

Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the etiological agent of a respiratory disease that has a 10% mortality rate. We previously showed that SARS-CoV lacking the E gene (SARS-CoV-ΔE) is attenuated in several animal model systems. Here, we show that absence of the E protein resulted in reduced expression of proinflammatory cytokines, decreased numbers of neutrophils in lung infiltrates, diminished lung pathology, and increased mouse survival, suggesting that lung inflammation contributed to SARS-CoV virulence. Further, infection with SARS-CoV-ΔE resulted in decreased activation of NF-κB compared to levels for the wild-type virus. Most important, treatment with drugs that inhibited NF-κB activation led to a reduction in inflammation and lung pathology in both SARS-CoV-infected cultured cells and mice and significantly increased mouse survival after SARS-CoV infection. These data indicated that activation of the NF-κB signaling pathway represents a major contribution to the inflammation induced after SARS-CoV infection and that NF-κB inhibitors are promising antivirals in infections caused by SARS-CoV and potentially other pathogenic human coronaviruses.
INTRODUCTION
Severe acute respiratory syndrome (SARS), caused by a coronavirus (SARS-CoV), affected approximately 8,000 individuals during the 2002-2003 epidemic and had a mortality rate of 10% (1, 2). SARS-CoV has not reappeared in humans since 2004, but SARS-like coronaviruses are present in bats circulating all over the world, making another SARS outbreak a possibility (3–6). Consistent with this prediction, a new coronavirus, Middle East respiratory syndrome coronavirus (MERS-CoV), emerged during the summer of 2012 in Saudi Arabia and is the etiological agent of severe pneumonia in patients from 10 Middle Eastern and European countries (7, 8). As of 22 October 2013, a total of 139 confirmed cases were reported, with a 43% mortality rate (http://www.cdc.gov/coronavirus/mers/). This novel coronavirus is closely related to the two bat coronaviruses Bt-CoV-HKU4 and Bt-CoV-HKU5 (8), suggesting that MERS-CoV, like SARS-CoV, originated from bats. MERS-CoV has probably been amplified in one or more intermediate hosts, including camels, and was then transmitted to humans (9). The fact that zoonotic coronaviruses frequently cross species barriers to infect humans makes the generation of vaccines and the identification of antivirals important goals.
SARS-CoV is an enveloped, positive-sense RNA virus, with a genome of 29.7 kb (10). The replicase is encoded within the 5′ two-thirds of the genome and includes two overlapping open reading frames (ORFs), named ORF1a and ORF1b. Translation of these ORFs results in the expression of two polyproteins, pp1a and pp1ab, which are processed by two viral proteinases to yield 16 nonstructural proteins (nsps) (11). These proteins participate in the replication of the viral genome and transcription of subgenomic messenger RNAs (sgmRNAs), which encode viral structural proteins (spike [S], envelope [E], membrane [M], and nucleoprotein [N]) (11). In addition, all coronaviruses encode a set of group-specific proteins whose sequence and number vary among the different coronavirus species (12). In the case of SARS-CoV, the genes 3a, 3b, 6, 7a, 7b, 8a, 8b, and 9b encode group-specific proteins (13).
SARS-CoV E protein contains 76 amino acids and is composed of a short hydrophilic amino terminus followed by a hydrophobic region and a hydrophilic carboxy terminus (14). The hydrophobic region forms at least one amphipathic α helix that oligomerizes to form an ion-conductive pore in membranes (14). The ion conductance and selectivity of this ion channel are influenced by the lipid charge of the membranes (15, 16). The requirement for E protein in virus production varies among coronaviruses. Thus, E protein is essential for the generation of propagation-competent viruses in cells infected with transmissible gastroenteritis coronavirus (TGEV) or MERS-CoV (17–20) but not for mouse hepatitis virus (MHV) or SARS-CoV (21–23). SARS-CoV lacking E protein is attenuated in hamsters, transgenic mice expressing the SARS-CoV receptor human angiotensin converting enzyme 2 (hACE2), and BALB/c mice infected with a mouse-adapted SARS-CoV (21, 22, 24). We also showed that SARS-CoV E protein reduced apoptosis and the stress response induced after SARS-CoV infection (25).
Reduced survival in SARS is associated with male gender, age over 60 years, neutrophilia, and severe biochemical abnormalities, such as the upregulation of proinflammatory cytokines (26–30). Several species of animals can be infected with SARS-CoV, but the model that best fits human SARS is infection of conventional mice with mouse-adapted SARS-CoVs (31–33). This model reproduces many aspects of the human disease, such as rapid and high viral replication in lungs, viremia, dissemination of virus to extrapulmonary sites accompanied by lymphopenia and neutrophilia, pathological changes in the lungs, and lethality (31–33). Furthermore, the disease is more severe in aged mice, as occurs in humans (28, 34).
The elevated cytokine/chemokine response in severe SARS reflects a dysregulated immune response. Activation of NF-κB is a hallmark of most infections, including those caused by viruses, leading to protective and pathological responses. Accordingly, the role of NF-κB as a potential therapeutic target in microbial diseases has been extensively studied (35), and more than 700 NF-κB inhibitors have been described (36). SARS-CoV proteins nsp1, nsp3a, nsp7a, spike, and nucleocapsid promote NF-κB activation, possibly contributing to SARS-CoV-induced pathogenesis (37–42). Aged macaques develop a more severe pathology than younger animals, with an increase in expression of genes associated with inflammation linked to a central role of NF-κB and a reduction in expression of type I interferon (IFN-β) (34).
We studied the effect of deleting SARS-CoV E protein in inflammatory responses and pathogenesis in cell cultures and mice. A mouse-adapted SARS-CoV lacking E protein induced less lung pathology, mediated by decreased expression of inflammation mediators, lower numbers of neutrophils in lung infiltrates, and limited NF-κB activation. Drugs that specifically inhibit NF-κB signaling led to a reduction in inflammation and lung pathology and increased mouse survival after infection with SARS-CoV. These results indicated that NF-κB activation is partly pathogenic in SARS and that NF-κB inhibitors are promising antivirals for treatment of this infection and perhaps for other infections caused by human coronaviruses, such as MERS-CoV.
MATERIALS AND METHODS
Reagents.
The NF-κB inhibitors caffeic acid phenethyl ester (CAPE), resveratrol, Bay11-7082, and parthenolide were purchased from Tocris Biosciences, Sigma, Calbiochem, and Enzo Life Sciences, respectively.
Virus.
The mouse-adapted MA15 viruses (32), recombinant SARS-CoV-MA15 (rSARS-CoV-MA15) and rSARS-CoV-MA15-ΔE were rescued from infectious cDNA clones as previously described (21, 24). All work with infectious viruses and infected animals was performed in biosafety level 3 (BSL3) facilities by personnel wearing positive-pressure air-purifying respirators (3M HEPA AirMate; 3M, St. Paul, MN).
Mice.
Pathogen-free 8-week-old BALB/c Ola Hsd or BALB/c AnNCr female mice were obtained from Harlan (The Netherlands) or from the National Cancer Institute (Bethesda, MD, USA), respectively. BALB/c mice were infected at the age of 8 (AnNcr) or 16 weeks (Ola Hsd), with 6,000 PFU or 100,000 PFU, respectively. All protocols were approved by the University of Iowa Institutional Animal Care and Use Committee and by the Ethical Review Committee at the Centro de Investigación en Sanidad Animal of the Instituto Nacional de Investigaciones Agrarias (CISA-INIA). Infected mice were housed in a ventilated rack (animal transport unit-biocontainment unit [ATU-BCU], Allentown, Inc., Allentown, NJ).
Cells.
African green monkey kidney-derived Vero E6 cells were kindly provided by Eric Snijder (Medical Center, University of Leiden, The Netherlands). Mouse delayed brain tumor (DBT) cells, expressing the SARS-CoV receptor mouse ACE2 (DBT-mACE2 cells), were generated in our laboratory as previously described (43). In all cases, cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 25 mM HEPES and 10% fetal bovine serum (Biowhittaker). Virus titrations were performed in Vero E6 cells following standard procedures using closed flasks or plates sealed in plastic bags, as previously described (21).
Clinical disease in BALB/c mice.
Eight-week-old or 16-week-old mice were lightly anesthetized with isoflurane and inoculated intranasally with 6,000 or 100,000 PFU of rSARS-CoV-MA15 or rSARS-CoV-MA15-ΔE virus, respectively, in 50 μl of DMEM. Infected mice were examined and weighed daily.
Histopathological examination of lungs from infected mice.
Eight-week-old or 16-week-old mice were infected as described above with 6,000 or 100,000 PFU, respectively, of rSARS-CoV-MA15 or rSARS-CoV-MA15-ΔE. Lungs were removed from mice at 2 and 4 days postinfection (dpi), fixed in zinc formalin, and processed as described previously (44). For routine histology, sections were stained with hematoxylin and eosin.
Evaluation of pulmonary infiltrates by flow cytometry.
Eight-week-old or 16-week-old mice were infected as above with 6,000 or 100,000 PFU, respectively, of virus. Lungs were removed from mice at 2, 4, and 6 dpi, and cells were prepared as previously described (45). Macrophages were CD45+ CD11b+ Ly6C+ Ly6G−. Neutrophils were CD45+ CD11b+ Ly6C+ Ly6G+. The following monoclonal antibodies were used: rat anti-mouse CD11b (M1/70), rat anti-mouse CD16/32 (2.4G2), rat anti-mouse Ly6G (1A8), and rat anti-mouse Ly6C (AL-21) all from BD Biosciences (San Diego, CA), and anti-mouse F4/80 (BM8) from eBioscience (San Diego, CA). For surface staining, 106 cells were blocked with 1 μg of anti-CD16/32 antibody and 1% rat serum, stained with the CD11b-, Ly6G-, Ly6C-, and F4/80-specific antibodies, as indicated above, and then fixed using Cytofix Solution (BD Biosciences).
RNA analysis by RT-qPCR.
Total RNA from DBT-mACE2 cells was extracted using an RNeasy Minikit (Qiagen). Mice were inoculated intranasally with 100,000 PFU of rSARS-CoV-MA15 or rSARS-CoV-MA15-ΔE in 50 μl of DMEM. Lungs were removed from mice at 2 and 4 dpi and incubated in RNA Later (Ambion) at 4°C for 24 h, prior to being frozen at −80°C. Then, lungs were thawed and homogenized in RLT buffer (Qiagen) using a gentleMACS Dissociator (Miltenyi Biotec), and total RNA was extracted using an RNeasy Minikit (Qiagen). For quantitative reverse transcription-PCR (RT-qPCR) of cellular genes, cDNA was prepared using a High-Capacity cDNA transcription kit (Applied Biosystems), followed by PCR using TaqMan gene expression assays (Applied Biosystems) specific for murine genes (Table 1). Data were acquired with an ABI Prism 7000 sequence detection system (Applied Biosystems) and analyzed using ABI Prism 7000 SDS, version 1.0, software. Gene expression relative to mock-infected samples is shown.
TABLE 1.
TaqMan assays used to analyze the expression of cellular genes by RT-qPCR
| Gene product | TaqMan assaya | Description |
|---|---|---|
| 18S | Mm03928990-g1 | Ribosomal RNA 18S |
| TNF | Mm00443258-m1 | Tumor necrosis factor |
| CCL2/MCP-1 | Mm00441242-m1 | Monocyte chemotactic protein 1 |
| CCL5/RANTES | Mm01302428-m1 | Regulated upon activation, normal T-cell expressed, and secreted |
| CXCL1/NAP-3 | Mm04207460-m1 | Neutrophil-activating protein 3 |
| CXCL2/MIP-2 | Mm00436450-m1 | Macrophage inflammatory protein 2 |
| CXCL10/IP-10 | Mm00445235-m1 | Interferon-inducible protein 10 |
| IL-6 | Mm00446190-m1 | Interleukin-6 |
Mm, Mus musculus.
Cytokine multiplex analysis.
Lungs were homogenized, and nuclear, cytoplasmic, and extracellular proteins were extracted using a Nuclear Extract Kit (Active Motif, Carlsbad, CA). Supernatant proteins (cytoplasmic and extracellular proteins) were diluted 1:4 in assay diluent (Millipore) prior to analysis. The expression of CCL2/MCP-1, and CXCL10/IP-10 was measured using Luminex technology and a mouse cytokine antibody bead kit (Milliplex map kit; Millipore) according to the manufacturer's instructions.
Luciferase assays.
To analyze the induction of NF-κB, nuclear factor of activated T cells (NF-AT), AP-1, and the cis-acting replication element (CRE), luciferase reporter assays were used in DBT-mACE2 cells (Cignal Reporter Assay Kits CCS-013L for NF-κB, CCS-015L for NF-AT, CCS-011L for AP-1, and CCS-002L for CRE; Qiagen). To this end, plasmids encoding the firefly luciferase ORF under the control of the different promoters were cotransfected with a plasmid encoding the Renilla luciferase to normalize levels of transfection. Separately, a plasmid encoding a noninducible firefly luciferase reporter and the plasmid encoding the Renilla luciferase were cotransfected as negative controls. In addition, plasmids constitutively expressing green fluorescent protein (GFP), firefly luciferase, and Renilla luciferase were cotransfected as positive controls.
To analyze the activation of interferon regulatory factor 3 (IRF3) and IRF7, three plasmids were cotransfected: (i) the reporter plasmid p55UASGluc encoding the firefly luciferase gene under the control of the GAL4 upstream activation sequence (UAS); (ii) plasmid pGAL4DBD-IRF3 or pGAL4DBD-IRF7, encoding the IRF3 or IRF7 transactivation domains fused to the GAL4 DNA binding domain (DBD), kindly provided by T. Fujita (Kyoto University, Kyoto, Japan) (46); and (iii) the control plasmid pRL-SV40 (where SV40 is simian virus 40), encoding Renilla luciferase (Promega). Firefly and Renilla luciferase expression levels were monitored using a Dual Glo assay luciferase system (Promega) and a luminometer.
DBT-mACE2 cells were cotransfected with the plasmids, using a three-dimensional (3D) Nucleofector (Lonza) according to the manufacturer's instructions. Then, 24 h later, the cells were infected with rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15 at a multiplicity of infection (MOI) of 0.05. Firefly and Renilla luciferase expression levels were measured at 48 h postinfection (hpi) and normalized to Renilla luciferase levels.
NF-κB activity in tissue culture and in vivo.
Cells were infected with the virus rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15 at an MOI of 0.05 and collected at 48 hpi. Mice were infected intranasally as described above with 100,000 PFU of rSARS-CoV-MA15 or rSARS-CoV-MA15-ΔE virus in 50 μl of DMEM or treated with DMEM alone. Lungs were removed from mice at 2 and 4 dpi and homogenized. Nuclear extracts from infected cells and homogenized lungs were obtained using a nuclear extraction kit as described above. NF-κB DNA-binding activity in infected cells was measured using an enzyme-linked immunosorbent assay (ELISA)-based assay and a chemiluminescent kit (TransAM p65 Transcription Factor Assay kit; Active Motif). NF-κB in lung nuclear extracts was analyzed by Western blotting using antibodies specific for NF-κB (rabbit polyclonal antibody) (ab7970; Abcam) and histone H3 (rabbit polyclonal antibody) (39164; Active Motif). Bound antibodies were detected with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody and the Immobilon Western chemiluminescent substrate (Millipore), according to the manufacturer's recommendations. Densitometric analysis of NF-κB and histone H3 bands from mock-, rSARS-CoV-MA15-, and rSARS-CoV-MA15-ΔE-infected mice was performed using Quantity One, version 4.5.1, software (Bio-Rad). In each case, the levels of NF-κB were normalized to the levels of histone H3. Three different experiments and appropriate gel exposures were used in all cases with similar results. In addition, different exposures of the same experiment were analyzed to ensure that data obtained were within linear range.
NF-κB inhibitor treatment of SARS-CoV-infected cells.
DBT-mACE2 cells were infected at an MOI of 0.05 with rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15. At 43 hpi, half of the culture medium was removed and replaced with medium containing CAPE (50 μg/ml), resveratrol (40 μg/ml), Bay11-7082 (40 μM), or parthenolide (24 μM). At 48 hpi, supernatants were collected, and total cellular RNA was extracted using an RNeasy Minikit (Qiagen). In parallel, cells were previously cotransfected with luciferase plasmids to measure NF-κB activity, as described above.
NF-κB inhibitor treatment of SARS-CoV-infected BALB/c mice.
Sixteen-week-old mice were infected with 100,000 PFU of rSARS-CoV-MA15 as described above. At 4 hpi and every 24 h thereafter, from days 1 to 4, mice were treated intraperitoneally with the NF-κB inhibitors CAPE and parthenolide at 20 mg/kg of body weight/day, either alone or in combination or with vehicle (phosphate-buffered saline [PBS] containing 1.2% dimethyl sulfoxide [DMSO] and 2.5% Tween 20). Survival was analyzed in three independent experiments, with six mice per group. In addition, three mice per group were sacrificed at day 4 postinfection (p.i.), and lungs were collected to evaluate the expression of cytokines by RT-qPCR and to determine viral titers. To evaluate virus titers, right lungs were homogenized in phosphate-buffered saline (PBS) containing 100 UI/ml penicillin, 0.1 mg/ml streptomycin, 50 μg/ml gentamicin, and 0.5 μg/ml amphotericin B (Fungizone), using a gentleMACS Dissociator (Miltenyi Biotec). Virus titrations were performed in Vero E6 cells according to standard procedures using closed flasks or plates sealed in plastic bags, as previously described (21).
RESULTS
Pathology induced by rSARS-CoV-MA15-ΔE in BALB/c mice.
We previously showed that mouse-adapted rSARS-CoV-MA15-ΔE is attenuated in young (6 weeks old) and aged (12 and 18 months old) BALB/c mice, leading to no significant weight losses and no mortality (24). To identify mechanisms leading to rSARS-CoV-MA15-ΔE attenuation, pulmonary pathology associated with rSARS-CoV-MA15 and rSARS-CoV-MA15-ΔE infection was evaluated in 8-week-old mice intranasally infected with 6,000 PFU, the dose used for immunization in our previous studies (24). Infection of BALB/c mice with rSARS-CoV-MA15 caused intense lung inflammation, characterized by peribronchiolar and interstitial inflammatory infiltrates at 4 and 6 dpi (Fig. 1). In contrast, lung infiltrates were reduced in mice infected with rSARS-CoV-MA15-ΔE (Fig. 1), suggesting that the presence of E protein, either by a direct or an indirect effect, contributed to induce a robust inflammatory immune response. Similar results in terms of weight loss, virus titer, and pathology were obtained with 8- and 16-week-old mice infected with 6,000 and 100,000 PFU, respectively. The dose of 100,000 PFU of rSARS-CoV-MA15 was lethal in 16-week-old mice and was used in most of the experiments described below. Quantification of total lung leukocytes, macrophages, and neutrophils indicated that the total number of leukocytes and neutrophils was reduced in mice infected with either dose of rSARS-CoV-MA15-ΔE at 2, 4, and 6 dpi (Fig. 2), whereas the total number and percentage of macrophages were increased compared to amounts in mice infected with the corresponding dose of rSARS-CoV-MA15 (Fig. 2, shown for 6,000 PFU). Furthermore, the percentage of lung neutrophils was also decreased in rSARS-CoV-MA15-ΔE-infected mice. These results indicated that the presence of E protein led to a specific increase in neutrophil influx.
FIG 1.

Lung pathology associated with rSARS-CoV-MA15-ΔE infection in BALB/c mice. Eight-week-old BALB/c mice were infected with 6,000 PFU of rSARS-CoV-MA15-ΔE (ΔE) or wild-type (wt) rSARS-CoV-MA15. After 4 (A) and 6 (B) dpi, mice were sacrificed, and lungs were processed for histopathological examination. Original magnifications were ×4 and ×40 in upper and lower images, respectively, of each panel.
FIG 2.

Analysis of lung inflammatory cell infiltrates during rSARS-CoV-MA15-ΔE infection of BALB/c mice. Leukocytes from lungs of 8-week-old BALB/c mice infected with 6,000 PFU of rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15 were isolated at 2, 4, and 6 dpi, and the inflammatory cell populations were identified by flow cytometry as described in Materials and Methods. The total numbers of leukocytes, macrophages and neutrophils and the percentage of macrophages and neutrophils were determined. Error bars represent the means from six mice per group.
A property of the E protein is that it is involved in virus assembly, and we previously showed that rSARS-CoV-ΔE grew to lower titers in tissue culture cells than rSARS-CoV, reflecting this role. To determine whether rSARS-CoV-MA15-ΔE grew to lower titers in mice, 16-week-old BALB/c mice were infected with 100,000 PFU of rSARS-CoV-MA15-ΔE and rSARS-CoV-MA15, and virus titers in lungs were determined at 2 and 4 dpi. Titers were 1 × 106 PFU/g of tissue and 4 × 107 PFU/g at day 2 and 1 × 106 PFU/g and 3 × 107 PFU/g at day 4 for rSARS-CoV-MA15-ΔE and rSARS-CoV-MA15, respectively, confirming that rSARS-CoV-MA15-ΔE replicated less robustly in mice.
Proinflammatory response induced by rSARS-CoV-MA15-ΔE in BALB/c mice.
To determine whether the limited inflammation induced by rSARS-CoV-MA15-ΔE was associated with a reduction in proinflammatory cytokines and with a decreased clinical disease and mortality, mice were infected with rSARS-CoV-MA15 or rSARS-CoV-MA15-ΔE. Weight loss and survival were evaluated daily (Fig. 3A). As expected, whereas mice infected with rSARS-CoV-MA15-ΔE did not significantly lose weight and all of them survived, mice infected with wild-type rSARS-CoV-MA15 started to lose weight at day 2 p.i., and 80% of the mice died by day 6 (Fig. 3A). RNA and protein were extracted from lung samples at days 2 and 4 p.i. to measure proinflammatory cytokine mRNA levels. In rSARS-CoV-MA15-infected mice, the expression of tumor necrosis factor (TNF), CCL2/MCP-1, CCL5/RANTES, CXCL1/neutrophil activating protein-3 (NAP-3), CXCL2/MIP-2, CXCL10/IP-10, and interleukin-6 (IL-6) was clearly induced, particularly at 2 dpi, to higher levels than in mice infected with rSARS-CoV-MA15-ΔE (Fig. 3B). In fact, the induction of proinflammatory cytokines was up to 70-fold higher in mice infected with rSARS-CoV-MA15 than in those infected with the deletion mutant lacking E protein (Fig. 3B). As a control, expression levels of the gene encoding the 18S rRNA did not change in mock-, rSARS-CoV-MA15-ΔE-, and rSARS-CoV-MA15-infected mice (Fig. 3B). To evaluate whether the differential induction of cytokine mRNAs in rSARS-CoV-MA15-ΔE- and rSARS-CoV-MA15-infected mice correlated with protein levels, CCL2 and CXCL10 protein levels were determined in the lungs of control and infected mice. In agreement with the results shown in Fig. 3, CCL2 and CXCL10 protein levels were also increased in rSARS-CoV-MA15-infected mice compared to those in mice infected with rSARS-CoV-MA15-ΔE (Fig. 4).
FIG 3.

Effect of SARS-CoV E protein on clinical disease and proinflammatory response in vivo. Sixteen-week-old BALB/c mice were infected with 100,000 PFU of rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15. (A) Weight loss and survival were evaluated daily in mice infected with rSARS-CoV-MA15-ΔE (ΔE) or rSARS-CoV-MA15 (wt) (10 mice per group). (B) Lung RNAs were extracted at 2 and 4 dpi, and the expression levels of cellular mRNAs corresponding to proinflammatory cytokines (CCL2, CCL5, TNF, CXCL1, CXCL2, CXCL10, and IL-6) and 18S rRNA were measured by RT-qPCR. Numbers indicate the levels of gene expression in rSARS-CoV-MA15-ΔE- or rSARS-CoV-MA15-infected mice compared to mock-infected mice. Error bars represent the means of three mice analyzed for each point.
FIG 4.
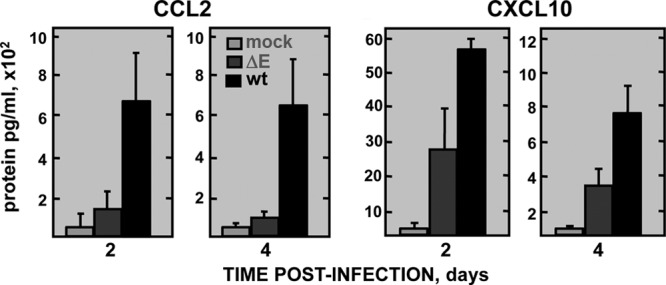
Expression of proinflammatory cytokines in rSARS-CoV-MA15-ΔE-infected mice. Sixteen-week-old BALB/c mice were infected with 100,000 PFU of rSARS-CoV-MA15-ΔE (ΔE) or rSARS-CoV-MA15 (wt) or were mock infected (mock). Lung proteins were extracted at 2 and 4 dpi, and the accumulation of CCL2 and CXCL10 was measured as described in Materials and Methods. Concentrations of protein are expressed as picograms per milliliter of lung tissue extract. Error bars represent the means of three mice analyzed for each point.
Proinflammatory cytokines induced by rSARS-CoV-MA15-ΔE in cell culture.
To analyze whether rSARS-CoV-MA15-ΔE also induced a decreased proinflammatory response in cultured cells, mouse DBT-mACE2 cells (43) were infected with rSARS-CoV-MA15-ΔE and rSARS-CoV-MA15 at MOIs of 0.5 and 0.05. The expression of the proinflammatory cytokines CCL2, CXCL1, and CXCL2 was evaluated by RT-qPCR at 24, 48, and 72 hpi (Fig. 5A). rSARS-CoV-MA15 virus induced the expression of higher levels of proinflammatory cytokines, particularly at 48 hpi (MOI of 0.5) and 48 to 72 hpi (MOI of 0.05) than the E deletion mutant. Therefore, similarly to the expression in mice lungs, rSARS-CoV-MA15-ΔE also induced a decreased proinflammatory response compared to that of rSARS-CoV-MA15 in infected cells (Fig. 5A). To analyze whether rSARS-CoV-MA15-ΔE growth in mouse DBT-mACE2 cells was compromised, virus titers in rSARS-CoV-MA15-ΔE- and wild-type rSARS-CoV-MA15-infected cell supernatants were compared (Fig. 5B). rSARS-CoV-MA15-ΔE grew to lower titers (from 15- to 600-fold lower than those observed for wild-type rSARS-CoV-MA15 at 24, 48, and 72 hpi), as happens in mice (Fig. 5B).
FIG 5.

Expression kinetics of proinflammatory cytokines in rSARS-CoV-MA15-ΔE-infected cells. DBT-mACE2 cells were infected at MOIs of 0.5 and 0.05 with rSARS-CoV-MA15-ΔE (ΔE) and rSARS-CoV-MA15 (wt). (A) Cellular RNAs were extracted at 24, 48, and 72 hpi. The expression of the indicated cytokines was determined by RT-qPCR. In each case, the corresponding mRNA expression levels in rSARS-CoV-MA15-ΔE- or rSARS-CoV-MA15-infected cells were plotted as fold change relative to expression levels in uninfected cells. (B) Virus titers in supernatants of rSARS-CoV-MA15-ΔE-infected and rSARS-CoV-MA15-infected DBT-mACE2 cells. Error bars represent standard deviations of the means from three experiments.
rSARS-CoV-MA15 induced the activation of proinflammatory transcription factors.
The most important signal transduction pathways activated by viruses leading to the expression of proinflammatory cytokines are mediated by transcription factors IRF3, IRF7, CRE, AP-1, NF-κB, and NF-AT. To analyze the mechanism by which rSARS-CoV-MA15-ΔE induced a limited proinflammatory response, DBT-mACE2 cells (43) were cotransfected with plasmids expressing the firefly luciferase under the control of each of these transcription factors and with a plasmid expressing the Renilla luciferase. Cells were then mock infected or infected with rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15. Firefly luciferase levels were measured at 48 h postinfection (Fig. 6A) and normalized to those of Renilla luciferase. Transcription factors IRF3 and NF-κB were activated in rSARS-CoV-MA15-infected cells compared to levels in mock-infected cells. Interestingly, the activation of NF-κB was significantly higher (3-fold) in rSARS-CoV-MA15-infected cells than in rSARS-CoV-MA15-ΔE-infected cells, whereas IRF3 was not differentially activated (Fig. 6A). We confirmed this finding by measuring nuclear levels of NF-κB using an ELISA. The levels of p65 were measured as p50-p65 heterodimers, which are in general the most abundant forms of NF-κB (47). NF-κB was activated to higher levels in rSARS-CoV-MA15-infected cells than in rSARS-CoV-MA15-ΔE-infected cells (Fig. 6B). Virus titers at the time point the experiment was analyzed were 6 × 105 PFU/ml and 4 × 107 PFU/ml for rSARS-CoV-MA15-ΔE and wild-type rSARS-CoV-MA15 virus in cell culture supernatants, respectively. These data suggested that transcription factors IRF3 and NF-κB were responsible for the greater inflammation observed in infected cells and mice, with NF-κB being more important in the increased inflammation observed after rSARS-CoV-MA15 infection.
FIG 6.

Activation of proinflammatory pathways in rSARS-CoV-MA15-ΔE-infected cells. (A) DBT-mACE2 cells were transfected with plasmids expressing firefly and Renilla luciferase, as described in Materials and Methods. Cells were mock infected (mock) or infected with rSARS-CoV-MA15-ΔE (ΔE) or rSARS-CoV-MA15 (wt) at an MOI of 0.05 at 24 h posttransfection. Renilla and firefly luciferase expression levels were quantified 48 h later, and the expression of firefly luciferase was normalized to that of Renilla luciferase. The level of firefly luciferase expression in cells transfected with a plasmid expressing the firefly luciferase under the control of a basal, noninducible promoter (Pr−) was also determined. (B) Cells were infected with rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15 at an MOI of 0.05. At 48 hpi, nuclear extracts were prepared, and the presence of NF-κB was quantified by ELISA as described in Materials and Methods. Error bars represent the means of three independent experiments.
NF-κB activation in the lungs of rSARS-CoV-infected mice.
To determine whether NF-κB was also differentially activated in the lungs of rSARS-CoV-MA15-ΔE- and rSARS-CoV-MA15-infected mice, 16-week-old mice were infected with these viruses using 100,000 PFU and analyzed for NF-κB expression by Western blotting using a p65-specific antibody, as described in Materials and Methods. Levels of NF-κB increased about 4-fold in rSARS-CoV-MA15-infected lungs compared to mock-infected ones at day 2 p.i. (Fig. 7), with levels up to 3.1-fold higher in rSARS-CoV-MA15-infected lungs than in rSARS-CoV-MA15-ΔE-infected lungs (Fig. 7), consistent with the results observed in infected cells.
FIG 7.

Activation of NF-κB in rSARS-CoV-MA15-ΔE-infected mice. BALB/c mice were infected with 100,000 PFU of rSARS-CoV-MA15-ΔE or rSARS-CoV-MA15. Nuclear lung proteins were extracted at 2 dpi. (A) The accumulation of NF-κB and histone H3 in the nucleus of three infected mice per condition was evaluated by Western blotting. (B) NF-κB and histone H3 amounts were quantified by densitometric analysis. The graph shows the NF-κB/histone H3 ratio in mock-, rSARS-CoV-MA15-ΔE (ΔE)-, and rSARS-CoV-MA15 (wt)-infected mice at 2 dpi. Error bars represent the means of three mice analyzed for each time point.
Effect of NF-κB on rSARS-CoV-induced inflammation.
The results described above suggest that NF-κB is key in the differential development of inflammation in cells and in mice infected with rSARS-CoV-MA15 and rSARS-CoV-MA15-ΔE. However, virus replication was diminished by the absence of E protein in both cells and mice, possibly confounding interpretation. To address directly the role of NF-κB in inflammation, we treated rSARS-CoV-MA15-infected cells with four NF-κB inhibitors, CAPE, resveratrol, Bay11-7082, and parthenolide (48–51). Four inhibitors were used to minimize the likelihood of off-target effects. NF-κB activity decreased in both mock- and rSARS-CoV-MA15-infected cells treated with the four NF-κB inhibitors compared to activity in nontreated cells (Fig. 8A). Interestingly, none of these inhibitors affected virus titers (Fig. 8B), indicating that the treatment with the inhibitors did not compromise the cell viability and that the effect observed was due to a decrease in NF-κB activation and was not simply due to a significant inhibition of viral replication by the NF-κB inhibitors. The expression levels of TNF, CCL2, and CXCL2, which were upregulated during SARS-CoV infection and which are induced by NF-κB (52), were clearly decreased (from 2- to 5-fold) in rSARS-CoV-MA15-infected cells treated with the NF-κB inhibitors (Fig. 8C). These data indicated that NF-κB activation induces the expression of inflammatory cytokines after SARS-CoV infection.
FIG 8.

Effect of NF-κB on proinflammatory cytokine induction after rSARS-CoV-MA15 infection. DBT-mACE2 cells were infected at an MOI of 0.05 with rSARS-CoV-MA15 for 48 h. Mock-infected (mock) or rSARS-CoV-MA15-infected (wt) cells were treated at 43 hpi with the NF-κB inhibitors CAPE, resveratrol, Bay 11–7082, and parthenolide or left untreated (white bars). (A) NF-κB activation was analyzed in cells transfected with luciferase plasmids as described in the legend of Fig. 6. (B) Virus titers in cell culture supernatants were determined by plaque assay in Vero E6 cells. (C) Expression of TNF, CCL2, and CXCL2 and that of 18S rRNA as a control was evaluated by RT-qPCR. The expression levels of the different genes were normalized to the expression of nontreated, mock-infected cells. Error bars represent the means of three independent experiments.
Effect of NF-κB inhibitors on the survival of rSARS-CoV-MA15-infected mice.
To determine whether NF-κB inhibitors increased survival after SARS-CoV infection, rSARS-CoV-MA15- and mock-infected mice were treated intraperitoneally with CAPE, parthenolide, or both. All mock-infected BALB/c mice treated with inhibitors showed no signs of clinical disease or weight loss and no mortality (data not shown). Of rSARS-CoV-MA15-infected mice, 16.7% survived, whereas 44.4 and 33.3% of CAPE- and parthenolide-treated mice survived, respectively (Fig. 9). Interestingly, in the case of mice simultaneously administered both NF-κB inhibitors, survival increased to 55.6%, suggesting complementary effects of the drugs.
FIG 9.

Effect of NF-κB inhibitors in rSARS-CoV-MA15-infected mice. Sixteen-week-old mice were intranasally infected with 100,000 PFU of rSARS-CoV-MA15 or mock infected. At 4 hpi and every 24 h from days 1 to 4, mock-infected and wild-type (wt)-infected mice were intraperitoneally injected with CAPE (wt, +C), parthenolide (wt, +P), both CAPE and parthenolide (wt, +C +P), or vehicle (wt, −). Animals were monitored daily for mortality. Data are representative of three independent experiments, comprising 6 mice per experiment and group (data for treated mock-infected mice are not shown). Differences in survival between nontreated mice and mice treated with CAPE, parthenolide, and CAPE plus parthenolide were statistically significant (P < 0.02).
Effect of NF-κB inhibitors on rSARS-CoV-induced inflammation in mice.
To analyze whether treatment with NF-κB inhibitors reduced the expression of proinflammatory cytokines in mouse lungs, as was observed in tissue culture cells, total lung RNA from nontreated or treated mice was extracted, and expression of TNF, CCL2, CXCL2, and a control RNA (18S rRNA) was evaluated by RT-qPCR. Levels of the three cytokine mRNAs were significantly reduced (from 2.2- to 4.5-fold) in all groups of mice treated with the different drugs compared to levels in nontreated mice. As a control, the expression of the 18S rRNA did not change in nontreated and drug-treated mice (Fig. 10A). Virus titers in lung homogenates were similar in both untreated and treated animals (Fig. 10B), suggesting that the reduction of proinflammatory cytokines after treatment with NF-κB inhibitors was not a consequence of reduced virus replication. To evaluate whether NF-κB inhibitors reduced lung pathology induced after SARS-CoV infection, lung sections were analyzed (Fig. 11). In nontreated infected mice, inflammatory infiltrates and lung edema were clearly observed. Interestingly, pulmonary pathology was clearly lower in infected mice treated with each of the NF-κB inhibitors. Furthermore, higher reduction of pathology was observed in the mice treated with both inhibitors at the same time (Fig. 11). These results indicated that NF-κB inhibitors also reduced the expression of proinflammatory cytokines and lung pathology in vivo, suggesting that they may be useful for SARS therapy.
FIG 10.

Effect of NF-κB inhibitors on proinflammatory cytokine induction after rSARS-CoV-MA15 infection in vivo. Mice were infected and treated as described in the legend of Fig. 9. (A) Four days after infection, total RNA from lungs was extracted, and the expression of TNF, CCL2, CXCL2, and 18S rRNA was quantified. The expression levels of the different genes were normalized to levels in nontreated, mock-infected mice. Error bars represent the means of three independent mice per group. (B) Four days after infection, virus titers in lung homogenates were determined. Error bars represent the means of three independent mice per group. Black bars, untreated.
FIG 11.

Lung pathology induced after rSARS-CoV-MA15 infection in the presence of NF-κB inhibitors. Mice were mock infected or infected and treated as indicated in the legend of Fig. 9. Four days after infection, lung sections from mock-infected mice (A) and from rSARS-CoV-MA15-infected mice, which were left untreated (B) or treated with CAPE (C), parthenolide (D), or both (E), were evaluated. Three independent mice per group were analyzed. Representative images are shown.
DISCUSSION
SARS-CoV lacking E gene is attenuated (21, 22, 24). Here, we showed that rSARS-CoV-MA15-ΔE induced less inflammation in lungs than rSARS-CoV-MA15. This decrease in inflammation correlated with decreased expression of proinflammatory mediators, such as cytokines and chemokines, with lower numbers of neutrophils in infected lungs and with a reduction in NF-κB activation. Some of the decreased inflammation observed in rSARS-CoV-MA15-ΔE-infected mice may have resulted from a reduction of virus titers after mouse infection by rSARS-CoV-MA15-ΔE compared to rSARS-CoV-MA15.
At the same time, an alternative explanation has been proposed as complementary results also suggested that E protein contributed to the extent of the inflammatory response, independently of its role in virus replication. For instance, rSARS-CoV-ΔE grew to lower titers than rSARS-CoV, but, in spite of this, it induced an increased stress response and apoptosis (25). In addition, we have shown that virus titers in the lungs of mice infected with recombinant SARS-CoVs with point mutations or small deletions in the E protein were the same as in mice infected with rSARS-CoV-MA15. However, these viruses induced limited pathological changes and were attenuated (J. L. Nieto-Torres, J. M. Jimenez-Guardeño, J. A. Regla-Nava, M. L. DeDiego, and L. Enjuanes, unpublished results). Furthermore, SARS-CoV E protein is a viroporin with ion channel activity (15, 53), and we showed that SARS-CoV in which this activity was abrogated lost its virulence without reductions in virus titer, again indicating that an intrinsic property of E protein is responsible for virus virulence and that changes in virus titer were not necessary for increased attenuation (54). Similar results have been reported for other respiratory viruses expressing active viroporins, such as M2 and PB1-F2 of influenza virus, SH of Rous sarcoma virus (RSV), and 2B of rhinovirus. Inflammasome activation was observed in cells infected by these viruses when the viroporin displayed ion channel activity but not in its absence (55–57). Also, expression of E protein from porcine reproductive and respiratory syndrome virus (PRRSV), in the absence of virus infection, altered ion concentrations within the cell, leading to inflammasome activation, indicating that the viroporin itself was responsible for the activation (58). More importantly, when mice were infected with influenza viruses in which the ion channel activity of PB1-F2 protein was intact or abrogated, virus titers were similar, and a virulent or attenuated phenotype was observed, respectively (59). Overall, the data indicate that the intrinsic activities of these viroporins may also be responsible for virus virulence.
After infection of mice with rSARS-CoV-MA15, increases in the expression of the inflammatory cytokine TNF, of chemokines CCL2, CCL5, CXCL1, CXCL2, and CXCL10, and of IL-6 and in the migration of neutrophils to infected lungs were observed compared to levels in rSARS-CoV-MA15-ΔE-infected mice. In the lungs of human patients with fatal SARS, elevated levels of IL-6 and of chemokines such as CCL2 and CXCL10 were also detected (60–62). Similarly, during influenza virus and RSV infections, increased numbers of neutrophils in the lungs contributed to worse outcomes (63–67). Neutrophil-mediated inflammation also contributes to bronchiolitis after RSV infection, and antioxidant treatment, which reduced neutrophil influx to the lungs and proinflammatory cytokine expression, also reduced clinical illness in RSV-infected mice (68). The increase in neutrophil influx in rSARS-CoV-MA15-infected mice compared to that in mice infected with rSARS-CoV-MA15-ΔE was not associated with elevated numbers of macrophages, suggesting that the neutrophil increase was cell type specific.
NF-κB was differentially activated following rSARS-CoV-MA15-ΔE and rSARS-CoV-MA15 infection in cell cultures and in vivo. This transcription factor regulated the expression of proinflammatory mediators such as TNF, CCL2, and CXCL2 after SARS-CoV infection. Administration of two inhibitors of NF-κB, CAPE and parthenolide, increased mouse survival after infection and was associated with reduced expression of proinflammatory cytokines in the lungs. These drugs showed complementary effects as treatment with both of them increased survival to a higher extent than treatment with either one alone. Therefore, NF-κB inhibitors may be useful antivirals in SARS therapy. Similarly, NF-κB inhibitors may be effective as antivirals against influenza virus (69–71) and RSV (72).
NF-κB is activated following signaling events leading to activation of mitogen-activated protein kinase kinase kinases (MAP3Ks), which activate IKKα and -β, leading to IκB phosphorylation. Phosphorylated IκB is degraded through the ubiquitin-dependent 26S proteasome pathway. Degradation of IκB unmasks the nuclear NF-κB-mediated transcription. In addition to the inhibitors used in this study, other inhibitors of the signaling pathway leading to NF-κB activation could be considered to treat SARS patients. For example, inhibitors that (i) block the binding of cytokines to their receptors, such as anti-TNF antibodies, (ii) inhibit IKK activation and function, (iii) interfere with IκB degradation, such as ubiquitination and proteasome inhibitors, and (iv) impede nuclear translocation, DNA binding, and transcriptional activation of NF-κB (reviewed in references 35 and 36) could be assessed. In addition to NF-κB, IRF3 was also activated in SARS-CoV-infected cells and most probably in vivo. As this transcription factor is also implicated in the upregulation of proinflammatory cytokines after infection (73), IRF3 inhibitors could also be evaluated in SARS-CoV-infected mice. One advantage of antivirals that target cellular protein activity rather than that of viral proteins is that their effect is not likely to be negated by mutations in the virus genome.
In summary, we identify promising antivirals for SARS-CoV therapy by applying basic knowledge of the signaling pathways altered during infection by this virus. This strategy complements the development of candidate vaccines previously reported by our group (24, 74, 75). The next step will be to confirm the efficacy and absence of side effects in other animal models, such as macaques, which are closer to humans.
ACKNOWLEDGMENTS
This work was supported by grants from the Ministry of Science and Innovation of Spain (BIO2010-16705), the Seventh Framework Programme (FP7/2007-2013) of the European Commission (EC) under the project EMPERIE (EC grant agreement number 223498), and the U.S. National Institutes of Health (2P01AI060699-06A1 and CRIP-HHSN266200700010C). M.L.D. received a contract from the project EMPERIE (EC grant agreement number 223498), and J.A.R.-N. and C.C.-R. received a contract from Fundación La Caixa.
We thank Marga Gonzalez for technical assistance.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Peiris JS, Guan Y, Yuen KY. 2004. Severe acute respiratory syndrome. Nat. Med. 10:S88–S97. 10.1038/nm1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peiris JS, Yuen KY, Osterhaus AD, Stohr K. 2003. The severe acute respiratory syndrome. N. Engl. J. Med. 349:2431–2441. 10.1056/NEJMra032498 [DOI] [PubMed] [Google Scholar]
- 3.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045. 10.1073/pnas.0506735102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. 10.1126/science.1118391 [DOI] [PubMed] [Google Scholar]
- 5.Lau SK, Li KS, Huang Y, Shek CT, Tse H, Wang M, Choi GK, Xu H, Lam CS, Guo R, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Ecoepidemiology and complete genome comparison of SARS-related Rhinolophus bat coronavirus in China reveal bats as reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84:2808–2819. 10.1128/JVI.02219-02209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hon CC, Lam TY, Shi ZL, Drummond AJ, Yip CW, Zeng F, Lam PY, Leung FC. 2008. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82:1819–1826. 10.1128/JVI.01926-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 8.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. 10.1128/mBio.00473-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, Muth D, Raj VS, Vries LS, Corman VM, Drexler JF, Smits SL, El Tahir YE, De Sousa R, van Beek J, Nowotny N, van Maanen K, Hidalgo-Hermoso E, Bosch BJ, Rottier P, Osterhaus A, Gortazar-Schmidt C, Drosten C, Koopmans MP. 2013. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 13:859–866. 10.1016/S1473-3099(1013)70164-70166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rota PA, Oberste MS, Monroe SS, Nix WA, Campganoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen M-H, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rassmussen M, Fouchier R, Gunther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. 10.1126/science.1085952 [DOI] [PubMed] [Google Scholar]
- 11.Ziebuhr J. 2005. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 287:57–94. 10.1007/3-540-26765-4_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enjuanes L, Gorbalenya AE, de Groot RJ, Cowley JA, Ziebuhr J, Snijder EJ. 2008. The Nidovirales, p 419–430 In Mahy BWJ, Van Regenmortel M, Walker P, Majumder-Russell D. (ed), Encyclopedia of virology, 3rd ed. Elsevier Ltd., Oxford, United Kingdom [Google Scholar]
- 13.Gorbalenya AE, Snijder EJ, Spaan WJ. 2004. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J. Virol. 78:7863–7866. 10.1128/JVI.78.15.7863-7866.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres J, Parthasarathy K, Lin X, Saravanan R, Liu DX. 2006. Model of a putative pore: the pentameric alpha-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys. J. 91:938–947. 10.1529/biophysj.105.080119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, Dediego ML, Torres J, Aguilella VM, Enjuanes L. 2012. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 432:485–494. 10.1016/j.virol.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, Dediego ML, Enjuanes L, Aguilella VM. 2013. Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. Biochim. Biophys. Acta 1828:2026–2031. 10.1016/j.bbamem.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtis KM, Yount B, Baric RS. 2002. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 76:1422–1434. 10.1128/JVI.76.3.1422-1434.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ortego J, Ceriani JE, Patino C, Plana J, Enjuanes L. 2007. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 368:296–308. 10.1016/j.virol.2007.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortego J, Escors D, Laude H, Enjuanes L. 2002. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 76:11518–11529. 10.1128/JVI.76.22.11518-11529.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almazan F, DeDiego ML, Sola I, Zuñiga S, Nieto-Torres JL, Marquez-Jurado S, Andres G, Enjuanes L. 2013. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio 4(5):e00650–13. 10.1128/mBio.00650-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeDiego ML, Alvarez E, Almazan F, Rejas MT, Lamirande E, Roberts A, Shieh WJ, Zaki SR, Subbarao K, Enjuanes L. 2007. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 81:1701–1713. 10.1128/JVI.01467-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeDiego ML, Pewe L, Alvarez E, Rejas MT, Perlman S, Enjuanes L. 2008. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology 376:379–389. 10.1016/j.virol.2008.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo L, Masters PS. 2003. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 77:4597–4608. 10.1128/JVI.77.8.4597-4608.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fett C, DeDiego ML, Regla-Nava JA, Enjuanes L, Perlman S. 2013. Complete protection against severe acute respiratory syndrome coronavirus-mediated lethal respiratory disease in aged mice by immunization with a mouse-adapted virus lacking E protein. J. Virol. 87:6551–6559. 10.1128/JVI.00087-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, Regla-Nava JA, Alvarez E, Oliveros JC, Zhao J, Fett C, Perlman S, Enjuanes L. 2011. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS pathog. 7:e1002315. 10.1371/journal.ppat.1002315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt G, Ahuja A, Yung MY, Leung CB, To KF, Lui SF, Szeto CC, Chung S, Sung JJY. 2003. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348:1986–1994. 10.1056/NEJMoa030685 [DOI] [PubMed] [Google Scholar]
- 27.Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, Chan PK, Ng MH, Yu LM, Hui DS, Tam JS, Cheng G, Sung JJ. 2003. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326:1358–1362. 10.1136/bmj.326.7403.1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagata N, Iwata-Yoshikawa N, Taguchi F. 2010. Studies of severe acute respiratory syndrome coronavirus pathology in human cases and animal models. Vet. Pathol. 47:881–892. 10.1177/0300985810378760 [DOI] [PubMed] [Google Scholar]
- 29.Leung CW, Kwan YW, Ko PW, Chiu SS, Loung PY, Fong NC, Lee LP, Hui YW, Law HK, Wong WH, Chan KH, Peiris JS, Lim WW, Lau YL, Chiu MC. 2004. Severe acute respiratory syndrome among children. Pediatrics 113:e535–543. 10.1542/peds.113.6.e535 [DOI] [PubMed] [Google Scholar]
- 30.Manocha S, Walley KR, Russell JA. 2003. Severe acute respiratory distress syndrome (SARS): a critical care perspective. Crit. Care Med. 31:2684–2692. 10.1097/01.CCM.0000091929.51288.5F [DOI] [PubMed] [Google Scholar]
- 31.Day CW, Baric R, Cai SX, Frieman M, Kumaki Y, Morrey JD, Smee DF, Barnard DL. 2009. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology 395:210–222. 10.1016/j.virol.2009.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3:e5. 10.1371/journal.ppat.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagata N, Iwata N, Hasegawa H, Fukushi S, Harashima A, Sato Y, Saijo M, Taguchi F, Morikawa S, Sata T. 2008. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am. J. Pathol. 172:1625–1637. 10.2353/ajpath.2008.071060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smits SL, de Lang A, van den Brand JM, Leijten LM, van IJcken WF, Eijkemans MJ, van Amerongen G, Kuiken T, Andeweg AC, Osterhaus AD, Haagmans BL. 2010. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 6:e1000756. 10.1371/journal.ppat.1000756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitiello M, Galdiero M, Finamore E, Galdiero S, Galdiero M. 2012. NF-kappaB as a potential therapeutic target in microbial diseases. Mol. Biosyst. 8:1108–1120. 10.1039/c2mb05335g [DOI] [PubMed] [Google Scholar]
- 36.Gilmore TD, Herscovitch M. 2006. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 25:6887–6899. 10.1038/sj.onc.1209982 [DOI] [PubMed] [Google Scholar]
- 37.Law AH, Lee DC, Cheung BK, Yim HC, Lau AS. 2007. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 81:416–422. 10.1128/JVI.02336-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanzawa N, Nishigaki K, Hayashi T, Ishii Y, Furukawa S, Niiro A, Yasui F, Kohara M, Morita K, Matsushima K, Le MQ, Masuda T, Kannagi M. 2006. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett. 580:6807–6812. 10.1016/j.febslet.2006.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao QJ, Ye LB, Timani KA, Zeng YC, She YL, Ye L, Wu ZH. 2005. Activation of NF-κB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim. Biophys. Sin. 37:607–612. 10.1111/j.1745-7270.2005.00082.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Wu K, Wang D, Yue X, Song D, Zhu Y, Wu J. 2007. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-κB. Virology 365:324–335. 10.1016/j.virol.2007.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Ye L, Ye L, Li B, Gao B, Zeng Y, Kong L, Fang X, Zheng H, Wu Z, She Y. 2007. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 128:1–8. 10.1016/j.virusres.2007.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dosch SF, Mahajan SD, Collins AR. 2009. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 142:19–27. 10.1016/j.virusres.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Regla-Nava JA, Jimenez-Guardeno JM, Nieto-Torres JL, Gallagher TM, Enjuanes L, DeDiego ML. 2013. The replication of a mouse adapted SARS-CoV in a mouse cell line stably expressing the murine SARS-CoV receptor mACE2 efficiently induces the expression of proinflammatory cytokines. J. Virol. Methods 193:639–646. 10.1016/j.jviromet.2013.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCray PB, Jr, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S. 2007. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 81:813–821. 10.1128/JVI.02012-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, Van Rooijen N, Perlman S. 2009. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 5:e1000636. 10.1371/journal.ppat.1000636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17:1087–1095. 10.1093/emboj/17.4.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. 2000. Mechanism of κB DNA binding by Rel/NF-κB dimers. J. Biol. Chem. 275:24392–24399. 10.1074/jbc.M003784200 [DOI] [PubMed] [Google Scholar]
- 48.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc. Natl. Acad. Sci. U. S. A. 93:9090–9095. 10.1073/pnas.93.17.9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hehner SP, Hofmann TG, Droge W, Schmitz ML. 1999. The anti-inflammatory sesquiterpene lactone parthenolide inhibits NF-κB by targeting the IκB kinase complex. J. Immunol. 163:5617–5623 [PubMed] [Google Scholar]
- 50.Holmes-McNary M, Baldwin AS., Jr 2000. Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IκB kinase. Cancer Res. 60:3477–3483 [PubMed] [Google Scholar]
- 51.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. 1997. Novel inhibitors of cytokine-induced IκBαphosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272:21096–21103. 10.1074/jbc.272.34.21096 [DOI] [PubMed] [Google Scholar]
- 52.Ali S, Mann DA. 2004. Signal transduction via the NF-κB pathway: a targeted treatment modality for infection, inflammation and repair. Cell Biochem. Funct. 22:67–79. 10.1002/cbf.1082 [DOI] [PubMed] [Google Scholar]
- 53.Torres J, Maheswari U, Parthasarathy K, Ng L, Liu DX, Gong X. 2007. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 16:2065–2071. 10.1110/ps.062730007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Alcaraz A, Aguilella VM, Enjuanes L. 2013. Relevance of SARS-CoV E protein ion channel activity in virus fitness and virulence. 32nd Annu. Meet. Amer. Soc. Virol. The Pennsylvania State University, State College, PA [Google Scholar]
- 55.Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M. 2013. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax 68:66–75. 10.1136/thoraxjnl-2012-202182 [DOI] [PubMed] [Google Scholar]
- 56.Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. 1 July 2013. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 10.1165/rcmb.2013-0032OC [DOI] [PubMed] [Google Scholar]
- 57.Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11:404–410. 10.1038/ni.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang K, Hou Q, Zhong Z, Li X, Chen H, Li W, Wen J, Wang L, Liu W, Zhong F. 2013. Porcine reproductive and respiratory syndrome virus activates inflammasomes of porcine alveolar macrophages via its small envelope protein E. Virology 442:156–162. 10.1016/j.virol.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 59.McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A. 2013. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 9:e1003392. 10.1371/journal.ppat.1003392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang NL, Chan PK, Wong CK, To KF, Wu AK, Sung YM, Hui DS, Sung JJ, Lam CW. 2005. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin. Chem. 51:2333–2340. 10.1373/clinchem.2005.054460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J, Luo W, Chen T, Qin Q, Deng P. 2005. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 171:850–857. 10.1164/rccm.200407-857OC [DOI] [PubMed] [Google Scholar]
- 62.Cameron MJ, Ran L, Xu L, Danesh A, Bermejo-Martin JF, Cameron CM, Muller MP, Gold WL, Richardson SE, Poutanen SM, Willey BM, DeVries ME, Fang Y, Seneviratne C, Bosinger SE, Persad D, Wilkinson P, Greller LD, Somogyi R, Humar A, Keshavjee S, Louie M, Loeb MB, Brunton J, McGeer AJ, Kelvin DJ. 2007. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 81:8692–8706. 10.1128/JVI.00527-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. 2012. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 8:e1002641. 10.1371/journal.ppat.1002641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. 2011. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179:199–210. 10.1016/j.ajpath.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brandes M, Klauschen F, Kuchen S, Germain RN. 2013. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154:197–212. 10.1016/j.cell.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bataki EL, Evans GS, Everard ML. 2005. Respiratory syncytial virus and neutrophil activation. Clin. Exp. Immunol. 140:470–477. 10.1111/j.1365-2249.2005.02780.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yasui K, Baba A, Iwasaki Y, Kubo T, Aoyama K, Mori T, Yamazaki T, Kobayashi N, Ishiguro A. 2005. Neutrophil-mediated inflammation in respiratory syncytial viral bronchiolitis. Pediatr. Int. 47:190–195. 10.1111/j.1442-200x.2005.02039.x [DOI] [PubMed] [Google Scholar]
- 68.Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, Casola A. 2006. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 174:1361–1369. 10.1164/rccm.200603-319OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiesener N, Zimmer C, Jarasch-Althof N, Wutzler P, Henke A. 2011. Therapy of experimental influenza virus infection with pyrrolidine dithiocarbamate. Med. Microbiol. Immunol. 200:115–126. 10.1007/s00430-010-0182-x [DOI] [PubMed] [Google Scholar]
- 70.Pinto R, Herold S, Cakarova L, Hoegner K, Lohmeyer J, Planz O, Pleschka S. 2011. Inhibition of influenza virus-induced NF-kappaB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res. 92:45–56. 10.1016/j.antiviral.2011.05.009 [DOI] [PubMed] [Google Scholar]
- 71.Palamara AT, Nencioni L, Aquilano K, De Chiara G, Hernandez L, Cozzolino F, Ciriolo MR, Garaci E. 2005. Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 191:1719–1729. 10.1086/429694 [DOI] [PubMed] [Google Scholar]
- 72.Zang N, Xie X, Deng Y, Wu S, Wang L, Peng C, Li S, Ni K, Luo Y, Liu E. 2011. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. J. Virol. 85:13061–13068. 10.1128/JVI.05869-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mogensen TH, Paludan SR. 2001. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 65:131–150. 10.1128/MMBR.65.1.131-150.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lamirande EW, DeDiego ML, Roberts A, Jackson JP, Alvarez E, Sheahan T, Shieh WJ, Zaki SR, Baric R, Enjuanes L, Subbarao K. 2008. A live attenuated SARS coronavirus is immunogenic and efficacious in golden Syrian hamsters. J. Virol. 82:7721–7724. 10.1128/JVI.00304-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Netland J, DeDiego ML, Zhao J, Fett C, Alvarez E, Nieto-Torres JL, Enjuanes L, Perlman S. 2010. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 399:120–128. 10.1016/j.virol.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]