

Abstract
B-cell lymphomas comprise an increasing number of clinicopathological entities whose characterization has historically been based mainly on histopathological features. In recent decades, the analysis of chromosomal aberrations as well as gene and miRNA expression profile studies have helped distinguish particular tumor types and also enabled the detection of a number of targets with therapeutic implications, such as those activated downstream of the B-cell receptor. Our ability to identify the mechanisms involved in B-cell lymphoma pathogenesis has been boosted recently through the use of Next Generation Sequencing techniques in the analysis of human cancer. This work summarizes the recent findings in the molecular pathogenesis of B-cell neoplasms with special focus on those clinically relevant somatic mutations with the potential to be explored as candidates for the development of new targeted therapies. Our work includes a comparison between the mutational indexes and ranges observed in B-cell lymphomas and also with other solid tumors and describes the most striking mutational data for the major B-cell neoplasms. This review describes a highly dynamic field that currently offers many opportunities for personalized therapy, although there is still much to be gained from the further molecular characterization of these clinicopathological entities.
Introduction
B-cell lymphoma classification comprises a relatively large number of well-defined entities whose characterization has historically been based on the histopathological features of these tumors. In recent decades, many chromosomal translocations involving oncogenes and tumor suppressor genes have been described, among which C-MYC (MYC hereafter), CCND1 (CYCLIN-D1 hereafter), BCL2, BCL6, to name but a few, have given rise to a scenario in which these diseases (e.g. mantle cell lymphoma and follicular lymphoma) carry specific molecular alterations that can allow individual lymphoma types to be distinguished, with advanced stages of progression being recognizable because of the accumulation of alterations in tumor suppressor genes, such as TP53 or CDKN2A.1 Moreover, specific gene expression and miRNA profiling signatures have also helped to distinguish between particular tumor types as the consequence of the deregulation of specific genes or pathways.2 In addition to this, we now know that signaling from the B-cell receptor3,4 and co-receptors (CD19, Toll-like receptors)5,6 can be considered as triggers for activating critical signaling pathways that regulate a range of biological activities in B-cell lymphomas, including altered metabolism,7 enhanced proliferation and survival,1 and cell migration in response to soluble factors.8 These highlight the increasing interest in developing BCR downstream signaling inhibitors with the potential for use in clinical practice. Despite the great efforts made towards understanding the critical mechanisms governing B-cell lymphomagenesis, and the amount of data already generated, we are still faced with an imperfect scenario in which many well-defined clinicopathological entities still lack specific genetic markers with a pathogenic role, and where the clinical diversity of many of these conditions remains partially unexplained. More importantly, the genetic changes identified so far have not enabled targeted therapies to be assigned, so cytotoxic and immune therapies continue to be the main therapeutic tools for treating B-cell lymphoma.
Our ability to identify the mechanisms involved in B-cell lymphoma pathogenesis has been boosted recently through the use of Next Generation Sequencing (NGS) techniques in the analysis of human cancer. This has revealed an unsuspected degree of complexity in the cancer cell machinery.9,10 The application of these high-throughput sequencing techniques to the study of different lymphoma types has yielded a considerable amount of whole genome and exome/transcriptome sequencing data for the most frequent B-cell mature neoplasms, including diffuse large B-cell lymphoma (DLBCL),11–13 Burkitt Lymphoma (BL),14 multiple myeloma (MM),15 chronic lymphocytic leukemia (CLL),16–18 mantle cell lymphoma (MCL),19,20 follicular lymphoma (FL),21 hairy cell leukemia (HCL),22 lymphoplasmacytic lymphoma (LPL)23 and splenic marginal zone lymphoma (SMZL).24,25 Although the work continues, the data generated so far are helping us revise our understanding of the molecular pathogenesis of these lymphomas through the revelation of new oncogenic mechanisms and mutated genes. These results are providing us with a more solid rationale for the therapeutic targeting of these tumors.
Mutation frequency in B-cell lymphoma
Solid tumors have a notably variable mutation rate, from the high mutational index (MI; number of mutations/megabase (Mb)) of tobacco-associated lung cancer and UV-induced melanoma (MI > 7), to the lower rates for the non-hypermutated form of colorectal carcinoma (MI = 3.2) and breast cancer (MI = 1.4).26–31 Despite there being no established correlation between mutational load and clinical outcome, and considering that the current data have been generated using different NGS platforms, including whole genome (WGS), whole exome (WES) and whole transcriptome (mRNA-seq) approaches, we have compared the mutational index data of B-cell lymphomas obtained from independent NGS studies (Figure 1 and Table 1) and found that, in general, B-cell lymphomas have a mutational load that is lower than in UV-induced melanomas or tobacco-associated lung cancer, and roughly comparable to that seen in solid tumors in adults. Of the B-cell lymphomas, aggressive B-cell lymphomas (i.e. BL: MI = 4.2; DLCBL: MI = 1.7–3.2) have a higher MI than the other low-grade B-cell lymphomas analyzed (MM, SMLZ, CLL, MCL and HCL: MI ≤ 1). Analyzing the nucleotide substitutions, the rate between transitions (A/G or C/T base changes) versus transversions (C,T/A,G) (Figure 1), we observed that percentage differences between these nucleotide substitutions seem to be more restricted (mostly not exceeding 65% of transitions) in B-cell lymphomas than in the solid tumors induced by tobacco or UV radiation (melanoma > 72%) (Figure 1 and Table 1).
Figure 1.

Somatic mutations affecting B-cell lymphomas. The bar graph shows the mutational indexes (MIs) of B-cell lymphoma studies compared with selected studies reflecting those of solid tumours. MI indicates the number of mutations (possibly affecting protein activity) per Megabase (Mb), assuming the size of the human exome to be 30 Mb. The table shows the MI, range, percentage of transitions and transversions, sample size and type of mutational analysis: whole exome sequence (WES), whole genome sequence (WGS), mRNA-seq. (c) indicates cell lines used. (u) indicates untreated. Table 1. Data supporting Figure 1. Mutational Indexes (MI), range, percentage of transitions and transversions, sample size and type of mutational analysis: whole exome sequence (WES), whole genome sequence (WGS), mRNA-seq. (c) indicates cell lines used. *Focused on mutations affecting protein function.
Table 1.
Mutations affecting B-cell lymphoma main subtypes. Most frequent genetic alterations and mutations affecting B-cell lymphomas. Selection of important genetic events affecting specific types of B-cell lymphomas and those mutations now considered being important in the pathogenesis of each entity. Percentages are for the cohorts in each study. Other: multiple molecular events that can occur alongside mutations; N/D: not determined; N/A: not applicable.
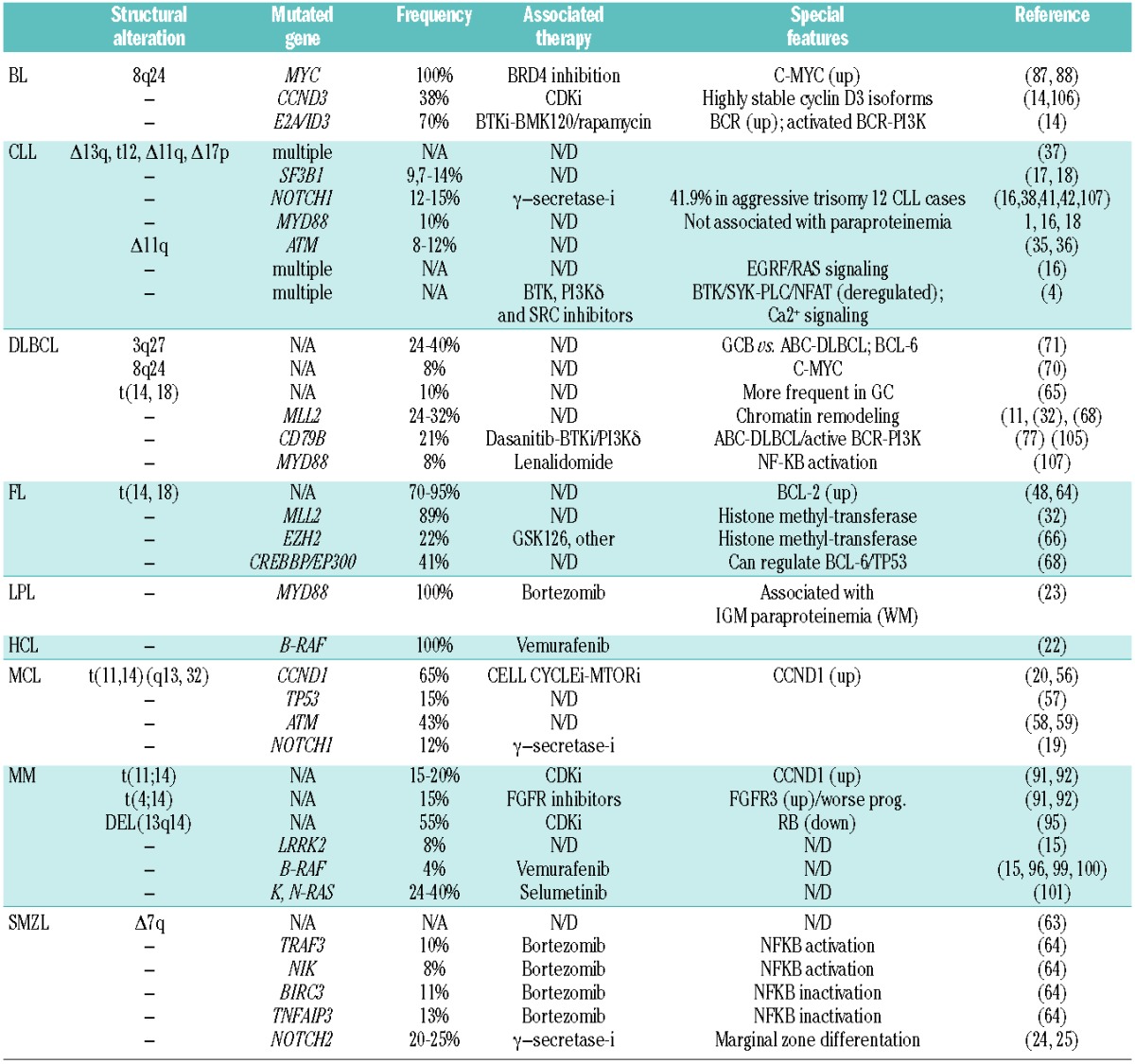
More remarkable results are summarized below.
Chronic lymphocytic leukemia
Cell survival in chronic lymphocytic leukemia (CLL) seems to depend on the integrated signaling derived from the B-cell receptor coupled with signaling from other surface receptors such as chemokine receptors (CXCR5/CXCL13, CXCR4/SDF1), TOLL-like receptors, the co-stimulators CD40/CD40L and integrins.33–35 Copy number studies have found multiple copy number variations in CLL, including deletions in 13q (33–64%) and 11q22.3(ATM) (10–20%) and trisomy 12 (15–25%), which have been associated with different probabilities of disease progression and survival.36 Amongst them, mutations and/or deletions in 17p affecting TP53 can be found in 3–12% of newly diagnosed CLL cases, with patients presenting del17p being highly resistant to conventional therapies.37
NGS analyses have recently revealed a relatively high number of recurrent somatic mutations within CLL cases. The most frequent mutation (4 – 15%) affects NOTCH1, and generates a premature stop codon, resulting in a C-terminal truncated NOTCH1 protein lacking the C-terminal domain, which contains a PEST sequence that, upon removal, results in the accumulation of an active protein isoform.16 Interestingly, NOTCH1 mutations are more frequent in IGVH-unmutated cases and have been associated with disease progression.16,38–41 More specifically, the NOTCH1 mutations have been found in up to 42% of trisomy 12 CLL cases.42
Additionally, SF3B1, a ubiquitously expressed splicing factor, has been found mutated in 5–17% of CLL cases, in which it is also associated with a poor prognosis, reduced overall survival and resistance to treatment with fludarabine, through a mechanism that still requires further attention since those genes targeted by the deregulation of this splicing factor remain to be fully identified.17 A variable proportion (3–10%) of CLL patients also carry the recurrent mutation L265P in the MYD88 gene; in these cases, MyD88 immunoprecipitation identified IRAK1 as binding MyD88. MyD88 plays an active part in the signaling pathways of interleukin-1 and Toll-like receptors during the immune response.16,18
Molecular prognosis in CLL has benefited from all these changes.43 Therefore, Rossi and co-workers have stratified CLL patients into subgroups: high-risk, with TP53 and/or BIRC3 abnormalities (10-year survival: 29%); intermediate-risk, with NOTCH1 and/or SF3B1 mutations and/or del11q22–q23 (10-year survival: 37%); low-risk, with +12 or normal cytogenetics (10-year survival: 57%); and very low-risk, with only del13q14, whose 10-year survival (69.3%) was comparable with that of the matched general population.43 There is a growing expectation that the mutational landscape and other genomic analysis in CLL may enable a more personalized targeted therapy. Thus, increased expression of BCR downstream effectors such as Bruton tyrosine kinase (BTK), SYK or PLC (Figure 2) has been correlated with a poorer prognosis and shorter time to progression,4 this association favoring a therapy based on BCR/BTK inhibitors. Additional evidence in favor of targeting BCR signaling is found in the mutational studies showing clustering of somatic mutations in genes belonging to pathways involved in BTK downstream effectors, such as those found in PLC/Ca2+ signaling, including PLC, CAMK and NFAT (6 of 105 of the patients analyzed by Quesada et al.).17 This is also supported by the BCR downstream pathway mutations detected in the series by Domenech et al.,44 which affects KRAS, SMARCA2, NFK-BIE and PRKD3, all of which have been shown to play a role in BCR, NF-kB and related signaling pathways.44 Additionally, members of the MAPK signaling pathway have been found mutated (e.g. EGFR, FGFR2, KRAS, BRAF)16,17 or with increased mRNA expression (e.g. increased expression of SOS1, an RAS upstream regulator and CYCLIN-D2, a cell cycle promoting protein).4
Figure 2.

Principal inhibitors of BCR downstream signaling. Principal signaling pathways activated downstream of BRC and its associated co-receptors. Brown: CD19-mediated PI3K/mTOR activity; Blue: MAPK activation; Red: BTK/PLC downstream activity and signaling through secondary messengers (Ca2+ and DAG); Purple: Toll-like receptors/Myd88 activation of NFKB; Pink: direct specific inhibitors of potential use in targeted therapy for B-cell lymphomas.
Different therapeutic agents (e.g. fosfomatininb, a SYK inhibitor; GS-1101, an inhibitor of PI3Kδ; ibrutinib (PCI-32765), a BTK inhibitor; and navitoclax (ABT-263), an inhibitor of BCL2 and related proteins) have shown promising results in in vitro studies and early clinical trials, although the association between response and specific mutational changes has still to be established.45–49 CLL sequencing projects have also illustrated the fascinating phenomenon of subclonal dynamics, which is partially shaped by the treatment received by the patients,50 a finding that clinical trials targeting mutated genes should take into account.
Hairy cell leukemia
Hairy cell leukemia (HCL) molecular analysis and targeted therapy is a success story in cancer genomic research.22 The use of a genome-wide sequencing approach has enabled the detection of a gain of function oncogenic mutation in BRAF (BRAF-V600E), which was previously identified as playing a critical role in other human cancers such as malignant melanoma and colorectal adenocarcinoma.51 Mutated cases expressed phosphorylated MEK and ERK (the downstream targets of the BRAF kinase), indicating a constitutive activation of the RAF-MEK-ERK pathway.22 This oncogenic alteration is highly specific to this type of lymphoma since it has only rarely been detected in other B-cell lymphomas such as SMZL, BL, follicular lymphoma (FL), DLBCL, MCL and CLL.22,52 Moreover, mutations in BRAF can be found in almost 100% of the cases and can be considered a driver mutation of this disease.
Thus, it is possible that HCL patients may benefit from a targeted inhibition of activated BRAF, following the recently described example of a refractory HCL treated with vemurafenib.53 In this respect, downstream of BRAF-V600E, high levels of phosphorylated MEK and ERK proteins in this pathway were detected in FFPE as well as purified HCL cell samples by IHC and IF,22 corroborating previous results from HCL cells.54 Moreover, since the BRAF-V600E mutation may largely explain the activation of MEK/ERK observed in HCL, it is possible that the detection of this mutation using a specific antibody55 may serve to discriminate qualitative (HCL from HCL-like) and quantitative (percentage of positive BRAF cells) differences between those cases of non-responsive HCL that may require alternative therapy.
Mantle cell lymphoma
Mantle cell lymphoma (MCL) is a clinically challenging B-cell lymphoma that responds poorly to conventional chemotherapy. Malignant B cells present in this lymphoma are CD5-positive and have a higher level of expression of CYCLIN-D1 due to the chromosomal translocation t(11,14)(q13, 32), which is present in up to 95% of cases and usually co-exists with a number of other oncogenic molecular features, including enhanced expression of MYC, inactivation of the cell cycle inhibitors p16/INK4A and p14/ARF, or gains of 3q, 12q and losses of 9p, 9q and 17p.56 Mutations in TP53 affect up to 15% of MCL cases, usually giving rise to high-risk variants with high proliferative activity and poor prognosis.57 Alongside this, truncating or missense mutations involving the PI3K domain in the ATM gene have also been found in 40–45% of MCL cases.58,59
More recently, mutations in NOTCH1, which produce a truncated protein in the C-terminal with increased oncogenic activity, were detected in up to 12% of the clinical samples analyzed,19 and those patients harboring this mutation had a shorter OS than controls (1.43 vs. 3.86 years, respectively). This mutation can have therapeutic implications since γ-secretase inhibitors preventing the generation of the oncogenic domain of Notch and suppressing the Notch activity are under development60 and could be used in NOTCH1-positive MCL cases. A global NGS analysis of somatic mutations in MCL samples has recently been published.20 The results obtained showed up to 34.5% of the samples harboring mutations in CYCLIN-D1. Moreover a number of mutated genes that could potentially co-operate with CYCLIN-D1 in the pathogenesis of MCL were also detected. Among these, WHSC1 and MLL2, two genes with epigenetic activity as histone methyltransferases, carried mutations in 10% and 14% of the samples. In addition, a number of genes involved in intracellular signaling have been found recurrently mutated, like the anti-apoptotic gene BIRC3 (6%) or NOTCH1 (5.2%) and NOTCH2 (4.7%). These results are pointing at multiple potential therapeutic applications ready to be explored. In this respect, MCL researchers have identified the loss of 9p, 19p and 6q24/25, leading to the reduced expression of several members (MOBKL2A, MOBKL2B and LATS2, respectively) of the Hippo pathway,61 a finding that has been associated with poorer MCL survival.62
Splenic marginal zone lymphoma
Although splenic marginal zone lymphomas (SMZL) lack recurrent translocations, up to 45% of cases have 7q deletions, which are considered to be their main cytogenetic abnormality.63 Genomic analysis using NGS approaches have identified multiple mutations in the NF-kB signaling pathway that activate this pathway in up to 58% of SMZL cases.64 Of the latter, inactivating mutations can be detected in negative regulators of NF-kB activity such as BIRC3 (11%), IKBKB (3%) and TNFAIP3 (13%), whereas activating mutations may be present in other molecules such as TRAF3 (10%) and NIK (8%).25 More recently, NOTCH2, a gene required for marginal zone differentiation, was found to be mutated in 20–25% of SMZL cases.24,25 This NOTCH2-Q2325X mutant generates a gain of function C-terminal truncated mutant that lacks degradation signals and hence increases protein accumulation and the subsequent activation of this signaling pathway. The clinical relevance of NOTCH2 mutations is still under debate,24,25 and the role of γ-secretase inhibitors remains to be explored.
Follicular lymphoma
Most follicular lymphoma (FL) cases are characterized by the hallmark t(14;18)(q32;q21) translocation, which brings about constitutive overexpression of the apoptosis inhibitor BCL2 protein.65 Mutational profile studies of FL patients now reveal a number of abnormalities mostly affecting chromatin remodeling proteins that are being explored for their suitability as FL therapeutic targets.66 Thus, FL samples harbor mutations in chromatin regulatory genes such as MLL2 (in 89% of cases reported by Morin et al.32), EZH2, a histone methyl transferase (22% of cases presenting activating mutations in codon 641,21,67) or the closely related histone acetyltransferases CREBBP (36%) and EP300 (8%).21 Importantly, mutant CREBBP and EP300 proteins were shown to be deficient in acetylating BCL6 and p53, leading to constitutive BCL6 activation and to a decreased p53 tumor suppressor activity.68 In the light of these data, it is conceivable that drugs targeting EZH2 such as those already described (GSK126)66 or acetylation/deacetylation mechanisms can be considered good candidates for combination FL therapy. In addition, FL samples contain mutations in TNFRSF14 (a member of the superfamily of TNF receptors) and other genes.69
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) molecular pathogenesis divides it into GCB-DLCBL (germinal center B cell), the more aggressive ABC-DLBCL (activated B cell),70 and primary mediastinal (PM) DLBCL, a tumor displaying some Hodgkin-like molecular traits. Molecular alterations, like translocations involving BCL2 oncogene loci, have been found in 30–40% of cases of GCB-DLBCL,70 whereas translocations affecting BCL6 seem to occur more frequently in the ABC-subtype.71 Genetic changes and sensitivity to targeted drugs seem to be essentially related with this molecular subclassification.
More than 300 genes, with a long tail distribution and strong variations between patients, have been found to be recurrently mutated in DLBCL, an enormous diversity that should be taken into account when selecting possible targets.72 The list of more frequently mutated genes includes TP53, MYD88, PIM1, CARD11, BCL6, CREBBP, EZH2 and others.72
A significant number of these somatic mutations involve members of the NF-kB pathway, including MYD88. Thus, 29% of ABC-DLBCL have been shown to carry an activating mutation (MYD88-L265P) that can promote cell survival by assembling a protein complex containing IRAK1 and IRAK4 which leads to IRAK4 kinase activity towards IRAK1 phosphorylation, and a subsequent activation of NF-κB signaling, secretion of IL-6, IL-10 and interferon-β which have been shown to elicit a latter auto-paracrine activation of JAK kinases and STAT proteins, hence making MYD88 a major downstream effector of BTK signaling (39% of the total number of DLBCL cases analyzed) towards NF-KB activation.11,72–77 In general, mutations in signaling pathways converging on NF-kB activation have been detected in 63% of the activated ABC cases compared with 31% of the GCB-DLBCL samples analyzed.11 The ABC signature in DLBCL75,77 is associated with a more aggressive phenotype, a finding that has led to the establishment of a mouse model consisting of activated NF-kB signaling against a BLIMP knock-out background that causes an ABC-DLBCL-like disease.74 This model may help develop new therapeutic alternatives for the treatment of ABC-DLBCL, including combination strategies using NF-kB inhibitors in conjunction with other agents (Figure 2).
A complex biological interrelationship has been shown between STAT3 and NF-kB pathways in ABC-DLBCL.73 JAK-STAT pathway can be considered actionable in DLBCL, since its inactivation using pharmacological or genetic inhibition in ABC-DLBCL cells has been shown to inhibit cell proliferation and trigger apoptosis. This observation is accompanied with an increased expression and activation of important oncogenes, such as C-JUN and MYC, and decreased expression of p27/KIP2 (an anti-oncogene that regulates cell cycle and differentiation programs), which have been found deregulated in ABC-DLBCL samples.78
BCR signaling is critical for B-cell lymphomagenesis and, therefore, can be considered a major therapeutic target (see Discussion) (Figure 2).79 Using a combinatory approach of DLBCL biopsies and cell lines, it has been shown that chronically active BCR signaling in ABC-DLBCL can be dependent on BTK-dependent signaling or can be elicited by somatic mutations affecting the immunoreceptor tyrosine-based activation motif (ITAM) signaling modules of CD79B and CD79A and also can be influenced by mutations in the signaling adaptor CARD11 which can lead to enhanced NF-kB signaling in ABC-DLBCL.77 These results highlight the therapeutic potential of targeting BCR downstream effectors with specific pharmacological agents like dasatinib, for the SRC family of tyrosine kinase inhibitors,80 SYK inhibitors,8 ibrutinib (PCI-32765), a selective BTK inhibitor of proven value in the treatment of relapsed/refractory B-cell malignancies,47,81 or CAL-101, a PI3Kδ inhibitor.82
Our perspective on the mutational landscape governing DLBCL pathogenesis has improved after learning that a number of nuclear proteins that control chromatin structure and gene expression harbor mutations like MLL2, a histone methyltransferase that is inactivated in 24–32% of cases,32,68 MEF2B (15.7% reported by Morin et al.32) and EZH2, a protein that forms part of the repressive PRC2 (polycomb repressive complex 2) and has been found bearing activating mutations in 32% of DLBCL cases,32 or CREBBP/EP300 mutations, as present in 39% of DLBCLs.68 This observation may be clinically relevant in the near future since selective inhibition of EZH2 activity using a pharmacological inhibitor (GSK126) has been shown to be efficient in anti-DLBCL cell growth activity both in vitro and in vivo.66
Opportunities for targeting GC-DLBCL have also emerged from the study by Amengual and co-workers, which recently demonstrated that treatment of DLBCL cell lines with pan-DAC inhibitors in combination with niacinamide produces synergistic cytotoxicity in GC-DLBCL, correlated with acetylation of both Bcl6 and p53.83
Rational targeting of actionable mutated genes in DLBCL is nevertheless a challenge, particularly after the discovery of an unexpected high grade of mutational/genetic heterogeneity found in the different NGS studies of human DLBCLs,72 and following the observations that ongoing acquisition of mutations and intratumoral clonal heterogeneity are frequent findings in DLBCL.13 Both the prognostic and predictive value of these mutations is still a matter of active research.
Burkitt lymphoma
Rearrangements in 8q24 occur in almost all cases of Burkitt lymphoma (BL), leading to a deregulated expression of the MYC oncogene.84 This abnormal MYC/MAX activity directly regulates the expression of multiple sets of genes85 hence controlling a plethora of cellular activities such as the activation of proliferation/metabolism and inhibition of differentiation and apoptosis.86 However, current BL therapies are still far from targeting this activity effectively, since MYC has not been found to be an actionable target so far. Nevertheless, it has been recently proposed that indirect targeting of MYC via BRD4, a transcriptional regulatory protein known to up-regulate MYC, may help target human cancers (like BL) highly dependent on MYC activity.87,88
Newly generated NGS data are improving our understanding of BL, enabling the detection of multiple mutations in genes with the potential to co-operate with MYC in B-cell lymphomagenesis. In this regard, two recent studies showed that up to 70% of BLs had mutations in the transcription factor TCF3 (E2A) or its negative regulator ID3.14,89,90 These findings may have important implications for therapy since E2A/ID3 mutations elicited a concomitant activation of BCR/PI3K signaling, and raised the possibility of using targeted inhibition of BCR downstream signaling through the usage of BTK inhibitors47 or PI3K/mTOR inhibitors (including BMK120 or rapamycin/rapalogs).91 Other mutated genes were CYCLIN-D3 (in 38% of sporadic cases) and members of the E2F family of transcription factors (in 11%), playing a role in the progression of cell cycle and potential indicator for the targeted inhibition of CDK/cell cycle progression.92 These findings are consistent with a recent publication showing that transgenic mice with deregulated MYC/PI3K activity in their B cells develop BL tumors that are phenotypically and genetically similar to those of humans, with multiple somatic mutations in CYCLIN-D3, similar to those found in human BL specimens.93 Finally, the most frequently mutated gene in BL is MYC14 (up to 70% of cases), although the functional relevance of these mutations remains to be fully clarified.
Multiple myeloma
Multiple myeloma (MM) is a complex malignancy characterized by a range of structural chromosomal changes, including multiple recurrent IGH reciprocal translocations such as t(11;14)(q13;q32), t(4;14)(p16;q32), t(14;16)(q32;q23) and del(13q14), which result in the increased expression of CYCLIN-D1, the membrane receptor FGFR3, the oncogene C-MAF and the loss of RB expression, respectively.94,95 It has been proposed that other secondary alterations can occur as this tumor progresses, including somatic mutations like those known to activate the RAS family of monomeric GTPases (24–50% of MMs) and the acquisition of t8q24 (50% of advanced MMs), which increases MYC expression.96,97 RAS downstream signaling also seems to be central to the pathogenesis of MM, since RAS itself and other downstream RAS effectors (i.e. BRAF) bear activating mutations that can be found in up to 50% of MM cases.15,98
Previous observations highlighted NF-kB signaling as a critical survival mechanism for MM cells, with copy number variations and mutations affecting up to 20% of the primary MM cases.99,100 Sequencing studies have confirmed and extended these observations through the identification of multiple mutations in NF-kB-associated genes that may affect up to one-third of the cases.15 These findings have provided a rationale for the successful usage of NF-kB inhibitors, including drugs such as thalidomide, lenalidomide and bortezomib.101 Other genes found mutated are DIS3, a gene involved in mRNA processing, and a number of genes with epigenetic activities such as MLL, MLL2, MLL3 and UTX.15 Therapeutic targeting of MM patients will nevertheless need to address the dynamic changes being described through the life of the tumor.98,102,103
Discussion
The mutational analysis presented in this review indicates that the total number of somatic mutations in BCL is much lower than in UV-induced or tobacco-induced solid tumors, but is still in the range of other solid adult tumors like breast or colorectal cancer (rates of 0.5–4 mutations/Mb).28 Within B-cell lymphomas, it seems that CLL and HCL carry a lower proportion of mutated genes, with some CLL cases even failing to show exomic mutations in potentially relevant genes. These findings could have beneficial consequences for targeted therapy, since it might be easier to detect key targets for the development of specific personalized therapies in tumors with lower mutational indexes. Despite the relatively low MI observed, the collected data strikingly confirm that some entities previously defined by precise histological features have underlying specific molecular alterations, as seems to be the case for LPL (MYD88), HCL (BRAF) and SMZL (NOTCH2), thus confirming the existence of these entities and indicating that these tumor type-specific events could be used for molecular diagnosis of individual tumor types.
In addition, sequencing data also show that not only specific genes, but also a number of signaling pathways are recurrently mutated in various lymphoma types. For instance, mutations in genes and pathways already suspected to participate in B-cell lymphoma pathogenesis, like those elicited by BCR/BTK, PI3K, RAS/MAPK, TLR or NF-kB, have been confirmed, but sequencing studies also show that other unsuspected pathways, like those affecting chromatin remodeling and transcriptional control of gene expression, are deregulated as a consequence of very frequent mutational events that occur in almost every type of lymphoma.
Mutated genes and pathways seem to point towards strong links between precise B-cell differentiation program alterations and specific tumor types. Thus, MZL cases carry mutations in genes involved in normal marginal zone differentiation (NOTCH2 and others), FL and GC-DLBCL show mutations in EZH2,104 MZL and post-GC tumors display mutation in NF-kB genes, while ABC DLBCL and MZL cases carry changes in genes involved in chronic active BCR signaling.
The mutational analysis of B-cell lymphomas highlights the role of deregulated BCR signaling in B-cell lymphoma-genesis. The most relevant BCR and co-receptor signaling pathways, together with the specific inhibitors that are currently available or under clinical development for specifically targeting deregulated BCR-downstream pathways, depending on the molecular context, are summarized in Table 1, illustrated in Figure 2, and described below.
Downstream of BCR, BTK can activate PLC-gamma and generate secondary messenger signaling that triggers the activation of important effectors such as PKC (in response to DAG) and CaM/Calcineurin (in response to transient Ca2+ release). Deregulated expression of multiple members of this pathway, as well as multiple somatic mutations, have been detected in different B-cell lymphoma types. This finding is of particular interest since a number of pharmacological agents have been developed to target BCR downstream activity (Figure 2), being of particular interest the data obtained from the BTK inhibitor PCI32765 (ibrutinib) in multiple B-cell lymphomas including MCL, a lymphoma known not to be responsive to current therapies and that showed improved clinical responses to this inhibitor.47 Along these lines of evidence, it has recently been shown that a group of relapsed/refractory ABC DLBCL patients with enhanced/chronically active BCR signaling can specifically benefit from receiving ibrutinib,105 thus highlighting the use of molecular characterization as a powerful tool for guiding new targeted therapies. In addition, downstream of BCR/SYK-LYN tyrosine kinases, the RAS-MAPK pathway is frequently altered in B-cell lymphomas (e.g. HCL or MM) and probably plays a role in promoting a more transformed phenotype, possibly through the MAPK-mediated activation of highly oncogenic transcription factors such as AP-1 or MYC.108 Activating mutations directly affecting RAS or RAF protein activities have been found in most HCL (BRAF) and in MM (N- and K-RAS), but only rarely in other lymphopro-liferative lesions. This contrasts with the findings in solid tumors and AML, where activating BRAF and RAS mutations are relatively common events.
As part of this intricate signaling network, deregulated PI3K/mTOR signaling can sustain the metabolic requirements of highly proliferative lymphomas, such as BL,110 thereby possibly co-operating with MYC, which is typically over-expressed in all BL cases and targets the expression of a number of metabolism genes.111 In this respect, mutations that give rise to activated E2A-mediated transcription and elevated PI3K signaling have been found recurrently in BL, highlighting a possible requirement for PI3K/mTOR in early stages of Burkitt lymphomagenesis. It seems that highly proliferative lymphomas like BL or specific cases of MM112 may be excellent case scenarios for therapeutic targeting of this pathway with rapamycin or its derivatives.113
Playing a key role in downstream signaling from BCR, TLR and other co-receptors, NF-kB activity seems to be deregulated in the majority of B-cell lymphomas, although specific features of activation and potential inhibition may differ among specific entities and even tumor subgroups. Deregulated NF-kB can be elicited by the numerous activating mutations found in NF-kB upstream regulators including those found in MYD88 (in CLL, DLBCL, LPL) or in NIK/TNFAIP3 in SMLZ, to name but a few. Through these mutations, B-cell lymphoma cells can acquire survival advantages and also promote oncogenic interactions with the microenvironment. These biological activities can be elicited through the transcriptional activation of multiple cytokines and soluble factors such as IL-6, IL-4 and IL-10, which also control normal B-cell activities.106 Promising pre-clinical data show that the novel IRAK4 inhibitors (ND-2110 and ND-2158), when combined with the BTK-inhibitor, ibrutinib, work synergistically, inducing apoptosis in B-cell lymphoma with the L265 MYD88 mutation.76
In addition to considering BCR downstream signaling as a mayor target for the development of new and future therapeutic strategies, NGS approaches have enabled the detection of a number of somatic mutations affecting chromatin remodeling proteins. These include MLL2, EZH2, MEF2B, SMARCA2, ARID1A, EP300 and CREBBP, and splicing factors such as SF3B1.17,18 This has broadened our mechanistic perspective on the molecular pathogenesis of B-cell lymphomas and is likely to have an important influence on the direction of future clinical studies.
Analyzing the current mutational data from a broader perspective, the results of NGS projects in B-cell lymphoma have essentially confirmed the taxonomy we currently apply in diagnosis, but at the same time, several layers of complexity are being added to our understanding of lymphoma pathogenesis. Mutational rate and specific mutated pathways and genes may differ markedly within individual entities, as has recently been shown in DLBCL,72 hence suggesting that therapeutic indications may require mutational signatures to be established case by case. In addition, initial data are starting to show that at least some BCL may carry a large number of micro-clones with rich complex dynamics throughout the life of the tumor,13,21,98,102,103,107 which may imply that mutational signatures need to be re-investigated at different times during the life of the tumor in order to capture its changing nature.
Lessons learned from solid and hematopoietic tumors suggest that the likely scenario for a more successful targeted therapy will be based on a combination of drugs targeting actionable mutated genes or deregulated pathways detected in major or minor clones within the tumor. The information we now have available suggests that a great effort should be made to understand better the inhibition of genes regulating chromatin conformation and together with other better characterized targets.
Finally, together with the somatic mutations described above, B-cell lymphomas harbor multiple structural aberrations, including CNVs and translocations, whose role in the pathobiology, characterization and clinical significance has been extensively studied (Table 1). Nevertheless, NGS analysis may still enable the detection of multiple structural changes affecting yet-to-be-determined coding and non-coding regions of the genome that may have critical roles in cancer (e.g. ENCODE project).
Acknowledgments
This work has been supported by grants from the Asociación Española contra el Cáncer (AECC), the Spanish Ministerio de Educación y Ciencia (SAF2008-03871) and the Sociedad para el Desarrollo Regional de Cantabria (Gobierno de Cantabria-SODERCAN). MSB currently holds a Miguel Servet contract from the Fondo de Investigación Sanitaria (Instituto de Salud Carlos III, FIS), Spain.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101(4):1220–35 [DOI] [PubMed] [Google Scholar]
- 2.Di Lisio L, Martinez N, Montes-Moreno S, Piris-Villaespesa M, Sanchez-Beato M, Piris MA. The role of miRNAs in the pathogenesis and diagnosis of B-cell lymphomas. Blood. 2012;120(9):1782–90 [DOI] [PubMed] [Google Scholar]
- 3.Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–20 [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez A, Villuendas R, Yanez L, Gomez ME, Diaz R, Pollan M, et al. Molecular heterogeneity in chronic lymphocytic leukemia is dependent on BCR signaling: clinical correlation. Leukemia. 2007;21(9):1984–91 [DOI] [PubMed] [Google Scholar]
- 5.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. 2012; 12(4):282–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012;122(6):2257–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22(4):547–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoellenriegel J, Coffey GP, Sinha U, Pandey A, Sivina M, Ferrajoli A, et al. Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia. 2012;26(7):1576–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239): 719–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109(10):3879–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339): 467–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012; 44(1):47–52 [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26): 2497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–71 [DOI] [PubMed] [Google Scholar]
- 20.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121(9):1604–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011; 364(24):2305–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–33 [DOI] [PubMed] [Google Scholar]
- 24.Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209 (9):1553–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490 (7418):61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44(10):1104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485 (7399):502–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K, Aoude LG, et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet. 2012; 44(2):165–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hillmen P. Using the biology of chronic lymphocytic leukemia to choose treatment. Hematology Am Soc Hematol Educ Program. 2011;2011:104–9 [DOI] [PubMed] [Google Scholar]
- 34.Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010;115(22): 4497–506 [DOI] [PubMed] [Google Scholar]
- 35.Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489 (7415):309–12 [DOI] [PubMed] [Google Scholar]
- 36.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000; 343(26):1910–6 [DOI] [PubMed] [Google Scholar]
- 37.Foa R, Del Giudice I, Guarini A, Rossi D, Gaidano G. Clinical implications of the molecular genetics of chronic lymphocytic leukemia. Haematologica. 2013;98(5):675–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Giudice I, Rossi D, Chiaretti S, Marinelli M, Tavolaro S, Gabrielli S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3): 437–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012; 119(2):521–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balatti V, Lerner S, Rizzotto L, Rassenti LZ, Bottoni A, Palamarchuk A, et al. Trisomy 12 CLLs progress through NOTCH1 mutations. Leukemia. 2013;27(3):740–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oscier DG, Rose-Zerilli MJ, Winkelmann N, Gonzalez de Castro D, Gomez B, Forster J, et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood. 2013;121(3):468–75 [DOI] [PubMed] [Google Scholar]
- 42.Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119(2):329–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121(8):1403–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Domenech E, Gomez-Lopez G, Gzlez-Pena D, Lopez M, Herreros B, Menezes J, et al. New mutations in chronic lymphocytic leukemia identified by target enrichment and deep sequencing. PLoS One. 2012;7(6): e38158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Has Significant Activity in Patients With Relapsed/Refractory B-Cell Malignancies. J Clin Oncol. 2012;31(1)88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1): 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54 [DOI] [PubMed] [Google Scholar]
- 52.Tiacci E, Schiavoni G, Forconi F, Santi A, Trentin L, Ambrosetti A, et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood. 2012;119(1):192–5 [DOI] [PubMed] [Google Scholar]
- 53.Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366(21):2038–40 [DOI] [PubMed] [Google Scholar]
- 54.Kamiguti AS, Harris RJ, Slupsky JR, Baker PK, Cawley JC, Zuzel M. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene. 2003;22(15):2272–84 [DOI] [PubMed] [Google Scholar]
- 55.Andrulis M, Penzel R, Weichert W, von Deimling A, Capper D. Application of a BRAF V600E mutation-specific antibody for the diagnosis of hairy cell leukemia. Am J Surg Pathol. 2012;36(12):1796–800 [DOI] [PubMed] [Google Scholar]
- 56.Jares P, Campo E. Advances in the understanding of mantle cell lymphoma. Br J Haematol. 2008;142(2):149–65 [DOI] [PubMed] [Google Scholar]
- 57.Greiner TC, Moynihan MJ, Chan WC, Lytle DM, Pedersen A, Anderson JR, et al. p53 mutations in mantle cell lymphoma are associated with variant cytology and predict a poor prognosis. Blood. 1996;87(10):4302–10 [PubMed] [Google Scholar]
- 58.Camacho E, Hernandez L, Hernandez S, Tort F, Bellosillo B, Bea S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood. 2002;99(1): 238–44 [DOI] [PubMed] [Google Scholar]
- 59.Fang NY, Greiner TC, Weisenburger DD, Chan WC, Vose JM, Smith LM, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci USA. 2003;100(9):5372–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pui CH. T cell acute lymphoblastic leukemia: NOTCHing the way toward a better treatment outcome. Cancer Cell. 2009;15(2):85–7 [DOI] [PubMed] [Google Scholar]
- 61.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hartmann EM, Campo E, Wright G, Lenz G, Salaverria I, Jares P, et al. Pathway discovery in mantle cell lymphoma by integrated analysis of high-resolution gene expression and copy number profiling. Blood. 2010; 116(6): 953–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salido M, Baro C, Oscier D, Stamatopoulos K, Dierlamm J, Matutes E, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group. Blood. 2010;116(9):1479–88 [DOI] [PubMed] [Google Scholar]
- 64.Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, et al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118(18):4930–4 [DOI] [PubMed] [Google Scholar]
- 65.Negrini M, Silini E, Kozak C, Tsujimoto Y, Croce CM. Molecular analysis of mbcl-2: structure and expression of the murine gene homologous to the human gene involved in follicular lymphoma. Cell. 1987;49(4):455–63 [DOI] [PubMed] [Google Scholar]
- 66.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427)108–12 [DOI] [PubMed] [Google Scholar]
- 67.Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS One. 2011;6(12):e28585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011; 471(7337):189–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70(22):9166–74 [DOI] [PubMed] [Google Scholar]
- 70.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(25): 1937–47 [DOI] [PubMed] [Google Scholar]
- 71.Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia. 2007;21(11):2332–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110 (4):1398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111(7): 3701–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18(6):580–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111(3):1515–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Witzig TE, Gupta M. Signal transduction inhibitor therapy for lymphoma. Hematology Am Soc Hematol Educ Program. 2010;2010:265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carbox-amide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49(23):6819–32 [DOI] [PubMed] [Google Scholar]
- 81.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1): 58–61 [DOI] [PubMed] [Google Scholar]
- 82.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110 delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amengual JE, Clark-Garvey S, Kalac M, Scotto L, Marchi E, Neylon E, et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013; 122(12):2104–13 [DOI] [PubMed] [Google Scholar]
- 84.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1982;79 (24):7824–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci USA. 2003;100(14):8164–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lutz W, Leon J, Eilers M. Contributions of Myc to tumorigenesis. Biochim Biophys Acta. 2002;1602(1):61–71 [DOI] [PubMed] [Google Scholar]
- 87.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011; 478(7370):524–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012; 44(12)1316:20. [DOI] [PubMed] [Google Scholar]
- 90.Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012; 44(12):1321–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Limon JJ, Fruman DA. Akt and mTOR in B Cell Activation and Differentiation. Front Immunol. 2012;3:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, et al. Therapeutic Targeting of the Cyclin D3:CDK4/6 Complex in T Cell Leukemia. Cancer Cell. 2012;22(4):452–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22(2): 167–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20(40):5611–22 [DOI] [PubMed] [Google Scholar]
- 95.Schmidt-Hieber M, Gutierrez ML, Perez-Andres M, Paiva B, Rasillo A, Tabernero MD, et al. Cytogenetic profiles in multiple myeloma and monoclonal gammopathy of undetermined significance: a study in highly purified aberrant plasma cells. Haematologica. 2013;98(2):279–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ortega MM, Faria RM, Shitara ES, Assis AM, Albuquerque DM, Oliveira JS, et al. N-RAS and K-RAS gene mutations in Brazilian patients with multiple myeloma. Leuk Lymphoma. 2006;47(2):285–9 [DOI] [PubMed] [Google Scholar]
- 97.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA. 2000;97(1):228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120(5): 1077–86 [DOI] [PubMed] [Google Scholar]
- 99.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mohty B, El-Cheikh J, Yakoub-Agha I, Avet-Loiseau H, Moreau P, Mohty M. Treatment strategies in relapsed and refractory multiple myeloma: a focus on drug sequencing and ‘retreatment’ approaches in the era of novel agents. Leukemia. 2012;26(1):73–85 [DOI] [PubMed] [Google Scholar]
- 102.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120(5):1060–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42(2):181–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson VH, Gerecitano JF, Goy A, de Vos S, Kenkre VP, Barr PM, et al. The Bruton’s Tyrosine Kinase (BTK) Inhibitor, Ibrutinib (PCI-32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multicenter, Open-Label, Phase 2 Study 54th ASH Annual Meeting and Exposition, Atlanta, GA, USA 2012 [Google Scholar]
- 106.Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–59 [DOI] [PubMed] [Google Scholar]
- 107.Yang Y, Shaffer AL, 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21(6):723–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26(22):3240–53 [DOI] [PubMed] [Google Scholar]
- 109.Dominguez-Sola D, Dalla-Favera R. Burkitt lymphoma: much more than MYC. Cancer Cell. 2012;22(2):141–2 [DOI] [PubMed] [Google Scholar]
- 110.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci USA. 2006;103(47):17834–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maiso P, Liu Y, Morgan B, Azab AK, Ren P, Martin MB, et al. Defining the role of TORC1/2 in multiple myeloma. Blood. 2011;118(26):6860–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Acosta-Rodriguez EV, Merino MC, Montes CL, Motran CC, Gruppi A. Cytokines and chemokines shaping the B-cell compartment. Cytokine Growth Factor Rev. 2007;18(1–2):73–83 [DOI] [PubMed] [Google Scholar]