

Abstract
Sin Nombre virus (SNV) is a rodent-borne hantavirus that causes hantavirus pulmonary syndrome (HPS) predominantly in North America. SNV infection of immunocompetent hamsters results in an asymptomatic infection; the only lethal disease model for a pathogenic hantavirus is Andes virus (ANDV) infection of Syrian hamsters. Efforts to create a lethal SNV disease model in hamsters by repeatedly passaging virus through the hamster have demonstrated increased dissemination of the virus but no signs of disease. In this study, we demonstrate that immunosuppression of hamsters through the administration of a combination of dexamethasone and cyclophosphamide, followed by infection with SNV, results in a vascular leak syndrome that accurately mimics both HPS disease in humans and ANDV infection of hamsters. Immunosuppressed hamsters infected with SNV have a mean number of days to death of 13 and display clinical signs associated with HPS, including pulmonary edema. Viral antigen was widely detectable throughout the pulmonary endothelium. Histologic analysis of lung sections showed marked inflammation and edema within the alveolar septa of SNV-infected hamsters, results which are similar to what is exhibited by hamsters infected with ANDV. Importantly, SNV-specific neutralizing polyclonal antibody administered 5 days after SNV infection conferred significant protection against disease. This experiment not only demonstrated that the disease was caused by SNV, it also demonstrated the utility of this animal model for testing candidate medical countermeasures. This is the first report of lethal disease caused by SNV in an adult small-animal model.
INTRODUCTION
Sin Nombre virus (SNV) and Andes virus (ANDV), both members of the genus Hantavirus within the family Bunyaviridae, are the predominant etiological agents of hantavirus pulmonary syndrome (HPS) in North and South America, respectively (1–3). Hantaviruses are negative-strand, single-stranded RNA viruses with three segments, denoted small (S), medium (M), and large (L). The S segment encodes the nucleoprotein (N), the M segment encodes the glycoproteins (Gn and Gc), and the L segment encodes the RNA-dependent RNA polymerase (RdRp) (4). Hantaviruses predominantly infect microvascular endothelial cells and create a vascular leakage-based disease by altering the barrier properties of the endothelium (5). This nonlytic infection renders the endothelium unable to regulate tissue fluid accumulation, leading to pulmonary edema, tachycardia, shock, and cardiac failure (6). The mechanism underlying this endothelium dysfunction and related pathogenic effects remains unknown.
Currently, Syrian hamsters infected with ANDV remain the only small-animal lethal disease model for hantaviruses that cause HPS (7). This disease model closely mimics human disease in incubation time, infection of the endothelium, and rapid onset of disease. Hamsters develop respiratory distress in the final 24 h prior to death approximately 10 to 14 days postinfection when challenged with 200 to 2,000 PFU of ANDV. Maporal virus, not known to cause disease in humans, causes a disease similar to HPS in Syrian hamsters, but with lower morbidity and mortality (8). Along with SNV, infection of hamsters with other hantaviruses (e.g., Hantaan [HTNV], Puumala [PUUV], Dobrava [DOBV], and Seoul [SEOV]) results in an asymptomatic infection that is rapidly cleared and is distinguishable only by the subsequent presence of neutralizing antibodies (9–12). The lack of disease associated with hamster infection with the aforementioned hantaviruses, except ANDV, limits our understanding of the pathogenesis of these viruses. Development of disease models for these viruses is an important step to expand the capability of evaluating potential therapeutics for hantavirus disease.
Cyclophosphamide is a chemotherapeutic agent that suppresses B cell and T cell function (13–15) and also causes apoptosis in other cell types, including neutrophils, macrophages, and dendritic cells (16, 17). Dexamethasone is a glucocorticosteroid that acts as an immunosuppressant by interfering with NF-κB-dependent gene activation, inhibiting lymphocyte proliferation, and reducing inflammatory responses by reducing proinflammatory gene expression and inducing anti-inflammatory genes (18). Both cyclophosphamide and dexamethasone have been used as immunosuppressive agents to alleviate or create disease in animal models of infectious diseases, namely, West Nile virus (19), severe acute respiratory syndrome (SARS) (20), lymphocytic choriomeningitis virus (21), and adenovirus (22).
In the current study, we take an alternative approach to create a model of lethal hantavirus disease by evaluating the pathogenesis of a normally infectious yet nonpathogenic hantavirus in immunocompetent hamsters and in immunocompromised hamsters treated with dexamethasone and cyclophosphamide individually and in combination. This model of SNV-associated lethal disease in Syrian hamsters is the first report of an HPS-like disease caused by SNV in an adult small-animal model and should improve efforts to develop vaccines and therapeutics to prevent and treat hantavirus disease in humans.
MATERIALS AND METHODS
Virus, cells, and medium.
SNV strain CC107 (23) was propagated in Vero E6 cells (Vero C1008, ATCC CRL 1586). Preparation of twice-plaque-purified SNV stock has been described previously (7). Cells were maintained in Eagle's minimum essential medium with Earle's salts containing 10% fetal bovine serum, 10 mM HEPES, pH 7.4, penicillin-streptomycin (Invitrogen) at 1×, and gentamicin sulfate (50 μg/ml) at 37°C in a 5% CO2 incubator.
Dexamethasone and cyclophosphamide administration.
Water-soluble dexamethasone and cyclophosphamide monohydrate were purchased from Sigma-Aldrich. On the indicated days, anesthetized hamsters were injected intraperitoneally (i.p.) with the indicated dosages per kilogram of body weight of drug diluted in sterile phosphate-buffered saline (PBS), pH 7.4.
Challenge with hantavirus.
Female Syrian hamsters 6 to 8 weeks of age (Harlan, Indianapolis, IN) were anesthetized by inhalation of vaporized isoflurane using an IMPAC 6 veterinary anesthesia machine. Once anesthetized, hamsters were injected with 2,000 PFU of virus diluted in PBS. Intramuscular (i.m.) (caudal thigh) injections consisted of 0.2 ml delivered with a 1-ml syringe with a 25-gauge, five-eighths-inch needle.
Blood chemistries.
Blood samples were collected in lithium heparin capillary blood collection tubes. Concentrations of alanine aminotransferase (ALT), alkaline phosphatase (ALP), and aspartate aminotransferase (AST) were determined using a comprehensive metabolic reagent disc and an Abaxis Piccolo xpress chemistry analyzer.
Hematology.
Blood samples collected in lithium heparin capillary blood collection tubes were analyzed using an Advia 120 hematology analyzer using proprietary software version 3.1.8.0-MS. The dog setting was used for the complete blood count (CBC), and the guinea pig setting was used for the differential.
Plaque assay.
Hantavirus plaque assays were performed as previously described (12).
PRNT.
Plaque reduction neutralization tests (PRNT) were performed as previously described (7). Serum samples were gamma irradiated on dry ice with 3 × 106 rads from a 60C source.
N-specific ELISA.
The enzyme-linked immunosorbent assay (ELISA) used to detect N-specific antibodies (N-ELISA) was described previously (10, 24). The endpoint titer was determined as the highest dilution that had an optical density (OD) greater than the mean OD for serum samples from negative-control wells plus 3 standard deviations. The PUUV N antigen was used to detect SNV N-specific antibodies as previously reported (7).
Isolation of RNA and real-time PCR.
Approximately 250 mg of lung tissue was homogenized in 1.0 ml TRIzol reagent using gentleMACS M tubes and a gentleMACS dissociator on the RNA setting. Serum samples were added directly to TRIzol reagent. RNA was extracted from TRIzol samples as recommended by the manufacturer. The concentration of the extracted RNA was determined using a NanoDrop 8000 instrument and raised to a final concentration of 10 ng/μl. Real-time PCR was conducted on a Bio-Rad CFX thermal cycler using an Invitrogen Power SYBR green RNA-to-CT one-step kit according to the manufacturer's protocols. Primer sequences are as follows (26): SNV S 26F, 5′-CTA CGA CTA AAG CTG GAA TGA GC-3′; SNV S 96R, 5′-GAG TTG TTG TTC GTG GAG AGT G-3′. Cycling conditions were 30 min at 48°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. Data acquisition occurred following the annealing step.
Preparation of tissues for histology.
Tissues were fixed in 10% neutral buffered formalin, trimmed, processed, embedded in paraffin, cut at 5 to 6 μm, and stained with hematoxylin and eosin (H&E). Immunolocalization of SNV in tissues was performed with an immunoperoxidase procedure (horseradish peroxidase EnVision system; Dako, Glostrup, Denmark) according to the manufacturer's directions. The primary antibody was an anti-SNV nucleocapsid rabbit polyclonal antibody diluted 1:3,000 (provided by Diagnostic Service Division, U.S. Army Medical Research Institute of Infectious Disease [USAMRIID], Fort Detrick, MD). Negative controls included naive hamster tissue incubated with nonimmune rabbit IgG in place of the primary antibody and naive hamster tissue exposed to the primary antibody and negative serum. After deparaffinization and peroxidase blocking, tissue sections were pretreated with proteinase K for 6 min at room temperature, rinsed, and then covered with primary antibody and incubated at room temperature for 1 h. They were rinsed, and then the peroxidase-labeled polymer (secondary antibody) was applied for 30 min. Slides were rinsed, and a substrate-chromogen solution (3,3′-diaminobenzidine; Dako, Glostrup, Denmark) was applied for 5 min. The substrate-chromogen solution was rinsed off the slides, and the slides were stained with hematoxylin and rinsed. The sections were dehydrated and cleared with xylitol (Xyless), and then a coverslip was placed on top.
Statistical analysis.
Comparison of white blood cells (WBC), lymphocytes, neutrophils, ALT, AST, and ALP was done using a paired t test. Survival curves were compared with Kaplan-Meier survival analysis with log-rank comparisons and Dunnett's correction. Comparison of the viral genome and infectious virus was done using a one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test. P values of less than 0.05 were considered significant. Analyses were conducted using GraphPad Prism (version 5).
Ethics statement.
All work involving the use of SNV in animals was performed in USAMRIID's biosafety level 4 laboratory. Animal research was conducted under an institutional animal care and use committee (IACUC)-approved protocol at USAMRIID (USDA registration number 51-F-00211728 and Office of Laboratory Animal Welfare [OLAW] assurance number A3473-01) in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International, and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals (27).
RESULTS
Dexamethasone and cyclophosphamide immunosuppress Syrian hamsters.
In order to develop an immunosuppressed hamster model, groups of three hamsters were administered dexamethasone and cyclophosphamide, alone or in combination, according to the dosing schedule outlined in Table 1. On day 0, all hamsters were infected with 2,000 PFU of SNV i.m. WBC counts were monitored prior to and after virus infection to confirm immunosuppression in treatment groups (Fig. 1A). Dexamethasone and cyclophosphamide, alone and in combination, resulted in statistically significant reductions in WBC counts compared to no-treatment controls (for dexamethasone, P = 0.0428; for cyclophosphamide, P = 0.0010; and for dexamethasone and cyclophosphamide, P = 0.0020). The combination of dexamethasone and cyclophosphamide elicited the most immunosuppressive activity of the three treatment groups (3- to 4-fold reduction across all time points) based on WBC counts. Similarly, the combination of dexamethasone and cyclophosphamide also resulted in consistently reduced lymphocyte counts (for dexamethasone, P = 0.0155; for cyclophosphamide, P = 0.0115; for dexamethasone and cyclophosphamide, P = 0.0020) (Fig. 1B). This combination did not reduce the number of peripheral blood neutrophils but did prevent neutrophil counts from increasing late after infection (Fig. 1C). In addition, liver panels demonstrate (based on the lack of a statistically significant increase in measured liver enzymes) that neither dexamethasone nor cyclophosphamide nor the combination caused toxicity in the hamster model compared to no-treatment controls (Fig. 1D to F).
TABLE 1.
Dexamethasone and cyclophosphamide dosing schedulesa
| Treatment group | Dose(s) (mg/kg) (day[s] postinfection) |
|
|---|---|---|
| Loading | Maintenance | |
| Dex | 16 (−3), 8 (−2, −1) | 4 (0–13) |
| CyP | 140 (−3) | 100 (−1, 1, 4, 7, 10, 13) |
| Dex/CyP | Same dosing as Dex and CyP aloneb | Same dosing as Dex and CyP alone |
| No treatment | NA | NA |
Dexamethasone (Dex) and cyclophosphamide (CyP) were administered by i.p. injection. NA, not applicable.
On days when both compounds were administered to the dexamethasone/cyclophosphamide group, compounds were combined into a single injection.
FIG 1.

Dexamethasone and cyclophosphamide immunosuppress Syrian hamsters without toxicity. Groups of 3 hamsters were treated with dexamethasone and cyclophosphamide, alone or in combination, according to the schedule outlined in Table 1. Whole blood collected was tested for WBC (A), lymphocytes (B), neutrophils (C), ALT (D), AST (E), and ALP (F). Asterisks indicate that results were statistically significant compared to no-treatment controls, as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant.
SNV-infected, immunosuppressed hamsters exhibited clinical signs and mortality similar to what is exhibited by ANDV-infected hamsters.
Hamsters in all groups were challenged with 2,000 PFU of SNV i.m. on day 0 (Fig. 2A). A single hamster succumbed on day 5 in the dexamethasone-treated group (suspected to be from complications due to repeated blood draws; no apparent toxicity was measured or observed); the remaining dexamethasone-treated hamsters survived to day 28 (P = 0.8900). Hamsters in the cyclophosphamide-treated and dexamethasone/cyclophosphamide-treated groups displayed clinical signs including staggered gait, tachypnea, dyspnea, lethargy, and severe pulmonary edema. Clinical signs of disease in terminal hamsters were most notable within the 24 h preceding death. Clinical signs of disease were not observed in any surviving hamster. SNV was uniformly lethal for hamsters in the dexamethasone/cyclophosphamide-treated group (P < 0.0001), resulting in a mean time to death of 13 days and a range of 10 to 14 days postchallenge. Half of the hamsters receiving cyclophosphamide alone developed HPS approximately 16 days postchallenge, with a range of 12 to 19 days postchallenge (P = 0.2034). Of note in the cyclophosphamide-treated group, two hamsters developed neurological symptoms, including holding their head to one side and circling inside the pan repeatedly. As previously reported, immunocompetent hamsters infected with SNV developed no clinical signs and survived to the end of the study (7). Serum from all surviving hamsters on day 28 was subjected to an N-ELISA to confirm infection (Fig. 2B). Survivors in the dexamethasone-treated and no-treatment groups all had ELISA titers of ≥3 log10, confirming a productive infection and humoral response. Not surprisingly, the four surviving hamsters in the cyclophosphamide-treated group were unable to mount a robust immune response and had ELISA titers that were either at or below the level of detection for the assay (P < 0.0001).
FIG 2.
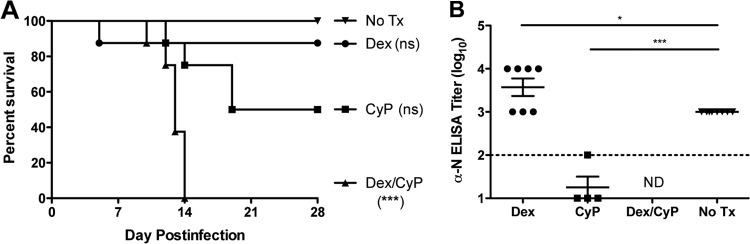
Dexamethasone/cyclophosphamide immunosuppression allows SNV to be uniformly lethal in hamsters. Groups of 8 hamsters were treated with dexamethasone and cyclophosphamide, alone or in combination, according to the schedule outlined in Table 1. (A) Hamsters were challenged with 2,000 PFU SNV i.m. on day 0 and observed for survival. (B) Sera collected from surviving hamsters were tested by ELISA for evidence of infection. *, P < 0.05; ***, P < 0.001 (compared to no-treatment controls). ns, not significant; ND, no surviving hamsters in this group.
Immunosuppression of SNV-infected hamsters allows detection of viremia.
We have previously reported that SNV-infected hamsters had undetectable levels of infectious virus in both the serum and whole blood and viral genome was not detectable in peripheral blood mononuclear cells (PBMCs) (12). In the current study, serum and lung tissue were collected from hamsters 10 days postinfection and evaluated for the presence of viral genome and infectious virus (Fig. 3). Increased levels of viral genome were detectable in the serum of immunosuppressed hamsters compared to those in the serum of immunocompetent hamsters (P = 0.0867) (Fig. 3A), and levels were highest, by approximately 2-fold, in hamsters treated with both dexamethasone and cyclophosphamide. Similarly, approximately 4-fold more infectious virus was detected in the serum of cyclophosphamide- and dexamethasone/cyclophosphamide-treated hamsters than in that of hamsters treated with dexamethasone and untreated hamsters (P = 0.0003) (Fig. 3B). In the lung, slightly higher, although not significantly different, levels of viral genome were detected in immunosuppressed hamsters than in immunocompetent hamsters, with SNV genome levels ranging from 105 to 106 (P = 0.0721) (Fig. 3C). However, there was a statistically significant increase in infectious virus in the cyclophosphamide and dexamethasone/cyclophosphamide hamster groups compared to untreated controls (P < 0.0001) (Fig. 3D).
FIG 3.

Immunosuppression of SNV-infected hamsters allows viremia to be detected. Serum and lung isolated from dexamethasone-, cyclophosphamide-, and dexamethasone/cyclophosphamide-treated, SNV-infected hamsters on day 10 postinfection were evaluated for viral genome (A and C) and infectious virus (B and D) by RT-PCR and plaque assay, respectively. Asterisks indicate that results were statistically significant compared to no-treatment controls, as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant.
SNV infection of immunosuppressed hamsters results in marked pathology and widespread dissemination of virus in lung tissues.
Ten days postinfection, three hamsters per treatment group were euthanized and lung tissues were collected for histologic analysis and immunohistochemistry. H&E staining of lung sections showed minimal differences in dexamethasone-treated (Fig. 4A) and untreated (Fig. 4D) hamsters compared to uninfected controls (Fig. 4E). However, cyclophosphamide-treated (Fig. 4B) and dexamethasone/cyclophosphamide-treated (Fig. 4C) hamster lungs exhibited interstitial inflammation and vascular leakage consistent with changes normally seen in hamsters infected with ANDV (Fig. 4F). Immunohistochemistry performed on lung sections from each of the treatment groups showed widespread immunoreactivity in the alveolar septa and endothelial cells of larger vessels, with the most prominent staining noted in lung sections from the dexamethasone/cyclophosphamide treatment group (Fig. 5A to C). Hamsters in the untreated, SNV-infected group showed scattered, minimal immunoreactivity of infected endothelial cells, a finding which is consistent with previous results (Fig. 5D) (12).
FIG 4.
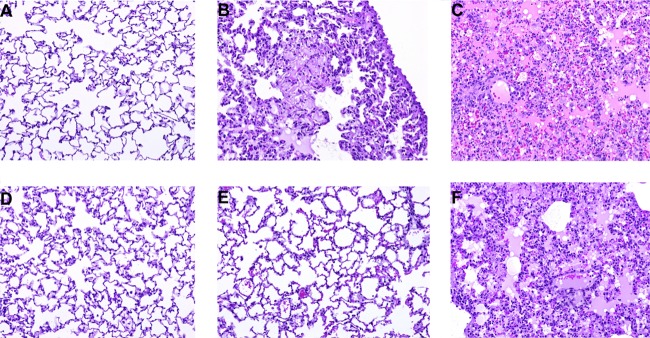
Dexamethasone/cyclophosphamide immunosuppression of SNV-infected hamsters creates lung pathology similar to that of ANDV-infected hamsters. H&E staining of lungs collected on day 10 from hamsters infected with SNV and treated with dexamethasone (A), cyclophosphamide (B), or dexamethasone/cyclophosphamide (C) or untreated (D). An uninfected control (E) and ANDV-infected hamster tissue (F) (12) are shown for comparison. All images were acquired at a magnification of ×20.
FIG 5.
SNV infection of immunosuppressed hamsters allows increased infection of pulmonary endothelial cells detected by immunohistochemistry. Immunohistochemistry using an α-nucleocapsid antibody was performed on lung sections collected on day 10 from hamsters infected with SNV and treated with dexamethasone (A), cyclophosphamide (B), or dexamethasone/cyclophosphamide (C) or untreated. Images were acquired at a magnification of ×20 (inset magnification, ×40).
SNV isolated from immunosuppressed hamsters remains nonpathogenic in immunocompetent hamsters.
Despite the fact that repeated serial passaging of SNV through hamsters does not increase its pathogenicity in immunocompetent hamsters (28), we were interested in determining if passaging SNV through an immunocompromised hamster can alter the virus to the point of creating disease in an immunocompetent host. To this end, 2,000 PFU of SNV isolated from the pleural effluent of a dexamethasone/cyclophosphamide-treated, SNV-infected hamster was used to infect immunocompetent hamsters. These hamsters all survived to day 28 with no signs of disease, but all developed α-N antibodies to the SNV challenge as measured by ELISA, indicating productive but asymptomatic infection (data not shown).
Passive transfer of α-SNV antibodies demonstrates viral specificity in the development of HPS.
To confirm that SNV was the etiologic agent responsible for disease in immunosuppressed hamsters, we showed that SNV neutralizing antibodies can prevent or delay disease in these animals. Two groups of 8 hamsters each were immunosuppressed with dexamethasone and cyclophosphamide in combination according to the strategy detailed in Table 1. All hamsters were then infected with 2,000 PFU of SNV i.m. on day 0. Subsequently, on day 5 postchallenge, one group was administered 12,000 neutralizing antibody units (NAU)/kg of α-SNV antibodies in the form of sera from rabbits vaccinated with an SNV DNA vaccine (Fig. 6A) (29). All hamsters not receiving α-SNV antibodies developed disease and succumbed 12 to 13 days postchallenge. Three hamsters receiving α-SNV antibodies either succumbed or were euthanized on days 17 to 18, with the remaining 5 hamsters surviving to day 28 (P = 0.0001). SNV neutralizing antibodies delayed or prevented lethal disease in hamsters, confirming that HPS disease is specific to SNV infection. Lungs collected on day 28 from surviving hamsters were evaluated for the presence of viral genome by real-time PCR (RT-PCR) (Fig. 6B). Surviving hamsters had comparable levels of viral genome in lung tissue isolated on day 10 postinfection (Fig. 3C).
FIG 6.
HPS disease is specifically caused by SNV infection in immunosuppressed hamsters. Groups of 8 hamsters were immunosuppressed with the combination of dexamethasone and cyclophosphamide according to Table 1. (A) On day 5, a single group of 8 hamsters was injected with 12,000 NAU/kg α-SNV antibodies. Both groups of hamsters were observed for survival. (B) Lung tissue isolated on day 28 postinfection was evaluated for viral genome by RT-PCR. ***, P value of <0.001 compared to immunosuppressed, infected controls.
DISCUSSION
In this report, we describe the first SNV lethal disease small-animal model. To date, evaluations of potential SNV vaccines and immunotherapeutics have involved using either the deer mouse or hamster SNV infection model (29, 30). Attempts to develop an SNV disease model have included experimental infections of rodents (i.e., deer mice and hamsters) using serially passaged virus that has resulted in increased viral replication in tissues, including the lung, liver, and spleen, but no signs of disease (28, 31). In contrast, the immunosuppression methodology incorporated in this study allowed SNV to replicate and disseminate in the hamster host, creating an HPS disease similar to the disease observed in the ANDV hamster disease model and in human HPS cases.
Both SNV and ANDV are New World hantaviruses that cause HPS in humans, and the kinetics and pathology of SNV in an immunocompromised hamster and ANDV in an immunocompetent hamster are remarkably similar. ANDV-infected hamsters will succumb to HPS disease in 10 to 14 days when challenged i.m. with 200 to 2,000 PFU of virus, and this is essentially the same time to death that was observed in the immunocompromised hamsters challenged i.m. with 2,000 PFU of SNV. Viremia can be detected in immunocompromised, SNV-infected hamsters, and these hamsters develop lung pathology similar to the pathology exhibited by immunocompetent, ANDV-infected hamsters.
Infection of immunocompetent Syrian hamsters with cell culture-passaged SNV leads to only low levels of viral dissemination (12). SNV antigen was able to be found in scattered endothelial cells in the kidney, heart, lung, and brain, but the detection of these cells was rare and viremia was never detected. Moreover, only modest changes in neutrophil and lymphocyte cell numbers were observed. In contrast, infection of immunocompetent Syrian hamsters with Syrian hamster-passaged SNV results in widespread and prolonged viral dissemination to endothelial cells in multiple tissues, approaching 108 S-segment RNA copies in lung tissue that were detectable by PCR (28). However, in both cases, infection did not result in disease. In the study presented here, approximately 106 S-segment RNA copies were detected in lung tissue 10 days postinfection in hamsters treated with dexamethasone, cyclophosphamide, or the combination (Fig. 3) but only hamsters receiving cyclophosphamide, alone or in combination with dexamethasone, showed signs of pathology (Fig. 2 and 4). Interestingly, higher titers of infectious virus were also found in the lungs of these animals. This may suggest that certain components of the cellular immune response (limited by cyclophosphamide treatment) regulate pathogenesis following SNV infection.
Cyclophosphamide has been used to create viral disease models in multiple rodent species, including Syrian hamsters (20, 21, 32–37). Cyclophosphamide is an alkylating agent which intercalates into the DNA of actively dividing cells, causing apoptosis resulting in lymphopenia, suppressed B cell activity and activation, suppressed regulatory T cell function, neutropenia, and decreases in macrophage and dendritic cell numbers (14–17, 38, 39). When used in viral infection models, this often leads to increased morbidity and mortality and enhanced viral replication and dissemination. Correspondingly, we did see significant decreases in total WBC counts (Fig. 1) as well as consistently significant decreases in lymphocyte cell numbers and increased titers of infectious virus in the serum and lungs of cyclophosphamide-treated animals. Somewhat surprisingly, the numbers of peripheral blood neutrophils in cyclophosphamide-treated animals were not dramatically different from numbers in untreated animals even though neutrophils, as well as macrophages, are sensitive to apoptosis due to cyclophosphamide (16, 17). However, the activation status of neutrophils, specifically NF-κB activation, can greatly influence neutrophil sensitivity to apoptosis-inducing stimuli (40). This argues that the decrease in WBC counts was a more direct result of decreased lymphocyte numbers (Fig. 1B). Other than a slight increase in neutrophil numbers on day 3 postinfection, treatment of animals with cyclophosphamide did not result in increased numbers of neutrophils compared to untreated animals (Fig. 1C).
Dexamethasone is commonly used to treat or prevent disease in animal models (41–44) and has been used, along with the related glucocorticosteroid methylprednisone, in attempts to treat human hantavirus disease (45–47). Dexamethasone is a glucocorticosteroid which can downregulate proinflammatory cytokine expression, induce anti-inflammatory cytokines and proliferation, and induce apoptosis in multiple cell types (48, 49). Dexamethasone has been shown to be effective in downregulating type I interferon responses by regulating STAT1 activation (50); therefore, we hypothesized that treatment of hamsters with dexamethasone would allow SNV to disseminate and cause disease. Indeed, SNV was able to disseminate following dexamethasone treatment, but dexamethasone treatment alone resulted in a lack of viremia detected 10 days postchallenge and was insufficient to allow SNV to cause disease (Fig. 2 and 4). Dexamethasone alone also resulted in smaller decreases in lymphocyte numbers around the time of virus challenge and failed to control neutrophil proliferation on days 5 and 10. Increased neutrophil numbers may be a result of the inability of steroids to block NF-κB-induced neutrophil activation and proliferation resulting from a strong viral stimulus or the ability of glucocorticoids to inhibit neutrophil apoptosis. Several studies have demonstrated that dexamethasone delays or even prevents neutrophil apoptosis, thus prolonging neutrophil survival and functional responsiveness (51–54), due in part to the stabilization of the anti-apoptotic Mcl-1L protein (55). This, along with increased expression of neutrophil survival factors, such as granulocyte colony-stimulating factor (G-CSF) (56) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (57), which are often induced during viral infections, may explain why dexamethasone-treated animals did not exhibit a more significant decrease in neutrophil numbers. Moreover, treatment of neutrophils with dexamethasone does not appear to inhibit their ability to degranulate and release various antimicrobial reactive oxygen species (51, 58). Earlier studies of ANDV infection of hamsters suggest that hamsters become viremic approximately 6 to 7 days postchallenge (12), and the kinetics of disease are similar between immunocompetent hamsters infected with ANDV and immunocompromised hamsters infected with SNV. Correspondingly, we observed an increase in the number of circulating neutrophils beginning 5 days postchallenge. The increase in neutrophil numbers corresponds with the lower titers of infectious virus found in dexamethasone-treated animals and suggests the possibility that increases in activated neutrophils may protect against hantavirus disease by reducing live virus, possibly via the expression of proinflammatory cytokines, phagocytosis, or activation of endothelial cell responses.
Interestingly, the combination of cyclophosphamide and dexamethasone was uniformly lethal in hamsters infected with SNV whereas cyclophosphamide alone resulted in only 50% mortality and a prolonged disease course. This suggests that the presence of dexamethasone at the time of challenge probably allows the virus a better opportunity to replicate and disseminate by reducing both innately (e.g., skeletal muscle, smooth muscle, and endothelial cells) derived and cellularly (e.g., dendritic cells, NK cells, neutrophils) derived sources of type I interferon. The fact that cyclophosphamide alone still resulted in 50% mortality suggests that cellular sources of type I interferon are important in early clearance of SNV in immunocompetent hamsters.
While liver panels did not show any toxicity in any of the treatment groups in the hamster, demonstrating that the disease is specific to SNV is a key experiment in this study. Using passively transferred α-SNV neutralizing antibodies administered 5 days postinfection, we were able to show protection from lethal disease. This demonstrated that SNV was the etiologic agent that caused the lethal disease in the treated hamsters. This supports the idea that while neutralizing antibodies are not required for protection, they are sufficient to protect (59–65). Furthermore, this experiment demonstrates the practical use of this new animal model for evaluating candidate medical countermeasures to prevent and treat hantavirus disease.
A key limitation to the use of this model is that immunosuppression precludes studying the contribution of the immune system in protection from hantavirus infection and vaccine efficacy in that dexamethasone and cyclophosphamide inhibit a wide range of immune cell types. This makes it difficult to directly evaluate a potential vaccine since these immune cells will be unable to secrete antibody or proliferate upon recognition of the viral antigen. Nevertheless, it is possible to indirectly evaluate vaccines that produce neutralizing antibodies, as illustrated in the passive transfer experiment shown in Fig. 6. Another limitation of the immunosuppression approach is that targeting immune modulation to enhance or prevent specific components of the immune response to hantavirus disease would not be possible due to this general immunosuppression methodology. However, this model would be invaluable for evaluating candidate antiviral therapies and investigating nonimmune mechanisms of disease pathogenesis. Disease models created by immunosuppressing animals also typically limit the extent to which disease pathogenesis can be studied, especially when disease is hypothesized to be mediated by components of the immune system. An interesting observation from this study is that lymphocyte numbers are significantly reduced in animals treated with the combination of cyclophosphamide and dexamethasone. Much has been made regarding the role of T cells in the pathogenesis of hantavirus disease. Indeed, activated hantavirus-specific T cells are found in human HPS cases (6, 66, 67), and correlations have been drawn between disease severity and T cell numbers (68). We and others have demonstrated that T cells are not required for disease pathogenesis in the hamster model of HPS (69, 70), in part due to the observation that cyclophosphamide treatment of ANDV-infected hamsters significantly reduced the number of T cells in the blood and lungs of hamsters but did not alter the course of disease in hamsters. Similarly, here we demonstrate that the combination of dexamethasone and cyclophosphamide significantly reduces lymphocyte numbers, and only when hamsters are immunocompromised can SNV uniformly cause disease. This argues for a disease mechanism that is independent of T cells.
Animal models of infectious disease are invaluable tools to study pathogenesis and to discover and test candidate medical countermeasures. Here, we have used a strategy whereby an immunologically competent animal (i.e., capable of mounting a normal immune response to a vaccine) can be transiently immunosuppressed to allow SNV to disseminate and cause disease that mimics HPS in humans. Further studies will be needed to elucidate the mechanism underlying the pathophysiology in hamsters and then to determine if the same mechanism accounts for the development of HPS in humans. The dexamethasone/cyclophosphamide approach described here differs from the use of transgenic knockout animals with defective innate and/or adaptive immune systems because the animals have a normal immune system up until the time when immunosuppression is induced. Furthermore, immunosuppression is transient and can be restored upon cessation of treatment. It will be of interest to determine if this same approach can be used to develop disease models for hantaviruses that cause hemorrhagic fever with renal syndrome (e.g., Hantaan virus) or for other viruses within the Bunyaviridae family for which there are no practical disease models. It will also be of interest to investigate whether this dexamethasone/cyclophosphamide approach can be used to develop disease models in other animal species that have useful attributes, such as mice for the abundance of immune reagents or nonhuman primates for their more human-like physiology.
ACKNOWLEDGMENTS
We thank Chris Mech, Matthew Josleyn, and Andrew Chrovian for technical assistance and Steven Kern for performing statistical analyses.
This work was funded by the U.S. Army Medical Research and Material Command, Military Infectious Disease Research Program, Program Area T. Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI098933.
Opinions, interpretations, conclusions, and recommendations are ours and are not necessarily endorsed by the U.S. Army or the Department of Defense.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Khan AS, Young JC. 2001. Hantavirus pulmonary syndrome: at the crossroads. Curr. Opin. Infect. Dis. 14:205–209 10.1097/00001432-200104000-00016 [DOI] [PubMed] [Google Scholar]
- 2.Schmaljohn C. 2009. Vaccines for hantaviruses. Vaccine 27(Suppl 4):D61–D64. 10.1016/j.vaccine.2009.07.096 [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention 1993. Update: outbreak of hantavirus infection—southwestern United States, 1993. MMWR Morb. Mortal. Wkly. Rep. 42:441–443 [PubMed] [Google Scholar]
- 4.Schmaljohn C, Nichol ST. 2006. Bunyaviridae, p 1741–1789 In Knipe DM, Howley PM. (ed), Fields virology. Lippincott, Williams, and Wilkins, Philadelphia, PA [Google Scholar]
- 5.Gavrilovskaya I, Gorbunova E, Matthys V, Dalrymple N, Mackow E. 2012. The role of the endothelium in HPS pathogenesis and potential therapeutic approaches. Adv. Virol. 2012:467059. 10.1155/2012/46705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS, Rollin PE, Ksiazek TG, Nichol ST, Mahy BWJ, Peters CJ. 1995. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 7.Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14. 10.1006/viro.2001.1133 [DOI] [PubMed] [Google Scholar]
- 8.Milazzo ML, Eyzaguirre EJ, Molina CP, Fulhorst CF. 2002. Maporal viral infection in the Syrian golden hamster: a model of hantavirus pulmonary syndrome. J. Infect. Dis. 186:1390–1395. 10.1086/344735 [DOI] [PubMed] [Google Scholar]
- 9.Brocato RL, Josleyn MJ, Wahl-Jensen V, Schmaljohn CS, Hooper JW. 2013. Construction and nonclinical testing of a Puumala virus synthetic M gene-based DNA vaccine. Clin. Vaccine Immunol. 20:218–226. 10.1128/CVI.00546-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooper JW, Kamrud KI, Elgh F, Custer D, Schmaljohn CS. 1999. DNA vaccination with hantavirus M segment elicits neutralizing antibodies and protects against Seoul virus infection. Virology 255:269–278. 10.1006/viro.1998.9586 [DOI] [PubMed] [Google Scholar]
- 11.Spik KW, Badger C, Mathiessen I, Tjelle T, Hooper JW, Schmaljohn C. 2008. Mixing of M segment DNA vaccines to Hantaan virus and Puumala virus reduces their immunogenicity in hamsters. Vaccine 26:5177–5181. 10.1016/j.vaccine.2008.03.097 [DOI] [PubMed] [Google Scholar]
- 12.Wahl-Jensen V, Chapman J, Asher L, Fisher R, Zimmerman M, Larsen T, Hooper JW. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81:7449–7462. 10.1128/JVI.00238-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cupps TR, Edgar LC, Fauci AS. 1982. Suppression of human B lymphocyte function by cyclophosphamide. J. Immunol. 128:2453–2457 [PubMed] [Google Scholar]
- 14.Ikezawa Y, Nakazawa M, Tamura C, Takahashi K, Minami M, Ikezawa Z. 2005. Cyclophosphamide decreases the number, percentage and the function of CD25+ CD4+ regulatory T cells, which suppress induction of contact hypersensitivity. J. Dermatol. Sci. 39:105–112. 10.1016/j.jdermsci.2005.02.002 [DOI] [PubMed] [Google Scholar]
- 15.Zhu LP, Cupps TR, Whalen G, Fauci AS. 1987. Selective effects of cyclophosphamide therapy on activation, proliferation, and differentiation of human B cells. J. Clin. Invest. 79:1082–1090. 10.1172/JCI112922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santosuosso M, Divangahi M, Zganiacz A, Xing Z. 2002. Reduced tissue macrophage population in the lung by anticancer agent cyclophosphamide: restoration by local granulocyte macrophage-colony-stimulating factor gene transfer. Blood 99:1246–1252. 10.1182/blood.V99.4.1246 [DOI] [PubMed] [Google Scholar]
- 17.Zuluaga AF, Salazar BE, Rodriguez CA, Zapata AX, Agudelo M, Vesga O. 2006. Neutropenia induced in outbred mice by a simplified low-dose cyclophosphamide regimen: characterization and applicability to diverse experimental models of infectious diseases. BMC Infect. Dis. 6:55. 10.1186/1471-2334-6-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fauci AS, Dale DC, Balow JE. 1976. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann. Intern. Med. 84:304–315. 10.7326/0003-4819-84-3-304 [DOI] [PubMed] [Google Scholar]
- 19.Mateo R, Xiao SY, Guzman H, Lei H, Da Rosa AP, Tesh RB. 2006. Effects of immunosuppression on West Nile virus infection in hamsters. Am. J. Trop. Med. Hyg. 75:356–362 [PubMed] [Google Scholar]
- 20.Schaecher SR, Stabenow J, Oberle C, Schriewer J, Buller RM, Sagartz JE, Pekosz A. 2008. An immunosuppressed Syrian golden hamster model for SARS-CoV infection. Virology 380:312–321. 10.1016/j.virol.2008.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovesi EV, Peters CJ. 1987. Immunosuppression-induced susceptibility of inbred hamsters (Mesocricetus auratus) to lethal-disease by lymphocytic choriomeningitis virus infection. Arch. Virol. 97:61–76. 10.1007/BF01310734 [DOI] [PubMed] [Google Scholar]
- 22.Toth K, Spencer JF, Dhar D, Sagartz JE, Buller RM, Painter GR, Wold WS. 2008. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc. Natl. Acad. Sci. U. S. A. 105:7293–7297. 10.1073/pnas.0800200105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmaljohn AL, Li D, Negley DL, Bressler DS, Turell MJ, Korch GW, Ascher MS, Schmaljohn CS. 1995. Isolation and initial characterization of a newfound hantavirus from California. Virology 206:963–972. 10.1006/viro.1995.1019 [DOI] [PubMed] [Google Scholar]
- 24.Elgh F, Lundkvist A, Alexeyev OA, Stenlund H, Avsic-Zupanc T, Hjelle B, Lee HW, Smith KJ, Vainionpaa R, Wiger D, Wadell G, Juto P. 1997. Serological diagnosis of hantavirus infections by an enzyme-linked immunosorbent assay based on detection of immunoglobulin G and M responses to recombinant nucleocapsid proteins of five viral serotypes. J. Clin. Microbiol. 35:1122–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundstrom JB, McMullan LK, Spiropoulou CF, Hooper WC, Ansari AA, Peters CJ, Rollin PE. 2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 75:6070–6085. 10.1128/JVI.75.13.6070-6085.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trombley AR, Wachter L, Garrison J, Buckley-Beason VA, Jahrling J, Hensley LE, Schoepp RJ, Norwood DA, Goba A, Fair JN, Kulesh DA. 2010. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am. J. Trop. Med. Hyg. 82:954–960. 10.4269/ajtmh.2010.09-0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 28.Safronetz D, Prescott J, Haddock E, Scott DP, Feldmann H, Ebihara H. 2013. Hamster-adapted Sin Nombre virus causes disseminated infection and efficiently replicates in pulmonary endothelial cells without signs of disease. J. Virol. 87:4778–4782. 10.1128/JVI.03291-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hooper JW, Josleyn M, Ballantyne J, Brocato R. 2013. A novel Sin Nombre virus DNA vaccine and its inclusion in a candidate pan-hantavirus vaccine against hantavirus pulmonary syndrome (HPS) and hemorrhagic fever with renal syndrome (HFRS). Vaccine 31:4314–4321. 10.1016/j.vaccine.2013.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medina RA, Mirowsky-Garcia K, Hutt J, Hjelle B. 2007. Ribavirin, human convalescent plasma and anti-β3 integrin antibody inhibit infection by Sin Nombre virus in the deer mouse model. J. Gen. Virol. 88:493–505. 10.1099/vir.0.82459-0 [DOI] [PubMed] [Google Scholar]
- 31.Botten J, Mirowsky K, Kusewitt D, Bharadwaj M, Yee J, Ricci R, Feddersen RM, Hjelle B. 2000. Experimental infection model for Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus). Proc. Natl. Acad. Sci. U. S. A. 97:10578–10583. 10.1073/pnas.180197197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blandford G, Charlton D. 1977. Studies of pulmonary and renal immunopathology after nonlethal primary Sendai viral infection in normal and cyclophosphamide-treated hamsters. Am. Rev. Respir. Dis. 115:305–314 [DOI] [PubMed] [Google Scholar]
- 33.Johnson RA, Prince GA, Suffin SC, Horswood RL, Chanock RM. 1982. Respiratory syncytial virus infection in cyclophosphamide-treated cotton rats. Infect. Immun. 37:369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kercher L, Mitchell BM. 2000. Immune transfer protects severely immunosuppressed mice from murine cytomegalovirus retinitis and reduces the viral load in ocular tissue. J. Infect. Dis. 182:652–661. 10.1086/315781 [DOI] [PubMed] [Google Scholar]
- 35.Kong X, Hellermann GR, Patton G, Kumar M, Behera A, Randall TS, Zhang J, Lockey RF, Mohapatra SS. 2005. An immunocompromised BALB/c mouse model for respiratory syncytial virus infection. Virol. J. 2:3. 10.1186/1743-422X-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sidwell RW, Bailey KW, Morrey JD, Wong MH, Baldwin TJ, Smee DF. 2003. Inhibition of influenza virus infections in immunosuppressed mice with orally administered peramivir (BCX-1812). Antiviral Res. 60:17–25. 10.1016/S0166-3542(03)00113-X [DOI] [PubMed] [Google Scholar]
- 37.Smee DF, Burger RA, Coombs J, Huffman JH, Sidwell RW. 1991. Progressive murine cytomegalovirus disease after termination of ganciclovir therapy in mice immunosuppressed by cyclophosphamide treatment. J. Infect. Dis. 164:958–961. 10.1093/infdis/164.5.958 [DOI] [PubMed] [Google Scholar]
- 38.Mackall CL, Fleisher TA, Brown MR, Magrath IT, Shad AT, Horowitz ME, Wexler LH, Adde MA, McClure LL, Gress RE. 1994. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood 84:2221–2228 [PubMed] [Google Scholar]
- 39.Nakahara T, Uchi H, Lesokhin AM, Avogadri F, Rizzuto GA, Hirschhorn-Cymerman D, Panageas KS, Merghoub T, Wolchok JD, Houghton AN. 2010. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood 115:4384–4392. 10.1182/blood-2009-11-251231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hotta K, Niwa M, Hara A, Ohno T, Wang X, Matsuno H, Kozawa O, Ito H, Kato K, Otsuka T, Matsui N, Uematsu T. 2001. The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-kappa B activation. Eur. J. Pharmacol. 433:17–27. 10.1016/S0014-2999(01)01480-7 [DOI] [PubMed] [Google Scholar]
- 41.Li C, Yang P, Zhang Y, Sun Y, Wang W, Zou Z, Xing L, Chen Z, Tang C, Guo F, Deng J, Zhao Y, Yan Y, Tang J, Wang X, Jiang C. 2012. Corticosteroid treatment ameliorates acute lung injury induced by 2009 swine origin influenza A (H1N1) virus in mice. PLoS One 7:e44110. 10.1371/journal.pone.0044110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Londono P, Komura A, Hara N, Zipris D. 2010. Brief dexamethasone treatment during acute infection prevents virus-induced autoimmune diabetes. Clin. Immunol. 135:401–411. 10.1016/j.clim.2010.01.007 [DOI] [PubMed] [Google Scholar]
- 43.Matsuse H, Kondo Y, Machida I, Kawano T, Saeki S, Tomari S, Obase Y, Fukushima C, Mizuta Y, Kohno S. 2006. Effects of anti-inflammatory therapies on recurrent and low-grade respiratory syncytial virus infections in a murine model of asthma. Ann. Allergy Asthma Immunol. 97:55–60. 10.1016/S1081-1206(10)61370-1 [DOI] [PubMed] [Google Scholar]
- 44.Wigenstam E, Jonasson S, Koch B, Bucht A. 2012. Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology 301:66–71. 10.1016/j.tox.2012.06.020 [DOI] [PubMed] [Google Scholar]
- 45.Singh AE, Werker DH, Boychuk LR, Miedzinski LJ. 1995. Hantavirus pulmonary syndrome: report of four Alberta cases. Can. J. Infect. Dis. 6:184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castillo C, Naranjo J, Sepulveda A, Ossa G, Levy H. 2001. Hantavirus pulmonary syndrome due to Andes virus in Temuco, Chile: clinical experience with 16 adults. Chest 120:548–554. 10.1378/chest.120.2.548 [DOI] [PubMed] [Google Scholar]
- 47.Vial PA, Valdivieso F, Ferres M, Riquelme R, Rioseco ML, Calvo M, Castillo C, Diaz R, Scholz L, Cuiza A, Belmar E, Hernandez C, Martinez J, Lee SJ, Mertz GJ. 2013. High-dose intravenous methylprednisolone for hantavirus cardiopulmonary syndrome in Chile: a double-blind, randomized controlled clinical trial. Clin. Infect. Dis. 57:943–951. 10.1093/cid/cit394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnes PJ. 2006. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br. J. Pharmacol. 148:245–254. 10.1038/sj.bjp.0706736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R. 2013. Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS One 8:e53936. 10.1371/journal.pone.0053936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. 2011. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl. Acad. Sci. U. S. A. 108:9554–9559. 10.1073/pnas.1017296108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cox G. 1995. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 154:4719–4725 [PubMed] [Google Scholar]
- 52.Liles WC, Dale DC, Klebanoff SJ. 1995. Glucocorticoids inhibit apoptosis of human neutrophils. Blood 86:3181–3188 [PubMed] [Google Scholar]
- 53.Meagher LC, Cousin JM, Seckl JR, Haslett C. 1996. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 156:4422–4428 [PubMed] [Google Scholar]
- 54.Nittoh T, Fujimori H, Kozumi Y, Ishihara K, Mue S, Ohuchi K. 1998. Effects of glucocorticoids on apoptosis of infiltrated eosinophils and neutrophils in rats. Eur. J. Pharmacol. 354:73–81. 10.1016/S0014-2999(98)00426-9 [DOI] [PubMed] [Google Scholar]
- 55.Sivertson KL, Seeds MC, Long DL, Peachman KK, Bass DA. 2007. The differential effect of dexamethasone on granulocyte apoptosis involves stabilization of Mcl-1L in neutrophils but not in eosinophils. Cell. Immunol. 246:34–45. 10.1016/j.cellimm.2007.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Raam BJ, Drewniak A, Groenewold V, van den Berg TK, Kuijpers TW. 2008. Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood 112:2046–2054. 10.1182/blood-2008-04-149575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi SD, Voyich JM, Whitney AR, DeLeo FR. 2005. Spontaneous neutrophil apoptosis and regulation of cell survival by granulocyte macrophage-colony stimulating factor. J. Leukoc. Biol. 78:1408–1418. 10.1189/jlb.0605289 [DOI] [PubMed] [Google Scholar]
- 58.Jaovisidha P, Peeples ME, Brees AA, Carpenter LR, Moy JN. 1999. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J. Immunol. 163:2816–2820 [PubMed] [Google Scholar]
- 59.Brocato R, Josleyn M, Ballantyne J, Vial P, Hooper JW. 2012. DNA vaccine-generated duck polyclonal antibodies as a postexposure prophylactic to prevent hantavirus pulmonary syndrome (HPS). PLoS One 7:e35996. 10.1371/journal.pone.0035996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Custer DM, Thompson E, Schmaljohn CS, Ksiazek TG, Hooper JW. 2003. Active and passive vaccination against hantavirus pulmonary syndrome with Andes virus M genome segment-based DNA vaccine. J. Virol. 77:9894–9905. 10.1128/JVI.77.18.9894-9905.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hooper JW, Ferro AM, Wahl-Jensen V. 2008. Immune serum produced by DNA vaccination protects hamsters against lethal respiratory challenge with Andes virus. J. Virol. 82:1332–1338. 10.1128/JVI.01822-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klingstrom J, Stoltz M, Hardestam J, Ahlm C, Lundkvist A. 2008. Passive immunization protects cynomolgus macaques against Puumala hantavirus challenge. Antivir. Ther. 13:125–133 [PubMed] [Google Scholar]
- 63.Liang M, Chu YK, Schmaljohn C. 1996. Bacterial expression of neutralizing mouse monoclonal antibody Fab fragments to Hantaan virus. Virology 217:262–271. 10.1006/viro.1996.0113 [DOI] [PubMed] [Google Scholar]
- 64.Xu Z, Wei L, Wang L, Wang H, Jiang S. 2002. The in vitro and in vivo protective activity of monoclonal antibodies directed against Hantaan virus: potential application for immunotherapy and passive immunization. Biochem. Biophys. Res. Commun. 298:552–558. 10.1016/S0006-291X(02)02491-9 [DOI] [PubMed] [Google Scholar]
- 65.Zhang XK, Takashima I, Hashimoto N. 1989. Characteristics of passive immunity against hantavirus infection in rats. Arch. Virol. 105:235–246. 10.1007/BF01311360 [DOI] [PubMed] [Google Scholar]
- 66.Mori M, Rothman AL, Kurane I, Montoya JM, Nolte KB, Norman JE, Waite DC, Koster FT, Ennis FA. 1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179:295–302. 10.1086/314597 [DOI] [PubMed] [Google Scholar]
- 67.Nolte KB, Feddersen RM, Foucar K, Zaki SR, Koster FT, Madar D, Merlin TL, McFeeley PJ, Umland ET, Zumwalt RE. 1995. Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum. Pathol. 26:110–120. 10.1016/0046-8177(95)90123-X [DOI] [PubMed] [Google Scholar]
- 68.Kilpatrick ED, Terajima M, Koster FT, Catalina MD, Cruz J, Ennis FA. 2004. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 172:3297–3304 [DOI] [PubMed] [Google Scholar]
- 69.Prescott J, Safronetz D, Haddock E, Robertson S, Scott D, Feldmann H. 2013. The adaptive immune response does not influence hantavirus disease or persistence in the Syrian hamster. Immunology 140:168–178. 10.1111/imm.12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hammerbeck CD, Hooper JW. 2011. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 85:9929–9944. 10.1128/JVI.05356-11 [DOI] [PMC free article] [PubMed] [Google Scholar]