

Recently, major advances in our understanding of the pathogenesis of sporadic myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) have been made through unbiased gene discovery approaches. Similar advances have been made in understanding the genetic basis of rare cases of familial MDS and AML. Currently, germline mutations in genes encoding transcription factors, including RUNX1, CEBPA, and more recently GATA2,1–8 have been identified in patients with familial MDS/AML. In patients with familial MDS/AML, however, there is great heterogeneity in the age of disease onset as well as the clinical characteristics of the myeloid malignancy which develops in affected members of such families. For instance, in the largest single survey of disease phenotypes in individuals with germline GATA2 mutations, 50% of patients were without symptoms at the age of 20 and 16% continued to remain without symptoms by the age of 40.7 In this issue of Haematologica, West et al. begin to unravel the genetic alterations that frequently occur together and collaborate with germline GATA2 mutations to promote the development of MDS and AML.8
In 2011, four papers were published identifying heterozygous germline GATA2 mutations as the cause of four previously described clinical syndromes: primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome),1 RUNX1/CEBPA wild-type familial AML/MDS,2 monocytopenia and mycobacterial infections (MonoMAC syndrome),9 and the dendritic cell, monocyte, B and NK lymphoid deficiency syndrome (DCML deficiency).10 Since then, approximately 200 patients with germline GATA2 mutations have been described (Table 1), each presenting with variety of clinical presentations but all with high risk of developing MDS/AML. In a summary of these studies, approximately 70% of GATA2-deficient individuals appear to develop MDS or AML in their lifetime.
Table 1.
Development and characteristics of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) in individuals with germline GATA2 mutations.1
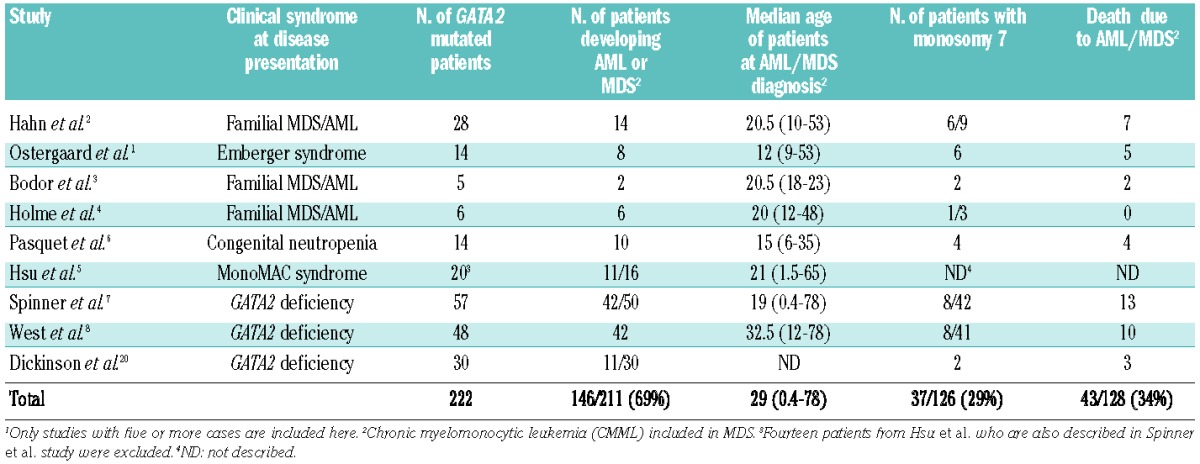
Interestingly, attempts have been made to correlate the risk and outcomes of myeloid malignancy in patients with germline GATA2 mutations with the genotype of the GATA2 mutation present, but this has been limited by the number of patients.7 Although monosomy 7 clearly appears to be enriched in GATA2-deficient individuals who develop MDS and AML (30% of individuals; Table 1), the first clue to a specific molecular abnormality which might be an important collaborating genetic event for the development of overt myeloid malignancy in GATA2 mutant families came from recent work by Bodor et al.3 In this prior study of a germline GATA2-mutant kindred, somatic ASXL1 mutations were present exclusively in the two members of the family who developed MDS/AML. This finding strongly suggested that ASXL1 mutations might be an important trigger for the development of overt disease in GATA2-mutated patients.
West et al. performed targeted sequencing of ASXL1 in 48 patients with germline GATA2 mutations and identified heterozygous ASXL1 mutations in 14 of them (29%). Given the rarity of ASXL1 mutations in individuals with myeloid malignancies less than 60 years old, the high frequency of ASXL1 mutations in GATA2-deficient individuals developing MDS/AML is remarkable. Eight different ASXL1 mutations were seen in ten different GATA2-mutant backgrounds. Similar to the pedigree studied by Bodor et al., in this study one pair of sisters had the same GATA2 mutation but only the sister who had an ASXL1 mutation actually developed clinically evident MDS. This finding further underscores the likely collaboration between GATA2 and ASXL1 mutations in promoting the development of MDS. In another informative pedigree here, two sisters with the same GATA2 mutation had discordant ASXL1 genotypes but both developed chronic myelomonocytic leukemia (CMML). This finding further validates the already strong link between the presence of ASXL1 mutations and the clinical phenotype of CMML.11
Although this study had relatively limited number of patients due to the rarity of germline GATA2 mutant patients, the authors were able to determine that overall survival in ASXL1-mutant/GATA2-mutant patients was worse than that in patients with an ASXL1 wild-type/GATA2 mutant genotype.8 This finding is quite consistent with those of larger studies in MDS patients revealing an unwavering association between the presence of ASXL1 mutations and adverse outcome.11,12
In contrast to de novo MDS, MDS occurring in patients with GATA2 germline mutations are usually hypocellular for age with increased reticulin fibrosis.7 Interestingly, results from the study by West et al. suggest that the development of ASXL1 mutations coincides with progression from the hypoplastic MDS characteristic of GATA2 deficiency to a more proliferative disease.8 More sensitive quantitative sequencing, comparing samples during the hypoplastic MDS phase of disease and during acute transformation will be needed to understand how early the ASXL1 mutations occur in the pathogenesis of myeloid disease in these individuals. This is especially relevant since most individuals with co-occurring GATA2 and ASXL1 mutations in this study had additional cytogenetic abnormalities such as monosomy 7.
Further unbiased genome-wide sequencing studies, currently being undertaken by this group and others, are needed to understand the full spectrum of somatic mutations in hematopoietic cells in individuals with disease evolution. For instance, recent whole exome sequencing of serial samples over a 17-year period from a single patient with severe congenital neutropenia progressing to AML showed a number of early and late genetic defects associated with leukemic progression.13 A nonsense mutation in the gene encoding for granulocyte colony-stimulating factor receptor, CSF3R, appeared to be a clear, early event in the severe congenital neutropenia phase of the disease. In contrast, mutations arising later in the development of AML included mutations in ASXL1, SUZ12, EP300, RUNX1, and an additional mutation in CSF3R. Based on the work by West et al., it is quite plausible that the development of MDS and AML in GATA2-deficient individuals might be similarly driven by a stepwise accumulation of genetic mutations with clonal expansion and selection, with ASXL1 seeming to play a central role.
The strong genetic link between GATA2 and ASXL1 mutations in patients with this rare germline disorder raises the question of the frequency of ASXL1 mutations in diseases marked by somatic GATA2 mutations. For instance, somatic GATA2 mutations have been described in Philadelphia chromosome-positive chronic myeloid leukemia patients at transformation to myeloid blast crisis.14 Most of these cases were associated with a gain-of-function mutation in GATA2 (Leu359Val) in contrast to GATA2 mutations seen in patients with GATA2-mutant germline syndromes. Somatic GATA2 mutations have also been identified in de novo AML. In such cases, GATA2 mutations tend to be enriched in normal karyotype AML patients with biallelic CEBPA mutations. From the limited data published on such patients, GATA2 mutant de novo AML does not appear to be significantly associated with ASXL1 mutations. Moreover, GATA2 mutations in de novo AML appear to be associated with a relatively favorable prognosis.15
The significant co-occurrence of GATA2 deficiency with ASXL1 mutations at development of MDS/AML strongly suggests a cooperative interaction of these genetic events in promoting hematopoietic transformation. ASXL1 is a Polycomb associated protein which has been shown to affect transcription through effects on the ability of the Polycomb repressive complex 2 to perform histone H3 lysine 27 methylation16 and also potentially by interacting with the histone H2A lysine 119 deubiquitinase enzyme BAP1.17 Genome-wide localization studies of ASXL1 by chromatin immunoprecipitation followed by next-generation sequencing recently showed that ASXL1 localizes strongly to promoter regions of the genome.18 Moreover, ASXL1 binding strongly overlaps with that of ETS transcription factors.18 It is now well understood that deletion of key ETS transcription factors, such as PU.1, promotes aggressive myeloid malignancies in vivo. Interestingly even lowering levels of PU.1 to 20% of normal levels promotes leukemogenesis. This observation highlights the importance of transcriptional regulation of ETS target genes in the pathogenesis of myeloid malignancy. Theses facts, taken together with the knowledge that GATA2 interacts with and represses PU.1,19 possibly suggest that the explanation for the genetic interaction between ASXL1 and GATA2 may lie in the intersection of these factors in transcriptional regulation of key PU.1 target genes. Given the importance of ASXL1 mutations in myeloid malignancies and the development of molecular knowledge regarding GATA2 function in hematopoiesis, future functional work dissecting the interaction of ASXL1 mutations and GATA2 haploinsufficiency may address this hypothesis. It is hoped that further in vitro and in vivo work will elucidate this fascinating genetic interaction identified by West et al.8
Acknowledgements
J-BM is supported by a grant from the Fondation de France. OA-W is supported by an NIH K08 Clinical Investigator Award (1K08CA160647-01), a U.S. Department of Defense Postdoctoral Fellow Award in Bone Marrow Failure Research (W81XWH-12-1-0041), the Josie Roberston Clinical Investigator Program, and a Damon Runyon Clinical Investigator Award with Support from the Evans Foundation.
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43(10):929–31 [DOI] [PubMed] [Google Scholar]
- 2.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bodor C, Renneville A, Smith M, Charazac A, Iqbal S, Etancelin P, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica. 2012;97(6):890–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I. Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. Br J Haematol. 2012;158(2):242–8 [DOI] [PubMed] [Google Scholar]
- 5.Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(19):3830–7, S1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasquet M, Bellanne-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2013;121 (5):822–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics and immunity. Blood. 2013. November 13 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2014;99(2):276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36 [DOI] [PubMed] [Google Scholar]
- 12.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119(22):5071–7 [DOI] [PubMed] [Google Scholar]
- 14.Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105 (6):2076–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2013. September 13 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 16.Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210(12):2641–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.May G, Soneji S, Tipping AJ, Teles J, McGowan SJ, Wu M, et al. Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell. 2013;13(6):754–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2013. December 17 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]