

Abstract
Soluble forms of the human immunodeficiency virus type 1 (HIV-1) primary receptor CD4 (soluble CD4 [sCD4]) have been extensively characterized for a quarter of a century as promising HIV-1 inhibitors, but they have not been clinically successful. By combining a protein cavity-filling strategy and the power of library technology, we identified an engineered cavity-altered single-domain sCD4 (mD1.22) with a unique combination of excellent properties, including broad and potent neutralizing activity, high specificity, stability, solubility, and affinity for the HIV-1 envelope glycoprotein gp120, and small molecular size. To further improve its neutralizing potency and breadth, we generated bispecific multivalent fusion proteins of mD1.22 with another potent HIV-1 inhibitor, an antibody domain (m36.4) that targets the coreceptor-binding site on gp120. The fusion proteins neutralized all HIV-1 isolates tested, with potencies about 10-, 50-, and 200-fold higher than those of the broadly neutralizing antibody VRC01, the U.S. FDA-approved peptide inhibitor T20, and the clinically tested sCD4-Fc fusion protein CD4-Ig, respectively. In addition, they exhibited higher stability and specificity and a lower aggregation propensity than CD4-Ig. Therefore, mD1.22 and related fusion proteins could be useful for HIV-1 prevention and therapy, including eradication of the virus.
INTRODUCTION
Soluble forms of human CD4 (sCD4) comprising all four (D1 to D4) or the first two (D1D2) extracellular domains are potent inhibitors of the human immunodeficiency virus type 1 (HIV-1) in vitro (1, 2). Several promising monomeric (3–5), dimeric (6–8), and tetrameric (9–11) sCD4 derivatives have been tested in animal models and in human clinical trials, but they exhibited modest and transient antiviral activities. Previously, we demonstrated that decreasing the molecular size of D1D2 to a single domain, D1, significantly increased its antiviral activity and reduce its nonspecificity, i.e., interactions with molecules other than the HIV-1 envelope glycoprotein (Env) gp120; a D1 variant (mD1.2) was identified that is also more soluble than D1D2 (12). However, mD1.2 still binds to human B cells and CD4+ T cells without HIV-1 Env expression, although it binds more weakly than D1D2 and its stability is comparable to that of D1D2, which is relatively low (12).
It has been shown previously that some proteins exhibit poor hydrophobic packing, leading to low stability and solubility due to the presence of cavities within or on the surfaces of proteins that are either empty or hydrated (13, 14). Identification of such cavities and filling them with bulkier hydrophobic amino acid side chains have proven effective in improving stability and other properties of proteins (15). By combining this cavity-filling strategy with the power of library technology, we identified an mD1.2 mutant, designated mD1.22, that has significantly higher soluble expression, thermal stability, and specificity than mD1.2. Bispecific multivalent fusion proteins of mD1.22 with m36.4, an engineered human antibody domain targeting a CD4-induced (CD4i) epitope overlapping the HIV-1 coreceptor-binding site (CoRbs) on gp120 (16–18), exhibited remarkable neutralizing activity against HIV-1 as well as higher stability and specificity and a lower aggregation propensity than CD4-Ig, a clinically tested D1D2-Fc fusion protein (6, 7). Therefore, mD1.22 and related fusion proteins are promising drug candidates for HIV-1 prevention and therapy, including eradication of the virus.
MATERIALS AND METHODS
Cells, viruses, plasmids, proteins, and other reagents.
BJAB cells were a gift from Anu Puri (National Cancer Institute, Frederick, MD). We purchased 293T and SUPT1 cells from ATCC, and 293 FreeStyle cells were obtained from Invitrogen. Other cell lines and plasmids used for expression of various HIV-1 Envs were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (ARRRP). gp140SC, gp140MS, gp120MS, D1D2, CD4-Ig, mD1.2Fc, IgG1 m102.4, IgG1 m909, m36.4, and m36h1Fc were produced in our laboratory as described previously (12, 16, 17, 19, 20). gp140Con-s (21), gp140CH12.0544.2, and gp14089.6 were gifts from Barton F. Haynes (Duke University Medical Center, Durham, NC). IgG1s VRC01, b12, and 2G12 and Fab b12 were obtained from the ARRRP. Human serum was purchased from Invitrogen.
Computational analysis for identification of cavities and D1 mutagenesis.
The atomic coordinates of D1 were extracted from the crystal structure of a ternary complex of HIV-1 gp120 with D1D2 and the antibody 17b (PDB entry 2NY1). To identify cavities in D1, we used the Hollow program (22) with a grid spacing of 0.25 Å to probe inside the D1 structure, and this generated a casting of the interior volume of the protein filled with dummy atoms. The interior cavities of the D1 structure were located by concomitantly visualizing the dummy atoms and the amino acid residues at the protein core by using the PyMOL molecular graphics system (version 1.5.0.4; Schrödinger, LLC). The approximate volume of the cavity was calculated by using the normal Voronoi method with Chothia radii (23). The single point mutation A55V was modeled by using the PyMOL Mutagenesis Wizard with an appropriate side chain rotamer, and its effect on D1 stability was predicted using the Site-Directed Mutator (SDM) server (24).
Library construction and panning.
The phage display libraries of mD1.2 mutants were constructed by random mutagenesis. We used the following primers: SfiF, 5′-GAGGAGGAGGAGGAGGAGGCGGGGCCCAGGCGGCC-3′ (sense); mD1.2R, 5′-CTGGTCCCACAGGCTCCGCCGGCTGTCWNNCCTGTCGTTCAG-3′ (antisense); mD1.2F, 5′-CTGTGGGACCAGGGAAACTTCCCANNSATCATCAAGAAC-3′ (sense); FlagR, 5′-CTGGGGACTGCCCTTATCGTCATCGTCCTTG-3′ (antisense); mD1.22F, 5′-AGCTTCCTGACCAAGGGACCTAGCAAGNNSAACGACAGGGTTGAC-3′ (sense); mD1.22R, 5′-GGTCAGGAAGCTGCCCTGGTTGCCCAGWNNCTTGATCTGGTTGCT-3′ (antisense).
To randomize the D1 residues A55 and L69, two gene fragments were first amplified by PCR with an mD1.2-carrying plasmid as the template and primer pairs SfiF/mD1.2R and mD1.2F/FlagR, respectively. The PCR products were gel purified and used as templates for amplification of full-length mD1.2 mutants. Overlapping PCR was performed in a volume of 50 μl by using both fragments at the same molarities for 7 cycles in the absence of primers and 15 additional cycles in the presence of 500 pM primers SfiF and FlagR. The overlapping PCR product was gel purified, digested with SfiI, and gel purified again. The purified fragments were then cloned into phagemid pComb3X linearized by using SfiI. A phage library was prepared by electroporation of Escherichia coli strain TG1 electroporation-competent cells (Stratagene, La Jolla, CA) with desalted and concentrated ligation, as described previously (25). For randomization of the D1 residues I36 and L51, a second library was constructed in the same way except for the use of mD1.22, which was identified from the first library, as a template and primer pairs SfiF/mD1.22R and mD1.22F/FlagR for amplification of mD1.22 gene fragments.
The phage libraries were used for selection of mD1.2 mutants against HIV-1 Envs (in 96-well plates coated with the Envs, as described previously [26]). For sequential panning, 50 and 5 ng of gp140SC (clade B) and gp120MS (clade A) were used in the first and second rounds, respectively. To identify individual mutants that specifically bound to the Envs and were soluble in the periplasm of E. coli cells, clones were randomly picked from the second rounds of panning, inoculated into 96-well plates, and induced for protein expression by using isopropyl β-d-1-thiogalactopyranoside (IPTG). To select those clones that had high yields of soluble expression and/or high stability against degradation, the supernatants of individual clones were incubated at 4°C for 3 days and then screened with a soluble expression-based monoclonal enzyme-linked immunosorbent assay (semELISA) as described previously (27).
Cloning of mD1.22Fc and mCD4-Ig.
The following primers were used for cloning of mD1.22Fc and mCD4-Ig: D1-53F, 5′-TGACGCGGCCCAGCCGGCCAAGAAGGTGGTGTACGGC-3′ (sense); D1-53FcR, 5′-TTTGTCGGGCCCGCCTACCACTACCAGCTG-3′ (antisense); D1D2mF, 5′-GACAGGGTTGACAGCCGGCGG-3′ (sense); D1D2mR, 5′-GCTGTCAACCCTGTCGTTCAG-3′ (antisense); D1D2FcF, 5′-TGGTTTCGCTACCGTGGCCCAGCCGGCCAAGAAGGTGGTGCTGGGC-3′ (sense); D1D2FcR, 5′-GTGAGTTTTGTCGGGCCCGGCCAGCACCACGATGTC-3′ (antisense).
To clone mD1.22Fc, an mD1.22 gene fragment was PCR amplified with primers D1-53F and D1-53FcR. The product appended with SfiI and ApaI restriction sites on both sides was digested and cloned into pSecTagB-Fc. For construction of mCD4-Ig, two gene fragments were amplified with a D1D2-carrying plasmid as the template and primer combinations D1D2FcF/D1D2mR and D1D2mF/D1D2FcR, respectively. The PCR products were gel purified and joined together by overlapping PCR using primers D1D2FcF and D1D2FcR, as described above. The overlapping PCR product was digested with SfiI and ApaI and cloned into pSecTagB-Fc.
Cloning of 2Dm2m, 4Dm2m, and 6Dm2m.
The following primers were used for cloning of 2Dm2m, 4Dm2m, and 6Dm2m: mD1.22H2, 5′-ACTACAGGTGTCCACTCCAAGAAGGTGGTGTACGGC-3′ (sense); mD1.22H4, 5′-CCTTGGAGCTCGATCCGCCACCGCCAGAGCCACCTCCGCCTGAACCGCCTCCACCGCCTACCACTACCAGCTG-3′ (antisense); m36.4L2, 5′-CTTACAGATGCCAGATGTCAGGTGCAGCTGGTGCAG-3′ (sense); m36.4L4, 5′-AGAGCCACCTCCGCCTGAACCGCCTCCACCTGAGGAGACGGTGACCAG-3′ (antisense); bnIgG20H1, 5′-GTGTTCTAGAGCCGCCACCATGGAATGGAGCTGGGTCTTTCTCTTC-3′ (sense); bnIgG20H3, 5′-GGAGTGGACACCTGTAGTTACTGACAGGAAGAAGAGAAAGAC-3′ (antisense); bnIgG20L1, 5′-GTGTAAGCTTACCATGGGTGTGCCCACTCAGGTCCTGGGGTTGCTG-3′ (sense); bnIgG20L3, 5′-ACATCTGGCATCTGTAAGCCACAGCAGCAGCAACCCCAGGAC-3′ (antisense); bnIgG20L4, 5′-GTGTGAATTCATTAACACTCTCCCCTGTTGAA-3′ (antisense); CLF, 5′-TCAGGCGGAGGTGGCTCTGGCGGTGGCGGATCACGAACTGTGGCTGCACCA-3′ (sense); HleaderF, 5′-TAATTCTCTAGAGCCGCCACCATG-3′ (sense); CH3R, 5′-AGAGCCACCTCCGCCTGAACCGCCTCCACCTTTACCCGGAGACAGGGA-3′ (antisense); AAAF, 5′-TGAGTGCGACGGCCGGCA-3′ (sense); AAAR, 5′-CCCGAGGTCGACGCTCTC-3′ (antisense); CLDmF, 5′-AGCGGTGGCGGGGGAAGTGGCGGTGGAGGGAGCAAGAAGGTGGTGTACGGC-3′ (sense); D1mRR, 5′-ATCAATGAATTCATTAGCCTACCACTACCAGCTG-3′ (antisense); CLR, 5′-ACTTCCCCCGCCACCGCTGCCACCCCCTCCACACTCTCCCCTGTTGAA-3′ (antisense).
2Dm2m was constructed by cloning mD1.22 and m36.4 into the plasmid vector pDR12, which contains human IgG1 constant region-encoding gene cassettes and allows simultaneous expression of the heavy and light chains, by using protocols and reagents similar to those we used previously (28). To fuse mD1.22 to the N terminus of the human IgG1 heavy chain constant region, mD1.22 and the heavy chain leader peptide (Hleader) gene fragments were PCR amplified with primer pairs mD1.22H2/mD1.22H4 and bnIgG20H1/bnIgG20H3, respectively. Hleader was joined to mD1.22 by overlapping PCR with both templates (at the same molarities) for 7 cycles in the absence of primers and 15 additional cycles in the presence of primers bnIgG20H1 and mD1.22H4. The product appended with XbaI and SacI restriction sites on both sides was digested and cloned into pDR12. To fuse m36.4 to the N terminus of the human IgG1 light chain constant region, gene fragments encoding the light chain leader peptide (Lleader), m36.4, and the human IgG1 kappa light chain constant region (CK) were amplified by PCR with primer pairs bnIgG20L1/bnIgG20L3, m36.4L2/m36.4L4, and CLF/bnIgG20L4, respectively. Lleader was linked to m36.4 and CK by overlapping PCR with primers bnIgG20L1 and bnIgG20L4, as described above. The Lleader-m36.4-CK fragment was then digested with EcoRI and HindIII and cloned into the pDR12 construct containing mD1.22 in the heavy chain. To generate 4Dm2m, Hleader-mD1.22-Fc, mD1.22, and poly(A) signal gene fragments were PCR amplified by using 2Dm2m as a template and primer pairs HleaderF/CH3R, mD1.22mF/mD1.22mR, and AAAF/AAAR, respectively. Hleader-mD1.22-Fc, mD1.22, and poly(A) were joined to each other by overlapping PCR with primers HleaderF and AAAR. The product was digested with XbaI and SalI and cloned into 2Dm2m. To clone 6Dm2m, Lleader-m36.4-CK and mD1.22 gene fragments were amplified with primer pairs bnIgG20L1/CLR and CLDmF/D1mRR, respectively. Lleader-m36.4-CK was fused to the N terminus of mD1.22 by overlapping PCR using primers bnIgG20L1 and D1mRR. The product was digested with EcoRI and HindIII and cloned into 4Dm2m.
Protein expression and purification.
D1 mutants were expressed in E. coli HB2151; all Fc-fusion proteins were produced in 293 FreeStyle cells, as described previously (17). D1 mutants, which were tagged with six-histidine at their C terminus, were purified from the soluble fraction of HB2151 periplasm via immobilized metal ion affinity chromatography (IMAC) with Ni-nitrilotriacetic acid resin (Qiagen, Valencia, CA) according to the manufacturer's protocols. Fc-fusion proteins were purified from 293 FreeStyle cell culture supernatants by using protein A-Sepharose 4 Fast Flow (GE Healthcare, Piscataway, NJ) column chromatography.
Size exclusion chromatography.
A Superdex75 10/300 GL column (GE Healthcare, Piscataway, NJ) was calibrated with protein molecular mass standards of 14-kDa RNase A, 25-kDa chymotrypsin, and 44-kDa ovalbumin. Purified D1 mutants and D1D2 in phosphate-buffered saline (PBS) were loaded onto the preequilibrated column and eluted with PBS at 0.5 ml/min. Fc-fusion proteins were analyzed using a Superdex200 10/300 GL column (GE Healthcare, Piscataway, NJ) calibrated with protein molecular mass standards of 14-kDa RNase A, 25-kDa chymotrypsin, 44-kDa ovalbumin, 67-kDa albumin, 158-kDa aldolase, 232-kDa catalase, 440-kDa ferritin, and 669-kDa thyroglobulin.
Dynamic light scattering.
Proteins concentrated to 10 mg/ml in 220 μl were stored at −80°C and slowly thawed on ice before measurements. Samples were then incubated at 4 or 37°C. On day 0, 1, 3, and 7, 50-μl samples were collected and centrifuged at 18,000 × g for 10 min. The supernatants were diluted to 1 mg/ml, and 400 μl was used for dynamic light scattering (DLS) measurements (Zetasizer Nano ZS 3600; Malvern Instruments Ltd., Westborough, MA) according to the manufacturer's instructions.
ELISAs.
ELISAs were performed as described previously (12, 17). Bound D1 mutants were detected by horseradish peroxidase (HRP)-conjugated anti-FLAG tag antibodies (Sigma-Aldrich, St. Louis, MO) or by using the Penta-His HRP conjugate kit from Qiagen. Bound Fc-fusion proteins were detected with HRP-conjugated anti-human IgG (Fc-specific) antibodies (Sigma-Aldrich, St. Louis, MO). Half-maximal binding (the 50% effective concentration [EC50]) was calculated by fitting the data to the Langmuir adsorption isotherm.
Surface plasmon resonance.
Binding kinetics of D1 mutants with HIV-1 gp140 were assessed by using surface plasmon resonance (SPR) analysis on a Biacore X100 apparatus (GE Healthcare) and using a single-cycle approach according to the manufacturer's instructions. Briefly, purified HIV-1 gp140 was diluted in sodium acetate (pH 5.0) and immobilized directly onto a CM5 sensor chip via the standard amine coupling method. The reference cell was injected with N-hydroxysuccinimide–1-ethyl-3-(3-dimethyaminopropy)carbodiimide and ethanolamine without injection of gp140. D1 mutants and D1D2 were diluted with running buffer HBS-EP (100 mM HEPES [pH 7.4], 1.5 M NaCl, 30 mM EDTA, 0.5% surfactant 20). All analytes were tested at 500, 100, 20, 4, and 0.8 nM concentrations. Kinetic constants were calculated from the sensorgrams fitted with the monovalent binding model of the BiacoreX100 Evaluation software 2.0.
Flow cytometry.
For detection of interactions with human blood cell lines, approximately 106 BJAB or SUPT1 cells in 200 μl PBS containing 0.1% bovine serum albumin (PBSA) were incubated with sCD4-Fc fusion proteins at a final concentration of 1 μM on ice for 1 h. Cells were washed twice and resuspended in 200 μl PBSA and 1 μl goat F(ab′)2 anti-human IgG (γ-specific)–fluorescein isothiocyanate conjugate (Sigma-Aldrich, St. Louis, MO) was added. Following a 30-min incubation on ice, cells were washed twice and then subjected to flow cytometry analysis.
Solubility measurements.
Proteins in PBS were concentrated using an ultrafiltration method and then quantified as described previously (12).
Circular dichroism spectroscopy.
Secondary structures and thermal stability of D1 mutants were determined by circular dichroism (CD) spectroscopy as described previously (29).
Serum stability measurement.
Proteins in PBS were mixed at a 1:1 (vol/vol) ratio with human serum or PBS to give a final concentration of 500 nM in a total volume of 50 μl. After incubation at 37°C, reactions were stopped by freezing the samples at −80°C. After all samples were collected, 50 μl of 4% milk in PBS were added to each sample, and diluted proteins were evaluated in ELISAs with gp140Con-s. Standard curves were generated by using the original protein stocks to quantify functional proteins that survived the different periods of incubation.
Neutralization of historical HIV-1 isolates in HOS-CD4-CCR5 cell-based assays.
HIV-1 pseudoviruses were generated, and a HOS-CD4-CCR5 cell-based neutralization assay was performed as described previously (17).
Neutralization of more recent HIV-1 isolates in TZM-bl cell-based assays.
HIV-1 pseudoviruses were generated and the TZM-bl cell-based neutralization assay was performed as described previously (30).
Neutralization of HIV-1 isolates predominantly circulating in China in TZM-bl cell-based assays.
A panel of pseudoviruses with Envs derived from HIV-1 isolates predominantly circulating in China was generated, and previously described neutralization assay protocols were used (31, 32).
Neutralization of HIV-1 in a human peripheral blood mononuclear cell-based assay.
HIV-1 infectious molecular clones carrying the Renilla luciferase reporter gene were tested, and a neutralization assay of HIV-1 in human peripheral blood mononuclear cells (PBMC) was performed as described previously (33).
Inhibition of HIV-1 Env-mediated cell-cell fusion.
Cell-cell fusion assays were performed as described previously (34), except that in our study we used HOS-CD4-CCR5 and 293T cells.
Toxicity test.
Antibody toxicity on PBMC and TZM-bl cells was evaluated with the CellTiter 96 AQueous One Solution cell proliferation assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] system (Promega, Madison, WI) according to the manufacturer's instructions.
RESULTS
Identification of a cavity in D1 and selection of a cavity-altered mD1.2 variant (mD1.22) with improved soluble expression and thermal stability.
Our computational analysis with the Hollow program identified a cavity (volume, approximately 57 Å3) in D1 lined by four hydrophobic residues (I36, L51, A55, and L69) (Fig. 1A). Accordingly, we chose these residues for random mutagenesis and construction of phage display libraries of mD1.2 mutants. For simplicity and convenience in PCR amplification, we first generated a library in which the residues A55 and L69 were randomized by using the degenerate codon NNS, which encodes the complete set of standard amino acids. Panning the library sequentially against two HIV-1 Envs from different clades and screening for high Env-binding activity, soluble expression, and stability against prolonged incubation resulted in the identification of mD1.22, which has a single amino acid substitution (A55V) compared to mD1.2 (Fig. 1B). We then constructed a second library by using mD1.22 as a template for randomization of the residues I36 and L51. Panning and screening the second library did not lead to the identification of mD1.22 variants with improved properties, and so our further characterization was focused on mD1.22.
FIG 1.

Identification of a cavity in the hydrophobic core of D1 (A) and a cavity-altered D1 variant, mD1.22 (B). (A) The void filled with dummy atoms (the irregular sphere) depicts the presence of a cavity in the interior of D1 that was identified by the Hollow program. The hydrophobic residues lining the cavity (I36, L51, A55, and L69) are indicated, and their side chains are shown in stick representation. (B) The amino acid sequence of mD1.22 is aligned with those of mD1.2 and the wild-type D1. The sequences are numbered. The residues in the D1 mutants that are identical to those of the wild-type D1 are indicated by dots.
mD1.22 was expressed as a soluble protein in E. coli with a yield (5 mg/liter) that was about 7-fold higher than that of mD1.2 (0.75 mg/liter). It was monomeric in PBS (pH 7.4), as measured by size exclusion chromatography, with an apparent molecular weight (aMW) that was slightly higher than its calculated molecular weight (cMW) (13,484, including the six-histidine and FLAG tags at its C terminus) (Fig. 2A). It was concentrated to 175 mg/ml in PBS without visible precipitation after high-speed centrifugation, suggesting high solubility of the protein. mD1.22 (melting temperature [Tm], 68.3°C) was thermally more stable than mD1.2 (Tm, 60.7°C) (Fig. 2B). Similarly to mD1.2, mD1.22 consists primarily of a β-strand secondary structure at 25°C (Fig. 2C).
FIG 2.

Oligomeric state (A), thermal stability (B), and secondary structure (C) of mD1.22. (A) The oligomeric state of mD1.22 was measured by size exclusion chromatography. The arrows at the top indicate the elution volumes of the molecular mass standards in PBS: RNase A (14 kDa), chymotrypsin (25 kDa), and ovalbumin (44 kDa). The thermal stability and secondary structure of mD1.22 were measured by CD. The fraction folded (ff) of the proteins was calculated as follows: ff = ([θ] − [θM])/([θT] − [θM]). [θT] and [θM] are the mean residue ellipticities at 225 nm of the folded state at 25°C and the unfolded state at 90°C. Tm values were determined based on the first derivative [d(fraction folded)/dT] with respect to temperature (T). The measurement of the secondary structure was performed at 25°C.
Affinity, neutralizing activity, and specificity of mD1.22.
Binding of mD1.22 to genetically diverse HIV-1 Envs was analyzed by ELISA and SPR. In an SPR analysis with four HIV-1 Env gp140s, mD1.22 showed affinities comparable to or higher than those of mD1.2 (Table 1). Like D1D2 and mD1.2, mD1.22 enhanced binding of a CD4i antibody-Fc fusion protein (m36h1Fc) (17) to a consensus gp140 (gp140Con-s) designed by aligning >1,000 sequences of HIV-1 group M (21) in an ELISA (data not shown). In contrast, b12, a broadly neutralizing antibody (bnAb) that targets the CD4-binding site (CD4bs) on gp120 (35), did not show enhancement effects. mD1.22 neutralized two R5-tropic HIV-1 primary isolates from clade B, Bal and JRFL, with a potency severalfold higher than those of D1D2 and mD1.2 (Fig. 3). m102.4, a negative-control antibody specific to Nipah and Hendra viruses (19), did not significantly inhibit any isolate tested.
TABLE 1.
Binding kineticsa of mD1.2 and mD1.22 with HIV-1 gp140 measured by SPR
| HIV-1 gp140 | Clade | mD1.2 |
mD1.22 |
||||
|---|---|---|---|---|---|---|---|
| Ka (M−1 s−1) | Kd (s−1) | KD (nM) | Ka (M−1 s−1) | Kd (s−1) | KD (nM) | ||
| gp140Con-s | Consensusb | 1.7 × 105 | 9.3 × 10−4 | 5.4 | 1.7 × 105 | 7.7 × 10−4 | 4.4 |
| gp140MS | A | 4.2 × 105 | 1.0 × 10−4 | 0.24 | 3.8 × 105 | 7.3 × 10−5 | 0.19 |
| gp14089.6 | B | 3.4 × 105 | 4.0 × 10−4 | 1.2 | 2.5 × 105 | 2.1 × 10−4 | 0.87 |
| gp140CH12.0544.2 | B | 1.5 × 105 | 1.1 × 10−4 | 0.72 | 1.2 × 105 | 2.0 × 10−5 | 0.17 |
Ka, association rate constant; Kd, dissociation rate constant; KD, equilibrium dissociation constant.
A consensus gp140 designed by aligning >1,000 sequences of HIV-1 group M.
FIG 3.
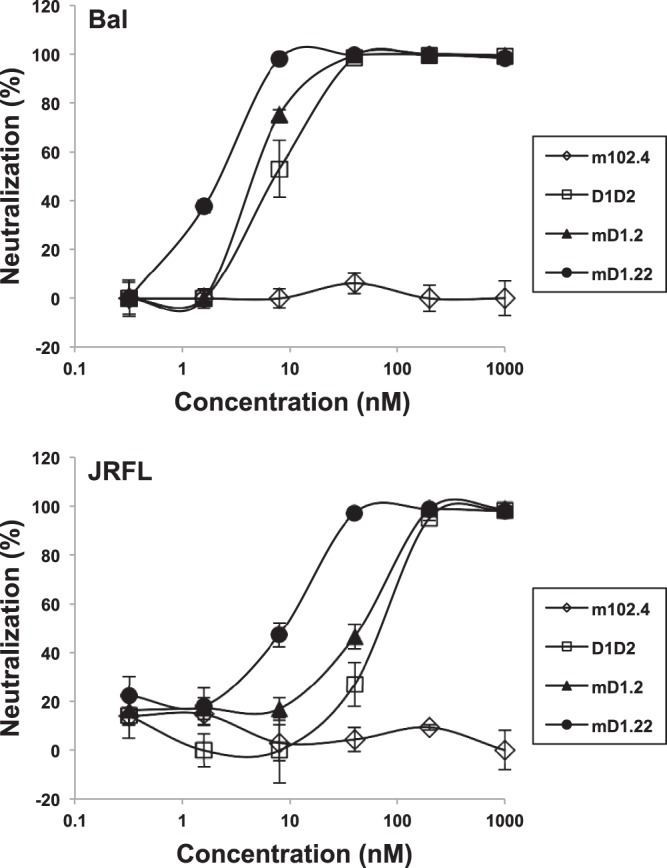
Neutralization of HIV-1 pseudoviruses by mD1.22. Bal and JRFL are two R5-tropic HIV-1 primary isolates from clade B. The neutralization assay was performed in duplicate with HOS-CD4-CCR5 cells as target cells. m102.4, a negative-control antibody specific to Nipah and Hendra viruses, is in the IgG1 format.
CD4 also interacts with the major histocompatibility complex class II (MHC-II) molecules that assist T cell receptors in activating T cells and presumably other targets (36). To briefly evaluate the nonspecificity of mD1.22 in vivo, we used a human B cell line that expresses high level of MHC-II (BJAB) and a human T cell line with no detectable MHC-II expression (SUPT1) (12). Because oligomerization of CD4 is required for a stable interaction with MHC-II (37), we generated Fc-fusion proteins of D1D2 (CD4-Ig), mD1.2 (mD1.2Fc), and mD1.22 (mD1.22Fc). Our results showed that CD4-Ig at a concentration of 1 μM bound strongly to both cell lines; mD1.2Fc also bound to the cell lines, although to a lesser extent (Fig. 4). In contrast, the negative-control m36h1Fc and, unexpectedly, mD1.22Fc, did not significantly interact with the cells at the same concentration, suggesting that mD1.22 might have a higher specificity to HIV-1 than do mD1.2 and D1D2 in vivo.
FIG 4.

Binding of mD1.22Fc to human blood cell lines as measured by flow cytometry. Closed and open diagrams are for reference cells and cells incubated with proteins at a final concentration of 1 μM, respectively.
In an attempt to elucidate possible mechanisms underlying the remarkable differences between mD1.2 and mD1.22, we identified a panel of CD4-specific antibodies from our yeast display libraries, one (m1006) of which is sensitive to D1 conformational changes. In an ELISA, the A55V mutation resulted in a dramatic decrease of m1006 binding to mD1.22Fc compared to mD1.2Fc (Fig. 5A), suggesting that the single amino acid substitution induces conformational changes in D1. m1006 also exhibited lower binding to mCD4-Ig, a CD4-Ig variant with the same mutation, than to CD4-Ig, suggesting that the ability of the A55V mutant to induce D1 conformational changes is D2 independent. As a consequence, mCD4-Ig bound to gp140Con-s more strongly (Fig. 5B) and to BJAB and SUPT1 cells more weakly (Fig. 5C) than CD4-Ig.
FIG 5.

Effects of the A55V mutation on D1 conformations. (A) ELISA binding of the anti-D1 antibody m1006 to the Fc-fusion proteins of D1D2, mD1.2, and their mutants. Ninety-six-well plates were coated with the Fc-fusion proteins, and bound m1006 was detected by HRP-conjugated anti-FLAG tag antibodies. (B) ELISA binding of mCD4-Ig to 96-well plates coated with gp140Con-s. Bound Fc fusion proteins were detected by HRP-conjugated anti-human IgG (Fc-specific) antibodies. (C) Binding of mCD4-Ig to human blood cell lines as measured by flow cytometry. Closed diagrams are for reference cells. Open diagrams with light and dark curves are for cells incubated with CD4-Ig and cells incubated with mCD4-Ig at a final concentration of 1 μM, respectively.
Generation and initial characterization of bispecific multivalent fusion proteins of mD1.22 with m36.4.
To generate highly potent and cross-reactive HIV-1 inhibitors, we combined mD1.22 with m36.4, a broadly neutralizing antibody domain that targets the CoRbs on gp120 (16), and we used the human IgG1 constant region as a scaffold for multimerization (Fig. 6A). 2Dm2m, a bispecific tetravalent construct, was generated by fusing mD1.22 and m36.4 to the N termini of the human IgG1 heavy (CH1) and kappa light (CK) chain constant regions, respectively, via a polypeptide linker composed of three repeats of the G4S motif. Based on the finding that the CD4bs and CoRbs on gp120 are in close proximity (38), we assumed that a similar orientation of the two binding moieties would favor simultaneous targeting of the same gp120 molecule, although such a construct as 2Dm2m could also build inter-gp120 or interspike cross-linking. For possibly higher avidity, we covalently linked another mD1.22 to the C terminus of 2Dm2m Fc to generate a hexavalent construct, 4Dm2m. Fusion of another mD1.22 to the C terminus of 4Dm2m CK resulted in an octavalent construct, 6Dm2m. In a parallel experiment, we used m36.4 to make constructs with higher valences. However, the constructs containing additional m36.4 at the C termini did not show increased binding activity or neutralization potency than 2Dm2m, likely because the polypeptide linker could interrupt the interaction of m36.4 with gp120 in such configurations, in agreement with our previous study (16). Therefore, our further characterizations were focused on 2Dm2m, 4Dm2m, and 6Dm2m.
FIG 6.

Design, generation, and initial characterization of bispecific multivalent fusion proteins of mD1.22 with m36.4. (A) Schematic representation of the fusion proteins. Calculated molecular masses are shown in parentheses. (G4S)3 denotes a polypeptide linker composed of three repeats of the G4S motif. Fc, human IgG1 crystallizable fragment; CH1, human IgG1 heavy chain constant region 1; CK, human antibody kappa light chain constant region. (B) Reducing SDS-PAGE results. Molecular masses of standards are shown on the left. (C) Size exclusion chromatography results. The arrows at the top indicate the elution volumes of the molecular mass standards in PBS: RNase A (14 kDa), chymotrypsin (25 kDa), ovalbumin (44 kDa), albumin (67 kDa), aldolase (158 kDa), catalase (232 kDa), ferritin (440 kDa), and thyroglobulin (669 kDa).
2Dm2m, 4Dm2m, and 6Dm2m were expressed in transiently transfected 293 FreeStyle cells and secreted into shaking culture supernatants at yields of approximately 8, 10, and 6 mg/liter, respectively. They ran on a reducing SDS-PAGE with aMWs larger than their cMWs (147,600, 171,900, and 196,300 for 2Dm2m, 4Dm2m, and 6Dm2m, respectively), likely due to glycosylation (Fig. 6B). They were mostly (>95%) monomers in PBS, as demonstrated via size exclusion chromatography (Fig. 6C).
Exceptionally potent and broad neutralizing activities of the bispecific multivalent fusion proteins.
In the experiments with two HIV-1 primary isolates (Bal and JRFL), we found that the inhibitory activities of the mD1.22-m36.4 fusion protein against virus-cell interactions and Env-mediated cell-cell fusion were generally positively correlated with their valences (Tables 2 and 3). Therefore, 4Dm2m and 6Dm2m, the two proteins with the highest valences, were extensively characterized. Previous studies had shown that HIV-1 antibodies may exhibit various levels of neutralizing activity in assays where different cell types or the same type of cells with different surface receptor concentrations are used (34, 39). A recent study demonstrated that HIV-1 became increasingly resistant to the newly identified potent bnAbs, including VRC01 (40) and PG9 and PG16 (41) over the course of the epidemic (42, 43). We therefore tested our proteins with different target cells and panels of HIV-1 isolates for a relatively comprehensive evaluation of their neutralizing potencies and breadths of activities.
TABLE 2.
Neutralization of HIV-1 by mD1.22 of different valences in HOS-CD4-CCR5 cell-based assays
| HIV-1 isolate | Clade | Tropism | IC50 (nM)a |
IC90 (nM)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m36.4 | mD1.22 | mD1.22Fc | 2Dm2m | 4Dm2m | 6Dm2m | m36.4 | mD1.22 | mD1.22Fc | 2Dm2m | 4Dm2m | 6Dm2m | |||
| Bal | B | R5 | 16 ± 1 | 2.1 ± 0.1 | 0.40 ± 0.00 | 0.34 ± 0.15 | 0.12 ± 0.01 | 0.063 ± 0.004 | 75 ± 18 | 5.2 ± 0.2 | 1.9 ± 0.2 | 0.59 ± 0.05 | 0.36 ± 0.06 | 0.22 ± 0.02 |
| JRFL | B | R5 | 18 ± 5 | 2.9 ± 0.4 | 1.2 ± 0.1 | 0.090 ± 0.008 | 0.060 ± 0.014 | 0.043 ± 0.004 | 106 ± 4 | 25 ± 1 | 25 ± 6 | 0.59 ± 0.06 | 0.29 ± 0.09 | 0.21 ± 0.01 |
The assay was performed in duplicate. Results are means ± standard deviations.
TABLE 3.
Inhibition of HIV-1 Env-mediated cell-cell fusion by mD1.22 of different valences
| HIV-1 isolate | Clade | Tropism | IC50 (nM)a |
IC90 (nM)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m36.4 | mD1.22 | mD1.22Fc | 2Dm2m | 4Dm2m | 6Dm2m | m36.4 | mD1.22 | mD1.22Fc | 2Dm2m | 4Dm2m | 6Dm2m | |||
| Bal | B | R5 | 11 ± 1 | 21 ± 0 | 24 ± 4 | 21 ± 6 | 4.1 ± 0.1 | 3.0 ± 0.0 | 235 ± 49 | 108 ± 4 | 135 ± 21 | 113 ± 39 | 13 ± 3 | 5.0 ± 0.8 |
| JRFL | B | R5 | 128 ± 5 | 66 ± 1 | 66 ± 1 | 7.5 ± 0.0 | 1.6 ± 0.7 | 0.46 ± 0.07 | 1,075 ± 35 | 208 ± 4 | >222 | 80 ± 9 | 11 ± 2 | 10 ± 3 |
The assay was performed in duplicate. Results are means ± standard deviations.
In an assay with HOS-CD4-CCR5 cells and 13 historical HIV-1 isolates, 4Dm2m and 6Dm2m potently neutralized all viruses, with 50% inhibitory concentrations (IC50s) and IC90s in the picomolar range (Table 4). They were on average severalfold more potent than the bnAb VRC01 (P = 0.017, 0.009, 0.011, and 0.010 for the IC50s and IC90s of 4Dm2m and 6Dm2m versus those of VRC01, respectively; Student's paired t test) and were much more potent than the bnAb 2G12 (44), which did not neutralize all viruses. In another assay where TZM-bl cells and two more recent HIV-1 isolates randomly selected from each clade from A to D, AE, and AG (total n = 12) were used, 4Dm2m and 6Dm2m were much more potent than b12 and CD4-Ig and about 10-fold more potent than VRC01 when the geometric means of their IC50s and IC90s were compared (Table 5). Both b12 and VRC01 did not neutralize two of the isolates at the highest concentration tested. However, the higher potencies of 4Dm2m and 6Dm2m compared to VRC01 were not statistically significant (P = 0.131, 0.132, 0.062, and 0.070 for the IC50s and IC90s of 4Dm2m and 6Dm2m versus those of VRC01, respectively; Student's paired t test). In yet another assay, we compared 4Dm2m and 6Dm2m to T20 (Fuzeon, or enfuvirtide), a U.S. FDA-approved peptide inhibitor derived from the HIV-1 Env gp41 C-terminal heptad repeat (CHR), C34, another potent CHR peptide inhibitor, and CD4-Ig for neutralizing activities against a panel of 41 HIV-1 isolates that are predominantly circulating in China (Table 6). 4Dm2m and 6Dm2m neutralized all viruses, with IC50 geometric mean values of approximately 0.20 nM, which are about 50-fold (P = 0.002, Student's paired t test), 16-fold (P < 0.001, Student's paired t test), and 200-fold (P < 0.001, Student's paired t test) lower than those (11, 3.5, and 42 nM) of T20, C34, and CD4-Ig, respectively. The superior potencies of 4Dm2m and 6Dm2m were also observed when the IC90s were compared. Finally, these inhibitors were tested using PBMC as target cells. Both 4Dm2m and 6Dm2m neutralized all three HIV-1 isolates from clades B, C, and CRF01_AE, with potencies comparable to or greater than those of b12, VRC01, and CD4-Ig (data not shown). None of the fusion proteins neutralized pseudotyped murine leukemia viruses (MuLV), and no toxicity was observed with the compounds on PBMC or TZM-bl cells in a viability test. These results suggest that bispecific multivalent fusion proteins of mD1.22 with m36.4 could provide 100% neutralization coverage of HIV-1 at clinically achievable low concentrations.
TABLE 4.
Neutralization of historical HIV-1 isolates in HOS-CD4-CCR5 cell-based assays
| HIV-1 isolate | Clade | Tropism | IC50 (nM)a |
IC90 (nM)a |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2G12b | VRC01b | 4Dm2m | 6Dm2m | 2G12 | VRC01 | 4Dm2m | 6Dm2m | |||
| 92UG037.8 | A | R5 | 0.53 | 0.33 | 0.051 | 0.14 | 63 | 3.8 | 0.45 | 2.1 |
| Bal | B | R5 | 3.1 | 0.11 | 0.15 | 0.12 | 41 | 0.56 | 0.50 | 0.38 |
| JRFL | B | R5 | 3.0 | 0.31 | 0.14 | 0.10 | 13 | 0.82 | 0.39 | 0.43 |
| JRCSF | B | R5 | 0.58 | 0.28 | 0.29 | 0.21 | 9.3 | 5.6 | 3.0 | 1.0 |
| AD8 | B | R5 | 1.2 | 0.36 | 0.13 | 0.11 | 18 | 4.3 | 0.74 | 0.60 |
| R2 | B | R5 | 0.68 | 0.34 | 0.028 | <0.021 | 4.6 | 4.4 | 0.43 | 0.14 |
| IIIB | B | X4 | 1.5 | 0.26 | 0.037 | <0.021 | 9.3 | 0.83 | 0.10 | 0.086 |
| NL4-3 | B | X4 | 1.3 | 0.25 | 0.094 | 0.068 | 18 | 2.6 | 0.15 | 0.19 |
| 89.6 | B | R5X4 | 1.3 | 0.37 | <0.021 | <0.021 | 43 | 14 | <0.021 | <0.021 |
| GXC | C | R5 | >67 | 2.1 | 0.61 | 0.63 | >67 | 6.6 | 4.0 | 1.7 |
| Z2Z6 | D | R5 | 0.70 | 0.11 | 0.14 | 0.045 | 14 | 0.85 | 1.1 | 0.50 |
| CM243 | E | R5 | >67 | 0.53 | 0.29 | 0.15 | >67 | 9.5 | 1.2 | 1.8 |
| GXE | E | R5 | >67 | 2.0 | 0.83 | 0.83 | >67 | 26 | 8.2 | 6.3 |
| Arithmetic meanc | 24 | 0.57 | 0.22 | 0.19 | 41 | 6.1 | 1.6 | 1.2 | ||
| Geometric meanc | 3.2 | 0.37 | 0.12 | 0.083 | 26 | 3.4 | 0.56 | 0.46 | ||
The assay was performed in duplicate. Reported results are means (without standard deviations). Means were calculated only when the difference between results for the duplicate assays was within 3-fold.
The bnAbs 2G12 and VRC01 used in the neutralization assay were in the IgG1 format.
Arithmetic and geometric means were calculated for all viruses, including those with results of <0.021 nM, which were assigned a value of 0.01, and those with results of >67 nM, which were assigned a value of 100.
TABLE 5.
Neutralization of HIV-1 in TZM-bl cell-based assays
| HIV-1 isolate | Clade | Tropism | IC50 (nM)a |
IC90 (nM)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| b12b | VRC01b | CD4-Ig | 4Dm2m | 6Dm2m | b12 | VRC01 | CD4-Ig | 4Dm2m | 6Dm2m | |||
| KNH1088.EC5 | A | R5 | 117 | 3.0 | >250 | 0.48 | 1.0 | >167 | 18 | >250 | 18 | 33 |
| KNH1144.EC1 | A | R5 | >167 | 2.2 | 4.8 | 0.081 | 0.11 | >167 | 9.5 | 42 | 1.0 | 1.1 |
| Bal.EC1 TC | B | R5 | 1.2 | 0.39 | 0.60 | 0.057 | 0.045 | 3.6 | 5.2 | 3.2 | 0.22 | 0.20 |
| BZ167.ec9 | B | X4 | 14 | 6.4 | 13 | 0.077 | 0.10 | >167 | 31 | 60 | 0.50 | 0.76 |
| GS-015.EC12 | C | R5 | 0.085 | >167 | 0.20 | 0.022 | 0.027 | 1.6 | >167 | 3.2 | 0.17 | 0.17 |
| PBL286.VRC36aPV | C | R5 | 4.0 | 2.1 | 87 | 2.0 | 2.1 | 19 | 10 | >250 | 14 | 12 |
| A07412VRC12A | D | R5 | 12 | 1.3 | 57 | 0.75 | 0.76 | >167 | 6.0 | >250 | 5.7 | 6.1 |
| 57128.VRC18 | D | R5 | 2.3 | >167 | 5.4 | 0.40 | 0.44 | 30 | >167 | 41 | 2.8 | 2.6 |
| CM240.EC1 | AE | R5 | 82 | 0.66 | 20 | 0.45 | 0.40 | >167 | 11 | >250 | 3.7 | 3.6 |
| N1 1046.e3 PV | AE | R5 | 138 | 4.2 | 183 | 3.5 | 3.1 | >167 | 18 | >250 | 27 | 30 |
| CAM0015BBY.EC3 | AG | R5 | 81 | 21 | 41 | 0.72 | 1.4 | >167 | >167 | >250 | 7.4 | 7.6 |
| 55815.EC3 | AG | R5 | >167 | 0.33 | 6.2 | 0.084 | 0.077 | >167 | 2.6 | 116 | 0.61 | 0.67 |
| Arithmetic meanc | 71 | 37 | 60 | 0.72 | 0.80 | 138 | 59 | 172 | 6.8 | 8.2 | ||
| Geometric meanc | 17 | 4.2 | 14 | 0.28 | 0.32 | 67 | 21 | 82 | 2.4 | 2.6 | ||
The assay was performed in duplicate. Reported results are means (without standard deviations). Means were calculated only when the difference between results for the duplicate assays was within 3-fold.
The bnAbs b12 and VRC01 used in the neutralization assay were in the IgG1 format.
Arithmetic and geometric means were calculated for all viruses, including those with results of >167 nM, which were assigned a value of 200, and those with results of >250, which were assigned a value of 300.
TABLE 6.
Neutralization in TZM-bl cell-based assays of HIV-1 isolates circulating predominantly in China
| HIV-1 isolate | Clade | Tropism | IC50 (nM)a |
IC90 (nM)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T20 | C34 | CD4-Ig | 2Dm2m | 4Dm2m | 6Dm2m | T20 | C34 | CD4-Ig | 2Dm2m | 4Dm2m | 6Dm2m | |||
| CH64 | BC | R5 | 3.1 | 1.2 | 48 | 1.7 | 0.17 | 0.13 | 25 | 6.7 | >250 | 14 | 1.6 | 0.80 |
| CH70 | BC | R5 | 65 | 7.6 | 7.3 | 0.40 | 0.11 | 0.09 | >750 | 84 | 109 | 3.1 | 1.2 | 0.67 |
| CH91 | BC | R5 | 8.6 | 2.3 | 35 | 3.7 | 0.39 | 0.27 | 310 | 12 | >250 | 56 | 10 | 3.5 |
| CH110 | BC | R5 | 4.3 | 0.78 | 28 | 0.93 | 0.11 | 0.09 | 262 | 6.3 | >250 | 8.7 | 0.83 | 0.58 |
| CH114 | BC | R5 | 102 | 5.4 | 23 | 1.5 | 0.11 | 0.18 | >750 | 25 | 198 | 19 | 4.0 | 1.9 |
| CH117 | BC | R5 | 2.3 | 0.60 | 27 | 5.4 | 0.33 | 0.22 | 34 | 3.2 | >250 | 62 | 15 | 5.1 |
| CH119 | BC | R5 | 2.0 | 3.3 | 85 | 2.7 | 0.17 | 0.13 | 25 | 20 | >250 | 25 | 3.4 | 1.1 |
| CH120 | BC | R5 | 11 | 4.6 | 104 | 4.3 | 0.33 | 0.27 | 443 | 39 | >250 | 46 | 6.6 | 3.5 |
| CNE7 | BC | R5 | 4.3 | 6.4 | 19 | 1.5 | 0.18 | 0.14 | 58 | 22 | 161 | 8.1 | 1.8 | 1.2 |
| CNE15 | BC | R5 | 14 | 3.8 | 106 | 3.0 | 0.29 | 0.17 | 157 | 22 | >250 | 20 | 2.6 | 1.3 |
| CNE16 | BC | R5 | 14 | 11 | 11 | 0.77 | 0.08 | 0.07 | 169 | 51 | 229 | 8.5 | 1.2 | 0.71 |
| CNE20 | BC | R5 | 5.5 | 3.3 | 65 | 11 | 0.05 | 0.29 | 41 | 14 | >250 | >100 | 5.3 | 2.9 |
| CNE23 | BC | R5 | 15 | 2.1 | >250 | 5.4 | 0.49 | 0.48 | 228 | 18 | >250 | 35 | 4.9 | 9.9 |
| CNE30 | BC | R5 | 26 | 14 | 17 | 0.58 | 0.07 | 0.06 | 285 | 110 | 211 | 3.6 | 0.44 | 0.31 |
| CNE40 | BC | R5 | 4.9 | 0.77 | 0.06 | 0.04 | 0.01 | 0.01 | 108 | 7.8 | 0.50 | 0.33 | 0.11 | 0.09 |
| CNE46 | BC | R5 | 26 | 6.4 | 99 | 3.8 | 0.27 | 0.21 | 743 | 115 | >250 | 81 | 9.4 | 2.8 |
| CNE47 | BC | R5 | 4.9 | 1.9 | 152 | 1.5 | 0.11 | 0.12 | 42 | 14 | >250 | 30 | 2.8 | 1.2 |
| CNE49 | BC | R5 | 9.1 | 2.3 | 21 | 0.40 | 0.05 | 0.05 | 236 | 12 | 224 | 2.6 | 0.56 | 0.44 |
| CNE53 | BC | R5 | 51 | 2.0 | 14 | 2.8 | 0.24 | 0.47 | 721 | 23 | >250 | 95 | 5.5 | 12 |
| CNE68 | BC | R5 | 32 | 2.2 | 9.7 | 0.40 | 0.11 | 0.04 | 321 | 12 | 133 | 4.5 | 0.78 | 0.89 |
| CNE1 | B′ | X4 | 1.6 | 0.73 | 197 | 0.47 | 0.11 | 0.22 | 17 | 6.8 | >250 | 4.1 | 0.61 | 2.2 |
| CNE4 | B′ | R5 | 1.7 | 6.3 | 3.9 | 0.40 | 0.11 | 0.13 | 14 | 23 | 24 | 0.40 | 2.4 | 0.80 |
| CNE6 | B′ | R5 | 1.4 | 1.6 | 68 | 1.7 | 0.33 | 0.22 | 15 | 15 | 192 | 6.9 | 2.3 | 1.4 |
| CNE9 | B′ | R5 | 1.7 | 0.87 | 9.9 | 1.1 | 0.22 | 0.22 | 14 | 5.8 | 63 | 7.0 | 3.2 | 1.7 |
| CNE11 | B′ | R5 | 3.5 | 4.6 | >250 | 1.8 | 0.33 | 0.36 | 36 | 42 | >250 | 11 | 2.6 | 3.4 |
| CNE14 | B′ | R5 | 9.7 | 3.9 | 105 | 0.80 | 0.17 | 0.18 | 305 | 17 | 248 | 4.13 | 2.4 | 1.6 |
| CNE57 | B′ | X4 | 2.5 | 2.0 | 250 | 2.1 | 1.2 | 0.71 | 24 | 14 | >250 | >100 | 19 | 22 |
| CNE64 | B′ | R5 | 1.9 | 5.1 | 12 | 1.4 | 0.56 | 0.22 | 16 | 21 | 113 | 6.5 | 3.2 | 1.5 |
| CNE17 | C | R5 | 42 | 3.2 | 11 | 2.4 | 0.11 | 0.13 | 676 | 82 | 231 | 36 | 2.4 | 0.84 |
| CNE58 | C | R5 | 54 | 2.0 | >250 | 10 | 2.1 | 1.1 | 745 | 13 | >250 | >100 | >28 | >28 |
| CNE65 | C | R5 | 5.9 | 3.1 | 15 | 1.1 | 0.28 | 0.22 | 78 | 16 | 115 | 12 | 3.3 | 1.5 |
| CNE66 | C | R5 | 129 | 2.1 | 159 | 1.2 | 0.17 | 0.09 | 729 | 11 | >250 | >100 | 7.7 | 3.1 |
| CNE3 | AE | R5 | 5.6 | 7.1 | >250 | 1.9 | 0.39 | 0.31 | 38 | 71 | >250 | 11 | 2.8 | 2.0 |
| CNE5 | AE | R5 | 2.1 | 5.9 | 94 | 1.8 | 0.67 | 0.53 | 19 | 63 | 236 | 15 | 8.7 | 5.9 |
| CNE55 | AE | R5 | 26 | 7.9 | 213 | 5.9 | 0.45 | 0.46 | 549 | 103 | >250 | 42 | 10 | 5.1 |
| CNE59 | AE | R5 | 1.4 | 3.4 | 10 | 0.93 | 0.17 | 0.18 | 17 | 28 | 220 | 16 | 2.7 | 3.8 |
| CNE107 | AE | X4 | 3.8 | 2.8 | 135 | 3.7 | 0.52 | 0.89 | 45 | 32 | >250 | 33 | 18 | 21 |
| AE20 | AE | R5 | 212 | 7.4 | 50 | 1.5 | 0.72 | 0.49 | 572 | 69 | >250 | 10 | 5.8 | 4.1 |
| AE03 | AE | R5 | 304 | 24 | >250 | 5.7 | 0.67 | 0.40 | 510 | 85 | >250 | 58 | 12 | 15 |
| YN192.31 | AE | R5 | 42 | 27 | 155 | 5.7 | 0.89 | 2.4 | 336 | 225 | >250 | 65 | 21 | 19 |
| GX2010.36 | AE | R5 | 255 | 15 | 12 | 4.7 | 0.11 | 0.13 | >750 | 165 | >250 | 90 | 18 | 6.8 |
| Arithmetic meanb | 37 | 5.3 | 94 | 2.6 | 0.34 | 0.32 | 277 | 42 | 242 | 38 | 6.2 | 5.0 | ||
| Geometric meanb | 11 | 3.5 | 42 | 1.7 | 0.22 | 0.20 | 123 | 25 | 195 | 17 | 3.5 | 2.4 | ||
The assay was performed in duplicate. Reported results are means (without standard deviations). Means were calculated only when the difference between results for the duplicate assays was within 3-fold.
Arithmetic and geometric means were calculated for all viruses, including those with results of >750 nM, which were assigned a value of 800, those with results of >250 nM, which were assigned a value of 300, those with results of >100, which were assigned a value of 150, and those with results of >28 nM, which were assigned a value of 30.
At similarly low concentrations (0.032 to 10 nM), D1D2 enhanced Bal infectivity (data not shown), in agreement with a previous study (45). mD1.22 also exhibited enhancement effects at low concentrations (0.032 to 2 nM). In contrast, 4Dm2m and 6Dm2m did not significantly enhance Bal infectivity at any concentration tested. Similar results were obtained with the JRFL isolate.
Solubility, stability, and aggregation propensity of the bispecific multivalent fusion proteins.
To evaluate the potential of these potent inhibitors for further development as drugs, we tested several drug-related properties, including solubility, stability, and aggregation propensity. 4Dm2m and 6Dm2m in PBS were concentrated to 25.0 and 25.9 mg/ml, respectively, without visible precipitation after high-speed centrifugation. They were stored at 4°C for 2 weeks, and no additional precipitation was observed, suggesting high solubility of the proteins.
To test whether soluble aggregates of 4Dm2m and 6Dm2m formed during prolonged incubation, we used DLS (Fig. 7). Proteins concentrated to 10 mg/ml in PBS were stored at −80°C and slowly thawed on ice before measurements. Our results showed that the particles of 4Dm2m and m909, a control human antibody in the IgG1 format (20), were predominantly small (average diameters, 16.3 and 12.7 nm, respectively). The minor aggregates of 4Dm2m disappeared within the first day of incubation at both 4°C and 37°C, while m909 aggregation continued to occur following the incubation at 37°C. In comparison, about 30 to 50% of the CD4-Ig particles were large, with average diameters of approximately 100 nm, and the aggregation persisted during the whole period of incubation. 6Dm2m partially aggregated after the freeze-thaw cycle; although the aggregation was weakened following the incubation, it led to a relatively wide particle size distribution. These results suggest that like typical IgG1s, 4Dm2m has a low aggregation propensity, lower than that of CD4-Ig.
FIG 7.

Aggregation propensities of 4Dm2m and 6Dm2m. Proteins at a concentration of 10 mg/liter in PBS were analyzed via DLS after incubation at either 4 or 37°C for different periods of time. m909, a folate receptor β-specific human antibody in the IgG1 format, was used as a control. The three curves in each histogram that almost overlap with each other represent three individual measurements.
The proteins were then assessed for their stability against degradation during 15 days of incubation with PBS or human serum at 37°C (Fig. 8). Incubation in PBS for 10 days caused no significant decrease in the amount of functional 4Dm2m, whereas about 20% and 30%, respectively, of 6Dm2m and CD4-Ig lost binding activity to gp140Con-s. 4Dm2m began to be degraded and 6Dm2m continued to be degraded thereafter, while CD4-Ig appeared to be stable. Finally, about 90% of 4Dm2m remained functional, slightly more than those (80%) of 6Dm2m and CD4-Ig. With human serum, levels of all proteins declined at similar rates during the first 5 days of incubation. However, degradation of 4Dm2m and 6Dm2m became slower than that of CD4-Ig during the next 5 days of incubation. At the end of the measurement period, the level of CD4-Ig that retained binding activity was slightly higher than that for functional 4Dm2m and 6Dm2m, but the differences in the amounts were not statistically significant.
FIG 8.
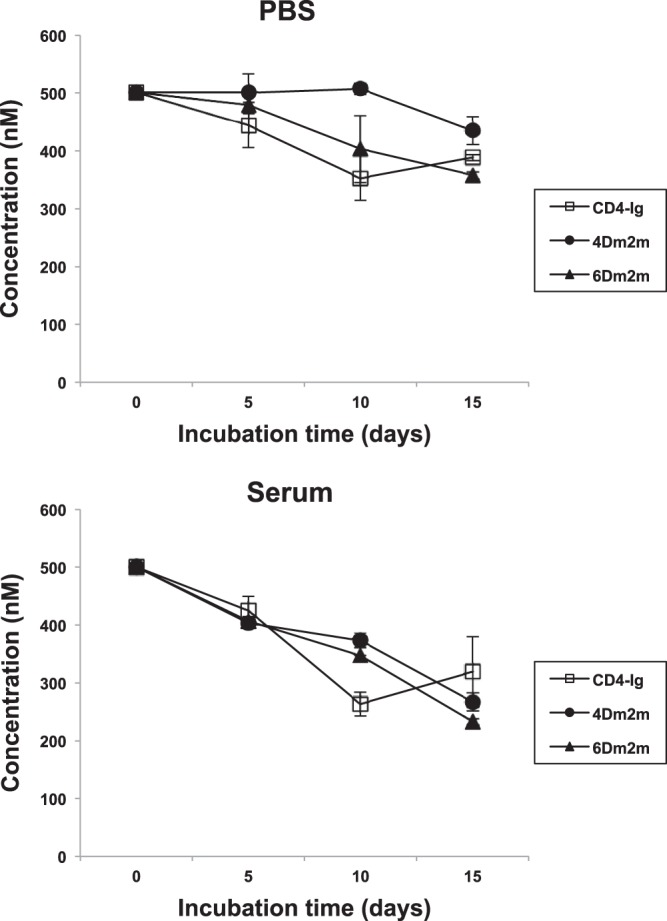
Stability of 4Dm2m and 6Dm2m against degradation in PBS and in human serum. Functional proteins following incubation with PBS or human serum at 37°C for different periods of time were quantified by using ELISA and standard curves made with untreated proteins. In the ELISAs, 96-well plates were coated with gp140Con-s, and bound Fc-fusion proteins were detected by using HRP-conjugated anti-human IgG (Fc-specific) antibodies.
DISCUSSION
A major finding of this study is the identification of a cavity-altered single CD4 domain with a unique combination of properties, including broad and potent neutralizing activity, high specificity, stability, solubility, and affinity for gp120 and a small molecular size. The substitution of a single amino acid residue (A55V) lining a cavity structure in D1 improved the soluble expression, thermal stability, and specificity of mD1.2. The difference between the van der Waals volumes of valine and alanine residues is about 50 Å3 (46), which is similar to the volume (approximately 57 Å3) of the cavity observed in D1. This suggests that the introduction of a couple of methylene groups into the cavity might contribute to an increase in hydrophobic interactions in the D1 core and thereby improve protein stability without causing any steric clashes. It was previously shown that mutations designed to fill cavities may be effective in improving protein stability in some cases (47), but in other cases the expected hydrophobic stabilization is offset by the strains introduced by mutations (48, 49). The SDM method (24) predicted that the A55V mutation is stabilizing, based on calculation of the difference in free energy of unfolding between the wild-type and modeled A55V mutant CD4 D1 structures (ΔΔG = 1.02). However, the effects of the point mutation on other hydrophobic residues in the vicinity of the cavity and/or other interior residues in the hydrophobic network layers may not be predictable.
The A55V substitution does not reduce the binding of mD1.22 to HIV-1 Envs but results in decreased mD1.22 interactions with the human blood cell lines BJAB and SUPT1. The crystal structures of D1D2-gp120 and D1D2–MHC-II complexes show that CD4 binds similarly to both targets, and only the D1 domain makes contacts (38, 50, 51). Monomeric human sCD4 exhibits nanomolar affinities for HIV-1 gp120 (12); however, oligomerization of CD4 is required for stable interaction with MHC-II (37), suggesting that the binding interfaces of gp120 and MHC-II on D1 are substantially different. CD4-Ig strongly bound to not only BJAB cells but also SUPT1 cells, which do not express MHC-II, suggesting multitarget and/or nonspecific interactions of CD4 in vivo. Interestingly, mD1.2Fc showed largely decreased interactions with the cells compared to CD4-Ig, and very weak or no interactions were observed for mD1.22Fc (Fig. 4). In mD1.2, the mutation is on the surface areas (mainly the D1 interface with D2) that are not involved in the CD4 interaction with MHC-II (12, 50, 51). The side chain of the amino acid residue 55 is buried, according to the D1D2 crystal structure (38, 50), so it is not supposed to directly contact MHC-II and other targets. It is therefore most likely that the decreased interactions of mD1.2 and mD1.22 with BJAB and SUPT1 cells are due to possible conformational changes induced by the mutagenesis. Indeed, the anti-D1 antibody m1006 bound to CD4-Ig, mD1.2Fc, and mD1.22Fc to different extents (Fig. 5A), indicating such a possibility. Moreover, the A55V substitution had similar although relatively modest effects on CD4-Ig (Fig. 5), suggesting that its ability to induce conformational changes in D1 is not necessarily dependent on D2 but could be affected by D2 and/or other factors. In all cases, the mutagenesis did not lead to reduced affinities or cross-reactivity of D1 with HIV-1 Envs, providing supporting evidence for our hypothesis that decreasing the molecular size of D1D2 to D1 and altering D1 structures through mutagenesis can significantly reduce the nonspecificity of D1 while preserving its binding and cross-reactivity with HIV-1 Envs. One can also hypothesize that the cavity in the hydrophobic core of D1 was generated during human evolution for specific biological functions related to binding to MHC-II and other possible targets of CD4 (52). Interestingly, previous studies showed that monomeric sCD4 did not inhibit immune functions in vitro (53) or in monkeys (54) and that dimeric (8) and tetrameric (11) sCD4 derivatives were well tolerated in humans. Therefore, the polyspecificity of CD4 may not lead to significant side effects of sCD4-based HIV-1 inhibitors.
Small-size CD4 mimics have been previously reported. For example, the 27-amino-acid peptides CD4M3, CD4M8, and CD4M9 were generated by transferring 9 residues of the D1 CDR2 loop to a scorpion toxin scaffold (55). They bind to HIV-1 gp120 and inhibit virus infection, with IC50s in the micromolar range. To optimize interactions with gp120, nonstandard (nonproteinogenic) amino acid residues were introduced into CD4M9 (56, 57). The optimized peptides CD4M33, CD4M47, and [Phe23]M47 had CD4-like affinities and the ability to neutralize diverse HIV-1 primary isolates. [Phe23]M47 was recently further improved by replacing the phenylalanine at position 23 with cyclohexylmethoxy phenylalanine to increase the flexibility of inserts into the interfacial Phe43 cavity of gp120 (58). The new variant M48U1 achieved picomolar affinities and neutralization of all 180 HIV-1 isolates tested except those from clade AE with noncanonical Phe43 cavities. Another example is the small-molecule organic compounds NBD-556 and NBD-557, for which potent virus-cell and cell-cell fusion inhibitory activities have been shown at low micromolar levels (59). These CD4 mimics are potentially useful as anti-HIV-1 agents, especially as topical microbicides. However, the usefulness of the peptides as candidate therapeutics or components of vaccine immunogens could be limited due to their short half-lives in vivo, as well as the presence of nonstandard amino acid residues and a nonhuman scaffold that could be immunogenic in humans.
Another major finding was that the bispecific multivalent fusion proteins composed of the unique single human CD4 domain mD1.22 and the engineered CD4i antibody domain m36.4 exhibited exceptionally potent and broad inhibitory activities, as well as excellent drug-related properties. HIV-1 entry relies on CD4 and a coreceptor (either CCR5 or CXCR4), which on cell surfaces are thought to be clustered (60, 61). Poor competition with the clustered receptors could lead to relatively weak neutralizing activity of HIV-1 inhibitors targeting the CD4bs and the CoRbs, which form monomers or low-order oligomers. This could be particularly true if HIV-1 Env-mediated cell-cell transmission needs to be interrupted because it occurs via formation of virological synapses, which are organized contact areas with concentrated receptors and Envs (62, 63). In agreement with this line of reasoning are the findings of a recent study that showed that bnAbs to the CD4bs such as VRC01 and b12 dramatically lost potency during cell-cell transmission, although they potently blocked infection by free viruses (64). Our results also showed that mD1.22, m36.4, and their fusion proteins neutralized free viruses more potently than cell-cell fusion (Tables 2 and 3). Moreover, we found that 2Dm2m did not inhibit Bal Env-mediated cell-cell fusion more strongly than m36.4, mD1.22, or mD1.22Fc, while 4Dm2m and 6Dm2m did exhibit higher inhibitory activities than 2Dm2m; a similar trend was observed with the JRFL isolate (Table 3). These results suggest the possible existence of a valency threshold (the number of valencies ≥6, if both m36.4 and mD1.22 are considered) for this class of multivalent inhibitors.
Previously, a polyvalent sCD4 construct (D1D2Ig-αtp) was designed in which D1D2 was fused through a hinge to the human IgG1 Fc; the latter was further covalently linked to an 18-amino-acid human IgAα secretory tailpiece (45). Each D1D2Ig-αtp molecule carries ≥12 D1D2 units and has molecular sizes ranging from 600 to 1,200 kDa, with a 12-nm average hydrodynamic radius. Cryo-electron tomographic analysis showed that D1D2Ig-αtp cross-links the viral spikes on the same virus particle and on neighboring viruses, resulting in inactivation of not only the bound spikes but also those on the closely apposed surfaces of cross-linked viruses (65). The novel mode of action leads to the very potent neutralizing activity (IC90s, <3 nM) of D1D2Ig-αtp (66). 4Dm2m and 6Dm2m could have a similar mechanism of neutralization, in which cross-linking may be achieved not only through mD1.22, which targets the CD4bs, but also via m36.4, which is directed against the CoRbs. Monomeric sCD4 has an intrinsic capacity to enhance HIV-1 entry at low concentrations while D1D2Ig-αtp does not, likely due to the high avidity of D1D2Ig-αtp for HIV-1 Envs, which leads to efficient competition with cell membrane-associated clustered CD4 (45). Our results also showed that 4Dm2m and 6Dm2m did not promote infection by Bal and JRFL at suboptimal concentrations, while D1D2 and mD1.22 did. It is conceivable that in addition to the avidity of mD1.22, m36.4 could further decrease the probability of HIV-1 infectivity enhancement by interrupting the gp120-coreceptor interactions. However, if the CD4 element of our constructs alone promotes infection in some cases, their neutralization potencies and breadths would be limited by that of m36.4. Another major advancement with 4Dm2m and 6Dm2m compared to D1D2Ig-αtp is their much smaller IgG1-like molecular sizes and shapes, which are expected to result in favorable diffusion and retention in vivo, as well as stability and homogeneity, which are important considerations in biotherapeutics development. In addition, 4Dm2m and 6Dm2m bound to MHC-II-expressing BJAB and CD4+ SUPT1 cells much more weakly than did CD4-Ig (data not shown), suggesting that they could be even more specific to HIV-1 than D1D2Ig-αtp in vivo.
Virus eradication has been a major goal of HIV-1 research. A possible key determinant of success is discovery of molecules that exhibit high affinity, cross-reactivity, and specificity to HIV-1 Envs and could therefore guide toxic drugs or killer cells for specific killing of potentially all HIV-1-infected cells expressing the Envs. mD1.22 and m36.4, alone or in combination, are promising candidates as such molecules. They could be used to generate antibody-drug conjugates (67), bispecific killer cell engagers (68), or chimeric antigen receptors (69), which have been highly successful in cancer therapy. Their Fc-fusion proteins could be engineered to enhance Fc-mediated immune effector mechanisms for clearance of infected cells, such as antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity (70). Some HIV-1-infected cells express low levels of Envs on their cell surface. One can hypothesize that due to avidity and/or synergistic effects, bispecific multivalent Env-binding molecules such as 4Dm2m could confer increased sensitivity of targeting those infected cells with low Env expression. HIV-1 latently infected cells are a long-lived viral reservoir that must be eliminated or reduced to a very small size in order to achieve a functional cure for HIV-1 infection. However, there is no or only minimal expression from the the HIV-1 genome in latently infected cells. It is therefore most likely that the above-proposed strategies need to be combined with agents capable of activating HIV-1 latently infected cells so that they can be specifically targeted and killed (71).
Aggregation and immunogenicity are of significant concern for the development of biotherapeutics. The DLS analysis revealed a low level of soluble aggregates of 4Dm2m after a freeze-thaw cycle, but the aggregation was reversible, disappearing within the first day of incubation, and did not recur during the 7 days of incubation at either 4°C or 37°C (Fig. 7), suggesting a low aggregation propensity of the bispecific multivalent construct. The engineering of mD1.22, m36.4, and their fusion proteins might lead to immunogenicity, and only human clinical trials can definitely evaluate such a possibility. The relatively complex structures of the fusion proteins might pose potential challenges for large-scale production. Other drug-related properties, such as pharmacokinetics, also need to be investigated in detail if the proteins are to be further developed as HIV-1 therapeutics.
ACKNOWLEDGMENTS
We thank Barton F. Haynes and Anu Puri for providing reagents and cells and John Owens, Mary Bryson, and Sebastian Molnar for technical assistance. We also acknowledge Christina Ochsenbauer and Agnes Chenine for provision of LucR infectious molecular clones.
This project was supported by the Intramural AIDS Targeted Antiviral Program (IATAP) of the National Institutes of Health (NIH), the Intramural Research Program of the NIH, National Cancer Institute (NCI), Center for Cancer Research, federal funds from the NIH NCI under contract number NO1-CO-12400, the U.S.—China Program for Biomedical Research Cooperation, and grants to Y.H. (81025009 and 81271830) from the Natural Science Foundation of China.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Dimitrov DS, Willey RL, Martin MA, Blumenthal R. 1992. Kinetics of HIV-1 interactions with sCD4 and CD4+ cells: implications for inhibition of virus infection and initial steps of virus entry into cells. Virology 187:398–406. 10.1016/0042-6822(92)90441-Q [DOI] [PubMed] [Google Scholar]
- 2.Smith DH, Byrn RA, Marsters SA, Gregory T, Groopman JE, Capon DJ. 1987. Blocking of HIV-1 infectivity by a soluble, secreted form of the CD4 antigen. Science 238:1704–1707. 10.1126/science.3500514 [DOI] [PubMed] [Google Scholar]
- 3.Kahn JO, Allan JD, Hodges TL, Kaplan LD, Arri CJ, Fitch HF, Izu AE, Mordenti J, Sherwin JE, Groopman JE, Volberding PA. 1990. The safety and pharmacokinetics of recombinant soluble CD4 (rCD4) in subjects with the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex: a phase 1 study. Ann. Intern. Med. 112:254–261 [DOI] [PubMed] [Google Scholar]
- 4.Schacker T, Collier AC, Coombs R, Unadkat JD, Fox I, Alam J, Wang JP, Eggert E, Corey L. 1995. Phase I study of high-dose, intravenous rsCD4 in subjects with advanced HIV-1 infection. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 9:145–152 [PubMed] [Google Scholar]
- 5.Schooley RT, Merigan TC, Gaut P, Hirsch MS, Holodniy M, Flynn T, Liu S, Byington RE, Henochowicz S, Gubish E, et al. 1990. Recombinant soluble CD4 therapy in patients with the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex: a phase I-II escalating dosage trial. Ann. Intern. Med. 112:247–253 [DOI] [PubMed] [Google Scholar]
- 6.Langner KD, Niedrig M, Fultz P, Anderson D, Reiner G, Repke H, Gelderblom H, Seed B, Hilfenhaus J, Zettlmeissl G. 1993. Antiviral effects of different CD4-immunoglobulin constructs against HIV-1 and SIV: immunological characterization, pharmacokinetic data and in vivo experiments. Arch. Virol. 130:157–170 [DOI] [PubMed] [Google Scholar]
- 7.Meng TC, Fischl MA, Cheeseman SH, Spector SA, Resnick L, Boota A, Petrakis T, Wright B, Richman DD. 1995. Combination therapy with recombinant human soluble CD4-immunoglobulin G and zidovudine in patients with HIV infection: a phase I study. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 8:152–160 [PubMed] [Google Scholar]
- 8.Chamow SM, Duliege AM, Ammann A, Kahn JO, Allen JD, Eichberg JW, Byrn RA, Capon DJ, Ward RH, Ashkenazi A. 1992. CD4 immunoadhesins in anti-HIV therapy: new developments. Int. J. Cancer Suppl. 7:69–72 [PubMed] [Google Scholar]
- 9.Allaway GP, Davis-Bruno KL, Beaudry GA, Garcia EB, Wong EL, Ryder AM, Hasel KW, Gauduin MC, Koup RA, McDougal JS, et al. 1995. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res. Hum. Retroviruses 11:533–539. 10.1089/aid.1995.11.533 [DOI] [PubMed] [Google Scholar]
- 10.Fletcher CV, DeVille JG, Samson PM, Moye JH, Jr, Church JA, Spiegel HM, Palumbo P, Fenton T, Smith ME, Graham B, Kraimer JM, Shearer WT. 2007. Nonlinear pharmacokinetics of high-dose recombinant fusion protein CD4-IgG2 (PRO 542) observed in HIV-1-infected children. J. Allergy Clin. Immunol. 119:747–750. 10.1016/j.aci.2006.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobson JM, Israel RJ, Lowy I, Ostrow NA, Vassilatos LS, Barish M, Tran DN, Sullivan BM, Ketas TJ, O'Neill TJ, Nagashima KA, Huang W, Petropoulos CJ, Moore JP, Maddon PJ, Olson WC. 2004. Treatment of advanced human immunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542. Antimicrob. Agents Chemother. 48:423–429. 10.1128/AAC.48.2.423-429.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Feng Y, Gong R, Zhu Z, Wang Y, Zhao Q, Dimitrov DS. 2011. Engineered single human CD4 domains as potent HIV-1 inhibitors and components of vaccine immunogens. J. Virol. 85:9395–9405. 10.1128/JVI.05119-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonavane S, Chakrabarti P. 2008. Cavities and atomic packing in protein structures and interfaces. PLoS Comput. Biol. 4(9):e1000188. 10.1371/journal.pcbi.1000188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubbard SJ, Argos P. 1994. Cavities and packing at protein interfaces. Protein Sci. 3:2194–2206. 10.1002/pro.5560031205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borgo B, Havranek JJ. 2012. Automated selection of stabilizing mutations in designed and natural proteins. Proc. Natl. Acad. Sci. U. S. A. 109:1494–1499. 10.1073/pnas.1115172109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Xiao X, Wang Y, Zhu Z, Dimitrov DS. 2010. Bifunctional fusion proteins of the human engineered antibody domain m36 with human soluble CD4 are potent inhibitors of diverse HIV-1 isolates. Antiviral Res. 88:107–115. 10.1016/j.antiviral.2010.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Zhu Z, Feng Y, Dimitrov DS. 2008. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc. Natl. Acad. Sci. U. S. A. 105:17121–17126. 10.1073/pnas.0805297105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyerson JR, Tran EE, Kuybeda O, Chen W, Dimitrov DS, Gorlani A, Verrips T, Lifson JD, Subramaniam S. 2013. Molecular structures of trimeric HIV-1 Env in complex with small antibody derivatives. Proc. Natl. Acad. Sci. U. S. A. 110:513–518. 10.1073/pnas.121480110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Z, Bossart KN, Bishop KA, Crameri G, Dimitrov AS, McEachern JA, Feng Y, Middleton D, Wang LF, Broder CC, Dimitrov DS. 2008. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J. Infect. Dis. 197:846–853. 10.1086/528801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng Y, Shen J, Streaker ED, Lockwood M, Zhu Z, Low PS, Dimitrov DS. 2011. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res. Ther. 13:R59. 10.1186/ar3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao HX, Sutherland LL, Xia SM, Brock ME, Scearce RM, Vanleeuwen S, Alam SM, McAdams M, Weaver EA, Camacho Z, Ma BJ, Li Y, Decker JM, Nabel GJ, Montefiori DC, Hahn BH, Korber BT, Gao F, Haynes BF. 2006. A group M consensus envelope glycoprotein induces antibodies that neutralize subsets of subtype B and C HIV-1 primary viruses. Virology 353:268–282. 10.1016/j.virol.2006.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho BK, Gruswitz F. 2008. Hollow: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8:49. 10.1186/1472-6807-8-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai J, Gerstein M. 2002. Calculations of protein volumes: sensitivity analysis and parameter database. Bioinformatics 18:985–995. 10.1093/bioinformatics/18.7.985 [DOI] [PubMed] [Google Scholar]
- 24.Topham CM, Srinivasan N, Blundell TL. 1997. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng. 10:7–21. 10.1093/protein/10.1.7 [DOI] [PubMed] [Google Scholar]
- 25.Chen W, Zhu Z, Feng Y, Xiao X, Dimitrov DS. 2008. Construction of a large phage-displayed human antibody domain library with a scaffold based on a newly identified highly soluble, stable heavy chain variable domain. J. Mol. Biol. 382:779–789. 10.1016/j.jmb.2008.07.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Y, Zhu Z, Xiao X, Choudhry V, Barrett JC, Dimitrov DS. 2006. Novel human monoclonal antibodies to insulin-like growth factor (IGF)-II that potently inhibit the IGF receptor type I signal transduction function. Mol. Cancer Ther. 5:114–120. 10.1158/1535-7163.MCT-05-0252 [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Zhu Z, Feng Y, Dimitrov DS. 2010. A large human domain antibody library combining heavy and light chain CDR3 diversity. Mol. Immunol. 47:912–921. 10.1016/j.molimm.2009.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Feng Y, Zhao Q, Zhu Z, Dimitrov DS. 2012. Human monoclonal antibodies targeting nonoverlapping epitopes on insulin-like growth factor II as a novel type of candidate cancer therapeutics. Mol. Cancer Ther. 11:1400–1410. 10.1158/1535-7163.MCT-12-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, Prabakaran P, Veenstra TD, Tarasov SG, Ishima R, Dimitrov DS. 2009. Engineered human antibody constant domains with increased stability. J. Biol. Chem. 284:14203–14210. 10.1074/jbc.M900769200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown BK, Wieczorek L, Sanders-Buell E, Rosa Borges A, Robb ML, Birx DL, Michael NL, McCutchan FE, Polonis VR. 2008. Cross-clade neutralization patterns among HIV-1 strains from the six major clades of the pandemic evaluated and compared in two different models. Virology 375:529–538. 10.1016/j.virol.2008.02.022 [DOI] [PubMed] [Google Scholar]
- 31.Yao X, Chong H, Zhang C, Waltersperger S, Wang M, Cui S, He Y. 2012. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J. Biol. Chem. 287:6788–6796. 10.1074/jbc.M111.317883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shang H, Han X, Shi X, Zuo T, Goldin M, Chen D, Han B, Sun W, Wu H, Wang X, Zhang L. 2011. Genetic and neutralization sensitivity of diverse HIV-1 Env clones from chronically infected patients in China. J. Biol. Chem. 286:14531–14541. 10.1074/jbc.M111.224527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edmonds TG, Ding H, Yuan X, Wei Q, Smith KS, Conway JA, Wieczorek L, Brown B, Polonis V, West JT, Montefiori DC, Kappes JC, Ochsenbauer C. 2010. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13. 10.1016/j.virol.2010.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choudhry V, Zhang MY, Harris I, Sidorov IA, Vu B, Dimitrov AS, Fouts T, Dimitrov DS. 2006. Increased efficacy of HIV-1 neutralization by antibodies at low CCR5 surface concentration. Biochem. Biophys. Res. Commun. 348:1107–1115. 10.1016/j.bbrc.2006.07.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roben P, Moore JP, Thali M, Sodroski J, Barbas CF, III, Burton DR. 1994. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J. Virol. 68:4821–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Germain RN. 1997. T-cell signaling: the importance of receptor clustering. Curr. Biol. 7:R640–R644 [DOI] [PubMed] [Google Scholar]
- 37.Sakihama T, Smolyar A, Reinherz EL. 1995. Oligomerization of CD4 is required for stable binding to class II major histocompatibility complex proteins but not for interaction with human immunodeficiency virus gp120. Proc. Natl. Acad. Sci. U. S. A. 92:6444–6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78:13232–13252. 10.1128/JVI.78.23.13232-13252.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861. 10.1126/science.1187659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. 10.1126/science.1178746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Euler Z, Bunnik EM, Burger JA, Boeser-Nunnink BD, Grijsen ML, Prins JM, Schuitemaker H. 2011. Activity of broadly neutralizing antibodies, including PG9, PG16, and VRC01, against recently transmitted subtype B HIV-1 variants from early and late in the epidemic. J. Virol. 85:7236–7245. 10.1128/JVI.00196-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouvin-Pley M, Morgand M, Moreau A, Jestin P, Simonnet C, Tran L, Goujard C, Meyer L, Barin F, Braibant M. 2013. Evidence for a continuous drift of the HIV-1 species towards higher resistance to neutralizing antibodies over the course of the epidemic. PLoS Pathog. 9(7):e1003477. 10.1371/journal.ppat.1003477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A, et al. 1994. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res. Hum. Retroviruses 10:359–369. 10.1089/aid.1994.10.359 [DOI] [PubMed] [Google Scholar]
- 45.Arthos J, Cicala C, Steenbeke TD, Chun TW, Dela Cruz C, Hanback DB, Khazanie P, Nam D, Schuck P, Selig SM, Van Ryk D, Chaikin MA, Fauci AS. 2002. Biochemical and biological characterization of a dodecameric CD4-Ig fusion protein: implications for therapeutic and vaccine strategies. J. Biol. Chem. 277:11456–11464. 10.1074/jbc.M111191200 [DOI] [PubMed] [Google Scholar]
- 46.Richards FM. 1974. The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol. 82:1–14 [DOI] [PubMed] [Google Scholar]
- 47.Ishikawa K, Nakamura H, Morikawa K, Kanaya S. 1993. Stabilization of Escherichia coli ribonuclease HI by cavity-filling mutations within a hydrophobic core. Biochemistry 32:6171–6178. 10.1021/bi00075a009 [DOI] [PubMed] [Google Scholar]
- 48.Karpusas M, Baase WA, Matsumura M, Matthews BW. 1989. Hydrophobic packing in T4 lysozyme probed by cavity-filling mutants. Proc. Natl. Acad. Sci. U. S. A. 86:8237–8241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka M, Chon H, Angkawidjaja C, Koga Y, Takano K, Kanaya S. 2010. Protein core adaptability: crystal structures of the cavity-filling variants of Escherichia coli RNase HI. Protein Pept. Lett. 17:1163–1169. 10.2174/09298661079160342 [DOI] [PubMed] [Google Scholar]
- 50.Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. 2001. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc. Natl. Acad. Sci. U. S. A. 98:10799–10804. 10.1073/pnas.191124098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin Y, Wang XX, Mariuzza RA. 2012. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc. Natl. Acad. Sci. U. S. A. 109:5405–5410. 10.1073/pnas.1118801109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogata K, Kanei-Ishii C, Sasaki M, Hatanaka H, Nagadoi A, Enari M, Nakamura H, Nishimura Y, Ishii S, Sarai A. 1996. The cavity in the hydrophobic core of Myb DNA-binding domain is reserved for DNA recognition and trans-activation. Nat. Struct. Biol. 3:178–187 [DOI] [PubMed] [Google Scholar]
- 53.Berger EA, Chaudhary VK, Clouse KA, Jaraquemada D, Nicholas JA, Rubino KL, Fitzgerald DJ, Pastan I, Moss B. 1990. Recombinant CD4-Pseudomonas exotoxin hybrid protein displays HIV-specific cytotoxicity without affecting MHC class II-dependent functions. AIDS Res. Hum. Retroviruses 6:795–804. 10.1089/aid.1990.6.795 [DOI] [PubMed] [Google Scholar]
- 54.Bugelski PJ, Thiem PA, Truneh A, Morgan DG. 1991. Recombinant human soluble CD4 does not inhibit immune function in cynomolgus monkeys. Toxicol. Pathol. 19:580–588 [DOI] [PubMed] [Google Scholar]
- 55.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Menez A, Roumestand C, Yang YS, Ylisastigui L, Benjouad A, Gluckman JC. 1999. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 96:13091–13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Menez A, Lusso P, Veas F, Vita C. 2003. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 21:71–76. 10.1038/nbt768 [DOI] [PubMed] [Google Scholar]
- 57.Stricher F, Huang CC, Descours A, Duquesnoy S, Combes O, Decker JM, Kwon YD, Lusso P, Shaw GM, Vita C, Kwong PD, Martin L. 2008. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J. Mol. Biol. 382:510–524. 10.1016/j.jmb.2008.06.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acharya P, Luongo TS, Louder MK, McKee K, Yang Y, Do Kwon Y, Mascola JR, Kessler P, Martin L, Kwong PD. 2013. Structural basis for highly effective HIV-1 neutralization by CD4-mimetic miniproteins revealed by 1.5 Å cocrystal structure of gp120 and M48U1. Structure 21:1018–1029. 10.1016/j.str.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. 2005. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339:213–225. 10.1016/j.virol.2005.06.008 [DOI] [PubMed] [Google Scholar]
- 60.Krummel MF, Sjaastad MD, Wulfing C, Davis MM. 2000. Differential clustering of CD4 and CD3ζ during T cell recognition. Science 289:1349–13452. 10.1126/science.289.5483.1349 [DOI] [PubMed] [Google Scholar]
- 61.Singer II, Scott S, Kawka DW, Chin J, Daugherty BL, DeMartino JA, DiSalvo J, Gould SL, Lineberger JE, Malkowitz L, Miller MD, Mitnaul L, Siciliano SJ, Staruch MJ, Williams HR, Zweerink HJ, Springer MS. 2001. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J. Virol. 75:3779–3790. 10.1128/JVI.75.8.3779-3790.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293. 10.1084/jem.20030648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Earl LA, Lifson JD, Subramaniam S. 2013. Catching HIV ‘in the act' with 3D electron microscopy. Trends Microbiol. 21:397–404. 10.1016/j.tim.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 8(4):e1002634. 10.1371/journal.ppat.1002634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett A, Liu J, Van Ryk D, Bliss D, Arthos J, Henderson RM, Subramaniam S. 2007. Cryoelectron tomographic analysis of an HIV-neutralizing protein and its complex with native viral gp120. J. Biol. Chem. 282:27754–27759. 10.1074/jbc.M702025200 [DOI] [PubMed] [Google Scholar]
- 66.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682. 10.1038/nature01188 [DOI] [PubMed] [Google Scholar]
- 67.Sievers EL, Senter PD. 2013. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 64:15–29. 10.1146/annurev-med-050311-201823 [DOI] [PubMed] [Google Scholar]
- 68.Choi BD, Cai M, Bigner DD, Mehta AI, Kuan CT, Sampson JH. 2011. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin. Biol. Ther. 11:843–853. 10.1517/14712598.2011.572874 [DOI] [PubMed] [Google Scholar]
- 69.Turtle CJ, Hudecek M, Jensen MC, Riddell SR. 2012. Engineered T cells for anti-cancer therapy. Curr. Opin. Immunol. 24:633–639. 10.1016/j.coi.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, Vielmetter J, Carmichael DF, Hayes RJ, Dahiyat BI. 2006. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U. S. A. 103:4005–4010. 10.1073/pnas.0508123103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen W, Ying T, Dimitrov DS. 2013. Antibody-based candidate therapeutics against HIV-1: implications for virus eradication and vaccine design. Expert Opin. Biol. Ther. 13:657–671. 10.1517/14712598.2013.761969 [DOI] [PMC free article] [PubMed] [Google Scholar]