

Abstract
In Staphylococcus aureus, the low-molecular-weight thiol called bacillithiol (BSH), together with cognate S-transferases, is believed to be the counterpart to the glutathione system of other organisms. To explore the physiological role of BSH in S. aureus, we constructed mutants with the deletion of bshA (sa1291), which encodes the glycosyltransferase that catalyzes the first step of BSH biosynthesis, and fosB (sa2124), which encodes a BSH-S-transferase that confers fosfomycin resistance, in several S. aureus strains, including clinical isolates. Mutation of fosB or bshA caused a 16- to 60-fold reduction in fosfomycin resistance in these S. aureus strains. High-pressure liquid chromatography analysis, which quantified thiol extracts, revealed some variability in the amounts of BSH present across S. aureus strains. Deletion of fosB led to a decrease in BSH levels. The fosB and bshA mutants of strain COL and a USA300 isolate, upon further characterization, were found to be sensitive to H2O2 and exhibited decreased NADPH levels compared with those in the isogenic parents. Microarray analyses of COL and the isogenic bshA mutant revealed increased expression of genes involved in staphyloxanthin synthesis in the bshA mutant relative to that in COL under thiol stress conditions. However, the bshA mutant of COL demonstrated decreased survival compared to that of the parent in human whole-blood survival assays; likewise, the naturally BSH-deficient strain SH1000 survived less well than its BSH-producing isogenic counterpart. Thus, the survival of S. aureus under oxidative stress is facilitated by BSH, possibly via a FosB-mediated mechanism, independently of its capability to produce staphyloxanthin.
INTRODUCTION
Staphylococcus aureus, a leading cause of bacterial infections (1), is normally a commensal organism in healthy individuals that can become pathogenic. Over the course of infection, the bacterium encounters metabolic and environmental stresses within the host environment, including reactive oxygen species (ROS) generated by host neutrophils (2) and macrophages (3). Without an effective response to the damage caused by oxidative stress, the bacterium is at risk of accumulating deleterious mutations, damaged proteins (4), or lipid peroxides that may lead to membrane degradation, lysis, and cell death.
One strategy employed by the bacteria to mitigate the effects of oxidative stress revolves around the use of low-molecular-weight (LMW) thiols to maintain the cell's intracellular reducing environment. In many prokaryotes, including Gram-negative rods, the active LMW thiol is glutathione (GSH), which has been implicated in bacterial responses to oxidative stress (5, 6), antibiotic resistance (7), and survival during the course of infection (8, 9).
Bacillithiol (BSH), first characterized in 2009, is a LMW thiol that is an α-anomeric glycoside of l-cysteinyl-d-glucosamine conjugated to l-malic acid and is produced by some genera of Firmicutes, including S. aureus (10). The first gene of the BSH biosynthetic pathway, bshA, is responsible for conjugating l-malic acid to UDP-GlcNAc to form N-acetylglucosaminylmalate and is necessary for the synthesis of BSH (11, 12). The next gene, bshB, encodes an enzyme responsible for deacetylating GlcNAc-Mal, and the last gene, bshC, codes for a cysteine ligase (11). While multiple bshB paralogs occur in Bacillus species (11, 12), there is only one, bshB2, in S. aureus (11). Interestingly, laboratory strains of S. aureus in the 8325 lineage, such as SH1000 and 8325-4, contain an 8-bp insertion in bshC which renders the protein inactive and, hence, prevents the synthesis of BSH (13, 14).
The enzyme FosB has been shown to confer resistance to fosfomycin in S. aureus (15, 16). In vitro, FosB purified from S. aureus has been reported to function as a BSH-S-transferase (BST) to inactivate fosfomycin by opening up the epoxide ring of fosfomycin through the conjugation of BSH to the compound and, hence, inactivating the drug (17–19). While factors such as importers (e.g., GlpT [16] and UhpT [20]) have been connected to fosfomycin resistance, their exact contribution to resistance in S. aureus depends on transport of the drug rather than direct inactivation of FosB.
The role of BSH in S. aureus clinical isolates has not been previously examined; therefore, we undertook the current study to decipher the role of BSH in several methicillin-resistant S. aureus (MRSA) isolates. Furthermore, the function of FosB outside antibiotic resistance has not been ascertained. We report here on the role of BSH in the oxidative stress response of MRSA, as well as identify a novel role for FosB in the oxidative stress response. Collectively, our data suggest that BSH and FosB are crucial for the survival of S. aureus when encountering oxidative stresses. In particular, BSH is required for improved bacterial survival after exposure to professional macrophages and neutrophils, and this effect is independent of staphyloxanthin production.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The wild-type and mutant S. aureus strains as well as the plasmids used in this study are listed in Table 1. All S. aureus precultures were grown in tryptic soy broth (TSB; Teknova) at 37°C with shaking. After overnight growth, the precultures were diluted to an optical density at 650 nm (OD650) of 0.1 in 5 ml of fresh TSB, as measured by a Spectronic 20D+ spectrophotometer (Spectronic Inc.), using 18-mm borosilicate glass tubes. Escherichia coli strains DH5α and DC10B (21) were grown at 37°C in Luria-Bertani (LB) broth or on LB agar (Becton, Dickinson). When needed for selection, antibiotics were used at the following final concentrations: 100 μg/ml of ampicillin (Amp) for E. coli and 10 μg/ml of chloramphenicol (Cm) for S. aureus.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| RN4220 | MSSA cloning intermediate derived from 8325-4 | 60 |
| USA300 CDC | CA-MRSA, wild-type reference strain | 61 |
| ALC7450 | USA300 CDC fosB mutant | This study |
| ALC7574 | USA300 CDC fosB mutant complemented with pALC7576 | This study |
| WI-2335 | USA300 CA-MRSA, clinical isolate, spa type t008 | Sanjay Shukla |
| ALC6573 | WI-2335 fosB mutant | This study |
| ALC6575 | WI-2335 fosB mutant complemented with pALC6574 | This study |
| ALC7578 | WI-2335 fosB mutant complemented with pALC7576 | This study |
| ALC7262 | WI-2335 bshA mutant | This study |
| ALC7263 | WI-2335 fosB bshA mutant | This study |
| TCH959 | USA300 CA-MSSA, clinical isolate | 62 |
| ALC7224 | TCH959 fosB mutant | This study |
| ALC7579 | TCH959 fosB mutant complemented with pALC7576 | This study |
| COL | Archaic HA-MRSA strain | 63 |
| ALC7069 | COL fosB mutant | This study |
| ALC7070 | COL fosB mutant complemented with pALC6574 | This study |
| ALC7071 | COL fosB mutant complemented with pALC7576 | This study |
| ALC7247 | COL bshA mutant | This study |
| ALC7267 | COL bshA mutant complemented with pALC7266 | This study |
| Mu50 | VISA | 64 |
| ALC7441 | Mu50 fosB mutant | This study |
| ALC7506 | Mu50 fosB mutant complemented with pALC6574 | This study |
| ALC7432 | Mu50 bshA mutant | This study |
| ALC7568 | Mu50 bshA mutant complemented with pALC7266 | This study |
| SH1000 | MSSA derivative of 8325-4 with a functional rsbU | 65 |
| ALC7446 | SH1000 with bshC repaired | This study |
| Plasmids | ||
| pMAD-Cm | Allelic replacement vector containing Cm resistance marker | 23 |
| pEPSA5 | S. aureus-E. coli shuttle vector with a xylose-inducible promoter | 24 |
| pKOR1 | Allelic replacement vector containing Cm resistance marker and secY counterselection | 22 |
| pALC6572 | pMAD-Cm for deletion of fosB | This study |
| pALC6574 | pMAD-Cm for the complementation of the fosB deletion | This study |
| pALC7576 | pEPSA5 vector for fosB expression | This study |
| pALC7246 | pKOR1 for deletion of bshA | This study |
| pALC7266 | pKOR1 for the complementation of the bshA deletion | This study |
| pALC7588 | pEPSA5 vector for bshA expression | This study |
| pALC7561 | pKOR1 for the repair of bshC in the 8325 lineage | This study |
CA, community acquired; HA, hospital acquired; MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus; VISA, vancomycin-intermediate S. aureus.
DNA manipulations.
A QIAprep Spin Miniprep kit (Qiagen) was used to prepare plasmid DNA from E. coli. Plasmid DNA from S. aureus was isolated by lysostaphin digestion, followed by plasmid extraction with the QIAprep kit (Qiagen). Chemically competent DH5α or DC10B (21) strains of E. coli or electrocompetent S. aureus cells were used for transformations. The restriction enzymes, ligases, and molecular weight markers used in this study were purchased from New England BioLabs. An Invitrogen TOPO TA cloning kit with pCR2.1 was used in an intermediate step in the construction of plasmids pALC6572, pALC6574, and pALC7576 (Table 1) following the manufacturer's instructions. For the construction of pALC7246 and pALC7266, a Gateway BP Clonase II enzyme mix kit (Life Technologies) was used to introduce the DNA fragments of choice into pKOR1 (22). ExTaq DNA polymerase (Takara) was used to amplify DNA fragments for all constructs. DNA sequencing was performed using fluorescently labeled dideoxynucleotide sequencing (BigDye terminators; PE Applied Biosystems) at the Dartmouth College Molecular Biology and Proteomics Core Facility.
Construction of fosB and bshA deletion mutants and complemented strains.
The fosB mutants in various S. aureus strains—COL, Mu50, USA300 TCH959, USA300 WI-2335, and the USA300 CDC reference strain—were generated by in-frame deletion of fosB (sa2124) via allelic replacement using published protocols for the temperature-sensitive plasmid pMAD-Cm (23). The primers used for the construction of the mutants are available upon request. The chromosomal deletion of fosB in putative mutants was verified by PCR and DNA sequencing. The mutants were subsequently complemented by using the same pMAD-Cm system to reintroduce an intact copy of fosB into the native fosB locus of the mutants.
We also complemented the fosB mutants in trans. In this case, we transformed pALC7576, a plasmid consisting of a recombinant pEPSA5 (24) containing a xylose-inducible promoter and the ribosomal binding site followed by the cognate fosB coding region, into E. coli DC10B, followed by electroporation directly into the cognate mutants.
The bshA deletion mutants were generated in several strain backgrounds through in-frame deletion of bshA (sa1291) using pKOR1, a temperature-sensitive plasmid with a Cm resistance selection marker (22). Chromosomal deletion of bshA was verified by PCR and DNA sequencing. The complemented strains were constructed using the same pKOR1 system to reintroduce an intact copy of bshA into the native locus on the chromosome of the mutants. In trans complementation was done by introducing pALC7588, another plasmid consisting of a pEPSA5 backbone containing a xylose-inducible promoter, followed by the ribosomal binding site and the cognate bshA coding region, into the bshA mutants.
Determination of LMW thiol levels.
For determination of LMW thiol levels, cultures were diluted 1:100 from precultures into 130 ml TSB in a 500-ml sidearm flask and grown at 37°C with shaking until they reached an OD650 of 1.1 (late exponential phase). Cultures were divided into 3 samples of 40 ml each (in separate 300-ml sidearm flasks)—with one treated with 10 mM H2O2, another treated with 1 mM diamide, and the third treated with medium alone—and grown for an additional 20 min at 37°C with shaking. Individual cultures were then divided into two samples, each of which was harvested by centrifugation at 3,200 × g at 4°C for analysis. Intracellular reduced thiols (BSH, Cys, and coenzyme A [CoA] sulfhydrate [CoASH]) were derivatized, in triplicate, with monobromobimane (mBBr). Samples for disulfide analysis were first treated with N-ethylmaleimide (NEM), followed by dithiothreitol (DTT) reduction and labeling with mBBr of reduced thiols and oxidized disulfides (BSSB and cystine), and were analyzed by fluorescent high-pressure liquid chromatography (HPLC), as previously described (18, 25). However, it is not possible to accurately quantify CoA disulfide because the DTT treatment used in the disulfide analysis also cleaves thioesters such as acetyl-CoA into CoA, resulting in an overestimate of CoA disulfide (26). Standards of known concentration were used to quantify bimane-derivatized thiols from cellular samples.
Antibiotic susceptibility assays.
The determination of the MICs for fosfomycin, oxacillin, penicillin G, d-cycloserine, vancomycin, imipenem, and bacitracin (Sigma) in a broth microdilution format for various S. aureus strains was performed according to the Clinical and Laboratory Standards Institute (CLSI) protocol (27). Xylose was added to a final concentration of 1% to cultures that contained strains complemented with pALC7576 or pALC7588, as well as to their respective isogenic mutant and parental strains. In some experiments, exogenous cysteine was added to a final concentration of 500 μM, a concentration which was previously shown to rescue cysM mutants in S. aureus (28).
Growth of S. aureus bshA and fosB mutants in the presence of hydrogen peroxide.
For growth in 10 mM H2O2, a nonlethal concentration that triggers alteration of gene expression (29), cultures were diluted as described above in 5 ml TSB, with compounds added in addition to the H2O2 at the following final concentrations: 4 mM diamide was added immediately, and 50 mM thiourea was added 5 min later. Hourly readings of the OD650 were taken to monitor growth.
For overnight stress assays, cultures were diluted as described above in 5 ml TSB and grown in triplicate, with one tube containing 25 mM H2O2, the other containing 5 mM diamide, and the last tube containing both compounds at the stated concentrations.
Detoxification of H2O2 by S. aureus.
For assay of the detoxification of H2O2 by S. aureus, precultures diluted to an OD650 of 0.1 in 10 ml of fresh TSB were exposed to 10 mM H2O2. Serial samples (1 ml each) were taken from the culture at the desired time points for analysis. An OxiSelect hydrogen peroxide assay kit (Cellbio labs) was used to quantify H2O2 in the culture supernatant following the instructions in the manufacturer's insert, after the sample was processed by centrifugation and filtration to remove any residual bacteria.
Assays for peroxidase activity.
Three different assays were utilized to assess the potential peroxidase activity of FosB. First, 5 μM S. aureus FosB, overexpressed and purified as recently described (18), was incubated at 22°C in the presence of 250 μM BSH and 0.5 mM MgCl2 in 100 mM HEPES, pH 7. H2O2 was added to 200 μM, and the BSH concentration was monitored over 15 min by titrating reaction aliquots with 2 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellman's reagent) in 50 mM sodium phosphate buffer, pH 7.4, and measuring the absorbance at 412 nm using an extinction coefficient of 14,150 M−1 cm−1 (30, 31). Control reactions included assays without either H2O2 or FosB.
Second, horseradish peroxidase (HRP) was used to monitor the H2O2 concentration in the presence of FosB. The assay mixtures consisted of 1 μM FosB, 250 μM BSH, 0.5 mM MgCl2, and 100 mM HEPES, pH 7, in a final volume of 50 μl. Reactions were initiated with the addition of 250 μM H2O2 and monitored over time by quenching with 450 μl of 0.5 mM tetramethylbenzidine and 0.15 mU HRP in 0.1 M sodium phosphate, pH 7.4. After 5 min, an equal volume of 1 M phosphoric acid was added and the absorbance was read at 450 nm. The H2O2 concentration was determined using a standard curve of known H2O2 concentrations. The control reactions did not include either FosB or BSH.
Third, FosB peroxidase activity was assessed using the FOX1 assay (32). The reaction mixtures, consisting of 0, 1, or 10 μM FosB, 100 mM HEPES, pH 7, 0.5 mM MgCl2, and 100 μM BSH, were initiated with 100 μM H2O2 and monitored over 30 min by quenching with the FOX1 assay reagent (100 μM xylenol orange, 250 μM ammonium ferrous sulfate, 100 mM sorbitol, 25 mM H2SO4). After incubation at room temperature for 30 min, the samples were centrifuged to pellet the debris and the absorbance of the supernatant was read at 560 nm and compared to a standard curve of known H2O2 concentrations. The control reactions did not include either FosB or BSH.
NADP+/NADPH quantification.
The protocol for the quantification of NADP+/NADPH was conducted using an NADPH/NADP+ kit from Biovision Inc. modified for S. aureus. As NADPH can be unstable, the following steps were carried out as quickly as possible. H2O2 (10 mM) was added to a 10-ml S. aureus culture at an OD650 of 0.1 as described above. At the indicated time point, cells were pelleted at 3,220 × g at 4°C, washed with cold sterile phosphate-buffered saline (PBS), resuspended in 400 μl of NADP/NADPH extraction buffer (Biovision Inc.) within 10 min of the initial pelleting of the culture, and lysed with 0.1 mm glass/silica beads in a BeadBeater apparatus (BioSpec). The resultant supernatant, obtained by centrifuging samples at 20,000 × g for 15 min at 4°C, was filtered through a 10-kDa-cutoff filter (Amicon), as suggested by the assay manufacturer, to remove enzymes which utilize NADPH.
Samples (50 μl) were transferred in triplicate to a 96-well plate, followed by the addition of 100 μl of NADP cycling mix to each well. After incubation in the dark at room temperature for 5 min and the addition of 10 μl of NADPH developer, the plate was shaken for 2 h and the OD450 was measured. The concentration of each well was derived from the standard curve prepared using standards provided by the manufacturer. The amounts of NADP+ and NADPH were normalized to the amount of total cellular proteins.
Total antioxidant capacity.
Diluted precultures were grown as described above to an OD650 of 0.7, incubated with or without H2O2 for 20 min, and harvested by pelleting. Cells were washed thrice with ice-cold PBS and resuspended in 600 μl of PBS. Cells were then freeze-thawed twice before mechanical lysis. The supernatant was harvested after centrifugation at 4°C and 5,000 × g for 10 min. The protein concentrations of the samples (30 μl each) were determined using a 660-nm protein assay kit (Pierce). The remaining portions of the samples were assayed using the OxiSelect total antioxidant capacity assay kit (Cell Biolabs), with the standard curve constructed following the instructions in the manufacturer's insert.
RNA extraction.
RNA extractions were carried out as previously described (33), with some modifications. In brief, cells were inoculated into 30 ml TSB in 300-ml sidearm flasks (Bellco) and grown to an OD650 of 1.1. The culture was divided into 2 separate 50-ml tubes, one with TSB containing 1 mM diamide and the other with TSB alone as a control, grown for an additional 20 min, and centrifuged at 3,220 × g and 4°C. The resulting cell pellets were resuspended in 1 ml of TRIzol reagent (Invitrogen) and mechanically lysed using 0.1-mm glass/silica beads in a BeadBeater apparatus. The RNA in the aqueous phase was precipitated with ethanol. The RNA used for the microarray analysis was kept in ethanol until further processing, while the samples used in quantitative reverse transcription-PCR (qRT-PCR) were resuspended in 50 μl of diethylpyrocarbonate-treated water. The concentration and purity of the total RNA were determined by measuring the absorbance at 230 nm, 260 nm, and 280 nm using a Nanodrop1000 spectrophotometer (Thermo Fisher).
Microarray analysis.
The microarray was manufactured by in situ synthesis of 15,600 60-base-long oligonucleotide probes (Agilent), selected as previously described (34). The probe set covered multiple genomes and plasmids, as previously published (35).
Total RNA purified from strain COL and the bshA mutant strains as described above was treated with DNase using an Ambion DNA-free kit. The removal of DNA was confirmed by quantitative PCR (Mx3005P; Agilent) with primers specific for the 16S rRNA gene (36). Five micrograms of total S. aureus RNA was then reverse transcribed and labeled by Cy3-dCTP using SuperScript II reverse transcriptase (Invitrogen) and purified on QIAquick columns (Qiagen). Cy5-dCTP, in the presence of the Klenow fragment of DNA polymerase I (Invitrogen), was used to label genomic DNA from strains used to design the arrays, which were used for the normalization process (36). Hybridization, washings, and scanning were performed as previously described (35).
Fluorescence intensities were extracted using Feature Extraction software (version 9; Agilent). Local background-subtracted signals were corrected for unequal dye incorporation or an unequal load of the labeled product. Data from three independent biological experiments were expressed as log10 ratios and analyzed using GeneSpring software, version 8.0 (Silicon Genetics). The statistical significance of differentially expressed genes was calculated by analysis of variance (ANOVA) including a Benjamini and Hochberg false discovery rate correction of 5% (P value cutoff = 0.05) and an arbitrary cutoff of 2.0 for expression ratios.
qRT-PCR analysis.
qScript cDNA Supermix (Quanta Biosciences) was used to reverse transcribe DNase I-treated RNA to yield cDNA. Following the manufacturer's instructions, PerfeCTa SYBR green FastMix (Quanta Biosciences) was used to generate standard curves of the cDNA concentration/crossing point (Cp) for the target genes and the reference gene, gyrB. We analyzed the expression of the target genes using the dilution for which the qRT-PCRs had the highest efficiency and reproducibility. Roche's LightCycler 480 SW (version 1.5) software was used to analyze the qRT-PCRs. Control reaction mixtures containing master mix and primers but no cDNA were also analyzed.
Staphyloxanthin extraction.
Staphyloxanthin was extracted from cultures using a modified version of a previously published protocol (37). Briefly, S. aureus precultures were diluted to an OD650 of 0.1 in 10 ml of fresh TSB, grown at 37°C for 72 h in triplicate, centrifuged, and washed in PBS. Staphyloxanthin was extracted with methanol treatment for 20 min at 42°C, and the OD450 was determined. Cultures that were induced with 10 mM H2O2 or 1 mM diamide were grown as described above, with the exception that the oxidants were added at the beginning of incubation and every 12 h until extraction.
Human whole-blood survival assay.
Overnight cultures grown in TSB were harvested by centrifugation and washed three times with PBS. Then, 2 × 105 bacterial cells were placed in 1 ml of fresh whole human blood containing heparin, and the initial inoculum was determined by serial dilution. Cultures were rotated at 37°C for 5 h, with aliquots removed hourly, serially diluted in PBS, plated on tryptic soy agar, and incubated at 37°C overnight. Assays with all samples were performed in triplicate.
Statistical analysis.
Data are expressed as the mean ± standard deviation. For some assays, an unpaired Student's t test was used to compare the wild types to the respective fosB or bshA mutants, with P values of ≤0.05 considered significant. In the analysis of LMW thiol levels, an ANOVA of thiol content and thiol ratios was carried out, with post hoc testing being done if the probability of the null hypothesis being true was less than 0.1; a P value of <0.05 was considered significant.
Microarray data accession number.
The complete microarray data set has been posted on the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GPL11157 for the platform design and accession number GSE46885 for the original data set.
RESULTS
FosB utilizes BSH as a substrate to confer fosfomycin resistance.
In previous studies, FosB was shown to mediate fosfomycin resistance in S. aureus through the use of BSH to disrupt the epoxide ring of fosfomycin (18, 19, 38). We investigated here the role of BSH in fosfomycin resistance mediated by FosB in a wide variety of methicillin-sensitive and methicillin-resistant S. aureus strains, including clinical isolates. Mutants with clean gene deletions, complemented mutants, and wild-type strains were used for this purpose. As expected from previous reports, the fosB mutants of USA300 CDC, USA300 WI-2335, USA300 TCH959, COL, and Mu50 all had a fosfomycin MIC of 39.1 μg/ml, whereas each corresponding wild-type strain exhibited significantly higher fosfomycin resistance levels (Table 2). Complementation of fosB in the mutants restored the level of fosfomycin resistance to wild-type levels.
TABLE 2.
Fosfomycin susceptibility of S. aureus strains and isogenic mutants
| Straina | MIC (μg/ml) |
|---|---|
| USA300 CDC | 625 |
| ALC7450 (ΔfosB) | 39.1 |
| ALC7574 (ΔfosB/pEPSA5::fosB) | 625 |
| WI-2335 | 1,250 |
| ALC6573 (ΔfosB) | 39.1 |
| ALC6575 (ΔfosB::fosB) | 1,250 |
| ALC7578 (ΔfosB/pEPSA5::fosB) | 1,250 |
| ALC7262 (ΔbshA) | 39.1 |
| ALC7262 (ΔbshA) + 500 μM Cys | 39.1 |
| ALC7263 (ΔfosB ΔbshA) | 39.1 |
| TCH959 | 625 |
| ALC7224 (ΔfosB) | 39.1 |
| ALC7579 (ΔfosB/pEPSA5::fosB) | 625 |
| COL | 312.5 |
| ALC7069 (ΔfosB) | 39.1 |
| ALC7070 (ΔfosB::fosB) | 625 |
| ALC7071 (ΔfosB/pEPSA5::fosB) | 625 |
| ALC7247 (ΔbshA) | 39.1 |
| ALC7247 (ΔbshA) + 500 μM Cys | 39.1 |
| ALC7267 (ΔbshA::bshA) | 625 |
| Mu50 | ≥2,500 |
| ALC7441 (ΔfosB) | 39.1 |
| ALC7506 (ΔfosB::fosB) | ≥2,500 |
| ALC7432 (ΔbshA) | 39.1 |
| ALC7432 (ΔbshA) + 500 μM Cys | 39.1 |
| ALC7568 (ΔbshA::bshA) | ≥2,500 |
| SH1000 | 19.3 |
| ALC7446 (BSH positive) | 625 |
Isogenic strains with the respective mutations are indicated under the parental strain name.
Importantly, the bshA mutation also affects fosfomycin resistance. The deletion of bshA alone and also that of bshA and fosB together led to the same MIC (39.1 μg/ml) as that for the corresponding isogenic fosB mutants. Complementation of bshA in the ΔbshA strains restored wild-type levels of fosfomycin resistance. Cysteine (Cys) has been suggested to be a possible substrate for FosB (19); therefore, we also tested the ability of Cys to restore fosfomycin resistance in bshA mutants. The addition of 500 μM Cys to the culture medium did not increase the fosfomycin resistance of the bshA mutants (Table 2). The laboratory strain SH1000 contains an intact fosB gene but does not produce BSH due to a missense mutation in the bshC coding region (13, 14). Following repair of the bshC gene in SH1000 to yield strain ALC7446, increased fosfomycin resistance was seen in ALC7446 compared to that seen in isogenic strain SH1000 (625 μg/ml versus 9.3 μg/ml, respectively) (Table 2). In S. aureus, the resistance mechanism by which FosB inactivates fosfomycin appears to require BSH, in line with detailed kinetic analysis of S. aureus FosB (17, 18).
BSH levels among S. aureus strains.
The amount of variance in fosfomycin MICs (Table 2) among wild-type S. aureus strains was of interest. One plausible explanation for the heterogeneity in the MICs of fosfomycin is that each strain may produce a different amount of BSH. To address this possibility, we quantified the levels of reduced thiols (BSH, Cys, and CoASH) and oxidized thiol (BSSB and cystine) in various clinical and laboratory isolates via HPLC analysis. The data confirmed that ALC7446 produces 0.51 μmol BSH per gram of cell dry mass (Table 3). Some variations in the reduced thiol (BSH, Cys, and CoASH) levels were observed among the strains. Cys and CoASH levels were not significantly different among the strains. The average level of BSH in the COL and WI-2335 strains was significantly above the average value among the strains tested, while that of Mu50 was below (Table 3). The variations in reduced thiol levels did not translate into differences in the ratios of the level of the reduced thiol to that of its oxidized form, as these were comparable among the strains (Table 3). Overall, with the exception of SH1000, the levels of BSH among the strains did not appear to correlate significantly with the observed levels of fosfomycin resistance.
TABLE 3.
Quantification of thiols in select S. aureus strains
| Strain | Amt (μmol/g)a |
Ratio |
|||
|---|---|---|---|---|---|
| BSH | Cys | CoASH | BSH/BSSB | Cys/Cystine | |
| COL | 1.07* ± 0.2 | 1.08 ± 0.4 | 0.89 ± 0.50 | 20.7 ± 16.3 | 23.6 ± 18.6 |
| Mu50 | 0.18 ± 0 | 0.88 ± 0.1 | 0.42 ± 0.08 | 26.0 ± 15.0 | 22.5 ± 9.7 |
| MW2 | 0.50 ± 0 | 1.00 ± 0.1 | 0.69 ± 0.08 | 18.0 ± 8.7 | 34.6 ± 17.4 |
| N315 | 0.45 ± 0.2 | 1.03 ± 0.2 | 0.30 ± 0.08 | 19.2 ± 6.6 | 59.1 ± 21.0 |
| SH1000 | 0* ± 0.0 | 0.5 ± 0.0 | 0.59 ± 0.25 | 0* ± 0.0 | 80.8* ± 30.1 |
| ALC7446 | 0.51 ± 0.1 | 1.23 ± 0.3 | 0.38 ± 0.19 | 28.0 ± 4.3 | 51.3 ± 23.4 |
| USA300 LAC | 0.42 ± 0.12 | 1.15 ± 0.3 | 0.47 ± 0.28 | 14.9 ± 8.2 | 35.8 ± 17.3 |
| USA300 WI-2335 | 1.63* ± 0.4 | 1.51 ± 0.1 | 0.40 ± 0.13 | 39.4 ± 13.4 | 19.5 ± 5.0 |
| WI-2335 ΔfosB | 0.61 ± 0.1 | 0.79 ± 0.0 | 0.34 ± 0.15 | 17.4 ± 3.7 | 12.8 ± 2.5 |
μmol thiol per g of cell dry mass. *, a significant difference of individual values from the mean of the entire column group using ANOVA post hoc testing, where P is <0.05. Due to the zero BSH value of SH1000, this value was not included in the calculation of the group mean.
Mutation of fosB in WI-2335 was accompanied by a 63% reduction in BSH levels (P = 0.0128), a 48% decrease in Cys levels (P = 0.0142), and a 15% decrease in CoASH levels relative to the corresponding levels in the parental strain (Table 3). These decreases suggest that the absence of FosB affects the redox state of the cell. However, the BSSB and cystine levels did not change significantly in the mutant compared to the wild type (data not shown), showing that the LMW thiol pool in the fosB mutant is not overly oxidized. The reason for the decreases in the thiol pool remains unclear and requires further investigations.
Growth of the bshA and fosB mutants under different laboratory conditions.
The role of BSH and of FosB in the physiology of S. aureus has not been well examined. The effect of the bshA and fosB deletions on the growth rate was determined under a variety of conditions. To focus our study, we chose to explore the effects of these mutations on strains COL and WI-2235 because these strains represent archaic and more recent MRSA isolates, respectively. When grown at 25°C, 37°C, or 42°C, the growth rates of the mutants did not differ significantly from those of the wild-type strains. Mutations of bshA and fosB also had no effect on the observed MIC values for several cell wall-active antibiotics, including oxacillin, penicillin G, d-cycloserine, vancomycin, imipenem, and bacitracin.
Growth delay of the bshA and fosB mutants after oxidative stress.
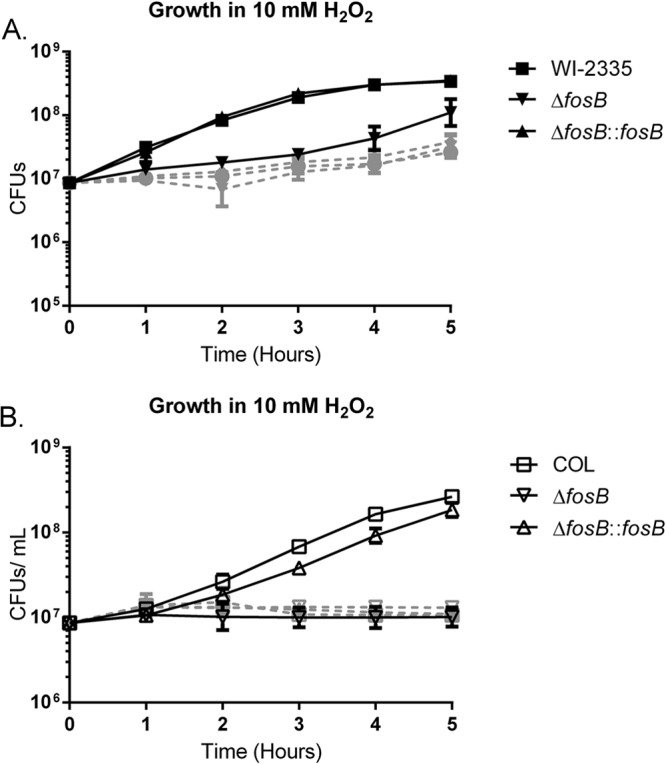
Given the reported role of related LMW thiols, such as GSH, in the protection of cells from oxidative stress (6, 39, 40), we evaluated the growth of strains USA300 WI-2335 and COL, their isogenic bshA and fosB mutants, and complemented mutant strains in the presence of 10 mM H2O2 (Fig. 1). Both the bshA and fosB mutants of COL and WI-2335 exhibited a growth delay for up to 4 h when grown in medium containing H2O2 compared to the growth of the isogenic parent and complemented mutants. To determine if this delay was due to oxidative stress caused by H2O2, 50 mM thiourea, a scavenger for superoxide radicals, hydroxyl radicals, and H2O2 (41), was added to the cultures 5 min after H2O2 treatment. The addition of thiourea to the medium restored the growth of the fosB and bshA mutants of WI-2335 and COL to the respective wild-type levels, confirming that the observed growth delay was due to oxidative stress (Fig. 1, solid lines). These data also highlight differences that can occur across S. aureus strains. For example, wild-type COL appears to be more susceptible to the effects of 10 mM H2O2 than wild-type WI-2335 in the time frame tested (Fig. 1).
FIG 1.

Growth of isogenic strains in TSB with 10 mM H2O2. Overnight cultures were diluted to an OD650 of 0.1 before the addition of 10 mM H2O2. The growth of wild-type strains WI-2335 (A and B) and COL (C and D) as well as the isogenic bshA (A and C) and fosB (B and D) mutants and complemented strains in the medium was monitored via hourly reading of the OD650. Solid lines, the presence of 50 mM thiourea.
Response of fosB mutants to H2O2.
While the defect observed in the bshA mutant grown in TSB with 10 mM H2O2 was expected (42), the linkage of FosB to the S. aureus oxidative stress response was not anticipated. To explore whether the observed growth delay of the fosB mutant in response to H2O2 was attributable to the decreased ability of the mutant to degrade H2O2, the degradation of H2O2 was assayed in WI-2335 (chosen for its high BSH content), its isogenic fosB mutant, and the complemented strain. After 10 min, all the strains had degraded approximately 50% of the exogenously added H2O2, showing no difference in the ability of the fosB mutant and the parental strain to rapidly degrade H2O2. At 20 min after the addition of 10 mM H2O2, the concentrations of H2O2 in the cultures fell to 77.3 ± 9.15 μM, 24.4 ± 3.6 μM, and 37.6 ± 13.1 μM for the wild-type strain WI-2335, its isogenic fosB mutant, and the complemented mutant, respectively, indicating that all strains were able to degrade more than 99% of the added H2O2 within 20 min. This result indicates that the mutant and the parent had a similar ability to degrade H2O2.
Although the decomposition of H2O2 by S. aureus is largely attributed to catalase activity (43, 44), there still remains a question as to whether a small amount of H2O2 degradation can be catalyzed by a peroxidase. For example, GSH-S-transferases exhibit peroxidase activity (45, 46), thus alerting us to consider FosB to be a BSH-dependent peroxidase in S. aureus. However, over the course of three independent peroxidase assays, purified FosB was not found to catalyze the degradation of H2O2 in vitro. Therefore, the role of FosB in protecting S. aureus from oxidative stress does not appear to involve peroxidative ability.
A third explanation for the observed growth delay is that the fosB mutants might be more susceptible to killing by H2O2. Viability after an initial encounter with H2O2 was assessed by determining the number of CFU in a culture immediately before and after the addition of 10 mM H2O2. Comparisons of the numbers of CFU revealed comparable killing of the cultures at 45%, 52%, and 55% for the wild type, the ΔfosB strain, and the complemented mutant, respectively, upon exposure to 10 mM H2O2. The cause of the growth deficiency observed in the fosB mutant after encountering H2O2 is unknown, but the growth deficiency does not appear to be caused by the inability of the mutant to degrade H2O2 or survive exposure to H2O2.
Effects of combined H2O2 and diamide stress.
To assess if thiol stress might play a role in the growth delay observed in the bshA and fosB mutants of COL and WI-2335 exposed to H2O2, experiments that combined the thiol oxidant diamide and H2O2 were conducted. The growth defect of the mutants obtained in the first 4 h with H2O2 could be recapitulated in both the wild type and complemented strains of WI-2335 and COL by adding 4 mM diamide to the culture medium containing H2O2 (Fig. 2). Notably, strains grown in the presence of 4 mM diamide alone grew at a rate similar to that for strains grown without diamide.
FIG 2.
Effect of diamide on growth in H2O2-stressed cells. The growth of cultures of strains WI-2335 (A) and COL (B) with 10 mM H2O2 and with 4 mM diamide (gray, dashed lines) or without 4 mM diamide (black, solid lines) was determined at 1-h intervals.
The effects of both H2O2 and diamide on overnight growth were also examined to establish if this combined thiol and oxidative stress could affect the growth of the bshA and fosB mutants over a long period. The combination of 25 mM H2O2 and 5 mM diamide severely disrupted the overnight growth of the fosB and bshA mutants of COL and WI-2335, while the wild type and the complemented mutants of both strains could grow in the presence of this combination (Fig. 3). Only the bshA mutant of COL had impaired growth in 5 mM diamide alone (Fig. 3A). In the presence of 25 mM H2O2, the bshA mutant of WI-2335 failed to grow overnight (Fig. 3B). These differences in growth again highlight strain-specific differences which affect the ability of the fosB and bshA mutants to respond to H2O2 and diamide during overnight growth.
FIG 3.

Overnight growth of isogenic strains under oxidizing conditions. Wild-type strains COL (A) and WI-2335 (B) as well as their isogenic fosB and bshA mutants and complemented mutant strains were diluted to an OD650 of 0.1 in TSB before the addition of 5 mM diamide, 10 mM H2O2, or both compounds and grown at 37°C for 24 h.
NADPH levels in the mutant strains.
In Gram-negative bacteria, oxidative stress has been shown to affect the cellular NADPH levels (47), as NADPH-dependent disulfide reductases reduce oxidized GSH (48, 49). There are four putative oxidoreductases (YpdA, YqiW, YphP, and YtxJ) that have been postulated to reduce oxidized BSH (BSSB) (10, 11, 38). Given the presence of these genes in S. aureus (e.g., a ypdA homolog [GenBank accession number YP_008485371]), we explored whether mutation of fosB or bshA led to changes in NADPH levels in the mutants relative to the wild type. In TSB and TSB supplemented with 10 mM H2O2, wild-type COL and complemented mutants had larger amounts of NADPH than the isogenic fosB or bshA mutants (Fig. 4). In fosB and bshA mutants of COL, there was no measurable NAPDH detected in samples obtained from bacteria grown in TSB (Fig. 4A) or in TSB with 10 mM H2O2 (Fig. 4B). The fosB and bshA mutants of COL also had significantly higher levels of NADP+ than the wild type in TSB (Fig. 4A).
FIG 4.
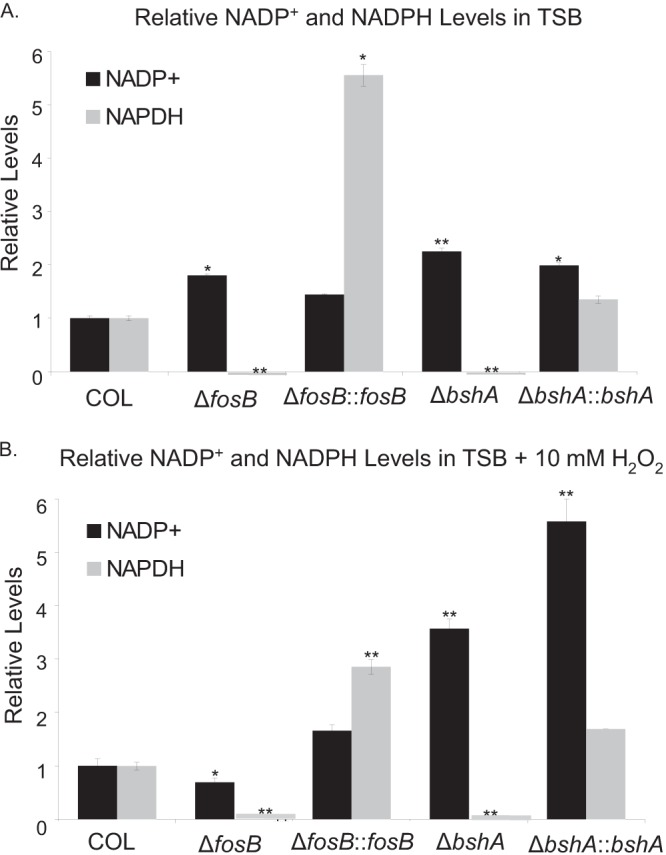
Relative levels of NADPH in isogenic COL strains. Strains were diluted to an OD650 of 0.1 in TSB (A) or TSB with 10 mM H2O2 (B) and grown for 4 h before NADP and NADPH levels per μg of total cellular protein were determined; values were normalized to those for the wild type under the same conditions. Statistics were calculated relative to the results for the wild type under the same conditions. *, P < 0.05; **, P < 0.001.
NADPH levels were also examined in strain WI-2335 and its isogenic mutants. Unlike COL, however, we were able to complement the bshA mutant of WI-2335 in trans only with a multicopy plasmid, as complementation at the chromosomal level was unsuccessful. Accordingly, NADPH levels in the complemented strain are not presented due to magnified results as a result of elevated induction from a multicopy plasmid. The level of NADPH in the WI-2335 fosB mutant was 35% of that of the parental strain in TSB and 1% of that of the wild type in 10 mM H2O2 (P < 0.0001). The WI-2335 bshA mutant did not have any measurable NADPH, like the COL bshA mutant. In TSB alone, the WI-2335 parental strain contained 77% less NADP+ than the isogenic fosB mutant (P ≤ 0.0004) and 35% less than the bshA mutant (P = 0.0007). Together, these results reveal that both mutants have lower NADPH levels than the parent, suggesting a reduced redox capacity due to a relative paucity of the NADPH needed to reduce oxidized thiols in the fosB and bshA mutants in COL and WI-2335.
Total antioxidant capacity of isogenic fosB and bshA strains.
The lower levels of NADPH in the fosB and bshA mutants highlight the possibility that the mutants may have a diminished capacity to reduce oxidants. The total antioxidant capacity, which is proportional to copper-reducing equivalents and can be determined by the ability of the sample to maintain reduced Cu2+ (see Material and Methods), was assayed in the fosB and bshA mutants as well as their isogenic wild-type and complemented strains. The mutants displayed a lower antioxidant capacity than the wild type and the complemented mutant strains (Fig. 5). This reduced capacity also occurred in the presence of H2O2 (Fig. 5). Furthermore, the bshA and fosB mutants exhibited a lower antioxidant capacity when grown with H2O2 than without, indicating that the oxidative stress imposed by H2O2 further reduced the total antioxidant capacity of the mutants (Fig. 5).
FIG 5.
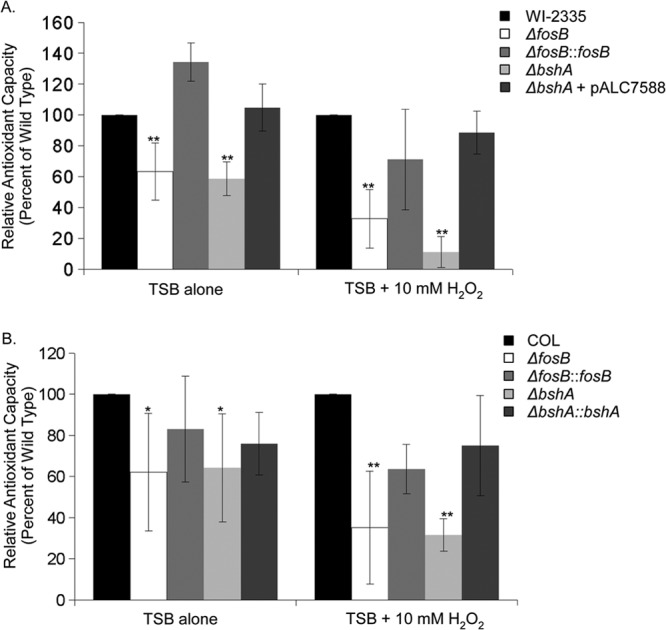
Total antioxidant capacity in COL and WI-2335 isogenic strains. The total antioxidant capacity for isogenic strains of WI-2335 (A) and COL (B), including their isogenic bshA and fosB mutants, was determined for cultures grown to an OD650 of 0.7 and induced for an extra 20 min with and without H2O2. The copper-reducing equivalent of each strain was determined and normalized as a percentage of the antioxidant capacity of the wild-type strain. P values were calculated relative to the values for the wild type under the same conditions. *, P < 0.05; **, P < 0.001.
Microarray analysis of gene expression in the COL bshA mutant.
To understand the global transcriptional changes associated with the absence of BSH in S. aureus, microarray analyses were performed with wild-type COL and its isogenic bshA mutant in TSB with and without diamide at a concentration (1 mM) previously shown to induce BSH synthesis in Bacillus subtilis (11). We found that 5% of the genome in the bshA mutant was either up- or downregulated compared with the level of expression of the isogenic parent in TSB (Table 4). The absence of bshA alone had only a modest effect on the transcriptional response of S. aureus to diamide.
TABLE 4.
Genes differentially expressed in ΔbshA COL strains in TSBa
| Gene functional group and gene/locus | Function | Fold change in expressionb |
|---|---|---|
| Transporters | ||
| brnQ1 | Branched-chain amino acid transport system II carrier | −2.14 |
| cbiO | Cobalt transporter ATP-binding subunit | −2.30 |
| citM | Citrate transporter | −3.66 |
| copA | Copper-transporting ATPase | 2.70 |
| cycA-aapA | d-Serine/d-alanine/glycine transporter | −2.09 |
| feoA | Ferrous iron transport protein A | 5.00 |
| feoB | Ferrous iron transport protein B | 3.33 |
| fhuA | Ferrichrome transport ATP-binding protein | −2.07 |
| fhuB | Ferrichrome transport permease protein | −2.17 |
| ribU | Riboflavin transporter | −2.04 |
| SACOL0665 | Iron compound-binding ABC transporter, putative | −2.90 |
| SACOL1367 | Amino acid permease | −2.66 |
| SACOL2521 | MFS transporter family member, putative | −2.19 |
| sirA | Iron compound ABC transporter | −3.10 |
| uhpT | Hexose phosphate transport protein | 20.00 |
| Amino acid metabolism and biosynthesis | ||
| arcB2 | Ornithine carbamoyltransferase | 11.11 |
| argG | Argininosuccinate synthase | 2.63 |
| femC | Glutamine synthetase | −2.99 |
| glmS | Glucosamine:fructose-6-phosphate aminotransferase | −2.18 |
| glnA | Glutamine synthetase | −2.89 |
| glnR | Glutamine synthetase repressor | −3.60 |
| ipdC | Indole-3-pyruvate decarboxylase | 2.04 |
| purB | Adenylosuccinate lyase | −2.13 |
| putA | Proline dehydrogenase | 4.17 |
| SACOL1058 | Class 1 aminotransferase involved in lysine biosynthesis | −2.02 |
| SACOL1801 | Dipeptidase involved in lysine biosynthesis | −2.05 |
| SACOL1976 | Nitric oxide synthase, oxygenase subunit | 2.13 |
| Carbohydrate metabolism and TCA cycle | ||
| gutB | Sorbitol dehydrogenase | −2.44 |
| lacG | 6-Phospho-beta-galactosidase | −2.46 |
| ldh1 | l-Lactate dehydrogenase | 2.38 |
| manA | Mannose-6-phosphate isomerase | −2.02 |
| pckA | Phosphoenolpyruvate carboxykinase | 2.33 |
| pflA | Formate acetyltransferase | 12.50 |
| pflB | Formate acetyltransferase | 16.67 |
| SACOL0235 | Hexitol dehydrogenase | −2.10 |
| Virulence factors | ||
| agrB | Accessory gene regulator protein B | 4.76 |
| cidA | Holin-like protein | 2.08 |
| coa | Staphylocoagulase precursor | 2.86 |
| ebh | Cell wall-associated fibronectin-binding protein | −4.78 |
| hla | Alpha-hemolysin precursor | 2.17 |
| isdA | LPXTG cell wall surface anchor protein | −2.87 |
| isdC | Iron-regulated cell surface protein | −2.35 |
| lipA | Lipase/esterase | −2.45 |
| mntC | Iron-repressed lipoprotein | 2.08 |
| sarX | Regulator of agr and exoprotein synthesis | −2.18 |
| sdrC | Ser-Asp-rich fibrinogen-binding protein | 3.70 |
| spa | Protein A precursor | 4.17 |
| ssaA | Secretory antigen precursor | −2.32 |
| Transcriptional regulators | ||
| nirR | Transcriptional regulator | 2.50 |
| SACOL0218 | MarR family member | 3.85 |
| SACOL1296 | GntR family transcriptional regulator | −3.95 |
| SACOL2349 | TetR family transcriptional regulator | −2.08 |
| SACOL2732 | Transcriptional regulator, putative | 2.00 |
| Other | ||
| acpD | FMN-dependent NADH azoreductase | 2.50 |
| bshA | Glycosyltransferase involved in BSH biosynthesis | −135.15 |
| bshB2 | Deacetylase involved in BSH biosynthesis | −2.02 |
| clpB | ATP-dependent protease | 2.56 |
| cmk | Cytidylate kinase | −2.10 |
| coaBC | Phosphopantothenoylcysteine decarboxylase | −2.39 |
| cspB | Cold shock protein | −3.42 |
| cydA | Cytochrome d ubiquinol oxidase, subunit I | 2.13 |
| gatC | Aspartyl/glutamyl-tRNA amidotransferase subunit C | −2.73 |
| glpK | Glycerol kinase | −2.11 |
| greA | Transcription elongation factor | −2.01 |
| lytH | N-Acetylmuramoyl-l-alanine amidase | −20.99 |
| mnmA | tRNA-specific 2-thiouridylase | −2.36 |
| murI | Glutamate racemase | −2.51 |
| nanA | N-Acetylneuraminate lyase | 2.38 |
| nuc | Thermonuclease precursor | −2.68 |
| pepF | Oligoendopeptidase F | −3.70 |
| pnbA | para-Nitrobenzyl esterase | 2.50 |
| prmA | Ribosomal protein L11 methyltransferase | −2.08 |
| queC | ExsB protein | 2.78 |
| queD | 6-Pyruvoyl tetrahydrobiopterin synthase | 2.78 |
| queE | Radical-activating enzyme | 3.13 |
| SACOL0107 | Pyridoxal-dependent decarboxylase | −2.03 |
| SACOL2121 | Acetyltransferase | −2.00 |
| sodM | Manganese/iron superoxide dismutase | 4.55 |
| tagA | Teichoic acid biosynthesis protein | −2.78 |
| treP | PTS system, IIBC components | 2.17 |
| ung | Uracil-DNA glycosylase | −2.11 |
| Hypothetical and putative proteins | ||
| SACOL0012 | Homoserine O-acetyltransferase, putative | −3.77 |
| SACOL0104 | LucA family siderophore biosynthesis protein | −2.36 |
| SACOL0108 | ParBC superfamily member | −2.52 |
| SACOL0223 | Hypothetical protein | 2.44 |
| SACOL0234 | Hypothetical protein | −2.17 |
| SACOL0303 | 5′-Nucleotidase | −3.53 |
| SACOL0395 | Glycine cleavage system H protein, putative | 3.23 |
| SACOL0403 | BglG family transcriptional antiterminator | 2.33 |
| SACOL0411 | Membrane-bound protein, putative | −2.27 |
| SACOL0449 | Lipoprotein, putative | 3.57 |
| SACOL0552 | RNA-binding protein, putative | −2.39 |
| SACOL0568 | UvrB/UvrC family member | 2.70 |
| SACOL0604 | Deoxynucleoside kinase family protein | −2.6 |
| SACOL0606 | HAD superfamily hydrolase | −3.44 |
| SACOL0613 | GTP cyclohydrolase, putative | −2.31 |
| SACOL0615 | Hypothetical protein | −2.83 |
| SACOL0625 | VraX | 2.70 |
| SACOL0632 | Hypothetical protein | −2.29 |
| SACOL0671 | Alpha/beta fold family hydrolase | 2.22 |
| SACOL0820 | LysM domain-containing protein | −2.25 |
| SACOL0939 | NifU domain-containing protein | −2.10 |
| SACOL1175 | Hypothetical protein | 2.04 |
| SACOL1226 | TM2 domain-containing protein | 2.78 |
| SACOL1294 | Metallo-beta-lactamase family protein | −2.34 |
| SACOL1331 | Hypothetical protein | −2.19 |
| SACOL1358 | Hypothetical protein | 2.27 |
| SACOL1359 | Hypothetical protein | 2.38 |
| SACOL1376 | Hypothetical protein | 2.13 |
| SACOL1544 | Hypothetical protein | −2.44 |
| SACOL1548 | AtsA/ElaC family protein | −2.39 |
| SACOL1601 | Competence protein, putative | −2.51 |
| SACOL1670 | Hypothetical protein | −2.46 |
| SACOL1675 | TPR domain-containing protein | −2.08 |
| SACOL1788 | Hypothetical protein | 2.33 |
| SACOL1825 | N-Acetylmuramoyl-l-alanine amidase, putative | −2.52 |
| SACOL1846 | Excalibur calcium-binding domain protein | 3.03 |
| SACOL1896 | Hypothetical protein | −2.53 |
| SACOL2020 | Nitroreductase, putative | 2.50 |
| SACOL2076 | Hypothetical protein | 2.94 |
| SACOL2136 | NAD/NADP epimerase, putative | 2.04 |
| SACOL2163 | Hypothetical protein | 3.23 |
| SACOL2247 | Hypothetical protein | 2.63 |
| SACOL2402 | S-Transferase, putative | −3.45 |
| SACOL2484 | Alkylhydroperoxidase, putative | 2.94 |
| SACOL2491 | Hypothetical protein | 3.23 |
| SACOL2528 | Hypothetical protein | −2.18 |
| SACOL2600 | Thioredoxin reductase, putative | −2.07 |
| SACOL2676 | LPXTG-containing protein | 2.33 |
| SACOL2733 | Hypothetical protein | 2.56 |
TCA, trichloroacetic acid; MFS, major facilitator superfamily; FMN, flavin mononucleotide; PTS, phosphotransferase system; HAD, haloacid dehalogenase; TPR, tetratricopeptide repeat.
Fold change in expression in COL ΔbshA mutants relative to the level of expression in the wild type.
As expected, in the bshA mutant grown in TSB, bshA was not expressed (Table 4). The expression of bshB2, the second enzyme of the BSH biosynthetic pathway, was also decreased in the bshA mutant in comparison to the level of expression in the parent (Table 4). The effect of thiol stress (i.e., diamide) on the BSH biosynthetic genes was also examined in the wild-type strain COL, showing an increase in the expression of both bshA and bshC, the third and final gene of the BSH biosynthetic pathway in the parental strain, in the presence of 1 mM diamide (Table 5; see Table S1 in the supplemental material). The results were confirmed by independent qRT-PCR analysis, showing that bshA was upregulated 2.31-fold and bshC was upregulated 5.2-fold in COL cells treated with diamide compared to the level of regulation in those grown in TSB alone.
TABLE 5.
Selected genes with altered expression in COL induced with 1 mM diamidea
| Gene functional group and gene/locus | Function | Fold change in expression |
|---|---|---|
| Transporters | ||
| feoA | Ferrous iron transport protein A | −2.76 |
| fhuA | Ferrichrome transport ATP-binding protein | 2.50 |
| fhuB | Ferrichrome transport permease | 2.61 |
| fhuD | Ferrichrome transport permease | 3.15 |
| nupC | Nucleoside permease | −10.83 |
| rarD | Superfamily drug/metabolite transporter | 4.58 |
| SACOL0261 | Putative drug transporter | 4.85 |
| sirA | Iron compound ABC transporter | −3.25 |
| tagH | Teichoic acid export protein ATP-binding subunit | 3.54 |
| vraD | ABC transporter ATP-binding protein | 2.99 |
| vraE | ABC transporter, permease protein | 2.29 |
| Carbohydrate metabolism and TCA cycle | ||
| ccpA | Catabolite control protein | 2.41 |
| panB | 3-Methyl-2-oxobutanoate hydroxymethyltransferase | 2.70 |
| panC | Pantoate-beta-alanine ligase | 2.67 |
| panD | Aspartate alpha-decarboxylase | 2.84 |
| Nucleotide excision and repair | ||
| mutS2 | Recombination and DNA strand exchange inhibitor protein | 2.71 |
| radA | DNA repair | 4.69 |
| radC | DNA repair protein | 4.68 |
| SACOL1954 | Exonuclease | −3.47 |
| uvrA | Excinuclease ABC subunit A | 2.67 |
| uvrB | Excinuclease ABC subunit B | 2.97 |
| uvrC | Excinuclease ABC subunit C | 3.37 |
| Virulence factors | ||
| crtM | Squalene desaturase | −3.50 |
| crtN | Squalene synthase | −3.87 |
| crtQ | Glycosyltransferase | −3.65 |
| ebh | Cell wall-associated fibronectin-binding protein | 5.99 |
| hla | Alpha-hemolysin precursor | −6.37 |
| mntC | Iron-repressed lipoprotein | −9.11 |
| SACOL1220 | Fibronectin/fibrinogen binding-related protein | 3.47 |
| spa | Immunoglobulin G binding protein A precursor | −3.56 |
| sspA | Serine protease | −2.28 |
| Transcriptional regulator, spxA | Transcriptional regulator | −2.33 |
| Other | ||
| adaB | Methylated DNA-protein cysteine methyltransferase | 2.33 |
| atpA | FoF1 ATP synthase subunit alpha | −3.15 |
| atpB | FoF1 ATP synthase subunit A | −3.34 |
| atpC | FoF1 ATP synthase subunit epsilon | −2.43 |
| atpD | FoF1 ATP synthase subunit beta | −2.62 |
| atpE | FoF1 ATP synthase subunit C | −2.91 |
| atpF | FoF1 ATP synthase subunit B | −3.05 |
| atpG | FoF1 ATP synthase subunit gamma | −2.86 |
| atpH | FoF1 ATP synthase subunit delta | −2.98 |
| bshA | Glycosyltransferase involved in BSH biosynthesis | 2.31 |
| bshC | BSH synthase | 5.20 |
| cinA | Competence/damage-inducible protein | 2.67 |
| clpB | ClpB chaperone-like protein | 34.39 |
| clpP | ATP-dependent Clp protease, proteolytic subunit | 5.35 |
| dnaK | Molecular chaperone | 7.93 |
| groEL | Chaperonin | 7.17 |
| groES | Cochaperonin | 7.25 |
| grpE | Heat shock protein | 6.76 |
| hslO | Hsp33-like chaperonin | 3.92 |
| lytH | N-Acetylmuramoyl-l-alanine amidase | −3.31 |
| pbp4 | Penicillin-binding protein 4 | −2.41 |
| recJ | Single-stranded-DNA-specific exonuclease | 2.52 |
| recX | Recombination regulator | 2.33 |
| rexA | Exonuclease | 2.51 |
| sigB | RNA polymerase sigma factor SigB | −3.29 |
| rsbV | Anti-sigma B factor | −2.30 |
| rsbW | Serine-protein kinase | −3.29 |
| zwf | Glucose-6-phosphate 1-dehydrogenase | 3.38 |
| Hypothetical and putative proteins | ||
| SACOL1553 | Glyoxalase family protein | 2.46 |
| SACOL1749 | NADP-dependent malic enzyme, putative | −2.20 |
| SACOL1753 | Universal stress protein | −2.92 |
| SACOL1835 | Aldo/keto reductase family oxidoreductase | 2.42 |
| SACOL2293 | NAD/NADP octopine/nopaline dehydrogenase family | −3.21 |
| sarV | Hypothetical protein | 2.42 |
| tal | Transaldolase, putative | 4.47 |
| tpx | Thiol peroxidase, putative | 2.34 |
For a complete listing of genes that showed altered expression in this analysis, see Table S1 in the supplemental material. TCA, trichloroacetic acid.
Before the discovery of BSH, CoASH was believed to be the major LMW thiol in S. aureus (50). Expression of the panBCD genes, which encode part of the pantothenate pathway that can lead to CoASH synthesis, was increased almost 3-fold in COL treated with diamide relative to that in the wild type in TSB (Table 5). In contrast to the increased panBCD expression in COL treated with diamide, both BSH (0.39 ± 0.15) and CoASH (0.11 ± 0.06) levels were lower in COL cells treated with diamide than those grown in TSB alone (Table 3). Just as BSH was found to directly reduce H2O2 in our peroxidase assays (data not shown), BSH and CoASH may be acting directly on the diamide, thus causing a decrease in thiol levels relative to those in the same strain grown in TSB, despite increases in expression of the respective biosynthetic genes.
The crtMNQ genes encode part of the biosynthetic pathway for staphyloxanthin, a virulence factor which has been linked to oxidative stress protection in S. aureus (37). Of particular interest was the observation that crtMNQ transcription was downregulated at least 3.5-fold in the COL strain exposed to diamide compared with the level of expression in the nontreated control (Table 5; see Table S1 in the supplemental material). The expression of crtM, the first gene of the staphyloxanthin operon, was also 2.84-fold lower in COL cells treated with diamide than those not treated with diamide. Independent qRT-PCR analyses confirmed this decrease in crtM transcript levels in diamide-treated cells. The expression of crtM in the bshA mutant treated with diamide relative to that in the bshA mutant grown in TSB alone was increased 2.41-fold. Importantly, under diamide induction, the crtM, crtN, and crtQ transcripts were upregulated 2.63-, 2.08-, and 2.5-fold, respectively, in the bshA mutant compared to the level of expression in the parent. The upregulation of crtM in the bshA mutant under diamide induction was confirmed by qRT-PCR to be 1.5 times higher than the level of expression in the wild type. This difference in expression of staphyloxanthin biosynthetic genes between the bshA mutant and the wild type was not seen when comparing the wild type and the bshA mutant in TSB alone (Table 4). Studies have suggested that the operon containing crtMNQ may be partially regulated by the small RNA ssrA (51); accordingly, we determined the relative transcript levels of ssrA in our isogenic strains by qRT-PCR, as ssrA was not part of the probe set in the microarray. No differences in relative transcript levels of ssrA were observed when comparing the transcript levels of COL to those of the isogenic bshA mutant grown with and without diamide. Similarly, there were no differences in relative transcript levels of ssrA in either strain grown with or without diamide.
Staphyloxanthin levels in isogenic strains.
To determine if alterations in the expression of crtMNQ translated into changes in carotenoid levels, staphyloxanthin was extracted from selected strains. When cultures were grown in TSB alone, there was no significant difference in staphyloxanthin production between COL and its isogenic bshA mutant or WI-2335 and its isogenic bshA mutant. These experiments were then attempted with H2O2 and diamide treatment, again, without major differences in staphyloxanthin levels between wild-type strains and the isogenic mutants being detected.
Given that decreased crtMNQ expression was accompanied by increased bshA transcription in strain COL treated with diamide, the effect of overexpression of bshA on staphyloxanthin production was explored in wild-type strains SH1000, COL, and WI-2335. Indeed, overexpression of bshA from a xylose-inducible multicopy plasmid was accompanied by a decrease in staphyloxanthin content in all three wild-type strains relative to that in the same strain with an empty vector (Fig. 6).
FIG 6.
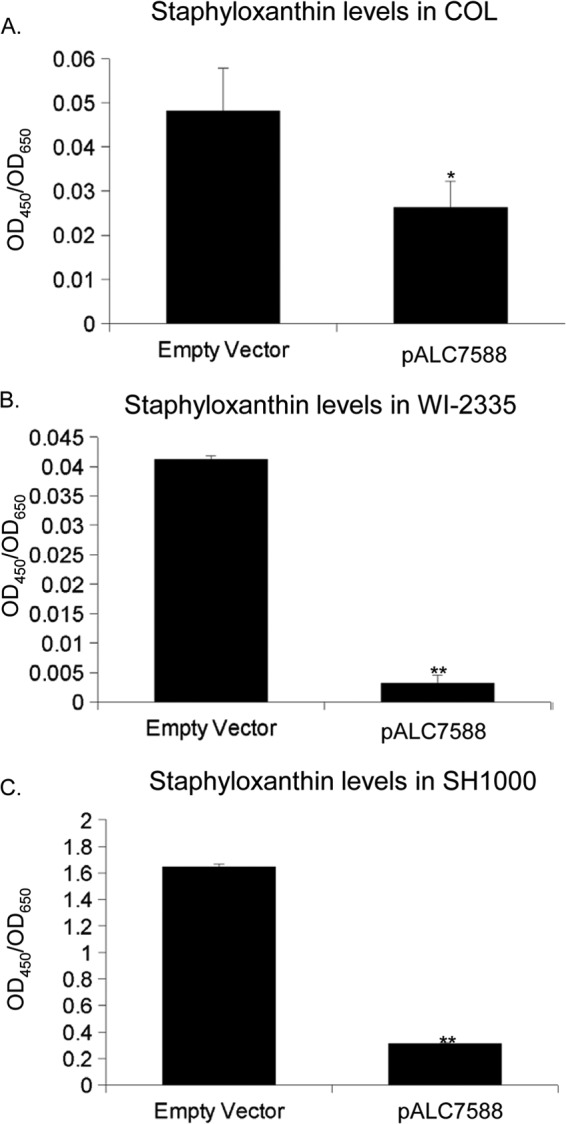
Staphyloxanthin levels in strains SH1000, WI-2335, and COL. Staphyloxanthin levels determined at an OD450 were normalized to those determined at an OD650 in wild-type strains COL (A), WI-2335 (B), and SH1000 (C) containing pEPSA5 (empty vector) or pALC7588 (pEPSA5 containing bshA) in TSB with 1% xylose. Statistics were calculated relative to the levels in the strain with pEPSA5. *, P < 0.05; **, P < 0.0001.
Survival of BSH-deficient strains in a whole-blood assay.
With data supporting the participation of BSH in the oxidative stress response of S. aureus, we then explored the role of BSH in the survival of S. aureus strains COL and SH1000 in whole blood containing neutrophils, macrophages, and complement. In whole blood, ALC7446 (a BSH-producing strain derived from SH1000) survived better than wild-type strain SH1000 (Fig. 7A), with a statistically significant difference in survival in the first hour. For strain COL, the bshA mutant survived significantly less well than the parental strain and the complemented mutant at 3 and 4 h after mixing the bacteria with whole blood (Fig. 7B). These studies indicate that BSH is important to S. aureus survival in an environment of oxidative stress in the presence of neutrophils and macrophages.
FIG 7.
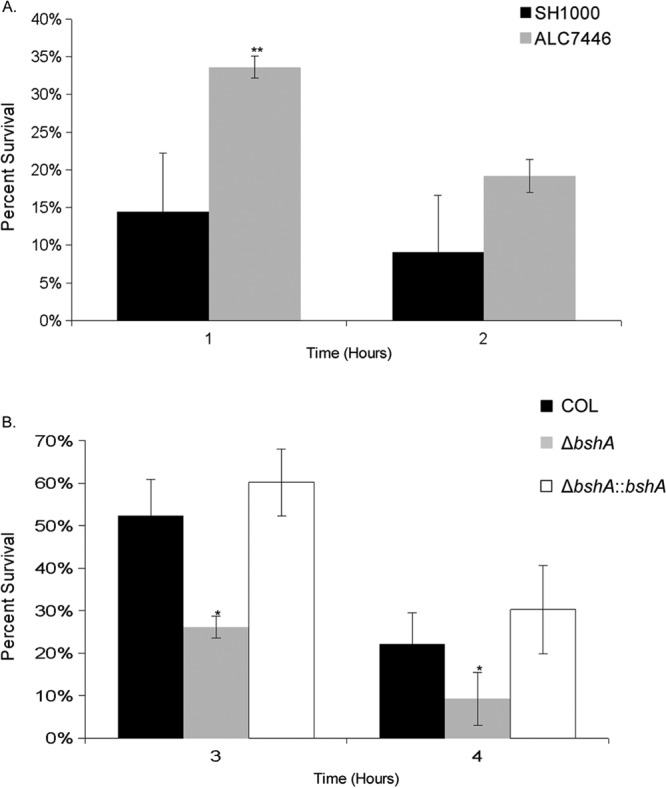
Survival of isogenic strains in a human whole-blood survival assay. The numbers of CFU of wild-type SH1000 or ALC7446 (SH1000 with a repaired bshC locus) (A) and the COL wild type, the ΔbshA mutant, and the complemented mutant (B) were determined hourly over the course of the human whole-blood killing assay and were normalized to the initial inoculum. Statistics were calculated relative to the values for the wild type. *, P < 0.05; **, P < 0.001.
DISCUSSION
Previous detailed kinetic studies of purified S. aureus FosB have demonstrated its function as a BSH-S-transferase when detoxifying fosfomycin via conjugation with BSH (18). However, the significance of this finding in clinically relevant MRSA isolates has not been studied. The importance of both BSH and FosB in conferring fosfomycin resistance across several S. aureus strains was confirmed in this study, as the disruption of fosB and bshA resulted in at least a 16- to 60-fold reduction in fosfomycin resistance (Table 2). The addition of exogenous Cys, a possible FosB substrate, did not increase the resistance of bshA mutants to fosfomycin, thus suggesting that FosB does not use Cys to detoxify the cell well-active antibiotic fosfomycin in the bacteria.
We recognize that the basal MIC of fosfomycin in the fosB and bshA mutants is relatively high at 39.1 μg/ml, despite marked differences in resistance in the corresponding parental and complemented strains (Table 2), thus implying that there might be other factors contributing to basal fosfomycin resistance in S. aureus. We have examined transporters (e.g., UhpT and GlpT) that may be involved in the uptake of fosfomycin into cells. There was no difference in glpT expression in the bshA and fosB mutants versus the parent. However, the uhpT transcript was upregulated 20-fold in the bshA mutant versus the level of expression in the parent (Table 4), but this increase was not found in the fosB mutant (data not shown). Despite this discrepancy in uhpT levels between the bshA and fosB mutants, there was no increase in sensitivity to fosfomycin resistance in the bshA mutant relative to that in the fosB mutant.
The difference in fosfomycin levels among different MRSA strains also cannot be explained by divergent BSH levels, as these levels have no correlation with the levels of fosfomycin resistance among these strains (Table 3). For example, strain Mu50 had the highest fosfomycin MIC of the strains tested, but it contained the second lowest BSH levels, indicating that other factors (e.g., the level of FosB or additional transporters) may play a role in the differences in fosfomycin resistance among S. aureus strains.
Of note, the fold changes in MICs reported here for the bshA and fosB mutants were higher than those previously reported in the literature (16- to 60-fold lower as opposed to 4 to 8 times lower) (18). This discrepancy can be explained by the use of a nonstandard protocol for MIC assays in the prior study (e.g., the use of TSB instead of Mueller-Hinton broth, as suggested in the CLSI protocol [27]). In addition, the previous BSH studies have utilized transposon mutants (18, 38) constructed in a strain cured of its plasmids (52); in addition, these studies did not complement the mutation to exclude any insertional polar effects that may have been caused by the transposon's insertion. We have confirmed here the contribution of both BSH and FosB to fosfomycin resistance in S. aureus using cis and trans complementation of our mutations.
We then examined whether BSH contributed to the response of S. aureus to oxidative stress and if FosB had a role in this response. Growth studies with 10 mM H2O2 showed a growth delay of the fosB and bshA mutants, suggesting that both BSH and FosB may play a role in the cell's response to oxidative stress. In the study carried out by Pöther et al. (14), strain 8325-4, a bshC mutant, was found to show equal resistance to 10, 20, and 40 mM H2O2 as the exogenously complemented, BSH-producing isogenic strains, divergent from what we have found in this study. In that study (14), LB medium was used, while we used TSB here. Pöther et al. (14) noted that LB medium can quench oxidants because of the peptides and amino acids present in the medium. In addition, unpublished work done in the Cheung lab has shown that the concentration of H2O2 necessary to kill S. aureus is dependent on the amount of cells present. For instance, 10 mM H2O2 can cause a growth delay in cultures of bshA mutants at an OD650 of 0.1 but has no effect on a culture of mutants at an OD650 of 0.5, presumably due to the various amounts of catalase present. Together, these differences may account for the divergent results on the effect of H2O2 on S. aureus BSH-deficient cells between our study and that of Pöther et al. (14).
The cause of the observed growth defect of the fosB mutant was not an inability to degrade the H2O2. We also established that purified FosB does not have peroxidase activity. Furthermore, viability studies indicated that the mutant cells were not killed by H2O2 but remained in a quiescent state in response to the initial oxidative stress. This effect of H2O2 on the growth of the bshA and fosB mutants could be replicated in wild-type cells when the oxidation caused by H2O2 was exacerbated by reversible thiol oxidation caused by diamide (Fig. 2). A recent study on the proteomic response of S. aureus to several stress conditions argues that S. aureus has a triphasic response to H2O2: an acute phase, a recovery phase, and a tolerance phase (53). The recovery phase is characterized by the resumption of growth and the repair of DNA damage. One explanation for the growth delay observed in the fosB and bshA mutants upon exposure to H2O2 may be related to the increased or sustained oxidation of cellular thiols present in proteins that are needed for growth during the recovery phase (4).
One function that has previously been attributed to BSH is the protection of protein thiols upon hypochlorite stress (25, 42). The results in this study indicate that thiol protection is also part of the oxidative stress response of S. aureus to H2O2. This protection may be dependent on FosB. The combined effect of diamide and H2O2 on the overnight growth of the fosB mutants further supports the notion that FosB is part of the S. aureus oxidative stress response. However, it should be noted that just as wild-type S. aureus showed a range of fosfomycin MICs, the mutation of bshA had different effects on the sensitivity of wild-type strains COL and WI-2335 to 25 mM H2O2 and 5 mM diamide, which may be due to inherent differences in the physiology of the strains.
An abnormal recovery from oxidative stress led to the analysis of the NADP+ and NADPH pools in the bshA and fosB mutants as well as the BSH levels in the fosB mutant. The decreased amount of measurable NADPH present in both mutants, despite the presence of higher levels of NADP+, suggested that NADPH is being utilized more quickly in the bshA and fosB mutants (Fig. 4). In the bshA and fosB mutants, larger amounts of NADPH could be used as a reducing agent by NADPH-dependent disulfide reductases. This explanation only partially answers the major question of why BSH levels are lower in the fosB mutant than in the parent, while BSSB levels are similar. Increased amounts of NADPH are needed to reduce BSSB when BSH levels are lower, therefore maintaining BSSB levels at the level observed in the parental strain and lower NADPH levels than in the parent. However, why the LMW thiol levels (BSH, Cys, and CoASH) are decreased in the fosB mutant relative to the WI-2335 wild type remains unknown.
While Upton and coworkers demonstrated in vitro that BSH inhibits BshA activity (54), they also proposed that the regulation of BSH biosynthesis might be multifactorial. The microarray comparing the bshA mutant to parent strain COL showed that in the absence of BshA, expression of bshB2 is downregulated. This downregulation suggests that bshB2 is responding to a signal caused by the mutation of bshA, as bshA and bshB2 are not on the same operon. The nature of the signal (divergent or shared, direct or indirect) is unclear and requires further investigation. One of the possible signals for bsh genes is probably connected to thiol stress, as diamide, which oxidizes thiols and decreases reduced thiol levels, increases expression of both bshA and bshC in the wild type (see Table S1 in the supplemental material). SpxA, which has been connected to both the diamide stress response (55) and the regulation of BSH synthesis (56) in B. subtilis, is upregulated in cells treated with diamide and may be the link connecting the disparate operons.
Of great interest was the indication that genes involved in the production of staphyloxanthin, which has been implicated in the oxidative stress response (37), are expressed differently between COL and its isogenic bshA mutant when both strains are grown in diamide. Although the bshA mutant exhibited increased crtM transcription, we were not able to correlate increased gene expression to elevated staphyloxanthin production in mutants grown in TSB alone, H2O2, or diamide. The reasons for this discrepancy are not clear, but we speculate that the extraction method may not be as sensitive as the gene expression assay. Alternatively, given the time frame of growth (72 h) necessary for the production and extraction of staphyloxanthin, the effect of oxidants may have dissipated by the time that the staphyloxanthin levels were analyzed. However, there was an observable decrease in staphyloxanthin production when bshA was overexpressed, even in the absence of BSH, thereby suggesting that an uncharacterized connection exists between these two factors. This observation was also of interest because staphyloxanthin has been shown to play a role in the survival of S. aureus in neutrophils (37, 57).
A previous investigation into the survival of S. aureus strains SH1000 and 8325-4 (both of which are bshC mutants) expressing nonphysiological levels of bshC exogenously on a plasmid showed that BSH in those laboratory strains supported growth in host cells (14). Here, we demonstrate that the inability to produce BSH through either the mutation of bshC (SH1000) or the deletion of bshA (COL) had significant effects on the ability of a strain to survive in a human whole-blood assay, strengthening the idea that BSH, and not the individual synthetic enzymes, is supporting the survival of S. aureus in phagocytes. More importantly, as the mutant possesses the capacity to produce more staphyloxanthin than the wild type under stress conditions, BSH appears to be playing an important role in the survival of S. aureus in the host, possibly independently of staphyloxanthin levels. The bshA and bshC mutants have a diminished capacity to respond to oxidative stress, such as that generated by professional phagocytes, perhaps explaining the increased susceptibility of BSH mutants to killing in the human whole-blood assay. However, the precise role of BSH, including the contribution of FosB and its newly observed role in the oxidative stress response, remains ill defined.
Many studies that proposed to identify and characterize major components in the oxidative stress response of S. aureus used SH1000 as their model strain (28, 43, 57–59). One consideration raised by our studies into the role of BSH in the oxidative stress response is whether the results of studies conducted in SH1000 or in strains of the 8325 lineage, such as RN6390 and RN4220, which do not produce BSH, are reflective of the results that would be obtained with clinical isolates. In light of the fact that BSH contributes to oxidative stress resistance, conclusions based on studies conducted with strains of this lineage may need to be reexamined.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sanjay Shukla for providing strain WI-2335 and the unpublished characterizations of WI-2335 and Eve-Julie Bonetti for technical assistance on the microarray analyses.
This work was supported by a T32 AI007519 predoctoral fellowship to A.C.P., NIH grant R01AI91801 to A.C., a CNPq-Brazil Scholarship to R.G.D., and BBSRC grant BB/H013504/1 to C.J.H.
Footnotes
Published ahead of print 28 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01074-13.
REFERENCES
- 1.Shorr AF, Tabak YP, Killian AD, Gupta V, Liu LZ, Kollef MH. 2006. Healthcare-associated bloodstream infection: a distinct entity? Insights from a large U.S. database. Crit. Care Med. 34:2588–2595 [DOI] [PubMed] [Google Scholar]
- 2.Babior BM, Kipnes RS, Cumvu JT. 1973. Biological defense mechanisms. J. Clin. Invest. 52:741–744. 10.1172/JCI107236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer GYN, Islam M, Quastel JH. 1961. Biochemical aspects of phagocytosis. Nature 192:535–541. 10.1038/192535a0 [DOI] [Google Scholar]
- 4.Weber H, Engelmann S, Becher D, Hecker M. 2004. Oxidative stress triggers thiol oxidation in the glyceraldehyde-3-phosphate dehydrogenase of Staphylococcus aureus. Mol. Microbiol. 52:133–140. 10.1111/j.1365-2958.2004.03971.x [DOI] [PubMed] [Google Scholar]
- 5.Kilili KG, Atanassova N, Vardanyan A, Clatot N, Al-Sabarna K, Kanellopoulos PN, Makris AM, Kampranis SC. 2004. Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 279:24540–24551. 10.1074/jbc.M309882200 [DOI] [PubMed] [Google Scholar]
- 6.Pastore A, Federici G, Bertini E, Piemonte F. 2003. Analysis of glutathione: implication in redox and detoxification. Clin. Chim. Acta 333:19–39. 10.1016/S0009-8981(03)00200-6 [DOI] [PubMed] [Google Scholar]
- 7.Rigsby RE, Fillgrove KL, Beihoffer LA, Armstrong RN. 2005. Fosfomycin resistance proteins: a nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Methods Enzymol. 401:367–379. 10.1016/S0076-6879(05)01023-2 [DOI] [PubMed] [Google Scholar]
- 8.Meury J, Kepes A. 1982. Glutathione and the gated potassium channels of Escherichia coli. EMBO J. 1:339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibayama K, Wachino J, Arakawa Y, Saidijam M, Rutherford NG, Henderson PJF. 2007. Metabolism of glutamine and glutathione via gamma-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol. Microbiol. 64:396–406. 10.1111/j.1365-2958.2007.05661.x [DOI] [PubMed] [Google Scholar]
- 10.Newton GL, Rawat M, La Clair JJ, Jothivasan VK, Budiarto T, Hamilton CJ, Claiborne A, Helmann J, Fahey RC. 2009. Bacillithiol is an antioxidant thiol produced in bacilli. Nat. Chem. Biol. 5:625–627. 10.1038/nchembio.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaballa A, Newton GL, Antelmann H, Parsonage D, Upton H, Rawat M, Claiborne A, Fahey RC, Helmann J. 2010. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in bacilli. Proc. Natl. Acad. Sci. U. S. A. 107:6482–6486. 10.1073/pnas.1000928107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsonage D, Newton GL, Holder RC, Wallace BD, Paige C, Hamilton CJ, Dos Santos PC, Redinbo MR, Reid SD, Claiborne A. 2010. Characterization of the N-acetyl-α-d-glucosaminyl l-malate synthase and deacetylase functions for bacillithiol biosynthesis in Bacillus anthracis. Biochemistry 49:8398–8414. 10.1021/bi100698n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton GL, Fahey RC, Rawat M. 2012. Detoxification of toxins by bacillithiol in Staphylococcus aureus. Microbiology 158:1117–1126. 10.1099/mic.0.055715-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pöther D-C, Gierok P, Harms M, Mostertz J, Hochgräfe F, Antelmann H, Hamilton CJ, Borovok I, Lalk M, Aharonowitz Y, Hecker M. 2013. Distribution and infection-related functions of bacillithiol in Staphylococcus aureus. Int. J. Med. Microbiol. 303:114–123. 10.1016/j.ijmm.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 15.Arca P, Reguera G, Hardisson C. 1997. Plasmid-encoded fosfomycin resistance in bacteria isolated from the urinary tract in a multicentre survey. J. Antimicrob. Chemother. 40:393–399. 10.1093/jac/40.3.393 [DOI] [PubMed] [Google Scholar]
- 16.Etienne J, Gerbaud G, Fleurette J, Courvalin P. 1991. Characterization of staphylococcal plasmids hybridizing with the fosfomycin resistance gene fosB. FEMS Microbiol. Lett. 68:119–122 [DOI] [PubMed] [Google Scholar]
- 17.Lamers AP, Keithly ME, Kim K, Cook PD, Stec DF, Hines KM, Sulikowski GA, Armstrong RN. 2012. Synthesis of bacillithiol and the catalytic selectivity of FosB-type fosfomycin resistance proteins. Org. Lett. 14:5207–5209. 10.1021/ol302327t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AA, Sharma SV, Strankman AW, Duran SR, Rawat M, Hamilton CJ. 2013. Mechanistic studies of FosB: a divalent metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem. J. 451:69–79. 10.1042/BJ20121541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma SV, Jothivasan VK, Newton GL, Upton H, Wakabayashi JI, Kane MG, Roberts AA, Rawat M, La Clair JJ, Hamilton CJ. 2011. Chemical and chemoenzymatic syntheses of bacillithiol: a unique low-molecular-weight thiol amongst low G + C Gram-positive bacteria. Angew. Chem. Int. Ed. Engl. 50:7101–7104. 10.1002/anie.201100196 [DOI] [PubMed] [Google Scholar]
- 20.Scortti M, Lacharme-Lora L, Wagner M, Chico-Calero I, Losito P, Vázquez-Boland JA. 2006. Coexpression of virulence and fosfomycin susceptibility in Listeria: molecular basis of an antimicrobial in vitro-in vivo paradox. Nat. Med. 12:515–517. 10.1038/nm1396 [DOI] [PubMed] [Google Scholar]
- 21.Monk I, Shah I, Xu M, Tan M, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3(2):e00277–11. 10.1128/mBio.00277-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. 10.1016/j.plasmid.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 23.Memmi G, Filipe SR, Pinho MG, Fu Z, Cheung AL. 2008. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother. 52:3955–3966. 10.1128/AAC.00049-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forsyth R, Haselbeck R. 2002. A genome wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 43:1387–1400. 10.1046/j.1365-2958.2002.02832.x [DOI] [PubMed] [Google Scholar]
- 25.Chi BK, Roberts AA, Huyen TT, Bäsell K, Becher D, Albrecht D, Hamilton CJ, Antelmann H. 2013. S-Bacillithiolation protects conserved and essential proteins against hypochlorite stress in Firmicutes bacteria. Antioxid. Redox Signal. 18:1273–1295. 10.1089/ars.2012.4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fenton SS, Fahey RC. 1986. Analysis of biological thiols: determination of thiol components of disulfides and thioesters. Anal. Biochem. 154:34–42. 10.1016/0003-2697(86)90492-6 [DOI] [PubMed] [Google Scholar]
- 27.Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard. M07-A8 29. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 28.Lithgow JK, Hayhurst EJ, Cohen G, Aharonowitz Y, Foster SJ. 2004. Role of a cysteine synthase in Staphylococcus aureus. J. Bacteriol. 186:1579–1590. 10.1128/JB.186.6.1579-1590.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang W, Small DA, Toghrol F, Bentley WE. 2006. Global transcriptome analysis of Staphylococcus aureus response to hydrogen peroxide. J. Bacteriol. 188:1648–1659. 10.1128/JB.188.4.1648-1659.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellman GL. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70–77. 10.1016/0003-9861(59)90090-6 [DOI] [PubMed] [Google Scholar]
- 31.Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol. 91:49–60. 10.1016/S0076-6879(83)91010-8 [DOI] [PubMed] [Google Scholar]
- 32.Wolff S. 1994. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 233:182–189. 10.1016/S0076-6879(94)33021-2 [DOI] [Google Scholar]
- 33.Tamber S, Schwartzman J, Cheung AL. 2010. Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect. Immun. 78:3637–3646. 10.1128/IAI.00296-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charbonnier Y, Gettler B, François P, Bento M, Renzoni A, Vaudaux P, Schlegel W, Schrenzel J. 2005. A generic approach for the design of whole-genome oligoarrays, validated for genomotyping, deletion mapping and gene expression analysis on Staphylococcus aureus. BMC Genomics 6:95. 10.1186/1471-2164-6-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Maréchal C, Seyffert N, Jardin J, Hernandez D, Jan G, Rault L, Azevedo V, François P, Schrenzel J, van de Guchte M, Even S, Berkova N, Thiéry R, Fitzgerald JR, Vautor E, Le Loir Y. 2011. Molecular basis of virulence in Staphylococcus aureus mastitis. PLoS One 6:e27354. 10.1371/journal.pone.0027354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talaat AM, Howard ST, Hale W, IV, Lyons R, Garner H, Johnston SA. 2002. Genomic DNA standards for gene expression profiling in Mycobacterium tuberculosis. Nucleic Acids Res. 30:e104. 10.1093/nar/30.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clauditz A, Resch A, Wieland K-P, Peschel A, Götz F. 2006. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 74:4950–4953. 10.1128/IAI.00204-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajkarnikar A, Strankman A, Duran S, Vargas D, Roberts AA, Barretto K, Upton H, Hamilton CJ, Rawat M. 2013. Analysis of mutants disrupted in bacillithiol metabolism in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 436:128–133. 10.1016/j.bbrc.2013.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cabiscol E, Tamarit J, Ros J. 2000. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int. Microbiol. 3:3–8 [PubMed] [Google Scholar]
- 40.Potter AJ, Trappetti C, Paton JC. 2012. Streptococcus pneumoniae uses glutathione to defend against oxidative stress and metal ion toxicity. J. Bacteriol. 194:6248–6254. 10.1128/JB.01393-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelner MJ, Bagnelli R, Welchl KJ. 1990. Thioureas react with superoxide radicals to yield a sulfhydryl compound. J. Biol. Chem. 265:1306–1311 [PubMed] [Google Scholar]
- 42.Chi BK, Gronau K, Mäder U, Hessling B, Becher D, Antelmann H. 2011. S-Bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell. Proteomics 10:M111.009506. 10.1074/mcp.M111.009506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cosgrove K, Coutts G, Jonsson I-M, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J. Bacteriol. 189:1025–1035. 10.1128/JB.01524-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 69:3744–3754. 10.1128/IAI.69.6.3744-3754.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cummins I, Cole D, Edwards R. 1999. A role for glutathione transferases functioning as glutathione peroxidases in resistance to multiple herbicides in blackgrass. Plant J. 18:285–292. 10.1046/j.1365-313X.1999.00452.x [DOI] [PubMed] [Google Scholar]
- 46.Prohaska J, Ganther H. 1976. Glutathione peroxidase activity of glutathione-S-transferases purified from rat liver. Biochem. Biophys. Res. Commun. 76:437–445. 10.1016/0006-291X(77)90744-6 [DOI] [PubMed] [Google Scholar]
- 47.Singh R, Mailloux RJ, Puiseux-Dao S, Appanna VD. 2007. Oxidative stress evokes a metabolic adaptation that favors increased NADPH synthesis and decreased NADH production in Pseudomonas fluorescens. J. Bacteriol. 189:6665–6675. 10.1128/JB.00555-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mapson L, Isherwood F. 1963. Glutathione reductase from germinated peas. Biochem. J. 86:173–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas EL. 1984. Disulfide reduction and sulfhydryl uptake by Streptococcus mutans. J. Antimicrob. Chemother. 157:240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.delCardayre SB. 1998. Coenzyme A disulfide reductase, the primary low molecular weight disulfide reductase from Staphylococcus aureus purification and characterization of the native enzyme. J. Biol. Chem. 273:5744–5751. 10.1074/jbc.273.10.5744 [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Wu N, Dong J, Gao Y, Zhang X, Shao N, Yang G. 2010. ssrA (tmRNA) acts as an antisense RNA to regulate Staphylococcus aureus pigment synthesis by base pairing with crtMN mRNA. FEBS Lett. 584:4325–4329. 10.1016/j.febslet.2010.09.024 [DOI] [PubMed] [Google Scholar]
- 52.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4(1):e00537–12. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuchs S, Zühlke D, Pané-Farré J, Kusch H, Wolf C, Reiß S, Binh LTN, Albrecht D, Riedel K, Hecker M, Engelmann S. 2013. Aureolib—a proteome signature library: towards an understanding of Staphylococcus aureus pathophysiology. PLoS One 8:e70669. 10.1371/journal.pone.0070669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Upton H, Newton GL, Gushiken M, Lo K, Holden D, Fahey RC, Rawat M. 2012. Characterization of BshA, bacillithiol glycosyltransferase from Staphylococcus aureus and Bacillus subtilis. FEBS Lett. 586:1004–1008. 10.1016/j.febslet.2012.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakano S, Küster-Schöck E, Grossman AD, Zuber P. 2003. Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 100:13603–13608. 10.1073/pnas.2235180100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaballa A, Antelmann H, Hamilton CJ, Helmann J. 2013. Regulation of Bacillus subtilis bacillithiol biosynthesis operons by Spx. Microbiology 159(Pt 10):2025–2035. 10.1099/mic.0.070482-0 [DOI] [PubMed] [Google Scholar]
- 57.Olivier A, Lemaire S, Van Bambeke F, Tulkens PM, Oldfield E. 2009. Role of rsbU and staphyloxanthin in phagocytosis and intracellular growth of Staphylococcus aureus in human macrophages and endothelial cells. J. Infect. Dis. 200:1367–1370. 10.1086/606012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uziel O, Borovok I, Schreiber R, Cohen G, Aharonowitz Y. 2004. Transcriptional regulation of the Staphylococcus aureus thioredoxin and thioredoxin reductase genes in response to oxygen and disulfide stress. J. Bacteriol. 186:326–334. 10.1128/JB.186.2.326-334.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vaish M, Singh VK. 2013. Antioxidant functions of nitric oxide synthase in a methicillin sensitive Staphylococcus aureus. Int. J. Microbiol. 2013:312146. 10.1155/2013/312146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kreiswirth BN, Betley M, O'Reilly M. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. 10.1038/305709a0 [DOI] [PubMed] [Google Scholar]
- 61.Tenover F, McDougal L, Goering R. 2006. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J. Clin. Microbiol. 44:108–118. 10.1128/JCM.44.1.108-118.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Highlander SK, Hultén KG, Qin X, Jiang H, Yerrapragada S, Mason EO, Shang Y, Williams TM, Fortunov RM, Liu Y, Igboeli O, Petrosino J, Tirumalai M, Uzman A, Fox GE, Cardenas AM, Muzny DM, Hemphill L, Ding Y, Dugan S, Blyth PR, Buhay CJ, Dinh HH, Hawes AC, Holder M, Kovar CL, Lee SL, Liu W, Nazareth LV, Wang Q, Zhou J, Kaplan SL, Weinstock GM. 2007. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7:99. 10.1186/1471-2180-7-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426–2438. 10.1128/JB.187.7.2426-2438.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225–1240. 10.1016/S0140-6736(00)04403-2 [DOI] [PubMed] [Google Scholar]
- 65.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ, England S. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467. 10.1128/JB.184.19.5457-5467.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.