

Abstract
Human cytomegalovirus (HCMV) employs numerous strategies to combat, subvert, or co-opt host immunity. One evolutionary strategy for this involves capture of a host gene and then its successive duplication and divergence, forming a family of genes, many of which have immunomodulatory activities. The HCMV US12 family consists of 10 tandemly arranged sequence-related genes in the unique short (US) region of the HCMV genome (US12 to US21). Each gene encodes a protein possessing seven predicted transmembrane domains, patches of sequence similarity with cellular G-protein-coupled receptors, and the Bax inhibitor 1 family of antiapoptotic proteins. We show that one member, US17, plays an important role during virion maturation. Microarray analysis of cells infected with a recombinant HCMV isolate with a US17 deletion (the ΔUS17 mutant virus) revealed blunted host innate and interferon responses at early times after infection (12 h postinfection [hpi]), a pattern opposite that previously seen in the absence of the immunomodulatory tegument protein pp65 (pUL83). Although the ΔUS17 mutant virus produced numbers of infectious particles in fibroblasts equal to the numbers produced by the parental virus, it produced >3-fold more genome-containing noninfectious viral particles and delivered increased amounts of pp65 to newly infected cells. These results suggest that US17 has evolved to control virion composition, to elicit an appropriately balanced host immune response. At later time points (96 hpi), ΔUS17 mutant-infected cells displayed aberrant expression of several host endoplasmic reticulum stress response genes and chaperones, some of which are important for the final stages of virion assembly and egress. Our results suggest that US17 modulates host pathways to enable production of virions that elicit an appropriately balanced host immune response.
INTRODUCTION
Human cytomegalovirus (HCMV) is a highly prevalent pathogen that has infected 50 to 90% of adults in many industrialized countries. HCMV infections, while life long, are typically of little proven consequence for immunocompetent individuals. However, HCMV can cause significant morbidity and mortality in immunocompromised individuals, such as HIV/AIDS patients and those on immune-suppressive drugs due to cancer treatment or organ transplantation. HCMV is one of the most frequent causes of congenital birth defects. It has the ability to cross the placenta during fetal development and can cause severe sequelae, such as microcephaly, hearing impairment, seizures, and low birth weight (1).
HCMV is a complex virus. With a double-stranded DNA genome of ∼236 kbp that encodes at least 170 proteins and 14 microRNAs, it has the largest genetic capacity of any human herpesvirus (2–5). Interestingly, only ∼40 proteins are essential for lytic replication in cell culture (6, 7). The remaining genes serve a variety of accessory functions, including host immunomodulation and cellular tropism determination. Although nonessential in cell culture systems, these accessory genes are highly conserved among clinical strains (2) and are likely to be important in human infections (8). The proteins encoded by many accessory genes group into families of related proteins, some of which can be evolutionarily traced to capture of spliced host mRNAs (4, 9, 10).
The US12 gene family consists of 10 tandemly arranged genes (US12 through US21) in the unique short (US) region of the HCMV genome (10). Conserved among primate cytomegaloviruses (CMVs; e.g., human, chimpanzee, rhesus, and gorilla CMVs), the US12 family is absent from CMVs of nonprimates, suggesting a relatively recent evolutionary emergence. All US12 family proteins share similar predicted six- or seven-transmembrane structures with traces of a G-protein-coupled receptor lineage (11). US14, US17, and US18 have been localized to various intracellular membranes of the endoplasmic reticulum (ER), Golgi apparatus, the trans-Golgi network, and early endosomes that are associated with the viral cytoplasmic virion assembly complex (cVAC) (12). US17 is expressed with late gene kinetics and can localize to the nuclei of infected fibroblasts (13).
US12 family members are highly conserved among clinical and laboratory strains of the virus (2). While this high level of conservation indicates important roles for US12 family members, little is known about the functions of US12 family members during infection. Identification of the functions or phenotypes associated with US12 family members is difficult because of potentially redundant functions among US12 family members and their individual and collective dispensability for virus growth in cultured primary human fibroblasts (6, 7, 14; F. Z. Wang, S. J. Gurczynski, and P. E. Pellett, unpublished data).
Viruses with deletions of individual US12 family genes can have cell type-specific phenotypes in different types of cells. Thus, deletion of US13 attenuated virus growth by 1 to 2 log units in human foreskin fibroblasts (HFFs) (7), and deletion of US18 completely abrogated virus growth in primary human gingival tissue (15). Interestingly, deletion of US16 or US19 augmented the growth of the laboratory-adapted Towne strain by 2 log units in human microvascular endothelial cells (7), indicating that under at least some circumstances, these proteins are some form of temperance factors. In contrast, when US16 was deleted from the clinical HCMV strain TR, the virus failed to grow in endothelial cells but grew to parental titers in fibroblasts (14).
The observations that deletion of some US12 family members can dramatically impact replication of HCMV in one cell type but have little impact in other cell types are important, because they provide evidence that US12 family members are indeed biologically active and serve important functions during lytic infection. In cells where deletion of either US18 or US16 abrogated virus replication, immediate early gene expression was completely absent (14, 15). Because the US16 mutant virions bound to endothelial and epithelial cells but did not deliver DNA or tegument proteins to their nuclei, the growth defect was attributed to a defect either in virion entry into the host cell or in the events immediately following entry, possibly due to misincorporation or modulation of levels of one or more virion protein constituents, such as envelope glycoproteins or tegument proteins.
Previously, we identified US17 to be a viral late gene that localizes to the nucleus in an infection-dependent manner (13). To further identify the biological activities of US17, we performed microarray analysis of cells infected with a recombinant HCMV isolate with a US17 deletion (the ΔUS17 mutant virus). In comparison to the responses of the parental virus, host innate and interferon responses were blunted at very early times postinfection (12 h postinfection [hpi]), a pattern opposite that seen in the absence of the immunomodulatory pp65 (pUL83) tegument protein (16, 17). Although the ΔUS17 mutant virus produced numbers of infectious particles in fibroblasts equal to the numbers produced by its parent, at equal multiplicities of infection (MOIs), it produced >3-fold more noninfectious viral particles and delivered increased amounts of pp65 to newly infected cells. Relative to its parent, at later time points (96 hpi) ΔUS17-infected cells displayed aberrant expression of several host ER stress response genes and chaperones, some of which are important in the final stages of virion assembly and egress. Our results suggest that US17 modulates host pathways to enable the production of virions that elicit an appropriately balanced host immune response.
MATERIALS AND METHODS
Cell culture and preparation of virus stocks.
All experiments in this study used normal HFFs. All cells were used between passages 10 and 15. Cells were cultured in Dulbecco modified Eagle medium (HyClone-Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum, 2 mM GlutaMAX (Life Technologies, Grand Island, NY), and 1% minimal nonessential amino acids (HyClone-Thermo Fisher Scientific, Rockford, IL). HCMV strain AD169 (ATCC), the AD169 bacterial artificial chromosome (BAC) pAD/Cre parental strain (provided courtesy of Dong Yu), and the US17 deletion (ΔUS17) virus were cultured by inoculating confluent HFF monolayers at an MOI of 0.001. Infected cells and supernatants were harvested at 14 days postinfection (dpi), and virus titers were determined by plaque assay on confluent HFF monolayers. For all experiments, at 72 h preinfection low-passage-number HFFs were seeded onto 35-mm dishes at 3 × 104 cells/cm2.
Virus purification.
For experiments involving analysis of purified virions, virus was concentrated by centrifugation of clarified supernatants through a 20% sorbitol cushion at 60,000 × g for 1 h in a Beckman SW 41 Ti rotor. Subsequent purification of virions was carried out by centrifuging concentrated virus through a 10 to 50% linear Nycodenz gradient for 2 h at 110,000 × g in a Beckman SW 41 Ti rotor (18).
Recombinant viruses.
A virus with deletion of the entire US17 open reading frame (ORF) was constructed using a BAC system (19). In short, Escherichia coli strain EL250, which harbors a temperature-inducible bacteriophage lambda prophage RED recombinase for homologous linear recombination and an additional arabinose-inducible FLP recombination target (FRT) recombinase, was transformed with a BAC of HCMV strain AD169 pAD/Cre DH18 (provided courtesy of Thomas Shenk), which is a full-length BAC clone of HCMV strain AD169 that harbors a transposon insertion cassette that disrupts the US17 open reading frame (6).
An insertion cassette was created by PCR amplification using a set of primers (forward primer, ATCGCCACCGCCGTCgaagttcctattctctagaaagtataggaacttcAGACGTCAGGTGGCACTTTT; reverse primer, AACGACGAGTTTTTCCGgaagttcctatactttctagagaataggaacttcAGCTCTTGATCCGGCAAAC) consisting of a 5′ portion encoding 15 to 17 bp of DNA directly flanking the US17 open reading frame upstream of the start codon or downstream of the stop codon (capital letters), an inner portion encoding an FLP recombinase site (lowercase letters), and 20 bp at their 3′ ends complementary to the ampicillin resistance gene of plasmid pPur (underlined letters) (Clontech, Mountain View, CA). A second round of amplification used the product of the first reaction as the template and a set of primers consisting of 50 bases flanking the US17 ORF (upstream primer, ACACTCTATAAACGGTTTCTCATACGCGCCTTTTGATCGCCACCGCCGTC; downstream primer, TTGGTGGAGACGGCCGGCGCGGCGGGTGGGGGAAACGACGAGTTTTTCCG). The resulting cassette thus consisted of an inner core ampicillin resistance gene flanked by two FLP recombinase sites with 50 bp of US17 ORF flanking DNA to facilitate linear recombination.
BAC-containing E. coli was shifted to 42°C for 15 min to activate the RED recombinase and then transformed with 300 ng of gel-purified PCR product. Recombinants were selected on LB agar plates containing 25 μg/ml ampicillin, and insertion of the cassette was confirmed by HCMV genome restriction digestion with HindIII and PCR from both within and outside the US17 ORF. To remove the cassette and generate the final in-frame deletion mutant, an overnight culture of the previously described E. coli strain carrying the HCMV BAC and ampicillin resistance gene cassette in place of the US17 ORF was subcultured 1/50 in fresh LB medium and incubated at 32°C until the culture reached an optical density at 600 nm of 0.5. Sterile arabinose was added to a final concentration of 0.1%, and the culture was incubated at 32°C for an additional hour to activate the FLP recombinase. Serial dilutions were plated on nonselective medium, colonies were picked and screened for sensitivity to ampicillin, and the deletion was verified by HindIII digestion. US17 mutants were further verified by PCR from both within and across the US17 ORF to confirm its absence. The resulting mutant had a deletion of the US17 ORF, leaving only a 34-bp FRT scar.
To construct the US17 repair virus with a C-terminal V5 epitope tag, a scarless GalK recombineering system was used (20). The US17 sequence from the AD169 genome was PCR amplified using a set of primers that added the V5 epitope tag to the C-terminal end (forward primer, TGTGGATCCATGTCTCCGAACTCA; reverse primer, TTCTCGAGTTACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCCGCCATGGTTCGCGTGAG, where bold text represents start or stop codons and underlined text represents the V5 epitope sequence.) The amplimer was cloned as a BamHI/XhoI fragment into the PCDNA3.1 vector (Life Technologies, Grand Island, NY). Next, a GalK kanamycin resistance (Kanr) cassette containing 50 bp of upstream and downstream homologous US17 sequence was PCR amplified from plasmid C255 (21) (provided courtesy of Dong Yu) (forward primer, TTGGTGGAGACGGCCGGCGCGGCGGGTGGGGGAAACGACGAGTTTTTCCGCCTGTTGACAATTAATCATCG; reverse primer, ACACTCTATAAACGGTTTCTCATACGCGCCTTTTGATCGCCACCGCCGTCCTCAGCAAAAGTTCGATTTA, where bold text represents homologous AD169 sequence, and nonbold text represents the GalK Kanr binding sequence). This sequence was recombined into the ΔUS17 locus using RED recombinase-mediated recombination, as described above for the deletion mutant. Finally, the GalK Kanr sequence was removed and replaced with the US17 C-terminal V5 sequence by constructing an amplimer consisting of the 17cV5 sequence with 50 bp of homologous US17 sequence at either end (forward primer, TTGGTGGAGACGGCCGGCGCGGCGGGTGGGGGAAACGACGAGTTTTTCCGTTACGTAGAATCGAGACCGAGGA; reverse primer, ACACTCTATAAACGGTTTCTCATACGCGCCTTTTGATCGCCACCGCCGTCATGTCTCCGAACTCAGAGGCCAC, where bold text represents homologous AD169 sequence).
Protein analysis: immunoblotting and silver staining.
At the indicated time points, infected cell monolayers (MOI = 3.0) were washed once with ice-cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay buffer (150 mM NaCl, 10 mM HEPES [pH 7.4], 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 1× protease inhibitors [Roche, Indianapolis, IN]). After incubation for 5 min on ice, lysates were clarified by centrifugation at 10,000 × g for 10 min at 4°C. Protein concentrations were measured by a bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA); equal amounts of total protein from each lysate or protein from gradient-purified virus fractions were run on 10% polyacrylamide gels. Silver staining of the polyacrylamide gels was carried out using a Pierce silver stain kit (Thermo Fisher Scientific, Waltham, MA). Immunoblots were transferred to a 0.1-μm-pore-size nitrocellulose membrane and then probed with antibodies against the following proteins: immediate early protein 1 (IE1), IE2, pp28, and gB (Virusys, Taneytown, MD), pp65 (Fitzgerald Industries, Acton, MA), glycoprotein H (gH; Santa Cruz Biotechnologies, Dallas, TX), and a polyclonal rabbit antibody against the C terminus of UL48 (a gift from Wade Gibson). Chemiluminescence was performed with SuperSignal West Pico substrate (Thermo Fisher Scientific, Waltham, MA) following the manufacturer's directions.
Isolation of viral DNA or total cellular RNA for genome quantitation, microarrays, and qRT-PCR analysis.
To ensure accurate multiplicities of infection, one dish of cells was trypsinized and cells were immediately counted prior to infection to gauge the final cell density. HFFs (p12) were then infected with either AD169 or pAD/Cre (the ΔUS17 mutant virus) (MOI = 3.0) for 12 or 96 h. Total RNA was extracted using the TRIzol reagent (Life Technologies, Grand Island, NY) following the manufacturer's directions. Briefly, cells were washed once with 1 ml PBS, after which 1 ml TRIzol reagent was added to each 35-mm dish. RNA was separated by addition of 200 μl chloroform to each 1-ml TRIzol sample, followed by centrifugation at 12,000 × g for 15 min at 4°C. The aqueous phase was transferred to a clean tube, and RNA was precipitated by addition of 500 μl of 100% isopropyl alcohol, followed by centrifugation at 12,000 × g for 15 min at 4°C. The RNA pellet was washed twice in 70% ethanol and resuspended in nuclease-free deionized water. DNase I treatment was conducted with 2 U of RNase-free DNase (New England BioLabs, Ipswich, MA) following the manufacturer's directions. The RNA concentration was measured by UV spectroscopy (260 nm, 280 nm) using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). RNA quality was assessed on an Agilent 2100 Bioanalyzer apparatus (Agilent Technologies, Santa Clara, CA); all samples had RNA integrity number values of 8 to 10. RNA was then hybridized on Illumina HT-12 (v4) human bead array chips. RNA quality assessment, chip hybridization, and array reading were performed at the Wayne State University Advanced Genomics Technology Center.
cDNA for quantitative reverse transcription-PCR (qRT-PCR) analysis was generated from 1 μg of total isolated RNA using an iScript first-strand cDNA synthesis kit (Bio-Rad, Hercules, CA). Equal volumes of cDNA were analyzed for all viruses using a custom TaqMan array (Life Technologies, Grand Island, NY) and predesigned primer/probe sets for the following: GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Hs02758991_g1), IFNB1 (beta 1 interferon; Hs01077958-s1), ISG15 (interferon-stimulated gene 15; Hs00192713_m1), CCL5 (Hs00174575_m1), CXCL10 (Hs00171042_m1), interleukin-6 (Hs00985639_m1), and TNFSF10 (tumor necrosis factor-like soluble factor 10 [TRAIL]; Hs00921974_m1). An ABI 7500 fast thermocycler (Life Technologies, Grand Island, NY) with the cycling protocol supplied with the custom array was used.
Isolation of viral genomic DNA from cell culture supernatants was carried out using a QIAamp MinElute virus spin kit following the manufacturer's direction. Supernatants were first clarified by centrifugation at 1,000 rpm for 10 min and treated with 2 U of DNase I (New England BioLabs, Ipswich, MA) per 200 μl supernatant for 10 min. Quantitation of the viral genomes was done using a SYBR green-based assay with primers specific for the HCMV UL83 ORF (forward primer, GCAGCCACGGGATCGTACT; reverse primer, GGCTTTTACCTCACACGAGCATT). Data were collected on a Bio-Rad MyIQ real-time thermocycler (40 cycles of 95°C for 15 s and 60°C for 1 min).
Bioinformatic analysis.
Microarray analyses were performed using BRB ArrayTools (v4.2.0 beta 2) developed by Richard Simon and the BRB ArrayTools Development Team. Differential gene expression analysis was conducted using the significance analysis of microarrays (SAM) (22) option in the BRB array and a false discovery rate (FDR) of 0.001. Lists of genes significantly differently expressed between each mutant and AD169 were generated at each time point, and expression values for these genes were then analyzed for other pairwise comparisons (mutant-infected versus mock-infected cells, AD169-infected versus mock-infected cells, etc.). Gene ontology categorization of differentially expressed genes was carried out using the Cytoscape (v2.8.1) platform and the biological gene ontology (GO) plug-in BiNGO (23). Gene ontology definitions and annotation files were downloaded from http://www.geneontology.org/ and were dated 19 January 2012. Genes were grouped on the basis of gene ontology biological function, and only overrepresented categories where P was <0.0001 were considered relevant for this study.
Immunofluorescence microscopy.
HFFs were seeded at 72 h preinfection onto gelatin-coated 8-well chamber slides (Thermo Fisher Scientific, Waltham, MA) and infected at an MOI of 3.0 with the indicated virus. At the times indicated below, cells were fixed in 4% paraformaldehyde for 10 min at room temperature (pp65 input assay) or ice-cold methanol for 15 min (interferon regulatory factor 3 [IRF3] nuclear localization assay). Paraformaldehyde-fixed cells were quenched in 50 mm NH4Cl for 10 min and permeabilized in blocking buffer (10% normal goat serum, 5% glycine in PBS, 0.2% Triton X-100). All cells were blocked for 1 h in blocking buffer without Triton X-100 before incubation with primary antibodies. Antibody staining was conducted using appropriately diluted primary antibody against pp65 (Fitzgerald Industries, Acton, MA), IRF3 (SL-12; Santa Cruz Biotechnologies, Dallas, TX) and Alexa Fluor 488-conjugated secondary antibody (Life Technologies, Grand Island, NY). Images were captured using a Nikon Eclipse microscope and equal exposure times for all paired samples. Fluorescence intensity was measured using ImageJ software. To calculate the nuclear fluorescence intensity of IRF3, binary masks were created using a threshold on the DAPI (4′,6-diamidino-2-phenylindole)-stained image for each sample. The mask was then applied to the IRF3 signal, and the mean IRF3 nuclear fluorescence signal of mock-infected cells was subsequently subtracted from the signal for every sample before quantitation.
Microarray data accession number.
All data collected from the bead array have been made publically available through the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) and were released on 30 November 2013 (accession number GSE50955).
RESULTS
Deletion of the HCMV AD169 US17 ORF does not significantly alter viral replication in primary fibroblasts.
A BAC mutant (the ΔUS17 mutant virus) in which the entire US17 ORF was replaced with a 32-bp FRT scar was constructed. This mutation did not alter the mapped polyadenylation signal shared by US18, US19, and US20 (24), and we verified US18 expression by immunofluorescence with a previously described antibody (13). The deletion is likely upstream of transcriptional signals for US16. This mutation deletes the C-terminal 68 amino acids of open reading frame cORF29 (RASCAL) (25). Expression of this protein was verified in strains Towne and TB40e, but not AD169. No evidence of RASCAL expression was found in a detailed translational analysis of cells infected with HCMV strain Merlin (5). In the same analysis, one ORF that is expressed from an alternative translation initiation codon within the US17 ORF and another that is internal to the US17 ORF but in the opposite orientation were identified. We cannot discount the possibility that the ΔUS17 phenotypes trace at least in part to effects on US16 or other proteins expressed from the US17 locus.
The construct was verified by viral genome restriction digestion and sequencing of PCR amplimers that spanned a region from 100 bp upstream to 100 bp downstream of the US17 ORF (Fig. 1A). One-step and multistep growth analyses of ΔUS17 were performed by infecting low-passage-number HFFs at an MOI of 3.0 or 0.01. Cell culture supernatants were sampled every 24 or 48 h and analyzed in triplicate by limiting dilution plaque assays. Consistent with the behavior of the HCMV Towne strain with a similar US17 ORF deletion (26) and strain AD169 with transposon insertions (6), from 2 to 11 dpi (low MOI, multistep) or 24 to 96 hpi (high MOI, one step) ΔUS17 grew to approximately the same titer as the parental BAC pAD/Cre strain (Fig. 1B and C). A revertant virus in which the US17 deletion was repaired by inserting the US17 sequence back into the mutant HCMV genome fused in frame with a C-terminal V5 epitope tag (Rev17v5) was also created. This mutant had growth characteristics similar to those of the deletion mutant and the parental virus. Immunoblotting for various kinetic classes of viral proteins (immediate early proteins 1 and 2 and late proteins pp65 and pp28) over a 96-h time course (MOI = 3.0) revealed no difference in the expression of viral proteins between the ΔUS17 mutant and the parental virus (Fig. 1D).
FIG 1.

Deletion of US17 does not significantly affect the production of infectious virus in fibroblasts. (A) Schematic detailing the US17 locus and recombinant viruses generated for use in this study. (B, C) Multistep (MOI = 0.1) (B) and single-step (MOI = 3.0) (C) growth curves comparing production of infectious virus using the parental virus pAD/Cre, the US17 deletion mutant (ΔUS17), and the repair virus 17cV5. (D) Immunoblot of cellular lysates from HFFs infected with either pAD/Cre (lanes P), the ΔUS17 mutant (lanes Δ17), or the repair virus 17cV5 (V5) at an MOI of 3.0 for 24 to 96 hpi.
Microarray experiment: design and quality control.
No specific biochemical function has been ascribed to any member of the HCMV US12 family, and viruses with deletions of individual US12 family members have diverse growth phenotypes in monocell culture systems. To identify the biological pathways affected by US17, we measured the global cellular transcriptional changes induced by a virus with a deletion of HCMV US17. We used Illumina HT-12 human bead arrays and RNA isolated from HFFs infected with either AD169 or ΔUS17 using an MOI of 6 at an early time point after infection (12 hpi) or a relatively late time point after infection (96 hpi). The 12-well design of the chips allowed multiple replicates per virus (triplicate biological specimens were used in this study). Each of the 47,213 unique probes for over 28,000 annotated genes is represented by an average of 30 individual beads per well. Together, the high number of replicates per probe and the number of biological replicates afforded a high level of statistical power that enabled robust discrimination of subtle differences in transcriptional profiles.
Raw expression data for the 47,213 probes from the arrays were imported and collated into BRB ArrayTools using a bead array-optimized robust spline normalization (27). A prefilter was applied to eliminate probes that showed no signal differences (P < 0.001) across the full set of viruses, time points, and technical replicates; the signal intensities of the probes for 10,672 unique transcripts were then passed along for downstream analysis.
To assess consistency across replicate arrays, we used hierarchical clustering to construct a dendrogram of the unique probes identified as described above (centered correlation, average linkage) (Fig. 2). Two major clades were apparent: one consisted of mock-infected cells collected at 12 hpi and cells that were infected with the two viruses and collected at 12 hpi, and the second one consisted of cells that were infected with the two viruses and collected at 96 hpi. The mock-infected cells and cells collected at 12 hpi clustered separately from the cells collected at 96 hpi, and the transcript profiles of all infected cells had no correlations with the profiles of the mock-infected cells, indicating greatly altered transcription profiles in cells infected with both the ΔUS17 mutant virus and AD169. The results for all sets of experimental triplicates clustered with themselves, indicating that the results obtained with the replicate arrays were in high agreement with each other (correlations, ≥0.80, except for the correlation for ΔUS17 at 12 hpi, which had a correlation between the replicate arrays of 0.75). At 96 hpi, the profiles of the mutant and AD169 were highly correlated (>0.90), indicating that most host transcripts were expressed at similar levels in infections with either virus. The dendrogram illustrates that while the transcript profile of ΔUS17 was highly similar to that of AD169, there were sufficient differences to classify them as distinct entities.
FIG 2.
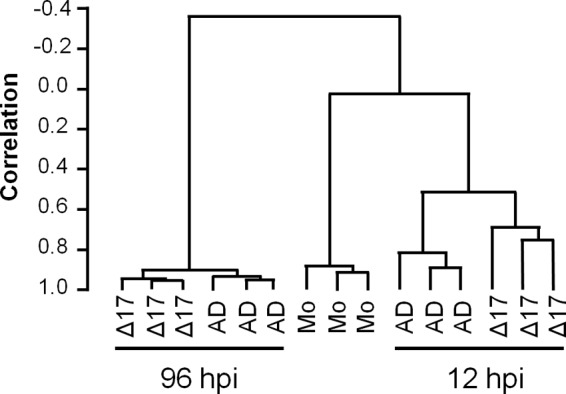
ΔUS17 alters small sets of host transcripts at both 12 and 96 hpi. Dendrograms showing the relationships of cellular transcript profiles generated by Illumina HT-12 bead array analysis. Three biological replicates were analyzed for each of the following viruses: AD169 (AD), ΔUS17 (Δ17), and no virus (mock infection [Mo]). Correlation scores closer to 1 denote a higher degree of similarity between two clades.
To analyze overall gene transcript profiles, scatterplots were constructed for both time points for all pairwise comparisons using the 10,672 probes that passed the filtering criteria (Fig. 3A and B); note that probes found by SAM to be significantly differentially expressed are indicated as either black or colored dots, while probes not found by SAM to be significantly differentially expressed are indicated as light gray dots (described in detail below). In comparison to mock infection, infection with both viruses induced numerous changes to the transcription profiles at either time point. At 12 hpi, both viruses modulated expression by ≥1.5-fold for over 1,800 probes versus the results for mock-infected cells (Fig. 3A). At 96 hpi, changes to the transcriptional profiles were even more pronounced. Both AD169 and the ΔUS17 mutant virus induced expression by ≥1.5-fold for over 3,000 probes compared with the results for mock-infected cells (Fig. 3B). In contrast, there were relatively few transcriptional differences between ΔUS17 and AD169, correlating with the similarity noted on the dendrogram in Fig. 2 and confirming that ΔUS17 modulated the expression of only a small percentage of transcripts relative to its parent.
FIG 3.

Transcription modulation by ΔUS17 at 12 and 96 hpi identified by SAM to be significant. (A and B) Pairwise scatterplot analysis of the 10,672 probes that passed the initial prescreen filter. Colored dots indicate GO biological themes of differentially regulated transcripts between ΔUS17 and AD169 (FDR = 0.001). Black dots, nonimmune genes found by SAM analysis to be significantly differentially expressed; gold dots, innate and intrinsic immune response-related genes found by SAM analysis to be significantly differentially expressed at 12 hpi; light blue dots, innate and intrinsic immune response-related genes found by SAM analysis to be significantly differentially expressed at 96 hpi; black lines, a ≥1.5-fold change in either direction. (C) Venn diagrams of total transcripts (left) and immune response-related transcripts (right) found by SAM analysis to be significantly differentially expressed at 12 and 96 hpi.
Differential expression of gene transcripts in cells infected with ΔUS17 versus those infected with AD169.
To analyze differentially expressed transcripts more robustly, we examined the specific differences in cellular transcript profiles between AD169 and ΔUS17 infections using SAM with a stringent false discovery rate (FDR) of 0.001. At 12 hpi, of the 10,672 probes that passed the filtering criteria, SAM indicated that only 278 were significantly differentially expressed between the two viruses (36 upregulated and 242 downregulated); at 96 hpi, 97 probes were differentially expressed (22 upregulated and 75 downregulated) (Fig. 3C; see Table S1 in the supplemental material).
To visualize the magnitude of the significant changes between AD169 and ΔUS17, probes found to be significantly differentially expressed by SAM were plotted as black or colored dots in the Fig. 3 scatterplots. At 12 hpi, a large number of genes found by SAM to be significantly differentially upmodulated in the data set for AD169-infected versus mock-infected cells were relatively attenuated in the data set for ΔUS17-infected versus mock-infected cells, thus resulting in the population of genes downmodulated in the comparison of ΔUS17 and AD169. In contrast, at 96 hpi, the genes that were downmodulated in the comparison of ΔUS17 and AD169 were the product of an amplification of downmodulations that occurred in the AD169 infections.
ΔUS17 modulates host innate and intrinsic immune responses early after infection.
The genes found by SAM analysis to be significantly differentially expressed between cells infected with AD169 and those infected with ΔUS17 were categorized using Cytoscape and the biological network GO plug-in (BiNGO). BiNGO searches for statistically significantly overrepresented GO categories among a set of genes and then maps those GO categories onto a visual network. This allows identification of clusters of GO categories that, when used in conjunction with the GO biological process hierarchy, highlights the major biological themes of the input list of genes. At 12 hpi, BiNGO was able to categorize 239/278 (86%) of the genes found by SAM analysis to be significantly differentially expressed into 259 statistically enriched biological process GO categories with a statistical threshold with a P value of <1 × 10−4 (see Table S2 in the supplemental material).
To further categorize this list of GO data, we utilized the orthogonal layout option within Cytoscape. This minimizes edge (the lines that connect the individual category bubbles on the network) overlap between GO categories and has the effect of grouping highly interconnected and, thus, closely related GO categories next to each other. The full visual network of all significantly enriched GO categories is presented in Fig. S1 in the supplemental material. The several clusters of GO categories relating to innate and intrinsic immunity, regulation of immune responses, apoptosis, and metabolism showed high levels of statistical enrichment.
Enrichment scores (ESs) were calculated for several of these clusters of highly interconnected GO categories from the data set for 12 hpi by calculating the inverse log of the geometric mean of the P values for each GO category within the cluster calculated by the use of BiNGO. Higher enrichment scores denote a lower average P value for the cluster and indicate a higher degree of average statistical significance (Fig. 4A). The most statistically significant clusters contained GO categories related to various aspects of innate and intrinsic immunity, especially type I interferon antiviral responses (41 categories, ES = 14.6). This cluster also contained the most significantly enriched category overall, response to virus (P = 2.63 × 10−40), which contained 40/239 (17%) of the genes found by SAM analysis to be significantly differentially modulated at 12 hpi. Genes in this category were associated with sensing and responding to viral infections, and many interferon-stimulated genes were included. Also noted were highly enriched clusters of GO categories related to the regulation of various immune system processes, such as leukocyte migration, production of type I interferon, regulation and production of cytokines, and regulation of NF-κB (67 categories, ES = 8.1), and apoptosis or regulation of apoptotic molecular function (32 categories ES = 7.2). Two less significant clusters of GO categories were also identified and contained GO categories related to blood vessel development and angiogenesis (13 categories, ES = 6.0) and metabolism (32 categories, ES = 5.7). Together, genes involved in these five biological themes comprised 174 of the 239 differentially expressed genes categorized by BiNGO at 12 hpi. The other 65 genes that were found by SAM analysis to be significantly differentially expressed at 12 hpi and that had gene ontology biological process information available showed no relationship to anything in the gene ontology hierarchy that passed our threshold of significance and thus were not considered for further analysis.
FIG 4.

Gene ontology analysis of transcripts found by SAM analysis to be significantly differentially expressed. (A) Table outlining the five biological themes identified in the comparison of transcript expression by ΔUS17 versus AD169 at 12 hpi. Listed are the numbers of individual gene ontology categories for each theme out of the 259 total GO categories identified to be statistically significantly differentially expressed (P < 0.0001). Also listed are the numbers of transcripts in each theme out of the 239 that were found by SAM analysis to be significantly differentially expressed and that could be categorized into a gene ontology biological process category; note that genes can simultaneously appear in multiple themes. The enrichment score denotes the average level of significance of all of the GO categories in a particular theme. Higher enrichment scores indicate a more significant, mean log-transformed P value for that theme. (B) Venn diagrams detailing the numbers of transcripts from ΔUS17 versus those from strain AD169 found by SAM analysis to be significantly differentially modulated at 12 hpi grouped into the five most highly significant biological categories identified by BiNGO gene ontology biological process clustering. (Left) Comparison of transcripts that grouped the three most significant gene ontology themes of innate immunity and interferon production, regulation of immune response, or apoptosis (the labeling for the groups follows the letter scheme used in panel A); (right) comparison of transcripts that grouped into either of the first three themes with those that grouped into the two minor identified gene ontology themes of blood vessel development and metabolism.
Venn diagrams were constructed to highlight the relationships of the individual genes in each of the five major biological process themes identified above. Of the 216 genes classified by BiNGO, 131 of them fell under the three most significant biological themes of innate and intrinsic immunity, regulation of immune processes, and apoptosis (Fig. 4B). Genes in these categories showed a large degree of overlap with each other, with few unique genes in each theme, with the exception of innate and intrinsic immunity, which had 44 genes that were not classified in any other category. We next compared the two less significant biological themes of metabolism and blood vessel development. Again, transcripts in these categories were found to have a high degree of overlap with GO categories related to the more statistically significant immune response and apoptosis GO clusters (Fig. 4). Taken together, the high degree of overlap between the five identified themes indicates that the most significant of them, innate and intrinsic immunity, represents the major biological theme of the genes differentially expressed at 12 hpi.
To put these results into the broader context of host immune responses during HCMV infection, we compared the expression of the 131 innate and intrinsic immunity-related transcripts found by SAM analysis to be significantly differentially regulated by infection with the ΔUS17 mutant virus with expression of the transcripts by mock-infected cells and visualized them by overlaying the genes on the Fig. 3 scatterplots. The subset of immune response/apoptosis-related transcripts identified above was readily apparent, showing a high degree of upmodulation in the pairwise comparison of AD169-infected versus mock-infected cells but a relative attenuation of upmodulation in the pairwise comparison of ΔUS17-infected versus mock-infected cells. The net result was an overall downmodulation of these transcripts between ΔUS17 and AD169 (Fig. 3A, gold dots). Of the 131 innate and intrinsic immunity-related transcripts, 123 were differentially downmodulated in this fashion by ΔUS17 compared to their modulation by AD169, with 54 of those transcripts being downregulated ≥2-fold. Many of the most highly downregulated genes encoded either interferon-stimulated genes or soluble factors, namely, CC and CXC chemokines and cytokines. These transcripts grouped into two of the highly significant gene ontology categories identified above: production of type I interferon (P = 6.22 × 10−32, 21 probes) and inflammatory response (P = 3.27 × 10−7, 8 probes). The expression levels of the genes in these two categories are illustrated in Fig. 5. Complete lists of all genes found by SAM analysis to be significantly differentially expressed at 12 hpi and 96 hpi and transcripts common to both time points are provided in Table S1 in the supplemental material.
FIG 5.
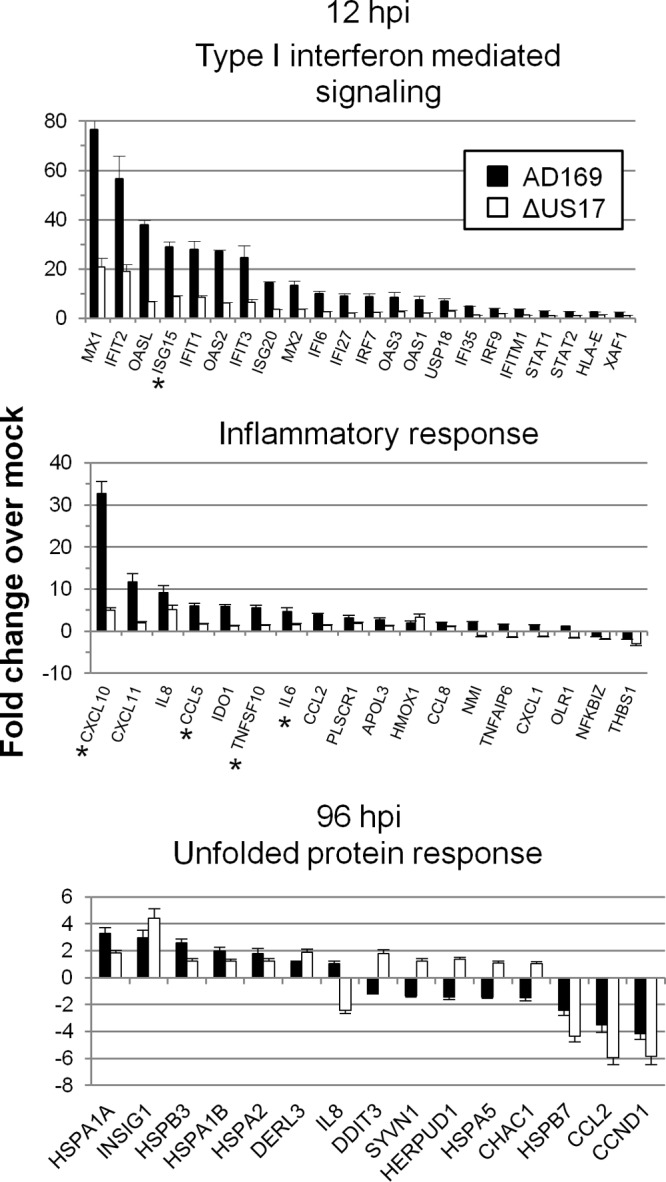
Transcript expression profiles for several GO categories highly significantly enriched at either 12 or 96 hpi. For 12 hpi, the categories included type I interferon-mediated signaling (P = 6.22 × 10−32), which mainly included antiviral interferon-stimulated transcripts, and inflammatory response (P = 3.27 × 10−7), mainly comprised of proinflammatory cytokine and chemokine transcripts. For 96 hpi, one GO category, response to unfolded protein, is highlighted (P = 2.53 × 10−8). Asterisks, genes validated by TaqMan qRT-PCR.
To further confirm the phenotype observed using the microarray, we employed a custom qRT-PCR array that targets six innate and intrinsic immune response-related transcripts that showed a high degree of downmodulation by mutant virus compared to their expression by AD169 at 12 hpi. We compared the parental virus pAD/Cre, a BAC version of AD169 which contains no deletion of the primary sequence but does contain a small 32-bp Lox scar between the US28 and US29 ORFs, two independently derived ΔUS17 mutant viruses (ΔUS17 and 2ΔUS17), and a US17 deletion repair virus (17cV5). The two independent ΔUS17 viruses showed the same downmodulation observed on the microarray (Fig. 6). Additionally, the repair virus restored the level of transcript expression to parental levels, indicating that deletion of the US17 locus had a specific effect on expression of immune response-related genes. Overall, these changes in gene expression at 12 hpi, a time that precedes de novo expression of most HCMV genes, suggest changes in the way in which ΔUS17 virions are sensed by the host cell.
FIG 6.
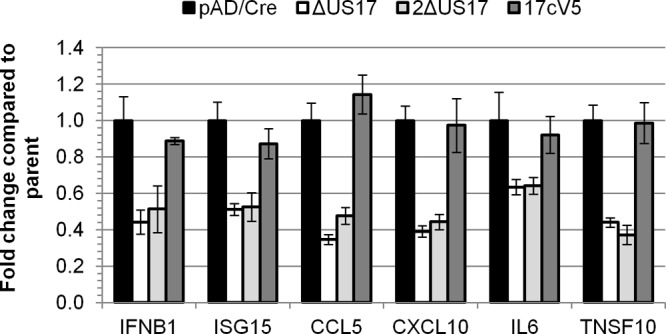
qRT-PCR verification of bead array-identified transcripts. HFFs were infected with either the parental BAC virus (pAD/Cre), either of two independently generated ΔUS17 viruses, or the US17 deletion repair virus (17cV5) for 12 h at an MOI of 3.0. TaqMan qRT-PCR was performed with primers specific for six different genes that were highly differentially regulated by ΔUS17 at 12 hpi. Shown are the averages of biological replicates (± 1 SEM), three for pAD/Cre, two for ΔUS17, three for 2ΔUS17, and one for 17cV5. Three technical replicates were performed for each experiment.
Gene ontology analysis at 96 hpi.
BiNGO categorized 86 of the 98 genes found by SAM analysis to be significantly differentially expressed at 96 hpi into 76 significantly enriched GO biological process categories (P < 1 × 10−4). As is evident in the scatterplots (Fig. 3B), the differences between ΔUS17 and AD169 at 96 hpi were generally fewer in number than those at 12 hpi. Fourteen of the overrepresented biological process categories were related to type I interferon, cytokine production, or various other aspects of immune responses, and another five categories were related to apoptosis (see Table S2 in the supplemental material). Together, these categories contained 43 differentially regulated transcripts, 22 of which were identified by SAM analysis as being differentially expressed between AD169 and ΔUS17 at both 12 hpi and 96 hpi (Fig. 3C). Interestingly, for most of these genes, although the change in magnitude relative to that for mock-infected cells decreased from 12 to 96 hpi, the directions of the transcriptional changes were consistent at both times, with the ΔUS17 mutant virus blunting the induction of certain immune response-related transcripts (predominantly interferon-responsive genes) while amplifying the suppression of other transcripts (predominantly, genes involved in tissue development). In sum, the differences between the effects of ΔUS17 and its parent on host immune response-related transcripts at 12 hpi persisted to at least 96 hpi.
Several ER stress-related and unfolded protein response-related transcripts were differentially regulated at 96 hpi but not at 12 hpi. This included transcripts for a number of 60-kDa heat shock proteins (HSP60), as well as transcripts for proteins involved in ER stress and the ER-associated degradation (ERAD) pathways. Positive and negative effects were seen among transcripts associated with these genes (Fig. 5). At least one of these genes (HSPA5, or BiP) is important for biogenesis and the function of the HCMV cytoplasmic virion assembly complex (cVAC) (28, 29). US17 shows its highest levels of expression from 96 to 120 hpi; thus, the modulation of transcription by the ΔUS17 mutant virus at late times is probably a more direct effect of the mutation, whereas the mutation has indirect effects at 12 hpi.
ΔUS17 induces changes in virion protein composition.
Changes in cellular gene expression at 12 hpi were unexpected, given that US17 is a late gene whose expression is dependent on viral DNA synthesis and is not detectably expressed in infected cells until at least 48 hpi (see the summary diagram in Fig. 11). We speculated that the changes in transcription of cellular interferon and innate immune response-related genes at 12 hpi were the product of changes in virion protein content caused by the absence of US17. To address this, we compared the protein profiles of gradient-purified virions by silver staining and immunoblotting for three tegument proteins (pp65/pUL83, pp28/pUL99, and the large tegument protein/pUL48) as well as two virion surface glycoproteins (gB/pUL55, and gH/pUL75). Differences in relative abundance were noted for several protein bands upon silver staining, indicating that the virions produced by ΔUS17 differ from those produced by the parental virus (Fig. 7A). No gross changes were seen by immunoblotting for any of the tegument proteins analyzed, pp65, pp28, or pUL48. However, for ΔUS17 virions there was an ∼2.8-fold decrease levels of the envelope glycoprotein, gH, while gB levels were unchanged (Fig. 7B). gH has a predicted size of ∼75 kDa, similar to the ∼75-kDa protein band that showed a reduced level in the silver-stained blot (Fig. 7A, middle arrow). This indicates that although equal levels of infectious virions are produced, some aspects of virion assembly are altered in the absence of the US17-coding region.
FIG 11.
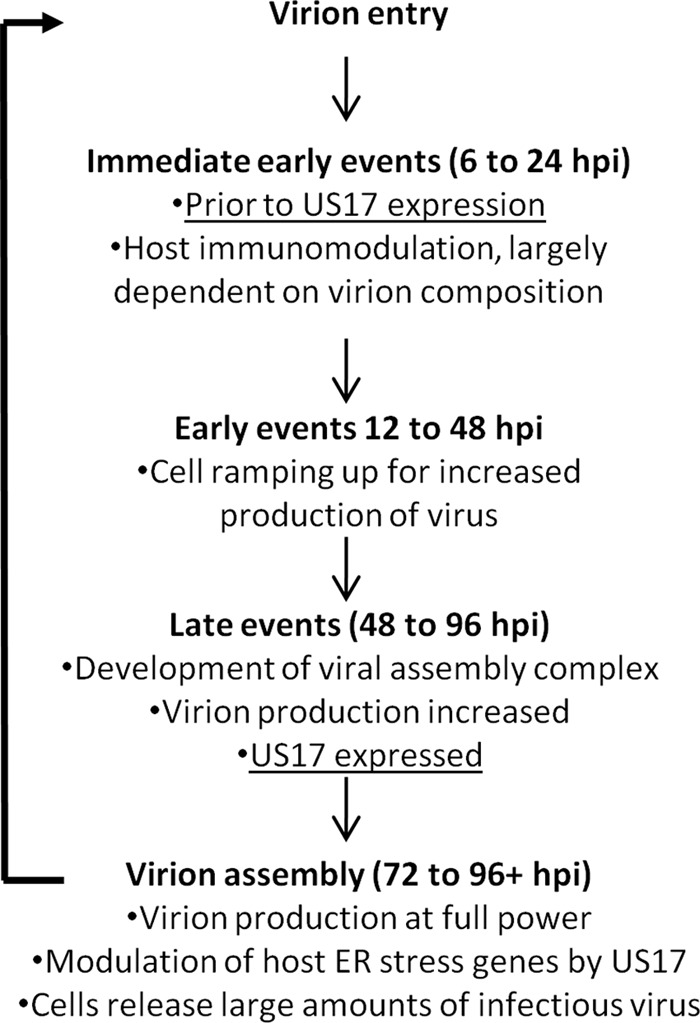
Proposed model for US17 direct and indirect functions. A timeline depicting US17's role as an indirect modulator of the host cell immune response at 12 hpi and a direct role for US17 at 96 hpi by way of modulation of host cell ER stress response genes is shown.
FIG 7.
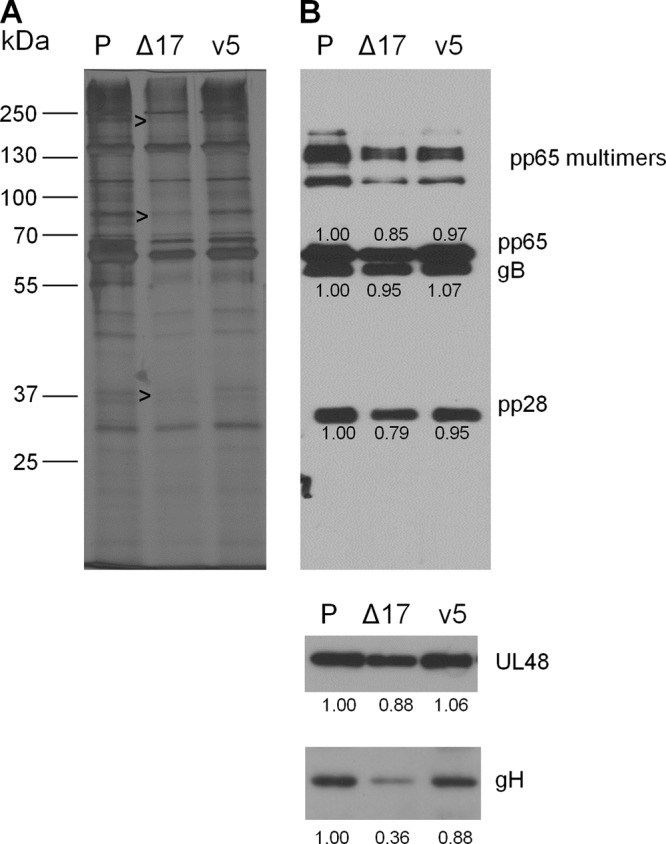
ΔUS17 changes virion composition. Supernatant-associated virus from either parental strain pAD/Cre (P), ΔUS17 (Δ17), or the 17cV5 repair virus (v5) was purified by centrifugation through a 10 to 50% Nycodenz gradient. Virion-containing fractions were subsequently dialyzed into PBS and analyzed for protein content. (A) Equal amounts of each sample were run on a 10% polyacrylamide gel and silver stained. Black arrowheads, subtle changes in the virion composition of ΔUS17. (B) Immunoblotting for various structural proteins: pp65 (pUL83), gB (pUL55), pp28 (pUL99), large tegument protein (pUL48), or gH (pUL75). The numbers underneath each blot are densitometric values, calculated using ImageJ software, normalized to the values for the parental sample.
ΔUS17 alters genome-to-PFU ratios.
Having shown in HFFs that the growth curves of viruses with deletions of the US17-coding region are indistinguishable from those of the parental virus (Fig. 1B), we analyzed the virions produced by ΔUS17 with respect to the ratio of the number of genomes to the number of PFU (genome-to-PFU ratio) and compared to the results to those for the virions produced by the parental virus. The levels of DNase-protected viral genomes in cell culture supernatants were quantified by quantitative PCR (qPCR) as a surrogate measurement for the total number of DNA-containing particles (infectious and noninfectious). At both 72 and 96 hpi, times when equal numbers of infectious virions were produced by both viruses, ΔUS17 produced 3.7- and 3.8-fold more genome-containing viral particles (Fig. 8B). To obtain the genome-to-PFU ratio, the fold changes in the levels of DNase-protected genomes were divided by the fold changes in infectious titer between ΔUS17 and pAD/Cre. ΔUS17 produced 3.4 genome-containing noninfectious particles for every such particle produced by the parental virus at 72 hpi (P = 0.001) and produced a similar increase at 96 hpi (3.3:1, P = 0.0003) (Fig. 8C). The repair virus had ratios similar to those of the parental virus (pAD/Cre).
FIG 8.

ΔUS17 alters the genome-to-PFU ratio of infected cell culture supernatants. HFFs were infected at an MOI of 3.0 with either the parental virus, ΔUS17 or the 17cV5 revertant virus. At 72 and 96 hpi (A), or 10 dpi (D), samples were plaque assayed for infectivity or assayed for total DNase-protected viral genomic content (B and E) by qPCR. In panel B, the results shown are normalized to those for the parent virus at 72 hpi to highlight the increase in the number of genomes produced at 96 hpi. (C and F) Genome-to-PFU ratios were calculated by dividing the ratios of the fold change (mutant versus parent) in the number of relative genomes and infectivity and are shown relative to the results for the parental virus at each time point.
The experiments described above employed clarified culture supernatants. To address the possibilities that the observed effects traced to soluble factors in the supernatants or to virus particles, such as dense bodies that are enriched for tegument proteins, such as pp65, and that have densities different from those of parental virions (30), we analyzed the genome-to-PFU ratios of the gradient-purified virions used in the protein analyses whose results are presented in Fig. 7A and B. In comparison to the parental virus or the repair virus, ∼3-fold fewer infectious virions were detected in the ΔUS17 fraction (Fig. 8D). Nonetheless, the ΔUS17 fraction contained 1.2-fold more DNase I-protected viral DNA than the parental virus (Fig. 8E) and 3.65 to 1 more noninfectious genome containing particles than the parental virus, while the US17 repair virus had a genome-to-PFU ratio of 1.65 to 1 (Fig. 8F). Thus, the genome-to-PFU ratio increase seen at 72 and 96 hpi during infection at a high MOI also occurred during infection at a low MOI and over a much longer infection time course of 10 days. Further, the particles produced by the ΔUS17 mutant virus had a density similar to that of the majority of infectious virions produced during HCMV infection in fibroblasts.
The elevated genome-to-PFU ratio of ΔUS17 was consistent over multiple experiments using a variety of biological conditions, i.e., MOIs ranging from 0.001 to 6.0 with time courses ranging from 3 dpi to 14 dpi. We also generated multiple stocks of both pAD/Cre and ΔUS17 and consistently detected higher genome-to-PFU ratios for ΔUS17, although its levels of infectious virus were always within 1- to 3-fold of those of the parent. Thus, under a variety of conditions, ΔUS17 produces a larger quantity of noninfectious genome-containing particles with densities similar to those of parental virions, while it produces the same number of infectious virions as the parental virus.
ΔUS17 virions deliver higher levels of pp65 (pUL83).
We studied the effects of this altered particle-to-PFU ratio on the initial stages of viral infection, specifically, delivery of tegument proteins to infected cells. We hypothesized that the larger number of genome-containing noninfectious viral particles delivered by the ΔUS17 mutant virus at a given multiplicity of infection would result in increased input levels of tegument proteins. We focused on the tegument protein pp65, as it is a major constituent of the virion structure and is delivered in sufficient quantities to infected cells to allow visualization of input protein immediately after infection. Also, pp65 antagonizes the nuclear translocation of interferon regulatory factor 3 (IRF3) (17) and downregulates expression of several interferon genes identified as being differentially regulated by ΔUS17 at 12 hpi. Thus, if the excess of noninfectious viral particles produced by ΔUS17 could enter into and deliver higher levels of pp65 and other tegument proteins to infected cells, this might explain the observed differences in immunomodulation by ΔUS17.
To directly visualize the amounts of pp65 delivered during virion entry, we employed immunofluorescence microscopy on HFFs infected at an MOI of 3 at 2 hpi, a time when little de novo viral protein production could have taken place. Localization of the pp65 signal was not altered in ΔUS17-infected cells, with the majority of the signal residing in the nucleoplasm and nucleoli and with small amounts remaining as punctate cytoplasmic staining (Fig. 9A). In comparison with the parental virus, ΔUS17 displayed a 2.6-fold (P = 0.001) increase in the level of intracellular pp65 delivered to each cell infected with ΔUS17 (Fig. 9B). While this result is possibly attributable, at least in part, to dense bodies, the increase in the amount of pp65 delivered to newly infected cells corresponds well with the ∼3-fold increase in the amount of noninfectious gradient-purified virions produced by ΔUS17 observed via qPCR.
FIG 9.
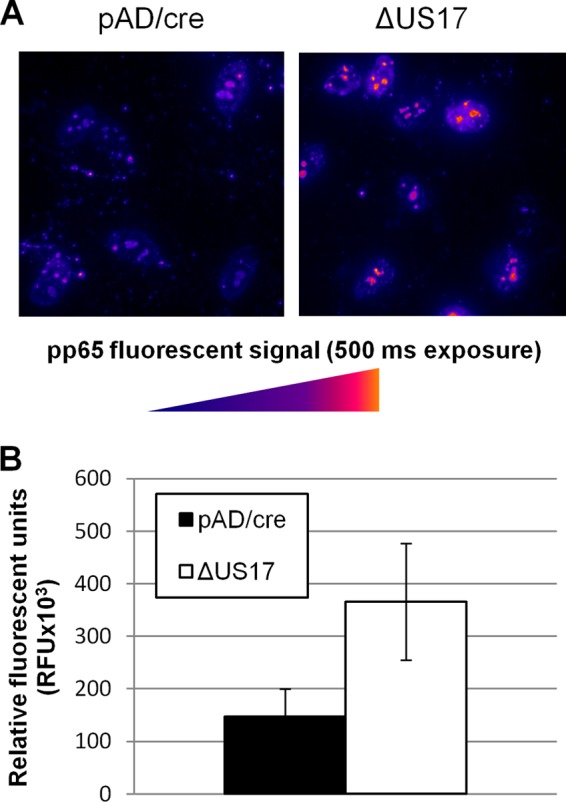
Higher levels of tegument proteins are delivered per PFU to ΔUS17 mutant virus-infected fibroblasts. HFFs were infected in 8-well chamber slides with the indicated virus at an MOI of 3.0. At 2 hpi, cells were washed with PBS, fixed in 4% paraformaldehyde, and stained with a pp65-specific monoclonal antibody. (A) Representative images showing pp65 signals. (B) Mean intensities (±1 SEM) of total pp65 fluorescent signals taken from three randomly chosen microscope fields from each of two independent biological experiments.
To learn whether the elevated levels of pp65 correlate with changes in interferon signaling, we assessed whether infection with ΔUS17 would result in a change in IRF3 expression or localization. An immunofluorescence assay for IRF3 was performed on HFFs infected at an MOI of 3 for either 3 or 12 h. Mock-infected cells displayed basal levels of expression of cytoplasmic IRF3 and very little nuclear expression at either time point (Fig. 10A and B). None of the viruses tested affected the total levels of IRF3 protein, in agreement with the results of microarray analysis, which showed no differences in expression of the IRF3 transcript. However, when the nuclear IRF3 fluorescence of ΔUS17 was compared to that of the parent, the amount for ΔUS17 was reduced by ∼2-fold (0.49 ± 0.09) at 3 hpi (Fig. 10C) and ∼3-fold (0.35 ± 0.17) at 12 hpi (Fig. 10D). The nuclear fluorescence of IRF3 was similar between the repair and parental viruses, indicating that although ΔUS17 does not alter the total level of IRF3 after infection, nuclear translocation is significantly inhibited. Although the amount of nuclear localization of IRF3 seen for the BAC variant of AD169 (pAD/Cre) used in this study was higher than that observed for several other strains of HCMV by Abate et al. (17), IRF3 nuclear localization was similarly inversely correlated with pp65 expression.
FIG 10.

ΔUS17 inhibits IRF3 nuclear translocation shortly after infection. HFFs were infected with the indicated virus at an MOI of 3.0 for 3 or 12 h and stained with a monoclonal antibody against IRF3. (A and B) Representative images showing IRF3 and DAPI fluorescent signals at either 3 or 12 h; (C and D) mean IRF3 nuclear fluorescence of three randomly chosen fields (±1 SEM) at 3 hpi (C) and 12 hpi (D). n, number of nuclei counted per condition.
DISCUSSION
HCMV employs numerous strategies to subvert or co-opt various aspects of the host cell biological machinery to benefit its protracted replication cycle (8, 31, 32). Here we identified the effects of US17 on the expression of cellular genes that regulate pathways important in infected cells. Specifically, microarray analysis revealed modulations of expression of cellular transcripts involved in innate and intrinsic defenses by ΔUS17 at both early and late times postinfection. At a later time point (96 hpi), ΔUS17 modulated the expression of transcripts associated with ER stress and the unfolded protein response. In addition, cells infected with ΔUS17 also produced >3-fold more noninfectious viral particles than the parental virus. When cells were infected at equal multiplicities of infection, the excess noninfectious particles entered host cells and delivered larger amounts of the immunomodulatory tegument protein pp65. This excess of pp65 and potentially other tegument proteins is likely responsible for the modulation of host immune transcripts at 12 hpi. Based on this, we propose a model wherein US17 (or other components of the US17 locus) contributes to the regulation of ER stress and unfolded protein response-related pathways that affect protein trafficking or folding, thereby influencing virion composition to fine-tune innate and intrinsic immune responses (Fig. 11).
ΔUS17 indirectly modulates host innate and intrinsic immune responses.
As for other infectious agents, modulation of immune responses is a fundamentally important aspect of HCMV biology. The process of HCMV host cell immunomodulation begins at the very earliest stages of viral attachment to the cell and is highly dependent on the structural components of the virion, i.e., envelope glycoproteins such as gB and gH and tegument proteins such as pp65 (16, 17, 33–35); reviewed in reference 36. We found that deletion of the US17 locus had little impact on the production of infectious viral particles in fibroblasts but nevertheless exerted a tangible influence over viral replication. ΔUS17 markedly blunted the host cell antiviral response at a very early time point (12 h) after infection. Many interferon-stimulated transcripts and transcripts encoding proinflammatory chemokines and cytokines were downmodulated by 3- to 5-fold by ΔUS17 compared to their level of expression in the parental virus when the viruses were used at an equal MOI.
We hypothesize that the differences in immunomodulation observed between ΔUS17 and its parent at equal MOIs result from the mutant delivering larger amounts of tegument proteins via the larger number of genome-containing noninfectious viral particles. One of the best characterized of these immunomodulatory structural proteins is the tegument protein UL83 (pp65), which blunts, but does not completely abrogate, the interferon response at very early times after infection (3 to 5 hpi). A recombinant virus that does not express pp65 elicited a vastly increased interferon response from host cells (16, 17). Complementary to that result, at equal multiplicities of infection, ΔUS17 delivered approximately 3-fold more pp65 than the parental virus. The additional pp65 and possibly other tegument proteins delivered by the larger amount of noninfectious viral particles produced by the ΔUS17 mutant virus likely explain the observed blunting of the host interferon response. We further tested this hypothesis by examining the expression of IRF3 at early times after infection. The induction of interferon-stimulated genes is largely controlled by interferon regulatory factors, such as IRF3, which are phosphorylated and translocated from the cytoplasm to the nucleus, where they act as transcription factors. The nuclear accumulation of IRF3 is inhibited by expression of pp65 even in the absence of infection, and a virus with a deletion of pp65 displayed much higher nuclear accumulation of IRF3, indicating that pp65 plays a significant role in modulating interferon signaling shortly after infection (17). Consistent with this, we found that ΔUS17 inhibited IRF3 nuclear translocation more robustly than the parental virus pAD/Cre and the repair virus Rev.cV5 at both 3 and 12 hpi, times that correspond to both the delivery of increased amounts of pp65 by ΔUS17 and the blunting of interferon-stimulated gene expression detected by microarray analysis at 12 hpi.
A role for US17-mediated manipulation of ER stress responses late in infection.
In contrast to the modulation of immune responses at 12 hpi, other effects were seen at 96 hpi, a time that corresponds to both the high level of expression of US17 and the formation of the cVAC (37). Specifically, expression of some ER stress response and chaperone transcripts was significantly altered by ΔUS17. By unknown mechanisms, HCMV modulates various pathways and genes involved in ER stress and the unfolded protein response (38, 39). One such gene, HSPA5 (GRP78/BiP), which was upregulated 1.6-fold by ΔUS17, is important for the formation of the cVAC and the production of infectious virions (28, 29, 40). Several other important regulators of ER stress were also modulated, including DDIT3 (also known as CHOP; 2.1-fold increase) and CHAC1 (1.6-fold increase). Relative to mock infection, infection with the ΔUS17 mutant virus and its parent altered regulation of most of these transcripts in opposite directions. This is in sharp contrast to the sets of immune response-related transcripts that at 12 hpi were modulated by ΔUS17 and its parent in the same direction relative to their expression in mock-infected cells.
The modulation of gene expression by ΔUS17 at later stages of infection provides insights into a potential mechanism of action for US17 and offers cellular targets that could be studied in this context. We note that US12 family members share sequence similarity with the transmembrane Bax inhibitor 1 (TMBIM) family of conserved eukaryotic antiapoptotic seven-transmembrane proteins (11). These proteins localize to the membranes of various cellular organelles, where they act as rheostats that modulate apoptotic and ER stress signaling by influencing cellular calcium levels (41–45). Taken together, it is possible that US17 is a Bax inhibitor 1 ortholog that shapes virion composition by modulating the levels of ER stress response and apoptosis genes involved in regulation of protein folding, trafficking, and cVAC function. Interestingly, the US17 transcript is expressed in latently infected CD34+ cells but not in latently infected monocytes (46). Thus, US17 may play roles in both the lytic and latent phases of HCMV replication.
HCMV virion composition and genome-to-PFU ratios.
HCMV has a high particle-to-PFU ratio, producing several hundred to several thousand noninfectious particles for every infectious particle (47). A recombinant of strain AD169 with a deletion of US24 produced an equal number of genome-containing particles but showed a 20- to 30-fold reduction in infectivity and an ∼10-fold higher genome-to-PFU ratio (48). Although no differences in the limited amount of viral proteins assayed were seen, the authors reasonably speculated that virion composition may have been altered in ΔUS24. Here we showed that ΔUS17 modulates the genome-to-PFU ratio of gradient-purified virions (with a relatively few dense bodies present) in a different manner. This virus did not affect the levels of infectious virions produced but produced larger numbers of genome-containing noninfectious particles (a net increase in total genome-containing particles).
In comparisons of ΔUS17 virions with those of its parent or the 17cV5 repair virus, the levels of the virion glycoprotein gH were reduced and other differences in virion composition were noted by silver staining. Glycoprotein H is part of the gH/gL/gO complex that is responsible for the binding and entry of HCMV into fibroblasts (49–51). Although we observed reduced levels of gH in ΔUS17 virions, entry of individual virions was not inhibited, as evidenced by the delivery of pp65 to host cells shortly after infection. Aside from their roles in virion entry, HCMV glycoproteins, including gB and gH, elicit immune responses through induction of both NF-κB and Sp1 pathways shortly after binding to the host plasma membrane (34, 35, 52). This virally regulated stimulation and redirection of the innate immune response immediately after infection creates a favorable environment inside the cell, facilitating the early stages of HCMV replication by upregulating expression of key cellular transactivators. Thus, while ΔUS17 mutant virus produces a net increase in the total amount of genome-containing particles compared to that for the parental virus, each of those particles was reduced in the levels of incorporated gH. The reduction was not drastic enough to hinder virion entry but may have increased the number of virions required to meet the level of initial signaling required to begin a productive infection. The exact molecular composition required for the infectivity of HCMV virions is not known; the ideal is likely to differ depending on the target cell or tissue and other physiological conditions. We are using mass spectroscopy to more precisely define the molecular properties of the ΔUS17 virions to further elucidate the relationship between virion composition and infectivity.
This work highlights the complex and incompletely understood process of HCMV virion assembly and shows that US17 (and possibly other US12 family members) shapes the virion composition to regulate host cell responses in newly infected cells. It is possibly counterintuitive that a virus would produce factors to increase its immunogenicity, thereby making it more easily recognized by the host's immune system. Nonetheless, the accumulated evidence is consistent with HCMV striking a complex finely tuned balance between stimulating, suppressing, and avoiding immune responses from the earliest stages of infection to ensure its reproduction and survival (53–56).
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Shenk (Princeton University), Dong Yu (Washington University), Wade Gibson (John Hopkins University), and Kezhong Zhang (Wayne State University School of Medicine) for generously sharing recombinant viruses, antibodies, and qRT-PCR facilities. We also thank the Applied Genomics Technology Center (AGTC) at Wayne State University for RNA analysis and hybridization onto the HT-12 bead arrays.
This work was supported by NIH grants 1 R56 AI099390-01, R03 AI076568-01, and R21 AI076591-01.
Footnotes
Published ahead of print 11 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02704-13.
REFERENCES
- 1.Mocarski ES, Jr, Shenk T, Griffiths PD, Pass RF. 2013. Cytomegaloviruses, p 1960–2014 In Knipe DM, Howley PM, Griffin DE, Cohen JI, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981. 10.1073/pnas.2136652100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dhuruvasan K, Sivasubramanian G, Pellett PE. 2011. Roles of host and viral microRNAs in human cytomegalovirus biology. Virus Res. 157:180–192. 10.1016/j.virusres.2010.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312. 10.1099/vir.0.79888-0 [DOI] [PubMed] [Google Scholar]
- 5.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. 10.1126/science.1227919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401. 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228. 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mocarski ES., Jr 2002. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 10:332–339. 10.1016/S0966-842X(02)02393-4 [DOI] [PubMed] [Google Scholar]
- 9.Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA, Kouzarides T, Martignetti JA, Preddie E, Satchwell SC, Tomlinson P, Weston KM, Barrell BG. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125–169 [DOI] [PubMed] [Google Scholar]
- 10.Weston K, Barrell BG. 1986. Sequence of the short unique region, short repeats, and part of the long repeats of human cytomegalovirus. J. Mol. Biol. 192:177–208. 10.1016/0022-2836(86)90359-1 [DOI] [PubMed] [Google Scholar]
- 11.Lesniewski M, Das S, Skomorovska-Prokvolit Y, Wang FZ, Pellett PE. 2006. Primate cytomegalovirus US12 gene family: a distinct and diverse clade of seven-transmembrane proteins. Virology 354:286–298. 10.1016/j.virol.2006.06.035 [DOI] [PubMed] [Google Scholar]
- 12.Das S, Pellett PE. 2007. Members of the HCMV US12 family of predicted heptaspanning membrane proteins have unique intracellular distributions, including association with the cytoplasmic virion assembly complex. Virology 361:263–273. 10.1016/j.virol.2006.11.019 [DOI] [PubMed] [Google Scholar]
- 13.Das S, Skomorovska-Prokvolit Y, Wang FZ, Pellett PE. 2006. Infection-dependent nuclear localization of US17, a member of the US12 family of human cytomegalovirus-encoded seven-transmembrane proteins. J. Virol. 80:1191–1203. 10.1128/JVI.80.3.1191-1203.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bronzini M, Luganini A, Dell'oste V, De Andrea M, Landolfo S, Gribaudo G. 2012. The US16 gene of human cytomegalovirus is required for efficient viral infection of endothelial and epithelial cells. J. Virol. 86:6875–6888. 10.1128/JVI.06310-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hai R, Chu A, Li H, Umamoto S, Rider P, Liu F. 2006. Infection of human cytomegalovirus in cultured human gingival tissue. Virol. J. 3:84. 10.1186/1743-422X-3-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. U. S. A. 100:11439–11444. 10.1073/pnas.1534570100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 78:10995–11006. 10.1128/JVI.78.20.10995-11006.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65. 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- 20.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian Z, Xuan B, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J. Virol. 82:3452–3465. 10.1128/JVI.01964-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98:5116–5121. 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maere S, Heymans K, Kuiper M. 2005. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21:3448–3449. 10.1093/bioinformatics/bti551 [DOI] [PubMed] [Google Scholar]
- 24.Guo YW, Huang ES. 1993. Characterization of a structurally tricistronic gene of human cytomegalovirus composed of U(s)18, U(s)19, and U(s)20. J. Virol. 67:2043–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller MS, Furlong WE, Pennell L, Geadah M, Hertel L. 2010. RASCAL is a new human cytomegalovirus-encoded protein that localizes to the nuclear lamina and in cytoplasmic vesicles at late times postinfection. J. Virol. 84:6483–6496. 10.1128/JVI.02462-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, Cosman D. 2003. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J. Exp. Med. 197:1427–1439. 10.1084/jem.20022059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du P, Kibbe WA, Lin SM. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24:1547–1548. 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 28.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J. Virol. 83:11421–11428. 10.1128/JVI.00762-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchkovich NJ, Maguire TG, Yu Y, Paton AW, Paton JC, Alwine JC. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 82:31–39. 10.1128/JVI.01881-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson W. 2008. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325:187–204 [DOI] [PubMed] [Google Scholar]
- 31.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6:266–275. 10.1038/nrmicro1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alwine JC. 2008. Modulation of host cell stress responses by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 325:263–279 [DOI] [PubMed] [Google Scholar]
- 33.Simmen KA, Singh J, Luukkonen BG, Lopper M, Bittner A, Miller NE, Jackson MR, Compton T, Fruh K. 2001. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 98:7140–7145. 10.1073/pnas.121177598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boehme KW, Guerrero M, Compton T. 2006. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 177:7094–7102 [DOI] [PubMed] [Google Scholar]
- 35.Yurochko AD, Hwang ES, Rasmussen L, Keay S, Pereira L, Huang ES. 1997. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J. Virol. 71:5051–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249–265. 10.1128/MMBR.00040-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869. 10.1128/JVI.01077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899. 10.1128/JVI.79.11.6890-6899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 83:3463–3474. 10.1128/JVI.02307-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchkovich NJ, Maguire TG, Alwine JC. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J. Virol. 84:7005–7017. 10.1128/JVI.00719-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojas-Rivera D, Armisen R, Colombo A, Martinez G, Eguiguren AL, Diaz A, Kiviluoto S, Rodriguez D, Patron M, Rizzuto R, Bultynck G, Concha ML, Sierralta J, Stutzin A, Hetz C. 2012. TMBIM3/GRINA is a novel unfolded protein response (UPR) target gene that controls apoptosis through the modulation of ER calcium homeostasis. Cell Death Differ. 19:1013–1026. 10.1038/cdd.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Mattia M, Gubser C, van Dommelen MM, Visch HJ, Distelmaier F, Postigo A, Luyten T, Parys JB, de Smedt H, Smith GL, Willems PH, van Kuppeveld FJ. 2009. Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol. Biol. Cell 20:3638–3645. 10.1091/mbc.E09-05-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HR, Lee GH, Ha KC, Ahn T, Moon JY, Lee BJ, Cho SG, Kim S, Seo YR, Shin YJ, Chae SW, Reed JC, Chae HJ. 2008. Bax inhibitor-1 is a pH-dependent regulator of Ca2+ channel activity in the endoplasmic reticulum. J. Biol. Chem. 283:15946–15955. 10.1074/jbc.M800075200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Xu W, Palmer AE, Reed JC. 2008. BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J. Biol. Chem. 283:11477–11484. 10.1074/jbc.M708385200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe N, Lam E. 2008. Arabidopsis Bax inhibitor-1: a rheostat for ER stress-induced programmed cell death. Plant Signal. Behav. 3:564–566. 10.4161/psb.3.8.5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14(+) monocytes and CD34(+) cells. PLoS Pathog. 9:e1003366. 10.1371/journal.ppat.1003366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benyesh-Melnick M, Probstmeyer F, McCombs R, Brunschwig JP, Vonka V. 1966. Correlation between infectivity and physical virus particles in human cytomegalovirus. J. Bacteriol. 92:1555–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng X, Schroer J, Yu D, Shenk T. 2006. Human cytomegalovirus pUS24 is a virion protein that functions very early in the replication cycle. J. Virol. 80:8371–8378. 10.1128/JVI.00399-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158. 10.1073/pnas.0509201102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldwin BR, Zhang CO, Keay S. 2000. Cloning and epitope mapping of a functional partial fusion receptor for human cytomegalovirus gH. J. Gen. Virol. 81:27–35 [DOI] [PubMed] [Google Scholar]
- 51.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123. 10.1073/pnas.0804365105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yurochko AD, Huang ES. 1999. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J. Immunol. 162:4806–4816 [PubMed] [Google Scholar]
- 53.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84:7803–7814. 10.1128/JVI.00139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viswanathan K, Smith MS, Malouli D, Mansouri M, Nelson JA, Fruh K. 2011. BST2/Tetherin enhances entry of human cytomegalovirus. PLoS Pathog. 7:e1002332. 10.1371/journal.ppat.1002332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo JY, Yaneva R, Hinson ER, Cresswell P. 2011. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332:1093–1097. 10.1126/science.1202007 [DOI] [PubMed] [Google Scholar]
- 56.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Fruh K, Picker LJ. 2013. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340:1237874. 10.1126/science.1237874 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.