

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) causes Kaposi's sarcoma and primary effusion lymphoma. KSHV-infected cells are predominantly latent, with a subset undergoing lytic reactivation. Rta is the essential lytic switch protein that reactivates virus by forming transactivation-competent complexes with the Notch effector protein RBP-Jk and promoter DNA. Strikingly, Rta homolog analysis reveals that prolines constitute 17% of conserved residues. Rta is also highly phosphorylated in vivo. We previously demonstrated that proline content determines Rta homotetramerization and function. We hypothesize that proline-directed modifications regulate Rta function by controlling binding to peptidyl-prolyl cis/trans isomerases (PPIases). Cellular PPIase Pin1 binds specifically to phosphoserine- or phosphothreonine-proline (pS/T-P) motifs in target proteins. Pin1 dysregulation is implicated in myriad human cancers and can be subverted by viruses. Our data show that KSHV Rta protein contains potential pS/T-P motifs and binds directly to Pin1. Rta transactivation is enhanced by Pin1 at two delayed early viral promoters in uninfected cells. Pin1's effect, however, suggests a rheostat-like influence on Rta function. We show that in infected cells, endogenous Pin1 is active during reactivation and enhances Rta-dependent early protein expression induced by multiple signals, as well as DNA replication. Surprisingly, ablation of Pin1 activity by the chemical juglone or dominant-negative Pin1 enhanced late gene expression and production of infectious virus, while ectopic Pin1 showed inhibitory effects. Our data thus suggest that Pin1 is a unique, dose-dependent molecular timer that enhances Rta protein function, but inhibits late gene synthesis and virion production, during KSHV lytic reactivation.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) (or human herpesvirus 8) is the etiological agent of Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL) (1). KS has gained clinical relevance due to its increased prevalence and virulence in human immunodeficiency virus type 1 (HIV-1)-infected patients, whose risk of KS is up to 20,000 times higher than that of non-KSHV-infected individuals (2). While treatment has reduced mortality, the virus remains a potent threat in developing nations (3).
KSHV, a member of the Gammaherpesviridae family, exists as a multicopy, double-stranded DNA episome in infected host cells (4, 5). While the majority of KSHV-infected cells contain latent virus, a small percentage of cells support “spontaneous” lytic reactivation (6–11), which produces viral oncoproteins and infectious virions essential for the growth and survival of KSHV tumors.
We and others have shown that KSHV protein Rta (replication and transcription activator, the ORF50 gene product) is the lytic switch necessary and sufficient for the onset of KSHV lytic reactivation in infected PEL cell models (12–14). Though Rta expression is sufficient to reactivate KSHV in a population of cells, single-cell assays suggest that it is not sufficient to reactivate the virus uniformly in every Rta-expressing cell (13, 15). Rta, a 120-kDa transcription factor, directly transactivates downstream viral and cellular genes through interactions with essential cofactors, including KSHV delayed early protein Mta (ORF57) (15–18) and cellular Notch pathway effector recombination-signal binding protein (RBP-Jk) (19–22).
Our previous data suggest that proline-directed modifications may be another significant mechanism for regulating Rta. We previously demonstrated that the proline content of the leucine heptapeptide repeat (LR) domain of Rta dramatically determines the oligomeric state of the cognate protein (23). In fact, mutating three leucines to prolines within the LR allowed Rta to almost exclusively form tetramers that functioned identically to wild-type (WT) Rta. In addition, 17% of conserved Rta residues in members of the Herpesviridae family are prolines. Many conserved prolines lie within critical functional domains of Rta, including regions that contribute to oligomerization, DNA binding, and RBP-Jk binding. Together with the observation that Rta is strongly phosphorylated in vivo (12), the extensive conservation of proline implies that proline-directed modifications may be important in regulating Rta function.
One potential proline-directed modification of Rta is prolyl isomerization. Rta contains 15 potential binding and regulatory motifs for the cellular peptidyl-prolyl cis/trans isomerase (PPIase) Pin1. Pin1 is a pleiotropic cell cycle regulator and tumor suppressor (24, 25). The 18-kDa protein has a WW DNA-binding domain containing two conserved tryptophans and a PPIase isomerization domain. Together, they target Pin1 to phosphoserine- or phosphothreonine-proline (pS/T-P) motifs in substrate proteins and catalyze the trans-to-cis conversion of proline (26–28). Pin1 prolyl isomerization can alter phosphoprotein function, cellular localization, or stability by rendering cis-form motifs resistant to kinase and phosphatase activity. Pin1's “locking” mechanism thereby can control the timing and amplitude of a specific process, such as protein folding or the G1/S checkpoint (27, 28). Overexpression of Pin1 has also been implicated in abnormal growth and pathophysiology of many tumor cell types that range from breast to prostate cancers (26). Pin1 function is often cell type and condition specific (24, 26, 29–33). For instance, in a study of Pin1 overexpression in tumor tissue types, the majority of uterine and prostate tumor samples had increases of Pin1, whereas pancreatic, endometrial, and most kidney tumors displayed no Pin1 overexpression. Prostate and colon cancer cell lines contain three- to fourfold-higher levels of Pin1 compared to those of healthy (noncancerous) cell types, which also can vary: low or undetectable in most nonactively dividing tissues, moderate in the pancreas and kidney, and strong in Fallopian tube ciliated cells and ovarian granulosa cells. Pin1 deficiency, conversely, is thought to be caused by a variety of aging phenotypes, including osteoporosis, telomere shortening, and neurodegenerative diseases such as Alzheimer's disease. Finally, changing conditions of healthy cells can affect Pin1 expression and activity, including cell cycle progression, oxidative stress, and postpregnancy breast development.
Importantly, various labs demonstrate that Pin1 can be subverted during viral infection. Pin1 downregulates expression of APOBEC3G (apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G), an HIV-1 replication antagonist (34). Pin1 stabilizes Tax oncoprotein of human T-cell leukemia virus type 1 (HTLV-1) (35). Pin1 destabilizes human interferon regulatory factor 3 (IRF3), which subsequently modulates the innate antiviral response (36). Pin1 enhances the function of the catalytic subunit of Epstein-Barr virus (EBV) viral DNA (vDNA) polymerase (37). Despite these examples, the literature is generally limited concerning Pin1's role in viral pathogenesis, and nothing has been reported on Pin1 function in KSHV-infected cells. It is therefore pertinent to examine a potential relationship between Pin1 activity and KSHV lytic reactivation.
In this report, we provide evidence that Pin1 is expressed and active in infected PEL cells and directly interacts with Rta protein. Our data show that while Pin1 promotes early events in reactivation, it inhibits late events. We found that Pin1 is a complex, time- and dosage-dependent stimulator of Rta transactivation of downstream promoters and of viral DNA replication. However, Pin1 inhibits production of mature viruses. Thus, Pin1 acts as a novel molecular timer that provides KSHV with a sustainable, highly adapted determinant of the initiation, progression, and completion of the productive reactivation cycle. We propose that Pin1 deregulation may promote viral delayed early oncogene expression while limiting host cell lysis.
MATERIALS AND METHODS
Cell culture, transfections, and luciferase assays.
A doxycycline (Dox)-inducible Rta cell line TREx BCBL-1-Rta (a gift of J. Jung, University of Southern California [USC] [38]), a wild-type, KSHV-infected B lymphoma cell line BCBL-1, and uninfected B lymphoma cell line BL-41 were maintained in RPMI 1640 supplemented with 12% fetal bovine serum (Sigma) as previously described (15, 22). For transfections, cells were seeded at 1 × 107 cells/transfection in 250 μl of incomplete RPMI 1640 in 4-mm cuvettes, electroporated at 975 μF and 200 V (BCBL-1) or 975 μF and 224 V (BL-41), and incubated in 10-ml complete medium.
Human cell line 293, wild-type and Pin1−/− murine embryonic fibroblasts (MEFs) (gifts of Kun Ping Lu [Harvard]), BAC16 (WT and Rta stop)-harboring cell line iSLK, and Vero rKSHV.219, Vero rKSHV.294, and 293 MSR tet-OFF cells (gifts of Jeff Vieira), were maintained as previously described (15, 22, 39–44). iSLK cell lines contain Dox-inducible Rta and were gifts of D. Ganem (42). The cells were seeded 1 day prior to transfection at 1 × 105 to 2 × 105/well in six-well plates or at 2.5 × 106/well in 100-mm plates. Transfections were performed by TransIT-LT1 transfection reagent (Mirus) with up to 5 μg of DNA according to the manufacturer's instructions.
All transfections were performed in duplicate or triplicate and included empty vector pcDNA3 to normalize the total amount of DNA per transfection. For luciferase reporter assays, up to 27 μg total DNA was used per transfection. pcDNA3.1-His-lacZ was cotransfected as an internal control, and normalization with β-galactosidase was performed as previously described (22). For viral antigen expression, at least 700 cells per technical replicate were counted.
Plasmids and bacmid.
All plasmids were amplified, purified, and sequenced as previously described (22). pcDNA3 is the empty vector control, and pcDNA3.1-His-lacZ, expresses His-tagged β-galactosidase (Invitrogen). pcDNA3-FLc50, pGem3-FLc50, and pcDNA3-FLg50 express wild-type Rta/ORF50 protein from cDNA (the “c” in FLc50) or genomic DNA (the “g” in FLg50) (12, 13). Plasmid pRSET 0.8, which expresses the His6-tagged Rta polypeptide C50 (amino acids [aa] 525 to 691), has been described previously (12–14). pGex3x expresses the glutathione S-transferase (GST) tag alone (GE Life Sciences). Promoter/reporter plasmids pFL57-GL3, pORF57 (−132)-GL3basic, pNut-1-GL3, pNut-1-GL3 (−706), and CMV-Luc (CMV stands for cytomegalovirus, and Luc stands for luciferase), were previously published (13, 15, 22). pH2b-GFP, a gift from G. Wahl, expresses green fluorescent protein (GFP)-tagged histone H2b (Addgene plasmid 11680) (45). pEGFP-C1-Pin1 (EGFP stands for enhanced GFP), which expresses GFP-tagged Pin1, and pGST-Pin1, which expresses GST-tagged, WT Pin1, were gifts from K. P. Lu (46). pcDNA3.1-Pin1 S16A expresses dominant-negative Pin1 and was a gift from Richard Venema (47). KSHV BAC16 was a gift from Jae Jung (39).
pGem3-Rta170-400 expresses Rta amino acids 170 to 400 (Rta 170-400) and was constructed by PCR amplification of the pGem3-FLc50 template using primers that introduced 5′ EcoRI and 3′ XbaI sites and translation initiation and termination codons. The amplicon was cloned into pGem3 (Promega) that had been digested with the same enzymes.
pcDNA3-Pin1 expresses WT Pin1, and it was constructed by PCR amplification of pEGFP-C1-Pin1 using primers that introduced 5′ BamHI and 3′ EcoRV sites and a translation initiation codon. The amplicon was cloned into pcDNA3 that had been digested with the same enzymes.
Protein expression and purification.
Glutathione S-transferase fusion protein production was conducted as described in reference 19 with the exceptions that single-colony inoculations contained 34 mg/ml chloramphenicol and 500-ml cultures did not contain glucose.
Whole-cell protein extracts from tissue cell cultures were prepared as previously described (23, 48) by lysis in radioimmunoprecipitation assay (RIPA) or reporter assay (Promega) buffers.
GST fusion protein pulldown assays.
Crude 1-ml Escherichia coli extracts containing GST fused to RBP-Jk (GST–RBP-Jk), GST fused to Pin1 (GST-Pin1), or GST alone were incubated with preswollen glutathione-Sepharose beads and l-[35S]methionine-labeled Rta or Pin1 proteins, programmed in rabbit reticulocyte lysates (RRL) (Promega TnT T7-coupled transcription/translation system), as previously described (48). Washes were performed with NETN+ (12). Up to 0.5 μl of each programmed RRL was put aside as protein input. Protein signals in polyacrylamide gels were amplified in 0.5 M salicylic acid, dried on Whatman paper in a gel drier, and visualized by Typhoon 9410 molecular imager (Amersham/GE) and autoradiography. Pulldowns conducted using PEL cell lysates were performed as described in reference 48.
Coimmunoprecipitations.
Total protein extracts were prepared from untreated or sodium butyrate (3 mM)-treated BCBL-1 cells in lysis buffer (150 mM NaCl, 1% NP-40, and 50 mM Tris [pH 8.0]) supplemented with dithiothreitol (DTT) and protease inhibitors. Total protein extracts (2,500 μg) were mixed with anti-Pin1 antibody (1:30 dilution; Epitomics) or IgG control and incubated with rotation for 2 h at 4°C, followed by 1 h in the presence of preequilibrated protein A Sepharose beads. The complexes were washed three times with lysis buffer, the beads were resuspended and boiled in 5× Laemmli buffer, and the proteins were separated by SDS-PAGE.
SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting were performed as previously described (23, 48). Gels were transferred onto nitrocellulose membranes overnight at 200 mA or for at least 2 h at 50 V. The following antibodies were used at the indicated dilutions: anti-ORF50 (13), 1:1,000; anti-GST (Sigma), anti-Pin1 and antibody against phosphorylated Pin1 (anti-phospho-Pin1) (S16), 1:1,000 (Cell Signaling); antitubulin and antiactinin, 1:5,000 (Sigma); and horseradish peroxidase (HRP)-conjugated anti-rabbit (ICN Biomedicals) or anti-mouse (Bethyl) secondary antibodies, 1:5,000. Phosphate-buffered saline (PBS) with 0.1% Tween 20 was used for washes. Enhanced chemiluminescence (Pierce) detecting HRP was performed according to the manufacturer's guidelines.
Viral reactivation assays and immunofluorescence.
Wild-type BCBL-1 and cells were reactivated using 40 ng/ml tetradecanoyl phorbol acetate (TPA), or 1 mM valproic acid (VPA). TREx BCBL-1-Rta and iSLK-BAC16 cells were induced with 1 μg/ml doxycycline; iSLK cells were seeded 1 day prior to treatment at 1 × 105 to 2 × 105/well. For immunofluorescence assays, experiments were performed in triplicate unless noted otherwise.
An indirect immunofluorescence assay (IFA) for PEL cells was conducted as described in references 13 and 15 with the following antibodies at the indicated dilutions: anti-ORF50 (13), 1:5,000; anti-K8.1, 1:1,000 (ABI) or 1:500 (SCBT); anti-Mta (15), 1:300; DyLight-labeled anti-mouse and anti-rabbit secondary antibodies, 1:2,000 (Thermo Scientific); and rhodamine-labeled anti-mouse, 1:500 (ICN). Images were captured using a Zeiss Axiovert 200M with Improvision Openlab 5.5.0 and a Nikon A1 point scanner with Nikon NIS Elements AR 3.22.14.
Intracellular viral DNA quantitation.
Total genomic DNA was isolated using the FlexiGene DNA kit (Qiagen) according to the manufacturer's instructions and quantitated using the NanoDrop spectrophotometer (Thermo Scientific).
To assess the relative amounts of viral DNA present, quantitative PCR (qPCR) was conducted using the QuantiTect SYBR green PCR kit (Qiagen) with a Bio-Rad C1000 touch thermal cycler per the manufacturer's suggestions. Curves were analyzed in CFX Manager (Bio-Rad), with single threshold cycle, baseline method set to subtracted curve fit, and baseline threshold set empirically for each experiment. qPCR primer sets for KSHV ORF20 and cellular 7SK, cycling conditions, and ΔΔCT quantitation were previously described (49).
Virion production assays.
Up to 3 ml of virus-containing medium from iSLK-BAC16 cells was collected in a 15-ml tube, centrifuged at 1,000 rpm for 10 min to clear debris, added into 6-well plates containing 293 cells, and quantitated 2 days postinfection.
Vero-rKSHV.294 cells were seeded at a density of 1 × 105 cells/well in a 6-well plate and transfected with up to 5 μg plasmid DNA using TransIT LT1 reagent (Mirus Bio). The following day, the cells were mock treated or treated with 0.5 μM juglone (5-hydroxynaphthoquinone) for 6 h, followed by mock treatment or addition of 1 mM VPA (as indicated in figures). The media were replaced with fresh supplemented DMEM and allowed to incubate for 96 h. Virus-containing media were collected and transferred to 293 MSR tet-OFF cells, which had been plated 24 h previously at a density of 2 × 105cells/well in a 6-well plate. After incubation for 72 h, media were transferred to a 96-well plate to measure secreted alkaline phosphatase (SEAP) using the Great EscAPe SEAP fluorescence detection kit (Clontech).
Fluorescence-activated cell sorting (FACS) analysis.
293 cells were collected, washed twice with PBS, and resuspended in up to 500 μl PBS. GFP-positive events were quantitated using an Accuri C6 cytometer according to the manufacturer's instructions. Events were analyzed using Accuri CFlow Plus software and gated on a subpopulation containing live, induced cells with a minimal fluorescence intensity of ∼104.
Pin1 chemical ablation.
Juglone (5-hydroxynaphthoquinone) was resuspended in ethanol to make 0.1 to 1 mM stock solutions; these were added to cell media at a 1:1,000 dilution per well. Juglone was applied only to cells below its maximal tolerated dose, which we determined by titration and measuring cellular growth.
Bioinformatics/BLAST analysis.
The BLASTp program (NCBI) was used to align the ORF50 gene product superfamily, TAF50, to the KSHV Rta amino acid sequence. The number, conservation, and relative position of aligned prolines adjacent to serines or threonines was determined.
RESULTS
Rta protein contains many pS/T-P motifs and directly interacts with Pin1 in vitro and in infected B cell lysates.
Our previous work showed that the proline content of Rta determines its oligomeric state (23). Overall, Rta is rich in proline, and proline constitutes 17% of its phylogenetically conserved amino acids. To determine whether Pin1 could be a candidate cellular cofactor of Rta-mediated lytic reactivation, we conducted in silico analysis of Rta's primary sequence to locate putative Pin1 interaction motifs (conserved prolines directly adjacent to serines and threonines). We found 15 potential pS/T-P motifs throughout Rta (Fig. 1).
FIG 1.
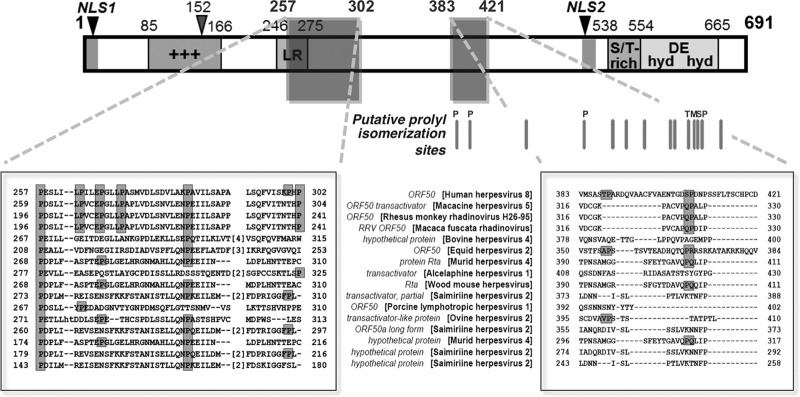
Rta protein is rich in proline and has putative, conserved Pin1 motifs. Of Rta's conserved amino acids, 17% are prolines. (Top) Schematic representation of the Rta protein. Regions of the Rta protein are shown as follows: +++, rich in positively charged amino acids; LR, leucine rich; S/T, serine/threonine, hyd DE hyd, hydrophobic/charged/hydrophobic amino acid rich; NLS, nuclear localization sequence. The gray bars below the map indicate 14 pS/T-P motifs, potential sites for Pin1 binding and isomerization; a 15th pSP motif is immediately adjacent to the motif labeled “M” and is not indicated on the chart by a bar. P, conserved proline; S, conserved serine; T, conserved threonine; M, fully conserved S/T-P motif. (Bottom) The two boxes show alignments of two proline-rich regions of Rta. In the right box, shaded letters indicate the amino acids that are part of the putative Pin1 motifs that are phylogenetically conserved with KSHV Rta. The conserved amino acids are shown on gray background. Gaps introduced to maximize the alignment of sequences are indicated by dashes. The numbers indicate amino acid position. T388 and S406 in the right-hand panel are putative phosphorylated residues that form part of potential Pin1 binding motifs. RRV, rhesus rhadinovirus.
To test whether Pin1 could directly bind to Rta, we used three complementary approaches. We immunoprecipitated Pin1 protein complexes from reactivated BCBL-1 protein lysates using Pin1-specific antisera or negative-control antibody. Western blotting demonstrated that Rta coprecipitated with Pin1, but not with nonspecific antibody (Fig. 2A). We also incubated whole-cell protein extracts from reactivated, KSHV-infected BCBL-1 cells with immobilized GST-Pin1. Rta bound to GST-Pin1, but not to GST alone (Fig. 2B), demonstrating a direct interaction of Pin1 with endogenous Rta.
FIG 2.
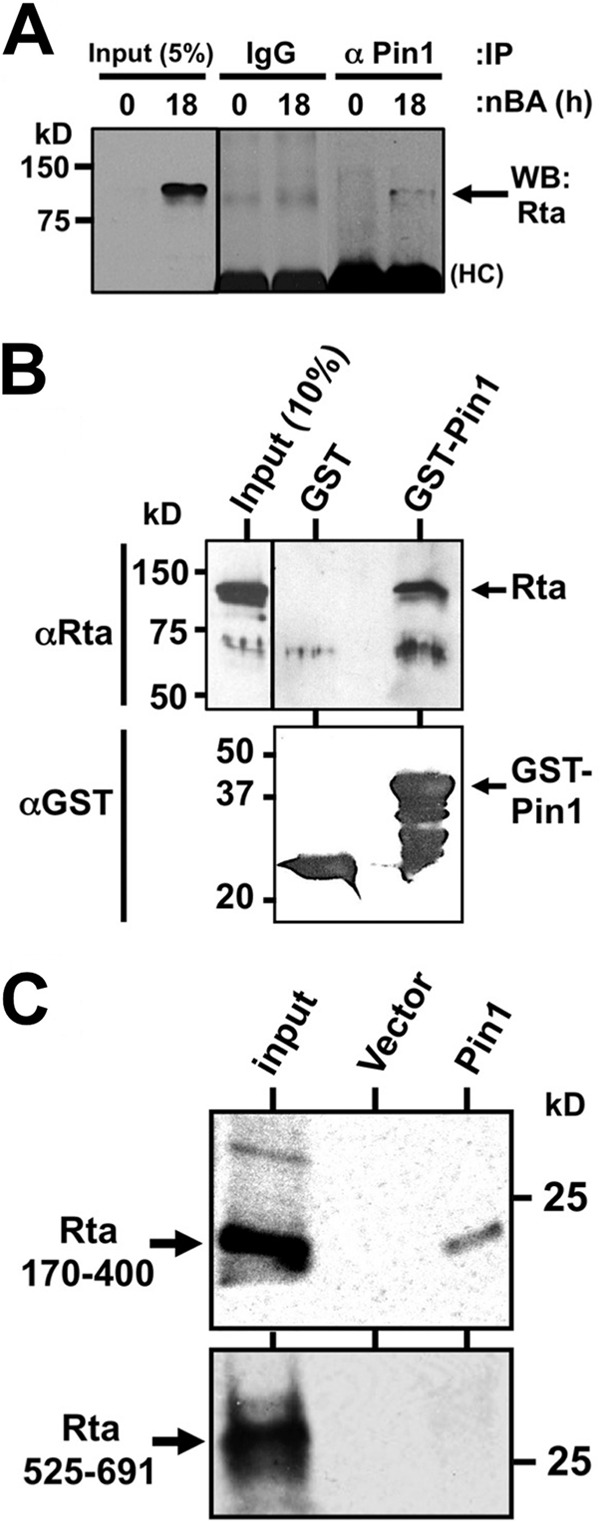
Rta and Pin1 physically interact. (A) Protein extracts from BCBL-1 cells that were untreated (0 h) or treated with sodium butyrate (nBA) (3 mM) for 18 h were analyzed by coimmunoprecipitations (IP) using the antibodies indicated above the gel (α Pin1, anti-Pin1 antibody). Protein complexes were analyzed by SDS-PAGE/Western blotting (WB) using Rta-specific antiserum. A short exposure of 5% of “input” protein extract was analyzed as a positive control. The molecular masses (in kilodaltons) of proteins are indicated to the left of the gel. HC, heavy chain. (B) GST pulldown assays were conducted using the indicated GST fusion proteins and total KSHV-infected PEL cell lysate. SDS-PAGE/Western blotting was performed using the primary antibodies listed and visualized by enhanced chemiluminescence (ECL). A short exposure of “input” protein extract was analyzed as positive control. (C) GST pulldown assays were performed as described above for panel B except that the indicated Rta polypeptides were labeled with 35S in RRL and visualized by autoradiography.
To confirm the interaction of Pin1 with Rta, we repeated this approach with WT and truncated recombinant Rta polypeptides labeled in programmed rabbit reticulocyte lysates (RRL). GST-Pin1 strongly and specifically pulled down full-length Rta (not shown), as well as a truncated Rta polypeptide comprising aa 170 to 400 (Fig. 2C). This region of Rta contains two putative Pin1 binding motifs at amino acids T388 and S406 (Fig. 1). Conversely, GST-Pin1 failed to interact with the C terminus of Rta (aa 525 to 691). These data show that the interaction of Pin1 with Rta depends upon specific Rta amino acids.
Pin1 relocalizes Rta in vivo.
To analyze interactions of Pin1 with Rta in situ, we visualized Rta and Pin1 in transfected Pin1−/− murine embryonic fibroblasts (MEFs). Rta formed several discrete, nuclear punctae in the absence of Pin1 (Fig. 3A, white arrows), while Pin1-GFP appeared diffuse but strong in the nucleus and weaker in the cytoplasm (Fig. 3). Coexpression of Rta and Pin1 resulted in colocalization of Rta to sites of Pin1 expression in 89% of coexpressing cells (Fig. 3A and B). Rta and Pin1 also colocalized in CV-1 and 293 cells (data not shown). These data suggest that Rta and Pin1 physically interact in vivo, and Pin1 redistributes Rta within the nucleus.
FIG 3.
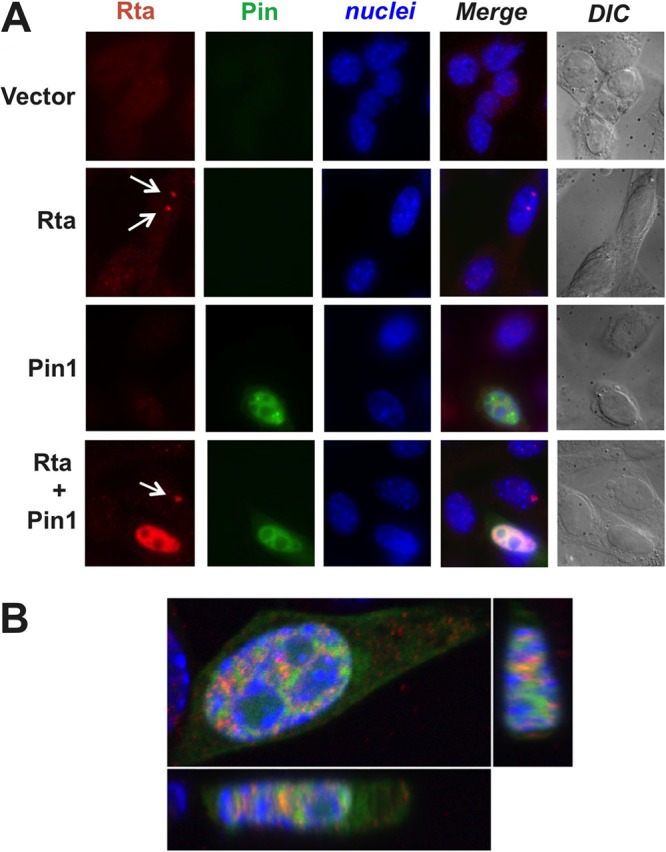
Pin1 alters Rta's subnuclear localization. (A) Pin1 knockout murine embryonic fibroblast (MEF) cells were transfected with Pin1-GFP and/or Rta expression vectors or empty vector, harvested after 24 h, fixed with 4% paraformaldehyde, permeabilized, and blocked. The cells were stained with Rta antibody (with DyLight secondary antibody) and 4′,6′-diamidino-2-phenylindole (DAPI) and analyzed by epifluorescence and confocal microscopy. The white arrows point to Rta punctae in Pin1-negative cells. DIC, differential interference contrast. (B) Confocal images of 3 axes of a typical Pin1-positive, Rta-positive cell showing that all Rta signal colocalizes with portions of Pin1 signal.
Pin1 enhances Rta transactivation of DE viral promoter targets.
To determine whether the physical interaction of Rta with Pin1 has functional consequences for Rta, we tested the effect of Pin1 expression on Rta transient transactivation of KSHV delayed early (DE) promoters. We previously demonstrated that Rta transactivated the Mta and nut-1/polyadenylated nuclear (PAN) promoters in KSHV-negative, BL-41 B lymphocytes (15, 19, 22, 23). In the absence of ectopic Pin1, Rta transactivated PAN about 10-fold in BL-41 cells (Fig. 4). Coelectroporation of Pin1 expression vector resulted in a dose-dependent increase in Rta-mediated transactivation of the polyadenylated nuclear (PAN) promoter, reaching a maximum of about 80-fold (Fig. 4). Conversely, ectopic Pin1 alone had no effect on the PAN promoter (Fig. 4).
FIG 4.
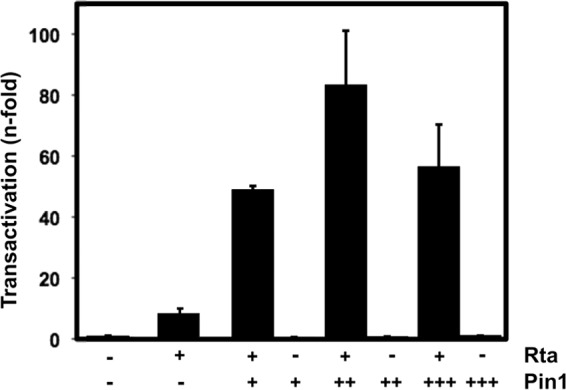
Pin1 enhances Rta transactivation of the nut-1/PAN promoter in uninfected B cells. BL-41 B lymphocytes were electroporated with 5 μg of the promoter-reporter plasmid pnut-1/GL3, together with empty expression vector, indicated amounts of Rta expression vector, or indicated amounts of Pin1 expression vector. Total DNA was normalized to 20 μg in each transfection by the addition of empty expression vector. Luciferase activity was measured from total protein extracts 2 days after transfection. Values were normalized to β-galactosidase expressed from a cotransfected control plasmid, and fold activation was calculated by comparison to empty vector. Symbols: −, vector not transfected; +, 1 μg plasmid; ++, 3 μg plasmid; +++, 9 μg plasmid.
To determine whether Pin1 was necessary for Rta transactivation, we repeated this approach in Pin1−/− MEF cells. Rta weakly transactivated the PAN (Fig. 5A) and Mta (Fig. 5B) promoters in the absence of Pin1. That result contrasts with robust transactivation of DE promoters by Rta in WT MEFs (20, 48; data not shown). As in BL-41 cells, Pin1 significantly increased Rta-mediated transactivation of both viral promoters in a dose-dependent fashion (Fig. 5A and B). Together, these data suggest that Pin1 is an Rta cofactor that enhances Rta transactivation of downstream viral target genes.
FIG 5.
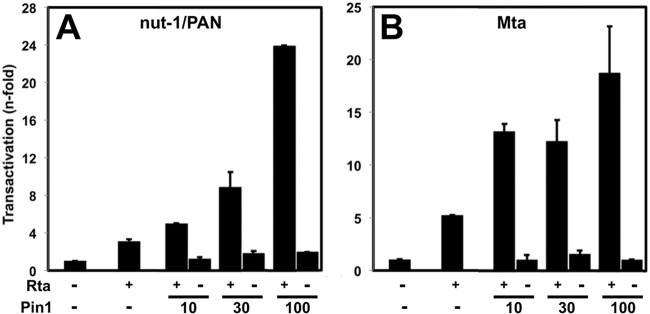
Pin1 enhances Rta transactivation of the nut-1/PAN and Mta promoters in Pin1 knockout MEF cells. (A and B) Pin1 knockout MEFs were transfected with plasmids expressing Rta (0.5 μg) (+) or Pin1 (nanogram amounts indicated) together with 0.5 μg of the promoter-reporter plasmid pNut-1-GL3 (A) or pORF57-GL3 (B), using the approach described in the legend to Fig. 4 (−, cells were not transfected with Rta or Pin1 plasmid, but received equal amount of empty expression vector). Normalization and fold transactivation were quantitated as described in the legend to Fig. 4.
In BL-41 and MEF transfections, Rta expression was linearly proportional to the amount of input plasmid (data not shown). However, cooperation between Pin1 and Rta could be attributable to the trivial explanation that Pin1 transactivates the cytomegalovirus (CMV) promoter that drives Rta transcription from its expression vector. To determine whether this was so, we replaced Rta with firefly luciferase. Titration of ectopic Pin1 expression vector had no effect on luciferase expressed from this vector (Fig. 6). Therefore, Pin1's enhancement of Rta transactivation was occurring posttranslationally.
FIG 6.
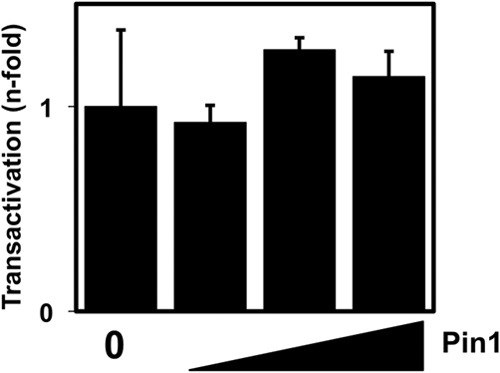
Pin1 does not transactivate the CMV promoter. Pin1 knockout MEFs were transfected with the promoter-reporter plasmid CMV-Luc, together with 10, 30, or 100 ng of Pin1 expression vector (amount of vector indicated by the height of the black triangle) or empty expression vector (0). Total DNA was normalized by the addition of empty expression vector. Luciferase activity was measured 2 days posttransfection, and fold activation was calculated by comparison to CMV-Luc cotransfected with the empty vector alone.
Endogenous Pin1 is expressed and phosphorylated in reactivating PEL cells.
To determine whether Pin1 could play a functional role in KSHV reactivation, we tested whether endogenous Pin1 was expressed and phosphorylated in infected BCBL-1 cells. We compared expression of total and phosphorylated Pin1 in latently infected cells or during viral reactivation for 2 days following treatment with phorbol ester TPA or histone deacetylase (HDAC) inhibitor VPA. Absolute Pin1 expression was largely unaltered during KSHV reactivation (Fig. 7A and B). Pin1 phosphorylation at Ser16, meanwhile, a known modulator of Pin1 substrate-binding activity, was significantly increased about 3-fold upon reactivation in a small subset of total Pin1 (Fig. 7A and B). These data suggest that Pin1 is active during KSHV reactivation.
FIG 7.

Pin1 is expressed constitutively and phosphorylated during KSHV reactivation. (A) BCBL-1 cells were treated with TPA (20 ng/ml) or VPA (1 mM) in duplicate cultures (cultures a and b) for the indicated time periods, and total cellular protein lysates (100 μg) were analyzed by SDS-PAGE/Western blotting using the indicated antibodies. pS16-Pin1, phosphorylated Pin1 (Ser16 phosphorylated). (B) Western blots were quantitated by densitometry, and fold changes in total Pin1 or phosphorylated Pin1 (phospho-Pin1) were calculated at each time point by comparison to latent expression (0 h). (C) TREx BCBL-1-Rta cells were treated with doxycycline (1 μg/ml) to induce Rta expression in duplicate cultures for the indicated time periods, and total cellular protein lysates were analyzed as described above for panel A. (D) Densitometric quantitation.
To determine whether increased Pin1 phosphorylation was attributable to viral reactivation or to a direct cellular effect by the reactivating chemicals, we performed a similar analysis of Pin1 expression in TREx BCBL-1-Rta cells. TREx BCBL-1-Rta cells are BCBL-1 cells stably transfected with an Rta cassette that is expressed uniformly in a doxycycline-dependent manner, leading to reactivation of KSHV in a high percentage of cells (38). As in wild-type BCBL-1 cells, Pin1 phosphorylation, but not expression (Fig. 7C and D), was significantly upregulated during reactivation in TREx BCBL-1-Rta cells. Notably, phosphorylated Pin1 (phospho-Pin1) expression increased more in TREx BCBL-1-Rta cells than in cognate PEL cells. These data indicate that lytic reactivation induced directly by Rta results in phosphorylation of a subset of Pin1.
Pin1 stimulates Rta transactivation of DE promoters and viral DNA replication in infected cells.
Having established Pin1 as an enhancer of Rta transactivation of DE promoters in reporter assays in uninfected cells, we tested whether Pin1 has the same effect during reactivation in infected cells. We used the Vero-rKSHV.219 cell line, which enables easy DE promoter quantitation by scoring induction of a PAN promoter reporter cassette that drives expression of the red fluorescent protein (RFP) (44). Similar to uninfected cells, transfection of the Pin1 expression vector significantly enhanced the percentage of RFP-positive (DE) cells induced by ectopic expression of an Rta expression vector (Fig. 8A).
FIG 8.
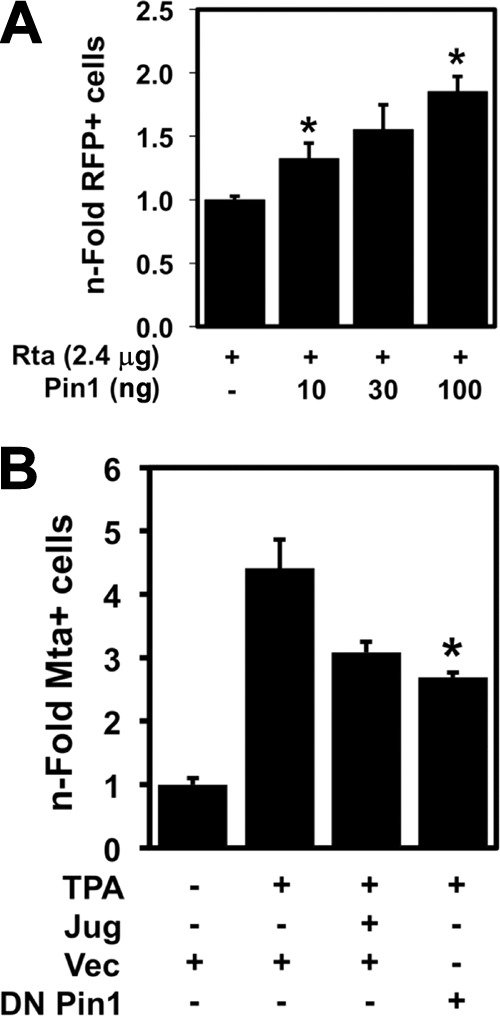
Pin1 enhances DE gene expression during KSHV reactivation. (A) Vero-rKSHV.219 cells were transfected with plasmids expressing indicated proteins (amounts indicated). Total DNA was normalized to 2.5 μg in each transfection by the addition of empty expression vector. The percentage of cells expressing RFP under the control of the DE nut-1/PAN promoter was counted by FACS 48 h posttransfection. The n-fold RFP-positive (RFP+) cells were graphed by normalizing cells cotransfected with Rta and Pin1 expression vectors to cells transfected with Pin1 expression vector and then comparing each value to cells transfected with Rta expression vector alone, which was set at 1. Values that are significantly different (P < 0.05) from the value for cells transfected with Rta expression vector alone are indicated by an asterisk. Symbols: −, plasmid not transfected; +, Rta plasmid transfected. (B) BCBL-1 cells were electroporated, in duplicate, with plasmid expressing the indicated protein (DN Pin1) or with empty vector (Vec) and then treated with 0.5 μM juglone (Jug) 6 h prior to the addition of 20 ng/ml TPA. Seventy-two hours after the addition of TPA, the percentage of cells expressing the DE protein Mta were quantitated manually by indirect immunofluorescence. The n-fold Mta+ cells were graphed similarly to panel A above; the values for untreated cells were set at 1. The value that is significantly different (P < 0.02) from the value for cells treated with TPA alone is indicated by an asterisk.
While Pin1 enhanced Rta-mediated transactivation in infected cells (Fig. 8A), Pin1's effect was not as dramatic as in uninfected cells. Interpreting experiments that use ectopic Pin1 expression is difficult, since endogenous Pin1 is abundantly expressed in most tissue culture cells, as we observed in PEL cells (Fig. 7). We reasoned that ablating Pin1 activity, in the context of chemical induction of Rta from the endogenous viral genome, might better elucidate the role of Pin1 in lytic reactivation. Therefore, we induced Rta expression and reactivation in the cells with TPA in the presence or absence of the specific Pin1 inhibitor juglone. This chemical irreversibly binds within Pin1's PPIase domain active site and prevents isomerization activity (50). TPA treatment alone induced a 4-fold induction of Mta expression in BCBL-1 cells (Fig. 8B). Juglone addition at the maximal tolerated dose (0.5 μM) reduced TPA-mediated Mta induction (Fig. 8B). As further proof that Pin1 regulates DE protein induction in PEL cells, ectopic expression of dominant-negative (DN) Pin1 also reduced TPA-mediated Mta induction (Fig. 8B).
Overall, these data showed that Pin1 enhances Rta transactivation of DE genes in uninfected and infected cells. Since Pin1 did not increase transcription of Rta driven by the CMV promoter (Fig. 6), Pin1 must enhance Rta transactivation posttranslationally. If Pin1-enhanced Rta transactivation was biologically significant, we reasoned that Pin1 should also enhance Rta-dependent vDNA replication. In our initial experiments, we found that ectopic Pin1 directly induced Rta expression from the latent viral genome in infected cells (J. Guito, D. Palmeri, and D. M. Lukac, unpublished data). Therefore, in order to unambiguously test Pin1's translational regulation of Rta in infected cells, we required an infection system in which all Rta expression was independent of Pin1. Thus, we employed an iSLK-BAC16-based system, in which the viral allele of Rta is rendered defective by insertion of a stop codon (BAC16 Rta-stop) and functionally replaced with a Dox-inducible allele of Rta integrated in the SLK cellular genome (43). We measured intracellular vDNA by qPCR in cells that were treated with Dox and/or juglone or not treated with Dox and/or juglone. Two or six days after Dox treatment, vDNA increased 15- to 80-fold, respectively (Fig. 9). Inhibition of Pin1 activity dramatically reduced vDNA accumulation by 40% after 2 days and to near-mock-treated levels after 6 days (Fig. 9). In agreement with Pin1's posttranslational enhancement of Rta transactivation, these data suggest that Pin1 posttranslationally enhances Rta-dependent vDNA replication.
FIG 9.
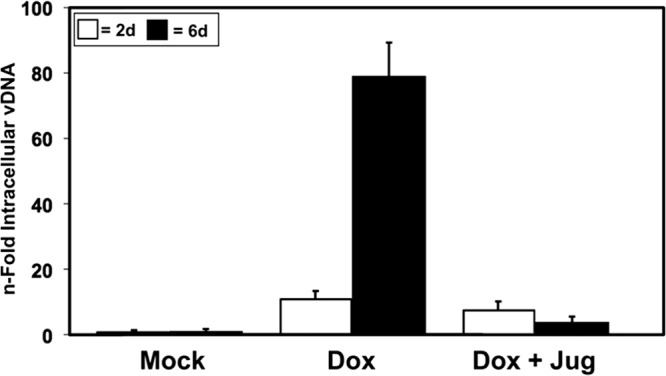
Pin1 inhibition decreases KSHV vDNA replication. iSLK cells infected with Rta null virus BAC16 Rta-stop were treated with 1 μg/ml doxycycline (Dox) or 0.5 μM juglone (Jug), and total intracellular viral DNA was quantitated 2 or 6 days (2d or 6d, respectively) after Dox treatment as described in Materials and Methods.
Pin1 inhibits production of mature, infectious KSHV.
We predicted that the dramatic decrease in vDNA replication when Pin1 was inhibited (Fig. 9) would be reflected in a concomitant reduction of infectious virus production. A very robust method for quantitating production of infectious, reactivated virus is based on a new reporter system developed by Gantt and colleagues (41). In this system, Vero cells are uniformly infected with recombinant KSHV that contains the secreted alkaline phosphatase (SEAP) gene under the control of a tetracycline-responsive promoter (41). If the complete viral reactivation program is induced, the cells release mature virus into the medium. The medium is then transferred to uninfected 293 reporter cells that constitutively express the tetracycline transactivator, and infectious virus is quantitated by measuring SEAP. We recently used this system to show that histone deacetylase 6 is required for high-level production of mature KSHV (H. Shin, J. DeCotiis, M. Giron, D. Palmeri, and D. M. Lukac, submitted for publication). In this system, treatment of the infected cells with VPA induced an ∼15-fold increase in infectious KSHV production (Fig. 10A). Surprisingly, inhibition of Pin1 by juglone treatment or ectopic expression of DN Pin1 dramatically enhanced production of virus stimulated by VPA (Fig. 10A); in the absence of VPA, juglone treatment, or transfection of DN Pin1 vector, had no effect on production of virus (Fig. 10A). We observed a similar, albeit less dramatic effect of juglone on virus production from the Rta null virus Dox-induced iSLK cells (Fig. 10B) under identical experimental conditions to those that showed reduced vDNA replication (Fig. 9). These data show that despite Pin1's enhancement of Rta transactivation and vDNA replication, Pin1 inhibits production of infectious virus.
FIG 10.
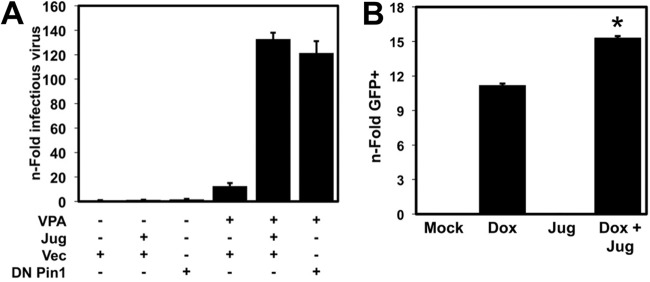
Pin1 inhibits production of infectious KSHV. (A) Vero-rKSHV.294 cells were transfected with the indicated plasmids and then mock treated or treated with 0.5 μM juglone (Jug) for 6 h, followed by mock treatment or 1 mM VPA. Infectious virions were quantitated 96 h after VPA addition, as described in Materials and Methods. (B) iSLK cells infected with Rta null virus BAC16 Rta-stop were treated with 1 μg/ml doxycycline (Dox) and/or 0.5 μM juglone (Jug), and infectious virions were quantitated 6 days after Dox treatment, as described in Materials and Methods. The value that is significantly different (P < 0.01) from the value for cells treated with Dox alone is indicated by an asterisk.
Pin1 inhibits KSHV late gene expression.
Our data suggested that Pin1's block to KSHV production occurs following vDNA replication but prior to release of mature virus from the cells. We previously demonstrated that the KSHV glycoprotein K8.1 is expressed with true late kinetics in response to ectopic expression of Rta in PEL cells; i.e., K8.1 expression requires Rta-dependent vDNA replication (13). We transfected BCBL-1 cells with ectopic Rta and/or Pin1 expression vectors, using the same amounts as in the BL-41 reporter assays (Fig. 5), and analyzed induction of K8.1 glycoprotein at 72 h postelectroporation. In agreement with Pin1's ability to inhibit infectious virus production, we found that Pin1 dramatically suppressed Rta-mediated stimulation of K8.1 expression (Fig. 11A). Similarly, inhibition of Pin1 with juglone enhanced both TPA- and VPA-stimulated reactivation in dose-dependent fashions (Fig. 11B). Under these experimental conditions, juglone's enhancing effect on VPA-stimulated reactivation increased up to the highest concentration, while its effect on TPA-stimulated reactivation peaked at 0.3 μM.
FIG 11.

Pin1 represses KSHV K8.1 expression. (A) KSHV-infected BCBL-1 cells were cotransfected in duplicate with 10 μg of Rta expression vector, the indicated amount (0, 3, or 10 μg) of Pin1 expression vector, or empty vector (−), together with plasmid H2b-GFP to mark electroporated cells. Total DNA was normalized by the addition of empty expression vector. Some samples were mock treated (−) or treated with 20 ng/ml TPA, as indicated. At 72 h after transfection, cells were stained with anti-K8.1 antibody and analyzed using epifluorescence microscopy to quantitate the percentage of GFP+/K8.1+ cells for each condition. The n-fold levels were normalized to the values for Pin1 alone at each concentration and compared to the value for empty vector. Values that are significantly different (P < 0.05) from the value for cells transfected with vector expressing Rta are indicated by an asterisk. (B) Duplicate cultures of KSHV-infected BCBL-1 cells were mock treated or treated with the indicated concentrations of juglone (Jug), together with TPA (20 ng/ml) or VPA (1 mM), and the percentages of K8.1+ cells were analyzed and quantitated at 72 h posttransfection. The n-fold levels were calculated by comparison to the values for cells treated with VPA or TPA alone. Values that are significantly different (P < 0.01) from the value for samples treated with TPA or VPA alone are indicated by an asterisk. (C) Duplicate cultures of KSHV-infected TREx-BCBL-1-Rta cells were treated with 1 μg/ml doxycycline (Dox), 20 ng/ml TPA, and 0.5 μM juglone (Jug), in the indicated combinations, and the percentage of K8.1+ cells were analyzed and quantitated. The n-fold levels were compared to the value for cells treated with TPA alone. Values that are significantly different (P < 0.02) from the value for cells treated with TPA alone or cells treated with TPA plus Dox alone are indicated by an asterisk. (D) BAC36-infected iSLK cells were mock treated or treated with 1 μg/ml doxycycline (Dox) and/or 0.5 μM juglone (Jug), as indicated, and the percentage of K8.1+ cells was quantitated at 2 or 4 days postinduction. Values that are significantly different (P < 0.03) from the value for cells treated with Dox alone are indicated by an asterisk.
A disadvantage of using BCBL-1 cells is that TPA- or VPA-induced reactivation occurs in less than 100% of cells, and both chemicals induce Rta simultaneously with off-target effects. We therefore sought additional confirmation of Pin1's effects in both TREx BCBL-1-Rta cells and infected iSLK cells, in which doxycycline (Dox) induces Rta expression in ∼100% of cells with minimal off-target effects. We first treated TREx BCBL-1-Rta cells with 0.5 μM juglone, with or without Dox induction of Rta, for 72 h. Similar to its effect in parental BCBL-1 cells, juglone treatment increased the percentage of cells expressing K8.1 glycoprotein (Fig. 11C). We also superinduced reactivation in these cells by combination treatment with both Dox and TPA. Juglone addition under those conditions also enhanced K8.1 induction (Fig. 11C). Meanwhile, in iSLK cells, juglone significantly increased K8.1 induction at 2 and 4 days after Dox addition (Fig. 11D). Together, our data show that Pin1 enhances Rta transactivation of DE genes and vDNA replication but inhibits Rta-dependent induction of K8.1 expression and production of infectious virus.
DISCUSSION
In this article, we demonstrate a direct interaction between cellular isomerase Pin1 and the KSHV lytic switch protein Rta (Fig. 2 and 3). We show that Pin1 is expressed and phosphorylated in infected cells after induction of the lytic cycle (Fig. 7) and that Pin1 directly interacts with Rta in vitro and in viral cell lysates (Fig. 2), most likely at one of Rta's putative conserved Pin1 recognition (pS/T-P) motifs (Fig. 1). Rta is expressed in a few nuclear punctae in the absence of Pin1 but is dramatically relocalized throughout nuclei in Pin1-expressing cells (Fig. 3). Pin1 enhances Rta transactivation of viral promoters in transient transfections (Fig. 4 and 5). In KSHV-infected cells, regardless of the method of reactivation, we show that Pin1 contributes to DE transactivation (Fig. 8) and vDNA replication (Fig. 9) but inhibits late K8.1 expression (Fig. 11) and production of mature infectious virus (Fig. 10).
While most Pin1 expressed in PEL cells remains unphosphorylated at Ser16, reactivation of virus with three inducers does result in significantly increased phosphorylation of a small proportion of Pin1 (Fig. 7). This increase is strongest with Rta-specific expression (Fig. 7C and D), likely a result of the much more robust reactivation efficiency in TREx BCBL-1-Rta cells. Phosphorylation of Pin1 is probably due to protein kinase A activation during reactivation, which phosphorylates Pin1 at Ser16 (46, 51). There is evidence that S16 phosphorylation of Pin1 can both positively and negatively regulate Pin1 activity. Some reports suggest that S16 phosphorylation prevents prolyl isomerases from binding their substrates (46, 52), yet others observed that Ser16 phosphorylation upon TPA treatment actually enables oncogenic interaction between Pin1 and Raf1 or RSK2 in a JB6 Cl41 mouse skin epidermal cell system (53–55). In light of this, it is not unreasonable to suggest that the small proportion of phosphorylated Pin1 in PELs is active in the context of Rta interaction. In this scenario, Rta production would increase phosphorylation of a very small amount of Pin1, which could then bind to Rta and exert its downstream effects. We cannot exclude the possibility that Pin1 also regulates Rta indirectly, however, through proteins that enhance or repress Rta function (56).
Pin1 clearly functions downstream of Rta expression during reactivation. In two different cell lines, we found that Pin1 greatly enhances Rta-mediated transactivation in a dose-dependent manner (Fig. 4 and 5). Pin1 is implicated in posttranscriptional processing, chromatin remodeling, target stability (p53, p65) or localization (β-catenin), oligomerization (IRF3), and protein folding for a variety of substrates (36, 57–60). It is telling that we observed significantly reduced levels of transactivation by Rta alone in the MEF cells compared to BL-41 cells (compare Fig. 4 to 5). As the MEF cells are Pin1 deficient, this could be a preliminary indication that Pin1, while potentially not required for Rta transactivation, instead optimizes Rta transactivation to stimulate downstream lytic cycle events. Together with Pin1's ability to activate Rta expression (J. Guito, D. Palmeri, and D. M. Lukac, unpublished), Pin1 and Rta seem to comprise a two-member regulatory loop in which Pin1 also stimulates Rta-mediated transactivation.
Rta contains multiple potential Pin1 recognition motifs and isomerization sites. Pin1 binds full-length Rta, as well as truncated mutant Rta 170-400, but not Rta 525-691 (Fig. 2). Intriguingly, Rta 170-400 contains a single Pin1 motif at T388 and a second one immediately C terminal to this domain at S406 (Fig. 1). Both of these putative Pin1 binding sites contain a proline that is conserved with Rta's homologs. This does not exclude other potential pS/T-P sites within Rta; many Pin1 substrates have multiple binding sites (57–59, 61, 62). Furthermore, it is possible that Pin1 binding to a site does not restrict its activity solely to that site, such that Pin1 might isomerize neighboring prolyl peptide bonds, such as those abundantly located in Rta's C terminus (Fig. 1). Considering the complex regulation by Pin1 reported here, multiple binding sites would provide Pin1 with a combinatorial influence on Rta function, further influenced by the phosphorylation status of particular Rta motifs.
Pin1 is abundantly expressed in many tissue culture cell lines (26, 46, 63), obfuscating the interpretation of Pin1 overexpression experiments. Given the pitfalls involved in analyses that rely on Pin1 overexpression, we used the irreversible Pin1 inhibitor juglone and the dominant-negative Pin1 (S16A) allele to inhibit Pin1 isomerization activity in infected cells. Our investigation of reactivation in the presence of the Pin1 inhibitors, in concert with TPA, VPA, or Dox treatment, supports the initial overexpression analyses (Fig. 10 and 11). The most dramatic effects of Pin1 inhibition were observed in infected Vero cells (Fig. 10A), with lesser effects in PEL cells (Fig. 10B and 11). The understated effects could be a consequence of incomplete inactivation of Pin1 throughout the cell at the tolerated dosages of juglone. Since inhibiting Pin1 with juglone in these experiments simultaneously reduces Rta expression, the relatively modest effects by juglone on K8.1 glycoprotein probably also reflect decreased Rta expression and decreased reactivation overall. Regardless of chemical treatment, Pin1 overexpression or inhibition support the conclusions that Pin1 stimulates Rta function but inhibits full reactivation, as measured by K8.1 expression or release of infectious virus.
While we were initially surprised that Pin1 inhibited viral production rather than enhanced it, the most dramatic phenotype of Pin1 ablation was the inhibition of vDNA replication (Fig. 9). Since this effect was observed in iSLK cells infected with Rta null virus, in which Rta expression is induced by Dox from an ectopic cassette carried in the cellular genome (42, 43), it proves that Pin1 does indeed interact with Rta protein posttranslationally in infected cells, rather than solely induce Rta expression during reactivation. We showed that Pin1 cooperated with Rta to transactivate DE genes (Fig. 4, 5, and 8), which would be a prerequisite to enhancing vDNA replication. Our data do not exclude the possibility that Pin1 enhances Rta function directly at ori-Lyt. For this scheme, successful replication requires Rta transactivation at ori-Lyt and recruitment of the viral origin recognition complex (vORC), including polymerase processivity protein ORF59 (64–66). Meanwhile, the iSLK data maintain the finding that Pin1 loses its enhancing effect on reactivation before production of viral late genes, such as the viral glycoprotein K8.1, culminating in reduction of mature virus production (Fig. 10). It is unknown what factor(s) causes Pin1's switch from enhancer to inhibitor, though one could speculate that a viral DE protein is involved, perhaps via direct complex formation with Pin1 that modulates function or competes with Pin1 for Rta binding. Pin1 stimulation of a sustained, high level of Rta expression might also be incompatible with successful productive reactivation.
A mounting body of recent literature implicates Pin1 in the pathogeneses of a wide variety of viruses, some of them oncogenic. For instance, in HIV-1 infections, Pin1 modulates the expression of the host HIV-1 inhibitor APOBEC3G and its incorporation into the virion (34). In hepatitis C virus (HCV) infections, Pin1 interacts with viral nonstructural proteins NS5A and NS5B to enhance HCV replication (67). In human T-cell leukemia virus type 1 (HTLV-1) infection, Pin1 stabilizes the Tax viral oncoprotein, allowing it to interact with IκB kinase gamma subunit (IKKγ) and contribute to NF-κB-mediated cell transformation (35). In human cytomegalovirus (HCMV) infection, Pin1 is recruited to reorganize nuclear lamin A/C upon phosphorylation of the lamina by viral kinase pUL97 and cellular protein kinase C (PKC) (68). In a very recent study, Pin1 was found to bind to Epstein-Barr virus (EBV) protein BALF5, the catalytic subunit of the viral DNA polymerase, and enhance EBV vDNA replication (37). Our current findings can now add KSHV to the list of viruses regulated by Pin1 isomerization. To our knowledge, this is the first discovery of an interaction between Pin1 and a DNA virus transcription factor.
Though Pin1 clearly plays a role in Rta-mediated lytic reactivation of KSHV, we have not yet elucidated the molecular mechanism by which Pin1 regulates Rta function. We believe, however, that Pin1 activity could be related to our lab's previous discovery that Rta functions as a tetramer (23). In that paper, we showed that the oligomerization of Rta depended on the proline content of aa 244 to 414 (23). Considering the necessity for proline on higher-order Rta structure, binding of Pin1 to Rta aa 170 to 400 and isomerization around prolines could significantly alter Rta's oligomeric state, stability, and interaction with positive and negative cofactors. We note the potential for cross talk of pathways that regulate other proline-directed modification, like the hypoxia response, with Pin1-mediated Rta isomerization.
We propose that Pin1 acts in a molecular timing capacity that cooperates in Rta transactivation ab initio, most likely by directly binding to Rta at a pS/T-P motif within aa 170 to 400. Later in reactivation, Pin1 tempers completion of the productive cycle and mature virus release. Framing Pin1 regulation as a molecular timer is not a new concept (27, 69) but fits intriguingly in this model of KSHV infection (Fig. 12). Our data suggest that physiologic Pin1 activity imposes an exquisite balance on initiation and completion of KSHV reactivation. Perturbation of this balance could promote conditions in which DE genes are strongly expressed and vDNA replication occurs, but is decoupled from late gene synthesis, resulting in reduction of virus release and cell lysis. Such a prosurvival scenario would facilitate participation of DE oncoproteins in KSHV tumorigenesis while safeguarding from the negative consequences incurred by host cell lysis and immune system activation.
FIG 12.
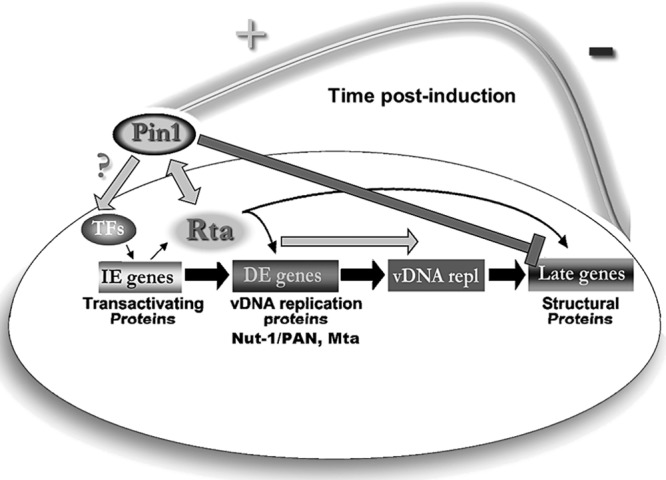
Pin1 acts as a novel lytic cycle timer. The majority of KSHV-infected cells exist in a latent program defined by limited viral gene expression and chronic cell growth stimulation. Disease models show that a small subpopulation of infected cells undergoing reactivation are essential for tumorigenesis. The lytic cycle cascade begins with Rta and other immediate early (IE) protein expression, followed by Rta-mediated transactivation of delayed early (DE) genes, such as Mta, and which include tumor seeding DE oncoproteins and viral DNA replication factors. Upon completion of Rta-dependent viral replication, late gene synthesis proceeds with structural protein and glycoprotein expression, such as K8.1 glycoprotein. Finally, there is assembly and egress of infectious virions that subsequently disseminate within the host and to other individuals. We hypothesize that Pin1 isomerase modulates Rta activity during reactivation. During early reactivation events (Rta DE transactivation and viral DNA replication [vDNA repl]), Pin1 strongly enhances Rta function (+). However, by an unknown mechanism within the lytic cascade, Pin1 transitions into an inhibitor of late events (late gene synthesis and infectious virus release), halting productive reactivation (−). Thus, Pin1 functions as a molecular timer. Pin1 is known to control strength and duration of an array of normal and pathological cellular signals, and we believe Pin1's timing activity is co-opted by KSHV to allow for an evolutionarily advantageous, nonproductive window that can allow for DE gene expression while protecting against cell lysis and immune response activation. TFs, transcription factors.
ACKNOWLEDGMENTS
We express our thanks to Kun Ping Lu, Don Ganem, Richard Venema, Jeff Vieira, and Jae Jung for reagents and cell lines; the UMDNJ FACS Core facility; Roxana Marin, Carlo Daep, Mahrukh Banday, Kalpana Dulal, Kamfai Chan, Bill Honnen, Sanjay Tyagi, Eliseo Eugenin, and past and present lab members for technical assistance, helpful discussions, and critical readings of the manuscript.
This research was supported by the NIH (R01A1078138 to D.M.L. and T32CA134268 to J.G.).
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Gantt S, Casper C. 2011. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr. Opin. Infect. Dis. 24:295–301. 10.1097/QCO.0b013e3283486d04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beral V, Peterman T, Berkelman R, Jaffe HW. 1990. Kaposi's sarcoma among persons with AIDS: a sexually transmitted infection? Lancet 335:123–127. 10.1016/0140-6736(90)90001-L [DOI] [PubMed] [Google Scholar]
- 3.Mosam A, Aboobaker J, Shaik F. 2010. Kaposi's sarcoma in sub-Saharan Africa: a current perspective. Curr. Opin. Infect. Dis. 23:119–123. 10.1097/QCO.0b013e328335b01a [DOI] [PubMed] [Google Scholar]
- 4.Renne R, Lagunoff M, Zhong W, Ganem D. 1996. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 70:8151–8154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867. 10.1073/pnas.93.25.14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, Van Marck E, Salmon D, Gorin I, Escande J-P, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551. 10.1073/pnas.96.8.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenner R, Alba M, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891–902. 10.1128/JVI.75.2.891-902.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 9.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun R, Lin S-F, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong W, Wang H, Herndier B, Ganem D. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U. S. A. 93:6641–6646. 10.1073/pnas.93.13.6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 14.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmeri D, Spadavecchia S, Carroll K, Lukac DM. 2007. Promoter and cell-specific transcriptional activation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 81:13299–13314. 10.1128/JVI.00732-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Z, Swaminathan S. 2006. Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol. 80:5251–5260. 10.1128/JVI.02570-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirshner JR, Lukac DM, Chang J, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 74:3586–3597. 10.1128/JVI.74.8.3586-3597.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majerciak V, Pripuzova N, McCoy JP, Gao SJ, Zheng ZM. 2007. Targeted disruption of Kaposi's sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J. Virol. 81:1062–1071. 10.1128/JVI.01558-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll KD, Bu W, Palmeri D, Spadavecchia S, Lynch SJ, Marras SA, Tyagi S, Lukac DM. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697–9709. 10.1128/JVI.00746-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang Y, Chang J, Lynch S, Lukac DM, Ganem D. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977–1989. 10.1101/gad.996502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y, Ganem D. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. U. S. A. 100:8490–8495. 10.1073/pnas.1432843100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmeri D, Carroll KD, Gonzalez-Lopez O, Lukac DM. 2011. Kaposi's sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jk/CSL. J. Virol. 85:11901–11915. 10.1128/JVI.05479=11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bu W, Carroll KD, Palmeri D, Lukac DM. 2007. The Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788–5806. 10.1128/JVI.00140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee TH, Pastorino L, Lu KP. 2011. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev. Mol. Med. 13:e21. 10.1017/S1462399411001906 [DOI] [PubMed] [Google Scholar]
- 25.Lu KP, Hanes SD, Hunter T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380:544–547. 10.1038/380544a0 [DOI] [PubMed] [Google Scholar]
- 26.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. 2004. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 164:1727–1737. 10.1016/S0002-9440(10)63731-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu KP, Finn G, Lee TH, Nicholson LK. 2007. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3:619–629. 10.1038/nchembio.2007.35 [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K, Uchida C, Shin RW, Shimazaki K, Uchida T. 2008. Prolyl isomerase, Pin1: new findings of post-translational modifications and physiological substrates in cancer, asthma and Alzheimer's disease. Cell. Mol. Life Sci. 65:359–375. 10.1007/s00018-007-7270-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atchison FW, Means AR. 2004. A role for Pin1 in mammalian germ cell development and spermatogenesis. Front. Biosci. 9:3248–3256. 10.2741/1476 [DOI] [PubMed] [Google Scholar]
- 30.Gallo G, Giordano A. 2005. Are RB proteins a potential substrate of Pin1 in the regulation of the cell cycle? J. Cell. Physiol. 205:176–181. 10.1002/jcp.20451 [DOI] [PubMed] [Google Scholar]
- 31.Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. 2002. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 22:5281–5295. 10.1128/MCB.22.15.5281-5295.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wulf G, Finn G, Suizu F, Lu KP. 2005. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Biol. 7:435–441. 10.1038/ncb0505-435 [DOI] [PubMed] [Google Scholar]
- 33.Wulf G, Ryo A, Liou YC, Lu KP. 2003. The prolyl isomerase Pin1 in breast development and cancer. Breast Cancer Res. 5:76–82. 10.1186/bcr572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watashi K, Khan M, Yedavalli VR, Yeung ML, Strebel K, Jeang KT. 2008. Human immunodeficiency virus type 1 replication and regulation of APOBEC3G by peptidyl prolyl isomerase Pin1. J. Virol. 82:9928–9936. 10.1128/JVI.01017-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peloponese JM, Jr, Yasunaga J, Kinjo T, Watashi K, Jeang KT. 2009. Peptidylproline cis-trans-isomerase Pin1 interacts with human T-cell leukemia virus type 1 Tax and modulates its activation of NF-kappaB. J. Virol. 83:3238–3248. 10.1128/JVI.01824-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. 2006. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 7:598–605. 10.1038/ni1347 [DOI] [PubMed] [Google Scholar]
- 37.Narita Y, Murata T, Ryo A, Kawashima D, Sugimoto A, Kanda T, Kimura H, Tsurumi T. 2013. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J. Virol. 87:2120–2127. 10.1128/JVI.02634-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220. 10.1128/JVI.77.7.4205-4220.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujimori F, Takahashi K, Uchida C, Uchida T. 1999. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem. Biophys. Res. Commun. 265:658–663. 10.1006/bbrc.1999.1736 [DOI] [PubMed] [Google Scholar]
- 41.Gantt S, Carlsson J, Ikoma M, Gachelet E, Gray M, Geballe AP, Corey L, Casper C, Lagunoff M, Vieira J. 2011. The HIV protease inhibitor nelfinavir inhibits Kaposi's sarcoma-associated herpesvirus replication in vitro. Antimicrob. Agents Chemother. 55:2696–2703. 10.1128/AAC.01295-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21. 10.1016/j.jviromet.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toth Z, Brulois KF, Wong LY, Lee HR, Chung B, Jung JU. 2012. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi's sarcoma-associated herpesvirus lytic gene expression. J. Virol. 86:9696–9707. 10.1128/JVI.01012-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. 10.1016/j.virol.2004.03.049 [DOI] [PubMed] [Google Scholar]
- 45.Kanda T, Sullivan KF, Wahl GM. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. 10.1016/S0960-9822(98)70156-3 [DOI] [PubMed] [Google Scholar]
- 46.Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP. 2002. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J. Biol. Chem. 277:2381–2384. 10.1074/jbc.C100228200 [DOI] [PubMed] [Google Scholar]
- 47.Ruan L, Torres CM, Qian J, Chen F, Mintz JD, Stepp DW, Fulton D, Venema RC. 2011. Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 31:392–398. 10.1161/ATVBAHA.110.213181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM. 2007. Direct interactions of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein Octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 81:8451–8467. 10.1128/JVI.00265-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bu W, Palmeri D, Krishnan R, Marin R, Aris VM, Soteropoulous P, Lukac DM. 2008. Identification of direct transcriptional targets of the Kaposi's sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 82:10709–10723. 10.1128/JVI.01012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, Grabley S, Kullertz G, Fischer G. 1998. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37:5953–5960. 10.1021/bi973162p [DOI] [PubMed] [Google Scholar]
- 51.Smet-Nocca C, Launay H, Wieruszeski JM, Lippens G, Landrieu I. 2013. Unraveling a phosphorylation event in a folded protein by NMR spectroscopy: phosphorylation of the Pin1 WW domain by PKA. J. Biomol. NMR 55:323–337. 10.1007/s10858-013-9716-z [DOI] [PubMed] [Google Scholar]
- 52.Eckert B, Martin A, Balbach J, Schmid FX. 2005. Prolyl isomerization as a molecular timer in phage infection. Nat. Struct. Mol. Biol. 12:619–623. 10.1038/nsmb946 [DOI] [PubMed] [Google Scholar]
- 53.Cho YS, Park SY, Kim DJ, Lee SH, Woo KM, Lee KA, Lee YJ, Cho YY, Shim JH. 2012. TPA-induced cell transformation provokes a complex formation between Pin1 and 90 kDa ribosomal protein S6 kinase 2. Mol. Cell. Biochem. 367:85–92. 10.1007/s11010-012-1322-y [DOI] [PubMed] [Google Scholar]
- 54.Khanal P, Choi HK, Namgoong GM, Ahn SG, Yoon JH, Sohn H, Choi HS. 2011. 5′-Nitro-indirubinoxime inhibits epidermal growth factor- and phorbol ester-induced AP-1 activity and cell transformation through inhibition of phosphorylation of Pin1. Mol. Carcinog. 50:961–971. 10.1002/mc.20761 [DOI] [PubMed] [Google Scholar]
- 55.Li M, Stukenberg PT, Brautigan DL. 2008. Binding of phosphatase inhibitor-2 to prolyl isomerase Pin1 modifies specificity for mitotic phosphoproteins. Biochemistry 47:292–300. 10.1021/bi701819k [DOI] [PubMed] [Google Scholar]
- 56.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front. Microbiol. 3:30. 10.3389/fmicb.2012.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, Lu KP. 2009. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat. Cell Biol. 11:97–105. 10.1038/ncb1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, Agarwal NK, Sarbassov D, Shi D, Yu D, Hung MC. 2009. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 28:2436–2445. 10.1038/onc.2009.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A, Kaplan F, Capobianco A, Pece S, Di Fiore PP, Del Sal G. 2009. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 11:133–142. 10.1038/ncb1822 [DOI] [PubMed] [Google Scholar]
- 60.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. 2002. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 277:47976–47979. 10.1074/jbc.C200538200 [DOI] [PubMed] [Google Scholar]
- 61.Le Clorennec C, Ouk TS, Youlyouz-Marfak I, Panteix S, Martin CC, Rastelli J, Adriaenssens E, Zimber-Strobl U, Coll J, Feuillard J, Jayat-Vignoles C. 2008. Molecular basis of cytotoxicity of Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J. Virol. 82:6721–6733. 10.1128/JVI.02250-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaw PE. 2007. Peptidyl-prolyl cis/trans isomerases and transcription: is there a twist in the tail? EMBO Rep. 8:40–45. 10.1038/sj.embor.7400873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, Kubota Y, Lu KP, Aoki I. 2005. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin. Cancer Res. 11:7523–7531. 10.1158/1078-0432.CCR-05-0457 [DOI] [PubMed] [Google Scholar]
- 64.Rossetto CC, Susilarini NK, Pari GS. 2011. Interaction of Kaposi's sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol. 85:3833–3841. 10.1128/JVI.02361-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615–8629. 10.1128/JVI.78.16.8615-8629.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Li H, Tang Q, Maul GG, Yuan Y. 2008. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: involvement of host cellular factors. J. Virol. 82:2867–2882. 10.1128/JVI.01319-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lim YS, Tran HT, Park SJ, Yim SA, Hwang SB. 2011. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J. Virol. 85:8777–8788. 10.1128/JVI.02533-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. 2010. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 285:13979–13989. 10.1074/jbc.M109.063628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nicholson LK, Lu KP. 2007. Prolyl cis-trans isomerization as a molecular timer in Crk signaling. Mol. Cell 25:483–485. 10.1016/j.molcel.2007.02.005 [DOI] [PubMed] [Google Scholar]