

Abstract
Histological and clinical investigations describe late stages of Legionnaires' disease but cannot characterize early events of human infection. Cellular or rodent infection models lack the complexity of tissue or have nonhuman backgrounds. Therefore, we developed and applied a novel model for Legionella pneumophila infection comprising living human lung tissue. We stimulated lung explants with L. pneumophila strains and outer membrane vesicles (OMVs) to analyze tissue damage, bacterial replication, and localization as well as the transcriptional response of infected tissue. Interestingly, we found that extracellular adhesion of L. pneumophila to the entire alveolar lining precedes bacterial invasion and replication in recruited macrophages. In contrast, OMVs predominantly bound to alveolar macrophages. Specific damage to septa and epithelia increased over 48 h and was stronger in wild-type-infected and OMV-treated samples than in samples infected with the replication-deficient, type IVB secretion-deficient DotA− strain. Transcriptome analysis of lung tissue explants revealed a differential regulation of 2,499 genes after infection. The transcriptional response included the upregulation of uteroglobin and the downregulation of the macrophage receptor with collagenous structure (MARCO). Immunohistochemistry confirmed the downregulation of MARCO at sites of pathogen-induced tissue destruction. Neither host factor has ever been described in the context of L. pneumophila infections. This work demonstrates that the tissue explant model reproduces realistic features of Legionnaires' disease and reveals new functions for bacterial OMVs during infection. Our model allows us to characterize early steps of human infection which otherwise are not feasible for investigations.
INTRODUCTION
Histopathologically, Legionnaires' disease, caused by the Gram-negative bacterium Legionella pneumophila, is an acute fibrinopurulent pneumonia. Since the first documented outbreak of Legionnaires' disease in 1976, several autopsy series have been published (1). Samples from patients who died from L. pneumophila pneumonia exhibit a massive infiltration of neutrophils and macrophages into the alveoli and destruction of alveolar septa. Moreover, the alveolar epithelium shows sloughs, and inflammatory cells exhibit intense necrosis. L. pneumophila is present mainly in alveoli and tends to cluster inside macrophages. In late infection stages, bacteria disseminate to the patient's spleen, kidneys, bone marrow, and lymph nodes (1–4).
Different models have been established to analyze specific aspects of infection. Besides human monocellular systems such as macrophages and epithelial cells, protozoa such as Acanthamoeba castellanii, Hartmannella vermiformis, and Dictyostelium discoideum were used to study the cellular and molecular pathogenicity of L. pneumophila (5–9). These studies revealed that L. pneumophila primarily enters phagocytes and resides within a unique membrane-bound compartment termed the Legionella-containing vacuole (LCV). The establishment of this replication niche requires the translocation of about 300 effector proteins into the host cell via a functional Dot/Icm type IV secretion (10–12). Studying transcriptional responses of L. pneumophila-infected macrophages and D. discoideum vegetative cells also shed light on the cellular mechanisms of Legionnaires' disease (13–16). Moreover, proteomic approaches were shown to be powerful tools to characterize both sides of the host-pathogen interaction (17–19). Mammalian models such as guinea pigs, mice, rhesus monkeys, and marmosets were used to address immunological, pathological, and pharmacological questions (20–22). Despite providing enormous progress in the knowledge about mechanisms of L. pneumophila infections, each of the current infection models has intrinsic limitations. Cell culture assays lack the complex interaction networks between the specialized cell types and extracellular components in the human lung. Guinea pig infections require intraperitoneal or intratracheal inoculation techniques, and owing to a different genetic and immunological background, the adequacy and transferability to humans can be questioned.
Given the different model-immanent limitations, numerous intra- and extracellular interactions of L. pneumophila factors with human lung tissue structures remain unknown. For example, early infection events appear to be underexplored, since histopathology studies were performed postmortem. Even conspicuous subcellular structures, such as the abundant outer membrane vesicles (OMVs) shed by L. pneumophila, have not yet been investigated in human lung tissue. OMVs contain large amounts of degradative enzymes and other virulence-related proteins, which could execute destructive and inflammatory activities (23, 24). The spectrum of underexplored interactions also includes pathogen-induced effects on the extracellular matrix (ECM), transcriptomic and proteomic responses of the pathogen and the infected tissue, and other, yet-unrecognized molecular processes which occur only in the context of human tissues.
In this study, we thoroughly analyzed L. pneumophila-infected human lung tissue explants (HLTEs) at multiple levels. We characterized the pathological features of infected tissues and determined the localization and growth kinetics of L. pneumophila wild-type and mutant strains in time course experiments with HLTEs. Moreover, we analyzed the contribution of OMVs to tissue destruction and demonstrated that the transcriptional response of L. pneumophila-infected HLTEs differs from previous results in monocellular models.
MATERIALS AND METHODS
Ethics statement.
For human lung tissue explants, written informed consent was obtained and all procedures were performed according to German national guidelines and approved by the Ethik-Kommission der Medizinischen Fakultät der Universität Lübeck (03/153).
Bacterial strains and culture.
L. pneumophila Corby and a DotA-negative strain (25, 26) (kindly provided by Antje Flieger, Robert-Koch-Institut, Wernigerode, Germany) were cultivated in yeast extract broth (YEB) (with 20 μg/ml kanamycin for the mutant) to the early stationary phase. For infection, the bacterial suspension was diluted to 107 bacteria/ml in RPMI 1640 (Gibco, Darmstadt, Germany) with 10% fetal calf serum (FCS), 20 mM HEPES, and 1 mM sodium pyruvate. OMVs were isolated from early-stationary-phase L. pneumophila cultures as described previously (27) and diluted to 100 μg/ml (total protein) in RPMI with supplements.
Human lung tissue explants and assessment of bacterial replication.
Tumor-free pulmonary tissue samples of approximately 1 cm3 were obtained from surgery patients as described previously (28). Samples were infected with the respective L. pneumophila strain and incubated at 37°C and 5% CO2 for up to 48 h. Microscopic inspection of untreated samples at different time points ensured tissue vitality (see below).
For CFU determination, triplicate samples from eight donors were infected. At the indicated time points, samples were weighed and homogenized in phosphate-buffered saline (PBS). Dilutions were plated on buffered charcoal-yeast extract (BCYE) and incubated at 37°C with 5% CO2 for 4 days. Extracellular replication of L. pneumophila in HLTEs was excluded by control experiments which showed that acellular tissue homogenate or tissue supernatant does not support bacterial growth. The CFU/g of tissue were determined; means and standard deviations of results for samples were compared by using Student's t test.
Tissue processing and histology analysis.
Tissue samples were fixed with the HEPES-glutamic acid organic solvent protection effect (HOPE) technique (DCS Diagnostics, Hamburg, Germany) (29). Briefly, samples were incubated in HOPE solution I at 4°C for 18 h and dehydrated in acetone at 4°C for 6 h. After overnight incubation in paraffin at 54°C, the samples were embedded in paraffin and stored at 4°C. Tissue blocks were cut on a microtome and deparaffinized as described previously (30). L. pneumophila was visualized by immunostaining with the anti-Mip antibody 2D8 (31) (diluted 1:50) or an anti-major outer membrane protein (anti-MOMP) antibody (diluted 1:100) (kindly provided by Joe Vogel, Washington University, St. Louis, MO, USA). Human alveolar epithelial cells (AEC) were visualized by immunostaining of Aquaporin 5 (diluted 1:100, clone EPR3747; Abcam, Cambridge, United Kingdom) and human alveolar macrophages by anti-CD68 (diluted 1:200; Dako Cytomation, Glostrup, Denmark). For detection, a horseradish peroxidase (HRP) polymer system (Zytomed Systems, Berlin, Germany) was applied according to the manufacturer's instructions with aminoethylcarbazole (AEC) as the chromogen. Slides were counterstained with Mayer's hemalaun, dehydrated in a graded ethanol series, and mounted with Pertex (Medite, Burgdorf, Germany). Images were taken with a Lumenera Infinity 4 digital charge-coupled device (CCD) camera on a Leica DMLB microscope.
Characterization of histological damage and statistical analysis.
Using hematoxylin-eosin (H&E)-stained slides, a qualitative tissue damage score was set up based on three criteria: protein exudate in the alveoli, epithelial delamination, and alveolar septum destruction. Damage severity was graded as 0 (no damage), 1 (little damage, distributed infrequently), 2 (damage, distributed frequently), or 3 (severe damage as the dominating pattern) and summed. Statistical analyses were performed with GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Medians and interquartile ranges of damage scores were compared by the Mann-Whitney test with a Bonferroni-corrected confidence interval of 98.3% (32). Affected cell types and compartments were identified and validated by trained pathologists.
Transcriptome analysis.
RNA was isolated from HOPE-fixed tissue samples as described previously (30). RNA quality and integrity were analyzed with the Agilent RNA 6000 Nano Assay on a Bioanalyzer (Agilent, Böblingen, Germany). Transcriptome analysis was conducted according to the manufacturer's instructions (Agilent one-color microarray-based gene expression analysis, low-input QuickAmp labeling kit, version 6.6). Cy3-labeled DNA (1,650 ng) of each sample was hybridized on one Agilent human gene expression 4×44K microarray. Tiff images of hybridized samples were obtained by scanning with an Agilent SureScan microarray scanner, and raw gene expression data were extracted using Agilent feature extraction software (v11.0.1.1). For hierarchical clustering, fold change analysis, and Gene Ontology (GO) term analysis, Agilent GeneSpring software (v12.1) was used. Quantile-normalized gene expression data were computed from raw data with Direct Array software (OakLabs, Hennigsdorf, Germany) as described previously (33).
qRT-PCR.
To validate the transcriptional analysis, 450 to 650 ng of total RNA from L. pneumophila-infected lungs and matched medium controls was isolated and reverse transcribed into cDNA (Maxima first-strand cDNA synthesis kit for quantitative real-time PCR [qRT-PCR]; Thermo Scientific, Schwerte, Germany). A DNase I digest to remove genomic DNA was included during synthesis; intron-spanning primers were designed with the Universal Probe Library (Roche Applied Science, Mannheim, Germany) assay design center to target mRNA of human uteroglobin (forward, CTCACCCTGGTCACACTGG; reverse, CTGAAAGCTCGGGCAGAT [probe no. 84]) and RPL32 as the reference gene (forward, CCACCGTCCCTTCTCTCTT; reverse, GGGCTTCACAAGGGGTCT [probe no. 10]) with NM_000994.3 and BC004481.2 as the input sequences, respectively. Samples were initially denatured at 95°C for 5 min, followed by 45 cycles of amplification (10 s at 95°C and 30 s at 60°C) on a LightCycler 480II (Roche Applied Science) using the Light Cycler 2× Probes Master Mix (Roche Applied Science), 0.4 μM oligonucleotides, and 0.2 μM 6-carboxyfluorescein (FAM)-labeled hydrolysis probes (Universal Probe Library; Roche Applied Science) in a final volume of 10 μl. Negative controls without cDNA templates were included. To omit differences in the amplification reaction, pooled cDNA from the investigated samples was used to produce standard dilutions (5-fold) and included in every run individually to determine the reaction efficiency. Data were processed for advanced relative quantification within the LightCycler 480 software version 1.5. and are shown as the normalized ratio adjusted for reaction efficiency.
Detection of uteroglobin in human HLTE supernatants.
The supernatants of human HLTEs infected with L. pneumophila Corby or the DotA-negative strain as well as matched medium controls were used for the enzyme-linked immunosorbent assay (ELISA) determination of secreted uteroglobin (human uteroglobin ELISA DuoSet; R&D Systems, Wiesbaden, Germany).
RESULTS
L. pneumophila causes tissue damage in infected human lung tissue explants.
To characterize the histopathological effects of L. pneumophila on HLTEs, infected and noninfected tissue sections were fixed by using the HOPE technique and stained with hematoxylin-eosin (Fig. 1A to G). Generally, the predominant observed damage phenotypes were protein exudate in the alveolar lumen (Fig. 1A, *), delamination of alveolar epithelial cells (Fig. 1G, arrow), and disintegrating connective tissue (Fig. 1A). Moreover, dead macrophages, identified by nuclear breakup, could be observed in infected tissue samples (see Fig. 5E). The damage increased over the course of infection (Fig. 1E to G).
FIG 1.
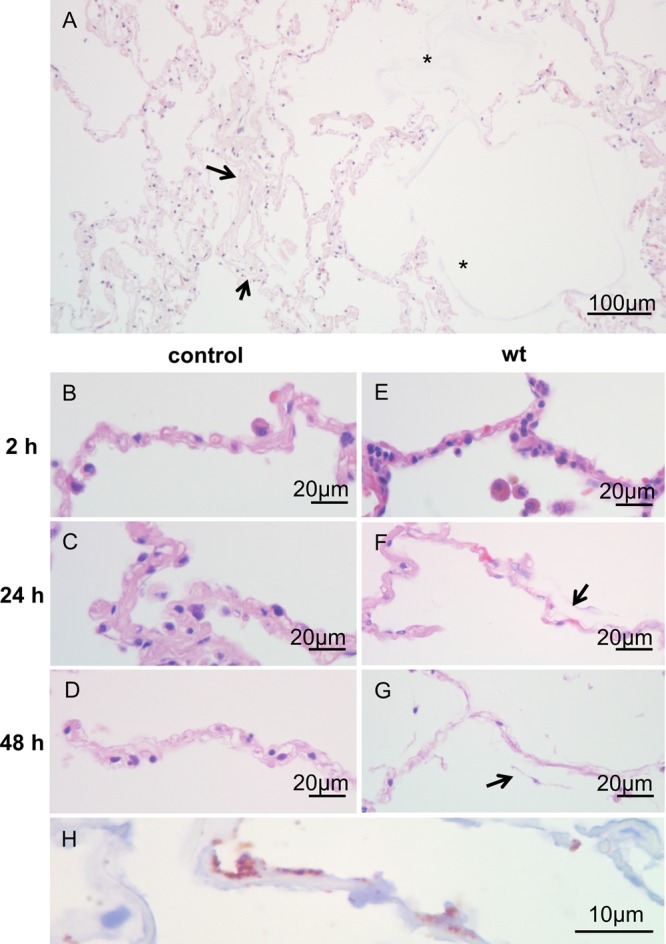
Progression of tissue damage caused by L. pneumophila within the human lung over time. (A) Hematoxylin-eosin-stained section of an HTLE challenged with L. pneumophila after 48 h at a magnification of ×20. Protein exudate into the alveolar lumen can be frequently observed in close proximity to sites of tissue damage (*). Collagen fibers within the alveolar septa appear loose (arrow), indicating decreasing septal integrity. (B, C, and D) Hematoxylin-eosin stains of uninfected control HTLEs at a magnification of ×40 after 2, 24, and 48 h of incubation, respectively. (E, F, and G) Hematoxylin-eosin stains of HTLEs challenged with L. pneumophila at 2, 24, and 48 h after infection, respectively. No damage could be observed at 2 h after infection (B and E). After 24 h, alveolar integrity is affected, shown by loosening of the collagen backbone in L. pneumophila-infected tissue (arrow in panel F) compared to uninfected controls (C). After 48 h of incubation, the integrity of uninfected tissue is moderately compromised (D), while L. pneumophila-infected tissue exhibits severe damage (G). Almost all septum-lining epithelial cells are delaminated (arrow in panel G), and the underlying scaffold appears to be disintegrated. (H) High-power magnification (×100) shows L. pneumophila targeted by immunohistochemistry with an anti-Mip antibody (red signal) at 48 h after infection. The pathogen adhered to alveolar septa and alveolar epithelial cells. At these sites, severe tissue damage was frequently observed. wt, wild type.
FIG 5.

L. pneumophila adheres extracellularly to the alveolar compartment and infects epithelial cells. Immunohistochemistry with an anti-Mip antibody on L. pneumophila-infected (A, B, and E) and uninfected (C and D) cells is shown (magnification, ×100). Colonization of alveolar epithelial cells and connective tissue was observed after 24 h (A) and 48 h (B). L. pneumophila-infected alveolar epithelial cells (asterisks in panels A and B) tended to form clusters over the course of stimulation (B). Furthermore, positive staining for L. pneumophila was frequently observed in dead alveolar macrophages (E).
No marked damage could be observed after 2 h of stimulation (Fig. 1B and E). After 24 h, the most abundant effect was the decreasing integrity of alveolar septa (arrow in Fig. 1F). At later time points, control samples were only slightly damaged, while L. pneumophila-infected HLTEs exhibited delamination of epithelial cells from the supporting connective tissue and shedding into the alveolar compartment (Fig. 1G, arrow). Furthermore, the normally compact ECM, including the collagen backbone of the remaining septa, appeared loose; even thicker connective tissue close to vessels or bronchioles disaggregated (Fig. 2C). Protein exudate was observed as a slightly reddish signal stained by H&E surrounding the alveolar septa (Fig. 1, *) and inside the alveolar lumen. Protein exudate was markedly more abundant in L. pneumophila-infected specimens than in controls and was repeatedly found adjacent to damaged tissue components such as affected septa (Fig. 1A, *). Importantly, L. pneumophila was frequently detected close to damaged tissue structures (Fig. 1H).
FIG 2.

Observed damage score of stimulated HLTEs compared to an uninfected control. (A) For each time point and condition, histological damage was classified in regard to protein exudate, delamination of epithelial cells, and integrity of alveolar septa on a scale from 0 to 3 and added up for each donor (n = 6 to 9 per condition and time point). Total damage score medians and interquartile ranges were calculated; values for infected or OMV-stimulated samples were compared to that for the uninfected control at the respective time point using the Mann-Whitney test with a Bonferroni-corrected confidence interval of 98.3%. *, P < 0.05; **, P < 0.01; ***, P < 0.001. n.s., not significant. (B to E) Hematoxylin-eosin-stained sections of HLTEs after 24 h of incubation. (B) Uninfected control; (C) L. pneumophila wild-type-infected HLTE; (D) sample infected with the DotA-deficient mutant; (E) sample incubated with 100 μg/ml OMVs. Infection with wild-type L. pneumophila leads to loosening of collagen backbones in alveolar septa and delamination of epithelial cells (C). The DotA-deficient mutant does not show a significant difference compared to the uninfected control (D). Incubation with OMVs resulted in severe tissue damage, including a widespread delamination of epithelial cells and loss of septal integrity (E).
Table 1 shows the distribution of damage scores of the individual phenotypes for each infection condition. In all categories, damage increased in the first 48 h, most strongly for wild-type-infected and OMV-stimulated samples (Fig. 2A to E). Incubation of tissue with L. pneumophila or isolated OMVs led to comparable damage scores, shown by a significant increase between 2 and 24 h compared to the control (Fig. 2A). Infection with a type IV secretion-deficient (DotA−) L. pneumophila mutant did not lead to strong epithelial delamination, and resulted in a total damage score comparable to that for uninfected controls. After 24 h, the damage score for wild-type-infected samples was significantly higher than that caused by DotA− (P = 0.0160; n = 7). Besides this difference and the aforementioned increase in total damage scores compared to those for uninfected controls, samples infected with wild-type or DotA− L. pneumophila or coincubated with OMVs did not differ significantly from each other at a given time point, assuming a confidence interval of 98.3%. To compare the tissue damage depending on the bacterial load, we challenged HLTEs of two different patients with 107, 108, and 109 bacteria/ml for 2 h to 48 h with wild-type L. pneumophila and the DotA-negative mutant. Higher concentrations (108 and 109 bacteria/ml) did not increase the tissue damage compared to that with 107 bacteria/ml (Fig. 3). The observed tissue damage caused by the DotA-negative strain was still less than that caused by the wild type, independent of the bacterial load and incubation time.
TABLE 1.
Damage score categories of HLTEsa
| Time (h) | Condition | Damage score (median ± interquartile range) |
|||
|---|---|---|---|---|---|
| Protein exudate | Epithelial delamination | Septal damage | Total damage | ||
| 2 | Control | 0 ± 1 | 0 ± 0 | 0 ± 0 | 1 ± 1 |
| Wild type | 1 ± 2 | 0 ± 1 | 0 ± 0 | 2 ± 2 | |
| DotA− | 1.5 ± 2.75 | 0 ± 0 | 0 ± 0 | 1.5 ± 1.75 | |
| OMVs | 0 ± 1 | 0 ± 0.5 | 0 ± 0.5 | 0 ± 2.5 | |
| 24 | Control | 1 ± 2 | 1 ± 1 | 0 ± 1 | 2 ± 2 |
| Wild type | 2 ± 2 | 1 ± 1.25 | 2 ± 1 | 5 ± 1.5 | |
| DotA− | 1 ± 1 | 1 ± 0 | 1 ± 0 | 2.5 ± 1.5 | |
| OMVs | 1 ± 1.5 | 2 ± 1 | 2 ± 1.5 | 6 ± 2 | |
| 48 | Control | 0.5 ± 1.25 | 0.5 ± 1 | 1 ± 0 | 3 ± 1.25 |
| Wild type | 2 ± 1 | 2 ± 1 | 3 ± 2 | 6 ± 1 | |
| DotA− | 1 ± 0.75 | 1 ± 0 | 1.5 ± 1 | 4 ± 0.75 | |
| OMVs | 1 ± 1 | 2.5 ± 2 | 2.5 ± 2 | 5.5 ± 1.75 | |
Human lung tissue explants (HTLEs) were incubated with medium, the indicated L. pneumophila strain, or 100 μg/ml outer membrane vesicles (OMVs) for the indicated time. Sample sizes were 6 to 9, depending on the infection condition. Per phenotype, the severity of damages was scaled from 0 to 3. The total damage score was calculated by adding the specific damage scores of each donor at a given time point.
FIG 3.

Comparison of tissue damage in HLTEs caused by different bacterial concentrations. HLTEs from two different patients were challenged with increasing concentrations of bacteria. (A) The damage score does not increase markedly with the number of infecting bacteria (median ± interquartile range [IQR]). (B and C) The tissue structure was visualized with the medium control (B) and increasing concentrations of the L. pneumophila wild-type and DotA-negative strains (C). Sections of paraffin-embedded HLTEs were stained with hematoxylin and eosin, and the damage score was evaluated. All images were taken at a magnification of ×40 at 48 h after infection.
L. pneumophila adheres to the alveolar lining and primarily infects alveolar macrophages.
In biopsy specimens from Legionnaires' disease patients, L. pneumophila is found predominantly in alveolar macrophages (1). In vitro data suggest that the pathogen also replicates within alveolar epithelial cells (6). In HTLEs, we detected L. pneumophila with anti-Mip and anti-MOMP antibodies. Clear signals were observed in alveolar macrophages (red color); the staining intensities varied depending on the expected antigen abundance (Fig. 4C and D). Interestingly, infected HLTEs displayed numerous L. pneumophila organisms adhering extracellularly to the alveolar surface (Fig. 5A and B). Where bacteria adhered to epithelial cells (see Fig. 4G for alveolar epithelial cells type I [AECI]) at septa and connective tissue, tissue damage and epithelial delamination increased locally (Fig. 1H). We confirmed that macrophages (see Fig. 4H for alveolar macrophages detected by anti-CD68 staining) are the major host cell in the alveolar compartment, since virtually all macrophages in affected alveoli were infected with L. pneumophila (Fig. 4C and D), while only a fraction of the epithelial cells had been invaded by the pathogen (Fig. 5A, *). Uninfected HLTEs did not yield a signal for any of the two antibodies (Fig. 5C and E). These observations demonstrate that the HLTE infection model produces representative results and that it allows us to describe early events of disease progression in a 48-h time frame.
FIG 4.

Detection of L. pneumophila and OMVs in the alveolar compartment. Immunohistochemistry with anti-Mip (A, C, and E) and anti-MOMP (B, D, and F) antibodies followed by visualization with HRP polymer and permanent AEC (red signals) is shown. Uninfected controls remain negative for both antibodies (A and B); L. pneumophila is observed mainly in alveolar macrophages with both antibodies (C and D). In contrast to the detection of the whole pathogen, OMVs were detected sufficiently only with the anti-MOMP antibody (F). Alveolar epithelial cells type I, which line the alveolar compartment (arrows) and the septa, are targeted by Aquaporin-5 (AQP5) in medium control tissue (G). Alveolar macrophages as the most abundant immune cells in the alveolar space (arrows) are targeted by anti-CD68 staining (H). All images are at a magnification of ×40 and all HLTEs after 24 h of incubation.
L. pneumophila replicates within HLTEs.
To assess the ability of HLTEs to support intracellular L. pneumophila replication, the bacterial loads of tissue samples were analyzed during 48 h after infection (Fig. 6). Colonies were visible on BCYE agar at 4 days after plating. Wild-type L. pneumophila multiplied by approximately 10-fold in 24 h, similarly to in previous studies in human macrophage-like cells (34). After 24 h, the bacterial load continued to increase at a lower rate. The amount of DotA-negative bacteria, which cannot replicate within host cells, did not increase significantly. These results revealed that HLTEs support L. pneumophila replication and suggest this infection model for the characterization of mutants on the tissue level.
FIG 6.
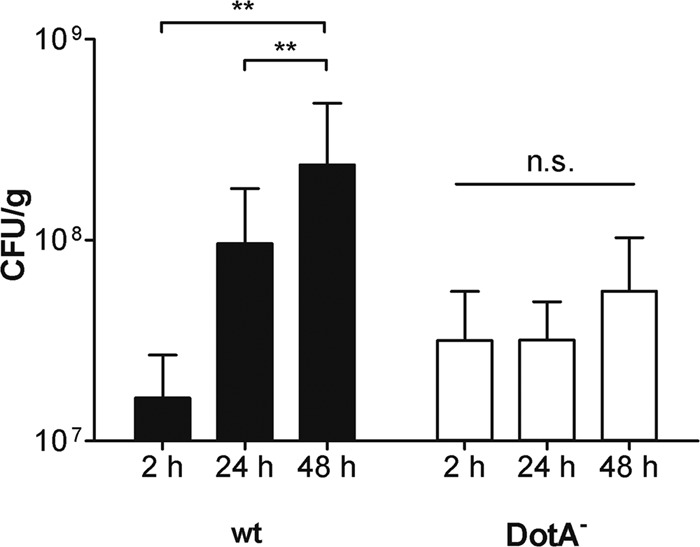
Replication of the L. pneumophila wild-type (wt) and DotA-deficient strains in HLTEs. Infected HTLEs were weighed, homogenized, and plated on BCYE agar at the indicated time points after infection. The graph shows means and standard deviations from triplicate experiments with tissue from eight donors. **, P < 0.01; n.s., not significant (P > 0.05) as determined by Student's t test for samples with identical variance.
L. pneumophila OMVs are located in alveolar macrophages.
L. pneumophila OMVs contain many virulence-related proteins, including degradative enzymes, and associate with alveolar epithelial cells (23, 35). Whether OMVs execute destructive activities and how they contribute to the infection on the tissue level is unknown.
Our results reveal a distinct localization of L. pneumophila OMVs in human lung tissue for the first time. Immunostaining showed that purified OMVs bind predominantly to the surfaces of alveolar macrophages and can be detected in their cytoplasm (Fig. 4E and F). Stimulating HLTEs with OMVs resulted in distinct tissue damage with epithelial cell delamination in affected alveoli and damage to collagen structures in septa and connective tissue fibers, starting approximately 24 h after infection. This damage is as severe as the effect observed in L. pneumophila-infected samples (Fig. 2A and D; Table 1) and has a comparable histological damage pattern.
Transcriptional response of HLTEs to L. pneumophila infection.
Previous transcriptional analyses identified host responses to L. pneumophila infection (14, 15, 36). To readdress this question on the tissue level, the transcriptome of L. pneumophila-infected HLTEs was compared to that of noninfected tissue from the same donors. A total of 2,499 genes were regulated with a fold change of ≥2.0 (Table 2) and were clustered hierarchically on entities (similarity of gene expression) and conditions (uninfected versus infected). The data from two independent experiments showed that distinct response levels could be observed (Fig. 7A). Gene Ontology analysis of the highly regulated genes revealed an enrichment of eight different terms among the 2,499 genes (Table 3).
TABLE 2.
Total number of differentially regulated genes in HLTEs at 24 h after infection with L. pneumophila
| Log2 fold change | No. of regulated genes |
|---|---|
| ≤2.0 | 1,986 |
| >2.0–3.0 | 156 |
| ≥3.0 | 20 |
| ≤−2.0 | 330 |
| ≥−2.0 | 7 |
FIG 7.

Transcriptional response of L. pneumophila-infected HLTEs and targeting of candidate genes on the protein level. (A) Heat map analysis and hierarchical clustering of 2,499 genes with a log2 fold change of ≥2. RNA was isolated from L. pneumophila wild-type-infected and uninfected HLTEs after 24 h of incubation (n = 2 each) to analyze the acute phase of infection. Distinct clusters were found to be differentially regulated after infection. (B to E) Immunohistochemistry against uteroglobin (B and C) and MARCO (D and E) on HLTEs after 24 h of incubation with medium (B and D) and L. pneumophila (C and E). Both proteins were detected mainly on alveolar macrophages. MARCO was observed to be downregulated over the course of stimulation at sites of tissue damage (asterisk in panel E). Images were taken at a magnification of ×40 with permanent AEC (red signals) as the chromogen. (F) Microarray data for uteroglobin were validated by qPCR normalized to ribosomal-like protein 32 (RPL32) from eight independent experiments. Uteroglobin is significantly upregulated after infection with L. pneumophila as determined by Student's t test (*, P < 0.05). (G) Secretion of uteroglobin in HLTE supernatants was quantified by ELISA from eight independent experiments; no significant difference was detected as computed with one-way analysis of variance (ANOVA).
TABLE 3.
Gene Ontology term analysis of genes differentially regulated after infection of HLTEs with L. pneumophilaa
| GO accession no. | GO term | Corrected P value |
|---|---|---|
| GO:0005576 | Extracellular region | 0.00003 |
| GO:0044421 | Extracellular region part | 0.00003 |
| GO:0005615 | Extracellular space | 0.00020 |
| GO:0002376 | Immune system process | 0.00854 |
| GO:0005929 | Cilium | 0.01026 |
| GO:0042953 | Lipoprotein transport | 0.01302 |
| GO:0035085 | Cilium axoneme | 0.02505 |
| GO:0005930 | Axoneme | 0.03395 |
A total of 2,499 genes with a fold change of ≥2 were used as the input list for Gene Ontology analysis. A Benjamini-Yekutieli correction was applied, and a P value of ≤0.05 was set as the cutoff.
Among the highly upregulated genes, we found that for uteroglobin, a protein secreted by Clara cells (log2 fold change, 2.18; Table 4). qRT-PCR with material from 8 experiments confirmed the significant regulation of uteroglobin by wild-type L. pneumophila (Fig. 7F). Targeting uteroglobin on the protein level via immunostaining did not reveal a marked difference between infected and uninfected samples. Alveolar macrophages were predominantly positive for uteroglobin (Fig. 7B and C). Since it is a secreted molecule, we further quantified by ELISA the amount of uteroglobin in HTLE supernatants. Interestingly, less uteroglobin was found in the supernatants of L. pneumophila wild-type- and DotA−-infected tissues than in medium controls (Fig. 7G).
TABLE 4.
Differential gene expression of uteroglobin and MARCO after infection with wild-type L. pneumophilaa
| Gene accession no. | Protein name | Donor | Relative expression level |
Log2 fold change | |
|---|---|---|---|---|---|
| Control | Wild type | ||||
| NM_003357 | Uteroglobin | 1 | 7,288.43 | 28,741.66 | |
| 2 | 1,157.49 | 6,897.58 | |||
| Mean | 4,222.96 | 17,819.62 | +2.18 | ||
| NM_006770 | MARCO | 1 | 3,190.08 | 958.49 | |
| 2 | 9,088.06 | 2,412.81 | |||
| Mean | 6,139.07 | 1,685.65 | −1.96 | ||
Quantile-normalized expression levels of uteroglobin and MARCO in two donors obtained from microarray raw data using Direct Array software.
Similarly, we targeted the macrophage receptor with collagenous structure (MARCO), a class A scavenger receptor. MARCO is strongly expressed in uninfected tissue and is 1.96 log2-fold downregulated after infection with L. pneumophila (Table 4). Immunostaining verified this finding, revealing a strong MARCO signal on alveolar macrophages in noninfected HLTEs (Fig. 7D) and a reduced expression on macrophages at sites of tissue destruction (Fig. 7E).
DISCUSSION
The histopathological descriptions of Legionnaires' disease are consistent but are restricted to the final stage of disease (1). Our understanding of L. pneumophila pneumonias is also limited by the poor amenability of infected human tissue. To overcome these restraints, powerful infection models ranging from monocellular host systems to mammals were developed. However, these models lack the communication between different cell types or have a nonhuman background, respectively. Data from animal models in particular cannot be generalized per se due to important differences in the expression, localization, and function of signaling molecules and receptors. Thus, not surprisingly, the function of L. pneumophila virulence factors varies considerably between host systems (37, 38).
In the present study, we established a novel L. pneumophila infection model involving human lung tissue explants (HTLEs). Particular aspects of infections with the pathogens Chlamydophila pneumoniae, Streptococcus pneumoniae, and Haemophilus influenzae were analyzed in similar systems (28, 39, 40). Although certain characteristics of HLTEs may depend on clinical parameters, the patients' medical conditions, and donor diversity, we obtained statistically robust, reproducible results. Thus, we utilized HTLEs with their multitude of cell types and extracellular components to investigate interactions between L. pneumophila and its human host at a unique level of complexity. The infection route is comparable to that in the human body, with the pathogen entering the alveoli via the bronchioli, albeit in a liquid phase rather than in aerosol droplets. The bacteria can reach the host cells and tissue structures from all sides, similarly as in the setup of a cell culture infection experiment.
Despite numerous sophisticated infection studies with L. pneumophila, the initial infection processes in the human lung remain unknown. Cell culture models suggest that alveolar macrophages are the most relevant cells for intracellular replication, while epithelial cells are infected to a minor degree. However, it is not clear whether these cells are relevant for the initial contact or if other cells or extracellular structures are also crucial for infection establishment. In the present study with HLTEs, we found that large numbers of L. pneumophila cells adhere extracellularly to the entire alveolar surface. With increasing incubation periods, the bacteria were detected primarily on and within alveolar macrophages, which are recruited to the alveolar space. Moreover, L. pneumophila could be detected in the connective tissue. Taken together, these results indicate that L. pneumophila initially binds to extracellular, yet-unidentified, alveolar tissue components and epithelial cells. The binding and invasion of alveolar macrophages recruited to the alveoli obviously represent a consecutive step of infection.
Histopathological analyses revealed that L. pneumophila-infected HLTEs are consistent with well-known features of Legionnaires' disease (41). This is not self-evident, since HLTEs do not include all immune system components which normally circulate through blood vessels and enter the tissue at infection sites. Infected HLTEs were characterized by specific damage to the tissue architecture. Alveolar septa were disrupted, alveolar epithelia appeared to be shaved off the underlying basal lamina, and protein exudate was detected. L. pneumophila colocalized with the damage to tissue structures at alveolar septa. This indicates that L. pneumophila causes the degradation of tissue barriers, possibly by destructive enzymes present on the surface or in the secretome of the bacterium (in the soluble fraction or in association with outer membrane vesicles) (23). This abolition of alveolar integrity, which could additionally be caused by the induction of tissue-destructive host molecules, likely contributes to the dissemination of L. pneumophila to neighboring alveoli and other organs (2, 3).
Interestingly, the tissue destruction in wild-type-infected HLTEs was markedly stronger than that in HLTEs infected with the DotA-negative strain. This is probably not due to larger amounts of bacteria and bacterial enzymes at later time points in wild-type-infected samples, as the DotA− L. pneumophila strain fails to cause higher tissue damage than the wild-type strain even if applied at 100-fold-higher numbers. However, it is conceivable that pulmonary cells secrete degradative enzymes or activate cell death pathways such as pyroptosis in response to wild-type, but not DotA−, L. pneumophila (42). The finding that the transcriptional response to these two strains is different in a human macrophage-like cell line supports this hypothesis (13).
To provide evidence that the HLTE model supports L. pneumophila replication and can identify attenuated mutants, we determined the growth kinetics of wild-type and DotA-deficient L. pneumophila strains in tissue samples. Importantly, in accordance with previous results in cell culture models (13, 43), we observed that wild-type L. pneumophila replicated within tissue samples, while DotA− bacteria, which are unable to multiply intracellularly, did not. Since the DotA-negative strain does not show growth defects in liquid media, the observed bacterial replication takes place within infected host cells, and not extracellularly, where there would be no growth disadvantage. Furthermore, we can conclude that the observed difference between the wild-type strain and the DotA− mutant can be explained by the intracellular growth of the wild type. Even if applied at 100-fold-higher numbers, the DotA− bacteria fail to cause remarkably higher tissue damage than the wild-type strain. These observations show that L. pneumophila-infected HLTEs yield reliable, robust results on bacterial replication which strongly correlate with current cell culture models. Moreover, these results pave the way for the characterization of L. pneumophila mutants under the complex conditions in the human lung.
The successful establishment of the HLTE infection model encouraged us to discover novel host-pathogen interactions in complex human tissue. Previously, L. pneumophila OMVs were shown to contain many virulence-related proteins, including proteases and lipases. Interestingly, OMVs do not kill host cells but specifically modulate the cytokine response of alveolar epithelial cells (23). To study the involvement of OMVs in infections of the human lung, we analyzed the localization and putative degradative effects of OMVs in HLTEs. We detected L. pneumophila and its OMVs at similar sites in the tissue. Since OMVs contain proteins involved in adhesion to host cells, such as MOMP and Hsp60 (23), our results indicate that the presence of these proteins is sufficient for the localization of these subcellular bacterial structures. Intriguingly, OMVs also caused histological tissue damage which was qualitatively and quantitatively similar to the destruction caused by L. pneumophila itself. The aforementioned degradative enzymes are likely responsible for this effect in OMV-treated and also in L. pneumophila-infected HLTEs. Following this notion, we propose that OMVs contribute to the extracellular pathogenicity of L. pneumophila, including the dissemination of the infection to other organs.
During acute L. pneumophila infections, dynamic and interrelated responses are triggered in the human lung at multiple levels. To describe such processes at a complexity above that of monocellular cultures, we recorded the transcriptional response of L. pneumophila-infected HLTEs. This approach revealed several new host factors which may be involved in the pathogenesis of Legionnaires' disease. We found 2,499 genes to be differentially expressed after infection, many of which have never been described to be related to L. pneumophila infections. A group of eight Gene Ontology terms is significantly regulated (P < 0.05) after infection, including a large number of extracellular proteins, components of the immune response, and, interestingly, lipoprotein transport proteins. We will characterize these data in the future to understand better how the pathogen establishes disease and how it modulates the host response.
Our analyses by microarray and validation by quantitative PCR revealed that uteroglobin mRNA was significantly upregulated at 24 h after infection. Uteroglobin, also termed Clara cell secretory protein or blastokinin, is a major constituent of the airway extracellular fluid (44). It inhibits immune cell recruitment both in vitro and after infection of mice with Pseudomonas aeruginosa (45, 46). Importantly, uteroglobin mRNA and protein levels decrease in lung epithelial cells after stimulation with P. aeruginosa or tumor necrosis factor alpha (TNF-α) (46, 47). It is conceivable that L. pneumophila follows a strategy similar to that of P. aeruginosa in regard to immune cell recruitment. Interestingly, we observed a reduced concentration of uteroglobin in the supernatants of HLTEs challenged with wild-type and DotA− L. pneumophila. This might be explained by an increased uptake of uteroglobin by alveolar macrophages upon stimulation, since these are shown as the main positive cells by immunostaining.
The downregulation of MARCO after an L. pneumophila infection is striking. MARCO is a class A scavenger receptor involved in the uptake of the bacterial pathogens Neisseria meningitidis, Clostridium sordellii, and Streptococcus mutans by macrophages and modulates cytokine responses (48–52). Intriguingly, S. mutans phagocytosis is partially mediated by MARCO, and the pathogen suppresses this function with a peptidyl-prolyl cis-trans isomerase (50). L. pneumophila features Mip, a virulence factor with the same enzymatic activity, on its surface and in association with OMVs (23, 53). Since the collagen-binding protein Mip is also involved in the extracellular pathogenicity of L. pneumophila, it will be interesting to investigate if Mip modulates MARCO activity and thereby contributes to bacterial dissemination within the lung.
In summary, the HLTE infection model narrows the gap between current infection models and actual human infections. It allows us to characterize tissue damage, bacterial dissemination, and the host's molecular response after an infection with L. pneumophila in great detail and will contribute to our understanding of the pathogen-host cross talk and to the development of interventional treatment strategies.
ACKNOWLEDGMENTS
This study was supported in part by a grant from the Deutsche Forschungsgemeinschaft (DFG) (STE 838/6-1), by a grant from the German Bundesministerium für Bildung und Forschung (BMBF) (LegioProTect 0315831), and by the Helmholtz International Graduate School for Infection Research.
We thank Antje Flieger (Robert Koch Institute, Wernigerode, Germany) for providing a mutant strain, Joe Vogel (Washington University, St. Louis, MO, USA) for providing an antibody, Andra Schromm and Ulrich Schaible for providing machinery and lab space, and Steffi Fox, Maria Lammers, and Jasmin Tiebach for their excellent technical assistance.
J.J., S.M., T.G., and M.S. conceived the experiments. J.J., S.M., J.T., J.R., and C.K. carried out the experiments. J.J., S.M., O.S., T.G., and M.S. analyzed data. J.J., S.M., T.G., and M.S. wrote the manuscript.
We declare that no conflict of interest exists.
Footnotes
Published ahead of print 28 October 2013
REFERENCES
- 1.Winn WC, Jr, Myerowitz RL. 1981. The pathology of the Legionella pneumonias. A review of 74 cases and the literature. Hum. Pathol. 12:401–422 [DOI] [PubMed] [Google Scholar]
- 2.Hambleton P, Baskerville A, Fitzgeorge RB, Bailey NE. 1982. Pathological and biochemical features of Legionella pneumophila infection in guinea-pigs. J. Med. Microbiol. 15:317–326. 10.1099/00222615-15-3-317 [DOI] [PubMed] [Google Scholar]
- 3.Watts JC, Hicklin MD, Thomason BM, Callaway CS, Levine AJ. 1980. Fatal pneumonia caused by Legionella pneumophila, serogroup 3: demonstration of the bacilli in extrathoracic organs. Ann. Intern. Med. 92:186–188. 10.7326/0003-4819-92-2-186 [DOI] [PubMed] [Google Scholar]
- 4.Theaker JM, Tobin JO, Jones SE, Kirkpatrick P, Vina MI, Fleming KA. 1987. Immunohistological detection of Legionella pneumophila in lung sections. J. Clin. Pathol. 40:143–146. 10.1136/jcp.40.2.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearlman E, Jiwa AH, Engleberg NC, Eisenstein BI. 1988. Growth of Legionella pneumophila in a human macrophage-like (U937) cell line. Microb. Pathog. 5:87–95. 10.1016/0882-4010(88)90011-3 [DOI] [PubMed] [Google Scholar]
- 6.Mody CH, Paine R, III, Shahrabadi MS, Simon RH, Pearlman E, Eisenstein BI, Toews GB. 1993. Legionella pneumophila replicates within rat alveolar epithelial cells. The J. Infect. Dis. 167:1138–1145. 10.1093/infdis/167.5.1138 [DOI] [PubMed] [Google Scholar]
- 7.Hagele S, Kohler R, Merkert H, Schleicher M, Hacker J, Steinert M. 2000. Dictyostelium discoideum: a new host model system for intracellular pathogens of the genus Legionella. Cell. Microbiol. 2:165–171. 10.1046/j.1462-5822.2000.00044.x [DOI] [PubMed] [Google Scholar]
- 8.Rowbotham TJ. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33:1179–1183. 10.1136/jcp.33.12.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fields BS, Sanden GN, Barbaree JM, Morrill WE, Wadowsky RM, White E, Feeley JC. 1989. Intracellular multiplication of Legionella pneumophila in amoebae isolated from hospital hot water tanks. Curr. Microbiol. 18:131–137. 10.1007/BF01570838 [DOI] [Google Scholar]
- 10.Bartfeld S, Engels C, Bauer B, Aurass P, Flieger A, Bruggemann H, Meyer TF. 2009. Temporal resolution of two-tracked NF-kappaB activation by Legionella pneumophila. Cell. Microbiol. 11:1638–1651. 10.1111/j.1462-5822.2009.01354.x [DOI] [PubMed] [Google Scholar]
- 11.Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Valero L, Rusniok C, Jarraud S, Vacherie B, Rouy Z, Barbe V, Medigue C, Etienne J, Buchrieser C. 2011. Extensive recombination events and horizontal gene transfer shaped the Legionella pneumophila genomes. BMC Genomics 12:536. 10.1186/1471-2164-12-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Losick VP, Isberg RR. 2006. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203:2177–2189. 10.1084/jem.20060766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fortier A, Faucher SP, Diallo K, Gros P. 2011. Global cellular changes induced by Legionella pneumophila infection of bone marrow-derived macrophages. Immunobiology 216:1274–1285. 10.1016/j.imbio.2011.06.008 [DOI] [PubMed] [Google Scholar]
- 15.Farbrother P, Wagner C, Na J, Tunggal B, Morio T, Urushihara H, Tanaka Y, Schleicher M, Steinert M, Eichinger L. 2006. Dictyostelium transcriptional host cell response upon infection with Legionella. Cell. Microbiol. 8:438–456. 10.1111/j.1462-5822.2005.00633.x [DOI] [PubMed] [Google Scholar]
- 16.Steinert M. 2011. Pathogen-host interactions in Dictyostelium, Legionella, Mycobacterium and other pathogens. Semin. Cell Dev. Biol. 22:70–76. 10.1016/j.semcdb.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 17.Shevchuk O, Batzilla C, Hagele S, Kusch H, Engelmann S, Hecker M, Haas A, Heuner K, Glockner G, Steinert M. 2009. Proteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int. J. Med. Microbiol. 299:489–508. 10.1016/j.ijmm.2009.03.006 [DOI] [PubMed] [Google Scholar]
- 18.Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, Hilbi H. 2009. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10:76–87. 10.1111/j.1600-0854.2008.00851.x [DOI] [PubMed] [Google Scholar]
- 19.Hayashi T, Nakamichi M, Naitou H, Ohashi N, Imai Y, Miyake M. 2010. Proteomic analysis of growth phase-dependent expression of Legionella pneumophila proteins which involves regulation of bacterial virulence traits. PLoS One 5:e11718. 10.1371/journal.pone.0011718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baskerville A, Fitzgeorge RB, Broster M, Hambleton P, Dennis PJ. 1981. Experimental transmission of legionnaires' disease by exposure to aerosols of Legionella pneumophila. Lancet ii:1389–1390 [DOI] [PubMed] [Google Scholar]
- 21.Blanchard DK, Djeu JY, Klein TW, Friedman H, Stewart WE., II 1987. Induction of tumor necrosis factor by Legionella pneumophila. Infect. Immun. 55:433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzgeorge RB, Baskerville A, Broster M, Hambleton P, Dennis PJ. 1983. Aerosol infection of animals with strains of Legionella pneumophila of different virulence: comparison with intraperitoneal and intranasal routes of infection. J. Hyg. (Lond.) 90:81–89. 10.1017/S0022172400063877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galka F, Wai SN, Kusch H, Engelmann S, Hecker M, Schmeck B, Hippenstiel S, Uhlin BE, Steinert M. 2008. Proteomic characterization of the whole secretome of Legionella pneumophila and functional analysis of outer membrane vesicles. Infect. Immun. 76:1825–1836. 10.1128/IAI.01396-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shevchuk O, Jager J, Steinert M. 2011. Virulence properties of the Legionella pneumophila cell envelope. Front. Microbiol. 2:74. 10.3389/fmicb.2011.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jepras RI, Fitzgeorge RB, Baskerville A. 1985. A comparison of virulence of two strains of Legionella pneumophila based on experimental aerosol infection of guinea-pigs. J. Hyg. (Lond.) 95:29–38. 10.1017/S0022172400062252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aurass P, Pless B, Rydzewski K, Holland G, Bannert N, Flieger A. 2009. bdhA-patD operon as a virulence determinant, revealed by a novel large-scale approach for identification of Legionella pneumophila mutants defective for amoeba infection. Appl. Environ. Microbiol. 75:4506–4515. 10.1128/AEM.00187-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jager J, Steinert M. 2013. Enrichment of outer membrane vesicles shed by Legionella pneumophila. Methods Mol. Biol. 954:225–230. 10.1007/978-1-62703-161-5_13 [DOI] [PubMed] [Google Scholar]
- 28.Dromann D, Rupp J, Rohmann K, Osbahr S, Ulmer AJ, Marwitz S, Roschmann K, Abdullah M, Schultz H, Vollmer E, Zabel P, Dalhoff K, Goldmann T. 2010. The TGF-beta-pseudoreceptor BAMBI is strongly expressed in COPD lungs and regulated by nontypeable Haemophilus influenzae. Respir. Res. 11:67. 10.1186/1465-9921-11-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vollmer E, Galle J, Lang DS, Loeschke S, Schultz H, Goldmann T. 2006. The HOPE technique opens up a multitude of new possibilities in pathology. Rom. J. Morphol. Embryol. 47:15–19. 10.1186/1471-2407-11-511 [DOI] [PubMed] [Google Scholar]
- 30.Marwitz S, Abdullah M, Vock C, Fine JS, Visvanathan S, Gaede KI, Hauber HP, Zabel P, Goldmann T. 2011. HOPE-BAL: improved molecular diagnostics by application of a novel technique for fixation and paraffin embedding. J. Histochem. Cytochem. 59:601–614. 10.1369/0022155411404417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helbig JH, Ludwig B, Luck PC, Groh A, Witzleb W, Hacker J. 1995. Monoclonal antibodies to Legionella Mip proteins recognize genus- and species-specific epitopes. Clin. Diagn. Lab. Immunol. 2:160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seneta E. 1992. Probability inequalities and Dunnett's test, p 29–45 In Hoppe FM. (ed), Multiple comparisons, selection, and applications in biometry. CRC Press, Boca Raton, FL [Google Scholar]
- 33.Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. 10.1093/bioinformatics/19.2.185 [DOI] [PubMed] [Google Scholar]
- 34.Juli C, Sippel M, Jager J, Thiele A, Weiwad M, Schweimer K, Rosch P, Steinert M, Sotriffer CA, Holzgrabe U. 2011. Pipecolic acid derivatives as small-molecule inhibitors of the Legionella MIP protein. J. Med. Chem. 54:277–283. 10.1021/jm101156y [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Moreira E, Helbig JH, Swanson MS. 2006. Membrane vesicles shed by Legionella pneumophila inhibit fusion of phagosomes with lysosomes. Infect. Immun. 74:3285–3295. 10.1128/IAI.01382-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jules M, Buchrieser C. 2007. Legionella pneumophila adaptation to intracellular life and the host response: clues from genomics and transcriptomics. FEBS Lett. 581:2829–2838. 10.1016/j.febslet.2007.05.026 [DOI] [PubMed] [Google Scholar]
- 37.Rossier O, Dao J, Cianciotto NP. 2009. A type II secreted RNase of Legionella pneumophila facilitates optimal intracellular infection of Hartmannella vermiformis. Microbiology 155:882–890. 10.1099/mic.0.023218-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tyson JY, Pearce MM, Vargas P, Bagchi S, Mulhern BJ, Cianciotto NP. 2013. Multiple Legionella pneumophila type II secretion substrates, including a novel protein, contribute to differential infection of amoebae Acanthamoeba castellanii, Hartmannella vermiformis, and Naegleria lovaniensis. Infect. Immun. 81:1399–1410. 10.1128/IAI.00045-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rupp J, Droemann D, Goldmann T, Zabel P, Solbach W, Vollmer E, Branscheid D, Dalhoff K, Maass M. 2004. Alveolar epithelial cells type II are major target cells for C. pneumoniae in chronic but not in acute respiratory infection. FEMS Immunol. Med. Microbiol. 41:197–203. 10.1016/j.femsim.2004.03.004 [DOI] [PubMed] [Google Scholar]
- 40.Xu F, Droemann D, Rupp J, Shen H, Wu X, Goldmann T, Hippenstiel S, Zabel P, Dalhoff K. 2008. Modulation of the inflammatory response to Streptococcus pneumoniae in a model of acute lung tissue infection. Am. J. Respir. Cell Mol. Biol. 39:522–529. 10.1165/rcmb.2007-0328OC [DOI] [PubMed] [Google Scholar]
- 41.Glavin FL, Winn WC, Craighead JE. 1979. Ultrastructure of lung in Legionnaires' disease. Observations of three biopsies done during the Vermont epidemic. Ann. Intern. Med. 90:555–559 [DOI] [PubMed] [Google Scholar]
- 42.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. U. S. A. 110:1851–1856. 10.1073/pnas.1211521110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy CR, Berger KH, Isberg RR. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28:663–674. 10.1046/j.1365-2958.1998.00841.x [DOI] [PubMed] [Google Scholar]
- 44.Singh G, Katyal SL, Brown WE, Kennedy AL, Singh U, Wong-Chong ML. 1990. Clara cell 10 kDa protein (CC10): comparison of structure and function to uteroglobin. Biochim. Biophys. Acta 1039:348–355. 10.1016/0167-4838(90)90270-P [DOI] [PubMed] [Google Scholar]
- 45.Vasanthakumar G, Manjunath R, Mukherjee AB, Warabi H, Schiffmann E. 1988. Inhibition of phagocyte chemotaxis by uteroglobin, an inhibitor of blastocyst rejection. Biochem. Pharmacol. 37:389–394. 10.1016/0006-2952(88)90204-3 [DOI] [PubMed] [Google Scholar]
- 46.Hayashida S, Harrod KS, Whitsett JA. 2000. Regulation and function of CCSP during pulmonary Pseudomonas aeruginosa infection in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L452–K459 [DOI] [PubMed] [Google Scholar]
- 47.Harrod KS, Jaramillo RJ. 2002. Pseudomonas aeruginosa and tumor necrosis factor-alpha attenuate Clara cell secretory protein promoter function. Am. J. Respir. Cell Mol. Biol. 26:216–223. 10.1165/ajrcmb.26.2.4718 [DOI] [PubMed] [Google Scholar]
- 48.Mukhopadhyay S, Chen Y, Sankala M, Peiser L, Pikkarainen T, Kraal G, Tryggvason K, Gordon S. 2006. MARCO, an innate activation marker of macrophages, is a class A scavenger receptor for Neisseria meningitidis. Eur. J. Immunol. 36:940–949. 10.1002/eji.200535389 [DOI] [PubMed] [Google Scholar]
- 49.Thelen T, Hao Y, Medeiros AI, Curtis JL, Serezani CH, Kobzik L, Harris LH, Aronoff DM. 2010. The class A scavenger receptor, macrophage receptor with collagenous structure, is the major phagocytic receptor for Clostridium sordellii expressed by human decidual macrophages. J. Immunol. 185:4328–4335. 10.4049/jimmunol.1000989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukouhara T, Arimoto T, Cho K, Yamamoto M, Igarashi T. 2011. Surface lipoprotein PpiA of Streptococcus mutans suppresses scavenger receptor MARCO-dependent phagocytosis by macrophages. Infect. Immun. 79:4933–4940. 10.1128/IAI.05693-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elomaa O, Kangas M, Sahlberg C, Tuukkanen J, Sormunen R, Liakka A, Thesleff I, Kraal G, Tryggvason K. 1995. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell 80:603–609. 10.1016/0092-8674(95)90514-6 [DOI] [PubMed] [Google Scholar]
- 52.Bowdish DM, Sakamoto K, Kim MJ, Kroos M, Mukhopadhyay S, Leifer CA, Tryggvason K, Gordon S, Russell DG. 2009. MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 5:e1000474. 10.1371/journal.ppat.1000474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner C, Khan AS, Kamphausen T, Schmausser B, Unal C, Lorenz U, Fischer G, Hacker J, Steinert M. 2007. Collagen binding protein Mip enables Legionella pneumophila to transmigrate through a barrier of NCI-H292 lung epithelial cells and extracellular matrix. Cell. Microbiol. 9:450–462. 10.1111/j.1462-5822.2006.00802.x [DOI] [PubMed] [Google Scholar]