

Abstract
Human cytomegalovirus-encoded UL92 plays an essential role in viral replication that has not been resolved. We show here that this gene controls the accumulation of true late (γ2) viral transcripts, a property shared with several other recently evaluated genes (UL79, UL87, UL91, and UL95) conserved among beta- and gammaherpesviruses. When the UL92 mutant virus was evaluated, function was fully complemented by either the natural protein or the homologous Rh127 protein from rhesus cytomegalovirus. N-terminal epitope-tagged UL92 protein is functional, follows complex early-late expression kinetics, and localizes in the nucleus within viral replication compartments. UL92 severely impacts the late (72-h postinfection) expression of nine genes encoding virion proteins (UL32, UL55, UL73, UL75, UL80, UL86, UL99, and UL115), as well as UL91 and itself, but does not influence the levels of UL44, UL82, or UL83 accumulation. Although viral DNA is made at normal levels, viral capsid accumulation in the nucleus is severely compromised in UL92 mutant virus-infected cells, and mature virions are not observed in the cytoplasm. Taken together, UL92 is a key regulator of late viral gene expression, apparently functioning with four other beta- or gammaherpesvirus gene products in a pattern that appears reminiscent of gene regulation in T4 DNA bacteriophage.
INTRODUCTION
Human cytomegalovirus (HCMV) causes significant disease in the developing fetus, as well as in immunocompromised individuals (1), exhibiting a broad tropism for differentiated myeloid, epithelial, endothelial, mesothelial, fibroblast, and neuronal cell types. In all of these settings, viral genes are expressed in a coordinated cascade that is characteristic of all herpesviruses. HCMV replication, although tightly regulated, is prolonged, such that viral late gene expression and progeny accumulate only after 2 to 3 days of infection. Like other herpesviruses, such as the alphaherpesvirus herpes simplex virus 1 (HSV-1), transcription is carried out by host RNA polymerase (Pol) II modified by viral proteins. Like HSV-1, HCMV relies on a virion tegument protein to transactivate immediate-early (IE; also called α) genes (2–4), relying on UL82-encoded pp71 to act on cellular Daxx/ATRX (5, 6). Once expressed, the IE gene products, IE2-p86 (7) and IE1-p72 (8, 9), coordinate (10, 11) the recruitment of host RNA Pol II to activate delayed early (DE; also called β) genes (1). HCMV mutants provided key evidence for a direct role of these IE genes in regulation of gene expression, while unveiling their role in modulating cytokine activation, programmed cell death, and chromatin remodeling (12–16). Once activated, viral UL112-113 gene products associate with specialized nuclear sites (17, 18), guided by IE2-p86, and this leads to recruitment of ppUL84 and ppUL44, the viral DNA Pol processivity (proc) factor into a complex that recognizes the origin of viral DNA replication, oriLyt, to initiate DNA synthesis (19–22), as well as the enhanced production of late (γ) viral gene products that are required for the assembly, maturation, and release of progeny (23).
Like other herpesviruses, HCMV encodes distinct categories of late genes, commonly referred to as leaky late (or γ1) and true late (or γ2); the former are expressed independent of viral DNA synthesis, and the latter are not expressed at all when viral DNA synthesis is blocked by specific inhibitors (1, 24). Genes in the true late category include UL99 (encoding pp28) (25, 26), UL94 (27), UL75 and UL115 (encoding gH/gL) (28–30), UL32 (encoding pp150) (31, 32), the middle transcription start site of UL44 (33–35), and a start site located within UL122 (IE2-p86) encoding IE2-p40 (also called L40) (10, 36). Many other viral transcripts are detected at late times (37). True late gene regulation in HCMV (35, 38, 39), like HSV-1 (40) is controlled by a small, TATA box-proximal region. An unusual, TATT sequence predominates in HCMV γ2 genes (27, 29, 33, 37, 39, 41).
Gammaherpesviruses, such as the mouse γMHV-68 and human Epstein-Barr virus (EBV), have provided direct insights into the function of a distinct set of genes that are conserved in betaherpesviruses but not in alphaherpesviruses, the so-called betagamma genes (42, 43). Open reading frames (ORFs) 18, 24, 30, 31, and 34 are homologs of HCMV UL79, UL87, UL91, UL92, and UL95, respectively, and all are involved in regulating late gene expression in γMHV-68 (44–47). EBV BcRF1, the homolog of γMHV-68 ORF24 and HCMV UL87, interacts with RNA Pol II and functions as a TATA-binding protein (TBP) with specificity for TATT (48, 49). All five HCMV betagamma genes are essential for replication (50, 51), suggesting that control of late transcription via RNA Pol II in this subfamily also relies on a late transcription complex (52) that may include UL79 (32), TBP-like UL87 (48), and UL95 and UL91 (53). In HCMV evidence suggests that this complex associates with the ppUL44 polymerase processivity sliding clamp independent of DNA synthesis (52). Reliance on a simple promoter, the presence of an unusual promoter element (TATT), and the requirement for specific viral genes to modify the core transcription machinery are reminiscent of bacteriophage T4, where the transcription machinery associates with the DNA Pol proc sliding clamp to regulate late genes (54).
Here, we evaluate HCMV UL92 gene function, completing the set of genes influencing late gene activation in γMHV-68 (44–47). Like the other betagamma genes, UL92 is conserved in HCMV strains, as well as in chimpanzee (42, 43), rhesus macaque (55, 56), and rodent (57, 58) cytomegaloviruses (CMVs), in addition to all characterized gammaherpesviruses. We show that full-length UL92 is necessary for the transcriptional activation of true late genes within the nucleus of infected cells. This characterization implicates conserved betagamma gene function controlling transcription of true late genes, findings that are consistent with a modification in host RNA Pol II machinery. (In an accompanying paper, D. Yu et al. [59] demonstrate the role of MCMV protein pM92 in gene expression and suggest that the function of UL92 is a potential target for therapeutic intervention in cases of CMV infection.)
MATERIALS AND METHODS
Plasmid construction.
To construct a plasmid expressing UL92 with three tandem FLAG epitope (3FLAG) tag at the N terminus, the full-length UL92 ORF was amplified by PCR from the HCMV Towne-BAC genome (GenBank accession no. AY315197) (60) and cloned in-frame into the HindIII-XbaI sites of p3FLAG-CMV10 vector (Sigma, St. Louis, MO) to produce p3FLAG-UL92. N-terminally 3FLAG-tagged rhesus macaque CMV (RhCMV) rh127 expression plasmid was constructed by substituting full-length rh127 ORF in p3FLAG-UL92. RhCMV strain 68-1 DNA (GenBank accession no. NC_006150) (61, 62) was generously provided by Peter Barry, University of California Davis. An influenza virus hemagglutinin (HA) epitope-tagged UL93 protein expression plasmid was constructed by inserting a PCR fragment containing the sequence of the UL93 ORF, along with a C-terminal HA epitope tag, into the HindIII-XbaI sites of pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA). For the construction of 3FLAG-UL92 expressing lentiviral vector, the 3FLAG-UL92 ORF was inserted into BamHI-NheI sites of pLV-EF1α-MCS-IRES-Puro lentiviral vector (Biosettia, San Diego, CA) to produce pLV-3FLAG-UL92.
Cells.
Primary human foreskin-derived fibroblasts (HFs) and 293T cells were cultured as described previously (53). HFs between passages 8 and 15 were used in all experiments. To generate HFs expressing 3FLAG-UL92, a lentivirus stock was prepared by transfecting pLV-3FLAG-UL92, psPAX2 (63), and VSV-G expressing plasmid into 293T cells. Low-passage HFs were transduced with lentiviral vector by spinoculation (1,000 × g for 1 h) and selected using 0.5 μg/ml of puromycin (Invitrogen, Carlsbad, CA.
BAC mutagenesis and recombinant viruses.
Bacterial artificial chromosomes (BACs) recombineering was carried out using GalK positive/negative selection protocols (64), as described for UL91 (53). To create UL92-GalK-BAC, double-stranded linear DNA carrying overhang of 50 bp of homology flanking both sides of the desired deletion region from Towne-BAC, in addition to the GalK cassette, was amplified by PCR using the pGalK plasmid (64) as a template with the primers 5′-GGCACGTCCAGAACGCGTTTACCGAGGAGATCCAGTTACACTCGCTCTAcctgttgacaattaatcatcggca-3′ and 5′-GGTCGAGCACCAGATGTAGAGGCAATTGCTCATCGTCAGCGAACCGCGCtcagcactgtcctgctcctt-3′. In the PCR primer sequences, nucleotides homologous to Towne-BAC DNA are indicated in uppercase letters, and nucleotides in the GalK cassette are indicated in lowercase letters. The double-stranded linear DNA was introduced into the SW102 Escherichia coli (64) carrying Towne-BAC by electroporation, and recombinants containing the GalK cassette were selected in galactose and chloramphenicol. UL921-28BAC was generated by replacing the GalK cassette using a linear fragment containing a single nucleotide insertion to introduce a translational stop plus frameshift at the Cys29 codon in UL92. The linear fragment was obtained by PCR from Towne-BAC DNA with the primer 5′-GGCACGTCCAGAACGCGTTTACCGAGGAGATCCAGTTACACTCGCTCTAGCGTAGCACGCGCTGCTTTCGCACG-3′, where the boldface “T” and the underlining indicate the insertion of a frameshift and the stop codon, respectively, and the primer 5′-GGTCGAGCACCAGATGTAGAGGCAATTGCTCATCGTCAGCGAACCGCGC-3′. The PCR-amplified DNA was transformed into SW102 E. coli containing UL92-GalK-BAC and selected on 2-deoxy-galactose minimal agar plates. UL921-167BAC was also generated in this manner by a single nucleotide insertion to introduce a translational stop plus frameshift at the Asp168 in UL92. To create a marker rescue virus (UL92-R-BAC), the UL921-28BAC was first replaced by a GalK cassette and then subjected to a second round of recombination by replacing GalK cassette with wild-type UL92. To make 3FLAG-UL92-BAC, the double-stranded linear fragment was amplified by PCR using the p3FLAG-UL92 plasmid as a template and the primers 5′-CCTGTCGTCCCTGCCCTTCTCCTGTTCCCTCCGCCCCCAAAACTGTCAGCTGACGCTCAgccaccatggactacaaagaccatgacgg-3′ and 5′-GGTCGAGCACCAGATGTAGAGGCAATTGCTCATCGTCAGCGAACCGCGCCTCAAACGCCAGATCCGAATACAGG-3′. In these PCR primer sequences, the boldface “T” and the underlining indicate the insertion of a frameshift and the stop codon at Asp78 in UL91 ORF, respectively, and nucleotides homologous to those in the Towne-BAC are shown in uppercase letters, and the nucleotides in the three tandem FLAG epitope tag are shown in lowercase letters. DNA was transformed into SW102 E. coli containing UL92-GalK-BAC and selected on 2-deoxy-galactose minimal agar plates. All generated BACs were assessed by fragment length polymorphism (RFLP) with restriction enzymes, PCR analysis, and DNA sequencing of the insert region. To generate recombinant viruses, BAC DNA and pp71 expression plasmid (53) were transfected into HFs or 3FLAG-UL92 expressing HFs. Infectious titers were determined by standard plaque assay. For the secondary spread assay, HFs were transfected with UL92-GalK-BAC, the pp71 expression plasmid, and one of the p3FLAG constructs in the presence or absence of UL93-HA and then analyzed as described previously (53). For live cell imaging and the quantification of fluorescence intensities, cell cultures were analyzed with a Live Cell Imaging System IncuCyte ZOOM (Essen Bioscience, Ann Arbor, MI) (53, 65).
Chemicals and antibodies.
Protein synthesis inhibitor cycloheximide (CHX) and HCMV DNA synthesis inhibitor phosphonoformate (PFA) were purchased from Sigma (St. Louis, MO). Proteasome inhibitor MG132 was obtained from Calbiochem (San Diego, CA). Mouse anti-FLAG (clone M2) monoclonal antibody was purchased from Sigma (St. Louis, MO). Mouse monoclonal antibodies against pp150 (clone 36-14) (31) and MCP (clone 28-4) (66) were kindly provided by William Britt, University of Alabama, Birmingham, AL. Other antibodies used in the present study were described previously (53).
Immunoblotting.
293T cells were transfected with the 3FLAG-UL92 or 3FLAG-rh127 constructs in Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and lysed in 2% sodium dodecyl sulfate (SDS)-containing sample buffer with β-mercaptoethanol at 48 h posttransfection. HFs infected with various viruses at the indicated multiplicity of infection (MOI) and times postinfection and processed as described previously (53). Extracellular virus particles were purified as described previously (67). Briefly, HFs were infected with Towne-BAC or 3FLAG-UL92-BAC virus (MOI of 3) and harvested at 4 dpi. Cellular debris was removed by low-speed centrifugation, and extracellular virus particles were collected on a 30% sucrose cushion (SW 41 rotor, Beckman L-80 ultracentrifuge, 24,000 rpm, 1 h) and resuspended and solubilized in 2% SDS-containing sample buffer in the presence of β-mercaptoethanol. Proteins were resolved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane (Immobilon; Millipore, Billerica, MA), probed with primary antibodies, and reacted with horseradish peroxidase-conjugated secondary antibody. A chemifluorescent signal was detected with an ECL Western blotting detection reagent (GE Healthcare, Buckinghamshire, United Kingdom).
Microscopy.
Immunofluorescence analysis (IFA) and transmission electron microscopy (TEM) were performed as described previously (53).
DNA and RNA analyses.
Accumulation of intracellular viral DNA or RNA was measured by quantitative PCR (qPCR) or reverse transcription coupled to qPCR (qRT-PCR) (53). Primers (5′-AGTACGACGCGTGTGTCATC-3′ and 5′-ATCCGAATACAGGTGCGTTT-3′) were used for detection of the UL92 transcript. Other primers used in the present study were described previously (53).
RESULTS
Construction of UL92 mutant BAC clones.
In order to study function of UL92 in HCMV replication, UL92 mutant viruses were constructed from Towne-BAC (60) in a manner that did not interfere with expression or functions of adjacent genes. Insertion and deletion mutagenesis has indicated that the highly conserved UL92 gene is essential for replication in both AD169 and Towne strains of HCMV (50, 51), but that overlap with the adjacent UL91 (53), as well as a UL93, makes it difficult to be confident in the independent importance of UL92. The overlapping C terminus of UL91 is dispensable (53), but the overlapping UL93 region is of unknown function. Alignment analysis revealed that UL92 homologous amino acid sequences from primate betaherpesviruses are highly conserved across the protein, and the N-terminal region of UL92 contains an N-terminal cysteine-rich region and a highly charged C terminus (Fig. 1A). Based on this information, Towne-BAC UL92 was substituted with a GalK cassette to generate UL92-GalK-BAC as an intermediate to introduce individual translational stop-frameshift mutations at Cys29 and Asp168 (UL921-28BAC and UL921-167BAC, respectively; Fig. 1A and B). In addition, a marker rescue virus (UL92-R-BAC) was generated from UL921-28BAC, as described in Materials and Methods. These mutant BACs were designed to disrupt UL92 within the cysteine-rich N terminus (UL921-28BAC), as well as to remove the highly charged C terminus (UL921-167BAC) without altering UL93. Two short ORFs (ORFL219C and ORFL220C) of unknown function are translated at low levels from the cDNA strand in this region (68). The 21-amino-acid (aa) ORFL219C remained intact in both mutant viruses, and the 50-aa ORFL220C was intact in UL921-28BAC but was mutated (C-terminal 6-aa YLLCFE to VLGLLV) in UL921-167BAC. The genomic integrity of all mutant and control BAC clones was verified by RFLP with restriction enzymes (Fig. 1C and data not shown), and the entire UL92 locus was sequenced to ensure that only the intended changes were introduced.
FIG 1.

Construction of UL92 mutant BAC clones. (A) Amino acid sequence alignment of UL92 homologs from HCMV (AY315197), RhCMV (NC_006150), CCMV (NC_003521), HHV6 (NC_001664), and HHV7 (NC_001716). Identical amino acids (black) and CMV conserved amino acids (gray) are highlighted. The positions of Cys29 and Asp168 where individual translational stop-frameshift mutations were introduced are indicated by black arrowheads. The N-terminal cysteine-rich region and a highly charged C terminus are indicated by gray bars. (B) Schematic diagrams of the UL89.2-to-UL89.1 region in Towne-BAC with other UL92 mutant BACs shown below. (C) Electrophoretic separation of BglII-digested Towne-BAC and UL92 mutant BACs on 0.8% agarose as a diagnostic for integrity of the genomes. Insertion of GalK cassette into the UL92 locus results in an ∼0.7-kb increase in the size of the BglII fragment (white arrowhead) compared to the 2.6-kb band (black arrowhead) in Towne-BAC and UL92 mutant BACs.
UL92 is essential and its C-terminal charged region is crucial for HCMV replication.
In order to characterize the replication properties of UL92 mutant virus without complementation, we first transfected each of the mutant BACs into HFs and identified transfected cells, as well as viral spread, by using green fluorescent protein (GFP) expression from a cassette within the viral genome (60). Towne-BAC and UL92-R-BAC generated similar sizes and morphologies of GFP-positive plaques, whereas UL921-28BAC and UL921-167BAC only produced single GFP-positive cells that remained at 10 days posttransfection (Fig. 2A to D). Thus, the replication defect observed in genome-wide mutagenesis (50, 51) was sustained. Replication of UL921-28BAC and UL921-167BAC was restored by transient expression of 3FLAG-UL92 (data not shown), a finding consistent with the direct role of this protein and not other potential gene products from this region. Because homologous recombination between the viral genome and 3FLAG-UL92-expressing plasmid might complicate this result, we performed additional complementation experiments with UL92-GalK-BAC using UL92 together with UL93 expression plasmids in a secondary spread assay. Replication of UL92-GalK-BAC was reconstituted when complemented by transfection with 3FLAG-UL92 in the presence but not in the absence of HA-tagged UL93 (Fig. 2E and F). These results reinforce earlier studies (50, 51) and show UL92 is necessary for viral replication. Finally, RhCMV rh127 also complemented replication (Fig. 2E and F), demonstrating functional conservation of this gene in another primate CMV.
FIG 2.
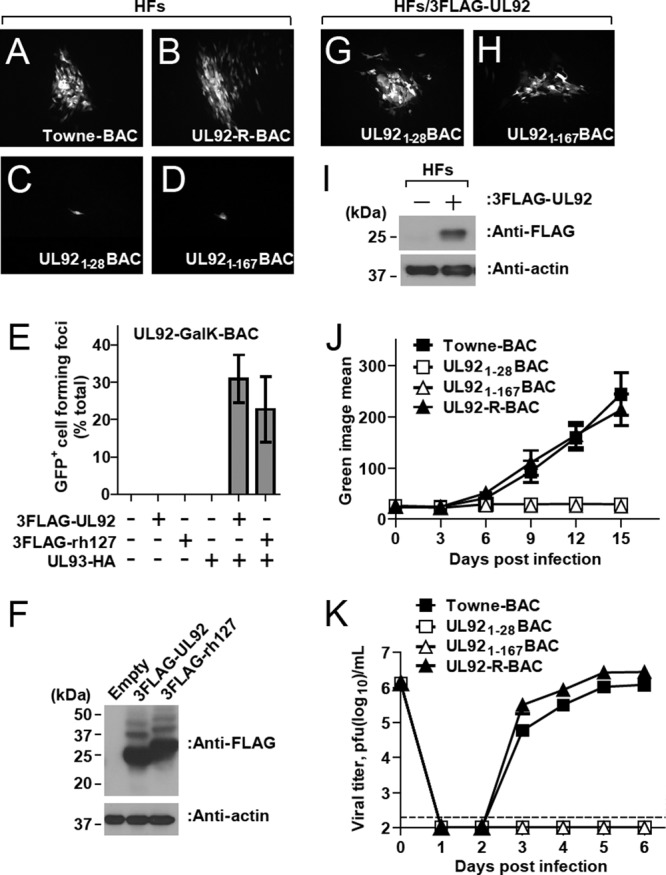
Replication properties of UL92 mutant BAC-derived viruses. (A to D) Typical GFP-positive plaque phenotypes at 10 days posttransfection of HFs with Towne-BAC (A), UL92-R-BAC (B), UL921-28BAC (C), and UL921-167BAC (D). Original magnification, ×100. (E) Secondary spread assay evaluation of plasmids 3FLAG-UL92 or 3FLAG-rh127 to complement UL92-GalK-BAC in the presence or absence of UL93-HA. (F) 293T cell lysates at 48 h posttransfection with indicated plasmids were subjected to immunoblotting with anti-FLAG antibody. The antibody against β-actin was used as a loading control. (G and H) Fluorescent images of GFP-positive cells at 10 days posttransfection of 3FLAG-UL92-expressing HFs with UL921-28BAC (G) and UL921-167BAC (H). Original magnification, ×100. (I) Lysates from 3FLAG-UL92-expressing HFs were subjected to immunoblotting with anti-FLAG antibody. The antibody against β-actin was used as a loading control. (J) Multistep growth curves (MOI of 0.01) of Towne-BAC and UL92 mutant viruses in HFs. Virus replication was quantified for 15 days based on the GFP signal intensity, which is expressed from a cassette within the viral genome. (K) Single-step growth curves (MOI of 3) of Towne-BAC- and UL92 mutant BAC-derived viruses in HFs. Virus titers were determined by plaque assays of supernatants collected at daily intervals. The data point at 0 dpi indicates the input virus dose. The detection limit of the plaque assay is indicated by a dashed line.
Based on the transient complementation results, we established 3FLAG-UL92 expressing HFs using lentivirus-mediated transduction for propagation of UL921-28BAC and UL921-167BAC viruses. 3FLAG-UL92 expressing HFs supported the replication of UL921-28BAC and UL921-167BAC (Fig. 2G and H), producing titers of mutant virus nearly identical to parental Towne-BAC. Expression of 3FLAG-UL92 in the transduced HFs was confirmed by immunoblotting (Fig. 2I). These results demonstrate that 3FLAG-tagged UL92 is fully functional and the phenotypes of UL921-28BAC and UL921-167BAC viruses was a direct result of UL92 disruption. In order to further access the growth defect of UL92 mutant viruses, HFs were infected with mutant viruses or controls (Towne-BAC and UL92-R-BAC) at an MOI of 0.01, and virus replication was quantified based on the GFP signal intensity, which is expressed from a cassette within the viral genome. UL921-28BAC and UL921-167BAC viruses failed to replicate, whereas UL92-R-BAC virus exhibited similar viral replication kinetics to parental Towne-BAC (Fig. 2J). When UL92 mutant viruses were evaluated under synchronous infection condition at an MOI of 3, neither mutant virus produced any detectable progeny where control viruses replicated well (Fig. 2K). Taken together, these data demonstrate that UL92 is essential for viral replication and a high MOI of UL92-null virus did not overcome a very tight replication block. This pattern is very similar to the phenotype of UL91 mutant virus (53). In addition, the highly charged aa 168 to 201 C-terminal region is crucial for UL92 function.
UL92 is expressed with complex early-late kinetics.
Although these results are consistent with UL92 encoding a protein, this gene product was not detected in a recent comprehensive translational profiling experiment (68). In order to analyze UL92 protein expression and localization, Towne-BAC was engineered to tag UL92 protein with three tandem FLAG epitope at the N terminus, based on the ability of this protein to fully complement mutant viruses when transduced into HFs (Fig. 2G and H). C-terminal tagged UL92 was not useful in the context of the viral genome because this disrupted the N terminus of UL93 (Fig. 1B), which is also an essential gene (50, 51). Guided by this information, we took advantage of observations showing that the region of UL91 downstream of aa 72 was completely dispensable for viral replication (53) and generated 3FLAG-UL92-BAC in a virus expressing UL911-97 (Fig. 3A). We introduced a translational stop frameshift within the Asp98 codon of UL91 to avoid translation of the dispensable region. This virus exhibited similar viral replication kinetics compared to Towne-BAC in HFs over a 15-day time course under low-MOI conditions (Fig. 3B).
FIG 3.
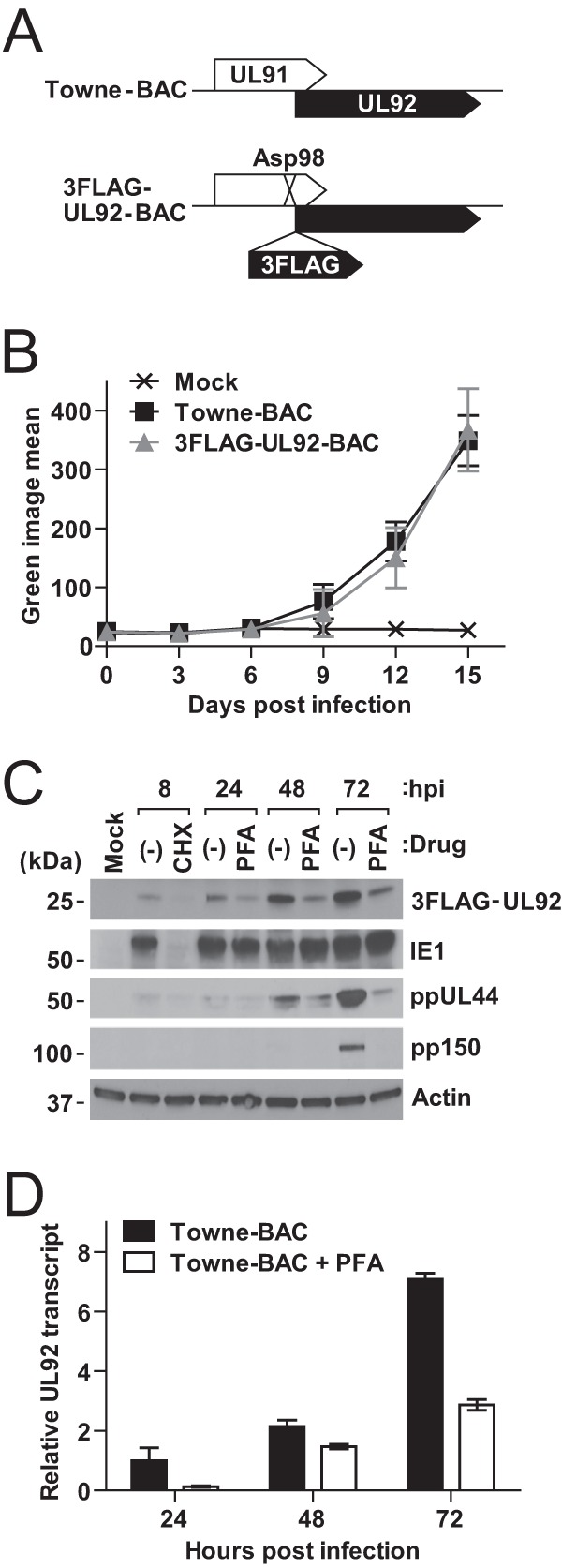
UL92 expression. (A) Schematic diagram of the genome structure of 3FLAG-UL92-BAC generated from Towne-BAC. Three tandem FLAG epitope tag and translational stop-frameshift mutations were introduced at N terminus of UL92 and Asp98 in UL91, respectively. (B) Multistep growth curves (MOI of 0.01) of Towne-BAC and 3FLAG-UL92-BAC viruses in HFs. Virus replication was quantified for 15 days based on the fluorescence intensity from virally encoded GFP. (C) Time course of 3FLAG-UL92 expression after 3FLAG-UL92-BAC virus infection of HFs at an MOI of 3 in the absence (−) or presence of CHX (50 μg/ml) or PFA (300 μg/ml). Shown are the accumulated levels of 3FLAG-UL92, along with IE1, ppUL44, pp150, or β-actin, at the indicated times as determined by immunoblot analysis. (D) UL92 transcript levels during Towne-BAC infection of HFs at an MOI of 3 in the presence or absence of PFA (300 μg/ml). Total RNA was isolated at the indicated times postinfection, and the amount of UL92 transcript was quantified by qRT-PCR using primers specific to the UL92 genes and normalized to β-actin. The normalized amount of UL92 transcripts in the cells infected with Towne-BAC in the absence of PFA at 24 hpi was set to 1.
In order to assess UL92 protein accumulation during infection, we next subjected 3FLAG-UL92-BAC to an immunoblot analysis. When infected at an MOI of 3, 3FLAG-UL92 protein was detected in HFs as early as 8 hpi and reached a steady state by 48 to 72 hpi (Fig. 3C). This tagged protein was detected in infected cells at expected size (∼25 kDa), as predicted from the amino acid sequence, indicating that we had included a bona fide start codon, along with the epitope tag insertion. The immunoblot signal at 8 hpi was dependent on de novo translation and was not detected when cells were pretreated (1 h), infected (1 h), and maintained (8 h) in CHX (50 μg/ml) (Fig. 3C). Signal was also absent in mock-infected cells. The accumulation of 3FLAG-UL92 protein was markedly decreased when infected cells were treated with 300 μg/ml of PFA (Fig. 3C), an expression pattern consistent with either DE (β) or leaky late (γ1) kinetics that is reminiscent of UL44 transcription where multiple promoters become activated throughout infection (33). Expression levels of representative IE (IE1), DE (UL44), and late (pp150) confirmed the desired impact of CHX and PFA treatment to the cells (Fig. 3C). To exclude the possibility that this pattern reflected the influence of the three tandem FLAG epitopes, HFs were infected with Towne-BAC at an MOI of 3 and UL92 transcript levels were quantified during infection. The UL92 transcript followed a similar complex pattern, accumulating abundantly at late times of infection with levels significantly, but not completely, reduced by treatment with 300 μg/ml of PFA (Fig. 3D). Thus, similar to the adjacent gene, UL91 (53), or to well-characterized UL44 (33), UL92 has a complex expression pattern that starts very early and continues to rise at late times in a pattern that is sensitive to inhibition of DNA synthesis.
DNA replication compartment formation and viral DNA synthesis are independent of UL92.
In order to determine whether UL92 function is required for formation of DNA replication compartments (RCs), HFs were infected with UL921-28 BAC or Towne-BAC at an MOI of 0.3. ppUL44, the virus-encoded DNA Pol proc factor, was used as a marker for RC formation. UL921-28 BAC infection resulted in formation of ppUL44-positive nuclear foci at 24 hpi that was comparable to Towne-BAC (Fig. 4A to D). The formation of RCs in UL921-28BAC-infected cells was also indistinguishable from parental Towne-BAC at 72 hpi (Fig. 4E to H). A similar result was obtained when HFs were infected with UL921-167BAC virus (data not shown), indicating that RCs form independently of UL92 function.
FIG 4.
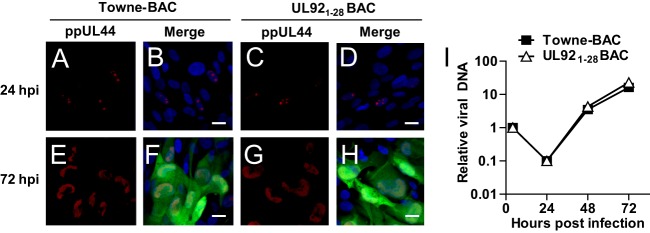
UL92 function and viral DNA synthesis. The immunofluorescence of HFs infected with Towne-BAC (A, B, E, and F) or UL921-28BAC (C, D, G, and H) viruses at an MOI of 0.3 was determined. Cells were fixed at 24 (A to D) or 72 (E to F) hpi for IFA. ppUL44 protein was visualized with mouse anti-ppUL44 monoclonal antibody and Alexa Fluor 568-conjugated anti-mouse IgG secondary antibody (red) (A, C, E, and G). The merged images (B, D, F, and H) include virus-encoded GFP fluorescence (green), DAPI (4′,6′-diamidino-2-phenylindole) staining, which identifies nuclei (blue). Original magnification, ×630. Bars, 20 μm. (I) Viral DNA replication in HFs infected with Towne-BAC and UL921-28BAC viruses at an MOI of 0.3. The accumulation of viral DNA at the indicated hpi was quantified by qPCR using primers specific for the IE1 gene and normalized to β-actin. The normalized amount of viral DNA in the cells infected with Towne-BAC at 4 hpi was set at 1.
Next, in order to compare viral DNA synthesis, total DNA was purified at the indicated times postinfection from HFs infected with UL921-28 BAC or Towne-BAC at an MOI of 0.3 and subjected to qPCR analysis (Fig. 4I). The accumulation of viral DNA in UL921-28BAC-infected cells was indistinguishable from parental Towne-BAC. A similar result was obtained when HFs were infected with UL921-167BAC virus (data not shown). These results demonstrate that, similar to the impact of UL91, UL79, UL87, and UL95 (32, 53, 69), robust viral DNA synthesis does not require UL92 function.
UL92 protein is recruited to the nucleus during infection and is associated with mature virus particles.
We first examined the intracellular localization of UL92 expressed alone in the absence of any other viral factors. Although no detectable FLAG staining occurred in nontransduced HFs (Fig. 5A and B), 3FLAG-UL92 showed a diffuse distribution in uninfected lentiviral vector-transduced HFs (Fig. 5C and D). In order to define the cellular distribution of UL92 during infection, HFs were infected with 3FLAG-UL92-BAC virus at an MOI of 3 and analyzed at 72 hpi by IFA with anti-FLAG antibody. Although FLAG staining was absent in HFs infected with Towne-BAC (Fig. 5E to H), FLAG epitope staining was detected in kidney-shaped nuclei of all 3FLAG-UL92-BAC-infected cells (Fig. 5I to L) according to an intranuclear distribution pattern reminiscent of ppUL44 and RCs (see Fig. 4A and B). Therefore, UL92 is recruited to the nucleus during infection and into RCs as they form over the course of infection.
FIG 5.

UL92 protein is recruited to the nucleus during infection and is virion-associated. Immunofluorescence of HFs (A and B) or 3FLAG-UL92-transduced HFs (C and D) fixed and stained with mouse anti-FLAG monoclonal antibody and Alexa Fluor 568-conjugated anti-mouse IgG secondary antibody (red) (A and C). The merged images (B and D) include DAPI staining which identifies nuclei (blue). HFs were infected with Towne-BAC (E to H) or 3FLAG-UL92-BAC (I to L) viruses at an MOI of 0.3. The cells were fixed at 72 hpi for IFA. 3FLAG-UL92 protein was visualized with mouse anti-FLAG monoclonal antibody and Alexa Fluor 568-conjugated anti-mouse IgG secondary antibody (red) (E and I). Shown are virally encoded GFP fluorescence (green) (F and J), DAPI staining which identifies nuclei (blue) (G and K) and the merged images (H and L). Original magnification, ×630. Bars, 20 μm. (M) UL92 protein is associates with viral particles. HF total cell lysate or culture supernatant was collected at 96 hpi from infected with Towne-BAC or 3FLAG-UL92-BAC (MOI of 3). Extracellular virus particles were collected by ultracentrifugation. All samples were subjected to SDS-PAGE, followed by immunoblotting with anti-MCP, anti-IE1/2, anti-ppUL44, or anti-FLAG antibodies. The arrow and asterisk indicate 3FLAG-UL92 proteins and nonspecific bands, respectively. ECV, extracellular viral particles.
In order to determine whether UL92 associates with mature virions and dense bodies, extracellular particles were collected from culture medium during Towne-BAC or 3FLAG-UL92-BAC infections by ultracentrifugation through a 30% sucrose cushion. These preparations were denatured, run on an SDS-polyacrylamide gel and immunoblotted to detect MCP, IE1, ppUL44, or FLAG epitope (3FLAG-UL92) (Fig. 5M). MCP, as expected, was readily detected in the sample of total cell and particle preparations. Particle preparations did not contain detectable nonstructural IE1 and ppUL44 antigens but did contain 3FLAG-UL92. Even though UL92 protein is difficult to detect immediately after infection (see Fig. 3C), it is present in extracellular virions and/or dense bodies. The levels of UL92 protein are low given the lack of detection in recent translational profiling of late-phase transcripts (68).
UL92 has a crucial role for viral replication before capsid assembly.
In order to directly assess the step in viral replication where UL92 acts, we used TEM analysis of cells infected with UL921-28BAC or Towne-BAC viruses. HFs were infected with viruses at an MOI of 3, and successful infection was confirmed by GFP expression under fluorescence microcopy. Whereas abundant virus particles and dense bodies associated with a juxtanuclear cytoplasmic assembly compartment (AC) was observed in Towne-BAC-infected cells, there were no obvious cytoplasmic particles or organized AC in UL921-28BAC virus-infected cells at 5 dpi (data not shown). In nuclei, all three types of capsid particles (A, lacking a scaffold or dense core; B, containing a scaffold; C, containing a dense core) (70) were detected in Towne-BAC-infected cells (Fig. 6A), but intranuclear capsid forms were difficult to find in UL921-28BAC-infected cells. A few examples of capsid forms (A, B, and C) were identified after careful scanning of several nuclei (Fig. 6B and data not shown). Thus, UL92 is necessary for efficient capsid assembly in the nucleus.
FIG 6.
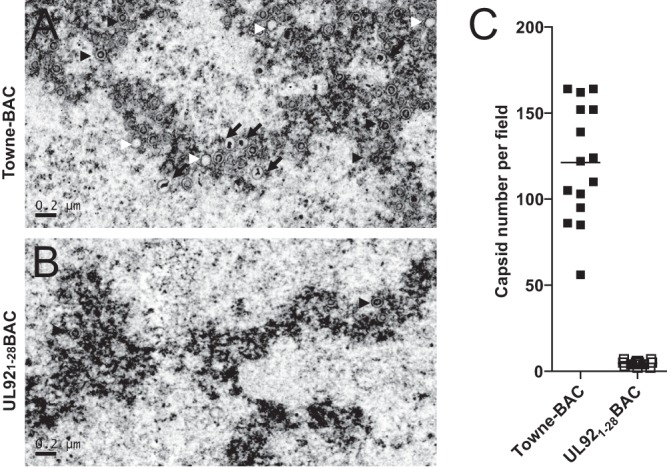
TEM of Towne-BAC or UL921-28BAC virus-infected cells. HFs were infected with Towne-BAC (A) or UL921-28BAC virus (B) at an MOI of 3 and fixed for TEM at 5 dpi. The white arrowheads point to A capsids, the black arrowheads point to B capsids, and the arrows point to C capsids. Scale bars are indicated. (C) Quantification of intranuclear capsids from electron micrographs. Five nuclei were randomly selected for each sample and three independent fields of image were counted from each nucleus at a magnification of ×10,000.
Next, we quantified capsid forms in randomly selected nuclei. The mean numbers of intranuclear capsids per nuclear field in UL921-28BAC-infected cells were much lower than in Towne-BAC-infected cells at 5 dpi (Fig. 6C). Cells infected with UL921-167BAC virus showed the same reduced levels of nuclear capsids as UL921-28BAC (data not shown). Together, these results indicate that the elimination of UL92 severely impacts an essential replication step after viral DNA replication but before capsid assembly during the viral replication cycle. Because HCMV DNA accumulated at normal levels here, as well as with other betagamma gene mutants (32, 53, 69), the accumulation and turnover of viral DNA appears to be completely independent of both capsid accumulation and the process of encapsidation of viral DNA. This surprising observation is supported by the UL91 mutant virus phenotype as well (53).
UL92 is required for accumulation of late viral proteins.
To better identify the steps during infection where UL92 functions, we used immunoblot analysis to assess the accumulation of representative viral proteins: IE (IE1), DE (ppUL44), and late (gB, pp28, pp150, and MCP). When HFs were infected with UL921-28BAC at an MOI of 3, the accumulation of pp28, pp150, and MCP were markedly reduced at 72 and 96 hpi compared to cell infected control Towne-BAC, whereas both IE1 and ppUL44 levels were comparable, and gB was modestly decreased in the cells infected with the mutant virus (Fig. 7A). Thus, UL92 is required for the accumulation of true late (γ2) proteins (pp28 and pp150) and also influences the levels of other late proteins (MCP and, possibly, gB) in a pattern reminiscent of UL91 mutants, as well as other betagamma gene mutants (32, 53, 69).
FIG 7.
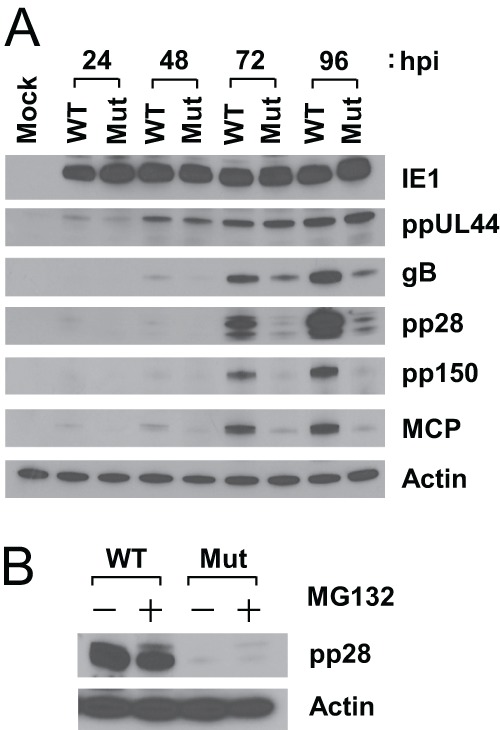
Immunoblot analysis of viral proteins from HFs infected with Towne-BAC or UL921-28BAC virus. (A) HFs were infected at an MOI of 3 and harvested at the indicated hpi. Shown are the accumulated levels of IE1, ppUL44, gB, pp28, pp150, MCP, or β-actin as determined by immunoblotting. (B) HFs were infected as described above and maintained in the absence (−) or presence (+) of 20 μM MG132 from 72 to 96 hpi. Cell lysates were prepared at 96 hpi and analyzed by immunoblotting with anti-pp28 antibody or with antibody against β-actin as a loading control. WT, Towne-BAC; Mut, UL921-28BAC.
Next, to test the possibility that UL92 has a role in reducing the degradation of viral late proteins, we assessed the impact of proteasome inhibitor on accumulation of pp28 protein, as a representative of late protein, in UL921-28BAC-infected cells. When infected cells are treated with 20 μM MG132 from 72 to 96 hpi, the accumulation of pp28 was not restored (Fig. 7B), suggesting that UL92 acts on the accumulation of late proteins independent of the proteasome.
UL92 is required for the efficient transcription of viral late genes.
In order to further define UL92 impacts on viral late gene transcript levels, we analyzed the accumulation of viral RNA for 14 characterized genes. Total RNA was extracted from HFs infected with Towne-BAC or UL921-28BAC viruses at an MOI of 3 and assessed by qRT-PCR at 24, 48, and 72 hpi (Fig. 8). Whereas IE1 (IE or α) or UL44 (DE or β) transcripts accumulated at comparable levels during mutant and control virus infection, gB (DE or β) levels were modestly decreased in UL92-null virus, a finding consistent with the impact on protein levels. The levels of leaky-late (γ1) transcripts, UL82 (pp71) and UL83 (pp65), were unchanged: however, UL91 and UL92 transcripts were reduced at 72 hpi when UL92 was abrogated, indicating that the transcription of both UL92 and the adjacent UL91 genes are upregulated by UL92 function. The accumulations of the true-late (γ2) transcripts UL32 (pp150), UL75 (gH), UL115 (gL) and UL99 (pp28) and the late (γ) transcripts UL73 (gN), UL80 (PR-AP), and UL86 (MCP) were all substantially reduced in the absence of UL92. Taken together, our results demonstrate that UL92 modestly impacts leaky late (γ1) genes but is absolutely crucial for efficient expression of true-late (γ2) viral genes, following a common pattern that seems to define the set of five betagamma genes (32, 53, 69).
FIG 8.
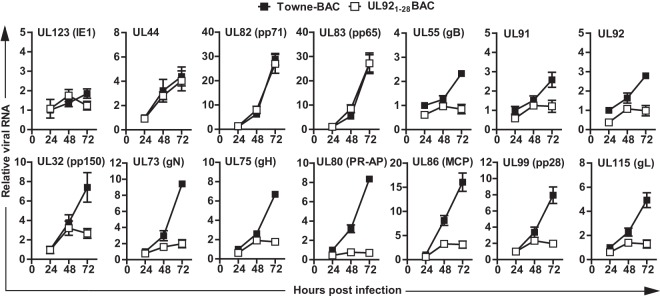
UL92 is required for the accumulation of viral late transcripts. HFs were infected with both UL921-28BAC and parental Towne-BAC viruses at an MOI of 3, total RNA was isolated at the indicated times postinfection, and viral RNA levels were quantified by qRT-PCR using primers specific to the indicated viral genes and normalized to β-actin. The normalized amount of viral transcripts in the cells infected with Towne-BAC at 24 hpi was set at 1.
DISCUSSION
In this study, we have dissected the essential functions of UL92 and shown that the 201-aa nuclear UL92 protein provides crucial late gene regulation during HCMV infection. UL92 is expressed with complex kinetics and is recruited into RCs at late times of infection. Without UL92, viral replication is blocked by the failure to accumulate late viral transcripts even though the formation of RCs and viral DNA synthesis proceed normally. UL92 is absolutely crucial for the accumulation of the true late (γ2) class of viral genes by supporting the efficient transcription rather than blocking protein degradation. As a consequence of low levels of late structural proteins, the production of viral capsids fails in UL92 mutant virus-infected cells. Given the replication defect of UL921-167BAC mutant virus, the highly charged aa 168 to 201 C-terminal region is crucial for function. Expression of the RhCMV homolog restored the replication defect of UL92-null virus, suggesting that other CMV homologs are likely to have similar functions. These observations place UL92 with other HCMV betagamma genes that have been characterized (32, 53, 69) and in line with hallmark work in gammaherpesviruses (44–49). The phenotype of HCMV UL92 mutants is reminiscent of the γMHV-68 ORF31 mutant (45), a sequence homolog of this gene. From this and previous studies on UL79, UL87, UL91, and UL95 (32, 53, 69), there appears to be common theme across the betagamma genes in activation of true late (γ2) gene transcription.
UL92 protein lacks an obvious nuclear localization signal and shows a diffuse distribution in uninfected HFs. This protein is specifically localized to the RC-like pattern in the nucleus at a late time of infection, suggesting that UL92 interacts with other RC gene products, potentially UL79 (32) or UL44 (69). The localization we observed for UL92 with the RC-like region in infected cells is similar to other betagamma late gene regulators, UL79, UL87, UL91, and UL95 (32, 53, 69), suggesting that all of these may assemble and function as a complex. Despite this pattern, viral DNA synthesis proceeds normally in the absence of UL92 or the other late gene regulators (32, 53, 69). Given that sufficient accumulation of viral late proteins is required for capsid formation, viral DNA synthesis and capsid assembly are completely uncoupled, reinforcing the observation made with UL91-null virus (53). On one hand, the initiation of viral late gene expression by viral and/or host factors remains largely unexplored. A reporter plasmid driven by the previously characterized (38) UL99 late promoter (positions −40 to +106) was not activated by transient expression of UL79, UL87, UL91, UL92, and/or UL95 (data not shown), suggesting that viral late gene expression requires additional viral factors or processes that accompany infection, such as oriLyt-dependent DNA synthesis. It is possible that HCMV UL79, UL87, and UL95 control of late transcription in a complex guided via the viral DNA Pol proc factor, ppUL44 (52), although there is currently no direct evidence that any of the betagamma gene products interact with ppUL44. In gammaherpesviruses, EBV BcRF1, the homolog of HCMV UL87 and γMHV-68 ORF24, acts as a TATA-binding protein, interacting preferentially with TATT to regulate late gene expression during infection (48, 49). In addition, γMHV-68 ORF30 and ORF34, homologs of HCMV UL91 and UL95, regulate the binding of RNA Pol II to the late gene promoters (47). This evidence all points to a direct contribution of betagamma late gene regulators in the assembly of a late gene RNA Pol II transcriptional complex. Further studies on the interaction partners of UL92 and the regulation of UL87 since a TBP will be important to begin to dissect the mechanism of HCMV late transcriptional regulation.
The body of evidence that both HCMV UL91 (53) and UL92 control viral late transcription broadens the relevance of this process in betaherpesviruses and, separately, raises the question of how the alphaherpesviruses regulate late gene expression can be effectively achieved without homologs of these proteins. Both UL91 and UL92 are essential for the expression of the same set of true late (γ2) genes UL32 (pp150), UL73 (gN), UL75 (gH), UL80 (PR-AP), UL86 (MCP) UL99 (pp28), and UL115 (gL), as well as leaky late (γ1) expression of UL55 (gB), in addition to UL91 and UL92 themselves. It seems likely all of these act via UL87, given its relationship to gammaherpesvirus late TBP with specificity for TATT (48, 49), the sequence that also characterizes HCMV true late promoters (27, 29, 33, 37, 39, 41). In HSV-1, the late gene expression is also controlled by a small, TATA box-proximal region: however, regulation does not involve novel TATT sequences, at least based on the characterized γ2 gC promoter (40). Even though there are some common features, alphaherpesviruses manage without a set of regulatory genes that are conserved in beta- and gammaherpesviruses. These viruses appear to have evolved a common late gene expression strategy. The accumulating evidence for a specific complex that coordinates host RNA Pol II in beta- and gammaherpesviruses brings to mind the precision with which bacteriophage T4 recognizes simple late transcriptional promoters with small proteins that link RNA Pol to the DNA Pol proc factor as a sliding clamp to provide access to promoters as viral DNA synthesis proceeds (54). It seems tempting to speculate that betagamma genes function is a related way, as a single complex or as alternative complexes to modulate RNA Pol II recognition of HCMV late promoters to assure appropriate levels of late transcripts.
This study advances the understanding of mechanism of viral late gene transcription, completing the set of genes influencing late gene activation in beta- and gammaherpesviruses. HCMV UL92 is required for viral late gene transcription at the RC-like region in the nucleus and thus, consequently, essential for viral replication, like the other betagamma genes, UL79, UL87, UL91, and UL95. How these viral late gene transactivators interact with viral and/or host factors and its relationship to core components of RNA Pol II transcriptional machinery, as well as viral DNA synthesis, will be the subject of future investigation.
ACKNOWLEDGMENTS
We thank Hong Yi and the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University for electron microscopy. Confocal microscopy was performed at the Cell Imaging and Microscopy Core at Emory Winship Cancer Institute. Peter Barry (University of California Davis) generously provided RhCMV genome DNA. William Britt (University of Alabama, Birmingham, AL) kindly provided pp150 and MCP antibodies. Linda Roback helped with the cell culture. We thank Mocarski lab members for critical reading of the manuscript as well as for useful discussions during this project. Finally, we appreciate helpful correspondence with Dong Yu (Washington University and Novartis), Hiroki Isomura (Gunma University), and Peter Geiduschek (UC San Diego) in this area.
Public Health Service grant RO1 AI020211 funded this research. S.O. is a visiting scholar at the Emory University supported by Shionogi & Co., Ltd., Japan.
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.Mocarski ES, Jr, Shenk T, Griffith P, Pass RF. 2013. Cytomegaloviruses, p 1960–2014 In Knipe DM, Howley PM. (ed), Fields virology, 6th ed. Lippincott/The Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 2.Liu B, Stinski MF. 1992. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 66:4434–4444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 79:7792–7802. 10.1128/JVI.79.12.7792-7802.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate-early viral genes in human cytomegalovirus-infected cells Proc. Natl. Acad. Sci. U. S. A. 97:14506–14511. 10.1073/pnas.97.26.14506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543–12554. 10.1128/JVI.01215-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavalai N, Stamminger T. 2010. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133 [DOI] [PubMed] [Google Scholar]
- 7.Stinski MF, Petrik DT. 2008. Functional roles of the human cytomegalovirus essential IE86 protein. Curr. Top. Microbiol. Immunol. 325:133–152. 10.1007/978-3-540-77349-8_8 [DOI] [PubMed] [Google Scholar]
- 8.Greaves RF, Mocarski ES. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72:366–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gawn JM, Greaves RF. 2002. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J. Virol. 76:4441–4455. 10.1128/JVI.76.9.4441-4455.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stinski MF, Meier JL. 2007. Immediate-early CMV gene regulation and function, p 241–263 In Arvin AM, Campadelli-Fiume G, Mocarski ES, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 11.White EA, Spector DH. 2007. Early CMV gene expression and function, p 264–294 In Arvin AM, Campadelli-Fiume G, Mocarski ES, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 12.Marshall EE, Bierle CJ, Brune W, Geballe AP. 2009. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 83:4112–4120. 10.1128/JVI.02489-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brune W. 2011. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 157:144–150. 10.1016/j.virusres.2010.10.012 [DOI] [PubMed] [Google Scholar]
- 14.McCormick AL, Mocarski ES. 2013. Cell death pathways controlled by cytomegaloviruses, p 263–276 In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol I Caister Scientific Press, Norfolk, United Kingdom [Google Scholar]
- 15.Reeves MB. 2011. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 157:134–143. 10.1016/j.virusres.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mocarski ES, Upton JW, Kaiser WJ. 2011. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 12:79–88. 10.1038/nrg2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishov AM, Maul GG. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134:815–826. 10.1083/jcb.134.4.815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishov AM, Stenberg RM, Maul GG. 1997. Human cytomegalovirus immediate-early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 138:5–16. 10.1083/jcb.138.1.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anders DG, Kerry JA, Pari GS. 2007. CMV DNA synthesis and late viral gene expression, p 295–310 In Arvin AM, Campadelli-Fiume G, Mocarski ES, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom [Google Scholar]
- 20.Pari GS. 2008. Nuts and bolts of human cytomegalovirus lytic DNA replication. Curr. Top. Microbiol. Immunol. 325:153–166. 10.1007/978-3-540-77349-8_9 [DOI] [PubMed] [Google Scholar]
- 21.Kim YE, Ahn JH. 2010. Role of the specific interaction of UL112-113 p84 with UL44 DNA polymerase processivity factor in promoting DNA replication of human cytomegalovirus. J. Virol. 84:8409–8421. 10.1128/JVI.00189-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagele D, Rossetto CC, Tarrant MT, Pari GS. 2012. Analysis of the interactions of viral and cellular factors with human cytomegalovirus lytic origin of replication, oriLyt. Virology 424:106–114. 10.1016/j.virol.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 20:392–401. 10.1016/j.tim.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mocarski ES. 2007. Betaherpes viral genes and their functions, p 204–230 In Arvin AM, Campadelli-Fiume G, Mocarski ES, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 25.Adam BL, Jervey TY, Kohler CP, Wright GL, Jr, Nelson JA, Stenberg RM. 1995. The human cytomegalovirus UL98 gene transcription unit overlaps with the pp28 true late gene (UL99) and encodes a 58-kilodalton early protein. J. Virol. 69:5304–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wing BA, Huang ES. 1995. Analysis and mapping of a family of 3′-coterminal transcripts containing coding sequences for human cytomegalovirus open reading frames UL93 through UL99. J. Virol. 69:1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wing BA, Johnson RA, Huang ES. 1998. Identification of positive and negative regulatory regions involved in regulating expression of the human cytomegalovirus UL94 late promoter: role of IE2-86 and cellular p53 in mediating negative regulatory function. J. Virol. 72:1814–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leatham MP, Witte PR, Stinski MF. 1991. Alternate promoter selection within a human cytomegalovirus immediate-early and early transcription unit (UL119-115) defines true late transcripts containing open reading frames for putative viral glycoproteins. J. Virol. 65:6144–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McWatters BJ, Stenberg RM, Kerry JA. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology 303:309–316. 10.1006/viro.2002.1614 [DOI] [PubMed] [Google Scholar]
- 30.Wang SK, Duh CY, Wu CW. 2004. Human cytomegalovirus UL76 encodes a novel virion-associated protein that is able to inhibit viral replication. J. Virol. 78:9750–9762. 10.1128/JVI.78.18.9750-9762.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986. 10.1128/JVI.74.2.975-986.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. Human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 85:4841–4852. 10.1128/JVI.02344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leach FS, Mocarski ES. 1989. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J. Virol. 63:1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isomura H, Stinski MF, Kudoh A, Nakayama S, Iwahori S, Sato Y, Tsurumi T. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 81:6197–6206. 10.1128/JVI.00089-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isomura H, Stinski MF, Kudoh A, Murata T, Nakayama S, Sato Y, Iwahori S, Tsurumi T. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638–1646. 10.1128/JVI.01917-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White EA, Del Rosario CJ, Sanders RL, Spector DH. 2007. The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. J. Virol. 81:2573–2583. 10.1128/JVI.02454-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760. 10.1073/pnas.1115861108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohler CP, Kerry JA, Carter M, Muzithras VP, Jones TR, Stenberg RM. 1994. Use of recombinant virus to assess human cytomegalovirus early and late promoters in the context of the viral genome. J. Virol. 68:6589–6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerry JA, Priddy MA, Kohler CP, Staley TL, Weber D, Jones TR, Stenberg RM. 1997. Translational regulation of the human cytomegalovirus pp28 (UL99) late gene. J. Virol. 71:981–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Homa FL, Glorioso JC, Levine M. 1988. A specific 15-bp TATA box promoter element is required for expression of a herpes simplex virus type 1 late gene. Genes Dev. 2:40–53. 10.1101/gad.2.1.40 [DOI] [PubMed] [Google Scholar]
- 41.Jahn G, Kouzarides T, Mach M, Scholl BC, Plachter B, Traupe B, Preddie E, Satchwell SC, Fleckenstein B, Barrell BG. 1987. Map position and nucleotide sequence of the gene for the large structural phosphoprotein of human cytomegalovirus. J. Virol. 61:1358–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312. 10.1099/vir.0.79888-0 [DOI] [PubMed] [Google Scholar]
- 43.Davison AJ, Bhella D. 2007. Comparative betaherpesviral genome and virion structure, p 177–203 In Arvin AM, Campadelli-Fiume G, Mocarski ES, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 44.Arumugaswami V, Wu TT, Martinez-Guzman D, Jia Q, Deng H, Reyes N, Sun R. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730–9740. 10.1128/JVI.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia Q, Wu TT, Liao HI, Chernishof V, Sun R. 2004. Murine gammaherpesvirus 68 open reading frame 31 is required for viral replication. J. Virol. 78:6610–6620. 10.1128/JVI.78.12.6610-6620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong E, Wu TT, Reyes N, Deng H, Sun R. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761–6764. 10.1128/JVI.02726-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu TT, Park T, Kim H, Tran T, Tong L, Martinez-Guzman D, Reyes N, Deng H, Sun R. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265–2273. 10.1128/JVI.01785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyrwicz LS, Rychlewski L. 2007. Identification of herpes TATT-binding protein. Antivir. Res. 75:167–172. 10.1016/j.antiviral.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 49.Gruffat H, Kadjouf F, Mariame B, Manet E. 2012. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J. Virol. 86:6023–6032. 10.1128/JVI.00159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228. 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401. 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, Tsurumi T. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 85:6629–6644. 10.1128/JVI.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Omoto S, Mocarski ES. 2013. Cytomegalovirus UL91 is essential for transcription of viral true late (γ2) genes. J. Virol. 87:8651–8664. 10.1128/JVI.01052-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geiduschek EP, Kassavetis GA. 2010. Transcription of the T4 late genes. Virol. J. 7:288. 10.1186/1743-422X-7-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Searles RP, Bergquam EP, Axthelm MK, Wong SW. 1999. Sequence and genomic analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, Desrosiers RC. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388–3398. 10.1128/JVI.74.7.3388-3398.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833–8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brocchieri L, Kledal TN, Karlin S, Mocarski ES. 2005. Predicting coding potential from genome sequence: application to betaherpesviruses infecting rats and mice. J. Virol. 79:7570–7596. 10.1128/JVI.79.12.7570-7596.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu D, Chapa TJ, Perng Y-C, French A. 2014. Murine cytomegalovirus protein pM92 is a conserved regulator of viral late gene expression. J. Virol. 88:131–142. 10.1128/JVI.02684-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75:1870–1878. 10.1128/JVI.75.4.1870-1878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW. 2003. Complete sequence and genomic analysis of rhesus cytomegalovirus. J. Virol. 77:6620–6636. 10.1128/JVI.77.12.6620-6636.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rivailler P, Kaur A, Johnson RP, Wang F. 2006. Genomic sequence of rhesus cytomegalovirus 180.92: insights into the coding potential of rhesus cytomegalovirus. J. Virol. 80:4179–4182. 10.1128/JVI.80.8.4179-4182.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A. 93:11382–11388. 10.1073/pnas.93.21.11382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCormick AL, Roback L, Wynn G, Mocarski ES. 2013. Multiplicity-dependent activation of a serine protease-dependent cytomegalovirus-associated programmed cell death pathway. Virology 435:250–257. 10.1016/j.virol.2012.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chee M, Rudolph SA, Plachter B, Barrell B, Jahn G. 1989. Identification of the major capsid protein gene of human cytomegalovirus. J. Virol. 63:1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tandon R, Mocarski ES. 2011. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J. Virol. 85:7129–7141. 10.1128/JVI.02549-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. 10.1126/science.1227919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Isomura H, Stinski MF. 2013. Coordination of late gene transcription of human cytomegalovirus with viral DNA synthesis: recombinant viruses as potential therapeutic vaccine candidates. Expert opinion on therapeutic targets. 17:157–166. 10.1517/14728222.2013.740460 [DOI] [PubMed] [Google Scholar]
- 70.Gibson W. 2006. Assembly and maturation of the capsid, p 231–244 In Reddehase MJ. (ed), Cytomegaloviruses: pathogenesis, molecular biology, and infection control. Caister Scientific Press, Norfolk, United Kingdom [Google Scholar]