

Abstract
Signaling through cAMP plays an important role in heart failure. Phosphorylation of cAMP response element binding protein (CREB) at serine-133 regulates gene expression in the heart. We examined the functional significance of CREB-S133 phosphorylation by comparing transgenic models in which a phosphorylation resistant CREB-S133A mutant containing either an intact or a mutated leucine zipper domain (CREB-S133A-LZ) was expressed in the heart. In vitro, CREB-S133A retained the ability to interact with wild-type CREB, whereas CREB-S133A-LZ did not. In vivo, CREB-S133A and CREB-S133A-LZ were expressed at comparable levels in the heart; however, CREB-S133A markedly suppressed the phosphorylation of endogenous CREB, whereas CREB-S133A-LZ had no effect. The one-year survival of mice from two CREB-S133A-LZ transgenic lines was equivalent to nontransgenic littermate control mice (NTG), whereas transgenic CREB-S133A mice died with heart failure at a median 30 wk of age (P < 0.0001). CREB-S133A mice had an altered gene expression characteristic of the failing heart, whereas CREB-S133A-LZ mice did not. Left ventricular contractile function was substantially reduced in CREB-S133A mice versus NTG mice and only modestly reduced in CREB-S133A-LZ mice (P < 0.02). When considered in light of other studies, these findings indicate that overexpression of the CREB leucine zipper is required for both inhibition of endogenous CREB phosphorylation and cardiomyopathy in this murine model of heart failure.
Keywords: adenosine; 3′,5′-cyclic; monophosphate; responsive; element; binding; protein
Signaling through cAMP plays an important role in the pathogenesis of human heart failure (30). One target of cAMP signaling in cardiac myocytes is transcriptional regulation mediated through the 43-kDa basic leucine zipper transcription factor cAMP responsive element (CRE) binding protein (CREB). Members of the CREB/CRE modulator (CREM)/activating transcription factor (ATF) family of transcription factors have important roles in cardiomyocyte biology and heart failure (6, 16, 26, 29). Myocardial target genes of CREB relevant to the pathogenesis of heart failure include Bcl2 (14, 32), inducible cAMP early repressor (3), and cardiotrophin-1 (9, 14).
The CREB/CREM/ATF family members contain a kinase-inducible transcriptional activation domain (10). Within this domain, CREB serine-133 is phosphorylated in cardiomyocytes following adrenergic stimulation (23) by cAMP-dependent activation of PKA (18) and by other kinase pathways relevant to cardiomyocyte survival and contractility, including Akt (4) and p38 mitogen- and stress-activated protein kinase-1 (18). cAMP-stimulated CREB serine-133 phosphorylation leads to its association with CREB binding protein (CBP) and p300 coactivator protein binding (20), activating gene expression selectively through an interaction with the TATA boxdependent basal transcriptional machinery (1, 2). In addition to the kinase-regulated transcriptional activation domain, the CREB protein includes a conserved COOH-terminal basic and leucine zipper domain responsible for protein dimerization and DNA binding (5). Through alternative splicing, the CREM gene produces two isoforms distinguished by unique leucine zipper domains that have 75% and 95% identity with CREB (8).
Compensation by CREM/ATF proteins may explain the lack of cardiac phenotype in CREB gene deletion models (13, 19). Instead, how CREB may regulate cardiomyocyte function has been explored in vivo in transgenic models. In particular, the CREB-S133A mutant, which cannot be phosphorylated (6, 17), can inhibit gene activation by CREB and other members of the CREB/CREM/ATF family by occupying CRE (34). Cardiomyocyte-targeted overexpression of the CREB-S133A mutant produces many features characteristic of human heart failure, including resistance to β-adrenergic stimulation, systolic and diastolic left ventricular (LV) dysfunction, depressed contractility, signs of chronic left and right heart failure, arrhythmia, and early mortality (7). Overexpression of a naturally occurring CREM splice variant including the basic leucine zipper (24) without the transcriptional activation domains [myosin heavy chain (MHC)-CREM-IbΔC-X] also produces heart failure, arrhythmia, and death despite enhanced contractility and systolic function (25).
A common molecular finding in MHC-CREB-S133A and MHC-CREM-IbΔC-X transgenic (TG) mice is a reduced level of the phosphorylated form of the endogenous CREB protein (6, 25). This suggests the hypothesis that inhibition of endogenous CREB phosphorylation is mediated through leucine zipper interactions and is responsible for a lethal heart failure phenotype. To examine this hypothesis, we therefore created TG mice overexpressing a CREB protein harboring both the nonphosphorylatable dominant-negative mutation (S133A) as well as loss-of-function leucine zipper mutations (L318V, L325V) targeted for expression in the adult heart by the mouse α-MHC gene promoter (MHC-CREB-S133A-LZ). Physiological and molecular analysis of these TG mice demonstrated that removing leucine zipper function normalizes levels of endogenous phospho-CREB and protects mice from a lethal heart failure phenotype. These data provide novel insights into the molecular basis by which perturbation of myocardial CREB/CREM signaling produces heart failure in vivo.
METHODS
Gene constructs
The hemagglutinin epitope-tagged CREB-S133A cDNA (6) was subcloned into pcDNA3 (Invitrogen) to generate pcDNA3-CREB-S133A. Site-directed mutagenesis (Stratagene) was used to introduce L318V and L325V mutations in the CREB leucine zipper domain, thus generating pcDNA3-CREB-S133A-LZ. Primers used for mutagenesis were 5′-GCA-GTG-GTT-GAA-AAT-CAA-AAC-AAG-ACA-GTG-ATT-GAG-GAG-3′ and 5′-CTC-CTC-CAT-CAC-TGT-CTT-GTT-TTG-ATT-TTC-AAC-CAC-TGC-3′, with the mutated codons substituting valine for leucine indicated in boldface. The Xho1 fragment from pcDNA3-CREB encoding the CREB leucine zipper (nucleotides 852 to 984) was subcloned to produce the pGEX-CREB-LZ plasmid.
Glutathione-S-transferase pull down
Glutathione-S-transferase (GST) and GST-CREB proteins expressed in BL21 cells from the pGEX and the pGEX-CREB-LZ plasmids, respectively, were purified on Glutathione Sepharose 4B (Amersham Biosciences). Recombinant CREB-S133A and CREB-S133A-LZ proteins were expressed and labeled with [35S]-methionine (Amersham Biosciences) from their respective pcDNA3 constructs using the TnT T7 Coupled Reticulocyte System (Promega). The radiolabeled proteins were mixed with GST fusion proteins immobilized on Glutathione Sepharose 4B in phosphate-buffered saline supplemented with 1% bovine serum albumen and 0.1% Nonidet P-40. After 20 min, the beads were washed with the binding buffer for 10 min with three buffer exchanges. Proteins eluted in sample buffer were separated by SDS-PAGE. The dried gel was exposed to Kodak BioMax MR autoradiography film.
Generation of TG mice
The CREB-S133A-LZ cDNA was subcloned downstream from the 5.8-kb α-MHC promoter fragment, including exons 1–3 and upstream of the SV40 polyadenylation site (11). Linearized plasmid was injected into the pronucleus of fertilized single-cell ICR (Taconic) embryos to produce MHC-CREB-S133A-LZ TG mice. Founders were identified by Southern blot analysis of tail DNA as previously described (6). All animal experimentation was performed in accordance with National Institutes of Health guidelines and according to protocols approved by the Animal Care and Use Committee of Tufts University and Medical College of Georgia. The generation of MHC-CREB-S133A mice was previously described (6). Male MHC-CREB-S133A mice had been crossed with wild-type ICR female mice for more than six generations; as such, all lines of mice described in this report shared the ICR background.
Western blot analysis
Hearts were rapidly frozen in liquid nitrogen. Heart tissue was homogenized (30 s) in buffer (1 mM Tris · HCl, pH 7.4) containing protease inhibitors (Roche complete protease inhibitor cocktail). The soluble lysates were separated by SDS-polyacrylamide gel electrophoresis under reducing conditions and electroblotted onto polyvinylidene difluoride membranes. Membranes were probed with anti-CREB (Cell Signaling), anti-CREB phosphoserine-133 (Cell Signaling), and anti-hemagglutinin (Covance) followed by goat anti-rabbit phosphatase (Amersham Biosciences). The chemifluorescent image was developed on a phosphorimager (Molecular Dynamics).
Real-time PCR
Total RNA was extracted from ventricles using TRIzol (Invitrogen) and treated with DNAseI (Roche Diagnostics) to remove genomic DNA contamination. cDNA synthesized from 1 μg total RNA using SuperScript III reverse transcriptase and oligo (dT) primers (Invitrogen) was diluted 1:100, and 1 μl was used for each PCR amplification, performed in triplicate, with QuantiTect SYBR Green MasterMix (Qiagen). Mouse transcripts were measured using custom primers for brain natriuretic peptide (BNP) (S-AGTCCTAGCCAGTCTCCAGAGCAA and AS-CAACTTCAGTGCGTTACAGCCCAA), β-MHC (S-CTGGAGAAGATCCGAAAGCA and AS-GTGTCCTTCAGCAAACTCTGG), sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2) (S-ATGACAATGGCACTTTCTGTTCTA and AS-AGGAGATTTTCAGCACCATCAG), and GAPDH as described (33). PCR products were verified by sequence analysis. Fluorescence data were analyzed with MJ Research Opticon sequence detection software (Bio-Rad), and PCR efficiency was calculated from slopes of standard curves. Fold changes in gene expression were determined using Q-gene (35) and normalized to GAPDH after correcting for differences in amplification efficiency.
Echocardiography
The day before the terminal hemodynamic study, 6-mo-old mice underwent transthoracic echocardiographic analysis as previously described (31). Briefly, mice were sedated with 1.5% inhaled isoflurane. A commercially available echocardiography system (HDI 5000, ATL, Bothell, WA) was utilized as previously described (31). Two-dimensional and M-mode images were recorded from the short axis at the high papillary muscle level as the mouse regained consciousness. Cardiac function was measured at the lowest levels of sedation that allowed accurate discrimination of the myocardial walls.
Hemodynamic evaluation
Mice were anesthetized with 2.0% inhaled isoflurane. Care was taken to maintain body temperature at 37.0 ± 0.5°C. The right cervical region was prepped, and following incision, the right common carotid artery was isolated by blunt dissection. A 1.4-Fr Millar pressure catheter was introduced into the right carotid artery and passed into the ascending aorta. After hemodynamic measurements were recorded in the aorta, the catheter was passed retrograde into the left ventricle. After a stable pressure tracing was achieved, intraventricular pressure recordings were made.
Electrophysiology studies
The MHC-CREB-S133A-LZ mice were evaluated in the same manner as described previously for the MHC-CREB-S133A mice (37). Briefly, a 2-Fr octapolar mouse electrophysiology catheter was advanced through the right external jugular vein, placing proximal electrodes in the right atrium and distal electrodes into the right ventricle. The midelectrodes were used to record the His-bundle electrogram. Surface ECG and intracardiac electrograms were recorded, and cardiac intervals were measured as previously described (37). Finally, programmed ventricular stimulation of the right ventricular apex was performed to assess the inducibility of ventricular arrhythmias as previously described (37).
Statistical analysis
Values are means ± SE. The significance of expression differences was determined by one-way ANOVA with pairwise multiple comparisons performed using the Holm-Sidak method as administered by the Sigma Stat program (Systat Software). When groups were compared with the NTG group and each other, ANOVA was performed using the Student-Neuman-Keuls test correction for multiple group comparisons. For all data, a P value of <0.05 was considered significant.
RESULTS
CREB-S133A-LZ does not bind the CREB leucine zipper
To explore the role of the CREB leucine zipper in CREB-S133A-induced cardiomyopathy, we first used a GST pull-down assay to compare the ability of mutant CREB proteins containing either just the S133A mutation (CREB-S133A) or both the S133A mutation and the leucine zipper mutations (CREB-S133A-LZ; Fig. 1A) to bind wild-type CREB leucine zipper. The CREB-S133A-LZ protein expressed by an in vitro transcription-translation system was confirmed to be a similar size as CREB-S133A. We then incubated [35S]-methionine-labeled recombinant proteins with GST or GST fused to the CREB leucine zipper (GST-CREB). The immobilized GST protein alone retained neither protein. Consistent with the known requirement of leucine-318 and -325 for CREB dimer formation, the CREB-S133A protein was retained by GST-CREB, whereas CREB-S133A-LZ was not (Fig. 1B). Thus the loss-of-function mutations introduced into the CREB leucine zipper domain in the CREB-S133A-LZ construct interfered with leucine zipper-mediated binding interactions.
Fig. 1.
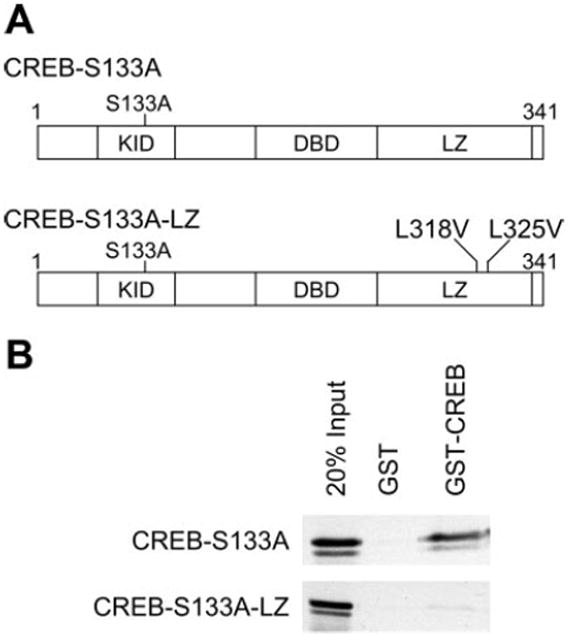
A: diagram of cAMP response element binding protein (CREB)-S133A and CREB-S133A-leucine zipper (LZ) protein structure. The amino acid location of each mutation is shown in relationship to the kinase inducible domain (KID), the DNA binding domain (DBD), and the LZ. B: CREB-S133A-LZ does not associate with the CREB LZ. Twenty percent of the [35S]-methionine-labeled in vitro translation product used for binding is shown in the first lane. After the radiolabeled proteins had been incubated with immobilized glutathione-S-transferase (GST) or GST-CREB, the beads were washed and the bound proteins were eluted and separated by 10% SDS-PAGE. CREB-S133A was selectively retained by the CREB LZ GST fusion protein and not by GST alone (B, top). The GST-CREB protein, which includes only the CREB LZ, did not retain the CREB-S133A-LZ mutant protein (B, bottom). Shown are representative results from 3 experiments.
Generation and characterization of CREB-S133A-LZ TG mice
Two lines of TG mice expressing CREB-S133A-LZ under the transcriptional control of the cardiac-restricted MHC promoter were created by pronuclear injection. Heart mass normalized to body weight was not significantly different in 10–12-wk-old MHC-CREB-S133A-LZ TG and NTG littermate females [mean ± SD, 3.66 ± 0.56 (n = 16) vs. 3.49 ± 0.69 (n = 6) mg/g, P = not significant (NS)] and male mice [4.29 ± 0.17 (n = 6) vs. 4.77 ± 1.86 (n = 7) mg/g, P = NS] from line 1.
Preserved phospho-CREB serine-133 in leucine zipper mutant TG mice
The CREB-S133A-LZ lines were compared with the same line of MHC-CREB-S133A line mice previously described (6). Expression of similar or greater amounts of recombinant MHC-CREB-S133A-LZ compared with CREB-S133A was confirmed by Western blot analysis (Fig. 2A). The abundance of cardiac serine-133 phosphorylated CREB is markedly reduced in CREB-S133A TG mice (6). To determine whether CREB-S133A-LZ also lowered endogenous CREB serine-133 phosphorylation levels, total ventricular protein extracts were probed with an antibody specific for serine-133 phosphorylated CREB. Western blot analysis confirmed markedly reduced phosphorylated CREB serine-133 in the setting of CREB-S133A overexpression, whereas phosphorylated CREB protein in CREB-S133A-LZ TG hearts was not different than NTG littermate control (Fig. 2B). These data demonstrate that an intact leucine zipper domain is required for inhibition of myocardial CREB phosphorylation in CREB-S133A TG mice.
Fig. 2.

A: cardiac expression of hemagglutinin (HA)-CREB mutant proteins. Equivalent amounts of whole heart lysates were separated and probed using an anti-CREB antibody (A, top) and an anti-HA antibody (A, bottom). Endogenous CREB protein was evident as 2 molecular species (top, solid arrows). The CREB transgene, which migrates more slowly because of the HA tag, was visible above the endogenous protein (top, open arrowhead). CREB-S133A expression was detected as a weak band by HA staining (bottom, second lane), whereas 2 lines of CREB-S133A-LZ mice showed more abundant transgene expression. B: CREB-S133A-LZ mice have preserved serine-133 phospho-CREB levels. Whole heart lysate immunoblots following SDS-PAGE were probed for serine-133 phospho-CREB levels. The separation of equivalent amounts of whole heart lysate is shown by Ponceau red staining (B, bottom). Abundant serine-133 phospho-CREB was found in nontransgenic (NTG) and transgenic CREB-S133A-LZ mice but not in CREB-S133A mice. Shown is a representative experiment of 3 from the LZ mutant line 1.
CREB-S133A-LZ mouse 1-yr survival is normal
Whereas MHC-CREB-S133A mice develop signs of advanced heart failure, leading to early mortality as previously described (6, 37), both lines of MHC-CREB-S133A-LZ TG mice appeared healthy and free of overt heart failure. To determine whether the cardiomyocyte effects of CREB-S133A-LZ expression were simply delayed, we analyzed survival of mice from the two TG lines over 1 yr. MHC-CREB-S133A mice died starting at 15 wk of age, and by 30 wk of age, half of the TG mice were dead (Fig. 3). By comparison, survival was not significantly reduced after 52 wk in both lines of MHC-CREB-S133A-LZ TG mice.
Fig. 3.

CREB-S133A-LZ mice survive normally. Graph shows Kaplan-Meyer curves of CREB-S133A (n = 118), CREB-S133A-LZ line 1 (n = 88), CREB-S133A-LZ line 2 (n = 93), and NTG (n = 149) littermate control mice. Although early death of CREB-S133A mice became apparent by 20 wk of age, no significant death of CREB-S133A-LZ mice was evident by 1 yr.
Gene expression in CREB-S133A-LZ TG hearts
Increased β-MHC and BNP expressions, along with reduced SERCA2 expression, are genetic changes characteristically found in human and experimental heart failure, including in MHC-CREB-S133A TG mice (6). To determine whether survival of mutant CREB TG mice correlated with ventricular gene expression, we tested the expression of these genes associated with the heart failure phenotype using quantitative PCR. There was no difference in β-MHC, BNP, and SERCA2 transcript levels, relative to GAPDH, in 6-mo-old CREB-S133A-LZ mouse hearts (n = 7) compared with NTG hearts (n = 5) (Fig. 4). By comparison, CREB-S133A mice (n = 2) had significantly greater expression of BNP and β-MHC and a trend toward reduced SERCA2 (P = 0.06) (Fig. 4), consistent with their known heart failure phenotype (6). The normal gene expression pattern found in CREB-S133A-LZ mice is consistent with the leucine zipper mutant TG mice having a markedly less severe phenotype than CREB-S133A mice.
Fig. 4.
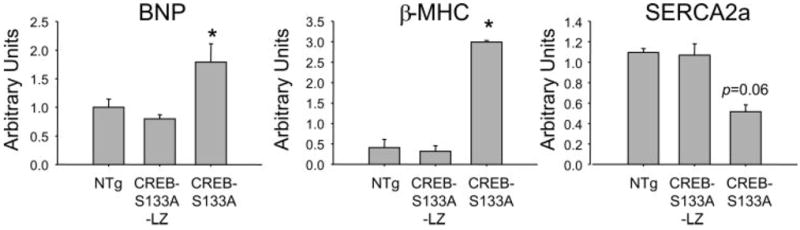
CREB-S133A-LZ hearts do not express markers of heart failure. Bars represent (means ± SE) brain natriuretic peptide (BNP; left), β-myosin heavy chain (β-MHC; middle), and sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2; right) transcript abundance relative to GAPDH. Whereas expression of the BNP and β-MHC transcripts was significantly different in CREB-S133A mice, the LZ mutant mice showed no difference compared with NTG controls. *P < 0.05 compared with NTG.
Cardiac function is markedly less impaired in CREB-S133A-LZ TG mice than in CREB-S133A mice
To determine the in vivo effects of the leucine zipper mutations on the generation of heart failure in the α-MHC-CREB-S133A model, we utilized echocardiography and LV catheterization to compare the cardiac function of 6-mo-old MHC-CREB-S133A-LZ mice with that of MHC-CREB-S133A mice and littermate NTG mice. Cardiac catheterization studies demonstrated that MHC-CREB-S133A-LZ TG mice have a similar heart rate and systolic blood pressure as NTG littermate control mice (Table 1). However, the systolic blood pressure was significantly lower in MHC-CREB-S133A mice. Systolic and diastolic cardiac performance is proportional to the change in the maximum and minimum first derivative of LV pressure during contraction (dP/dtmax) and relaxation (dP/dtmin), respectively. Consistent with the prior report (6), CREB-S133A mice were found to have a significantly reduced dP/dtmax and dP/dtmin (Table 1). dP/dtmax and dP/dtmin were also significantly blunted in CREB-S133A-LZ mice compared with NTG.
Table 1.
Hemodynamics in MHC-CREB-S133A and MHC-CREB-S133A-LZ NTG and TG mice
| NTG | CREB-S133A-LZ | CREB-S133A | |
|---|---|---|---|
| n | 7 | 6 | 5 |
| Heart rate, beats/min | 538±25 | 612±23 | 611±28 |
| Diastolic pressure, mmHg | 95.5±6.4 | 89.4±4.2 | 66.6±3.0*,† |
| Diastolic pressure, mmHg | 65.4±6.3 | 61.9±5.8 | 54.3±3.6 |
| dP/dtmax, mmHg/s | 11,676±532 | 7,527 ±501* | 6,480±189* |
| dP/dtmin, mmHg/s | 12,151±550 | 8,316±625* | 5,588±435*,† |
Values are means ± SE; n, number of animals. MHC, myosin heavy chain; TG, transgenic; NTG, nontransgenic; dP/dtmax and dP/dtmin, maximum and minimum first derivative of left ventricular (LV) pressure during contraction and relaxation, respectively.
P < 0.05 vs. NTG mice; and
P < 0.05 vs. cAMP response element binding protein (CREB)-S133A-leucine zipper (LZ) mice.
Echocardiography revealed that the LV end-diastolic diameter was similar in CREB-S133A-LZ and NTG animals, whereas the LV end-diastolic diameter was significantly greater in CREB-S133A mice (Table 2). LV end-systolic diameter was greater in MHC-CREB-S133A-LZ TG mice than in NTG mice; however, CREB-S133A mice demonstrated an even greater LV end-systolic diameter than did CREB-S133A-LZ mice (Table 2). Fractional shortening in MHC-CREB-S133A-LZ mice was significantly reduced compared with NTG mice; however, fractional shortening was reduced to a significantly greater extent in MHC-CREB-S133A mice than in MHC-CREB-S133A-LZ mice (Table 2). Taken together, these data support, therefore, that the introduction of the leucine zipper mutation partially reverses the dilated cardiomyopathic phenotype of the MHC-CREB-S133A mice.
Table 2.
Echocardiographic data in TG MHC-CREB-S133A and MHC-CREB-S133A-LZ TG vs. NTG mice
| NTG | CREB-S133A-LZ | CREB-S133A | |
|---|---|---|---|
| n | 7 | 6 | 5 |
| Heart rate, beats/min | 751±15 | 753±28 | 776±27 |
| LVEDD, mm | 2.5±0.1 | 2.7±0.2 | 3.9±0.1*,† |
| LVESD, mm | 1.0±0.1 | 1.4±0.1* | 3.0±0.1*,† |
| FS, % | 61.7±2.9 | 47.1±1.7* | 21.8±0.7*,† |
Values are means ± SE; n, number of animals. LVEDD and LVESD, LV end-diastolic and -systolic diameters, respectively; FS, fractional shortening.
P < 0.05 vs. NTG;
P < 0.05 vs. CREB-S133A-LZ.
Ventricular arrhythmias were not inducible in CREB-S133A-LZ mice
Because MHC-CREB-S133A TG mice can die suddenly and have significant tachy- and bradyarrhythmias (37), we tested the electrophysiological properties of MHC-CREB-S133A-LZ TG and NTG mice. The atrioventricular effective refractory period and the 2:1 atrioventricular cycle length (mean ± SD, 76.6 ± 5.8 vs. 80.0 ± 7.1, NTG vs. CREB-S133A-LZ, P = NS) were similar in MHC-CREB-S133A-LZ TG and NTG littermate control mice. Furthermore, whereas MHC-CREB-S133A mice were found to have significant spontaneous and inducible ventricular arrhythmias (37), no arrhythmias were induced by extra stimuli in TG MHC-CREB-S133A-LZ mice (n = 5) or NTG littermate control mice in this experiment (n = 3) using identical techniques.
DISCUSSION
When compared with MHC-CREB-S133A mice, which develop cardiomyopathy associated with overt heart failure, arrhythmia, and death (6, 7), MHC-CREB-S133A-LZ TG mice develop mild ventricular contractile dysfunction that is not associated with the expression of genetic heart failure markers, arrhythmia, or early mortality. Differences in contractile function between CREB-S133A and CREB-S133A-LZ mice were most apparent in echocardiography studies (Table 2) performed on lightly sedated awake mice. The deeper anesthesia required for cardiac catheterization studies may have limited our ability to detect significant differences other than dP/dtmin between CREB-S133A and CREB-S133A-LZ mice. The profound differences between these two TG models is unlikely to be a nonspecific effect (12), and it cannot be ascribed to differences in CREB protein expression because the CREB-S133A-LZ protein was as abundant, or more abundant, than the CREB-S133A protein (Fig. 2). These findings strongly suggest that the leucine zipper domain plays a critical role in the pathogenesis of severe cardiomyopathy and advanced heart failure in MHC-CREB-S133A mice. The less severe myopathic changes found in MHC-CREB-S133A-LZ TG mice may be caused by the inhibition of additional unknown transcriptional regulatory signals that are not dependent on intact leucine zipper function. However, because the leucine zipper mutant TG mice do not develop overt heart failure or premature death, these findings support the conclusion that the CREB leucine zipper is necessary for the severe heart failure phenotype caused by expression of the CREB-S133A mutant.
There are several possible mechanisms that may explain why CREB-S133A-dependent heart failure requires an intact leucine zipper. First, because the leucine zipper mutations used are known to significantly weaken DNA binding (21), CREB-S133A-LZ would not be expected to block p300/CBP-dependent transcription stimulated by the endogenous wild-type CREB protein (36). Second, transcriptional effects mediated by the interaction of CREB-S133A with the transducer of regulated CREB coactivator proteins (TORC) (15) would be unlikely because TORC protein binding requires the CREB basic/leucine zipper domain (15). Third, although CREB-S133A may broadly prevent downstream activation of CRE-regulated genes by forming complexes with endogenous CREB/CREM/ATF proteins, the leucine zipper mutant may be unable to sequester these proteins. An important contribution of this mechanism is supported by the GST pull-down protein-protein interaction studies which demonstrated that CREB-S133A, but not CREB-S133A-LZ, can bind endogenous CREB (Fig. 1B).
In addition to genomic mechanisms, the studies presented here suggest an important role of the CREB leucine zipper domain in regulating CREB phosphorylation. Numerous kinase signaling pathways, including PKA (18), Akt/PKB (4), and PKC (27), can phosphorylate CREB serine-133. Similar to the prior report (6), we found markedly reduced endogenous serine-133 phospho-CREB levels in the hearts of CREB-S133A TG mice (Fig. 2B). Conceivably, an overexpression of CREB protein with a mutated kinase inducible domain may competitively inhibit endogenous CREB phosphorylation. However, a direct inhibitory effect of the mutated kinase inducible domain is not likely because an overexpression of the CREM basic/leucine zipper domain alone, lacking the kinase inducible domain, also lowered phospho-CREB levels (25). Furthermore, our data show that overexpression of the mutated kinase inducible domain together with mutations in the leucine zipper does not lower endogenous phospho-CREB levels. Taken together, these findings strongly suggest that the CREB leucine zipper plays a critical role in regulating CREB phosphorylation.
Pharmacological kinase stimulants and inhibitors have a direct effect on cardiac function and heart failure. Overexpression of CREB/CREM/ATF family members in the heart has been found to lower endogenous phosphorylated CREB levels, and our studies demonstrate that the leucine zipper is required for this effect. The mechanism by which the CREB leucine zipper inhibits phosphorylation is not clear. The CREB leucine zipper is a strong interaction domain that forms homodimers; heterodimers are selectively formed with proteins containing a similar basic leucine zipper domain (28). PKA, the prototypical kinase that phosphorylates CREB (22), does not have a leucine zipper. However, PKA does form complexes with leucine zipper proteins (36), suggesting that either a direct or an indirect interaction may be possible. Changes in intracellular protein phosphatase activity induced by overexpression of CREB/CREB leucine zippercontaining proteins may also account for differences in endogenous CREB phosphorylation (25).
In conclusion, differences in cardiac CREB phosphorylation between CREB-S133A and CREB-S133A-LZ TG mice are associated with profound differences in heart failure severity seen in these two models. Future studies will help define whether the leucine zipper-dependent effects are mediated through a direct effect of CREB-S133A protein blocking p300/CBP-dependent gene expression at the genomic level or through a nongenomic effect that prevents CREB serine-133 phosphorylation. Taken together with prior reports, the data presented in this report demonstrate a critical role for the CREB leucine zipper in cardiomyocyte signaling and heart failure pathogenesis.
Acknowledgments
We thank Dr. Jeffrey M. Leiden for providing the MHC-CREB-S133A TG mouse model for this study and Jay Boltax for laboratory assistance.
GRANTS
These studies were supported by grants from the National Institutes of Health (G. S. Huggins and G. Reed) and the American Heart Association (G. S. Huggins).
References
- 1.Asahara H, Santoso B, Guzman E, Du K, Cole PA, Davidson I, Montminy M. Chromatin-dependent cooperativity between constitutive and inducible activation domains in CREB. Mol Cell Biol. 2001;21:7892–7900. doi: 10.1128/MCB.21.23.7892-7900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- 3.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci USA. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 5.Dwarki VJ, Montminy M, Verma IM. Both the basic region and the ‘leucine zipper’ domain of the cyclic AMP response element binding (CREB) protein are essential for transcriptional activation. EMBO J. 1990;9:225–232. doi: 10.1002/j.1460-2075.1990.tb08099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fentzke RC, Korcarz CE, Shroff SG, Lin H, Leiden JM, Lang RM. The left ventricular stress-velocity relation in transgenic mice expressing a dominant negative CREB transgene in the heart. J Am Soc Echocardiogr. 2001;14:209–218. doi: 10.1067/mje.2001.111473. [DOI] [PubMed] [Google Scholar]
- 8.Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell. 1991;64:739–749. doi: 10.1016/0092-8674(91)90503-q. [DOI] [PubMed] [Google Scholar]
- 9.Funamoto M, Hishinuma S, Fujio Y, Matsuda Y, Kunisada K, Oh H, Negoro S, Tone E, Kishimoto T, Yamauchi-Takihara K. Isolation and characterization of the murine cardiotrophin-1 gene: expression and nore-pinephrine-induced transcriptional activation. J Mol Cell Cardiol. 2000;32:1275–1284. doi: 10.1006/jmcc.2000.1161. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez GA, Yamamoto KK, Fischer WH, Karr D, Menzel P, Biggs W, 3rd, Vale WW, Montminy MR. A cluster of phosphorylation sites on the cyclic AMP-regulated nuclear factor CREB predicted by its sequence. Nature. 1989;337:749–752. doi: 10.1038/337749a0. [DOI] [PubMed] [Google Scholar]
- 11.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–9185. [PubMed] [Google Scholar]
- 12.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 13.Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, Labow MA. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isoda T, Paolocci N, Haghighi K, Wang C, Wang Y, Georgakopoulos D, Servillo G, Della Fazia MA, Kranias EG, Depaoli-Roach AA, Sassone-Corsi P, Kass DA. Novel regulation of cardiac force-frequency relation by CREM (cAMP response element modulator) FASEB J. 2003;17:144–151. doi: 10.1096/fj.01-0981com. [DOI] [PubMed] [Google Scholar]
- 17.Long F, Schipani E, Asahara H, Kronenberg H, Montminy M. The CREB family of activators is required for endochondral bone development. Development. 2001;128:541–550. doi: 10.1242/dev.128.4.541. [DOI] [PubMed] [Google Scholar]
- 18.Markou T, Hadzopoulou-Cladaras M, Lazou A. Phenylephrine induces activation of CREB in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J Mol Cell Cardiol. 2004;37:1001–1011. doi: 10.1016/j.yjmcc.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Matus M, Lewin G, Stumpel F, Buchwalow IB, Schneider MD, Schutz G, Schmitz W, Muller FU. Cardiomyocyte-specific inactivation of transcription factor CREB in mice. FASEB J. 2007;21:1884–1892. doi: 10.1096/fj.06-7915com. [DOI] [PubMed] [Google Scholar]
- 20.Mayr BM, Canettieri G, Montminy MR. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci USA. 2001;98:10936–10941. doi: 10.1073/pnas.191152098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayr BM, Guzman E, Montminy M. Glutamine rich and basic region/ leucine zipper (bZIP) domains stabilize cAMP-response element-binding protein (CREB) binding to chromatin. J Biol Chem. 2005;280:15103–15110. doi: 10.1074/jbc.M414144200. [DOI] [PubMed] [Google Scholar]
- 22.Meinkoth JL, Alberts AS, Went W, Fantozzi D, Taylor SS, Hagiwara M, Montminy M, Feramisco JR. Signal transduction through the cAMP-dependent protein kinase. Mol Cell Biochem. 1993;127-128:179–186. doi: 10.1007/BF01076769. [DOI] [PubMed] [Google Scholar]
- 23.Muller FU, Boknik P, Horst A, Knapp J, Linck B, Schmitz W, Vahlensieck U, Bohm M, Deng MC, Scheld HH. cAMP response element binding protein is expressed and phosphorylated in the human heart. Circulation. 1995;92:2041–2043. doi: 10.1161/01.cir.92.8.2041. [DOI] [PubMed] [Google Scholar]
- 24.Muller FU, Boknik P, Knapp J, Neumann J, Vahlensieck U, Oetjen E, Scheld HH, Schmitz W. Identification and expression of a novel isoform of cAMP response element modulator in the human heart. FASEB J. 1998;12:1191–1199. doi: 10.1096/fasebj.12.12.1191. [DOI] [PubMed] [Google Scholar]
- 25.Muller FU, Lewin G, Baba HA, Boknik P, Fabritz L, Kirchhefer U, Kirchhof P, Loser K, Matus M, Neumann J, Riemann B, Schmitz W. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem. 2005;280:6906–6914. doi: 10.1074/jbc.M407864200. [DOI] [PubMed] [Google Scholar]
- 26.Muller FU, Lewin G, Matus M, Neumann J, Riemann B, Wistuba J, Schutz G, Schmitz W. Impaired cardiac contraction and relaxation and decreased expression of sarcoplasmic Ca2+-ATPase in mice lacking the CREM gene. FASEB J. 2003;17:103–105. doi: 10.1096/fj.02-0486fje. [DOI] [PubMed] [Google Scholar]
- 27.Muthusamy N, Leiden JM. A protein kinase C-, Ras-, and RSK2-dependent signal transduction pathway activates the cAMP-responsive element-binding protein transcription factor following T cell receptor engagement. J Biol Chem. 1998;273:22841–22847. doi: 10.1074/jbc.273.35.22841. [DOI] [PubMed] [Google Scholar]
- 28.Newman JR, Keating AE. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science. 2003;300:2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 29.Okamoto Y, Chaves A, Chen J, Kelley R, Jones K, Weed HG, Gardner KL, Gangi L, Yamaguchi M, Klomkleaw W, Nakayama T, Hamlin RL, Carnes C, Altschuld R, Bauer J, Hai T. Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am J Pathol. 2001;159:639–650. doi: 10.1016/S0002-9440(10)61735-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- 31.Patten RD, Aronovitz MJ, Deras-Mejia L, Pandian NG, Hanak GG, Smith JJ, Mendelsohn ME, Konstam MA. Ventricular remodeling in a mouse model of myocardial infarction. Am J Physiol Heart Circ Physiol. 1998;274:H1812–H1820. doi: 10.1152/ajpheart.1998.274.5.H1812. [DOI] [PubMed] [Google Scholar]
- 32.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 33.Rube CE, Wilfert F, Uthe D, Schmid KW, Knoop R, Willich N, Schuck A, Rube C. Modulation of radiation-induced tumour necrosis factor alpha (TNF-alpha) expression in the lung tissue by pentoxifylline. Radiother Oncol. 2002;64:177–187. doi: 10.1016/s0167-8140(02)00077-4. [DOI] [PubMed] [Google Scholar]
- 34.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 35.Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 36.Tian L, Coghill LS, MacDonald SH, Armstrong DL, Shipston MJ. Leucine zipper domain targets cAMP-dependent protein kinase to mammalian BK channels. J Biol Chem. 2003;278:8669–8677. doi: 10.1074/jbc.M211661200. [DOI] [PubMed] [Google Scholar]
- 37.Zhu W, Lepore JJ, Saba S, Joseph S, Link MS, Homoud MK, Estes NA, Wang PJ, Leiden JM. Cardiac electrophysiologic abnormalities in the CREBA133 transgenic mouse model of idiopathic dilated cardiomyopathy. J Cardiovasc Electrophysiol. 2003;14:982–989. doi: 10.1046/j.1540-8167.2003.02002.x. [DOI] [PubMed] [Google Scholar]