

Abstract
Mycoplasma bovis is one of the major causative pathogens of bovine respiratory complex disease (BRD), which is characterized by enzootic pneumonia, mastitis, pleuritis, and polyarthritis. M. bovis enters and colonizes bovine respiratory epithelial cells through inhalation of aerosol from contaminated air. The nature of the interaction between M. bovis and the bovine innate immune system is not well understood. We hypothesized that M. bovis invades blood monocytes and regulates cellular function to support its persistence and systemic dissemination. We used bovine-specific peptide kinome arrays to identify cellular signaling pathways that could be relevant to M. bovis-monocyte interactions in vitro. We validated these pathways using functional, protein, and gene expression assays. Here, we show that infection of bovine blood monocytes with M. bovis delays spontaneous or tumor necrosis factor alpha (TNF-α)/staurosporine-driven apoptosis, activates the NF-κB p65 subunit, and inhibits caspase-9 activity. We also report that M. bovis-infected bovine monocytes do not produce gamma interferon (IFN-γ) and TNF-α, although the level of production of interleukin-10 (IL-10) is elevated. Our findings suggest that M. bovis takes over the cellular machinery of bovine monocytes to prolong bacterial survival and to possibly facilitate subsequent systemic distribution.
INTRODUCTION
Mycoplasma bovis is one of the major causative pathogens of bovine respiratory complex disease (BRD), which is characterized by enzootic pneumonia, pleuritis, and polyarthritis. Other pathogens in this group include Mannheimia haemolytica, Histophilus somni, Pasteurella multocida, bovine viral diarrhea virus (BVDV), bovine respiratory syncytial virus (BVRSV), and parainfluenza-3 virus (1). BRD is responsible for considerable economic losses to feedlot beef producers in Europe, Canada, and the United States (2, 3). Current control of M. bovis-associated BRD involves antimicrobial treatment as well as vaccination using M. bovis bacterins (4). Importantly, the absence of a cell wall in M. bovis restricts the spectrum of antibiotics that may be employed to treat these infections (2). Furthermore, successful antimicrobial treatment against M. bovis depends on early diagnosis and treatment. In addition, immune responses induced by bacterins have been shown to be ineffective (4).
To date, the interaction between M. bovis and different elements of host immunity are poorly understood. Observations from experimentally infected cattle have revealed stimulation of both innate and adaptive immunity (5, 6). The pulmonary bovine immune response to M. bovis is mainly anti-inflammatory, with high levels of interleukin-4 (IL-4) and moderate levels of gamma interferon (IFN-γ) and IgG1 antibodies (6, 7). Although M. bovis infects predominantly mucosal and serosal surfaces, live bacteria can be detected in brain of infected cattle (8). Bacterial antigen has also been observed in synovial membranes (9) and in liver and kidneys (10) of infected cattle. While M. bovis infects all subsets of bovine peripheral mononuclear cells (PBMC) and inhibits lymphocyte proliferation (11), interactions between M. bovis and a distinct PBMC population have not been characterized. Furthermore, the strategies used by M. bovis to achieve systemic dissemination have not been elucidated.
Kinase-mediated phosphorylation is the predominant mechanism for regulation of protein function (12). Furthermore, the conserved catalytic clefts of kinases make them highly attractive targets for drug therapy (13). Since the specificity of many kinases is determined by residues surrounding the phosphorylation site, peptides that are constructed to correspond to phosphorylation sites can serve as appropriate kinase substrates (14, 15) and have in fact been utilized in high-throughput arrays to quantify total cellular kinase activity (16–18). Bovine-specific peptide kinome arrays have been designed (19) and used to characterize signaling events in monocytes treated with bacterial lipopolysaccharide (LPS) and CpG oligodeoxy nucleotides (ODN) (20). These bovine-specific peptide arrays have also been used to characterize the host responses of bovine monocytes to infection with Mycobacterium avium subsp. paratuberculosis, revealing novel mechanisms utilized by the pathogen to subvert host innate immunity as well as identifying targets for therapeutic intervention (21, 22).
We previously demonstrated that apoptosis of bovine PBMC is delayed after in vitro infection with M. bovis (11). However, the mechanisms of this delay were not defined. Blood monocytes are an important subset of innate immune cells that perform multiple functions, including Toll-like receptor (TLR) recognition of pathogen-associated molecular patterns (PAMPs), phagocytosis of invading pathogens, antigen processing/presentation, and cytokine production (23). In addition, monocytes differentiate into mature dendritic cells (DCs) or macrophages and have the capacity to home in on tissues undergoing invasion and traffic between these tissues and lymphoid tissues (24). In this study, we isolated fresh bovine monocytes, infected them with M. bovis, and used kinome arrays to identify phosphorylation pathways modulated by the bacterium. Here, we demonstrate that upon in vitro infection of bovine monocytes with M. bovis, apoptosis is delayed, the p65 subunit of NF-κB is activated, and the initiator caspase 9 protease is blocked. We also show that M. bovis-infected monocytes produce neither IFN-γ nor tumor necrosis factor alpha (TNF-α) but induce the release of IL-10. Therefore, we present a novel strategy used by M. bovis to evade host innate immunity and suggest that this strategy favors the systemic dissemination of the bacterium.
MATERIALS AND METHODS
Bacterial strains and media.
All experiments were conducted by using M. bovis strain Mb1, which was previously isolated from the synovial fluid of a calf exhibiting signs of arthritis (25). Cultures were grown in modified Hayflick's medium at 37°C in a 5% CO2 atmosphere. At the exponential phase of growth, aliquots were collected, and bacterial cells were obtained by centrifugation (5,500 × g for 15 min) and then washed with minimum essential medium (MEM; Invitrogen, Burlington, ON, Canada). Bacteria were suspended to a cell density of 108 CFU/ml in MEM supplemented with 30% glycerol and stored at −70°C until use. MEM containing 10% fetal bovine serum (FBS), 0.05 mM 2-mercaptoethanol, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids (NEAA), and 10 mM HEPES buffer was used to culture monocytes for all subsequent assays.
Isolation and purification of monocytes from PBMC and infection with M. bovis.
Blood samples (300 ml from each animal) were obtained from 6 clinically healthy animals with no history of M. bovis infection. The buffy coat fraction (containing PBMC) was purified by Ficoll gradients (GE Healthcare, Mississauga, ON, Canada), as described previously (11). Magnetically activated cells sorting (MACS) was used to purify monocytes (CD14+ cells) by direct labeling with mouse monoclonal anti-human IgG2a CD14 microbeads (Miltenyi Biotec, Gladbach, Germany) and positive selection through an LS column (Miltenyi Biotec), according to the instructions of the manufacturer. Briefly, purified PBMC were enumerated by using a Coulter Counter, and viability was determined by trypan blue exclusion. PBMC were suspended to a cell density of 1.25 × 108 cells/ml in MACS buffer (0.5% bovine serum albumin [BSA] and 2 mM EDTA in phosphate-buffered saline [PBS]), and 2.4 ml was dispensed into each tube. Three hundred microliters of mouse anti-human CD14 microbeads was added to each tube, and tubes were placed at 4°C for 15 min before 3-fold washing with 40 ml of ice-cold MACS buffer by centrifugation at 300 × g for 8 min. Cells in each tube were suspended in 3 ml of MACS buffer and passed through an LS column mounted onto a magnetic field. The column was washed three times with 10 ml MACS buffer, and bound cells were eluted by filling the column with 5 ml of MACS buffer, forcing the cells down the column after removal from the magnetic field. The purity of monocytes was confirmed by flow cytometry after staining with goat anti-mouse IgG2a antibody conjugated to fluorescein isothiocyanate (FITC; Caltag Medsystems, Buckingham, United Kingdom). Typically, a purity of ≥97% was achieved for each sample. For all subsequent assays, unless otherwise stated, each well of a 6-well flat-bottom plate (Costar, Corning, NY) was seeded with 5 × 106 monocytes and left overnight at 37°C in a humidified incubator containing 5% CO2 before infection with 2.5 × 107 bacteria (multiplicity of infection [MOI] of 5:1). This MOI was used because it was optimized previously in our laboratory (11) and has been shown to diminish biological effects when reduced and to have no detectable change when increased.
Peptide arrays.
The design, construction, and application of the peptide arrays were based upon a previously reported protocol, with modifications (19). The kinome experiments for all the animals were performed simultaneously in a single run, minimizing the possibility of technical variances in the analysis. Briefly, approximately 1 × 107 cells were collected from 3 animals, pelleted, and lysed by addition of 100 μl lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mM NaF, 1 μg/ml leupeptin, 1 g/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF]) (all products were obtained from Sigma-Aldrich unless indicated otherwise). Cells were incubated on ice for 10 min and spun in a microcentrifuge for 10 min at 4°C. A 70-μl aliquot of this supernatant was mixed with 10 μl of activation mix (50% glycerol, 500 μM ATP [New England BioLabs, Pickering, ON, Canada], 60 mM MgCl2, 0.05% [vol/vol] Brij 35, 0.25 mg/ml BSA) and incubated on the array for 2 h at 37°C. Arrays were then washed with PBS–1% Triton. Slides were placed into phospho-specific fluorescent ProQ Diamond phosphoprotein stain (Invitrogen) with agitation for 1 h. Arrays were then washed three times in distain containing 20% acetonitrile (EMD Biosciences, VWR, Mississauga, ON, Canada) and 50 mM sodium acetate (Sigma) at pH 4.0 for 10 min. A final wash was done with distilled deionized H2O. Arrays were air dried for 20 min and then centrifuged at 300 × g for 2 min to remove any remaining moisture from the array. Arrays were read by using a GenePix Professional 4200A microarray scanner (MDS Analytical Technologies, Toronto, ON, Canada) at 532 to 560 nm with a 580-nm filter to detect dye fluorescence. Images were collected by using GenePix 6.0 software (MDS Analytical Technologies), and the spot intensity signal was collected as the mean of the pixel intensity using local feature background intensity background calculation with the default scanner saturation level.
Data sets for data analysis.
The data set contains the signal intensities associated with each of 300 peptides for the monocytes from 3 animals under the different treatment conditions. For each animal and each treatment, there were three intra-array replicates. All data processing and analyses were done as described previously (26), with the following study specifics.
(i) Animal-animal variability analysis.
For each of the 300 peptides, an F test was used to determine whether there were significant differences among the three animals under the same treatment conditions. Therefore, 300 F tests were carried out for a single treatment.
(ii) Treatment-treatment variability analysis.
Peptides identified by the F test as having consistent patterns of responses to the various treatments across the three animals were subjected to a paired t test to compare their signal intensities under a treatment condition with those under control conditions. For each animal-independent peptide, the responses from all three animals were pooled to increase the statistical confidence. Peptides with significant (P < 0.10) changes in phosphorylation were identified. This level of significance was chosen to retain as much data as possible and thus facilitate subsequent pathway analysis.
Analysis of differentially phosphorylated peptides.
A given peptide was selected for further analysis if two conditions were true: first, the peptide had to be consistently phosphorylated according to the chi-square test for both the treatment and the control conditions, and second, the P value resulting from a t test between the transformed treatment intensities and the transformed control intensities had to be <0.1. While 0.1 may seem like a liberal threshold, when performing pathway analysis, it is more important to avoid false-negative than false-positive results. This is due to the fact that several peptides are involved in the same biological pathway, and we are trying to identify as many of those peptides as possible. Even with a liberal P value threshold, it is unlikely that several peptides from the same biological pathway will be erroneously identified as being differentially phosphorylated when that pathway is in fact not affected by the treatment under investigation; however, it increases the chances that peptides from pathways that actually are affected by the treatment will be identified as being differentially phosphorylated. We have recently reported an analysis of the impact of different P value thresholds on false-negative probabilities (27). For peptides meeting the above-described two conditions, fold change (FC) values were calculated by using the formula 2d, where d = averagetreatment − averagecontrol, as previously described (26).
Pathway analysis of differentially phosphorylated peptides.
InnateDB is a publically available resource which, based on levels of either differential expression or phosphorylation, predicts biological pathways based on experiment fold change data sets (28). Pathways are assigned a probability value (P) based on the number of proteins present for a particular pathway as well as the degree to which they are differentially expressed or modified relative to a control condition.
Detection of activated NF-κB.
M. bovis-infected monocytes obtained from 6 animals were incubated for 24 h under conditions described above. Controls included 10 μg/well of Escherichia coli LPS (Sigma) and untreated monocytes. The NF-κB activation kit (Five Photon Biochemicals, San Diego, CA) was used to separate the cytoplasmic and nuclear fractions. Briefly, cells were centrifuged (150 × g) for 8 min, media were removed, and cells were washed with PBS. The cells were covered with 100 μl ice-cold cytoplasmic fractionation reagent (CER-1) containing protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM PMSF, and 1 mM dithiothreitol [DTT]), placed on ice for 5 min, and then scraped off and dispensed into 1.5-ml centrifuge tubes. The cells were centrifuged for 3 min at 2,500 rpm in a refrigerated microcentrifuge, and the supernatant was collected. The pellet was washed once with CER-1, suspended in 40 ml of nuclear fractionation reagent (NER-1), vortexed, and placed on ice for 10 min before centrifugation at 16,000 rpm and collection of the supernatant containing the nuclear fraction. Fractions from identical treatments from 3 animals were pooled, and the protein concentrations of both the cytoplasmic and nuclear fractions were determined by the Lowry method using the Coomassie blue reagent kit (Bio-Rad, Philadelphia, PA, USA). Fractions were stored at −80°C until use for Western blot analysis. To detect activated NF-κB, 10 μg of protein was subjected to 12.5% SDS-PAGE and electroblotted onto a nitrocellulose membrane (pore size, 0.45 μm) (Hybond; GE Healthcare, Mississauga, ON, Canada). The membrane was blocked with 5% BSA in TBS-T (Tris-buffered saline, 0.05% Tween 20) for 1 h and then incubated with rabbit anti-p65 (Five Photon Biochemical, San Diego, CA, USA) diluted 1/1,000. The membrane was incubated for 1 h, and after washes in TBS-T, bound antibody was detected with goat anti-rabbit IgG conjugated to Alexa 680 (Invitrogen, Camarillo, CA, USA) and read by using a phosphorimager at 700 nm. A rabbit antibody against the 15-kDa bovine H2b histone (Abcam, Cambridge, MA, USA) was used to demonstrate separation of nuclear and cytoplasmic fractions.
Caspase assays.
A caspase colorimetric protease assay sample kit (Aportaget; Invitrogen, Burlington, ON, Canada) was used to detect the activity of caspases 3, 6, and 9 in Mb1-infected monocytes. Monocytes obtained from 3 animals were infected with M. bovis and incubated for 24 h. Controls included monocytes incubated for 6 h with 20 μM/well staurosporine (STS; Sigma) and untreated monocytes. Cells were harvested and enumerated, and a pellet containing 5 × 106 cells was suspended in 50 μl of chilled cell lysis buffer provided in the kit and incubated on ice for 10 min before centrifugation at 10,000 × g. The supernatant containing the cytosolic fraction was transferred into a fresh tube, and the protein concentration was determined. The cytosolic fraction was adjusted to a concentration of 200 μg/ml in the cell lysis buffer provided in the kit, and 50 μl was aliquoted into 96-well round-bottom microtiter plates (Costar) before addition of 50 μl of 2× kit reaction buffer containing 10 mM DTT and 200 μM the respective substrate for caspase 3, 6, or 9 in a volume of 5 μl. The samples were incubated at 37°C for 2 h in the dark and then read at 405 nm with a 490-nm reference filter in a microplate reader (xMark microplate spectrophotometer; Bio-Rad, Philadelphia, PA, USA).
Cytokine ELISAs.
Sandwich enzyme-linked immunosorbent assays (ELISAs) for IFN-γ, TNF-α, and IL-10 were carried out to determine the cytokine profile of M. bovis-infected monocytes from six animals. Plates used for each assay were Immunolon 2 (Fisher, Ottawa, ON, Canada) for IFN-γ and TNF-α and Maxisorp (Fisher) for IL-10. Coating antibodies used were anti-bovine IFN-γ antibody 2-2-1A (29) at 0.125 μg/ml, mouse anti-bovine TNF-α antibody 1D11-13 (30) at 1 μg/ml, and mouse anti-bovine IL-10 (Cedarlane, Burlington, ON, Canada) at 0.5 μg/ml. Antibodies were diluted in carbonate coating buffer (50 mM Na2CO3, pH 9.6) and applied onto plates at a volume of 100 μl per well. Plates were incubated overnight at 4°C and washed four times with TBS-T. Undiluted supernatant samples were applied at 100 μl per well. Stocks of recombinant cytokines were prepared at the following dilutions: 1 ng/ml (IFN-γ), 1 ng/ml (TNF-α), and 75 units/ml (IL-10) in TBS-T–0.1% gelatin (TBST-g), TBS-T–0.1% casein (TBST-C), and TBS-T, respectively. Series of 2-fold dilutions were prepared from these stocks and used as standards. Plates were incubated for 2 h at room temperature and washed four times in TBS-T and appropriate secondary antibodies: rabbit anti-bovine IFN-γ (29) diluted 1/5,000, rabbit anti-bovine TNF-α (30) diluted 1/1,500, or biotinylated mouse anti-bovine IL-10 (Serotec) diluted to 0.125 μg/ml was added at 100 μl per well. Plates were incubated for 1 h at room temperature and then washed four times in TBS-T. For IFN-γ and TNF-α, 100 μl of biotinylated goat anti-rabbit IgG (Invitrogen) at a dilution of 1:10,000 was added to each well. Plates were incubated for 1 h at room temperature and then washed four times in TBS-T. Streptavidin alkaline phosphatase (Cedarlane Laboratories, Burlington, ON, Canada) was added to all plates (1:5,000 dilution) at 100 μl per well. Plates were incubated for 1 h at room temperature and then washed four times in TBS-T. Following washes, 100 μl of 1 mg/ml p-nitrophenyl phosphate in substrate buffer was added to each well. Plates were incubated at room temperature for 30 min and read by using a microtiter plate reader with the absorbance set at 405 nm and the reference set at 490 nm. Cytokine concentrations were calculated by using a regression model, which compared the absorbance of the samples with the standard curves.
Apoptosis assays.
The Alexa Fluor annexin V staining/Dead Cell apoptosis kit (Invitrogen) was used to determine whether M. bovis induces apoptosis in monocytes. Briefly, monocytes from six animals were incubated with M. bovis Mb1 for 24 h (MOI of 5:1). Controls included untreated monocytes and monocytes treated with staurosporine (20 μM for 6 h). To determine whether M. bovis affects TNF-α-driven apoptosis of bovine monocytes, cells were incubated with either TNF-α (5 μg) alone or M. bovis and TNF-α. After 24 h, the cells were washed twice with PBS and suspended at 2 × 106 cells/ml in 1× annexin V binding buffer. The cells were divided into 100-μl aliquots before addition of 5 μl of annexin V-FITC to each tube. Tubes were kept at room temperature in the dark for 15 min. Finally, 400 μl of annexin V binding buffer was added to each tube, and the cells were immediately analyzed by flow cytometry.
Statistical analyses.
Statistical analysis of kinome array data was performed by using InnateDB software (28). For kinome array data, statistical significance was assigned at a P value of ≤0.10. The statistical analyses of caspase activity, cytokine ELISAs, and relative percentages of cell survival were carried out by using one-way analysis of variance (ANOVA) with Prism version 5.04 for Windows (GraphPad Software, La Jolla, CA, USA). Data were considered statistically different if the P value was 0.05 or lower.
RESULTS
Signaling responses of bovine monocytes to infection with M. bovis.
Due to the capacity of bovine peptide kinome arrays to provide high-throughput data (19), we used this method to identify monocyte innate immune response signaling pathways after invasion by M. bovis. Pathways unrelated to innate immunity were ignored in this study. The criteria for selection of these pathways for validation were 2-fold: the presence of the pathway in all three animals under study and a significant (P ≤ 0.10) deviation from untreated monocytes. Kinome identification of significant phosphorylation patterns of M. bovis-infected monocytes was subjected to online algorithm searches (INOH, PID BIOCARTA, REACTOME, and PID NCI). These analyses revealed seven phosphorylation patterns involving NF-κB signaling, two pathways involving regulation of apoptosis, and one pathway that involved gene expression of suppressor of cytokine signaling 1 (SOCS1). The database and protein identification numbers, the number of genes affected in each pathway, P values for the deviation of each pathway from the background, as well as the cumulative gene fold change with reference to uninfected monocytes for each signaling pathway are shown in Table 1. The signaling responses implicated with the peptide arrays were then validated through independent techniques, including patterns of protein and functional assays.
TABLE 1.
Monocyte pathways regulated by M. bovis as determined by kinome arraysa
| Pathway | Database (protein identification) | P value of change (P ≤ 0.10) | Avg fold change |
|---|---|---|---|
| TLR (through NF-κB) | INOH (10068) | 0.02 | −1.6 |
| NF-κB | PID BIOCARTA (3978) | 0.02 | −1.6 |
| NF-κB activation | REACTOME (9204) | 0.05 | −1.7 |
| IKK–NF-κB | INOH (10150) | 0.05 | −1.7 |
| NF-κB activation | REACTOME (3767) | 0.01 | −1.8 |
| Atypical NF-κB | PID NCI (9435) | 0.05 | −2.1 |
| Canonical NF-κB | PID NCI (9358) | 0.01 | −1.5 |
| Caspase cascade, apoptosis | PID NCI (9311) | 0.04 | −2.9 |
| Apoptosis | REACTOME (1882) | 0.06 | −2.8 |
| Gene expression of SOCS1 | INOH (9782) | 0.06 | 1.3 |
| Gene expression SOCS1 | INOH (10060) | 0.06 | 1.3 |
Kinome arrays were performed as indicated in Materials and Methods.
Monocyte NF-κB expression is activated after infection with M. bovis.
Since NF-κB is an important regulator of both survival and apoptosis of host cells (31, 32), we tested whether infection of monocytes by M. bovis activates this transcription factor. Here, we compared the relative levels of the p65 subunit of NF-κB in the cytoplasmic and nuclear fractions of infected and uninfected monocytes, and the results are shown in Fig. 1. Western blot analysis showed higher levels of p65 in the nucleus for M. bovis-infected monocytes, as observed by the intensity of the bands in the nuclear fraction (Fig. 1A). Additionally, treatment of monocytes with LPS also activated NF-κB with levels comparable to those of M. bovis-treated monocytes (Fig. 1B). These results indicated that upon interaction with M. bovis, the activated portion of NF-κB is translocated to the nucleus of monocytes.
FIG 1.
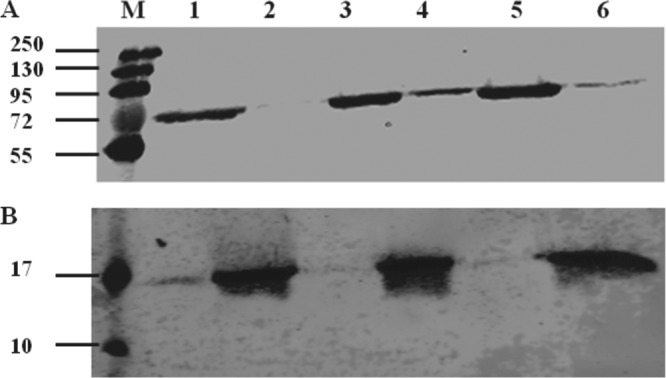
Translocation of the 65-kDa RelA (p65) NF-κB subunit to the cell nucleus. (A) Western blot of cytoplasmic (lanes 1, 3, and 5) and nuclear (lanes 2, 4, and 6) fractions (10 μg/lane) of M. bovis-infected monocytes pooled from 3 animals. Lanes: M, molecular mass marker; 1 and 2, untreated monocytes; 3 and 4, M. bovis-infected monocytes; 5 and 6, LPS-treated monocytes. (B) To confirm successful separation of nuclear and cytoplasmic fractions, the nuclear histones (15 kDa) are shown in the nuclear fractions (lanes 2, 4, and 6). The experiment was performed 3 times, and this figure is representative.
M. bovis delays monocyte apoptosis.
Considering that activation of NF-κB determines cellular survival or apoptosis (33) and the results of our kinome array identified this pathway (Table 1), we sought to find out whether activation of this transcription factor by M. bovis influences survival or apoptosis of bovine monocytes. After 24 h of incubation, an average of 2.5% of M. bovis-infected monocytes underwent apoptosis, compared to 20.2% of staurosporine (STS)-treated monocytes and 6% of untreated monocytes (Fig. 2). When monocytes were infected for 18 h followed by addition of STS and further incubation for 6 h, the percentage of monocytes undergoing STS-driven apoptosis was significantly (P ≤ 0.001) reduced to an average of 6.3% (Fig. 2A). Similarly, treatment of monocytes with TNF-α alone resulted in an average of 6.8% apoptotic cells, while M. bovis infection of TNF-α-treated monocytes reduced the mean percentage of apoptotic cells by half (3.6%). We therefore compared the relative proportions of live and dead cells from the three different treatments (Fig. 2B). As expected, the percentage of live M. bovis-infected monocytes was significantly higher than that of untreated cells (P ≤ 0.001) and TNF-α (P ≤ 0.01)- or STS (P ≤ 0.001)-treated monocytes. Interestingly, when monocytes were coincubated with M. bovis and STS or TNF-α, the presence of M. bovis significantly inhibited cell death associated with these stimuli (P ≤ 0.001 and P ≤ 0.05, respectively) (Fig. 2B). This observation was confirmed by a highly significant proportion of dead cells in STS- and TNF-α-treated cultures compared to very low percentages of dead cells in monocytes infected with M. bovis alone (P ≤ 0.001 and P ≤ 0.01, respectively) (Fig. 2B). M. bovis infection of STS- or TNF-α-treated monocytes reduced the proportion of dead monocytes, although this reduction was not significant for both stimuli (Fig. 2B). These results suggested that even in the presence of staurosporine or extrinsic TNF-α, M. bovis is capable of delaying monocyte apoptosis.
FIG 2.

Delay of apoptosis. (A) Representative dot plots showing apoptosis of CD14+ cells. Each well containing 5.0 × 106 cells was untreated (i), treated with TNF-α for 24 h (ii), untreated for 18 h before addition of staurosporine for 6 h without M. bovis infection (− M. bovis) (iii), infected with M. bovis only (iv), infected with M. bovis and simultaneously treated with TNF-α (v), and infected with M. bovis with addition of 20 μM staurosporine 18 h later (vi). For all plots, the cell populations are gated into 4 quadrants representing cells that are undergoing necrosis (L1), alive (L2), undergoing early apoptosis (R2), or undergoing late apoptosis or death (R1). (B) Summary of the delay of monocyte apoptosis results (6 animals) after treatments. Medium, untreated cells; M. bovis, M. bovis-infected cells; TNF-α, cells treated with TNF-α; STS, cells treated with staurosporine; M. bovis + TNF-α, cells treated with TNF-α and infected with M. bovis; M. bovis + STS, cells infected with M. bovis and treated with STS. Shown are apoptotic cells stained with annexin V (A), live cells (B), and dead cells (C) (measured by propidium iodide uptake). The bars on the y axis show the median percentages of cells. Significant differences between treatments are indicated by ∗ (P ≤ 0.05), ∗∗ (P ≤ 0.01), and ∗∗∗ (P ≤ 0.001).
M. bovis modulates caspase 9 production.
The caspase family of proteases coordinates many aspects of apoptosis (34, 35). Caspases are produced and retained intracellularly as inert proenzymes, each with two subunits: a large subunit and a small subunit with a variable amino-terminal prodomain (36). Activation of caspases is achieved by the loss of the prodomain by catalytic elimination of a carboxyl-terminal aspartate and subsequent heterodimerization of the large and small subunits into active enzymes (35, 36). Caspases can be divided into two classes: initiator caspases (caspases 8 and 9) act upstream of the effector caspases (caspases 3, 6, and 7) and respond to the TNF receptor family or Fas (caspase 8) (37, 38) or cytochrome c release following mitochondrial damage (caspase 9) (36). Once activated by initiator caspases, effector caspases cause cellular disintegration by cleavage of other death substrates and nucleoproteins with cellular DNA fragmentation and cell death as a result (36). Following kinome array identification of caspase signaling as a pathway regulated by M. bovis, as well as the observation that M. bovis inhibits monocyte apoptosis, we measured activation levels of one initiator caspase (caspase 9) and two effector caspases (caspases 3 and 6), and the results are shown in Fig. 3. There was no significant difference in activation of caspase 3 between uninfected cells, M. bovis-infected monocytes, and STS-treated cells (Fig. 3). While there was no significant difference in caspase 6 activation between uninfected cells and M. bovis-infected monocytes, this caspase was significantly activated (P ≤ 0.05) in staurosporine-treated monocytes compared to M. bovis-infected monocytes. In addition, caspase 9 activation was significantly increased (P ≤ 0.001) in untreated monocytes compared to M. bovis-infected monocytes. There was also a highly significant difference in caspase 9 activation between untreated monocytes and staurosporine-treated monocytes (Fig. 3). These results suggest that M. bovis is capable of modulating caspase 9 activity.
FIG 3.

Caspase activity of M. bovis-infected monocytes. Data for the activation of caspases 3, 6, and 9 in cytosolic fractions of monocytes after 24 h of incubation with M. bovis are shown. Activation is based on the cleavage of the corresponding synthetic substrates (caspase 3, DEVD; caspase 6, VEID; caspase 9, LEHD) labeled with para-nitroaniline, which is released after the substrate is cleaved by the specific caspase. All data were square root transformed before analysis and were generated from 3 animals and triplicate cultures for each treatment. Each bar represents the mean of 3 replicates for each treatment, and the error bars indicate the standard deviations from the means of each treatment. The x axis shows specific caspases (caspases 3, 6, and 9), and the y axis indicates the optical density at 405 nm (OD405) of the samples. Significant differences for each caspase activity between the treatments are indicated by ∗ (P ≤ 0.05), ∗∗ (P ≤ 0.01), and ∗∗∗∗ (P ≤ 0.0001).
M. bovis downregulates expression of proinflammatory cytokines.
The suppressor of cytokine signaling (SOCS) protein regulates Janus kinase (Jak)-signal transducer and activator of transcription (STAT) signaling (39). The observation that M. bovis infection of monocytes results in regulation of SOCS1 signaling (Table 1) as well as inhibition of apoptosis provoked our interest to determine how these events affect cytokine production by infected monocytes. There is evidence that SOCS1 inhibits the production of a broad spectrum of cytokines, including IFN-γ (40) and TNF-α (41), and could be involved in IL-10 suppression of IFN-γ (42). Using a sandwich ELISA, we measured the levels of production of IFN-γ, TNF-α, and IL-10 after 24 h of incubation of monocytes with M. bovis, and the results are shown in Fig. 4. Although untreated and M. bovis-infected monocytes did not produce detectable levels of IFN-γ and TNF-α, LPS treatment significantly (P ≤ 0.001) induced the production of these cytokines (Fig. 4A and B). On the other hand, M. bovis infection significantly (P ≤ 0.001) induced the production of large amounts of IL-10 in comparison to untreated cells (Fig. 4C). LPS-treated monocytes produced smaller amounts of IL-10 than did M. bovis-infected monocytes, although this difference was not significant (Fig. 4C). Here, we conclude that in vitro infection of monocytes by M. bovis regulates cytokine production away from a proinflammatory profile and stimulates the production of anti-inflammatory cytokines.
FIG 4.

Production of IFN-γ, TNF-α, and IL-10. The quantification of IFN-γ (A), TNF-α (B), and IL-10 (C) in supernatants from monocytes after 24 h of incubation with M. bovis is shown. Cytokines were quantified from undiluted supernatants. Each point represents the mean of triplicate wells for each animal, and the bar represents the median for all 6 animals for each cytokine. The treatment is indicated on the x axis. The left y axis shows IFN-γ and TNF-α levels in picograms/ml, and the right y axis shows the levels of IL-10 in units/ml. Significant differences in the levels of cytokines for each treatment are indicated by ∗∗∗ (P ≤ 0.001).
DISCUSSION
Mycoplasma bovis enters and colonizes respiratory epithelial cells of cattle when they inhale aerosol from contaminated air. This bacterium disseminates systemically, including the synovial membrane, liver, and kidneys (9, 10, 43). In addition, upon infection and establishment on bovine respiratory surfaces, M. bovis causes a chronic and sometimes asymptomatic state in the bovine host, which is characterized by intermittent shedding of the bacteria for months to years (44, 45). To date, the mechanisms by which M. bovis evades host immunity to establish persistent infection have not been elucidated. Using in vitro models of freshly isolated bovine PBMC and erythrocytes, we showed that M. bovis infects and persists in all PBMC subsets as well as erythrocytes (11). In addition, we showed that the ability of PBMC to release cytokines (IFN-γ) was not impaired, and M. bovis-infected PBMC were resistant to apoptosis (11). Since early establishment of M. bovis would depend largely on its ability to evade host innate immunity, we sought to characterize the interaction between M. bovis and bovine blood monocytes.
Bovine-specific kinome arrays have proven to be powerful tools to investigate signaling responses of bovine monocytes to immune stimulation as well as infection with bovine pathogens (19, 20, 22). The signaling responses identified through the peptide arrays were then validated through independent techniques. In the present study, we show for the first time that M. bovis-infected monocytes exhibit a delay in both spontaneous and TNF-α- or staurosporine-driven apoptosis (Fig. 2), activation of the p65 subunit of NF-κB (Fig. 1), and blockage of the initiator caspase 9 (Fig. 3). We also report that M. bovis infection of bovine monocytes does not induce proinflammatory cytokines (IFN-γ and TNF-α) but stimulates production of the anti-inflammatory cytokine IL-10 (Fig. 4). Taken together, these findings suggest that M. bovis infects and modifies the cellular machinery of bovine monocytes to prolong their survival and facilitate subsequent systemic distribution.
Inhibition of host cell apoptosis is gaining currency as a strategy for bacterial survival and pathogenesis (reviewed in reference 46). To inhibit apoptosis, three general mechanisms, which are likely not mutually exclusive, have been presented in various experimental models with bacterial pathogens. These mechanisms are the protection of the mitochondria and prevention of cytochrome c release, as observed for epithelial cells infected with Chlamydia (47–49) and Neisseria species; activation of cellular survival pathways (e.g., NF-κB) by various bacteria (46); and interaction of bacterial components with caspase proteases, as observed for Shigella flexneri (50) and Legionella pneumophila (51, 52). In our case, it appears that M. bovis employs a combination of these strategies to inhibit monocyte apoptosis.
The results of our study seem to contradict those of other studies, whereby mycoplasmas have been shown to either cause apoptosis or increase susceptibility of host cells to apoptosis (53–56). Those studies used mouse and human immortalized T and B cell lines (55), mouse splenic T cells (54), or freshly isolated PBMC (56). Quite likely, those reports may reflect the effect of mycoplasmas on these specific cell types but not on freshly isolated bovine monocytes. More recently, we have reported that M. bovis infection of bovine PBMC did not induce apoptosis and in fact appeared to prolong the longevity of these cells (11). Therefore, we conclude that the type of cell involved may define M. bovis interactions with host cells, although further experimentation is required to unravel how M. bovis modulates different lymphocyte subsets. One would also argue that in the face of the phenotypic plasticity displayed by mycoplasmas, the inhibition of apoptosis in our case might be strain specific rather than an attribute of M. bovis. However, we have tested this phenomenon in three other M. bovis isolates (2 from cattle and 1 from North American bison) and have observed results similar to those obtained from the Mb1 isolate (data not shown). While the relatively high level (20%) of constitutive death in untreated monocytes might be unexpected, we could not find data in the literature describing the in vitro life span of untreated bovine monocytes. Nonetheless, high levels (77%) of constitutive cell death in untreated human peripheral blood monocytes after 48 h despite the use of 10% fetal calf serum were reported previously (57). Since we also used 10% fetal calf serum in our medium, we postulate that the constitutive cell death observed is altogether not surprising and would be expected in unstimulated cells.
NF-κB is a crucial and ubiquitous transcriptional factor that regulates intercellular signaling, cell growth, cell migration, as well as augmentation of primary responses to pathogens (58, 59). In host eukaryotic cells, the NF-κB family of transcription factors is activated by numerous viral and bacterial products to induce the expression of early-response genes that subsequently shape the course of the overall response to the offending stimuli (60–62). Inactive NF-κB is retained in the cytoplasm by binding to inhibitor of NF-κB kinase subunit beta (IKK-β) proteins (63). Upon activation, IKK-β is phosphorylated, which facilitates its ubiquitination by a specific E3 ubiquitin complex (64). The polyubiquitination of IKK-β marks NF-κB for rapid proteasomal degradation, leaving NF-κB free to translocate into the nucleus and initiate gene expression (32, 65). Activation of NF-κB has been reported for other Mycoplasma species, including M. genitalium, M. fermentans, and M. pneumoniae (66–68). In those reports, lipoproteins from these mycoplasmas activated NF-κB following stimulation of TLR-2 and TLR-6 in in vitro cell models. It is likely that M. bovis activates NF-κB through the same pathway. The role of NF-κB in rescuing cells from apoptosis induced by TNF-α, radiation, and chemotherapy was described previously (69–71). Our results are consistent with observations for the intracellular Gram-negative bacterium Rickettsia rickettsii, whose in vitro invasion of vascular endothelial cells activates NF-κB, thus inhibiting apoptosis of host cells to enable the cell to persist as a site for bacterial replication (72).
The caspase family of proteases coordinates many aspects of apoptosis (34, 35). Caspases are produced and retained intracellularly as inert proenzymes, each with two subunits, a large subunit and a small subunit, with a variable amino-terminal prodomain (36). Activation of caspases is achieved by the loss of the prodomain by catalytic elimination of a carboxyl-terminal aspartate and subsequent heterodimerization of the large and small subunits into active enzymes (35, 36). Caspases can be divided into two classes: initiator caspases (caspases 8 and 9) act upstream of the effector caspases (caspases 3, 6, and 7) and respond to the TNF receptor family or Fas (caspase 8) (37, 38) or cytochrome c release following mitochondrial damage (caspase 9) (36). Once activated by initiator caspases, effector caspases cause cellular disintegration by cleavage of other death substrates and nucleoproteins, with cellular DNA fragmentation and cell death as a result (36). In the case of Rickettsia rickettsii, the blockage of NF-κB activation in infected endothelial cells results in apoptosis that is mediated by the initiator caspase 8 or 9 and the effector caspase 3 (58). In our case, diminished activation of caspase 9, but not caspase 3, was observed in M. bovis-infected monocytes. Upon initial observation, our results appear contradictory, since caspase 3 is an effector protease that exerts its effects downstream of caspase 9 and would be expected to be inactive upon inhibition of apoptosis (73). Nonetheless, whereas caspase 9 initiates the release of cytochrome c following mitochondrial damage, the existence of caspase 3 activation without the involvement of mitochondrial cytochrome c release has been reported (74). In this regard, those authors have shown the coexistence of parallel apoptotic pathways in mammalian cells, which are preferentially activated in a cell type- and stimulus-specific manner. Therefore, these discrepancies may reflect the distinct roles of multiple apoptotic pathways. In the present study, spontaneous apoptosis was observed in untreated monocytes (Fig. 2). This is in agreement with data from previous studies in mice and humans, which indicated that undifferentiated monocytes undergo spontaneous apoptosis in vitro (75, 76). Furthermore, the authors of those studies showed that this spontaneous apoptosis is mediated by Fas-FasL interactions, which cause apoptosis through the extrinsic pathway. On the other hand, staurosporine (STS) induces apoptosis through both caspase-dependent and caspase-independent mechanisms (77). Therefore, it is important to note that our data are not a direct comparison of apoptotic mechanisms between STS-treated monocytes or untreated monocytes on the one hand and M. bovis-infected monocytes on the other. However, abrogation of both spontaneous and STS/TNF-α-induced apoptosis by M. bovis suggests the involvement of multiple pathways that ensure prolonged survival of M. bovis-infected monocytes in vitro.
The profile of cytokines produced by M. bovis-infected monocytes (Fig. 4) indicates that the bacterium induces the production of anti-inflammatory cytokines, i.e., IL-10, but rather inhibits the production of potent proinflammatory cytokines (IFN-γ and TNF-α). IL-10 exerts a range of effects on various cell types by blocking their production of proinflammatory cytokines and chemokines (78). IL-10 is also involved in suppression of IFN-γ and IL-12 (Th1 cytokines) to shift the adaptive immune response to a Th2 one dominated by antibody responses (79). Interestingly, whereas IL-10 is known to impair the antigen-presenting capacity of monocytes and macrophages through decreased expression levels of surface major histocompatibility complex class II (MHC-II) and other costimulatory molecules, in our case, M. bovis-infected monocytes exhibited enhanced expression of MHC-II molecules (data not shown). Some reports have demonstrated that M. bovis preferentially induces a host Th2 immunological response characterized by strong IgG1 responses that do not confer protection in experimental vaccines (6, 7). The absence of IFN-γ and TNF-α from M. bovis-infected monocytes agrees with those observations. Nonetheless, several authors have reported the in vitro production of IFN-γ in M. bovis-infected PBMC, and it may be necessary to determine which lymphocyte subsets are responsible for the production of this cytokine. Our findings also raise the critical question of whether M. bovis-infected monocytes are able to differentiate into mature macrophages/DCs or if the infection arrests this progression. This is especially important given the previous demonstration that IL-10 prevents differentiation of monocytes into DCs but promotes their maturation into macrophages (80).
Previous work has implicated inhibition of lymphocyte proliferation (11, 81–83) as well as induction of apoptosis (53–56) as important strategies employed by M. bovis to evade host immunity. The current findings extend our understanding of the mechanisms employed by M. bovis to evade host innate immunity and establish itself systemically. Our findings also highlight the need to study how M. bovis interacts with distinct lymphocyte subsets, as it appears that the bacterium displays marked versatility to selectively exert different effects on distinct cell populations. For instance, it will be interesting to determine how M. bovis interacts with alveolar macrophages, as they are the principal immune-related cell population that initially comes into contact with respiratory epithelial surfaces. In conclusion, we have provided novel evidence that M. bovis prolongs the life of bovine monocytes in vitro and does not induce the production of IFN-γ and TNF-α but induces the release of IL-10. These findings could explain the systemic pathogenesis of a host of other mycoplasmal diseases and provide potential targets for intervention against these diseases.
ACKNOWLEDGMENTS
This work was made possible by financial support from the Saskatchewan Agriculture Development Fund (ADF), the Advancing Agriculture and Agri-Food (ACCAF) Program, and the Alberta Livestock and Meat Agency (ALMA).
We acknowledge Natasha Arsic and Yurij Popowych for their assistance with the fluorescence-activated cell sorter assays and Don Wilson and the whole VIDO Animal Care Unit for their invaluable help in obtaining cattle blood samples.
Footnotes
Published ahead of print 14 October 2013
This work was published with permission of the Director of VIDO as journal series number 662.
REFERENCES
- 1.Arcangioli MA, Duet A, Meyer G, Dernburg A, Bezille P, Poumarat F, Le Grand D. 2008. The role of Mycoplasma bovis in bovine respiratory disease outbreaks in veal calf feedlots. Vet. J. 177:89–93. 10.1016/j.tvjl.2007.03.008 [DOI] [PubMed] [Google Scholar]
- 2.Nicholas RA, Ayling RD. 2003. Mycoplasma bovis: disease, diagnosis, and control. Res. Vet. Sci. 74:105–112. 10.1016/S0034-5288(02)00155-8 [DOI] [PubMed] [Google Scholar]
- 3.Snowder GD, Van Vleck LD, Cundiff LV, Bennett GL. 2006. Bovine respiratory disease in feedlot cattle: environmental, genetic, and economic factors. J. Anim. Sci. 84:1999–2008. 10.2527/jas.2006-046 [DOI] [PubMed] [Google Scholar]
- 4.Maunsell FP, Donovan GA. 2009. Mycoplasma bovis infections in young calves. Vet. Clin. North Am. Food Anim. Pract. 25:139–177, vii. 10.1016/j.cvfa.2008.10.011 [DOI] [PubMed] [Google Scholar]
- 5.Howard CJ, Thomas LH, Parsons KR. 1987. Immune response of cattle to respiratory mycoplasmas. Vet. Immunol. Immunopathol. 17:401–412. 10.1016/0165-2427(87)90157-7 [DOI] [PubMed] [Google Scholar]
- 6.Vanden Bush TJ, Rosenbusch RF. 2003. Characterization of the immune response to Mycoplasma bovis lung infection. Vet. Immunol. Immunopathol. 94:23–33. 10.1016/S0165-2427(03)00056-4 [DOI] [PubMed] [Google Scholar]
- 7.Hermeyer K, Buchenau I, Thomasmeyer A, Baum B, Spergser J, Rosengarten R, Hewicker-Trautwein M. 2012. Chronic pneumonia in calves after experimental infection with Mycoplasma bovis strain 1067: characterization of lung pathology, persistence of variable surface protein antigens and local immune response. Acta Vet. Scand. 54:9. 10.1186/1751-0147-54-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda T, Shibahara T, Kimura K, Wada Y, Sato K, Imada Y, Ishikawa Y, Kadota K. 2003. Mycoplasma bovis-associated suppurative otitis media and pneumonia in bull calves. J. Comp. Pathol. 129:100–110. 10.1016/S0021-9975(03)00009-4 [DOI] [PubMed] [Google Scholar]
- 9.Haines DM, Martin KM, Clark EG, Jim GK, Janzen ED. 2001. The immunohistochemical detection of Mycoplasma bovis and bovine viral diarrhea virus in tissues of feedlot cattle with chronic, unresponsive respiratory disease and/or arthritis. Can. Vet. J. 42:857–860 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1476660/ [PMC free article] [PubMed] [Google Scholar]
- 10.Adegboye DS, Hallbur PG, Cavanaugh DL, Werdin RE, Chase CC, Miskimins DW, Rosenbusch RF. 1995. Immunohistochemical and pathological study of Mycoplasma bovis-associated lung abscesses in calves. J. Vet. Diagn. Invest. 7:333–337. 10.1177/104063879500700306 [DOI] [PubMed] [Google Scholar]
- 11.van der Merwe J, Prysliak T, Perez-Casal J. 2010. Invasion of bovine peripheral blood mononuclear cells and erythrocytes by Mycoplasma bovis. Infect. Immun. 78:4570–4578. 10.1128/IAI.00707-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. 10.1126/science.1075762 [DOI] [PubMed] [Google Scholar]
- 13.Cohen P. 2002. Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1:309–315. 10.1038/nrd773 [DOI] [PubMed] [Google Scholar]
- 14.Kemp BE, Graves DJ, Benjamini E, Krebs EG. 1977. Role of multiple basic residues in determining the substrate specificity of cyclic AMP-dependent protein kinase. J. Biol. Chem. 252:4888–4894 [PubMed] [Google Scholar]
- 15.Zetterqvist O, Ragnarsson U, Humble E, Berglund L, Engstrom L. 1976. The minimum substrate of cyclic AMP-stimulated protein kinase, as studied by synthetic peptides representing the phosphorylatable site of pyruvate kinase (type L) of rat liver. Biochem. Biophys. Res. Commun. 70:696–703. 10.1016/0006-291X(76)90648-3 [DOI] [PubMed] [Google Scholar]
- 16.Diks SH, Kok K, O'Toole T, Hommes DW, van Dijken P, Joore J, Peppelenbosch MP. 2004. Kinome profiling for studying lipopolysaccharide signal transduction in human peripheral blood mononuclear cells. J. Biol. Chem. 279:49206–49213. 10.1074/jbc.M405028200 [DOI] [PubMed] [Google Scholar]
- 17.Lowenberg M, Tuynman J, Bilderbeek J, Gaber T, Buttgereit F, van Deventer S, Peppelenbosch M, Hommes D. 2005. Rapid immunosuppressive effects of glucocorticoids mediated through Lck and Fyn. Blood 106:1703–1710. 10.1182/blood-2004-12-4790 [DOI] [PubMed] [Google Scholar]
- 18.van Baal JW, Diks SH, Wanders RJ, Rygiel AM, Milano F, Joore J, Bergman JJ, Peppelenbosch MP, Krishnadath KK. 2006. Comparison of kinome profiles of Barrett's esophagus with normal squamous esophagus and normal gastric cardia. Cancer Res. 66:11605–11612. 10.1158/0008-5472.CAN-06-1370 [DOI] [PubMed] [Google Scholar]
- 19.Jalal S, Arsenault R, Potter AA, Babiuk LA, Griebel PJ, Napper S. 2009. Genome to kinome: species-specific peptide arrays for kinome analysis. Sci. Signal. 2:pl1. 10.1126/scisignal.254pl1 [DOI] [PubMed] [Google Scholar]
- 20.Arsenault RJ, Jalal S, Babiuk LA, Potter A, Griebel PJ, Napper S. 2009. Kinome analysis of Toll-like receptor signaling in bovine monocytes. J. Recept. Signal Transduct. Res. 29:299–311. 10.3109/10799890903295127 [DOI] [PubMed] [Google Scholar]
- 21.Arsenault RJ, Li Y, Bell K, Doig K, Potter A, Griebel PJ, Kusalik A, Napper S. 2012. Mycobacterium avium subsp. paratuberculosis inhibits gamma interferon-induced signaling in bovine monocytes: insights into the cellular mechanisms of Johne's disease. Infect. Immun. 80:3039–3048. 10.1128/IAI.00406-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arsenault RJ, Li Y, Maattanen P, Scruten E, Doig K, Potter A, Griebel P, Kusalik A, Napper S. 2013. Altered Toll-like receptor 9 signaling in Mycobacterium avium subsp. paratuberculosis-infected bovine monocytes reveals potential therapeutic targets. Infect. Immun. 81:226–237. 10.1128/IAI.00785-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auffray C, Sieweke MH, Geissmann F. 2009. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27:669–692. 10.1146/annurev.immunol.021908.132557 [DOI] [PubMed] [Google Scholar]
- 24.Tacke F, Randolph GJ. 2006. Migratory fate and differentiation of blood monocyte subsets. Immunobiology 211:609–618. 10.1016/j.imbio.2006.05.025 [DOI] [PubMed] [Google Scholar]
- 25.Perez-Casal J, Prysliak T. 2007. Detection of antibodies against the Mycoplasma bovis glyceraldehyde-3-phosphate dehydrogenase protein in beef cattle. Microb. Pathog. 43:189–197. 10.1016/j.micpath.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Arsenault RJ, Trost B, Slind J, Griebel P, Napper S, Kusalik A. 2012. A systematic approach for approach analysis of peptide array kinome data. Sci. Signal. 5:pl2. 10.1126/scisignal.2002429 [DOI] [PubMed] [Google Scholar]
- 27.Maattanen P, Trost B, Scruten E, Potter A, Kusalik A, Griebel P, Napper S. 2013. Divergent immune responses to Mycobacterium avium subsp. paratuberculosis infection correlate with kinome responses at the site of intestinal infection. Infect. Immun. 81:2861–2872. 10.1128/IAI.00339-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan TH, Shah N, Lo R, Naseer M, Que J, Yau M, Acab M, Tulpan D, Whiteside MD, Chikatamarla A, Mah B, Munzner T, Hokamp K, Hancock RE, Brinkman FS. 2008. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 4:218. 10.1038/msb.2008.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baca-Estrada ME, Godson DL, Hughes HP, Van Donkersgoed J, Van Kessel A, Harland R, Shuster DE, Daley M, Babiuk LA. 1995. Effect of recombinant bovine interleukin-1 beta on viral/bacterial pneumonia in cattle. J. Interferon Cytokine Res. 15:431–439. 10.1089/jir.1995.15.431 [DOI] [PubMed] [Google Scholar]
- 30.Ellis JA, Godson D, Campos M, Sileghem M, Babiuk LA. 1993. Capture immunoassay for ruminant tumor necrosis factor-alpha: comparison with bioassay. Vet. Immunol. Immunopathol. 35:289–300. 10.1016/0165-2427(93)90040-B [DOI] [PubMed] [Google Scholar]
- 31.Campbell KJ, Perkins ND. 2006. Regulation of NF-kappaB function. Biochem. Soc. Symp. 2006:165–180 [DOI] [PubMed] [Google Scholar]
- 32.Karin M, Lin A. 2002. NF-kappaB at the crossroads of life and death. Nat. Immunol. 3:221–227. 10.1038/ni0302-221 [DOI] [PubMed] [Google Scholar]
- 33.Dev A, Iyer S, Razani B, Cheng G. 2011. NF-kappaB and innate immunity. Curr. Top. Microbiol. Immunol. 349:115–143. 10.1007/82_2010_102 [DOI] [PubMed] [Google Scholar]
- 34.Shi Y. 2002. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9:459–470. 10.1016/S1097-2765(02)00482-3 [DOI] [PubMed] [Google Scholar]
- 35.Thornberry NA, Lazebnik Y. 1998. Caspases: enemies within. Science 281:1312–1316. 10.1126/science.281.5381.1312 [DOI] [PubMed] [Google Scholar]
- 36.Riedl SJ, Shi Y. 2004. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 5:897–907. 10.1038/nrm1496 [DOI] [PubMed] [Google Scholar]
- 37.Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, Yonehara S. 1999. The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature 398:777–785. 10.1038/19709 [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Du F, Wang X. 2008. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133:693–703. 10.1016/j.cell.2008.03.036 [DOI] [PubMed] [Google Scholar]
- 39.Krebs DL, Hilton DJ. 2001. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19:378–387. 10.1634/stemcells.19-5-378 [DOI] [PubMed] [Google Scholar]
- 40.Song MM, Shuai K. 1998. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J. Biol. Chem. 273:35056–35062. 10.1074/jbc.273.52.35056 [DOI] [PubMed] [Google Scholar]
- 41.Morita Y, Naka T, Kawazoe Y, Fujimoto M, Narazaki M, Nakagawa R, Fukuyama H, Nagata S, Kishimoto T. 2000. Signals transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor alpha-induced cell death in fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 97:5405–5410. 10.1073/pnas.090084797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Y, Chen D, Tarcsafalvi A, Su R, Qin L, Bromberg JS. 2003. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J. Immunol. 170:1383–1391 [DOI] [PubMed] [Google Scholar]
- 43.Prysliak T, Van der Merwe J, Lawman Z, Wilson D, Townsend H, van Drunen Littel-van den Hurk S, Perez-Casal J. 2011. Respiratory disease caused by Mycoplasma bovis is enhanced by exposure to bovine herpes virus 1 (BHV-1) and not to bovine viral diarrhoea virus (BVDV) type 2. Can. Vet. J. 52:1195–1202 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3196011/ [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett RH, Jasper DE. 1977. Nasal prevalence of Mycoplasma bovis and IHA titers in young dairy animals. Cornell Vet. 67:361–373 [PubMed] [Google Scholar]
- 45.Punyapornwithaya V, Fox LK, Hancock DD, Gay JM, Alldredge JR. 2010. Association between an outbreak strain causing Mycoplasma bovis mastitis and its asymptomatic carriage in the herd: a case study from Idaho, USA. Prev. Vet. Med. 93:66–70. 10.1016/j.prevetmed.2009.08.008 [DOI] [PubMed] [Google Scholar]
- 46.Faherty CS, Maurelli AT. 2008. Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol. 16:173–180. 10.1016/j.tim.2008.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, Ying S, Hacker G. 2004. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J. Exp. Med. 200:905–916. 10.1084/jem.20040402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. 2006. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 281:31495–31501. 10.1074/jbc.M602796200 [DOI] [PubMed] [Google Scholar]
- 49.Xiao Y, Zhong Y, Greene W, Dong F, Zhong G. 2004. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect. Immun. 72:5470–5474. 10.1128/IAI.72.9.5470-5474.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark CS, Maurelli AT. 2007. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect. Immun. 75:2531–2539. 10.1128/IAI.01866-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. 2007. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell. Microbiol. 9:246–264. 10.1111/j.1462-5822.2006.00785.x [DOI] [PubMed] [Google Scholar]
- 52.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U. S. A. 103:18745–18750. 10.1073/pnas.0609012103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paddenberg R, Weber A, Wulf S, Mannherz HG. 1998. Mycoplasma nucleases able to induce internucleosomal DNA degradation in cultured cells possess many characteristics of eukaryotic apoptotic nucleases. Cell Death Differ. 5:517–528. 10.1038/sj.cdd.4400380 [DOI] [PubMed] [Google Scholar]
- 54.Shibata K, Watanabe T. 1997. Mycoplasma fermentans enhances concanavalin A-induced apoptosis of mouse splenic T cells. FEMS Immunol. Med. Microbiol. 17:103–109. 10.1111/j.1574-695X.1997.tb01002.x [DOI] [PubMed] [Google Scholar]
- 55.Sokolova IA, Vaughan AT, Khodarev NN. 1998. Mycoplasma infection can sensitize host cells to apoptosis through contribution of apoptotic-like endonuclease(s). Immunol. Cell Biol. 76:526–534. 10.1046/j.1440-1711.1998.00781.x [DOI] [PubMed] [Google Scholar]
- 56.Vanden Bush TJ, Rosenbusch RF. 2002. Mycoplasma bovis induces apoptosis of bovine lymphocytes. FEMS Immunol. Med. Microbiol. 32:97–103. 10.1111/j.1574-695X.2002.tb00540.x [DOI] [PubMed] [Google Scholar]
- 57.Heidenreich S, Schmidt M, August C, Cullen P, Rademaekers A, Pauels HG. 1997. Regulation of human monocyte apoptosis by the CD14 molecule. J. Immunol. 159:3178–3188 [PubMed] [Google Scholar]
- 58.Joshi SG, Francis CW, Silverman DJ, Sahni SK. 2003. Nuclear factor kappa B protects against host cell apoptosis during Rickettsia rickettsii infection by inhibiting activation of apical and effector caspases and maintaining mitochondrial integrity. Infect. Immun. 71:4127–4136. 10.1128/IAI.71.7.4127-4136.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tato CM, Hunter CA. 2002. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infect. Immun. 70:3311–3317. 10.1128/IAI.70.7.3311-3317.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofer S, Rescigno M, Granucci F, Citterio S, Francolini M, Ricciardi-Castagnoli P. 2001. Differential activation of NF-kappa B subunits in dendritic cells in response to Gram-negative bacteria and to lipopolysaccharide. Microbes Infect. 3:259–265. 10.1016/S1286-4579(01)01378-8 [DOI] [PubMed] [Google Scholar]
- 61.Li Q, Verma IM. 2002. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2:725–734. 10.1038/nri910 [DOI] [PubMed] [Google Scholar]
- 62.Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 22:2552–2560. 10.1093/emboj/cdg267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilmore TD. 2006. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25:6680–6684. 10.1038/sj.onc.1209954 [DOI] [PubMed] [Google Scholar]
- 64.Karin M, Delhase M. 2000. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin. Immunol. 12:85–98. 10.1006/smim.2000.0210 [DOI] [PubMed] [Google Scholar]
- 65.Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-kappaB activity. Annu. Rev. Immunol. 18:621–663. 10.1146/annurev.immunol.18.1.621 [DOI] [PubMed] [Google Scholar]
- 66.Into T, Kiura K, Yasuda M, Kataoka H, Inoue N, Hasebe A, Takeda K, Akira S, Shibata K. 2004. Stimulation of human Toll-like receptor (TLR) 2 and TLR6 with membrane lipoproteins of Mycoplasma fermentans induces apoptotic cell death after NF-kappa B activation. Cell. Microbiol. 6:187–199. 10.1046/j.1462-5822.2003.00356.x [DOI] [PubMed] [Google Scholar]
- 67.McGowin CL, Ma L, Martin DH, Pyles RB. 2009. Mycoplasma genitalium-encoded MG309 activates NF-kappaB via Toll-like receptors 2 and 6 to elicit proinflammatory cytokine secretion from human genital epithelial cells. Infect. Immun. 77:1175–1181. 10.1128/IAI.00845-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimizu T, Kida Y, Kuwano K. 2005. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF-kappa B through TLR1, TLR2, and TLR6. J. Immunol. 175:4641–4646 [DOI] [PubMed] [Google Scholar]
- 69.Beg AA, Baltimore D. 1996. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274:782–784. 10.1126/science.274.5288.782 [DOI] [PubMed] [Google Scholar]
- 70.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. 1996. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274:787–789. 10.1126/science.274.5288.787 [DOI] [PubMed] [Google Scholar]
- 71.Wang CY, Mayo MW, Baldwin AS., Jr 1996. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274:784–787. 10.1126/science.274.5288.784 [DOI] [PubMed] [Google Scholar]
- 72.Clifton DR, Goss RA, Sahni SK, van Antwerp D, Baggs RB, Marder VJ, Silverman DJ, Sporn LA. 1998. NF-kappa B-dependent inhibition of apoptosis is essential for host cell survival during Rickettsia rickettsii infection. Proc. Natl. Acad. Sci. U. S. A. 95:4646–4651. 10.1073/pnas.95.8.4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Porter AG, Janicke RU. 1999. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 10.1038/sj.cdd.4400476 [DOI] [PubMed] [Google Scholar]
- 74.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. 1998. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94:339–352. 10.1016/S0092-8674(00)81477-4 [DOI] [PubMed] [Google Scholar]
- 75.Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, Liles WC. 1997. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J. Exp. Med. 185:1511–1516. 10.1084/jem.185.8.1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perlman H, Pagliari LJ, Nguyen N, Bradley K, Liu H, Pope RM. 2001. The Fas-FasL death receptor and PI3K pathways independently regulate monocyte homeostasis. Eur. J. Immunol. 31:2421–2430. [DOI] [PubMed] [Google Scholar]
- 77.Belmokhtar CA, Hillion J, Segal-Bendirdjian E. 2001. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20:3354–3362. 10.1038/sj.onc.1204436 [DOI] [PubMed] [Google Scholar]
- 78.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. 10.1146/annurev.immunol.19.1.683 [DOI] [PubMed] [Google Scholar]
- 79.Couper KN, Blount DG, Riley EM. 2008. IL-10: the master regulator of immunity to infection. J. Immunol. 180:5771–5777 [DOI] [PubMed] [Google Scholar]
- 80.Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, Mantovani A. 1998. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur. J. Immunol. 28:359–369. [DOI] [PubMed] [Google Scholar]
- 81.Boothby JT, Jasper DE, Zinkl JG, Thomas CB, Dellinger JD. 1983. Prevalence of mycoplasmas and immune responses to Mycoplasma bovis in feedlot calves. Am. J. Vet. Res. 44:831–838 [PubMed] [Google Scholar]
- 82.Thomas CB, Mettler J, Sharp P, Jensen-Kostenbader J, Schultz RD. 1990. Mycoplasma bovis suppression of bovine lymphocyte response to phytohemagglutinin. Vet. Immunol. Immunopathol. 26:143–155. 10.1016/0165-2427(90)90063-X [DOI] [PubMed] [Google Scholar]
- 83.Vanden Bush TJ, Rosenbusch RF. 2004. Characterization of a lympho-inhibitory peptide produced by Mycoplasma bovis. Biochem. Biophys. Res. Commun. 315:336–341. 10.1016/j.bbrc.2004.01.063 [DOI] [PubMed] [Google Scholar]