

Abstract
Us3 is a serine-threonine protein kinase encoded by herpes simplex virus 1 (HSV-1). In this study, a large-scale phosphoproteomic analysis of titanium dioxide affinity chromatography-enriched phosphopeptides from HSV-1-infected cells using high-accuracy mass spectrometry (MS) and subsequent analyses showed that Us3 phosphorylated HSV-1-encoded dUTPase (vdUTPase) at serine 187 (Ser-187) in HSV-1-infected cells. Thus, the following observations were made. (i) In in vitro kinase assays, Ser-187 in the vdUTPase domain was specifically phosphorylated by Us3. (ii) Phosphorylation of vdUTPase Ser-187 in HSV-1-infected cells was detected by phosphate-affinity polyacrylamide gel electrophoresis analyses and was dependent on the kinase activity of Us3. (iii) Replacement of Ser-187 with alanine (S187A) in vdUTPase and an amino acid substitution in Us3 that inactivated its kinase activity significantly downregulated the enzymatic activity of vdUTPase in HSV-1-infected cells, whereas a phosphomimetic substitution at vdUTPase Ser-187 restored the wild-type enzymatic activity of vdUTPase. (iv) The vdUTPase S187A mutation as well as the kinase-dead mutation in Us3 significantly reduced HSV-1 replication in human neuroblastoma SK-N-SH cells at a multiplicity of infection (MOI) of 5 but not at an MOI of 0.01, whereas the phosphomimetic substitution at vdUTPase Ser-187 restored the wild-type viral replication at an MOI of 5. In contrast, these mutations had no effect on HSV-1 replication in Vero and HEp-2 cells. Collectively, our results suggested that Us3 phosphorylation of vdUTPase Ser-187 promoted HSV-1 replication in a manner dependent on cell types and MOIs by regulating optimal enzymatic activity of vdUTPase.
INTRODUCTION
Protein phosphorylation is one of the most common and effective posttranslational modifications by which a cell or virus regulates protein activity (1, 2). Many viruses have evolved mechanisms to utilize protein modification both for regulation of their own viral proteins and for establishment of a cellular environment for efficient viral replication. Phosphorylation in cells infected with herpesviruses is of particular interest because, unlike most other viruses, herpesviruses encode a virus-specific protein kinase(s) (3–5).
Herpes simplex virus 1 (HSV-1) is one of the best-characterized members in the subfamily Alphaherpesvirinae of the family Herpesviridae and is the etiologic agent of a variety of diseases in humans, such as mucocutaneous diseases, keratitis, skin diseases, and encephalitis (3). HSV-1 encodes at least two protein kinases, Us3 and UL13 (4–9). HSV-1 Us3 is a serine/threonine protein kinase with an amino acid sequence that is conserved in the subfamily Alphaherpesvirinae (6, 7). In vitro biochemical studies characterized the consensus target sequence of an HSV-1 Us3 homologue encoded by pseudorabies virus as RnX(S/T)YY, where “n” is greater than or equal to 2, “X” can be Arg, Ala, Val, Pro, or Ser, and “Y” can be any amino acid except an acidic residue (10–12). The phosphorylation target site specificity of HSV-1 and other alphaherpesvirus Us3 kinases has been reported to be similar to that of the pseudorabies virus homologue and to that of protein kinase A (PKA), a cellular cyclic AMP-dependent protein kinase (13), and Akt (14). The Us3 protein and its enzymatic activity have been suggested to play a critical role in HSV-1 replication and pathogenicity based on studies showing that recombinant Us3-null mutant viruses and recombinant viruses encoding catalytically inactive Us3 (Us3 kinase-dead mutant viruses) have impaired growth properties in cell cultures and reduced virulence, pathogenicity, and replication in mouse models (15–19).
HSV-1 Us3 has been considered to be a multifunctional protein regulating various aspects of cellular and viral functions by phosphorylating a number of cellular and viral substrates (4, 5). However, to date, although more than 15 putative HSV-1 Us3 substrates have been described (14, 20–29), only a few substrates, including gB, UL31, Us3 itself, UL47, and tuberous sclerosis complex 2, have been shown to be both physiological Us3 substrates in infected cells and directly linked with Us3 functions in infected cells (14, 19, 20, 22–24, 30). Therefore, there may be Us3 substrates other than those reported to date, and their identification and characterization are required to determine Us3 functions and understand their mechanisms.
As described above and elsewhere (3–5, 31), biological consequences and mechanisms of phosphorylation events in HSV-1-infected cells have gradually been elucidated. However, our knowledge of them remains limited and fragmented. In the present study, to close the knowledge gap, we carried out a large-scale phosphoproteomic analysis of titanium dioxide affinity chromatography-enriched phosphopeptides from HSV-1-infected cells using high-accuracy mass spectrometry (MS). In the phosphorylation status of viral and cellular proteins in HSV-1-infected cells determined by phosphoproteomic analysis, we focused on phosphorylation of HSV-1-encoded dUTPase (vdUTPase) at serine 187 (Ser-187) in this study.
dUTPases catalyze hydrolysis of dUTP to dUMP and pyrophosphate (32, 33). Since DNA polymerases are known to readily misincorporate dUTP into replicating DNA, which causes point mutations and strand breakage, dUTP hydrolysis by dUTPases is necessary for accurate replication of DNA genomes (32, 34–36). dUTPases are present in a wide variety of eukaryotic and prokaryotic organisms, including mammals, plants, Drosophila melanogaster, and Escherichia coli (32, 37). This ubiquity of dUTPase suggests its importance for DNA replication. Interestingly, dUTPases are also encoded by a number of viruses, including herpesviruses, poxviruses, adenoviruses, D-type retroviruses, and African swine fever virus (ASFV) (37, 38), and it has been suggested that strict control of dUTPase activity is critical in the replication of many viruses. HSV-1 vdUTPase, encoded by the UL50 gene, is one of the structural proteins in virion tegument, and its amino acid sequence is conserved throughout the Herpesviridae family (39–41). It has been reported that HSV-1 infection induced a high level of dUTPase activity in a manner dependent on vdUTPase (42), and purified recombinant vdUTPases catalyzed the hydrolysis of dUTP to dUMP and pyrophosphate in vitro (43). In agreement with findings for other dUTPases, HSV-1 vdUTPase has an antimutator function, based on studies showing that the null mutation in HSV-1 vdUTPase increased the mutation frequency in the viral genomes severalfold in cultured infected cells (44). It has also been reported that a recombinant HSV-1 virus carrying a null mutation in vdUTPase was less virulent than wild-type virus, replicated less well in the central nervous system (CNS), and reactivated less efficiently in a mouse model of HSV-1 infection (45), suggesting that vdUTPase was critical for viral replication and pathogenicity in vivo. Thus, the functional sequelae of HSV-1 vdUTPase have gradually been elucidated as described above. However, there is a lack of information on regulation of vdUTPase enzymatic activity in HSV-1-infected cells.
In the present study, the large-scale phosphoproteomic analysis and subsequent analyses showed that Us3 phosphorylated vdUTPase at Ser-187 in HSV-1-infected cells. Studies of the effects of Us3 phosphorylation of vdUTPase Ser-187 suggested that this phosphorylation contributed to efficient HSV-1 replication in a manner dependent on cell types and multiplicities of infection (MOIs) by regulating optimal enzymatic activity of vdUTPase.
MATERIALS AND METHODS
Cells and viruses.
Simian kidney epithelial Vero, human carcinoma HEp-2, human neuroblastoma SK-N-SH, and rabbit skin cells were described previously (46, 47), as was HSV-1 wild-type strain HSV-1(F) (48). Recombinant virus YK511, encoding an enzymatically inactive Us3 mutant in which the lysine at Us3 residue 220 was replaced with methionine (Us3K220M), and recombinant virus YK513, in which the Us3 K220M mutation in YK511 was repaired (Us3K220M-repair), were described previously (24).
Purification of virions.
Virions were purified as described previously (47).
Plasmids.
To generate a fusion protein of maltose binding protein (MBP) and a domain of vdUTPase, pMAL-vdUTPase-P1, pMAL-vdUTPase-P2, and pMAL-vdUTPase-P3 were constructed by cloning the domains of vdUTPase encoded by UL50 codons 1 to 225, 101 to 200, and 201 to 371, respectively, amplified by PCR from pYEbac102 (46), into pMAL-c (New England BioLabs) in frame with the MBP. To generate a fusion protein of MBP and the vdUTPase domain with the S187A mutation, pMAL-vdUTPase-P1-S187A was constructed as described previously (22). To generate a fusion protein of glutathione S-transferase (GST) and a domain of UL49A, pGEX-UL49A was constructed by cloning the domain encoding UL49A codons 24 to 91 amplified by PCR from pYEbac102 into pGEX4T-1 (GE Healthcare) in frame with GST.
Mutagenesis of viral genomes and generation of recombinant HSV-1.
The vdUTPase gene in the vdUTPase null mutant virus YK750 (ΔvdUTPase) was disrupted by insertion of a foreign gene cassette that contained an I-SceI site, a kanamycin resistance gene, and 37 bp of the vdUTPase sequence encoding codons 181 to 193 in which Ser-187 was replaced with alanine. YK750 (ΔvdUTPase) was generated by the Red-mediated mutagenesis procedure using E. coli GS1783 harboring pYEbac102, which contains a complete HSV-1(F) sequence with a bacterial artificial chromosome (BAC) sequence inserted into the HSV-1 intergenic region between UL3 and UL4 (46) as described previously (24, 49). Briefly, linear fragments containing the foreign gene cassette were generated by PCR using the primers shown in Table 1, with pEPkan-S as the template. The primers were designed to insert the foreign gene cassette into the Ser-187 locus of the vdUTPase gene. The linear PCR-generated fragments were electroporated into E. coli GS1783 containing pYEbac102, and the foreign cassette was inserted into the vdUTPase locus by induction of the Red recombination machinery. An E. coli clone (YEbac750) harboring the mutant HSV-1-BAC (pYEbac750), in which the foreign cassette was inserted into the vdUTPase locus, was selected in the presence of kanamycin. pYEbac750 was isolated from YEbac750, and YK750 (ΔvdUTPase) was generated by transfection of pYEbac750 into rabbit skin cells. Recombinant virus YK751 encoding vdUTPase with an alanine substituted for Ser-187 (vdUTPaseS187A) (Fig. 1) was constructed by excision of a part of the foreign gene cassette including the I-SceI site and the kanamycin resistance gene in the YK750 vdUTPase gene as described previously (24, 49). Briefly, part of the foreign gene cassette in pYEbac750 was excised by expressing the I-SceI restriction enzyme in YEbac750, followed by induction of Red recombination machinery. A kanamycin-sensitive E. coli clone (YEbac751) harboring the mutant HSV-1-BAC (pYEbac751) carrying the S187A mutation in the vdUTPase gene was selected and YK751 (vdUTPaseS187A) was generated as described above. Recombinant viruses YK753, encoding vdUTPase with an aspartic acid substituted for serine at residue Ser-187 (vdUTPaseS187D) (Fig. 1), and YK759, encoding vdUTPase with an alanine substituted for aspartic acid at residue Asp-97 (vdUTPaseD97A) (Fig. 1), were generated by the same procedure used to generate YK751 (vdUTPaseS187A) except that the primers listed in Table 1 were used. Recombinant viruses YK752, YK754, and YK760, in which the vdUTPaseS187A, vdUTPaseS187D, and vdUTPaseD97A mutations in YK751, YK753, and YK759, respectively (Fig. 1), were repaired, were generated by the same procedure used to generate YK751 (vdUTPaseS187A) except that the primers listed in Table 1 were used.
TABLE 1.
Primer sequences for construction of recombinant viruses
| Mutation | Sequence |
|---|---|
| ΔvdUTPase and vdUTPaseS187A | 5′-AAGCGTGACTCCGGCCCTACCGGCGCGACGCCGAGGGCGGGCCCTCGTCTATGCCGGCGAAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GTTCCGTCTGAACCGGCGTCAGCTCGCCGGCATAGACGAGGGCCCGCCCTCGGCGTCGCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| vdUTPaseS187D | 5′-AAGCGTGACTCCGGCCCTACCGGCGCGACGCCGAGGGCGGGACCTCGTCTATGCCGGCGAAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GTTCCGTCTGAACCGGCGTCAGCTCGCCGGCATAGACGAGGTCCCGCCCTCGGCGTCGCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| vdUTPaseΔ/SA-repair and vdUTPaseS187D-repair | 5′-AAGCGTGACTCCGGCCCTACCGGCGCGACGCCGAGGGCGGTCCCTCGTCTATGCCGGCGAAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GTTCCGTCTGAACCGGCGTCAGCTCGCCGGCATAGACGAGGGACCGCCCTCGGCGTCGCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| vdUTPaseD97A | 5′-GCTATCCAGCCCAGGGCACCACGTAATACTGGGTCTTATCGCTTCGGGGTACCGCGGAACAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GCGCTACGACCACGGCCATAACGGTTCCGCGGTACCCCGAAGCGATAAGACCCAGTATTACAACCAATTAACCAATTCTGATTAG-3′ | |
| vdUTPaseD97A-repair | 5′-GCTATCCAGCCCAGGGCACCACGTAATACTGGGTCTTATCGACTCGGGGTACCGCGGAACAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GCGCTACGACCACGGCCATAACGGTTCCGCGGTACCCCGAGTCGATAAGACCCAGTATTACAACCAATTAACCAATTCTGATTAG-3′ |
FIG 1.
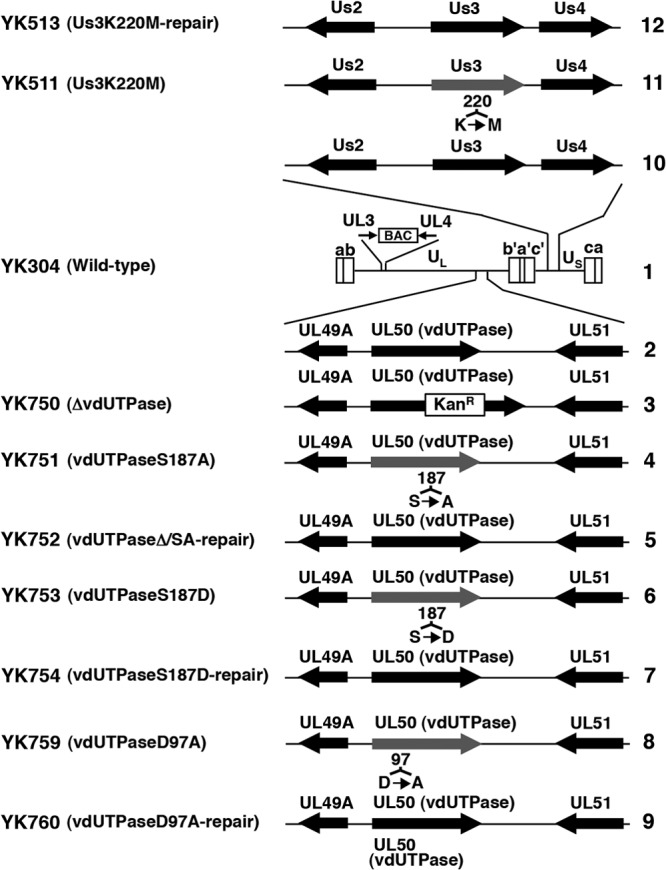
Schematic diagrams of the genome structure of wild-type and recombinant viruses used in this study. Line 1, Wild-type HSV-1(YK304) genome carrying a bacmid (BAC) in the intergenic region between UL3 and UL4. Line 2, domain carrying the UL49A, UL50 (vdUTPase), and UL51 open reading frames. Lines 3 to 9, recombinant viruses with a mutation in the UL50 (vdUTPase) gene. Line 10, domain carrying the Us2, Us3, and Us4 open reading frames. Lines 11 and 12, recombinant viruses with a mutation in the Us3 gene.
Production and purification of MBP and GST fusion proteins in E. coli.
A GST fusion protein, GST-UL49A, that had been transformed with pGEX-UL49A and purified using glutathione-Sepharose resin (GE Healthcare Life Sciences) as described previously (50) was expressed in E. coli XL1/blue. The MBP fusion proteins MBP-vdUTPase-P1, MBP-vdUTPase-P1-S187A, MBP-vdUTPase-P2, MBP-vdUTPase-P3, and MBP-LacZ were expressed in E. coli XL1/blue that had been transformed with pMAL-vdUTPase-P1, pMAL-vdUTPase-P1-S187A, pMAL-vdUTPase-P2, pMAL-vdUTPase-P3, and pMAL-c, respectively, and purified using amylose resin (New England BioLabs) as described previously (50).
Purification of GST fusion proteins from baculovirus-infected cells.
The GST-Us3 and GST-Us3K220M proteins were purified using glutathione-Sepharose resin (GE Healthcare Life Sciences) from the lysates of Sf9 cells infected with the recombinant baculoviruses Bac-GST-Us3 and Bac-GST-Us3K220M, respectively, as described previously (51).
Sample preparation for mass spectrometry (MS).
HEp-2 cells were infected at a multiplicity of infection (MOI) of 10 with wild-type HSV-1(F), harvested at 24 h postinfection, and suspended in 8 M urea containing PhosSTOP phosphatase inhibitor cocktail (Roche Diagnostics) and Benzonase nuclease (Novagen). The mixture was kept on ice for 1 h, and cellular debris was then pelleted by centrifugation at 15,000 rpm for 30 min. The cell lysate was reduced with 1 mM dithiothreitol (DTT) for 90 min and then alkylated with 5.5 mM iodoacetamide (IAA) for 30 min. After digestion with lysyl endopeptidase (Lys-C) (1:50 [wt/wt]) (Wako) at 37°C for 3 h, the resulting peptide mixtures were diluted with 10 mM Tris-HCl (pH 8.2) to a final urea concentration of <2 M and then digested with modified trypsin (1:50 [wt/wt]) (sequencing grade; Promega) at 37°C for 3 h. An equal amount of trypsin was then added for an overnight digestion. Phosphopeptides were enriched by using a Titansphere Phos-TiO kit (GL Sciences) according to the manufacturer's instructions. Briefly, after equilibration of the column, the peptide mixtures from HSV-1-infected cell lysates were applied to Spin Tips, mixed with buffer containing 2-hydroxypropanoic acid, and centrifuged. After the columns were washed, the captured peptides were eluted with a 5% ammonium solution and a 5% pyrrolidine solution. The enriched phosphopeptide solutions were acidified with 10% trifluoroacetic acid (TFA), desalted with ZipTip C18 resins (Millipore), and centrifuged in a vacuum concentrator.
Mass spectrometric analysis, protein identification, and determination of phosphorylated sites.
Shotgun proteomic analyses of the Titansphere eluates were performed by using a linear ion trap-orbitrap mass spectrometer (LTQ-Orbitrap Velos; Thermo Fisher Scientific) coupled with a nanoflow liquid chromatography (LC) system (Dina-2A; KYA Technologies). Peptides were injected into a 75-μm reversed-phase C18 column at a flow rate of 10 μl/min and eluted with a linear gradient of solvent A (2% acetonitrile and 0.1% formic acid in H2O) to solvent B (40% acetonitrile and 0.1% formic acid in H2O) at 300 nl/min. Peptides were sequentially sprayed from a nanoelectrospray ion source (KYA Technologies) and analyzed by collision-induced dissociation (CID). The analyses were carried out in the data-dependent mode, switching automatically between MS and tandem MS (MS/MS) acquisition. For CID analyses, full-scan MS spectra (from m/z 380 to 2,000) were acquired in the orbitrap with a resolution of 100,000 at m/z 400 after ion count accumulation to the target value of 1,000,000. The 20 most intense ions at a threshold above 2,000 were fragmented in the linear ion trap with a normalized collision energy of 35% for an activation time of 10 ms. The orbitrap analyzer was operated with the “lock mass” option to perform shotgun detection with high accuracy (52). Protein identification was conducted by analyzing the MS and MS/MS data against the RefSeq human protein (38,963 protein sequences) and nonredundant virus protein (1,034,534 protein sequences) databases (National Center for Biotechnology Information) by Mascot (Matrix Science). Carbamidomethylation of cysteine residues was set as fixed modifications, whereas methionine oxidation, protein N-terminal acetylation, pyroglutamination for N-terminal glutamine, and phosphorylation (Ser, Thr, and Tyr) were set as variable modifications. A maximum of two missed cleavages was allowed in the database search. The tolerance for mass deviation was set to 3 ppm for peptide masses and 0.8 Da for MS/MS peaks, respectively. In the process of peptide identification, we conducted decoy database searching by Mascot and applied a filter for a false-positive rate of <1%. Determination of phosphorylated sites in the peptides was performed using the software program Proteome Discoverer, version 1.3 (Thermo Fisher Scientific).
Phos tag SDS-PAGE analysis.
Phos tag (phosphate affinity) SDS-PAGE analysis was performed, following the standard protocol of Wako Chemicals. Briefly, Vero cells were mock infected or infected with HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 5 for 18 h or with HSV-1(F) at an MOI of 0.01 for 60 h and then harvested, solubilized, and analyzed by electrophoresis in a denaturing gel containing 200 μM MnCl2 and 100 μM Phos tag acrylamide (Wako). After electrophoresis, gels were soaked in standard transfer buffer (53) with 1 mM EDTA for 10 min to remove Mn2+. The separated proteins in the denaturing gels were then transferred to nitrocellulose membranes and analyzed by immunoblotting as described below.
Immunoblotting and immunofluorescence.
Immunoblotting and immunofluorescence were performed as described previously (47, 53). The amount of protein in immunoblot bands was quantitated using the Dolphin Doc image capture system with Dolphin-1D software (Wealtec).
In vitro kinase assays.
MBP fusion proteins were captured on amylose beads (New England BioLabs) and used as substrates in in vitro kinase assays with purified GST-Us3 and GST-Us3K220M as described previously (51).
Phosphatase treatment.
After in vitro kinase assays, MBP fusion proteins were treated with λ protein phosphatase (λ-PPase) (New England BioLabs) as described previously (51). Lysates of HSV-1(F)-infected Vero cells were treated with alkaline phosphatase (CIP) (New England BioLabs) as described previously (51).
dUTPase enzyme assay.
dUTPase activity was determined as described previously (45, 54), with minor modifications. Briefly, Vero cells in 6-well plates were mock infected or infected with each of the indicated viruses at an MOI of 5 for 18 h and then harvested and lysed in 200 μl NP-40 buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 1.0% Nonidet P-40 [NP-40]). After a brief centrifugation, 0.4 μl of each supernatant was mixed with 200 μl reaction buffer (50 mM Tris-HCl [pH 8.0], 2 mM β-mercaptoethanol, 1 mM MgCl2, 0.1% bovine serum albumin, 2 mM p-nitrophenylphosphate, and 0.24 nM [3H]dUTP [28.8 Ci/mmol; PerkinElmer]). The reaction was allowed to proceed for 30 min at 37°C and then terminated by spotting the reaction mixture onto DE81 circle discs (Whatman). The discs were washed three times for 5 min each with washing solution (1 mM ammonium formate and 4 M formic acid), followed by one wash with 95% ethanol for 3 min. The discs were air-dried and counted for radioactivity using an LS3801 scintillation counter (Beckman).
Antibodies.
To generate mouse polyclonal antibody to vdUTPase or HSV-1 UL49A, BALB/c mice were immunized once with purified MBP-vdUTPase-P2 and MBP-vdUTPase-P3 or with GST-UL49A with TiterMax Gold adjuvant liquid (TiterMax USA, Inc.), respectively. The sera of the immunized mice were used as anti-vdUTPase or anti-UL49A mouse polyclonal antibody. To generate rabbit polyclonal antibody to vdUTPase, rabbits were immunized with purified MBP-vdUTPase-P2 and MBP-vdUTPase-P3, following the standard protocol at MBL (Nagoya, Japan). Mouse monoclonal antibodies to gB (H1817) and VP5 (3B6) were purchased from Virusys. Rabbit polyclonal antibodies to UL49 and UL51 were described previously (55).
Statistical analysis.
Differences in relative dUTPase activities were statistically analyzed using the two-tailed Student t test.
RESULTS
Identification of phosphorylation sites on viral and cellular proteins in HSV-1-infected cells.
To analyze the large-scale phosphorylation status of viral and cellular proteins in HSV-1-infected cells, we carried out a phosphoproteomic analysis of titanium dioxide affinity chromatography-enriched phosphopeptides from HSV-1-infected cells by high-accuracy MS. Briefly, HEp-2 cells were infected at an MOI of 10 with wild-type HSV-1(F), harvested at 24 h postinfection, denatured, alkylated, and digested in-solution with a combination of endoproteinase Lys-C and trypsin. Resulting peptide mixtures were subjected to phosphopeptide enrichment, using titanium dioxide affinity chromatography, and enriched samples were analyzed using a high-accuracy LTQ-Orbitrap mass spectrometer. Peptide spectra were searched in sequence databases and validated as described in Materials and Methods. Using this approach, we detected 81 species of unique phosphopeptides with 65 unique phosphorylation sites on 25 distinct viral proteins from HSV-1(F)-infected cells. In addition to the phosphorylation sites in viral proteins, we also detected 655 species of unique phosphopeptides with 542 unique phosphorylation sites on 398 distinct cellular proteins from HSV-1(F)-infected cells.
Us3 phosphorylation of vdUTPase Ser-187 in vitro.
Among the MS-identified phosphorylation sites, phosphorylation sites in 4 viral and 19 host proteins completely or almost completely matched the consensus sequence of the Us3 phosphorylation site, and we focused on Ser-187 in HSV-1 dUTPase in this study. To examine whether HSV-1 Us3 can directly phosphorylate vdUTPase Ser-187 in vitro, we generated and purified a chimeric protein consisting of MBP fused to a peptide encoded by vdUTPase codons 1 to 225 (MBP-vdUTPase-P1) (Fig. 2A) and tested it as a substrate in in vitro kinase assays with purified wild-type GST-Us3 and the kinase-negative mutant GST-Us3K220M (51). As shown in Fig. 2B to G, MBP-vdUTPase-P1 (Fig. 2A) was labeled with [γ-32P]ATP by purified Us3 fused to glutathione S-transferase (GST) (GST-Us3), while mutant MBP-vdUTPase-P1-S187A, in which alanine was substituted for Ser-187 (Fig. 2A), and MBP-LacZ were not labeled. When the kinase-dead mutant GST-Us3K220M, in which the invariant lysine at Us3 residue 220 was replaced with methionine (Us3K220M), was used, none of the MBP fusion proteins were labeled (Fig. 2B to E). Furthermore, MBP-vdUTPase-P1 labeling by GST-Us3 was eliminated by phosphatase (λ-PPase) treatment, confirming that MBP-vdUTPase-P1 was labeled by phosphorylation (Fig. 2F and G). These results indicated that Us3 specifically and directly phosphorylated vdUTPase Ser-187 in vitro.
FIG 2.
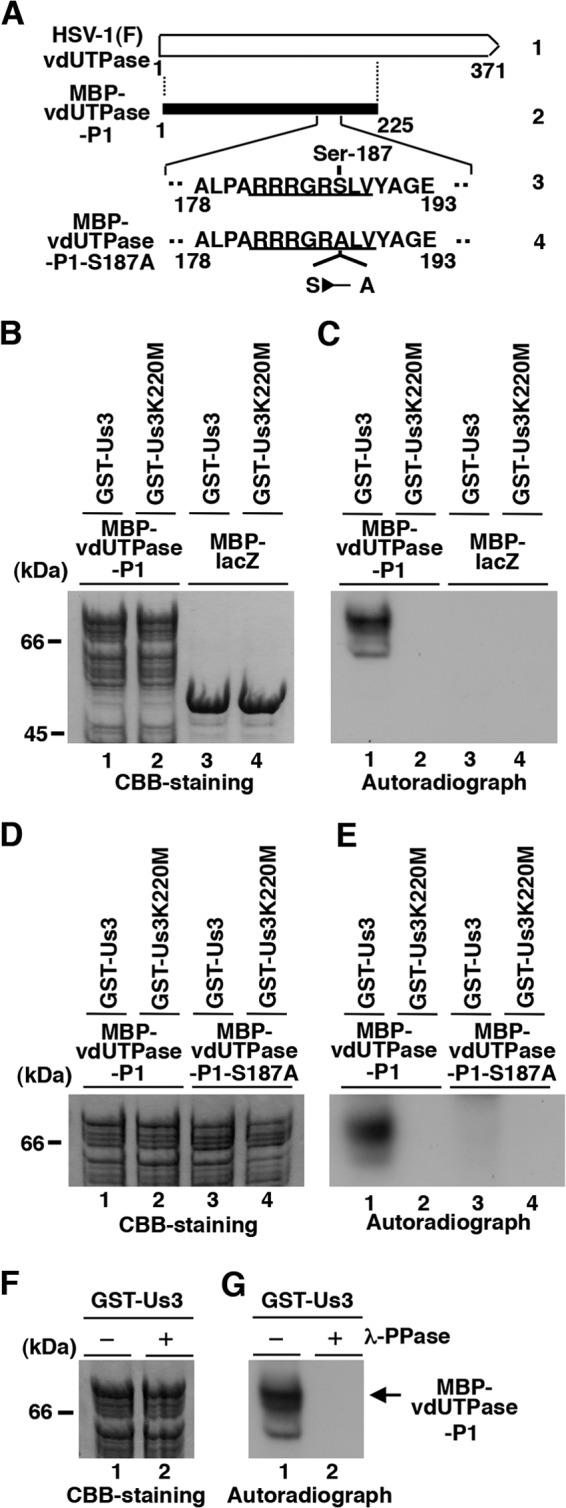
Us3 directly phosphorylated vdUTPase Ser-187 in vitro. (A) Schematic diagram of vdUTPase. Line 1, structure of the vdUTPase open reading frame. Line 2, domain of the vdUTPase gene encoding vdUTPase residues 1 to 225, which were used in these studies to generate the MBP-vdUTPase-P1 fusion protein. Line 3, amino acid sequence of vdUTPase residues 178 to 193. The site with the consensus sequence for phosphorylation by Us3 is underlined. Line 4, domain of the UL50 (vdUTPase) gene encoding vdUTPase residues 178 to 193 with the S187A mutation used in these studies to generate the MBP-vdUTPase-P1-S187A fusion protein. (B) Purified MBP-vdUTPase-P1 (lanes 1 and 2) and MBP-LacZ (lanes 3 and 4) incubated in kinase buffer containing [γ-32P]ATP and purified GST-Us3 (lanes 1 and 3) or GST-Us3K220M (lanes 2 and 4), separated on an SDS-PAGE gel, and stained with Coomassie brilliant blue (CBB). (C) Autoradiograph of the gel in panel B. (D) Purified MBP-vdUTPase-P1 (lanes 1 and 2) and MBP-vdUTPase-P1-S187A (lanes 3 and 4) incubated in kinase buffer containing [γ-32P]ATP and purified GST-Us3 (lanes 1 and 3) or GST-Us3K220M (lanes 2 and 4), separated on an SDS-PAGE gel, and stained with CBB. (E) Autoradiograph of the gel in panel D. (F) Purified MBP-vdUTPase-P1 incubated in kinase buffer containing [γ-32P]ATP and purified GST-Us3 and then either mock treated (lane 1) or treated with λ-PPase (lane 2), separated on an SDS-PAGE gel, and stained with CBB. (G) Autoradiograph of the gel in panel F. A molecular mass marker (in kilodaltons) is shown on the left.
Us3-dependent phosphorylation of vdUTPase Ser-187 in infected cells.
To examine whether Us3 mediated phosphorylation of vdUTPase Ser-187 in infected cells, we carried out phosphate affinity SDS-polyacrylamide gel electrophoresis (Phos tag SDS-PAGE) analyses of cells infected with wild-type and mutant HSV-1 viruses (Fig. 1). Phos-tag SDS-PAGE enables visualization of the phosphorylation status of a protein as a distinct band shift (56). For these analyses, we constructed the vdUTPase-null mutant virus YK750 (ΔvdUTPase), recombinant virus YK751 (vdUTPaseS187A), encoding a mutant vdUTPase in which vdUTPase Ser-187 was replaced with alanine, and their repaired virus, YK752 (vdUTPaseΔ/SA-repair) (Fig. 1). The viral growth curves of YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A) were similar to those of their repaired virus YK752 (vdUTPaseΔ/SA-repair) and wild-type HSV-1(F) in Vero cells infected at an MOI of 5 (Fig. 3A) or 0.01 (Fig. 3B). We have previously reported that the Us3 kinase-dead mutant virus YK511 (Us3K220M) grew as well as its repaired virus, YK513 (Us3K220M-repair), and wild-type HSV-1(F) in Vero cells (24). In addition, we noted that the null mutation in vdUTPase had no effect on expression of the neighboring UL49, UL49A, and UL51 genes (data not shown).
FIG 3.
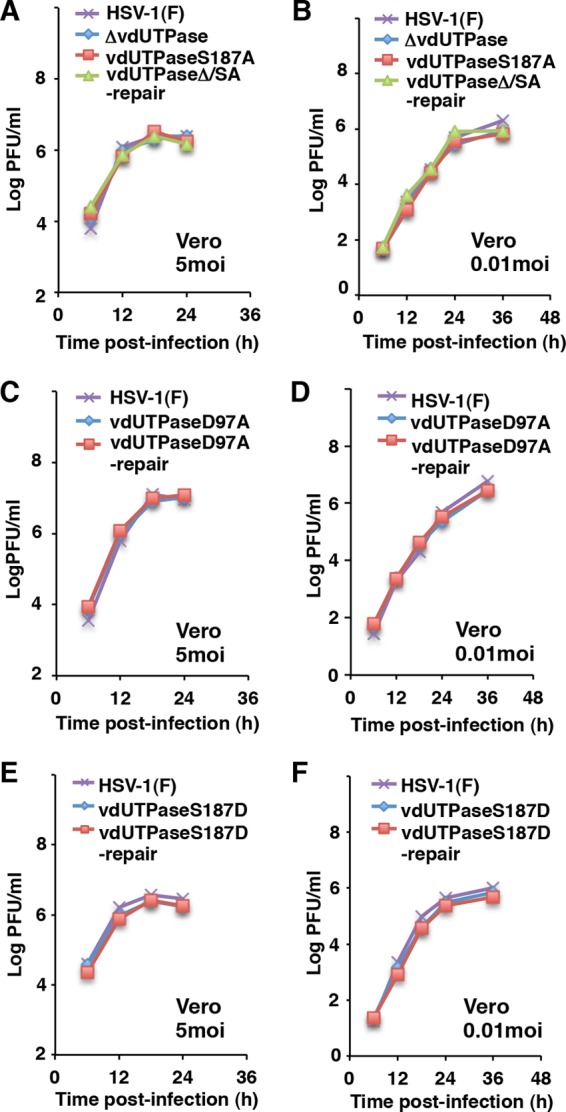
Growth curves of recombinant viruses in Vero cells used in this study. (A to F) Vero cells were infected at an MOI of 5 (left figures) or 0.01 (right figures) with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), and YK752 (vdUTPaseΔ/SA-repair) (A and B) or wild-type HSV-1(F), YK759 (vdUTPaseD97A), and YK760 (vdUTPaseD97A-repair) (C and D) or wild-type HSV-1(F), YK753 (vdUTPaseS187D), and YK754 (vdUTPaseS187D-repair) (E and F). Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and was assayed on Vero cells. Data are representative of three independent experiments.
Vero cells were infected with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 5, harvested at 18 h postinfection, and analyzed on a Phos tag-positive [Phos tag(+)] SDS-PAGE or Phos tag negative [Phos tag(−)] SDS-PAGE gel (Fig. 4A and C). vdUTPase proteins from Vero cells infected with wild-type HSV-1(F), YK752 (vdUTPaseΔ/SA-repair), and YK513 (Us3K220M-repair) were detected as two bands with large mobility differences in the Phos-tag(+) SDS-PAGE gel (Fig. 4A, upper panel, lanes 2 and 5, and C, upper panel, lanes 2 and 4). After phosphatase treatment of the lysate from wild-type HSV-1(F)-infected cells, the slower-migrating dUTPase band disappeared (Fig. 4B). The disappearance of the slower-migrating band after phosphatase treatment was also observed with vdUTPase proteins from Vero cells infected with YK751 (vdUTPaseS187A) (Fig. 4A, upper panel, lane 4) and YK511 (Us3K220M) (Fig. 4C, upper panel, lane 3). Taken together, these results indicated that the slower band was vdUTPase phosphorylated at Ser-187 and the faster band was vdUTPase unphosphorylated at Ser-187 and that Us3 mediated phosphorylation of vdUTPase Ser-187 in infected cells.
FIG 4.
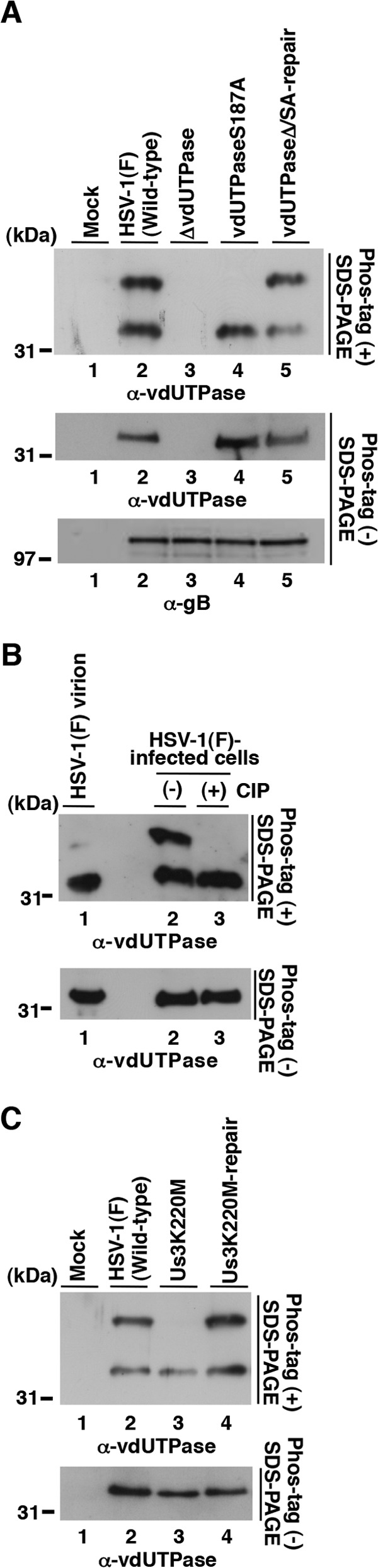
Us3 mediated phosphorylation of vdUTPase Ser-187 in infected cells. (A) Vero cells were either mock infected (lane 1) or infected with wild-type HSV-1(F) (lane 2), YK750 (ΔvdUTPase) (lane 3), YK751 (vdUTPaseS187A) (lane 4), or YK752 (vdUTPaseΔ/SA-repair) (lane 5) at an MOI of 5, harvested at 18 h postinfection, and analyzed on a Phos tag(+) SDS-PAGE gel (top) or Phos-tag(−) SDS-PAGE gel (center and bottom). The gels were immunoblotted with anti-vdUTPase mouse polyclonal antibody (top and center) or anti-gB mouse monoclonal antibody (bottom). (B) HSV-1(F) virions (lane 1) and lysates of Vero cells that were infected with wild-type HSV-1(F) (lanes 2 and 3). The infected Vero cell lysates were either untreated (lane 2) or treated with CIP (lane 3). The virion and cell lysates were then analyzed on a Phos tag(+) SDS-PAGE gel (top) or Phos-tag(−) SDS-PAGE gel (bottom) and immunoblotted with anti-vdUTPase mouse polyclonal antibody. (C) Vero cells were either mock infected (lane 1) or infected with wild-type HSV-1(F) (lane 2), YK511 (Us3K220M) (lane 3), or YK513 (Us3K220M-repair), analyzed on a Phos tag(+) SDS-PAGE gel (top) or Phos-tag(−) SDS-PAGE gel, and immunoblotted with anti-vdUTPase mouse polyclonal antibody. The experiments were performed as described for Fig. 4A.
We noted that about the same amounts of vdUTPase were detected in each of the two vdUTPase bands from wild-type HSV-1(F)-infected cells (Fig. 4A to C). Therefore, since almost all the vdUTPase protein from purified virions was in the faster-migrating band (Fig. 4B, upper panel, lane 1), vdUTPase with unphosphorylated Ser-187 was selectively packaged into virions. However, phosphorylation of vdUTPase Ser-187 in infected cells appeared to have no effect on vdUTPase packaging into virions, since the amount of vdUTPaseS187A incorporated into YK751 (vdUTPaseS187A) virions, produced in cells with no phosphorylated vdUTPase Ser187, was similar to amount of vdUTPase incorporated into wild-type HSV-1(F) virions, produced in cells with both phosphorylated and unphosphorylated vdUTPase (Fig. 5).
FIG 5.
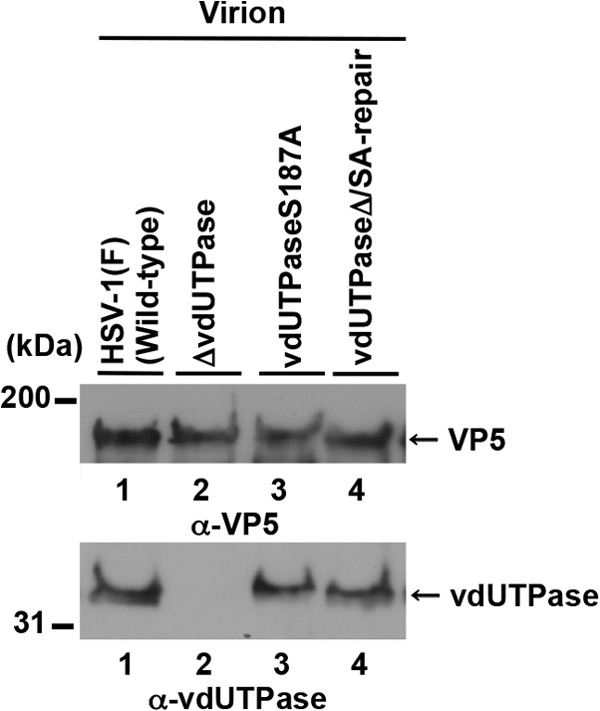
Incorporation of vdUTPase proteins into purifying virions. Immunoblots of electrophoretically separated virions of wild-type HSV-1(F) (lane 1), YK750 (ΔvdUTPase) (lane 2), YK751 (vdUTPaseS187A) (lane 3), or YK752 (vdUTPaseΔ/SA-repair) (lane 4) purified by sucrose gradient centrifugation and blotted with either anti-vdUTPase mouse polyclonal antibody or anti-VP5 mouse monoclonal antibody. A molecular mass marker (in kilodaltons) is shown on the left.
Effect of phosphorylation of vdUTPase Ser-187 on its enzymatic activity in infected cells.
To examine whether the enzymatic activity of vdUTPase in infected cells was regulated by phosphorylation of vdUTPase Ser-187, we constructed recombinant viruses YK759 (vdUTPaseD97A), encoding a mutant vdUTPase in which vdUTPase Asp-97 was replaced with alanine, YK753 (vdUTPaseS187D), encoding a mutant vdUTPase in which vdUTPase Ser-187 was replaced with aspartic acid, and their repaired viruses, YK760 (vdUTPaseD97A-repair) and YK754 (vdUTPaseS187D-repair) (Fig. 1). Asp-97 in HSV-1 vdUTPase is highly conserved in various dUTPases and has been reported to be crucial for dUTPase enzymatic activity (57). Replacement of the phosphorylation site with an acidic amino acid, such as glutamic acid or aspartic acid, mimics the negative charges produced by phosphorylation (20, 22, 23, 30). The growth curves of YK759 (vdUTPaseD97A) and YK753 (vdUTPaseS187D) were similar to those of their repaired viruses, YK760 (vdUTPaseD97A-repair) and YK754 (vdUTPaseS187D-repair), and of wild-type HSV-1(F) in Vero cells infected at an MOI of 5 (Fig. 3C and E) or 0.01 (Fig. 3D and F).
We then assayed the dUTPase activity in Vero cells mock infected or infected with each of these viruses (Fig. 6A). As reported previously (45, 54), infection of Vero cells with wild-type HSV-1(F), YK752 (vdUTPaseΔ/SA-repair), or YK760 (vdUTPaseD97A-repair) produced a similar high level of dUTPase enzymatic activity compared to findings for mock-infected cells (Fig. 6A). In contrast, there was little dUTPase enzymatic activity in cells infected with YK750 (ΔvdUTPase) or YK759 (vdUTPaseD97A) (Fig. 6A), confirming that the D97A mutation in vdUTPase abolished its enzymatic activity in infected cells. In cells infected with YK751 (vdUTPaseS187A), dUTPase enzymatic activity was significantly reduced compared to that in cells infected with wild-type HSV-1(F) or YK752 (vdUTPaseΔ/SA-repair) (Fig. 6A), indicating that vdUTPase Ser-187 was required for optimal vdUTPase activity in infected cells. In contrast, dUTPase enzymatic activity in cells infected with YK753 (vdUTPaseS187D) was similar to that in cells infected with wild-type HSV-1(F) or YK754 (vdUTPaseS187D-repair) (Fig. 6A), indicating that a negatively charged amino acid at vdUTPase Ser-187, due to either phosphorylation of Ser-187 or an S187D substitution, was required for optimal vdUTPase enzymatic activity in infected cells. Furthermore, dUTPase activity in cells infected with YK511 (Us3K220M) was significantly reduced compared to that in cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) (Fig. 6A), indicating that Us3 kinase activity was required for optimal vdUTPase enzymatic activity in infected cells. The amount of vdUTPase protein was also measured by immunoblotting, which showed no vdUTPase in mock- and YK750 (ΔvdUTPase)-infected cell reaction mixtures and a similar amount of vdUTPase in all other infected cell reaction mixtures (Fig. 6B). Taken together with the results above, that Us3 mediated phosphorylation of vdUTPase Ser-187 in infected cells, these data indicated that Us3 phosphorylated vdUTPase Ser-187 to regulate its enzymatic activity in infected cells.
FIG 6.
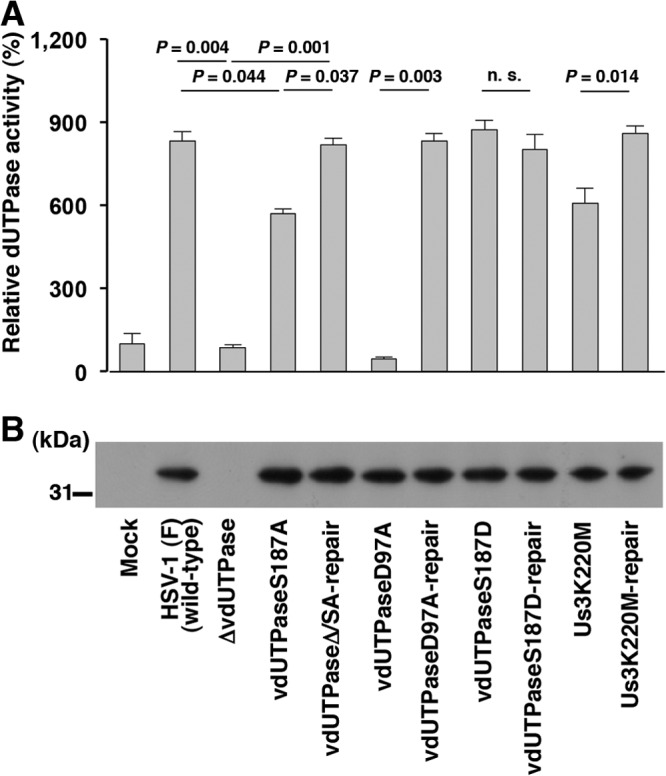
Us3-mediated phosphorylation of vdUTPase Ser-187 upregulated its enzymatic activity in infected cells. (A) Vero cells were mock infected or infected with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK759 (vdUTPaseD97A), YK760 (vdUTPaseD97A-repair), YK753 (vdUTPaseS187D), YK754 (vdUTPaseS187D-repair), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 5. At 18 h postinfection, the cells were solubilized and assayed for dUTPase activity. Each value is the mean ± standard error of the results of triplicate experiments and is expressed relative to the mean value of dUTPase activity in mock-infected cells, which was normalized to 100%. “P” represents the statistical significance value according to the two-tailed Student t test. n.s., not significant. Data are representative of three independent experiments. (B) Immunoblot of lysates prepared for the assays in panel A. The cell lysates were analyzed by immunoblotting with anti-vdUTPase mouse polyclonal antibody. A molecular mass marker (in kilodaltons) is shown on the left. Data are representative of three independent experiments.
Effect of phosphorylation of vdUTPase Ser-187 on HSV-1 replication in SK-N-SH and HEp-2 cells.
To investigate a role of phosphorylation of vdUTPase Ser-187 on HSV-1 replication in cell cultures, two sets of experiments were performed. In the first set of experiments, SK-N-SH or HEp-2 cells were infected with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK759 (vdUTPaseD97A), YK760 (vdUTPaseD97A-repair), YK753 (vdUTPaseS187D), or YK754 (vdUTPaseS187D-repair) at an MOI of 5, and viral titers were assayed at 36 h postinfection. As shown in Fig. 7A, the progeny virus titers of all the recombinant viruses tested were similar to that of wild-type HSV-1(F) in HEp-2 cells. In contrast, the virus titers of YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), and YK759 (vdUTPaseD97A) in SK-N-SH cells were significantly lower than those of their repaired viruses and wild-type HSV-1(F). Furthermore, the wild-type viral titer was restored in SK-N-SH cells infected with YK753 (vdUTPaseS187D) carrying a phosphomimetic mutation in vdUTPase Ser-187 (Fig. 7B). In agreement with findings for YK751 (vdUTPaseS187A), the titer of virus in cells infected with YK511 (Us3K220M) was significantly reduced compared to that in cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) (Fig. 7C), indicating that Us3 kinase activity was also required for efficient viral replication. In the second set of experiments, growth kinetics of each of the recombinant viruses in SK-N-SH cells at an MOI of 5 or 0.01 was analyzed. In agreement with results shown in Fig. 7B, YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), and YK759 (vdUTPaseD97A) replicated in SK-N-SH cells less efficiently than wild-type HSV-1(F), YK753 (vdUTPaseS187D), or their repair viruses at an MOI of 5 (Fig. 8A, C, and E). In contrast, the growth curves of wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK753 (vdUTPaseS187D), YK759 (vdUTPaseD97A), and their repaired viruses were similar in SK-N-SH cells at an MOI of 0.01 (Fig. 8B, D, and F). YK511 (Us3K220M) replicated in SK-N-SH cells less efficiently than wild-type HSV-1(F) or its repair virus at both MOIs (Fig. 8G and H). Taken together with results shown in Fig. 3, these results indicated that phosphorylation of vdUTPase Ser-187 and enzymatic activity of vdUTPase were required for efficient viral replication in SK-N-SH cells at an MOI of 5 but not in SK-N-SH cells at an MOI of 0.01 or HEp-2 and Vero cells at both MOIs and suggested that regulation of vdUTPase enzymatic activity by Us3 phosphorylation of vdUTPase at Ser-187 contributed to efficient HSV-1 replication in a manner dependent on cell types and MOIs.
FIG 7.
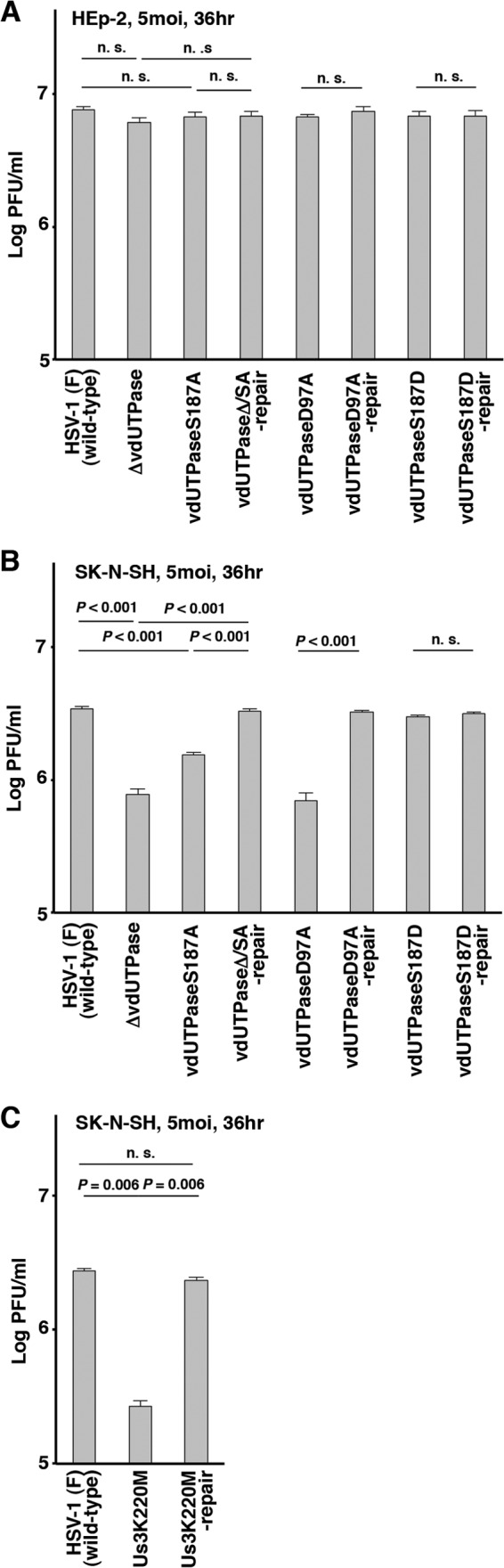
Effect of phosphorylation of vdUTPase Ser-187 on HSV-1 replication in SK-N-SH and HEp-2 cells. (A and B) HEp-2 (A) or SK-N-SH (B) cells were infected with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK759 (vdUTPaseD97A), YK760 (vdUTPaseD97A-repair), YK753 (vdUTPaseS187D), or YK754 (vdUTPaseS187D-repair) at an MOI of 5. Total virus from the cell culture supernatants and infected cells was harvested at 36 h postinfection, and virus titers were assayed. Each value is the mean ± standard error for six experiments. (C) SK-N-SH cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 5. The experiments were performed as described for panel B. Each value is the mean ± standard error for three experiments. “P” represents the statistical significance value according to the two-tailed Student's t test. Data are representative of three independent experiments.
FIG 8.
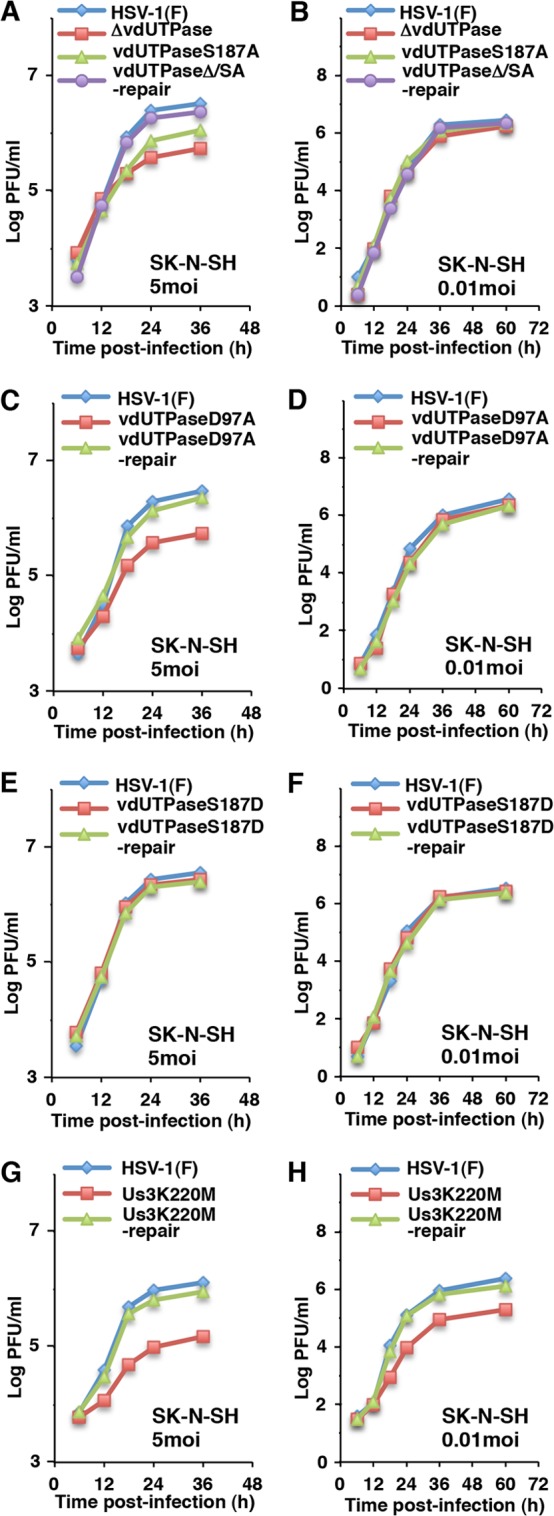
Growth curves of recombinant viruses in SK-N-SH cells used in this study. (A to H) Human neuroblastoma SK-N-SH cells were infected at an MOI of 5 (left figures) or 0.01 (right figures) with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), and YK752 (vdUTPaseΔ/SA-repair) (A and B), wild-type HSV-1(F), YK759 (vdUTPaseD97A), and YK760 (vdUTPaseD97A-repair) (C and D), wild-type HSV-1(F), YK753 (vdUTPaseS187D), and YK754 (vdUTPaseS187D-repair) (E and F), or wild-type HSV-1(F), YK511 (Us3K220M), and YK513 (Us3K220M-repair) (G and H). Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and was assayed on Vero cells. Data are representative of three independent experiments.
Effect of phosphorylation of vdUTPase Ser-187 on its localization in infected cells.
It has been reported that Us3 phosphorylation of known HSV-1 substrates, including UL31, UL47, and Us3 itself, was required for their proper localization in HSV-1-infected cells (20, 23, 24). To examine whether Us3 phosphorylation of vdUTPase at Ser-187 regulated localization of vdUTPase in HSV-1-infected cells as well, Vero cells were infected with wild-type HSV-1(F) or YK751 (vdUTPaseS187A) at an MOI of 5 and analyzed by immunofluorescence with confocal microscopy. As shown in Fig. 9A and C, vdUTPase localized predominantly in the nucleus of Vero cells infected with wild-type HSV-1(F) at 12 h postinfection. At 18 h postinfection, although still present in the nucleus, vdUTPase also became detectable in the cytoplasm, especially, in punctate structures in wild-type HSV-1(F)-infected Vero cells (Fig. 9E and G). In cells infected with YK751 (vdUTPaseS187A), the patterns of localization of vdUTPase at 12 and 18 h postinfection were similar to those in cells infected with wild-type HSV-1(F) (Fig. 9B, D, F, and H), indicating that phosphorylation of vdUTPase Ser-187 was not required for proper localization of vdUTPas in infected cells.
FIG 9.
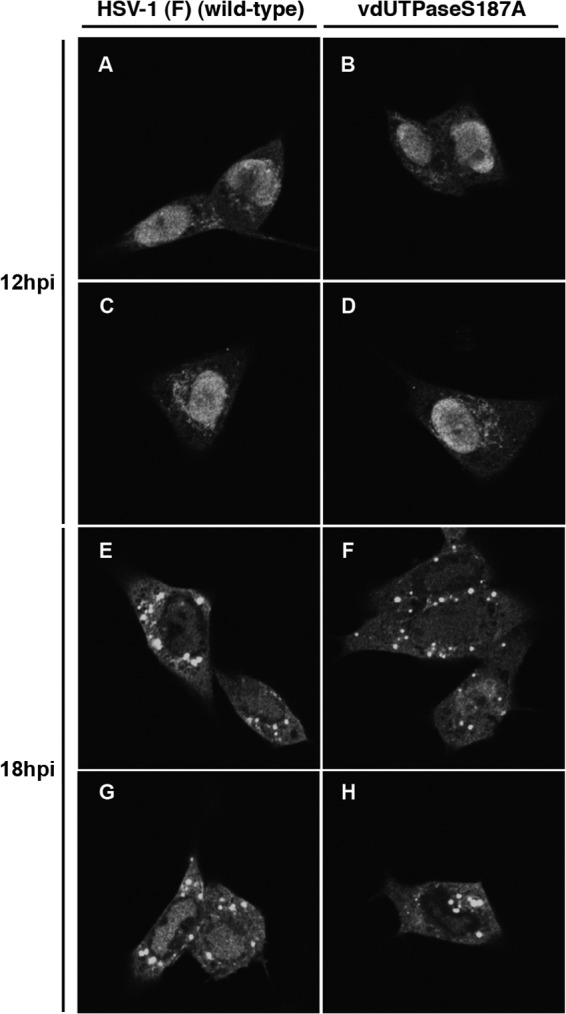
Digital confocal microscope images showing the localization of vdUTPase. Vero cells were infected with wild-type HSV-1(F) or YK751 (vdUTPaseS187A) at an MOI of 5. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an anti-vdUTPase rabbit polyclonal antibody, and examined by confocal microscopy.
DISCUSSION
The striking feature in this study was that vdUTPase was a novel physiological substrate of Us3, and this phosphorylation appeared to promote HSV-1 replication in a manner dependent on cell types or MOIs by regulating optimal enzymatic activity of vdUTPase in infected cells. This conclusion was based on the following series of observations: (i) MS analysis identified vdUTPase Ser-187 as a phosphorylation site; (ii) in vitro kinase assays showed that the Ser-187 in the vdUTPase domain was directly phosphorylated by Us3; (iii) phosphorylation of vdUTPase Ser-187 in infected cells was detected in Phos tag SDS-PAGE analysis and was dependent on Us3 kinase activity in infected cells; (iv) the S187A mutation in vdUTPase and kinase-dead mutation of Us3 significantly impaired the enzymatic activity of vdUTPase in infected cells and viral replication; and (v) a phosphomimetic mutation at the phosphorylation site in the vdUTPase restored wild-type vdUTPase activity and viral replication. From these observations, it seems reasonable to conclude that Us3 phosphorylation of vdUTPase at Ser-187 promoted HSV-1 replication by upregulating enzymatic activity of vdUTPase. However, there is an alternative interpretation of the data in this study. Based on the accumulating evidence suggesting that HSV-1 proteins thoroughly investigated so far turned out to perform multiple functions, there is a possibility that vdUTPase performs an entirely different function(s) unrelated to its enzymatic activity, probably in conjugation with a cellular and viral partner(s). Therefore, Us3 phosphorylation of vdUTPase at Ser-187 may not just alter the enzymatic activity of vdUTPase but may affect a vdUTPase function(s) unrelated to its enzymatic activity, such as its interaction with a cellular and/or viral partner(s). We noted that the S187A mutation in vdUTPase reduced viral replication in SK-N-SH cells at an MOI of 5 but not in SK-N-SH cells at an MOI of 0.01 or in HEp-2 and Vero cells at both MOIs, suggesting that the effect of Us3 phosphorylation of vdUTPase Ser-187 on viral replication was dependent on cell types and MOIs.
Us3 phosphorylation of vdUTPase Ser-187 was not essential for vdUTPase activity but appeared to optimize vdUTPase activity in infected cells, since the S187A mutation reduced vdUTPase enzymatic activity in infected cells only to 68% of that in wild-type virus-infected cells. In contrast, the data in this study indicated that this phosphorylation was required for efficient viral replication in SN-N-SH cells at an MOI of 5 but not at an MOI of 0.01. These results suggested that vdUTPase enzymatic activity in infected cells was tightly regulated by Us3 phosphorylation of vdUTPase Ser-187 and this strict regulation was involved in efficient viral replication in a specific cell type(s) and at a specific MOI(s). At present, the mechanism by which Us3 phosphorylation of vdUTPase Ser-187 is required for efficient HSV-1 replication only in the specific cell type (human neuroblastoma SK-N-SH cells) and at the specific MOI remains to be elucidated. It has long been assumed that viruses encode a dUTPase to compensate for low cellular dUTPase activity if that is the situation in their host cells, e.g., in resting and differentiated cells, such as neurons and macrophages, where cellular dUTPase activity has been suggested to be low (3, 58). In support of this hypothesis, it has been reported that replication of recombinant ASFV and D-type retroviruses (e.g., equine infectious anemia virus, feline immunodeficiency virus, and caprine arthritis encephalitis virus) carrying mutations in each of the viral dUTPases was severely affected in nondividing cells in vitro, whereas replication in actively dividing cells was only minimally decreased (58–62). In agreement with the hypothesis, HSV-1 vdUTPase was reported to be required for efficient viral replication and pathogenicity in the CNS as described above (45). Thus, there is a possibility that HSV-1 vdUTPase enzymatic activity may be upregulated by phosphorylation of vdUTPase Ser-187 by viral Us3 kinase to compensate for the low cellular endogenous dUTPase activity in SK-N-SH cells for efficient viral replication. The observation that the null, S187A, or D97A mutation in vdUTPase significantly impaired viral replication in SK-N-SH at an MOI of 5 but not at an MOI of 0.01 appeared to support this possibility, since it was conceivable that viral replications of the recombinant viruses carrying the mutations in vdUTPase at an MOI of 5 required much more cellular endogenous dUTPase activity than those at an MOI of 0.01. The cellular endogenous dUTPase activity in SK-N-SH cell may be too low to support optimal viral replications of the vdUTPase mutant viruses at an MOI of 5, whereas it may be sufficient for their efficient viral replication at an MOI of 0.01. By extension, this phosphorylation-mediated regulation of HSV-1 vdUTPase enzymatic activity might play a role in viral pathogenicity in the CNS in vivo. Further studies will be needed to clarify this subject and are in progress in this laboratory.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), a contract research fund for Program of Japan Initiative for Global Research Network on Infectious Diseases and Global COE Program Center of Education and Research for the Advanced Genome-Based Medicine—for personalized medicine and the control of worldwide infectious diseases, a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan, and grants from the Takeda Science Foundation, the Naito Foundation, and the Tokyo Biochemical Research Foundation. S.T. was supported by a fellowship from the Graduate Program for Leaders in Life Innovation from MEXT.
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Edelman AM, Blumenthal DK, Krebs EG. 1987. Protein serine/threonine kinases. Annu. Rev. Biochem. 56:567–613 [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. 10.1126/science.1075762 [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J. Virol. 85:1158–1173. 10.1128/JVI.01369-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deruelle MJ, Favoreel HW. 2011. Keep it in the subfamily: the conserved alphaherpesvirus US3 protein kinase. J. Gen. Virol. 92:18–30. 10.1099/vir.0.025593-0 [DOI] [PubMed] [Google Scholar]
- 6.McGeoch DJ, Davison AJ. 1986. Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res. 14:1765–1777. 10.1093/nar/14.4.1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frame MC, Purves FC, McGeoch DJ, Marsden HS, Leader DP. 1987. Identification of the herpes simplex virus protein kinase as the product of viral gene US3. J. Gen. Virol. 68(Part 10):2699–2704. 10.1099/0022-1317-68-10-2699 [DOI] [PubMed] [Google Scholar]
- 8.Chee MS, Lawrence GL, Barrell BG. 1989. Alpha-, beta- and gammaherpesviruses encode a putative phosphotransferase. J. Gen. Virol. 70(Part 5):1151–1160. 10.1099/0022-1317-70-5-1151 [DOI] [PubMed] [Google Scholar]
- 9.Smith RF, Smith TF. 1989. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 63:450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leader DP. 1993. Viral protein kinases and protein phosphatases. Pharmacol. Ther. 59:343–389. 10.1016/0163-7258(93)90075-O [DOI] [PubMed] [Google Scholar]
- 11.Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim. Biophys. Acta 1091:426–431. 10.1016/0167-4889(91)90210-O [DOI] [PubMed] [Google Scholar]
- 12.Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates indicate structural requirements distinct from other protein kinases. Biochim. Biophys. Acta 889:208–215 (Erratum, 927:303, 1987.) 10.1016/0167-4889(86)90106-0 [DOI] [PubMed] [Google Scholar]
- 13.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 101:9411–9416. 10.1073/pnas.0403160101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24:2627–2639. 10.1101/gad.1978310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. 10.1016/0042-6822(88)90417-5 [DOI] [PubMed] [Google Scholar]
- 16.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952. 10.1128/JVI.76.17.8939-8952.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412. 10.1128/JVI.78.1.399-412.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 83:11624–11634. 10.1128/JVI.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783. 10.1128/JVI.00103-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613. 10.1128/JVI.00845-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Patenode C, Roizman B. 2011. US3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 108:14632–14636. 10.1073/pnas.1111942108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250–261. 10.1128/JVI.01451-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191. 10.1128/JVI.00090-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, Roller RJ. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J. Virol. 81:10792–10803. 10.1128/JVI.00196-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470. 10.1128/JVI.00380-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munger J, Roizman B. 2001. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 98:10410–10415. 10.1073/pnas.181344498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. U. S. A. 90:6701–6705. 10.1073/pnas.90.14.6701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162. 10.1128/JVI.01447-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 32.Vertessy BG, Toth J. 2009. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42:97–106. 10.1021/ar800114w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shlomai J, Kornberg A. 1978. Deoxyuridine triphosphatase of Escherichia coli. Purification, properties, and use as a reagent to reduce uracil incorporation into DNA. J. Biol. Chem. 253:3305–3312 [PubMed] [Google Scholar]
- 34.Bessman MJ, Lehman IR, Adler J, Zimmerman SB, Simms ES, Kornberg A. 1958. Enzymatic synthesis of deoxyribonucleic acid. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acids. Proc. Natl. Acad. Sci. U. S. A. 44:633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sedwick WD, Brown OE, Glickman BW. 1986. Deoxyuridine misincorporation causes site-specific mutational lesions in the lacI gene of Escherichia coli. Mutat. Res. 162:7–20. 10.1016/0027-5107(86)90066-7 [DOI] [PubMed] [Google Scholar]
- 36.Kunz BA, Kohalmi SE. 1991. Modulation of mutagenesis by deoxyribonucleotide levels. Annu. Rev. Genet. 25:339–359. 10.1146/annurev.ge.25.120191.002011 [DOI] [PubMed] [Google Scholar]
- 37.McClure MA. 2001. Evolution of the DUT gene: horizontal transfer between host and pathogen in all three domains of life. Curr. Protein Pept. Sci. 2:313–324. 10.2174/1389203013381062 [DOI] [PubMed] [Google Scholar]
- 38.Baldo AM, McClure MA. 1999. Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J. Virol. 73:7710–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGeehan JE, Depledge NW, McGeoch DJ. 2001. Evolution of the dUTPase gene of mammalian and avian herpesviruses. Curr. Protein Pept. Sci. 2:325–333. 10.2174/1389203013380964 [DOI] [PubMed] [Google Scholar]
- 40.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preston VG, Fisher FB. 1984. Identification of the herpes simplex virus type 1 gene encoding the dUTPase. Virology 138:58–68. 10.1016/0042-6822(84)90147-8 [DOI] [PubMed] [Google Scholar]
- 42.Caradonna SJ, Cheng YC. 1981. Induction of uracil-DNA glycosylase and dUTP nucleotidohydrolase activity in herpes simplex virus-infected human cells. J. Biol. Chem. 256:9834–9837 [PubMed] [Google Scholar]
- 43.Bjornberg O, Bergman AC, Rosengren AM, Persson R, Lehman IR, Nyman PO. 1993. dUTPase from herpes simplex virus type 1; purification from infected green monkey kidney (Vero) cells and from an overproducing Escherichia coli strain. Protein Expr. Purif. 4:149–159. 10.1006/prep.1993.1021 [DOI] [PubMed] [Google Scholar]
- 44.Pyles RB, Thompson RL. 1994. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus type 1 strains in cultured murine cells. J. Virol. 68:4514–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyles RB, Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 66:6706–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 49.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 50.Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331. 10.1128/JVI.79.14.9325-9331.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. 2005. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4:2010–2021. 10.1074/mcp.T500030-MCP200 [DOI] [PubMed] [Google Scholar]
- 53.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams MV, Cheng Y. 1979. Human deoxyuridine triphosphate nucleotidohydrolase. Purification and characterization of the deoxyuridine triphosphate nucleotidohydrolase from acute lymphocytic leukemia. J. Biol. Chem. 254:2897–2901 [PubMed] [Google Scholar]
- 55.Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956. 10.1128/JVI.79.11.6947-6956.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5:749–757. 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 57.Barabas O, Pongracz V, Kovari J, Wilmanns M, Vertessy BG. 2004. Structural insights into the catalytic mechanism of phosphate ester hydrolysis by dUTPase. J. Biol. Chem. 279:42907–42915. 10.1074/jbc.M406135200 [DOI] [PubMed] [Google Scholar]
- 58.Payne SL, Elder JH. 2001. The role of retroviral dUTPases in replication and virulence. Curr. Protein Pept. Sci. 2:381–388. 10.2174/1389203013381008 [DOI] [PubMed] [Google Scholar]
- 59.Turelli P, Petursson G, Guiguen F, Mornex JF, Vigne R, Querat G. 1996. Replication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J. Virol. 70:1213–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH. 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U. S. A. 92:7480–7484. 10.1073/pnas.92.16.7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oliveros M, Garcia-Escudero R, Alejo A, Vinuela E, Salas ML, Salas J. 1999. African swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J. Virol. 73:8934–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Threadgill DS, Steagall WK, Flaherty MT, Fuller FJ, Perry ST, Rushlow KE, Le Grice SF, Payne SL. 1993. Characterization of equine infectious anemia virus dUTPase: growth properties of a dUTPase-deficient mutant. J. Virol. 67:2592–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]