

Abstract
A major challenge in the development of an HIV vaccine is that of contending with the extensive sequence variability found in circulating viruses. Induction of HIV-specific T-cell responses targeting conserved regions and induction of HIV-specific T-cell responses recognizing a high number of epitope variants have both been proposed as strategies to overcome this challenge. We addressed the ability of cytotoxic T lymphocytes from 30 untreated HIV-infected subjects with and without control of virus replication to recognize all clade B Gag sequence variants encoded by at least 5% of the sequences in the Los Alamos National Laboratory HIV database (1,300 peptides) using gamma interferon and interleukin-2 (IFN-γ/IL-2) FluoroSpot analysis. While targeting of conserved regions was similar in the two groups (P = 0.47), we found that subjects with control of virus replication demonstrated marginally lower recognition of Gag epitope variants than subjects with normal progression (P = 0.05). In viremic controllers and progressors, we found variant recognition to be associated with viral load (r = 0.62, P = 0.001). Interestingly, we show that increased overall sequence coverage, defined as the overall proportion of HIV database sequences targeted through the Gag-specific repertoire, is inversely associated with viral load (r = −0.38, P = 0.03). Furthermore, we found that sequence coverage, but not variant recognition, correlated with increased recognition of a panel of clade B HIV founder viruses (r = 0.50, P = 0.004). We propose sequence coverage by HIV Gag-specific immune responses as a possible correlate of protection that may contribute to control of virus replication. Additionally, sequence coverage serves as a valuable measure by which to evaluate the protective potential of future vaccination strategies.
INTRODUCTION
Observational and correlative studies strongly suggest a role for virus-specific cytotoxic T lymphocytes (CTLs) in the control of human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infection (1–7). Translating these observations into the development of a successful HIV vaccine has been incredibly challenging, in part due to the immense sequence diversity of HIV that must be covered by the induced immunity and to the difficulty in defining accurate correlates of protective immunity from disease progression. Several lines of evidence have indicated a superior role for Gag-specific CTL responses in control of viral replication (4, 5, 8). However, as individuals with progressive disease can also mount Gag-specific CTL responses, factors other than simple recognition must be involved that contribute to the in vivo effectiveness of some antiviral Gag-specific CTL responses.
One factor that has been proposed as an immune correlate of HIV control is an enhanced ability of Gag-specific CTLs to cross-react with epitope variants (9–12). In the majority of individuals, HIV infection leads to the induction of a great number of CTLs that are able to recognize and kill infected cells, but the extraordinary tolerance for sequence variability in the viral population can rapidly lead to the accumulation of escape mutations (13–15). In many cases, this immune adaptation renders the CTL responses ineffective and can lead to increasing viremia during the course of infection (16–18). The induction and maintenance of a broadly cross-reactive CTL repertoire may facilitate control of viral replication by suppressing the outgrowth of escape variants. Furthermore, broad epitope variant recognition has the potential to be beneficial in a vaccine setting, as recognition of epitope variants may afford targeting of a greater proportion of potentially infecting strains. Indeed, this rationale has already been translated into the design of innovative immunogens designed to incorporate as much sequence variability as feasible (19, 20). One approach currently moving into human phase I clinical trials includes polyvalent mosaic vaccines, which have been shown to improve epitope variant recognition in nonhuman primate (NHP) models compared to consensus or natural immunogens (21–23). At the same time, the opposite approach focusing variant responses on only highly conserved regions of HIV to provide recognition of more infecting strains is also being considered for HIV vaccine testing (11, 24–27).
It is important to consider that epitope variant recognition has been widely reported in the context of progressive infection (28–31). To date, variant recognition assessments have often focused on a limited number of epitopes and/or epitope variants, largely due to limited sample availability and the prohibitive cost of generating proteome-wide variant peptide sets. Most often, the effects of intraclade variation on T-cell recognition have been assessed in the setting of immune escape, and therefore the evaluation of variant-specific responses is limited to only those variants that arose in the autologous virus over time (15, 31–33). While greatly informative for our understanding of the dynamics of host-virus interactions, these types of approaches inevitably extend only incrementally our understanding of the potential for HIV-specific CTLs to recognize naturally occurring epitope variants. As a result, the relative contribution that broad epitope variant recognition may play in the control of HIV replication remains unclear.
Here we report a comprehensive evaluation of the cross-reactivity of Gag-specific T-cell responses to frequently found clade B variants in the Los Alamos National Laboratory HIV Sequence Database (HIVDB; http://www.hiv.lanl.gov/) in individuals with and without spontaneous control of viral replication. Contrary to our expectations, we observed extensive epitope variant recognition in progressors, which was associated with viral load. However, we found an inverse correlation between viral load and sequence coverage of frequently occurring variants, suggesting that it is the ability to target the most commonly occurring variants, rather than simply a large number of variants, that contributes to control of viral replication. These findings provide a greater understanding of epitope variant recognition during natural infection and offer important insight for informing the evaluation of variant-inclusive vaccines.
MATERIALS AND METHODS
Study subjects.
Thirty HIV-infected participants in the Seattle Long-Term Non-Progressor (34), Natural Progression, Primary Infection (35–37), and NIAID/NIH (38) cohorts were selected for this study. HIV controllers (n = 15) were defined by viral loads < 2,000 RNA copies/ml and CD4+ T-cell counts > 500. Elite controllers (EC) were further defined as having undetectable viral load (<50 RNA copies/ml), whereas viremic controllers (VC) had detectable viral loads of <2,000 RNA copies/ml. Progressors (n = 15) were defined as having median viral loads > 10,000 RNA copies/ml in the prior year. Three of the progressors were identified from the Seattle Primary Infection cohort and therefore had previously assigned publication identification numbers (P11 = 55097, P12 = 59530, and P15 = 75688). All subjects were studied in chronic infection and were antiretroviral therapy naive. The relevant institutional review boards approved all human subject protocols, and all subjects provided written informed consent before enrollment.
Peptide set.
A Gag peptide set of 11mer peptides overlapping by 10 amino acids (a.a.) was synthesized for use in this study (Sigma-Aldrich, St. Louis, MO) (see Table S1 in the supplemental material). All 11mers were generated to reflect any Gag variants present in at least 5% of clade B sequences (n = 1,621) in the HIVDB in 2010, resulting in a total of 1,300 peptides covering all 500 a.a. positions in Gag. Peptides were pooled based on each peptide's starting position in the alignment, resulting in a total of 489 pools.
IFN-γ/IL-2 FluoroSpot assay.
This gamma interferon and interleukin-2 (IFN-γ/IL-2) FluoroSpot assay was used for detection of Gag-specific T-cell responses (24). Cryopreserved peripheral blood mononuclear cells (PBMC) were thawed and incubated in R10 media (RPMI 1640 [GibcoBRL, Carlsbad, CA], 10% fetal bovine serum [FBS; Gemini Bioproducts, West Sacramento, CA], 2 mM l-glutamine [Gibco], 100 μg/ml streptomycin sulfate [Gibco], 100 U/ml penicillin G [Gibco]) overnight before stimulation. Between 70,000 and 100,000 PBMC/well were plated in a 96-well IPFL plate (Millipore, Bedford, MA), which had been precoated overnight with 1 μg/ml 1-D1K (Mabtech, Stockholm, Sweden) anti-IFN-γ monoclonal antibody and 1 μg/ml 2516KZ (BD Biosciences, Franklin Lakes, NJ) anti-IL-2 antibody. Peptide pools or individual 11mers were added at a final concentration of 10 μg/ml. Each plate contained cells stimulated with 1.8 μg/ml phytohemagglutinin (PHA) as a positive control and six negative-control wells of cells incubated with media alone. Plates were incubated overnight at 37°C in 5% CO2. Plates were developed using anti-IFN-γ 7-B6-1-FS-fluorescein isothiocyanate (FITC) antibody (Mabtech) diluted 1:400 and 0.1 μg/ml 2311KZ anti-IL-2 antibody (BD) for 2 h, followed by anti-FITC-green (Mabtech) diluted 1:400 and streptavidin-coupled Cy3 (Biolegend, San Diego, CA) diluted 1:1,000 for another hour. Plates were washed six times with phosphate-buffered saline (PBS) between all steps. Spots were counted using an AID iSpot FluoroSpot reader system.
All study subjects were screened for responses to the 489 peptide pools. For each positive pool, all containing 11mers (i.e., the different variants of the 11mer sequence) were tested individually in a confirmatory IFN-γ/IL-2 FluoroSpot assay. Positivity for both pooled and single-variant responses was defined as (i) >55 spot-forming cells (SFC)/million PBMC, (ii) >4 times the average of the contents of at least six negative-control wells from the same experiment, (iii) values > 3 standard deviations above the average of the negative-control well results, and (iv) the presence of at least 5 spots per well.
Gag sequencing.
A total of 10 to 25 gag sequences were obtained from plasma RNA from five VC and five progressors. For 8/10 study subjects, sequencing was performed at the same visit date used in the immunological assays. Because of plasma availability, clones from VCs C12 and C15 were sequenced from plasma at the next available visit dates (2 and 3 months later, respectively).
HIV-1 RNA was purified from plasma using a QIAamp Viral RNA Minikit (Qiagen, Valencia, CA). HIV-1-specific PCR was performed to amplify HIV-1 gag products with the use of endpoint-limiting dilution to obtain single templates. The HIV-1 gag gene (1,590-nucleotide [nt] fragment; the complete coding region of Gag and 103 nt of that of protease [PR]) was amplified by nested PCR with the following primer sets. For the first round PCR, nt 683 to 705 of HIV-1HXB2 (F683; 5′-CTC TCG ACG CAG GAC TCG GCT TG-3′) and nt 2484 to 2511 of HIV-1HXB2 (StepGR_1.0_2511; 5′-TTC CAA TTA TGT TGA CAG GTG TAG GTC C-3′) were used. For the second round, nt 762 to 786 of HIV-1HXB2 (F762; 5′-TTG ACT AGC GGA GGC TAG AAG GAG A-3′) and nt 2377 to 2403 of HIV-1HXB2 (StepGR_2.0_2403; 5′-CAA TTC CCC CTA TCA TTT TTG GTT TCC-3′) were used. The first round of PCR used Advantage 2 polymerase (Clontech, Mountain View, CA) and the following conditions: 95°C for 2 min, 35 cycles of 95°C for 30 s, 58°C for 30 s, and 68°C for 2 min, and a final extension step at 68°C for 10 min. The second round of PCR used Biolase Taq (Bioline, Taunton, MA) and the following conditions: 95°C for 2 min, 35 cycles of 95°C for 30 s, 62°C for 30 s, and 68°C for 2 min, and a final extension step at 68°C for 10 min.
PCR products were visualized using a QIAxcel DNA Fast Analysis kit (Qiagen), and PCR-positive samples were purified using a NucleoSpin Gel and PCR Clean-Up kit (Clontech) before being submitted directly for dideoxynucleotide chain termination (Sanger) sequencing. Chromatograms were assembled and edited using Geneious 5.6.5 (Biomatters Ltd., New Zealand). HIV populations from these sequenced 10 individuals were confirmed to represent clade B viruses (data not shown).
Data analysis.
The breadth of Gag-specific responses was calculated conservatively based on epitopic regions, which means that responses for up to 4 subsequent 11mer peptides (i.e., representing a potential shared 8mer) were counted as one epitopic region. Dually functional responses were determined by an IFN-γ plus IL-2 response to at least one 11mer peptide within a targeted epitopic region. The magnitude of the response to an epitopic region (in SFC/million PBMC) was defined as the highest response to a single peptide observed within the targeted epitopic region.
Variant recognition was calculated as the proportion of variants eliciting a response among all the 11mer variants tested in the targeted epitopic region, including overlapping peptide responses (see Table S2 in the supplemental material). For example, if a response is directed toward an epitopic region containing two overlapping peptides, each represented by four variants (i.e., 8 variants in total), and for both overlapping peptides two variants are recognized (i.e., a total of 4 variants recognized), variant recognition is calculated to be at 50%. Responses in which only a single sequence was tested (i.e., highly conserved, with no other variant present in ≥5% of sequences) were excluded from the variant recognition analysis as no variants were tested. Sequence coverage of frequently occurring Gag variants was calculated per epitopic region as the sum [frequency of targeted 11mers] over the sum [frequency of tested 11mers], where “frequency” refers to the proportion of the 11mer sequence in the clade B sequence alignment used to design the peptide set (see Table S2 in the supplemental material). We chose to normalize sequence coverage for each epitopic region by the sum [frequency of tested 11mers] to account for the fact that many epitopic regions had overlapping 11mers. Without normalization, the sum [frequency of targeted 11mers] would often be over 100%. In all targeted epitopic regions identified in this study, we found that the median value for the sum [frequency of tested 11mers] (i.e., the denominator in Table S2 in the supplemental material) that we used to normalize the coverage for each response was 81%. This highlights that although our peptide set does not cover all possible variations in clade B Gag sequences, the peptide design ensured that we were able to test for responses to a high proportion of circulating Gag variants.
Autologous gag sequences (whole-protein and viral portions corresponding to the targeted epitopic region) were aligned using the Muscle algorithm within Seaview v4.4.0 (39). The Le and Gascuel (LG) substitution model (40)-corrected pairwise diversity and divergence from clade B consensus were calculated using DIVEIN (41). Analyses were restricted to only those epitopic regions in which variant recognition was assessed. To generate the panel of founder viruses, we obtained previously generated sequences isolated from the first time point in early acute infection (median, 7 days post-onset of symptoms) from eight individuals (14). For each individual, a consensus sequence was generated from the first time point sequences using Seaview v4.4.0 and was used to represent the founder virus.
Statistical analysis.
For each study subject, the median variant recognition or median epitope coverage was calculated across all epitopes and used in comparisons between groups using a nonparametric Mann-Whitney test. Differences between pairwise distances between groups were assessed using a nonparametric Mann-Whitney test. Spearman rank testing was used to assess all correlations. GraphPad Prism X was used for all statistical analyses.
RESULTS
Significant differences in the quality but not the quantity of Gag-specific responses between controllers and progressors.
To assess whether broad recognition of Gag epitope variants was associated with control of viral replication, we screened for Gag-specific T-cell responses in 15 HIV controllers and 15 HIV progressors. The controller cohort was comprised of seven elite controllers (EC; viral load undetectable with a cutoff of 50 copies/ml) and eight viremic controllers (VC; viral load < 2,000 copies/ml) (Table 1). Approximately half of the controllers possessed HLA class I alleles previously described as associated with control of HIV replication (B*57 and/or B*27); however, these alleles did not segregate with EC over VC (Table 1). To reduce HLA bias, B*57-expressing progressors were included in this study (Table 1). All study subjects were antiretroviral therapy naive.
TABLE 1.
Subject characteristics
| Study subject | Statusa | CD4 countb (cells/μl) | Viral loadc (RNA copies/ml) | HLA class I designation (A1/A2/B1/B2/C1/C2)d |
|---|---|---|---|---|
| Controllers | ||||
| 1 | EC | 809 | <50 | A*03/A*31/B*27/B*57/C*02/C*12 |
| 2 | EC | 1,211 | <50 | A*02/A*03/B*13/B*18/C*06/C*07 |
| 3 | EC | 540 | <50 | A*02/A*11/B*35/B*57/C*04/C*06 |
| 4 | EC | 603 | <50 | A*02/ND/B*13/B*49/C*06/C*07 |
| 5 | EC | 643 | <50 | A*24/A*31/B*27/B*27/C*02/C*02 |
| 6 | EC | 1,389 | <50 | A*11/A*31/B*15/B*15/C*03/C*04 |
| 7 | EC | 1,916 | <50 | A*01/A*24/B*08/B*35/C*04/C*07 |
| 8 | VC | 523 | 246 | A*01/A*26/B*49/B*57/C*06/C*07 |
| 9 | VC | 784 | 125 | A*24/A*25/B*39/B*57/C*06/C*12 |
| 10 | VC | 580 | 432 | A*29/A*32/B*44/B*44/C*05/C*16 |
| 11 | VC | 747 | 250 | A*01/A*03/B*27/B*57/C*02/C*06 |
| 12 | VC | 570 | 386 | A*02/A*03/B*18/B*07/C*07/C*12 |
| 13 | VC | 702 | 163 | A*01/A*02/B*38/B*57/C*06/C*12 |
| 14 | VC | 790 | 191 | A*11/A*26/B*35/B*58/C*04/C*14 |
| 15 | VC | 1,181 | 430 | A*03/A*68/B*13/B*56/C*06/C*07 |
| Median | 747 | 125 | ||
| Progressors | ||||
| 1 | TP | 349 | 22,730 | A*01/A*01/B*08/B*08/C*07/C*07 |
| 2 | TP | 550 | 286,000 | ND |
| 3 | DP | 441 | 78,400 | A*01/A*02/B*15/B*57/C*03/C*06 |
| 4 | TP | 522 | 39,550 | A*03/A*24/B*35/B*44/C*02/C*04 |
| 5 | TP | 510 | 60,000 | A*02/A*03/B*07/B*44/C*05/C*07 |
| 6 | TP | 175 | 115,240 | A*24/A*25/B*15/B*58/C*04/C*07 |
| 7 | TP | 440 | 622,800 | A*01/A*01/B*08/B*40/C*03/C*07 |
| 8 | TP | 371 | 217,200 | A*11/A*31/B*08/B*08/C*07/C*07 |
| 9 | TP | 407 | 3,455 | A*02/A*33/B*35/B*44/C*04/C*16 |
| 10 | TP | 340 | 23,750 | ND |
| 11 | TP | 317 | 8,606 | A*23/A*30/B*08/B*42/C*07/C*17 |
| 12 | TP | 448 | 153,286 | A*01/A*01/B*08/B*08/C*07/C*07 |
| 13 | TP | 575 | 24,224 | A*03/A*30/B*35/B*57/C*04/C*07 |
| 14 | TP | 416 | 23,279 | A*68/A*74/B*14/B*57/C*07/C*08 |
| 15 | TP | 500 | 9,333 | A*02/A*32/B*08/B*55/C*03/C*07 |
| Median | 440 | 39,550 |
VC, viremic controller; EC, elite controller; TP, typical progressor; DP, delayed progressor. DP refers to an individual who previously was a controller and later progressed. At the time of study, this individual had a viral load > 10,000 copies/ml compared to that determined for the prior year.
Data represent CD4 counts within 4 weeks of sample collection.
Data represent viral loads on the day of sample collection for assays (progressors were defined as having a median viral load >10,000 in the past year).
B*27 and B*57 alleles are shown in bold. ND, not determined.
Study subjects were screened for cytokine-secreting T cells by IFN-γ/IL-2 FluoroSpot assays using a peptide set of 11mers overlapping by 10 amino acids (a.a.) spanning all 500 a.a. of Gag. The FluoroSpot assay uses unique fluorophore-conjugated antibodies to allow for simultaneous detection within the same well of IFN-γ, IL-2, and dual IFN-γ/IL-2 responses following peptide stimulation. Given the tight overlap of the peptide set, we took a conservative approach to calculating breadth in that responses of up to four sequential 11mers were counted as a response to a single epitopic region. We detected responses to a total of 347 epitopic regions. The breadths of Gag-specific responses were comparable between controllers and progressors, with controllers targeting a median of 12 epitopic regions and progressors targeting a median of 9 (P = 0.24; Fig. 1A). The peptide set used was very sensitive for the detection of HIV-specific T-cell responses, detecting more than double the number of responses previously described using other peptide sets (5, 42). This increase in sensitivity was likely due to the combination of the use of shorter peptides (11 rather than 15 a.a.) that more closely resemble the optimal HLA class I epitope and increased variant coverage relative to other peptide sets (11, 43).
FIG 1.
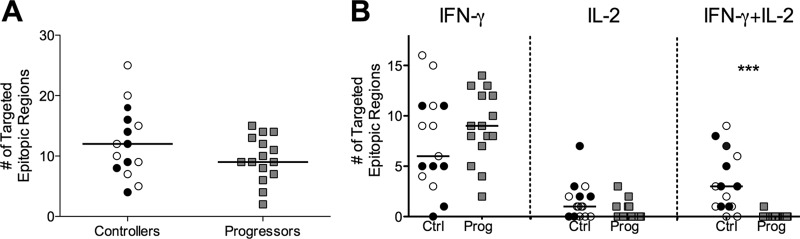
Breadth and functionality of Gag-specific T-cell responses in 15 controllers and 15 progressors. Controllers include seven elite controllers (viral load < 50 RNA copies/ml; black circles) and eight viremic controllers (viral load detectable but <2,000 RNA copies/ml; open circles). Progressors had median viral loads > 10,000 RNA copies/ml (gray squares). (A) Breadth was defined as the number of targeted epitopic regions, with responses to up to 4 consecutive 11mer peptides counted as a single epitopic region. Breadth values were calculated for all study subjects and compared between groups (P = 0.24). (B) Breadth of Gag-specific responses stratified by functionality and compared between groups for monofunctional IFN-γ (left column; P = 0.35) or IL-2 (middle column; P = 0.06) responses and dually functional IFN-γ/IL-2 responses (right column [***, P < 0.0001]). Responses were considered dually functional if any peptide in the epitopic region elicited a dual response. Ctrl, controllers; Prog, progressors.
While the magnitude of responses was variable both between and within individuals (see Fig. S1A and B in the supplemental material), there was no difference in the overall magnitude of responses between the two groups (P = 0.36; see Fig. S1C). However, we observed a significant qualitative difference between the two groups in the functionality of the detected responses: while progressors made predominantly single IFN-γ responses (P = 0.35 compared to controllers), controllers mounted a marginally greater number of IL-2 responses (P = 0.06) and a significantly larger amount of dual IFN-γ/IL-2 responses (P < 0.0001; Fig. 1B). CTL epitopes presented by HLA class I are typically 8 to 12 a.a. long, and therefore our peptide set preferentially detects CD8+ T-cell responses; nonetheless, as the FluoroSpot assay uses bulk PBMC to detect responses, we cannot rule out the possibility we were also detecting CD4+ T-cell responses.
Controllers and progressors target conserved and variable regions in Gag.
The overlapping design of our peptide set coupled with the extensive sequence representation of frequently occurring Gag variants allowed us to comprehensively detect Gag-specific responses without using autologous peptide sets. Overall, we found the results of targeting of Gag by T cells to be remarkably similar between controllers and progressors (Fig. 2). We found no evidence for differential targeting of variable regions and no significant differences overall in the number of responses to conserved epitopic regions (Fig. 2; see also Fig. S2 in the supplemental material). The only significant difference we observed between groups was the preferential targeting of residues 362 to 367 ([362VLAEAM367]) in controllers (P < 0.05 for all residues by Fisher's exact test; Fig. 2). These conserved residues lie at the proteolytic cleavage site between p24 and p2 and have recently been reported to be part of a highly constrained coevolving group of Gag residues with a low tolerance for mutation (44). We did not find evidence for an HLA bias in preferential targeting; however, study of larger cohorts would be needed to define a connection between these residues and a potential contribution to immune control. Given that half of our controllers possessed B*57 and/or B*27 alleles, we did observe more responders among controllers to residues 162 to 172 (P = 0.46) and residues 263 to 272 (P = 0.65), corresponding to the immunodominant B*57 KF11 and B*27 KK10 epitopes, respectively. However, these differences were not significant.
FIG 2.

Controllers and progressors target conserved and variable regions of Gag. Data represent the number of controllers and progressors that target each amino acid residue in Gag p17 (top), p24 (middle), and p15 (bottom panel), as well as the conservation score for each residue (gray line). *, P < 0.05.
Higher epitope variant recognition observed in progressors.
As the results determined for breadth and the targeting of Gag residues were comparable, we next determined if there were differences in the number of distinct sequence variances recognized (“variant recognition”) as shown by Gag-specific T-cell responses between the controller and progressor groups. The peptide set used in this study was designed to include any variant present in at least 5% of clade B Gag sequences from the HIVDB, allowing us to comprehensively identify responses to frequently occurring Gag variants. For each targeted epitope, we calculated variant recognition as the total number of 11mers recognized subtracted from the number of 11mers tested per targeted epitopic region (see Table S2 in the supplemental material). Both within and between individuals, the levels of variant recognition per epitopic region differed considerably (see Fig. S3A and B). Previous reports have suggested that B*57 and B*27 restricted T cells exhibit enhanced cross-reactivity capability (9, 10, 45). We did not detect a significant difference in overall variant recognition between individuals with (n = 10) or without (n = 20) B*57 and/or B*27 alleles (P = 0.61; see Fig. S4). Interestingly, the functionality of the responding T cells varied depending on the epitope variant. In both controllers and progressors, the majority of responses to epitope variants were detected primarily by IFN-γ secretion. Only in a few targeted epitopic regions did all recognized variants elicit an IFN-γ plus IL-2 response (e.g., epitopes 74 and 262 in C5; see Fig. S5). We observed marginally higher median epitope variant recognition in progressors (P = 0.05; Fig. 3). Interestingly, response magnitude was significantly correlated to variant recognition (r = 0.39, P = 0.03; see also Fig. S6 in the supplemental material).
FIG 3.
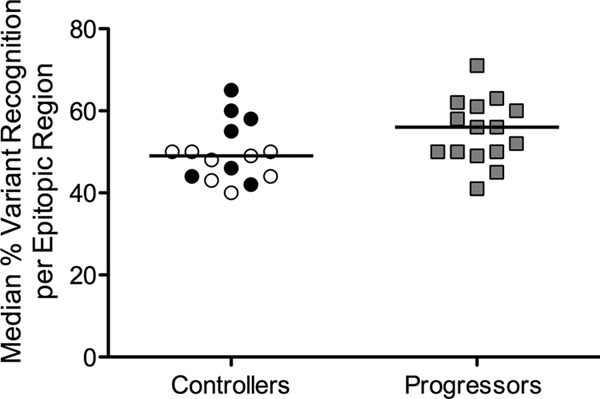
Higher variant recognition in progressors. Epitope variant recognition is measured as the number of 11mers recognized per number of 11mers tested in a targeted epitopic region. For each study subject, the median percentages of variant recognition were calculated across all targeted epitopic regions and compared between controllers and progressors (P = 0.05). EC, black circles; VC, open circles; progressors, gray squares.
Epitope variant recognition is associated with viral load in viremic controllers and progressors.
The extensive epitope variant recognition observed in progressors may be a consequence of persistently higher viral loads, which may sustain greater intrapopulation variance as well maintain high levels of antigen-specific T-cell responses. When we examined all study subjects, we observed a trend toward a correlation between epitope variant recognition and viral load (r = 0.33, P = 0.07; Fig. 4A). Exclusion of individuals with undetectable viral load from the analysis revealed a significant correlation between epitope variant recognition and viral load (r = 0.62, P = 0.001; Fig. 4B). We found no significant association between response magnitude and viral load (r = 0.24, P = 0.18; data not shown).
FIG 4.

Viral load and variant recognition in viremic subjects. (A and B) Median variant recognition per subject for EC (black circles), VC (open circles), and progressors (gray squares) is shown in relation to log10 viral load for all subjects (A) or excluding EC (B). (C) Variant recognition stratified by elite and viremic control compared to progressors (KW test P = 0.047; *, Dunn's posttest P < 0.05 for VC versus progressor). (D) Variant recognition in five progressors before (Pre-ARV) and after (Post-ARV) 6 months of treatment (P = 0.06).
Interestingly, when we segregated our controller cohort into elite controllers and viremic controllers, we found that progressors recognized significantly more variants only than viremic controllers (Kruskal-Wallis [KW] test P = 0.047, Dunn's posttest P < 0.05 for VC versus progressors; Fig. 4C). To further explore the relationship between viral load and variant recognition, we repeated the variant recognition assessment in five progressors who started antiretroviral therapy during the study period. Following 6 months of treatment, we observed a trend toward reduced epitope variant recognition in all five progressors (P = 0.06; Fig. 4D). Collectively, these data suggest that while viral load can explain the differential levels of variant recognition observed between viremic controllers and progressors, there are likely alternative mechanisms responsible for driving and maintaining the high levels of variant recognition observed in elite controllers.
Greater diversity in Gag epitopes found in progressors.
Persistence of viral antigen has been shown to promote proliferation and maintain antigen-specific CTL populations during chronic viral infection (46). Therefore, the high levels of variant-specific responses observed in progressive infection may be maintained due to the presence of highly diverse Gag variants in the viral population. To address the association between viral diversity and epitope variant recognition, we directly sequenced 10 to 25 individual Gag genes from plasma RNA from five viremic controllers and five progressors. We observed a significant correlation between viral load and mean pairwise distance (r = 0.75, P = 0.02; Fig. 5A). Diversity of the entire Gag protein was marginally higher in progressors than in controllers (P = 0.05, data not shown), even though controllers were on average infected for significantly longer (median, 20 years) than progressors (4 years, P = 0.008; data not shown), and diversity usually increases with duration of infection (47). This effect was more pronounced when examining pairwise distance only in viral regions corresponding to targeted epitopes (P = 0.0005; Fig. 5B).
FIG 5.

Viral diversity is higher in progressors than in VC. (A) Pairwise distance is correlated with viral load in this cohort. VC, open circles; progressors, gray squares. (B) From plasma viral RNA populations isolated from progressors and VCs, pairwise distances between Gag sequences corresponding to targeted epitopic regions were calculated and compared between groups (***, P = 0.0005). (C) Proportions of targeted epitopic regions for which recognized variants were present in the viral population (white), variants present in the viral population that were rare and not represented in the peptide set (gray), and variants present in the viral population that were tested but not recognized (black).
We next determined the relationship between the epitope variants that were recognized and the viral variants present in the autologous population. If the observed variant-specific T cells were effective at eliminating infected cells, we would not expect to see recognized epitope variants present in the plasma viral population. Within targeted epitopic regions, viremic controllers had more viral variants present in the plasma that were tested but not recognized than progressors (P = 0.008; Fig. 5C), suggesting greater evidence for escape in viremic controllers. In both controllers and progressors, we found targeted epitopic regions for which autologous viral variants were not contained in our peptide set (P = 0.34; Fig. 5C). These autologous variants represent uncommon sequences present in less than 5% of clade B sequences in the HIVDB. Although our data show that these regions are targeted, we are unable to address whether these autologous variants are recognized. Intriguingly, viral populations in progressors tended to more often match the recognized epitope variants than those in viremic controllers (P = 0.07; Fig. 5C).
Coverage of frequently occurring Gag variants is associated with control of viral replication.
An alternative approach to investigating the capacity of CTLs to cope with HIV sequence diversity is to evaluate recognized variants relative to the frequency with which they occur in circulating clade B sequences. For example, if an individual recognizes 3/5 tested variants (60%) but each of these variants is present in only 5% of circulating sequences, then the actual sequence coverage afforded through that variant recognition is quite poor (i.e., 5% each or 15% total). To assess the level of coverage in a given epitopic region, we summed the frequency of recognized 11mers versus the summed frequency of all tested 11mers in that region (see Table S2 in the supplemental material). Although our peptide set did not test every possible clade B Gag variant in the database, its design ensured that we were evaluating coverage of all frequently occurring Gag variants. For each targeted epitopic region, including both conserved and variable responses, we determined the level of sequence coverage through the recognized variants (see Fig. S7A and B). Controllers and progressors had comparable levels of overall sequence coverage through the Gag-specific repertoire (P = 0.29); however, elite controllers trended toward greater sequence coverage than both viremic controllers and progressors (KW test P = 0.09; Fig. 6A). Interestingly, although viremic controllers recognized significantly fewer variants than progressors (Fig. 4C), coverage levels were equivalent in the two groups, indicating that the number of variants recognized does not necessarily translate into broad sequence coverage. We found a significant inverse correlation between sequence coverage and viral load (r = −0.38, P = 0.03; Fig. 6B). There was no correlation between sequence coverage and response magnitude (r = −0.01, P = 0.95; data not shown).
FIG 6.

Increased sequence coverage is correlated with control of viral replication and recognition of founder viruses. (A) For each study subject, the median levels of coverage across targeted epitopic regions were calculated and compared between groups (KW test P = 0.09). (B) Coverage is inversely correlated with viral load. (C and D) Median recognition of founder viruses (y axis) is not correlated with median variant recognition (C) but does correlate with median coverage (D). EC, black circles; VC, open circles; progressors, gray squares.
Increased sequence coverage is correlated with recognition of more founder viruses.
Several vaccination strategies have designed multivalent immunogens with hopes of inducing T-cell responses that provide broad sequence coverage (19, 20, 24). Intuitively, increased sequence coverage should increase the number of potentially infecting sequences that would be recognized. To explore how the sequence coverage measured in this study can inform future vaccine evaluation, we analyzed all 347 responses relative to how often they matched a panel of eight founder viruses. This panel is a collection of clade B viruses isolated from recently infected individuals (median, 7 days post-onset of symptoms) in the same demographic population as the majority of participants in this study (14). For each targeted epitopic region, we determined what proportion of the founder viruses would be recognized given the epitope specificity of that individual's T-cell response (see Fig. S8A and B in the supplemental material). While we did not see a correlation between recognition of founder viruses and median epitope variant recognition (r = 0.22, P = 0.23; Fig. 6C), we did find a significant correlation between the number of founder viruses that were recognized and overall sequence coverage (r = 0.50, P = 0.004 Fig. 6D), suggesting that for vaccine-induced responses, a greater protective potential may be achieved by targeting the most commonly occurring variants rather than simply a large number of variants.
DISCUSSION
Our report provides the first investigation into the ability to recognize all frequently occurring Gag variants in subjects with and without spontaneous control of HIV replication. In agreement with a recent study by Mothe et al., the shorter peptide length, tight overlap, and vast sequence representation in our peptide set enabled us to detect a larger breadth of Gag-specific responses and that the targeting of the Gag protein by controllers is remarkably similar to the targeting of the Gag protein by progressors (11). We found that recognition of epitope variants was widespread in both controlled and progressive HIV infections. Interestingly, while responses to conserved epitopes and high numbers of HIV variants have each previously been associated with immune control of HIV (9, 11, 12, 48), our study identified the overall sequence coverage provided through the Gag-specific repertoire as a superior correlate of immune responses associated with virologic control.
Cross-reactivity has been proposed as an essential feature of T-cell receptors (TCR) (49, 50). Recognition of naturally occurring Gag variants can result either from a T cell expressing a TCR that is exceptionally flexible or through several different clonotypes, each recognizing one epitope variant or a range of epitope variants. As we identified responses by cytokine secretion using PBMC, our assay was unable to distinguish between these two possibilities. In viremic individuals, we found that contemporaneous viremia was associated with the extent of epitope variant recognition. Investigations into virus-host dynamics during acute HIV infection have shown that de novo responses to viral variants develop throughout infection (15, 31, 32, 51). Therefore, as different TCRs have differential abilities to tolerate variation within epitopes, sequential de novo responses associated with high viremia have the potential to increase the breadth and depth of responses, resulting in the extensive epitope variant recognition observed in progressive infection. Interestingly, we observed greater evidence for escape (i.e., fixation of a viral variant that was tested but not recognized) in epitopes targeted by viremic controllers than in those targeted by progressors. While these responses appear to be effective at driving escape, the lower level of variant recognition in viremic controllers may reflect that these individuals make fewer de novo responses to viral variants, possibly due to the weak potential of low viremia to prime new responses or to selection of escape mutations that abrogate epitope-HLA binding (52).
Constant antigenic stimulation has been shown to be necessary for long-term persistence of virus-specific CTLs during chronic viral infection (31, 46). The lack of IL-2 production in the majority of responses to epitope variants, the association between response magnitude and variant recognition, and the contraction of variant recognition upon ARV-mediated suppression of viral load support a model in which the maintenance of variant-specific responses is antigen dependent. Increasingly diverse viral quasispecies can also provide more opportunities for different TCRs to engage with a recognized antigen to stimulate T-cell proliferation. However, persistent antigenic stimulation can also promote T-cell exhaustion and functional impairment (53–57). Exhausted T cells characteristically exhibit weak cytotoxic potential and loss of polyfunctionality but maintain the ability to secrete IFN-γ (57). In the viral populations of progressors and, to a lesser extent, of viremic controllers, we found evidence for ineffective variant-specific responses, as many viral variants were present in the population despite being recognized. As this is a cross-sectional study, we cannot rule out the possibility that the viral populations were in the process of escaping (in the cases in which a recognized variant was a minor variant) or eventually would escape (in the cases in which a recognized variant was a major variant) in response to immune pressure. Given that the majority of responses to epitope variants detected in this analysis were monofunctional IFN-γ-positive responses, the ineffectiveness of these responses may have been a consequence of exhaustion. Our results are consistent with previous reports that have shown that the ability to recognize peptide variants as measured by IFN-γ secretion does not directly translate into an ability to inhibit replication of viruses harboring recognized variants (58, 59). Taken together, our data suggest that a perpetual cycle exists in viremic individuals by which the combination of high viral loads and diversity can drive the development and persistence of variant-specific responses while simultaneously rendering these responses ineffective at fully suppressing viral replication, ultimately permitting the emergence of new viral variants.
Elite controllers maintain levels of epitope variant recognition in the absence of antigenic stimulation comparable to those in progressors, suggesting that there may be alternative immunological mechanisms responsible for preserving the ability to recognize epitope variants. T-cell functional avidity has been associated with mediating epitope variant recognition (10, 11, 60). Given that functional avidity increases in the absence of antigen (55), the observed high levels of variant recognition in elite controllers may be due to the maintenance of a high-avidity Gag repertoire. Additionally, it has been reported that compared to typical progressors, B*27 and B*57 elite controllers possess distinct clonotypes that have greater cross-reactivity to epitope variants (45). Although we did not detect a significant difference in the overall variant recognition between individuals with and without B*57 and B*27 alleles, on a per-T-cell basis individual highly cross-reactive clonotypes may have been contributing to the observed levels of variant recognition found in elite controllers.
We found that increased sequence coverage by Gag-specific T-cell responses is associated with lower viral loads in infected subjects. The combination of all variant-specific and conserved responses contributes to overall sequence coverage, highlighting the importance of the collective Gag-specific repertoire in control of viral replication. On a single-epitope level, coverage of frequently occurring variants can lead to the selection of rare mutations to permit escape. Given the weak relationship between sequence conservation and viral fitness costs (61–64), even high sequence coverage of a single epitope may not translate directly to control. However, recent analyses have revealed extensive covariation and networks of interdependent amino acids within the Gag protein (44, 65, 66). Structural and functional constraints limit the tolerance of multiple simultaneous mutations within coevolving Gag sites (44). Additionally, accumulation of mutations within Gag has recently been shown to decrease HIV replicative capacity as well as to contribute to reducing the viral load set point when transmitted to a new recipient (67, 68). Therefore, broad sequence coverage afforded through a functionally effective Gag-specific repertoire has the potential to select for multiple rare mutations across the protein. Increasing the overall level of coverage increases the likelihood that the consequences of escape will compromise the viability of the virus and allow for immune control.
Increased sequence coverage through an individual's Gag-specific repertoire, but not variant recognition, correlated with higher recognition of different founder viruses. Although our study was limited to clade B infections, these results are encouraging for the protective potential of several vaccination strategies designed to elicit responses of broad coverage to global HIV sequences. Polyvalent mosaic vaccines and conserved-element vaccines both utilize multivalent immunogens to induce variant-specific responses that will ideally provide broad coverage either against whole proteins (19) or primarily against conserved regions (24). Our results suggest that the emphasis in the evaluation of these vaccines should be on the combined sequence coverage that can be achieved through all induced responses. However, our results were defined based on only Gag-specific responses, and it remains to be determined whether the protective potential of sequence coverage is applicable to other HIV proteins. Additionally, immunogenicity evaluation of these vaccines may depend on the identification of T-cell functional qualities that correlate with effective variant recognition. Given that dual IFN-γ/IL-2 responses to epitope variants were uncommon in our study, we were unable to evaluate whether the IFN-γ/IL-2 FluoroSpot assay better predicts effective variant recognition than the traditional IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay.
Taking these results together, our report provides a new potential correlate, “sequence coverage,” of protection from progression in HIV infection. Sequence coverage combines previously suggested markers for viral control (variant recognition and targeting of conserved epitopes) into a single measure that is more informative in predicting viral control. The inverse relationship between sequence coverage and viral load, as well as the positive correlation with the recognition of founder viruses, makes sequence coverage a candidate for assessment in novel vaccine strategies that aim at increasing the breadth and depth of HIV-specific immune responses, as well as those designed to focus the immune response on conserved regions.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R21AI078809, T32AI007140, P01AI057005, R01AI047086, and UM1AI068618.
The content of this publication is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
We thank all study participants for their participation in this research study as well as the clinic staff for their critical work and dedication. We thank Paula Walsh and Dave Friedrich for technical support and Stephen Voght for editorial assistance.
Footnotes
Published ahead of print 13 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02361-13.
REFERENCES
- 1.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitz J, Kuroda M, Santra S, Sasseville V, Simon M, Lifton M, Racz P, Tenner-Racz K, Dalesandro M, Scallon B, Ghrayeb J, Forman M, Montefiori D, Rieber E, Letvin N, Reimann K. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860 [DOI] [PubMed] [Google Scholar]
- 3.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405–411. 10.1038/nm0496-405 [DOI] [PubMed] [Google Scholar]
- 4.Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, Goulder PJ, Walker BD, Brander C, Mullins JI. 2008. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS One 3:e1424. 10.1371/journal.pone.0001424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53. 10.1038/nm1520 [DOI] [PubMed] [Google Scholar]
- 6.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, III, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak M, Jr, Haase AT, Lifson JD, Allen TM, Watkins DI. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133. 10.1038/nature11443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Julg B, Williams KL, Reddy S, Bishop K, Qi Y, Carrington M, Goulder PJ, Ndung'u T, Walker BD. 2010. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J. Virol. 84:5540–5549. 10.1128/JVI.02031-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillespie GM, Kaul R, Dong T, Yang HB, Rostron T, Bwayo JJ, Kiama P, Peto T, Plummer FA, McMichael AJ, Rowland-Jones SL. 2002. Cross-reactive cytotoxic T lymphocytes against a HIV-1 p24 epitope in slow progressors with B*57. AIDS 16:961–972. 10.1097/00002030-200205030-00002 [DOI] [PubMed] [Google Scholar]
- 10.Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, Pereyra F, Carrington M, Walker BD, Chakraborty AK. 2010. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature 465:350–354. 10.1038/nature08997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mothe B, Llano A, Ibarrondo J, Zamarreno J, Schiaulini M, Miranda C, Ruiz-Riol M, Berger CT, Herrero MJ, Palou E, Plana M, Rolland M, Khatri A, Heckerman D, Pereyra F, Walker BD, Weiner D, Paredes R, Clotet B, Felber BK, Pavlakis GN, Mullins JI, Brander C. 2012. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7:e29717. 10.1371/journal.pone.0029717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnbull EL, Lopes AR, Jones NA, Cornforth D, Newton P, Aldam D, Pellegrino P, Turner J, Williams I, Wilson CM, Goepfert PA, Maini MK, Borrow P. 2006. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J. Immunol. 176:6130–6146 [DOI] [PubMed] [Google Scholar]
- 13.Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O'Sullivan KM, Desouza I, Feeney ME, Eldridge RL, Maier EL, Kaufmann DE, Lahaie MP, Reyor L, Tanzi G, Johnston MN, Brander C, Draenert R, Rockstroh JK, Jessen H, Rosenberg ES, Mallal SA, Walker BD. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239–13249. 10.1128/JVI.79.21.13239-13249.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbeck JT, Rolland M, Liu Y, McLaughlin S, McNevin J, Zhao H, Wong K, Stoddard JN, Raugi D, Sorensen S, Genowati I, Birditt B, McKay A, Diem K, Maust BS, Deng W, Collier AC, Stekler JD, McElrath MJ, Mullins JI. 2011. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J. Virol. 85:7523–7534. 10.1128/JVI.02697-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, McNevin J, Cao J, Zhao H, Genowati I, Wong K, McLaughlin S, McSweyn MD, Diem K, Stevens CE, Maenza J, He H, Nickle DC, Shriner D, Holte SE, Collier AC, Corey L, McElrath MJ, Mullins JI. 2006. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J. Virol. 80:9519–9529. 10.1128/JVI.00575-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ammaranond P, van Bockel DJ, Petoumenos K, McMurchie M, Finlayson R, Middleton MG, Davenport MP, Venturi V, Suzuki K, Gelgor L, Kaldor JM, Cooper DA, Kelleher AD. 2011. HIV immune escape at an immunodominant epitope in HLA-B*27-positive individuals predicts viral load outcome. J. Immunol. 186:479–488. 10.4049/jimmunol.0903227 [DOI] [PubMed] [Google Scholar]
- 17.Feeney ME, Tang Y, Roosevelt KA, Leslie AJ, McIntosh K, Karthas N, Walker BD, Goulder PJ. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927–8930. 10.1128/JVI.78.16.8927-8930.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217. 10.1038/nm0297-212 [DOI] [PubMed] [Google Scholar]
- 19.Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, Kuiken C, Haynes B, Letvin NL, Walker BD, Hahn BH, Korber BT. 2007. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 13:100–106. 10.1038/nm1461 [DOI] [PubMed] [Google Scholar]
- 20.Nickle DC, Rolland M, Jensen MA, Pond SL, Deng W, Seligman M, Heckerman D, Mullins JI, Jojic N. 2007. Coping with viral diversity in HIV vaccine design. PLoS Comput. Biol. 3:e75. 10.1371/journal.pcbi.0030075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barouch DH, O'Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, Sun YH, La Porte A, Riggs AM, Lynch DM, Clark SL, Backus K, Perry JR, Seaman MS, Carville A, Mansfield KG, Szinger JJ, Fischer W, Muldoon M, Korber B. 2010. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 16:319–323. 10.1038/nm.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W, Theiler J, Szinger J, Balachandran H, Buzby A, Quinn D, Parks RJ, Tsao CY, Carville A, Mansfield KG, Pavlakis GN, Felber BK, Haynes BF, Korber BT, Letvin NL. 2010. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat. Med. 16:324–328. 10.1038/nm.2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santra S, Muldoon M, Watson S, Buzby A, Balachandran H, Carlson KR, Mach L, Kong WP, McKee K, Yang ZY, Rao SS, Mascola JR, Nabel GJ, Korber BT, Letvin NL. 2012. Breadth of cellular and humoral immune responses elicited in rhesus monkeys by multi-valent mosaic and consensus immunogens. Virology 428:121–127. 10.1016/j.virol.2012.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolland M, Nickle DC, Mullins JI. 2007. HIV-1 group M conserved elements vaccine. PLoS Pathog. 3:e157. 10.1371/journal.ppat.0030157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni V, Rosati M, Valentin A, Ganneru B, Singh AK, Yan J, Rolland M, Alicea C, Beach RK, Zhang GM, Le Gall S, Broderick KE, Sardesai NY, Heckerman D, Mothe B, Brander C, Weiner DB, Mullins JI, Pavlakis GN, Felber BK. 2013. HIV-1 p24(gag) derived conserved element DNA vaccine increases the breadth of immune response in mice. PLoS One 8:e60245. 10.1371/journal.pone.0060245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu L, Termini JM, Kanagavelu SK, Gupta S, Rolland MM, Kulkarni V, Pavlakis GN, Felber BK, Mullins JI, Fischl MA, Stone GW. 2011. Preclinical evaluation of HIV-1 therapeutic ex vivo dendritic cell vaccines expressing consensus Gag antigens and conserved Gag epitopes. Vaccine 29:2110–2119. 10.1016/j.vaccine.2010.12.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Létourneau S, Im EJ, Mashishi T, Brereton C, Bridgeman A, Yang H, Dorrell L, Dong T, Korber B, McMichael AJ, Hanke T. 2007. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS One 2:e984. 10.1371/journal.pone.0000984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoof I, Perez CL, Buggert M, Gustafsson RK, Nielsen M, Lund O, Karlsson AC. 2010. Interdisciplinary analysis of HIV-specific CD8+ T cell responses against variant epitopes reveals restricted TCR promiscuity. J. Immunol. 184:5383–5391. 10.4049/jimmunol.0903516 [DOI] [PubMed] [Google Scholar]
- 29.Malhotra U, Nolin J, Horton H, Li F, Corey L, Mullins JI, McElrath MJ. 2009. Functional properties and epitope characteristics of T-cells recognizing natural HIV-1 variants. Vaccine 27:6678–6687. 10.1016/j.vaccine.2009.08.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zembe L, Burgers WA, Jaspan HB, Bekker LG, Bredell H, Stevens G, Gilmour J, Cox JH, Fast P, Hayes P, Vardas E, Williamson C, Gray CM. 2011. Intra- and inter-clade cross-reactivity by HIV-1 Gag specific T-cells reveals exclusive and commonly targeted regions: implications for current vaccine trials. PLoS One 6:e26096. 10.1371/journal.pone.0026096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, McNevin JP, Holte S, McElrath MJ, Mullins JI. 2011. Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS One 6:e15639. 10.1371/journal.pone.0015639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 8:e1002529. 10.1371/journal.ppat.1002529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altfeld M, Addo MM, Shankarappa R, Lee PK, Allen TM, Yu XG, Rathod A, Harlow J, O'Sullivan K, Johnston MN, Goulder PJ, Mullins JI, Rosenberg ES, Brander C, Korber B, Walker BD. 2003. Enhanced detection of human immunodeficiency virus type 1-specific T-cell responses to highly variable regions by using peptides based on autologous virus sequences. J. Virol. 77:7330–7340. 10.1128/JVI.77.13.7330-7340.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horton H, Frank I, Baydo R, Jalbert E, Penn J, Wilson S, McNevin JP, McSweyn MD, Lee D, Huang Y, De Rosa SC, McElrath MJ. 2006. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J. Immunol. 177:7406–7415 [DOI] [PubMed] [Google Scholar]
- 35.Schacker T, Collier AC, Hughes J, Shea T, Corey L. 1996. Clinical and epidemiologic features of primary HIV infection. Ann. Intern. Med. 125:257–264. 10.7326/0003-4819-125-4-199608150-00001 [DOI] [PubMed] [Google Scholar]
- 36.Schacker TW, Hughes JP, Shea T, Coombs RW, Corey L. 1998. Biological and virologic characteristics of primary HIV infection. Ann. Intern. Med. 128:613–620. 10.7326/0003-4819-128-8-199804150-00001 [DOI] [PubMed] [Google Scholar]
- 37.Stekler J, Sycks BJ, Holte S, Maenza J, Stevens CE, Dragavon J, Collier AC, Coombs RW. 2008. HIV dynamics in seminal plasma during primary HIV infection. AIDS Res. Hum. Retroviruses 24:1269–1274. 10.1089/aid.2008.0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U. S. A. 97:2709–2714. 10.1073/pnas.050567397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le SQ, Dang CC, Gascuel O. 2012. Modeling protein evolution with several amino acid replacement matrices depending on site rates. Mol. Biol. Evol. 29:2921–2936. 10.1093/molbev/mss112 [DOI] [PubMed] [Google Scholar]
- 41.Deng W, Maust BS, Nickle DC, Learn GH, Liu Y, Heath L, Kosakovsky Pond SL, Mullins JI. 2010. DIVEIN: a web server to analyze phylogenies, sequence divergence, diversity, and informative sites. Biotechniques 48:405–408. 10.2144/000113370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frahm N, Korber BT, Adams CM, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel AG, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, St John A, Khatri A, Davis K, Mullins J, Goulder PJ, Walker BD, Brander C. 2004. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J. Virol. 78:2187–2200. 10.1128/JVI.78.5.2187-2200.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frahm N, Nickle DC, Linde CH, Cohen DE, Zuniga R, Lucchetti A, Roach T, Walker BD, Allen TM, Korber BT, Mullins JI, Brander C. 2008. Increased detection of HIV-specific T cell responses by combination of central sequences with comparable immunogenicity. AIDS 22:447–456. 10.1097/QAD.0b013e3282f42412 [DOI] [PubMed] [Google Scholar]
- 44.Dahirel V, Shekhar K, Pereyra F, Miura T, Artyomov M, Talsania S, Allen TM, Altfeld M, Carrington M, Irvine DJ, Walker BD, Chakraborty AK. 2011. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc. Natl. Acad. Sci. U. S. A. 108:11530–11535. 10.1073/pnas.1105315108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 13:691–700. 10.1038/ni.2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin H, Blackburn SD, Blattman JN, Wherry EJ. 2007. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 204:941–949. 10.1084/jem.20061937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489–10502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuñiga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122–3125. 10.1128/JVI.80.6.3122-3125.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mason D. 1998. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19:395–404. 10.1016/S0167-5699(98)01299-7 [DOI] [PubMed] [Google Scholar]
- 50.Sewell AK. 2012. Why must T cells be cross-reactive? Nat. Rev. Immunol. 12:669–677. 10.1038/nri3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen TM, Yu XG, Kalife ET, Reyor LL, Lichterfeld M, John M, Cheng M, Allgaier RL, Mui S, Frahm N, Alter G, Brown NV, Johnston MN, Rosenberg ES, Mallal SA, Brander C, Walker BD, Altfeld M. 2005. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J. Virol. 79:12952–12960. 10.1128/JVI.79.20.12952-12960.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL. 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J. Virol. 86:13202–13216. 10.1128/JVI.01998-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Appay V, Nixon DF, Donahoe SM, Gillespie GM, Dong T, King A, Ogg GS, Spiegel HM, Conlon C, Spina CA, Havlir DV, Richman DD, Waters A, Easterbrook P, McMichael AJ, Rowland-Jones SL. 2000. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192:63–75. 10.1084/jem.192.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. 10.1038/nature05115 [DOI] [PubMed] [Google Scholar]
- 55.Janbazian L, Price DA, Canderan G, Filali-Mouhim A, Asher TE, Ambrozak DR, Scheinberg P, Boulassel MR, Routy JP, Koup RA, Douek DC, Sekaly RP, Trautmann L. 2012. Clonotype and repertoire changes drive the functional improvement of HIV-specific CD8 T cell populations under conditions of limited antigenic stimulation. J. Immunol. 188:1156–1167. 10.4049/jimmunol.1102610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Streeck H, Brumme ZL, Anastario M, Cohen KW, Jolin JS, Meier A, Brumme CJ, Rosenberg ES, Alter G, Allen TM, Walker BD, Altfeld M. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. 5:e100. 10.1371/journal.pmed.0050100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennett MS, Ng HL, Ali A, Yang OO. 2008. Cross-clade detection of HIV-1-specific cytotoxic T lymphocytes does not reflect cross-clade antiviral activity. J. Infect. Dis. 197:390–397. 10.1086/525281 [DOI] [PubMed] [Google Scholar]
- 59.Valentine LE, Piaskowski SM, Rakasz EG, Henry NL, Wilson NA, Watkins DI. 2008. Recognition of escape variants in ELISPOT does not always predict CD8+ T-cell recognition of simian immunodeficiency virus-infected cells expressing the same variant sequences. J. Virol. 82:575–581. 10.1128/JVI.00275-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett MS, Joseph A, Ng HL, Goldstein H, Yang OO. 2010. Fine-tuning of T-cell receptor avidity to increase HIV epitope variant recognition by cytotoxic T lymphocytes. AIDS 24:2619–2628. 10.1097/QAD.0b013e32833f7b22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boutwell CL, Carlson JM, Lin TH, Seese A, Power KA, Peng J, Tang Y, Brumme ZL, Heckerman D, Schneidewind A, Allen TM. 2013. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. J. Virol. 87:3952–3965. 10.1128/JVI.03233-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rolland MMS, Swain JV, Lanxon-Cookson EC, Kim M, Westfall DH, Larsen BB, Gilbert PB, Mullins JI. 2013. HIV-1 conserved-element vaccines: relationship between sequence conservation and replicative capacity. J. Virol. 87:5461–5467. 10.1128/JVI.03033-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 9:e1003461. 10.1371/journal.ppat.1003461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manocheewa S, Swain JV, Lanxon-Cookson E, Rolland M, Mullins JI. 2013. Fitness costs of mutations at the HIV-1 capsid hexamerization interface. PLoS One 8:e66065. 10.1371/journal.pone.0066065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, Mullins JI, Walker BD, Harrigan PR, Goulder PJ, Heckerman D. 2008. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput. Biol. 4:e1000225. 10.1371/journal.pcbi.1000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rolland M, Carlson JM, Manocheewa S, Swain JV, Lanxon-Cookson E, Deng W, Rousseau CM, Raugi DN, Learn GH, Maust BS, Coovadia H, Ndung'u T, Goulder PJ, Walker BD, Brander C, Heckerman DE, Mullins JI. 2010. Amino-acid co-variation in HIV-1 Gag subtype C: HLA-mediated selection pressure and compensatory dynamics. PLoS One 5:e12463. 10.1371/journal.pone.0012463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJ, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009–1017. 10.1084/jem.20072457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W, Goepfert P, Price MA, Gilmour J, Mulenga J, Farmer P, Derdeyn CA, Tang J, Heckerman D, Kaslow RA, Allen SA, Hunter E. 2012. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog. 8:e1003041. 10.1371/journal.ppat.1003041 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.