significance to the current study, PRO 140 (Progenics Pharmaceuticals Inc.) is a humanized anti-CCR5 monoclonal antibody (MAb) that is in clinical development for the treatment of HIV infection. In a phase 2a study, PRO 140 was found to be well tolerated and, at the highest dose, provided a mean viral load reduction of 1.65 log (12). Similarly, HGS004, a fully humanized IgG4 monoclonal antibody, was found to be safe and, at the higher doses, gave a >1 log reduction in plasma HIV RNA levels in 54% of study subjects (59). If a favorable safety profile of these and later-generation antibodies emerges from larger phase 3 clinical trials, it would support the possibility that a CCR5 antibody-inducing vaccine could also be safely delivered. The cost of delivery would clearly favor a CCR5 vaccine over administration of anti-CCR5 MAb for use in the developing world. The development of such a vaccine also needs to address whether, if infection is not prevented, it would select for viral variants that use alternative coreceptors (such as CXCR4) as is observed upon treatment with maraviroc (60).
Because CCR5-specific antibodies are likely to be most effective at reducing viral infection and replication when also present at the initial site of viral entry, this experiment included two CCR5 vaccine arms. While the IM group received exclusively intramuscular immunizations, the IM/VAG group, after an initial intramuscular immunization (to induce systemic antibodies), received intravaginal booster immunizations also. In mice, a similar intravaginal aerosol immunization strategy had previously been found to induce IgG levels in the genital tract similar to those induced by the intramuscular immunization route (47). In the current nonhuman primate study, although the IM group developed slightly higher anti-CCR5 antibody titers in plasma than the IM/VAG group, we were not able to evaluate mucosal antibody levels, and there was no clear distinction between the outcomes after the vaginal SIVmac251 challenge, probably due to the small group sizes. Accordingly, for the remainder of this discussion, the data from the two groups are combined without further distinction.
The two CCR5 peptide vaccine constructs used in the current study were clearly immunogenic, as high titers of peptide-specific antibodies in plasma of both immunization groups (IM and IM/VAG) could be detected by ELISA methods. However, an in vitro viral inhibition assay, adapted from an established neutralization assay performed with TZM-bl cells, failed to detect significant inhibitory activity. Reasons for this can be multiple. It is unclear whether the anti-rhesus CCR5 antibodies cross-react with human CCR5 on TZM-bl cells; in this context, the 2D7 antibody recognizes human CCR5 (61) but not macaque CCR5 due to a single K-to-R change in this domain (62). In addition, as the level of CCR5 expression on TZM-bl cells is approximately 10-to-100-fold higher than on PBMC (48), this creates a higher barrier for in vitro inhibition and thus may lead to underestimation of the potential antiviral activity in vivo. However, in vitro viral inhibition assays that use primary rhesus PBMC are cumbersome and plagued by high variability. Thus, a novel assay(s) using macaque target cells may need to be developed to more closely mimic the antiviral activity of macaque anti-CCR5 antibody in vivo.
Following immunization, all animals were exposed intravaginally to a high dose of the very virulent SIVmac251 isolate. This challenge model is predicted to establish persistent viremia in nearly 100% of control animals. While the control animals and the majority of CCR5-immunized animals had the typical course of SIVmac251 infection with persistent viremia that correlated with disease progression, three immunized animals had a very unusual course of infection. These 3 animals showed an initial transient viremia (with detectable viral RNA at 4 consecutive time points) which after week 4 became undetectable.
We were not able to identify a clear correlate of protection for these 3 animals based on immunologic markers in peripheral blood. On the day of SIV challenge, the anti-CCR5 antibody titers in plasma of these 3 animals were in the higher range of their groups. Their preinfection CCR5 expression levels on peripheral blood lymphocytes were indistinguishable from those of the other animals. However, a limitation of our study is that we were not able to assess anti-CCR5 antibody levels and CCR5 receptor occupancy in the vaginal mucosa, where infection would have been initiated, or in the draining lymph nodes that facilitate viral dissemination.
The peak levels (∼103 to 104 SIV copies/ml plasma) and duration of the transient viremia in these 3 animals were indistinguishable from those observed following infection with some attenuated SIVs, such as the avirulent molecular clone SIVmac1A11. However, an important distinction is that SIVmac1A11-infected animals make SIV-specific antibodies within a few weeks of infection (63–65), while in the current study, the 3 protected animals did not mount any detectable anti-SIV antibody response. The lack of seroconversion, the undetectable virus levels in lymph node and colon biopsy specimens, the inability of the CD8+ depletion experiment to induce a viral rebound or seroconversion, and the high primary viremia upon stringent rechallenge suggest that the infection might have been cleared (i.e., might have been an abortive infection), rather than being a persistent, lingering infection of the kind associated with sustained antiviral immune responses. Similar observations suggestive of abortive infection have previously been described in preexposure prophylaxis studies where some drug-treated macaques exposed mucosally to SIVmac251 resulted in transient detection of low viremia and/or low antiviral antibody responses but afterward became negative by all markers, including no evidence of antiviral immunity upon rechallenge (66, 67). As hypothesized previously (66, 67), this phenomenon may be due to the many replication-defective or replication-impaired viral variants that are present in the uncloned SIVmac251 stocks, because in a high-dose challenge model, even minor variants are given the opportunity to initiate mucosal infection. Similar to the results seen with other in vitro-grown virus stocks, the amount of viral RNA greatly exceeds the levels of replication-competent virions, which suggests that most viral RNA copies represent noninfectious virus particles or infectious virus particles with defective genomes that, upon infection of target cells, can still induce a transient release of viral RNA into the circulation but cannot disseminate and establish persistent infection. In this context, we hypothesize that in the 3 protected animals in the current study, the anti-CCR5 antibodies reduced the effective infectious dose of the high-dose inoculum to below the threshold needed to establish a classical persistent infection.
Although the protective effect of the CCR5 vaccine constructs in the current study was limited to 3 of the 12 immunized animals (25%), it should be noted that we used a high-dose viral challenge model. As explained elsewhere, repeated low-dose viral challenge models are biologically very relevant and may be more relevant than high-dose challenge models to detect the protective effects of prophylactic strategies, as efficacy may be missed or underestimated with high-dose inoculation models (68). Accordingly, future studies aimed at further improving anti-CCR5 vaccines in nonhuman primate models can be performed best in such repeated low-dose challenge models that have been developed for every mucosal route.
More than 1 year after the initial SIVmac251 inoculation, the 3 animals were reexposed to an increasingly stringent SIVmac251 challenge by first the intravaginal and then the intravenous route of inoculation. Because all 3 animals became infected, this indicates that the resistance to infection induced by the CCR5 vaccine was, as expected, relative and became weaker when anti-CCR5 antibody numbers declined, especially against an intravenous high-dose challenge.
In conclusion, although the efficacy of the current vaccine constructs in a high-dose viral challenge model was only moderate, the data of our study offer a promising step forward and provide support for further research into anti-CCR5-targeting vaccines to reduce HIV transmission and replication. In particular, future research can focus on optimizing vaccine constructs, adjuvants, and immunization regimens to induce high-quality antibody responses that are durable and effective in blocking SIV and HIV infection at the sites of mucosal transmission and preventing systemic dissemination.
ACKNOWLEDGMENTS
We thank Linda Hirst, Joyce Lee, Colony Services, and the Pathology, Veterinary, and Clinical Laboratory staff of the California National Primate Research Center for expert technical assistance. The SIVmac239 Gag p27 peptides were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. The AT-2 inactivated SIVmac239 was provided by J. Lifson (SAIC Frederick, Inc., National Cancer Institute, Frederick, MD). We thank Gretta Borchard and David Watkins of the AIDS Vaccine Research Laboratory (University of Wisconsin, Madison) for the MHC typing and Leidos Biomedical Research, Inc., Frederick National Laboratory, Frederick, MD, for the viral RNA and DNA load determinations. The following reagent was obtained through the NIH Nonhuman Primate Reagent Resource: M-T807R1.
This work was supported by NIH grant 1RC2CA148982-01, grant RR-00169 from the National Center for Research Resources (NCRR; a component of the NIH) to the California National Primate Research Center, NIH grant 5R24RR016038 awarded to David Watkins, and HHSN27201100016C awarded to D.M. and by federal funds from the National Cancer Institute under contract HHSN26120080001E.
This work is solely our responsibility and does not necessarily represent the official views of the NCRR or NIH.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 4 December 2013
REFERENCES
- 1.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a Rantes, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. 10.1126/science.272.5270.1955 [DOI] [PubMed] [Google Scholar]
- 2.Deng HK, Unutmaz D, Kewalramani VN, Littman DR. 1997. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388:296–300. 10.1038/40894 [DOI] [PubMed] [Google Scholar]
- 3.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158. 10.1016/S0092-8674(00)81314-8 [DOI] [PubMed] [Google Scholar]
- 4.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557. 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Juarez J, Alali M, Dwyer D, Collman R, Cunningham A, Naif HM. 1999. Persistent CCR5 utilization and enhanced macrophage tropism by primary blood human immunodeficiency virus type 1 isolates from advanced stages of disease and comparison to tissue-derived isolates. J. Virol. 73:9741–9755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, Macdonald ME, Stuhlmann H, Koup RA, Landau NR. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86:367–377. 10.1016/S0092-8674(00)80110-5 [DOI] [PubMed] [Google Scholar]
- 7.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722–725. 10.1038/382722a0 [DOI] [PubMed] [Google Scholar]
- 8.Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, Van Der Ryst E. 2005. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 11:1170–1172. 10.1038/nm1319 [DOI] [PubMed] [Google Scholar]
- 9.Hunt JS, Romanelli F. 2009. Maraviroc, a CCR5 coreceptor antagonist that blocks entry of human immunodeficiency virus type 1. Pharmacotherapy 29:295–304. 10.1592/phco.29.3.295 [DOI] [PubMed] [Google Scholar]
- 10.Yost R, Pasquale TR, Sahloff EG. 2009. Maraviroc: a coreceptor CCR5 antagonist for management of HIV infection. Am. J. Health Syst. Pharm. 66:715–726. 10.2146/ajhp080206 [DOI] [PubMed] [Google Scholar]
- 11.Jacobson JM, Lalezari JP, Thompson MA, Fichtenbaum CJ, Saag MS, Zingman BS, D'ambrosio P, Stambler N, Rotshteyn Y, Marozsan AJ, Maddon PJ, Morris SA, Olson WC. 2010. Phase 2a study of the CCR5 monoclonal antibody PRO 140 administered intravenously to HIV-infected adults. Antimicrob. Agents Chemother. 54:4137–4142. 10.1128/AAC.00086-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson JM, Thompson MA, Lalezari JP, Saag MS, Zingman BS, D'ambrosio P, Stambler N, Rotshteyn Y, Marozsan AJ, Maddon PJ, Morris SA, Olson WC. 2010. Anti-HIV-1 activity of weekly or biweekly treatment with subcutaneous PRO 140, a CCR5 monoclonal antibody. J. Infect. Dis. 201:1481–1487. 10.1086/652190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chackerian B, Briglio L, Albert PS, Lowy DR, Schiller JT. 2004. Induction of autoantibodies to CCR5 in macaques and subsequent effects upon challenge with an R5-tropic simian/human immunodeficiency virus. J. Virol. 78:4037–4047. 10.1128/JVI.78.8.4037-4047.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chackerian B, Lowy DR, Schiller JT. 1999. Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. U. S. A. 96:2373–2378. 10.1073/pnas.96.5.2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chackerian B, Lowy DR, Schiller JT. 2001. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J. Clin. Invest. 108:415–423. 10.1172/JCI11849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chackerian B, Durfee MR, Schiller JT. 2008. Virus-like display of a neo-self antigen reverses B cell anergy in a B cell receptor transgenic mouse model. J. Immunol. 180:5816–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chackerian B, Lenz P, Lowy DR, Schiller JT. 2002. Determinants of autoantibody induction by conjugated papillomavirus virus-like particles. J. Immunol. 169:6120–6126 [DOI] [PubMed] [Google Scholar]
- 18.Chackerian B, Rangel M, Hunter Z, Peabody DS. 2006. Virus and virus-like particle-based immunogens for Alzheimer's disease induce antibody responses against amyloid-beta without concomitant T cell responses. Vaccine 24:6321–6331. 10.1016/j.vaccine.2006.05.059 [DOI] [PubMed] [Google Scholar]
- 19.Misumi S, Nakayama D, Kusaba M, Iiboshi T, Mukai R, Tachibana K, Nakasone T, Umeda M, Shibata H, Endo M, Takamune N, Shoji S. 2006. Effects of immunization with CCR5-based cycloimmunogen on simian/HIVSF162P3 challenge. J. Immunol. 176:463–471 [DOI] [PubMed] [Google Scholar]
- 20.Zuber B, Hinkula J, Vodros D, Lundholm P, Nilsson C, Morner A, Levi M, Benthin R, Wahren B. 2000. Induction of immune responses and break of tolerance by DNA against the HIV-1 coreceptor CCR5 but no protection from SIVsm challenge. Virology 278:400–411. 10.1006/viro.2000.0633 [DOI] [PubMed] [Google Scholar]
- 21.Bogers WM, Bergmeier LA, Ma J, Oostermeijer H, Wang Y, Kelly CG, Ten Haaft P, Singh M, Heeney JL, Lehner T. 2004. A novel HIV-CCR5 receptor vaccine strategy in the control of mucosal SIV/HIV infection. AIDS 18:25–36. 10.1097/00002030-200401020-00003 [DOI] [PubMed] [Google Scholar]
- 22.Barassi C, Soprana E, Pastori C, Longhi R, Buratti E, Lillo F, Marenzi C, Lazzarin A, Siccardi AG, Lopalco L. 2005. Induction of murine mucosal CCR5-reactive antibodies as an anti-human immunodeficiency virus strategy. J. Virol. 79:6848–6858. 10.1128/JVI.79.11.6848-6858.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chain BM, Noursadeghi M, Gardener M, Tsang J, Wright E. 2008. HIV blocking antibodies following immunisation with chimaeric peptides coding a short N-terminal sequence of the CCR5 receptor. Vaccine 26:5752–5759. 10.1016/j.vaccine.2008.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter Z, Smyth HD, Durfee P, Chackerian B. 2009. Induction of mucosal and systemic antibody responses against the HIV coreceptor CCR5 upon intramuscular immunization and aerosol delivery of a virus-like particle based vaccine. Vaccine 28:403–414. 10.1016/j.vaccine.2009.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sina ST, Ren W, Cheng-Mayer C. 2011. Coreceptor use in nonhuman primate models of HIV infection. J. Transl. Med. 9(Suppl 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veazey RS, Ketas TJ, Dufour J, Moroney-Rasmussen T, Green LC, Klasse PJ, Moore JP. 2010. Protection of rhesus macaques from vaginal infection by vaginally delivered maraviroc, an inhibitor of HIV-1 entry via the CCR5 co-receptor. J. Infect. Dis. 202:739–744. 10.1086/655661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Research Council 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 28.Marthas ML, Lu DL, Penedo MCT, Hendrickx AG, Miller CJ. 2001. Titration of an SIVmac251 stock by vaginal inoculation of Indian and Chinese origin rhesus macaques: transmission efficiency, viral loads, and antibody responses. AIDS Res. Hum. Retroviruses 17:1455–1466. 10.1089/088922201753197123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller CJ, Li Q, Abel K, Kim EY, Ma ZM, Wietgrefe S, La Franco-Scheuch L, Compton L, Duan L, Shore MD, Zupancic M, Busch M, Carlis J, Wolinsky S, Haase AT. 2005. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 79:9217–9227. 10.1128/JVI.79.14.9217-9227.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marthas ML, Van Rompay KK, Abbott Z, Earl P, Buonocore-Buzzelli L, Moss B, Rose NF, Rose JK, Kozlowski PA, Abel K. 2011. Partial efficacy of a VSV-SIV/MVA-SIV vaccine regimen against oral SIV challenge in infant macaques. Vaccine 29:3124–3137. 10.1016/j.vaccine.2011.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Rompay KKA, Singh RP, Pahar B, Sodora DL, Wingfield C, Lawson JR, Marthas ML, Bischofberger N. 2004. CD8+ cell-mediated suppression of virulent simian immunodeficiency virus during tenofovir treatment. J. Virol. 78:5324–5337. 10.1128/JVI.78.10.5324-5337.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Rompay KK, Trott KA, Jayashankar K, Geng Y, Labranche CC, Johnson JA, Landucci G, Lipscomb J, Tarara RP, Canfield DR, Heneine W, Forthal DN, Montefiori D, Abel K. 2012. Prolonged tenofovir treatment of macaques infected with K65R reverse transcriptase mutants of SIV results in the development of antiviral immune responses that control virus replication after drug withdrawal. Retrovirology 9:57. 10.1186/1742-4690-9-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shacklett BL, Cox CA, Sandberg JK, Stollman NH, Jacobson MA, Nixon DF. 2003. Trafficking of human immunodeficiency virus type 1-specific CD8+ T cells to gut-associated lymphoid tissue during chronic infection. J. Virol. 77:5621–5631. 10.1128/JVI.77.10.5621-5631.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaizu M, Borchardt GJ, Glidden CE, Fisk DL, Loffredo JT, Watkins DI, Rehrauer WM. 2007. Molecular typing of major histocompatibility complex class I alleles in the Indian rhesus macaque which restrict SIV CD8+ T cell epitopes. Immunogenetics 59:693–703. 10.1007/s00251-007-0233-7 [DOI] [PubMed] [Google Scholar]
- 35.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81:8827–8832. 10.1128/JVI.00895-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenizia C, Keele BF, Nichols D, Cornara S, Binello N, Vaccari M, Pegu P, Robert-Guroff M, Ma ZM, Miller CJ, Venzon D, Hirsch V, Franchini G. 2011. TRIM5alpha does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated Indian rhesus macaques following intrarectal challenge exposure. J. Virol. 85:12399–12409. 10.1128/JVI.05707-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cline AN, Bess JW, Piatak M, Jr, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34:303–312. 10.1111/j.1600-0684.2005.00128.x [DOI] [PubMed] [Google Scholar]
- 38.Venneti S, Bonneh-Barkay D, Lopresti BJ, Bissel SJ, Wang G, Mathis CA, Piatak M, Jr, Lifson JD, Nyaundi JO, Murphey-Corb M, Wiley CA. 2008. Longitudinal in vivo positron emission tomography imaging of infected and activated brain macrophages in a macaque model of human immunodeficiency virus encephalitis correlates with central and peripheral markers of encephalitis and areas of synaptic degeneration. Am. J. Pathol. 172:1603–1616. 10.2353/ajpath.2008.070967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Rompay KKA, Marthas ML, Ramos RA, Mandell CP, Mcgowan EK, Joye SM, Pedersen NC. 1992. Simian immunodeficiency virus (SIV) infection of infant rhesus macaques as a model to test antiretroviral drug prophylaxis and therapy: oral 3′-azido-3′-deoxythymidine prevents SIV infection. Antimicrob. Agents Chemother. 36:2381–2386. 10.1128/AAC.36.11.2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Rompay KKA, Singh R, Brignolo L, Lawson JR, Schmidt KA, Pahar B, Canfield DR, Tarara RP, Bischofberger N, Marthas M. 2004. The clinical benefits of tenofovir for simian immunodeficiency virus-infected macaques are larger than predicted by its effects on standard viral and immunological parameters. J. Acquir. Immune Defic. Syndr. 36:900–914. 10.1097/00126334-200408010-00003 [DOI] [PubMed] [Google Scholar]
- 41.Montefiori DC. 2004. Evaluating neutralizing antibodies against HIV, SIV and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. 64:12.11.1–12.11.17. 10.1002/0471142735.im1211s64 [DOI] [PubMed] [Google Scholar]
- 42.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125. 10.1128/JVI.79.16.10108-10125.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu L, Larosa G, Kassam N, Gordon CJ, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, Moore JP, Mackay CR. 1997. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J. Exp. Med. 186:1373–1381. 10.1084/jem.186.8.1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edinger AL, Amedee A, Miller K, Doranz BJ, Endres M, Sharron M, Samson M, Lu ZH, Clements JE, Murphey-Corb M, Peiper SC, Parmentier M, Broder CC, Doms RW. 1997. Differential utilization of CCR5 by macrophage and T cell tropic simian immunodeficiency virus strains. Proc. Natl. Acad. Sci. U. S. A. 94:4005–4010. 10.1073/pnas.94.8.4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farzadegan H, Henrard DR, Kleeberger CA, Schrager L, Kirby AJ, Saah AJ, Rinaldo CR, Jr, O'Gorman M, Detels R, Taylor E, Phair JP, Margolick JB. 1996. Virologic and serologic markers of rapid progression to AIDS after HIV-1 seroconversion. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13:448–455. 10.1097/00042560-199612150-00008 [DOI] [PubMed] [Google Scholar]
- 46.Misumi S, Nakajima R, Takamune N, Shoji S. 2001. A cyclic dodecapeptide-multiple-antigen peptide conjugate from the undecapeptidyl arch (from Arg(168) to Cys(178)) of extracellular loop 2 in CCR5 as a novel human immunodeficiency virus type 1 vaccine. J. Virol. 75:11614–11620. 10.1128/JVI.75.23.11614-11620.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunter Z, Tumban E, Dziduszko A, Chackerian B. 2011. Aerosol delivery of virus-like particles to the genital tract induces local and systemic antibody responses. Vaccine 29:4584–4592. 10.1016/j.vaccine.2011.04.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choudhry V, Zhang MY, Harris I, Sidorov IA, Vu B, Dimitrov AS, Fouts T, Dimitrov DS. 2006. Increased efficacy of HIV-1 neutralization by antibodies at low CCR5 surface concentration. Biochem. Biophys. Res. Commun. 348:1107–1115. 10.1016/j.bbrc.2006.07.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dykhuizen M, Mitchen JL, Montefiori DC, Thomson J, Acker L, Lardy H, Pauza CD. 1998. Determinants of disease in the simian immunodeficiency virus-infected rhesus macaque: characterizing animals with low antibody responses and rapid progression. J. Gen. Virol. 79(Pt 10):2461–2467 [DOI] [PubMed] [Google Scholar]
- 50.Smith SM, Holland B, Russo C, Dailey PJ, Marx PA, Connor RI. 1999. Retrospective analysis of viral load and SIV antibody responses in rhesus macaques infected with pathogenic SIV: predictive value for disease progression. AIDS Res. Hum. Retroviruses 15:1691–1701. 10.1089/088922299309739 [DOI] [PubMed] [Google Scholar]
- 51.Ma ZM, Keele BF, Qureshi H, Stone M, Desilva V, Fritts L, Lifson JD, Miller CJ. 6 July 2011. SIVmac251 is inefficiently transmitted to rhesus macaques by penile inoculation with a single SIVenv variant found in ramp-up phase plasma. AIDS Res. Hum. Retroviruses 10.1089/aid.2011.0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boyer JD, Kumar S, Robinson T, Parkinson R, Wu L, Lewis M, Watkins DI, Weiner DB. 2006. Initiation of antiretroviral therapy during chronic SIV infection leads to rapid reduction in viral loads and the level of T-cell immune response. J. Med. Primatol. 35:202–209. 10.1111/j.1600-0684.2006.00179.x [DOI] [PubMed] [Google Scholar]
- 53.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074–5077. 10.1128/JVI.80.10.5074-5077.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mothé BR, Weinfurter J, Wang C, Rehrauer W, Wilson N, Allen TM, Allison DB, Watkins DI. 2003. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 77:2736–2740. 10.1128/JVI.77.4.2736-2740.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lifson JD, Rossion JL, Piatak MJ, Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D. 2001. Role of CD8+ lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J. Virol. 75:10187–10199. 10.1128/JVI.75.21.10187-10199.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Rompay KK, Johnson JA, Blackwood EJ, Singh RP, Lipscomb J, Matthews TB, Marthas ML, Pedersen NC, Bischofberger N, Heneine W, North TW. 2007. Sequential emergence and clinical implications of viral mutants with K70E and K65R mutation in reverse transcriptase during prolonged tenofovir monotherapy in rhesus macaques with chronic RT-SHIV infection. Retrovirology 4:25. 10.1186/1742-4690-4-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. 2006. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203:35–40. 10.1084/jem.20051970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, Lindquist L, Lundkvist A, Svensson L. 2008. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J. Infect. Dis. 197:266–269. 10.1086/524709 [DOI] [PubMed] [Google Scholar]
- 59.Lalezari J, Yadavalli GK, Para M, Richmond G, Dejesus E, Brown SJ, Cai W, Chen C, Zhong J, Novello LA, Lederman MM, Subramanian GM. 2008. Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1-infected patients. J. Infect. Dis. 197:721–727. 10.1086/527327 [DOI] [PubMed] [Google Scholar]
- 60.Recordon-Pinson P, Raymond S, Bellecave P, Marcelin AG, Soulie C, Descamps D, Calvez V, Harrigan PR, Fleury H, Izopet J, Masquelier B; S AC11 Resistance Study Group 2013. HIV-1 dynamics and coreceptor usage in Maraviroc-treated patients with ongoing replication. Antimicrob. Agents Chemother. 57:930–935. 10.1128/AAC.02159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. U. S. A. 96:5215–5220. 10.1073/pnas.96.9.5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Rao E, Dioszegi M, Kondru R, Derosier A, Chan E, Schwoerer S, Cammack N, Brandt M, Sankuratri S, Ji C. 2007. The second extracellular loop of CCR5 contains the dominant epitopes for highly potent anti-human immunodeficiency virus monoclonal antibodies. Antimicrob. Agents Chemother. 51:1386–1397. 10.1128/AAC.01302-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marthas ML, Ramos RA, Lohman BL, Van Rompay KKA, Unger RE, Miller CJ, Banapour B, Pedersen NC, Luciw PA. 1993. Viral determinants of simian immunodeficiency virus (SIV) virulence in rhesus macaques assessed by using attenuated and pathogenic molecular clones of SIVmac. J. Virol. 67:6047–6055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marthas ML, Banapour B, Sutjipto S, Siegel ME, Marx PA, Gardner MB, Pedersen NC, Luciw PA. 1989. Rhesus macaques inoculated with molecularly cloned simian immunodeficiency virus. J. Med. Primatol. 18:311–319 [PubMed] [Google Scholar]
- 65.Van Rompay KK, Blackwood EJ, Landucci G, Forthal D, Marthas ML. 2006. Role of CD8+ Cells in controlling replication of nonpathogenic simian immunodeficiency virus SIVmac1A11. Virol. J. 3:22. 10.1186/1743-422X-3-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Rompay KKA, Mcchesney MB, Aguirre NL, Schmidt KA, Bischofberger N, Marthas ML. 2001. Two low doses of tenofovir protect newborn macaques against oral simian immunodeficiency virus infection. J. Infect. Dis. 184:429–438. 10.1086/322781 [DOI] [PubMed] [Google Scholar]
- 67.Van Rompay KKA, Marthas ML, Lifson JD, Berardi CJ, Vasquez GM, Agatep E, Dehqanzada ZA, Cundy KC, Bischofberger N, Pedersen NC. 1998. Administration of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) for prevention of perinatal simian immunodeficiency virus infection in rhesus macaques. AIDS Res. Hum. Retroviruses 14:761–773. 10.1089/aid.1998.14.761 [DOI] [PubMed] [Google Scholar]
- 68.Regoes RR, Longini IM, Feinberg MB, Staprans SI. 2005. Preclinical assessment of HIV vaccines and microbicides by repeated low-dose virus challenges. PLoS Med. 2:e249. 10.1371/journal.pmed.0020249 [DOI] [PMC free article] [PubMed] [Google Scholar]