

Abstract
A synthetic strategy has been developed culminating in stereoselective total syntheses of the small class of unusual monoterpenoid indole alkaloids exemplified by alstilobanine A (3) and E (2) and angustilodine (1). A pivotal step includes a novel intermolecular Michael-type addition of an indole ester dianion to a piperidine-derived nitrosoalkene to form the C15, C16 bond of the alkaloids. In addition, an application of the Romo protocol for effecting a stereoselective intramolecular nucleophile-assisted aldol-lactonization was employed, leading to a β-lactone incorporating the requisite cis-fused 2-azadecalin moiety and also setting the C15, C19, C20 relative stereochemistry of the metabolites. It was then possible to stereoselectively effect an aldolization of a dianion derived from this indole ester β-lactone intermediate with formaldehyde to introduce the requisite C16 hydroxymethyl group. Further manipulations of the system ultimately led to the three alkaloids in racemic form.
INTRODUCTION
Terpene-derived indole alkaloids are produced by several plant families, and are among the most abundant types of natural products with over 3000 such compounds currently known.1 These alkaloids encompass an extraordinarily diverse array of structural motifs and complexity. About ten years ago, Kam and Choo isolated angustilodine (1), a new skeletal type of monoterpene indole alkaloid, from the leaves of the Malayan plant Alstonia angustiloba (Figure 1).2 The structure of this unique metabolite was determined by detailed NMR spectroscopic analysis to be comprised of an indole appended to a cis-fused 2-azadecalin ring system bridged by an oxepane ether. In 2008, Morita and coworkers investigated alkaloids from the same plant and discovered the N-demethyl analog of angustilodine, alstilobanine E (2), along with alstilobanine A (3), which lacks the bridging oxepane ring found in compounds 1 and 2.3 Based on NMR NOESY correlations, it was proposed that alstilobanine A has the conformation shown in Figure 1, where the piperidine ring is in a chair conformation. On the other hand, the Morita group proposed that alstilobanine E has the piperidine ring in a boat conformation as shown in 2. Although the conformation of angustilodine was not explicitly discussed by Kam and Choo, it appears quite likely that this alkaloid has the same conformation as alstilobanine E. Alstilobanines A and E were found to possess moderate relaxant activity against phenylephrine-induced contractions of thoracic rat aortic rings. In a communication which appeared in 2012, we reported the first approach to these unusual alkaloids, resulting in a stereoselective total synthesis of racemic alstilobanine A (3).4,5 We now describe the details of our studies on alstilobanine A along with the extension of our synthetic strategy culminating in the first total syntheses of angustilodine and alstilobanine E.
Figure 1.

Structures and conformations of the alstilobanine/angustilodine alkaloids.
RESULTS AND DISCUSSION
Synthesis Plan
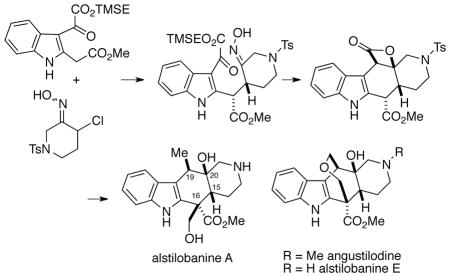
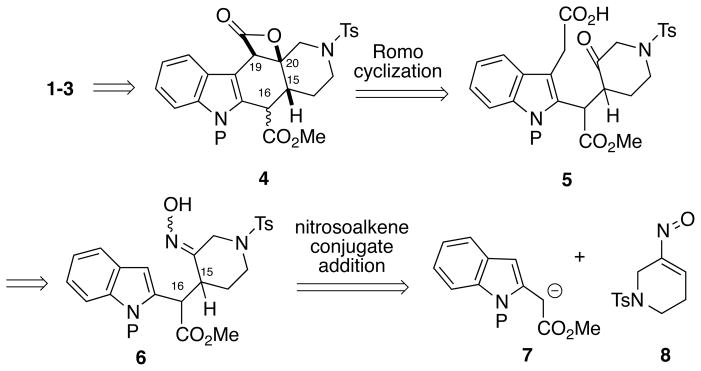
Our initial retrosynthetic analysis for synthesis of alkaloids 1-3 is outlined in Scheme 1. Thus, the plan was to prepare a key intermediate β-lactone 4 which incorporates the cis-2-azadecalin moiety found in the metabolites, and subsequently elaborate such a species into the three natural products by suitable manipulations using the carboxylate groups at C16 and C19. The pentacyclic β-lactone 4 would be prepared via a Romo-type intramolecular nucleophile-assisted aldol-lactonization of keto acid 5 which would generate the required alkaloid relative stereochemistry at C15, C19, and C20.6 Prior to the outset of our work, studies by the Romo group had shown that in simpler, all-carbon systems structurally related to intermediate 5, cis-decalins analogous to 4 can be formed with a high degree of stereoselectivity.6b We envisaged that the acetic acid unit of intermediate 5 would be installed by appropriate manipulations of indole 6. In turn, 6 would be produced via a conjugate addition of an indole-2-acetic ester enolate 7 with the in situ-generated nitrosoalkene 8 to form the C15, C16 bond of the natural products.7
Scheme 1.
First Generation Retrosynthetic Analysis of Alkaloids 1-3
Studies on Nitrosoalkene/Indole-2-acetic Ester Enolate Conjugate Additions
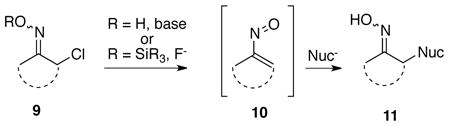
Our laboratory has recently been engaged in exploring the synthetic potential of conjugate additions of nucleophiles to transient, reactive nitrosoalkenes to effect the overall transformation of 9 to 11 as shown in Eq 1.8 There are presently two procedures normally used to generate nitrosoalkenes like 10. The most common method involves a base-promoted 1,4-elimination of an α-chlorooxime 9 (R = H) to produce the nitrosoalkene. One drawback of this methodology is that it requires at least two equivalents of a nucleophile, one of which acts as the base for the initial elimination step. Such a procedure, however, is inefficient when using valuable nucleophiles. A second, but to date less widely used procedure for nitrosoalkene generation developed by Denmark, et al. relies on treatment of an O-silyl-α-chlorooxime 9 (R = SiR3) with a fluoride source to form the nitrosoalkene 10.9 In principle, the Denmark methodology alleviates the problem of using excess nucleophile in a conjugate addition, and towards this end we have recently developed a general experimental procedure for effecting both inter- and intramolecular reactions of nitrosoalkenes formed in situ via this strategy with a variety of enolate nucleophile partners.8a–c
 |
(1) |
With the strategy in Scheme 1 in mind, we investigated the possibility of effecting the coupling of an ester enolate like 7 with piperidine-derived nitrosoalkene 8 to produce an intermdiate such as 6. Thus, N-Boc-protected indole 12, prepared from methyl indole-2-acetate,10 was converted to the enolate 14 with lithium hexamethyldisilazide (Scheme 2). Using the Denmark-based protocol which we previously devised,8a–c α-chloro-O-TBS-oxime 13, prepared from the corresponding α-chloroketone,11 was then added to this enolate, followed by tetrabutylammonium fluoride. Disappointingly, none of the adduct 16 resulting from addition of 14 to 8 was detected. As an alternative, the coupling was attempted by combining two equivalents of enolate 14 with one equivalent of the α-chlorooxime 15. In this case, a small amount of the desired product 16 was generated, but only in very poor yield.
Scheme 2.

Attempted Enolate/Nitrososalkene Conjugate Addition
In view of these problems, it was decided to form a monoanion from 2 equivalents of unprotected methyl indole-2-acetate (17)10 and attempt to couple this intermediate with one equivalent of α-chlorooxime 15 (Scheme 3). We were pleased to find that this reaction led to formation of Michael adduct 19 in an acceptable 59% yield, along with recovered starting ester 17 (66%) which could be recycled. Compound 19 is about a 3:1 mixture of C15, C16 diastereomers, but appears to be a single oxime geometric isomer which is probably E (vide infra). We believe that the anion formed in the deprotonation step probably exists as an equilibrium mixture of 18a and 18b. However, the alkylation with the transient nitrosoalkene 8 occurs only at the carbon adjacent to the ester, and no products resulting from N-alkylation (which may in fact be reversible), or alkylation at the indole C3 position were detected. We are not aware of any precedent for this transformation. It might also be noted that the same reaction could be effected using one equivalent each of α-chloro-O-TBS-oxime 13 and anion 18 via the Denmark protocol, but led to product 19 only in low yield.
Scheme 3.
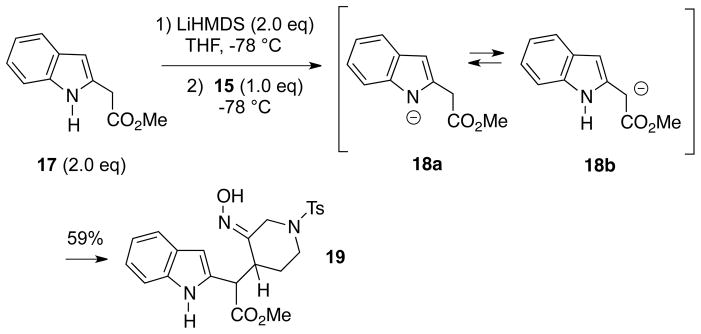
Successful Enolate/Nitrososalkene Conjugate Addition
Having the coupled product 19 in hand, we next examined a number of different homologations of oxime-protected versions of this intermediate in attempts to introduce an acetic acid unit at C3 of the indole (Cf. 5, Scheme 1). Unfortunately, none of these routes proved viable.5 However, one experiment performed on compound 19 did provide some useful information. Attempted reductive cleavage12 of the oxime functionality in 19 led to some of the desired ketone 20 along with tetracyclic indolopyrrole 21 as the major product (Eq 2). Thus, it appeared that protection of the indole nitrogen would probably be necessary prior to generating the 3-piperidone (vide infra).
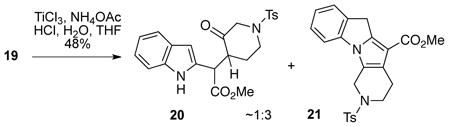 |
(2) |
Since it was not possible to introduce the requisite acetate unit into indole 19, we decided to explore a potentially more convergent route utilizing a C2, C3-disubstituted indole in the nitrosoalkene conjugate addition step. Therefore, indole 17 was first treated with oxalyl chloride, followed by addition of 2-trimethylsilylethanol/triethylamine to afford oxoacetate diester 22 in high yield having the two ester groups differentiated for later manipulations (Scheme 4).13 In order to improve the efficiency of our synthetic approach, we decided to explore a strategy which would potentially allow the use of equimolar equivalents of the coupling partners in the key nitrosoalkene conjugate addition step, thereby obviating the recovery/recycling of excess indole ester. Thus, indole oxoacetate derivative 22 (one equivalent) was first converted to the dianion 23 using two equivalents of lithium hexamethyldisilazide at low temperature. Subsequent addition of one equivalent of α-chlorooxime 15 to this dianion then led to the desired coupled product as a ~1.2:1 mixture (by NMR) of C15, C16 diastereomers 24a/b in nearly quantitative yield. These isomers could be separated chromatographically for characterization purposes, and each seemed to be a single E-oxime geometric isomer. The stereochemistry of 24a and 24b was established at a later stage of the synthesis (vide infra).
Scheme 4.
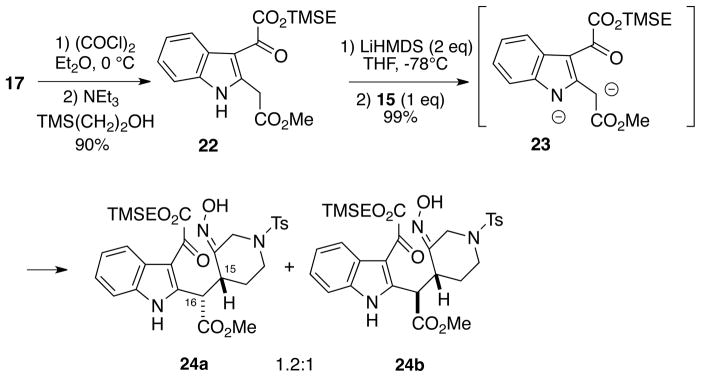
Efficient Nitrosoalkene Conjugate Addition to Form Adducts 24a/b
We believe that this transformation proceeds via an initial dehydrohalogenation of the α-chlorooxime 15 by the dianion 6 to generate the transient nitrosoalkene 8 along with a monoanion derived from the indole ester 22. As was observed with the anion 18 (Cf. Scheme 3), it seems likely that this intermediate is probably a similar equilibrium mixture of monoanions, but the conjugate addition to nitrosoalkene once again occurs exclusively at the carbon adjacent to the ester moiety to afford products 24a/b.
Elaboration of Adducts 24a/b to Ketoacid 34 and Subsequent Intramolecular Romo Cyclization
Since at this stage we believed that the C15, C16 stereochemistry was of no relevance to the synthesis, we opted to carry the mixture of diastereomers 24a/b on to the next series of transformations, which was aimed to remove the C19 carbonyl group to produce an acetate ester chain on the indole. It was eventually found that the best way to effect this deoxygenation process was by use of a modification of a sequence developed by Hlasta, et al.14 Thus, the oxime moiety was first protected as the TBS ether 25 (Scheme 5). The keto group of the oxoacetate mixture 25 was then reduced with sodium borohydride, and the resulting crude alcohol mixture was acylated with acetic anhydride/pyridine to give the corresponding acetates as a complex mixture of stereoisomers. This mixture, however, could be easily separated by flash chromatography to give 26 as a ~3:1 mixture of acetate isomers (55% from keto esters 25), and 28 as a ~2:1 mixture of acetates (36% from 25).
Scheme 5.
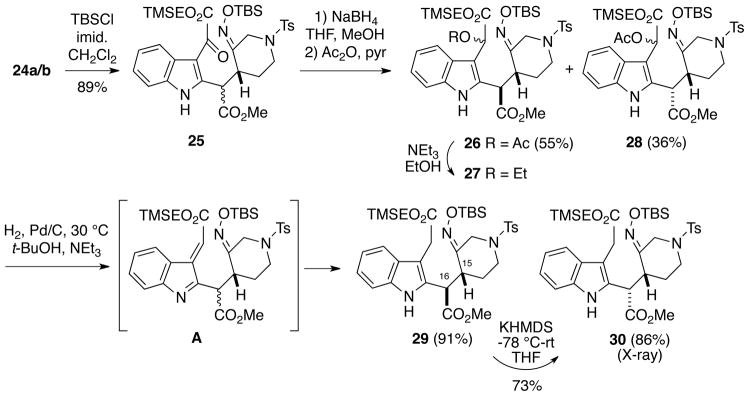
Elaboration of Adducts 24a/b into Intermediate 30
Hlasta and coworkers had previously found that it was necessary to run the catalytic hydrogenation of an acetate like 26 with 10% Pd/C in a 1:9 TEA/EtOH solvent mixture for reduction to occur, presumably via an intermediate azafulvene.14 Thus, treatment of compound 26 under these hydrogenation conditions provided the desired reduction product 29, albeit contaminated with an impurity which could not be separated by column chromatography. We suspected from 1H NMR analysis of the crude product that the impurity was ethoxy compound 27 resulting from the addition of ethanol to the azafulvene intermediate A. In fact, treatment of acetates 26 with ethanol/triethylamine indeed led to formation of 27 in good yield. We therefore investigated several other solvent combinations for the hydrogenation, and found that an alcohol is required for the reduction to occur. Ultimately it was determined that the reduction could be effected cleanly if carried out in t-butanol/triethylamine at 30 °C, although the hydrogenation is slow under these conditions, requiring four days to go to completion. However, using this approach, acetates 26 and 28 could be reduced to indole-3-acetates 29 and 30, respectively, in high isolated yields.
Although our initial assumption was that the C15, C16 stereochemistry would have no effect on any of the planned transformations of intermediates 29 and 30, the configuration at these centers in fact proved to have a critical role in the subsequent Romo cyclization (vide infra). Based on what was eventually learned in the work discussed below, it was determined that diastereomer 30 is the one required for the alkaloid synthesis. Fortunately, it was found that it is possible to easily equilibrate the undesired isomer 29 to 30. Therefore, treatment of ester 29 with potassium hexamethyldisilazide at −78 °C, followed by slowly warming to room temperature and then acid quenching resulted in the formation of what is apparently the thermodynamically more stable diastereomer 30 in 70% isolated yield. This compound proved amenable to X-ray crystallographic analysis, thereby confirming its configuration and E-oxime geometry. Although fortuitous, it is not clear to us why isomer 30 is the thermodynamic product.
With substantial quantities of intermediate 30 now in hand, it was possible to convert this compound in high overall yield into the ketoacid 34 necessary for the key intramolecular Romo cyclization (Scheme 6). Therefore, indole 30 was first N-protected as the Cbz derivative 31 in order to avoid the cyclization problem shown in Eq 2. Removal of the O-TBS protecting group yielded the free oxime 32, which could be hydrolyzed under acidic conditions to the piperidone 33.15 Attempts were then made to convert the β-trimethylsilylethyl ester to the corresponding acid with various fluoride sources but to no avail. However, the TMSE-ester of ketoacid 34 could be removed selectively from compound 33 using trifluoroacetic acid in methylene chloride.16
Scheme 6.
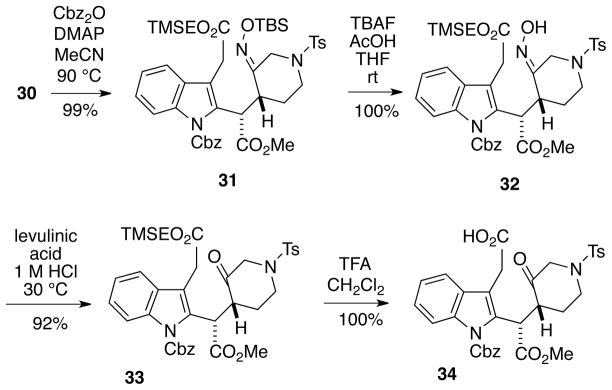
Preparation of Ketoacid 34 for the Romo Cyclization
The pivotal cyclization to form the cis-2-azadecalin system was then effected by applying the reaction conditions developed by Romo.6 Thus, treatment of ketoacid 34 with 4-pyrrolidinopyridine (PPY), 2-bromo-N-propylpyridinium triflate and diisopropylethylamine in methylene chloride, along with a small amount of acetic acid to suppress C16 ester epimerization, led to formation of an inseparable 97:3 mixture of the desired cis-azadecalin 35 and the trans-fused compound 37 in 93% total yield (Scheme 7). Hydrogenolysis of this mixture provided the unprotected indole β-lactones 36 (94%) and 38 (3%) which could be separated chromatographically.
Scheme 7.
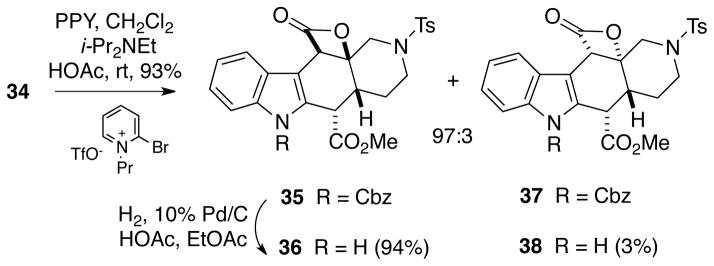
Intramolecular Romo Cyclization of Ketoacid 34
Interestingly, the Romo cyclization takes a different and more complicated stereochemical course when conducted in the C15, C16-isomeric series derived from diastereomer 29 (Scheme 8). TMSE-ester 39 can be prepared via the sequence shown in Scheme 6 starting from indole 29. However, when TMSE-ester 39 was treated under the same TFA cleavage conditions used to prepare ketoacid 34 from 33, partial C16 epimerization resulted to produce a mixture of ketoacids 40 whose diastereomeric composition could not be determined. Subsequent exposure of this mixture to the standard Romo conditions led to a complex mixture of products 41 and 42 in high overall yield, each as a mixture of C16 epimers. Notably, in this case the major products 42 have the undesired trans-2-azadecalin configuration. Clearly the configuration at C16 in the precursors to the Romo cyclization plays a key role in the stereochemical outcome of the process but at present we cannot assess exactly what factors are responsible for these results.
Scheme 8.

Romo Cyclization of the Ketoacid Diastereomer
Introduction of the C17 Hydroxymethyl Group
At this stage of the synthesis we began to investigate introduction of the C16 hydroxymethyl group into the system via an ester enolate aldolization or alkylation. To examine this process, the mixture of inseparable Cbz-protected indole β-lactones 35 and 37 was first selectively reduced with diisobutylaluminum hydride to afford the 1,3-diol 43a (66% isolated yield), which could be separated from a small amount of the corresponding diol produced from the trans-2-azadecalin (Scheme 9). Interestingly, extensive 2D NMR analysis (HMBC, HMQC, COSY and NOESY) indicated a conformation for the diol as shown in structure 43b, which is actually the cis-2-azadecalin invertamer of alstilobanine A (3) (Cf. Figure 1). This diol 43a could then be protected with acetaldehyde dimethyl acetal to afford acetal 44 as a single stereoisomer.
Scheme 9.

Hydroxymethylation of Dianion 46 to the Undesired Stereoisomer 45
A number of attempts were made to deprotonate ester 44 using a wide variety of amide bases, followed by alkylation with monomeric formaldehyde17,18,19 and several other electrophiles such as BOM-Cl and MOM-Cl, but in all cases only unchanged starting material was recovered. Based on these experiments, it appeared that an ester enolate was not being formed from 44. We attribute this problem to the indole Cbz protecting group, which for steric reasons may be causing the ester to assume a twisted conformation, precluding the proper alignment for deprotonation of the C16-H σ-bond with the carbonyl π-bond.
However, one experiment that was conducted pointed towards a possible solution to this enolization problem. Therefore, treatment of ester 44 with a mixture of sodium hydride and paraformaldehyde in DMF led to a high yield of the product 45 having the C16 hydroxymethyl moiety, but which also lacked the Cbz protecting group. Unfortunately, based on 2D NMR analysis, it was determined that 45 has the incorrect stereochemistry of the hydroxymethyl substituent at C16. We believe that in this transformation adventitious NaOH first promotes cleavage of the Cbz group, followed by subsequent ester deprotonation to afford a dianion 46. This dianion can exist in conformation 46a which is highly destabilized by A1,3-strain, or in the more stable conformation 46b. It appears that the reaction with formaldehyde probably occurs via the latter conformation from the face opposite to the bulky acetal ring.
Although these results indicated the feasibility of a C16 alkylation of a dianion derived from systems like 44, our major concern at this point was to find a way to introduce the hydroxymethyl group with the requisite stereochemistry. We reasoned that one way to control the configuration at C16 would be to effect an intramolecular alkylation. Therefore, diol 43 was selectively silylated at the primary alcohol to afford TBS ether 47 and the Cbz protecting group was then removed by catalytic hydrogenolysis to yield the NH indole 48 (Scheme 10). Silylation of the tertiary alcohol functionality of 48 was effected with known chloromethyl(dimethyl)silyl chloride20 to afford silyl ether 49. Finkelstein exchange of the chlorine in 49 for iodine produced iodomethyl compound 50.
Scheme 10.

Intramolecular Alkylation of a Silicon-Tethered Iodomethyl Compound
We were gratified to find that treatment of intermediate 50 with three equivalents of potasssium hexamethyldisilazide in THF at −78 °C and then warming to 0 °C resulted in an intramolecular alkylation of the ester enolate to produce the desired pentacyclic siloxane 51 in high yield. It was also possible to remove the TBS group from 51 to produce siloxane alcohol 52. With these two siloxanes in hand, a wide variety of conditions were attempted in order to effect a Tamao-Fleming oxidation to produce the triol 53.21 Under no circumstances, however, were we able to access this product. In general, siloxanes 51 and 52 were resistant to ring opening under both acidic and basic conditions, and under more forcing conditions only led to the product where the C16 hydroxymethyl group was lost from 53.5
In view of the problems discussed above we returned to the unprotected β-lactone ester 36 and explored its hydroxymethylation. We were pleased to find that intermediate 36 could indeed be deprotonated with two equivalents of lithium hexamethyldisilazide to generate the dianion 54, followed by alkylation with monomeric formaldehyde17,18 to produce α-hydroxymethyl ester 55 as a single diastereomer that has the correct configuration at C16 for the alkaloids 1-3 (Scheme 11). The stereochemistry of compound 55 was established by 2D NMR analysis and by its eventual conversion to the natural products 1-3. The β-lactone ring in dianion 54 appears to flatten the system sufficiently that the formadehyde alkylation occurs exclusively from the desired face in this instance. The stereochemistry of 55 was further confirmed by the fact that the β-lactone, upon treatment with triethylamine in methylene chloride, underwent acyl migration to afford the bridged ε-lactone 57. Since the hydroxymethyl group of 55 tends to be lost under a variety of reaction conditions (particularly basic ones) via a retro-aldol process, this functionality was protected as the TBS ether 56 for further use.
Scheme 11.

Stereoselective Hydroxymethylation of Dianion 54
Synthesis of (±)-Alstilobanine A (3)
The synthesis of alstilobanine A (3) from the intermediates in hand basically required a deoxygenation sequence to produce the C19 methyl group of the metabolite. The first route which we explored involved an initial partial reduction of ε-lactone 57 with diisobutylaluminum hydride/boron trifluoride etherate to provide aldehyde diol 58, albeit in only modest yield (Scheme 12). This compound was then converted to the dithiane 59 with 1,2-ethanedithiol promoted by boron trifluoride etherate. This dithiane could be directly reduced with W-2 Raney nickel to afford the desired methyl-containing compound 60. This sequence, however, suffered from a number of serious problems including low and/or highly irreproducible yields and unstable intermediates due to a tendency of the hydroxymethyl group to be lost by a retro-aldol reaction.19
Scheme 12.

Formation of a C18 Methyl Group
Thus, an alternative and much more efficient route to the alkaloid was devised. β-Lactone 56 was reduced with lithium borohydride to afford diol 61 (Scheme 13). Using a two-step sequence analogous to one reported by Huscroft, et al.22 diol 61 was first converted to iodo alcohol 62 via an Appel reaction. Catalytic hydrogenation of the iodomethyl compound 62 at atmospheric pressure using 10% Pd/C in a 1:1 mixture of ethyl acetate:t-butanol then led to the desired methyl compound 63. The N-tosyl protecting group of intermediate 63 was cleaved using magnesium metal turnings in methanol under sonication to afford piperidine 64 in good yield.23 The structure and stereochemistry of this intermediate were confirmed by 2D NMR analysis (see Supporting Information). Finally, removal of the TBS protecting group with HCl in methanol/chloroform afforded racemic alstilobanine A (3), isolated as its hydrochloride salt, having proton and carbon NMR spectra as reported for the alkaloid by Morita, et al.3,24
Scheme 13.

Completion of the Total Synthesis of Alstilobanine A
Syntheses of (±)-Alstilobanine E (2) and (±)-Angustilodine (1)
The next goal of this project was to utilize one of the intermediates prepared for alstilobanine A to construct the bridging oxepane ring of alkaloids 1 and 2, a task which proved considerably more difficult than originally envisioned. Towards this end, β-lactone 55 was reduced with DIBAL-H to afford diol 65 (Scheme 14). An attempt was then made to effect a Mitsunobu cyclization of this triol to generate the oxepane.25 Treatment of 65 with triphenylphosphine and DIAD (1 equivalent) in benzene at room temperature, however, did not afford any of the desired oxepane, but rather gave a chromatographically separable mixture of the tetrahydrofuranyl ether 6726 (51%) and oxetane 6827 (23%). Interestingly, using an excess of DIAD (5 equivalents) under the same reaction conditions produced the cyclopropane tetrahydropyranyl ether 66 in high yield.28 If DEAD was used instead of DIAD, under refluxing conditions the major cyclization product was oxetane 68 along with traces of tetrahydofuran 66 and cyclopropane 67.
Scheme 14.

Attempts to Form an Oxepane via a Mitsunobu Process
In another attempt to form an oxetane, intermediate iodomethyl compound 62, which was used in the synthesis of alstilobanine A (Cf. Scheme 13), was exposed to silver fluoroborate or silver triflate.29 In this case, however, only the cyclopropane 69 was formed in high yield (Scheme 15). Another variation which was tried with intermediate 62 was to treat it with tetrabutylammonium fluoride to promote cycloetherification. In this instance the only product formed was cyclopropane 70, where the hydroxymethyl group has been lost.
Scheme 15.

Additional Attempts at Oxepane Formation
In order to avoid cyclopropane formation, we decided to attenuate the reactivity of the indole at C3 by installing an electron withdrawing group on the nitrogen. Thus, indole β-lactone 56 was first protected as the Cbz derivative 71 (Scheme 16). The β-lactone could be reduced with lithium borohydride to give diol 72 which was then converted to bromomethyl compound 73 via an Appel reaction. Disappointingly, exposure of bromide 72 to silver triflate yielded only the oxetane 74 which was characterized by its conversion to oxetane 68, previously prepared as shown in Scheme 14. It might also be noted that a number of attempts were made to effect similar cycloetherifications in systems where the tertiary alcohol was protected, but these sequences were not fruitful.
Scheme 16.

Oxetane Formation from Bromomethyl Alcohol 73
In light of these failures, we turned to a different strategy to build the oxepane ring. Diol 61 could be cleanly oxidized with IBX30 to afford aldehyde 75 in excellent yield (Scheme 17). Removal of the TBS protecting group from 75 led to diol aldehyde 58 which had previously been prepared in low to moderate yield via partial reduction of ε-lactone 57 (Cf. Scheme 12). It was possible to convert compound 58 to dithioacetal diol 76 in high yield with ethanethiol catalyzed by titanium tetrachloride with no evidence of aldehyde epimerization.31 We were pleased to find that using a set of conditions developed by Nicolaou and coworkers for a related cyclization involving silver perchlorate catalysis,32 it was possible to cyclize dithioacetal alcohol 76 to the desired seven-membered ring hemithioacetals as a separable ~3:1 mixture of diastereomers 77 and 78 in high total yield. The stereochemistry of these hemithioacetals was established by 2D NMR NOESY analysis (see Supporting Information).
Scheme 17.

Formation of a Seven-Membered Hemithioacetal
With a seven-membered ring finally in hand, we next surveyed a number of methods and reagents for reductively removing the thioethyl group from 77/78. Raney nickel was investigated initially under several sets of conditions, but in general the major product from these reactions was the unsual oxetane acetal 79 along with only traces of the desulfurized product (Eq 3).33 Various hydride (ZnBH4, NaCNBH3/ZnCl2, etc) and free radical-based reductions (Bu3SnH, Ph3SnH, etc) also failed to give any of the desired oxetane.
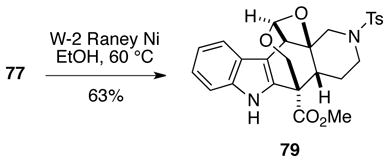 |
(3) |
It was eventually found that reduction of hemithioacetals 77 and 78 with triethylsilane catalyzed by 10% Pd/C afforded the requisite oxepane 80 in excellent yield (Scheme 18).34 The sulfonamide protecting group of 80 could then be removed with magnesium/methanol23 as was done for alstilobanine A to give racemic alstilobanine E (2) which had NMR spectra as described in the literature.3 Finally, reductive N-methylation of alstilobanine E with paraformaldehyde/sodium borohydride35 afforded racemic angustilodine (1) having proton and carbon NMR spectra identical to those provided by Professor Kam.2,24
Scheme 18.

Completion of the Total Syntheses of Alstilobanine E and Angustilodine
CONCLUSION
We have described the details of the first total syntheses of the structurally unique monoterpene indole alkaloids alstilobanine A (3), alstilobanine E (2) and angustilodine (1) from a common intermediate, indole β-lactone 56. We have devised a convergent route to this intermediate which includes as key steps (1) an unprecedented conjugate addition of the dianion of indole-2-acetate ester 23 to the piperidine-derived nitrosoalkene 8 to form the C15, C16 bond in adducts 24a/b and (2) a stereoselective intramolecular Romo nucleophile-assisted aldol-lactonization of ketoacid 34 to generate β-lactone 35 fused to the requisite cis-2-azadecalin needed for the alkaloid along with the attendant C15, C19, C20 relative stereochemistry. The vexing problem of introducing the C16 hydroxymethyl group needed for all three alkaloids was finally solved via an aldolization of a dianion derived from indole ester β-lactone intermediate 36 with monomeric formaldehyde. Some further manipulations eventually allowed conversion of the β-lactone carbonyl carbon into the C19 methyl group of alstilobanine A (3). Finally, a sequence was developed that allowed the silver-catalyzed cyclization of dithioacetal alcohol 76 into the seven-membered hemithioacetals 77/78, which could be reductively desulfurized and then subsequently converted into alstilobanine E (2) and angustilodine (1).
EXPERIMENTAL SECTION
General Methods
All non-aqueous reactions were carried out in oven- or flame-dried glassware under an argon atmosphere. All chemicals were purchased from commercial vendors and used as is, unless otherwise specified. Anhydrous tetrahydrofuran (THF) and dichloromethane (CH2Cl2) were obtained from a solvent purification system. Reactions were magnetically stirred and monitored by thin layer chromatography (TLC) with 250 μm precoated silica gel plates. Flash column chromatography was performed using silica gel (230–400 mesh). Chemical shifts are reported relative to chloroform (δ 7.26), methanol (δ 3.31), or DMSO (δ 2.50) for 1H NMR and chloroform (δ 77.2), methanol (δ 49.0), or DMSO (δ 39.5) for 13C NMR. High resolution mass spectra were obtained on a time of flight instrument using electrospray ionization.
4-Chloro-1-tosylpiperidin-3-one Oxime (15)
To a stirred solution of 4-chloro-1-tosylpiperidin-3-one 11 (5.40 g, 18.8 mmol) in DMSO (50 mL) was added H2NOH·HCl (1.46 g, 20.6 mmol) and the resulting solution was stirred at rt for 18 h. The reaction mixture was diluted with water (300 mL) and extracted repeatedly with CH2Cl2. The combined organic layers were then washed with water, brine, and then dried over MgSO4. Removal of the solvent in vacuo provided α-chlorooxime 15 (5.49 g, 97%) as a white solid which was sufficiently pure for use without further purification. 1H NMR (300 MHz, CDCl3, ~2:1 E/Z mixture of oxime isomers) δ 7.81-7.70 (m, 2H), 7.40-7.38 (d, J = 8.2 Hz, 2H), 5.53 (m, 0.33H), 5.15 (d, J = 14.2 Hz, 0.67H), 4.77 (t, J = 3.0 Hz, 0.67H), 4.30-4.27 (m, 0.33 H), 3.85-3.80 (m, 1H), 3.45 (d, J = 13.6 Hz, 0.33H), 3.23 (d, J = 14.5 Hz, 0.67H), 3.11 (td, J = 11.9, 3.0 Hz, 0.67H), 2.95 (m, 0.33H), 2.48 (m, 3H), 2.28-2.08 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 150.9, 150.4, 144.7 (2C), 133.6, 133.2, 130.4, 128.2, 128.0, 66.4, 61.1, 55.9, 45.7, 44.6, 41.3, 41.0, 38.6, 33.9, 32.8, 22.0. The compound was unstable on attempted mass spectrometric analysis.
Methyl 2-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-(1H-indol-2-yl)acetate (19)
To a stirred solution of indole ester 1710 (691 mg, 3.65 mmol) in THF (11 mL) at −78 °C was added LiHMDS (1.0 M in THF, 3.65 mL, 3.65 mmol). The resulting solution was stirred for 30 min and a solution of α-chlorooxime 15 (553 mg, 1.83 mmol, in 7 mL of THF) was added dropwise over 8 min. After stirring the mixture for an additional 90 min at −78 °C the reaction was quenched with aqueous NH4Cl. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 15–50% EtOAc/hexanes) to afford Michael adduct 19 (491 mg, 59%) as a ~3:1 mixture of diastereomers (foam) and recovered starting indole 17 (454 mg, 66%). 1H NMR (400 MHz, CDCl3, ~3:1 mixture of isomers) δ 8.83 (s, 1H), 8.60 (br s, 1H), 7.69-7.09 (m, 8H), 6.36 (s, 0.75 H), 6.29 (s, 0.25 H), 4.89 (d, J = 14.8 Hz, 0.75 H), 4.56 (d, J = 15.0 Hz, 0.25H), 3.98 (d, J = 10.3 Hz, 1 H), 3.69 (s, 0.75 H), 3.67 (s, 2.25 H), 3.60-3.46 (m, 1H), 3.20 (d, J = 14.4 Hz, 1H), 3.02 (td, J = 10.7, 4.7 Hz, 1H), 2.76-2.72 (m, 1H), 2.45-2.41 (m, 4H), 1.63-1.59 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 173.4, 153.4, 144.5, 136.7, 133.2, 132.5, 130.4, 130.3, 128.4, 128.1, 122.6, 120.7, 120.5, 111.5, 46.6, 46.5, 45.3, 42.3, 42.1, 28.1, 22.0; LRMS-ES+ m/z (relative intensity) 456; HRMS (m/z): [M+H]+ calcd for C23H26N3O5S 456.1593, found 456.1571.
2-(Trimethylsilyl)ethyl-2-(2-(2-methoxy-2-oxoethyl)-1H-indol-3-yl)-2-oxoacetate (22)
To a stirred solution of indole ester 1710 (3.50 g, 18.5 mmol) in Et2O (310 mL) at 0 °C was added oxalyl chloride (1.84 mL, 21.4 mmol). The resultant orange solution was stirred at rt for 16 h and then cooled to 0 °C. 2-Trimethylsilylethanol (8.0 mL, 56 mmol) was added, followed by slow addition of triethylamine (6.4 mL, 47 mmol) over 5 min. The resulting red suspension was stirred at 0 °C for 1.5 h and then diluted with water. The organic layer was separated and the aqueous layer extracted with Et2O. The combined organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo to give a viscous red oil which was purified by flash chromatography on silica gel (gradient 20–40% EtOAc/hexanes) to yield indole-3-oxoacetate 22 (purple solid, 6.03 g, 90%). 1H NMR (300 MHz, CDCl3) 10.50 (s, 1H), 7.83-7.78 (m, 1H), 7.45-7.40 (m, 1H), 7.30-7.23 (m, 2H), 4.56-4.50 (m, 2H), 4.32 (s, 2H), 3.82 (s, 3H), 1.22-1.16 (m, 2H), 0.10 (s, 9H); 13C NMR (75 MHz, CDCl3) 182.5; 171.3, 166.5, 142.3, 135.4, 126.1, 124.0, 123.4, 120.3, 112.3, 110.5, 65.2, 53.1, 32.9, 17.8, −1.1; LRMS-ES+ m/z (relative intensity) 362 (MH+, 80); HRMS (m/z): [M+H]+ calcd for C18H24NO5Si 362.1424, found 362.1416.
Methyl 2-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-2-(2-(trimethylsilyl)-ethoxy)acetyl)-1H-indol-2-yl)acetate (24a/b)
To a −78 °C solution of indole 22 (8.18 g, 22.6 mmol) was added LiHMDS (47.5 mL, 47.5 mmol, 1.0 M in THF) and the resulting solution was stirred for 30 min. A solution of α-chlorooxime 15 (9.6 g, 32 mmol) in THF (69 mL) was added via cannula over 10 min. The reaction mixture was stirred at −78 °C for 2 h and then diluted with aqueous NH4Cl. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 20–50% EtOAc/hexanes) to yield diastereomeric Michael adducts 24a and 24b (orange foam, 14.01 g, 99%, ~1.2:1 by 1H NMR) which were carried on to the next step without separation. 1H NMR (300 MHz, CDCl3) δ 10.31 (s, 0.5H), 10.23 (s, 0.5H), 8.90 (s, 0.5H), 8.66 (s, 0.5H), 7.79-7.58 (m, 3H), 7.39-7.17 (m, 5H), 5.59 (d, J = 5.9 Hz, 0.5H), 5.13 (d, J = 8.6 Hz, 0.5H), 4.93 (d, J = 14.5 Hz, 0.5H), 4.60-4.50 (m, 2H), 3.69-4.47 (m, 4H), 3.37 (p, J = 5.7 Hz, 0.5H), 3.20 (d, J = 14.6 Hz, 0.5 H), 3.10 (q, J = 7.3 Hz, 0.5H), 2.97-2.85 (m, 1H), 2.44-2.28 (m, 3H), 2.08-2.03 (m, 0.5H), 1.78-1.76 (m, 0.5H), 1.41-1.31 (m, 0.5H), 0.11-0.07 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 183.6, 183.0, 172.6, 171.9, 166.5, 166.5, 152.8, 152.7, 144.61, 144.57, 143.5, 143.4, 135.83, 135.80, 130.4, 130.3, 128.06, 127.96, 125.8, 125.6, 124.4, 124.2, 123.4, 123.2, 120.0, 119.6, 112.9, 112.7, 111.2, 111.1, 65.33, 65.29, 53.3, 53.2, 45.4, 44.9, 44.2, 43.4, 42.9, 42.7, 42.3, 41.9, 27.9, 27.8, 22.0, 21.9, 17.8, 14.6, 14.2, −1.1; LRMS-ES+ m/z (relative intensity) 666 (M+K+, 100); HRMS (m/z): [M+NH4]+ calcd for C30H41N4O8SSi 645.2414, found 645.2445.
For characterization of the isomers, a small amount of the mixture was separated by flash chromatography on silica gel (gradient 2–10% Et2O/CH2Cl2) to afford 24a (less polar) as an orange foamy solid. 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H, NH), 7.91 (s, 1H), 7.62 (d, J = 8.0 Hz, 3H), 7.38 (m, 1H), 7.26-7.19 (m, 4H), 5.52 (d, J = 6.1.1 Hz, 1H), 4.87 (d, J = 14.6 Hz, 1H), 4.50 (t, J = 8.2 Hz, 2H), 3.64 (s, 3H), 3.30 (m, 1H), 3.18 (d, J = 14.7 Hz, 1H), 2.82 (m, 1H), 2.43 (m, 1H), 2.34 (s, 3H), 2.02 (dd, J = 3.4, 12.8 Hz, 1H), 1.38 (m, 1H), 1.15 (t, J = 8.7 Hz, 2H), 0.07 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 183.2, 171.5, 166.0, 153.1, 144.2, 135.3, 133.0, 130.0, 127.7, 125.3, 123.9, 122.9, 119.6, 112.4, 111.1, 64.9, 52.9, 45.0, 43.0, 42.6, 42.0, 27.5, 21.6, 17.5, −1.4. 24b (more polar, brown foamy solid), 1H NMR (300 MHz, CDCl3) δ 9.84 (s, 1H), 7.97 (s, 1H), 7.61-7.57 (m, 3H), 7.29-7.23 (m, 2H), 1.17–7.14 (m, 3H), 5.00 (d, J = 8.4 Hz, 1H), 4.50-4.39 (m, 3H), 3.67 (s, 3H), 3.45-3.38 (m, 3H), 2.94 (m, 1H), 2.82 (m, 1H), 2.34 (s, 3H), 1.69 (m, 2H), 1.09-1.05 (m, 2H), 0.00 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 182.9, 172.8, 166.2, 152.9, 144.5, 143.5, 135.7, 133.3, 130.3, 128.1, 125.8, 124.4, 123.4, 120.2, 122.6, 111.2, 65.3, 53.3, 45.0, 44.1, 43.3, 42.0, 28.0, 22.0, 17.8, −1.1.
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)acetyl)-1H-indol-2-yl)acetate (25)
To a solution of ester oxime mixture 24a/b (5.66 g, 9.01 mmol) and imidazole (2.47 g, 36.3 mmol) in CH2Cl2 (240 mL) was added TBSCl (4.10 g, 27.2 mmol). The resulting suspension was stirred for 12 h at rt and then diluted with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel (30% EtOAc/hexanes) yielding O-TBS-oximes 25 (5.98 g, 89%) as an orange solid. 1H NMR (300 MHz, CDCl3) δ 9.91 (s, 0.5H), 9.79 (s, 0.5H), 7.77-7.61 (m, 3H), 7.41-7.21 (m, 5H) 5.56 (d, J = 5.7 Hz, 0.5H), 5.06-4.97 (m, 1H), 4.84 (d, J = 15.0 Hz, 0.5H), 4.54-4.45 (m, 2H), 3.69-3.60 (m, 4H), 3.41 (dt, J = 7.1, 12.6 Hz, 0.5H), 3.30 (d, J = 15.0 Hz, 0.5H), 3.18 (d, J = 14.9 Hz, 0.5H), 3.00-2.92 (m, 1H), 2.85-2.70 (m, 0.5H), 2.42 (s, 1.5H), 2.32 (s, 1.5H), 2.06-1.98 (m, 0.5H), 1.85-1.70 (m, 0.5H), 1.65-1.55 (m, 0.5H), 1.40-1.35 (m, 1H), 1.20-1.13 (m, 2H), 0.99 (m, 9H), 0.31-0.20 (m, 6H), 0.10-0.07 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 183.4, 182.8, 172.6, 171.0, 166.3, 166.1, 157.9, 156.8, 144.40, 144.37, 143.6, 143.4, 135.5, 135.4, 133.9, 133.6, 130.20, 130.15, 128.0, 127.9, 126.0, 125.7, 124.4, 124.2, 123.5, 123.2, 120.6, 120.0, 112.4, 112.2, 111.3, 65.1, 53.2, 53.0, 45.2, 44.1, 43.5, 43.3, 42.9, 28.1, 27.7, 26.34, 26.28, 21.93, 21.85, 18.3, 18.2, 17.79, 17.76, −1.1, −4.2, −4.6, −4.8; LRMS-ES+ m/z (relative intensity) 780 (M+K+, 100); HRMS (m/z): [M+H]+ calcd for C36H52N3O8SSi2 742.3014, found 742.3005.
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(1-hydroxy-2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetates (26 and 28)
To a stirred solution of α-ketoesters 25 (21.5 g, 29 mmol) in MeOH (55 mL) and THF (425 mL) cooled to 0 °C was added NaBH4 (1.32 g, 34.7 mmol). The resulting solution was stirred for 1 h at 0 °C, and then diluted with aqueous NH4Cl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with water and brine and then dried over MgSO4. Removal of the solvent in vacuo provided the indole-3-hydroxyacetate products as a white powder.
Without further purification, this mixture was dissolved in a 1:1 (v/v) mixture of Ac2O:pyridine (292 mL) and the solution was stirred at rt for 12 h. After removing the volatiles in vacuo, 26 and 28 were separated by flash chromatography on silica gel (gradient 15–40% EtOAc/hexanes). 26 (more polar isomer, orange foamy solid, 12.60 g, 55%, ~3:1 mixture of acetoxy diastereomers): 1H NMR (400 MHz, CDCl3) δ 9.07 (s, 0.75H), 9.03 (s, 0.25H), 7.75-7.71 (m, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.32-7.13 (m, 5H), 6.21 (s, 0.25H), 6.18 (s, 0.75H), 4.97-4.88 (m, 1H), 4.52 (d, J = 5.9 Hz, 0.25H), 4.48 (d, J = 5.5 Hz, 0.75H), 4.28-4.20 (m, 1H), 4.09-4.02 (m, 1H), 3.67-3.55 (m, 4H), 3.41-3.31 (m, 2H), 3.00-2.93 (m, 1H), 2.39-2.36 (m, 3H), 2.14 (s, 3H), 1.95-1.70 (m, 3H), 1.60-1.40 (m, 2H), 0.98 (s, 9H), 0.29-0.18 (m, 6H), 0.00–0.02 (m, 9H); 13C NMR (90 MHz, CDCl3) δ 171.0, 170.8, 170.6, 170.5, 169.1, 157.3, 144.0, 135.3, 135.2, 133.7, 133.5, 132.2, 131.7, 129.8, 127.6, 126.2, 126.1, 122.7, 120.3, 119.3, 111.2, 108.0, 107.8, 67.7, 64.2, 53.4, 52.5, 44.8, 43.2, 42.9, 42.7, 42.4, 42.1, 28.0, 26.0, 25.9, 21.5, 20.9, 20.7, 17.9, 17.3, −1.56, −1.59, −4.8, −5.1. 28 (less polar isomer, orange foamy solid, 8.29 g, 36%, ~2:1 mixture of acetoxy diastereomers): 1H NMR (300 MHz, CDCl3) δ 8.73-8.71 (m, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.35-7.14 (m, 5H), 6.24-6.22 (m, 1H), 5.02 (t, J = 7.1 Hz, 1H), 4.34-4.07 (m, 3H), 3.66-3.53 (m, 4H), 3.15-3.07 (m, 2H), 2.72 (td, J = 11.3, 3.2 Hz, 1H), 2.45 (s, 3H), 2.20-2.18 (m, 3H), 1.58-1.39 (m, 2H), 1.00-0.89 (m, 12H), 0.26 (s, 3H), 0.20 (s, 3H), 0.02–0.04 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 173.1, 173.0, 170.9, 170.8, 169.3, 169.2, 156.7, 144.4, 136.1, 136.0, 133.8, 133.6, 132.4, 132.2, 130.2, 128.1, 126.7, 123.5, 123.4, 121.1, 120.2, 111.5, 108.54, 108.50, 68.4, 68.0, 64.7, 64.5, 53.9, 53.0, 52.9, 45.4, 44.4, 44.1, 42.8, 42.6, 42.5, 28.5, 28.0, 26.4, 22.0, 21.2, 21.1, 18.4, 17.7, 17.6, −1.2, −4.75, −4.89, −4.92; LRMS-ES+ (mixture of diastereomers 26/28) m/z (relative intensity) 824 (M+K+, 25); HRMS (m/z): [M+H]+ calcd for C38H56N3O9SSi2 786.3276, found 786.3286.
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (29)
To a solution of acetates 26 (2.94 g, 3.75 mmol) in t-BuOH (80 mL) was added 10% Pd/C (1.20 g). The resulting mixture was evacuated and backfilled with H2 from a balloon and TEA (10.0 mL) was then added. The mixture was warmed to 30 °C and stirred for 4 days until all the starting material was consumed as judged by TLC. The reaction mixture was then diluted with EtOAc, filtered through a pad of Celite and concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 10–25% EtOAc/hexanes) to afford indole 29 (2.47 g, 91%) as an off-white foam. 1H NMR (360 MHz, CDCl3) δ 8.74 (br s, 1H), 7.62 (d, J = 7.2 Hz, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.28-7.24 (m, 3H), 7.17 (dd, J = 7.1, 7.5 Hz, 1H), 7.09 (dd, J = 7.3, 7.4 Hz, 1H), 4.82 (d, J = 15.1 Hz, 1H), 4.33 (d, J = 6.0 Hz, 1H), 4.11-4.07 (m, 2H), 3.64 (s, 3H), 3.60 (d, J = 4.4 Hz, 2H), 3.58-3.55 (m, 1H), 3.36 (d, J = 15.2 Hz, 1H), 3.30-3.25 (m, 1H), 2.90 (td, J = 4.0, 11.9 Hz, 1H), 2.36 (s, 3H), 1.89-1.84 (m, 1H), 1.59-1.54 (m, 1H), 0.94 (s, 9H), 0.21 (s, 3H), 0.15 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.7, 171.6, 157.2, 144.0, 135.4, 133.7, 130.0, 129.9, 129.5, 128.9, 127.7 (2C), 122.3, 119.7, 118.9, 111.0, 107.6, 63.2, 52.5, 44.7, 43.0, 42.7, 42.3, 30.8, 27.6, 26.0, 21.6, 18.0, 17.4, −1.4, −4.8, −5.0; LRMS-ES+ m/z (relative intensity) 766 (M+K+, 75); HRMS (m/z): [M+NH4]+ calcd for C36H57N4O7SSi2 745.3487, found 745.3478.
Synthesis of Indole Diester 30
Following a similar procedure described for preparation of 29, acetates 28 (3.95 g, 5.03 mmol) were reduced by catalytic hydrogenation to afford indole diester 30 as an off white foamy solid (3.16 g, 86%). Slow evaporation of this compound from EtOAc gave crystals suitable for X-ray crystallography (see Supporting Information).36 Mp 128–130 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.65 (d, J = 8.5 Hz, 2H), 7.57 (d, J = 7.7 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.17 (dd, J = 7.0, 7.1 Hz, 1H), 7.11 (dd, J = 7.1, 7.2 Hz, 1H), 5.03 (d, J = 14.4 Hz, 1H), 4.17-4.13 (m, 2H), 4.06 (d, J = 10.7 Hz, 1H), 3.64 (app. s, 2H), 3.62 (s, 3H), 3.57 (m, 1H), 3.06-3.00 (m, 2H), 2.66 (td, J = 3.8, 11.5 Hz, 1H), 2.44 (s, 3H), 1.46 (m, 1H), 1.39 (m, 1H), 0.97 (s, 9H), 0.23 (s, 3H), 0.16 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 173.0, 171.4, 156.5, 144.0, 135.7, 133.4, 129.8, 128.0 (2C), 127.8, 122.7, 120.1, 119.2, 111.0, 108.3, 63.3, 52.5, 45.2, 43.8, 42.5, 42.0, 30.6, 27.8, 26.1, 25.9, 21.7, 18.1, 17.5, −1.4, −5.1, −5.2; HRMS (m/z): [M+H]+ calcd for C36H54N3O7SSi2 728.3221, found 728.3224.
Epimerization of Ester 29 to Ester 30
To a solution of diester 29 (4.45 g, 6.12 mmol) in THF (81 mL) cooled to −78 °C was added dropwise KHMDS solution (0.5 M in toluene, 13.5 mL, 6.73 mmol, 1.1 equiv.). After the addition was complete, the cooling bath was removed and the reaction mixture was stirred at rt for 30 min. To the resulting olive green solution was added glacial acetic acid (0.40 mL) followed by aqueous NH4Cl. The mixture was extracted with EtOAc. The organic phase washed with brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography on silica gel (30 % EtOAc/hexanes) to afford diester 30 as a light yellow foam (3.23 g, 73%), which was identical to material prepared from hydrogenation of 28.
Benzyl 2-(1-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (31)
A stirred solution of indole 30 (1.05 g, 1.45 mmol) in acetonitrile (40 mL) was heated to 90 °C. Dibenzyl dicarbonate (2.07 g, 7.24 mmmol) was added, followed immediately by DMAP (0.53 g, 4.33 mmol). After gas evolution stopped (~1 min), the reaction mixture was heated at 90 °C for another 2 min, and cooled to rt. The solvent was removed in vacuo to give a pale brown residue, which was purified by flash chromatography on silica gel (gradient 5–20% EtOAc/hexanes) to afford 31 as a white foam (1.23 g, 99%, ~2:1 Cbz rotamers). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.6 Hz, 0.7H), 8.00 (m, 0.3H), 7.64 (d, J = 6.9 Hz, 2H), 7.57 (d, J = 6.4 Hz, 0.7H), 7.46-7.26 (m, 9H), 5.68 (d, J = 10.1 Hz, 0.3H), 5.17 (d, J = 11.7 Hz, 0.7H), 5.52-5.46 (m, 0.7H), 5.29 (d, J = 13.1 Hz, 0.7H), 5.16 (d, J = 9.3 Hz, 1.7H), 4.14-4.10 (m, 2H), 3.90 (d, J = 15.90 Hz, 0.3H), 3.65-3.53 (m, 2.7H), 3.48 (s, 3H), 3.11 (m, 1H), 2.90 (d, J = 13.2 Hz, 0.3H), 2.68 (d, J = 13.2 Hz, 0.7H), 2.60 (m, 0.3H), 2.49-2.44 (m, 3H), 2.17 (m, 0.7H), 1.40 (m, 0.3H), 1.12 (m, 1.7H), 0.98 (m, 9H), 0.22 (s, 2H), 0.19-0.17 (m, 4H), 0.02 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 171.7, 171.1, 170.8, 170.1, 155.8, 155.6, 151.8, 151.3, 143.8, 135.9, 135.6, 134.8, 133.4, 132.5, 132.2, 129.8, 129.4, 129.2, 129.0, 128.8, 127.9, 125.3, 123.5, 119.3, 118.9, 118.6, 117.2, 115.8, 69.3, 68.6, 63.6, 63.4, 52.1, 45.7, 42.8, 42.4, 42.0, 41.3, 40.7, 30.9, 30.2, 28.6, 27.5, 26.2, 25.6, 21.7, 18.3, 17.5, −1.5, −5.1; LRMS-ES+ m/z (relative intensity) 862 (MH+, 90); HRMS (m/z): [M+H]+ calcd for C44H60N3O9SSi2 862.3589, found 862.3585.
Benzyl 2-(1-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (32)
To a stirred solution of O-TBS-oxime 31 (12.9 g, 14.9 mmol) in THF (600 mL) was added AcOH (6.6 mL) followed by a solution of TBAF in THF (1 M, 22.1 mL, 22.1 mmol). The resulting solution was stirred at rt overnight and then diluted with aqueous NH4Cl. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash column chromatography on silica gel (30% EtOAc/hexanes) to yield free oxime 32 as a white foam (11.1 g, 100%, ~2:1 Cbz rotamers). 1H NMR (400 MHz, CDCl3) δ 8.10 (m, 0.7H), 8.0-7.8 (m, 1.3H), 7.65 (d, J = 7.1 Hz, 2H), 7.54 (m, 0.7H), 7.45 (m, 3.5H), 7.35-7.26 (m, 6H), 5.68 (m, 0.3H), 5.57 (d, J = 11.5 Hz, 0.7H), 5.47 (s, 0.7H), 5.18 (m, 1.4H), 5.07 (m, 1H), 4.13 (m, 2H), 3.87 (m, 0.3H), 3.65-3.59 (m, 3H), 3.48 (s, 3H), 3.09 (m, 1H), 2.91 (0.3H), 2.68 (d, J = 13.1 Hz, 0.7H), 2.60 (m, 0.3H), 2.48-2.44 (m, 3H), 2.19 (m, 0.7H), 1.64 (m, 0.4H), 1.40 (m, 0.3H), 1.17 (m, 1.6H), 0.94 (t, J = 8.8 Hz, 2H), 0.01 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 172.0, 171.5, 170.8, 170.5, 152.9, 151.8, 151.4, 143.9, 135.8, 135.5, 134.7, 133.0, 132.3, 132.0, 129.8, 129.4, 129.2, 129.1, 129.0, 128.8, 128.6, 127.9, 127.8, 127.1, 125.3, 124.9, 123.5, 123.2, 119.3, 118.6, 117.2, 116.1, 115.9, 69.5, 68.7, 65.4, 64.5, 63.8, 63.4, 52.6, 52.3, 45.7, 42.4, 41.8, 41.0, 40.7, 40.5, 31.0, 30.7, 30.3, 28.5, 27.6, 21.6, 21.1, 19.2, 17.3, 13.8, −1.5; LRMS-ES+ m/z (relative intensity) 748 (MH+, 75); HRMS (m/z): [M+H]+ calcd for C38H46N3O9SSi 748.2724, found 748.2718.
Benzyl 2-(2-Methoxy-2-oxo-1-(3-oxo-1-tosylpiperidin-4-yl)ethyl)-3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (33)
Oxime 32 (11.14 g, 14.9 mmol) was added to a mixture of levulinic acid and 1 M HCl (334 g, 9:1 v/v) and the mixture was stirred at 30 °C for 4.5 h. The reaction mixture was diluted with water (1 L), and was extracted with dichloromethane. The organic layer was washed with aqueous NaHCO3, water and brine, dried over Na2SO4, and concentrated. The residue was purified by flash column chromatography on silica gel (gradient, 20–30% EtOAc/hexanes) to afford ketone 33 as a slightly pink foam (10.04 g, 92%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 7.6 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.55-7.26 (m, 10H), 5.54 (d, J = 11.8 Hz, 1H), 5.20 (d, J = 11.6 Hz, 1H), 4.91 (d, J = 6.6 Hz, 1H), 4.18-4.07 (m, 2H), 4.21 (d, J = 13.6 Hz, 1H), 3.62 (s, 2H), 3.48 (s, 3H), 3.32 (q, J = 9.3 Hz, 1H), 3.20 (d, J = 13.6 Hz,, 1H), 2.48 (s, 3H), 2.46 (m, 1H), 1.46-1.42 (m, 2H), 0.95 (t, J = 8.6 Hz, 2H), 0.02 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 201.6, 171.2, 170.2, 151.5, 144.3, 135.4, 134.7, 132.2, 131.7, 130.0, 129.3, 129.1, 128.9, 128.0, 125.4, 123.7, 119.3, 117.4, 115.9, 68.9, 63.8, 56.1, 52.4, 48.8, 45.3, 40.0, 30.8, 27.5, 21.7, 17.4, −1.5; LRMS-ES+ m/z (relative intensity) 750 (M+NH4+, 75); HRMS (m/z): [M+NH4]+ calcd for C38H48N3O9SSi 750.2881, found 750.2878.
2-(1-((Benzyloxy)carbonyl)-2-(2-methoxy-2-oxo-1-(3-oxo-1-tosylpiperidin-4-yl)ethyl)-1H-indol-3-yl)acetic Acid (34)
To a stirred solution of TMSE-ester 33 (3.95 g, 5.38 mmol) in CH2Cl2 (138 mL) was added TFA (34 mL). The resulting solution was stirred at rt for 9 h and the solvent was removed in vacuo to give a residue which was purified by flash column chromatography on silica gel (40% EtOAc/hexanes + 1% AcOH) to give keto acid 34 as an off-white foam (3.39 g, 100%). 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 6.9 Hz, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.50-7.16 (m, 10H), 5.54 (d, J = 11.8 Hz, 1H), 5.21 (d, J = 11.1 Hz, 1H), 4.87 (d, J = 6.2 Hz, 1H), 4.00 (13.4 Hz, 1H), 3.68 (s, 2H), 3.46 (s, 3H), 3.36-3.29 (m, 1H), 3.18 (d, J = 13.9 Hz, 1H), 2.48 (s, 3H), 1.41 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 201.6, 175.3, 171.1, 151.4, 144.3, 137.9, 135.4, 134.5, 132.2, 131.9, 129.9, 129.3, 129.1, 128.8, 128.2, 127.9, 125.5, 125.3, 123.7, 119.1, 116.6, 115.9, 68.9, 56.0, 52.4, 48.7, 45.2, 40.0, 30.0, 27.5, 21.6; LRMS-ES+ m/z (relative intensity) 633 (MH+, 100); HRMS (m/z): [M+NH4]+ calcd for C33H36N3O9S 650.2172, found 650.2142.
8-Benzyl 7-Methyl 1-Oxo-4-tosyl-3,4,5,6,6a,7-hexahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7,8(12cH)-dicarboxylate (35) and trans-2-Azadecalin 37
To a stirred suspension of 4-pyrrolidinopyridine (1.09 g, 7.35 mmol), 2-bromo-N-propylpyridinium triflate (2.57 g, 7.35 mmol) and DIPEA (1.7 mL, 9.8 mmol) in CH2Cl2 (79 mL) at rt was added a solution of ketoacid 34 (3.00 g, 4.90 mmol) and glacial acetic acid (0.35 mL, 6.1 mmol) in CH2Cl2 over 1 h via a syringe pump. The resulting orange solution was stirred for another 3 h at rt. Solvent was then removed in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 30–40% EtOAc/hexanes) affording an inseparable mixture of β-lactones 35 and 37 as an off white solid (2.71 g, 93%). FTIR (film) 1835 cm−1; 1H NMR (400 MHz, CDCl3, ~97:3 mixture of diastereomers determined by 1H NMR, only the major diastereomer peaks are reported) δ 8.02 (d, J = 7.2 Hz, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 7.4 Hz,, 1H), 7.45 (m, 2H), 7.42-7.31 (m, 7H), 5.46 (d, J = 10.8 Hz, 1H), 5.32 (d, J = 12.0 Hz, 1H), 4.76 (s, 1H), 4.41 (d, J = 5.7 Hz, 1H), 3.88 (d, J = 12.0 Hz, 1H), 3.50 (m, 1H), 3.50 (s, 3H), 2.95 (d, J = 11.8 Hz, 1H), 2.55 (dd, J = 7.3, 14.5 Hz, 1H), 1.60 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 170.7, 165.4, 151.8, 144.5, 136.6, 134.5, 132.3, 130.1, 129.9, 129.0, 128.9, 128.8, 127.8, 126.9, 125.8, 123.8, 118.8, 115.6, 110.6, 75.6, 69.4, 53.0, 52.2, 50.9, 44.8, 43.3, 38.5, 25.1, 21.7; LRMS-ES+ m/z (relative intensity) 615 (MH+, 100); HRMS (m/z): [M+NH4]+ calcd for C33H34N3O8S 632.2067, found 632.2053.
Methyl 1-Oxo-4-tosyl-3,4,5,6,6a,7,8,12c-octahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7-carboxylate (36) and trans-2-Azadecalin 38
10% Pd/C (0.54 g) was suspended in a solution of the mixture of N-Cbz β-lactones 35 and 37 (2.71 g, 4.41 mmol) in EtOAc (280 mL). One drop of glacial acetic acid was added to the mixture, followed by three evacuation-backfill cycles with hydrogen gas from a balloon. The reaction mixture was stirred under a balloon of H2 for 1.5 h at rt. The mixture was filtered through a pad of Celite, concentrated and the residue was purified by flash chromatography on silica gel (gradient 2–5% Et2O/CH2Cl2) to afford β-lactone 36 (1.99 g, 94%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 9.50 (br s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.25-7.18 (m, 2H), 4.86 (s, 1H), 4.21 (d, J = 4.5 Hz, 1H), 4.09 (d, J = 11.6 Hz, 1H), 3.86 (s, 3H), 3.72 (dd, J = 2.0, 9.6 Hz, 1H), 2.78 (d, J = 11.6 Hz, 1H), 2.71 (dt, J = 4.5, 12.6 Hz, 1H), 2.46 (s, 3H), 2.30 (td, J = 2.5, 12.0 Hz, 1H), 1.58 (m, 1H), 1.40 (qd, J = 4.5, 13.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 171.5, 166.1, 144.5, 136.1, 132.6, 130.1, 129.1, 127.9, 125.7, 122.8, 120.7, 118.3, 111.7, 101.4, 53.6, 53.0, 51.7, 45.4, 39.5, 39.1, 24.5, 21.7; HRMS (m/z): [M+NH4]+calcd for C25H28N3O6S, 498.1699; found 498.1693. β-Lactone 38 (65 mg, 3%) was obtained as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 7.69 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.0 Hz, 1H), 7.15 (dd, J = 7.1, 7.4 Hz, 1H), 7.08 (dd, J = 7.4, 7.5 Hz, 1H), 4.84 (s, 1H), 4.03-4.00 (m, 2H), 3.51 (br s, 4H), 2.94 (dd, J = 3.6, 12.6 Hz, 1H), 2.88 (d, J = 11.6 Hz, 1H), 2.46-2.43 (m, 1H), 2.41 (s, 3H), 1.95 (br d, J = 10.1 Hz, 1H), 1.08 (qd, J = 4.3, 12.9 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 170.9, 166.9, 143.9, 136.2, 132.6, 130.0, 127.5, 125.4, 121.6, 119.3, 117.9, 111.5, 100.2, 76.8, 52.4, 52.1, 50.7, 44.8, 42.8, 38.8, 30.7, 28.7, 21.0; HRMS (m/z): [M + H]+ calcd for C25H25N2O6S, 481.1428; found 481.1426.
6-Benzyl 5-Methyl 11a-Hydroxy-11-(hydroxymethyl)-2-tosyl-3,4,4a,5,11,11a-hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (43a)
To a 0 °C solution of a mixture of β-lactones 35 and 37 (62.8 mg, 0.102 mmol) was added DIBAL-H (1.0 M in PhMe, 511 μL, 0.511 mmol). The resulting solution was stirred for 1.5 h, then poured into a stirring solution of aqueous NH4Cl and the mixture was stirred for an additional 2 h. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by preparative thin layer chromatography on silica gel (50% EtOAc/hexanes) to afford 1,3-diol 43a (white foam, 41.8 mg, 66%) and the corresponding diol derived from the trans-2-azadecalin 37 (5.6 mg, 9%). 1,3-diol 43a: 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 7.8 Hz, 1H), 7.67-7.64 (m, 3H), 7.49-7.47 (m, 2H), 7.43-7.28 (m, 7H), 5.47 (d, J = 12.0 Hz, 1H), 5.29 (d, J = 12.1 Hz, 1H), 4.80-4.75 (m, 1H), 4.68 (d, J = 4.0 Hz, 1H), 4.50-4.45 (m, 1H), 3.77 (br s, 1H), 3.65 (s, 1H), 3.50 (s, 3H), 2.47 (s, 3H), 2.30-2.22 (m, 2H), 2.15-2.10 (m, 1H), 1.99 (br s, 1H), 1.67 (s, 1H), 1.60-1.54 (m, 1H), 1.30-1.26 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 172.9, 152.0, 144.3, 137.1, 135.1, 133.3, 130.3, 130.2, 129.2, 129.1, 128.5, 128.1, 128.0, 125.2, 123.8, 119.4, 116.4, 114.0, 73.1, 69.3, 60.7, 54.3, 52.1, 46.1, 44.6, 43.6, 36.7, 24.9, 22.0; LRMS-ES+ m/z (relative intensity) 619 (MH+, 100); HRMS-ES+ (C33H35N2O8S) calcd 619.2114 (MH+), found 619.2111. Minor 1,3-diol (white foam): 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 7.9 Hz, 1H), 7.67-7.55 (m, 3H), 7.48-7.28 (m, 9H), 5.43 (q, J = 8.4 Hz, 2H), 4.65-4.53 (m, 2H), 4.33-4.30 (m, 1H), 4.10 (br s, 1H), 3.54 (s, 2H), 3.51 (s, 3H), 2.46-2.43 (m, 4H), 2.42-2.38 (m, 2H), 2.36-2.26 (m, 1H), 1.81-1.76 (m, 1H), 1.70-1.65 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 173.6, 151.9, 144.3, 137.0, 135.1, 133.3, 130.3, 129.30, 129.25, 129.2, 128.3, 128.2, 128.0, 125.4, 123.9, 119.5, 116.6, 72.5, 71.1, 69.3, 61.3, 57.5, 54.2, 52.7, 45.4, 25.7, 22.0; LRMS-ES+ m/z (relative intensity) 619 (MH+, 100); HRMS-ES+ (C33H35N2O8S) calcd 619.2114 (MH+), found 619.2138.
6-Benzyl 5-Methyl 11a-Hydroxy-11-(hydroxymethyl)-2-tosyl-3,4,4a,5,11,11a-hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (44)
To a solution of 1,3-diol 43a (12.0 mg, 0.019 mmol) in MeCH(OMe)2 (1 mL) was added a catalytic amount of p-TsOH·H2O (<1 mg) and the resulting mixture was placed in a preheated oil bath at 80 °C. After stirring for 30 min, the solution was concentrated in vacuo to give a residue which was purified by column chromatography on silica gel (30% EtOAc/hexanes) to yield the acetal 44 (pale gum, 11.6 mg, 93%). 1H NMR (300 MHz, CDCl3) δ 8.12 (br d, J = 6.1 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.52-7.48 (m, 3H), 7.46-7.38 (5H), 7.31-7.27 (m, 2H), 5.50 (d, J = 12.1 Hz, 1H), 5.34 (d, J = 10.3 Hz, 1H), 5.07 (q, J = 5.0 Hz, 1H), 4.78-4.66 (m, 3H), 4.36 (dd, J = 12.3, 3.0 Hz, 1H), 3.84-3.80 (m, 1H), 3.52 (s, 3H), 3.40 (br s, 1H), 2.49 (s, 3H), 2.27 (td, J = 12.2, 2.9 Hz, 1H), 2.19-2.06 (m, 2H), 1.70-1.55 (m, 1H), 1.31-1.23 (m, 1H), 1.20 (d, J = 4.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.9, 152.3, 144.5, 135.4, 133.5, 131.5, 130.4, 129.3, 129.13, 129.06, 128.3, 127.9, 124.8, 123.4, 119.5, 116.5, 116.3, 93.3, 72.5, 69.1, 65.3, 53.9, 52.1, 50.4, 46.2, 44.5, 43.3, 29.7, 23.7, 22.0, 21.6; LRMS-ES+ m/z (relative intensity) 645 (MH+, 50).
Methyl 10-(Hydroxymethyl)-3-methyl-7-tosyl-3,4,6,7,8,9,9a,10,11,15c-decahydro-1H-[1,4]dioxepino[5,6-c]pyrido[4,3-b]carbazole-10-carboxylate (45)
To a stirred suspension of paraformaldehyde (4.3 mg, 0.140 mmol) and ester 44 (6.0 mg, 0.009 mmol) in DMF (0.5 mL) was added NaH (60% dispersion on mineral oil, 1.9 mg, 0.005 mmol) and the resulting mixture was stirred for 2 h at rt. The reaction mixture was diluted with brine and EtOAc. The organic layer was separated and washed with water and then brine. The organic layer was dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash column chromatography on silica gel (75% EtOAc/hexanes) to give hydroxymethyl ester 45 (foam, 4.7 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 7.70 (d, J = 6.1 Hz, 2H), 7.57 (d, J = 6.0 Hz, 1H), 7.40-7.36 (m, 3H), 7.20 (t, J = 5.5 Hz, 1H), 7.12 (t, J = 5.8 Hz, 1H), 5.03 (q, J = 3.7 Hz, 1H), 4.83 (d, J = 9.0 Hz, 1H), 4.67 (d, J = 8.1 Hz, 1H), 4.37 (dd, J = 9.1, 2.1 Hz, 1H), 3.86-3.78 (m, 2H), 3.68 (s, 3H), 3.39-3.37 (m, 1H), 2.49 (s, 3H), 2.39-2.26 (m, 2H), 2.21-2.16 (m, 2H), 1.54-1.41 (m, 2H), 1.10 (d, J = 3.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 175.9, 144.5, 137.3, 133.3, 132.8, 130.3, 128.0, 126.0, 122.3, 119.7, 119.4, 111.8, 108.2, 93.1, 73.6, 68.7, 65.8, 52.8, 50.4, 49.5, 47.7, 46.2, 30.0, 22.7, 22.0, 21.3; LRMS-ES+ m/z (relative intensity) 541 (MH+, 30); HRMS-ES+ (C28H36N3O7S) calcd 558.2274 (M+NH4+), found 558.2265.
6-Benzyl 5-Methyl 11-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-2-tosyl-3,4,4a,5,11,11a-hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (47)
To a solution of 1,3-diol 43 (39.7 mg, 0.0642 mmol) and 2,6-lutidine (75 μL, 0.64 mmol) in CH2Cl2 (3 mL) at 0 °C was added TBSOTf (61 μL, 0.32 mmol). The resulting mixture was stirred for 1 h and then washed with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel (30% EtOAc/hexanes) to afford O-TBS-ether 47 (white foam, 45 mg, 96%). 1H NMR (300 MHz, CDCl3) δ 8.14 (d, J = 9.2 Hz, 1H), 7.70-7.67 (m, 2H), 7.59-7.56 (m, 1H), 7.48-7.34 (m, 7H), 7.30-7.25 (m, 2H), 5.50 (d, J = 7.8 Hz, 1H), 5.29 (d, J = 12.2 Hz, 1H), 4.73-4.69 (m, 2H), 4.51 (dd, J = 11.2, 1.7 Hz, 1H), 4.15 (d, J = 10.2 Hz, 1H), 3.77 (br d, J = 11.6 Hz, 1H), 3.66-3.64 (m, 1H), 3.50 (s, 3H), 2.47 (s, 3H), 2.22-2.17 (m, 2H), 2.10-2.03 (m, 1H), 1.63-1.53 (m, 2H), 0.60 (s, 9H), 0.04-0.03 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 173.1, 152.0, 144.3, 135.4, 130.2, 129.1, 129.0, 128.9, 128.6, 128.0, 124.7, 123.1, 119.9, 116.1, 115.6, 72.8, 69.0, 61.5, 54.7, 52.0, 46.2, 45.1, 43.5, 36.1, 30.1, 26.1, 25.7, 24.7, 22.0, 18.0, 1.4, −5.2, −5.7; LRMS-ES+ m/z (relative intensity) 733 (MH+, 100); HRMS-ES+ (C39H49N2O8SSi) calcd 733.2979 (MH+), found 733.2961.
Methyl 11-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-(((chloromethyl)dimethylsilyl)oxy)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (49)
To a solution of N-Cbz-indole 47 (45.0 mg, 0.061 mmol) in MeOH (1.5 mL) and EtOAc (1.5 mL) under an argon atmosphere was added 10% Pd/C (6.5 mg). The resulting mixture was evacuated and backfilled three times with H2 from a balloon and the resulting suspension was stirred for 2.5 h. The reaction mixture was then filtered through a pad of Celite with EtOAc and the filtrate was concentrated in vacuo to give NH indole 48 (colorless gum), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 7.8 Hz, 1H), 7.40-7.33 (m, 3H), 7.18 (t, J = 6.2 Hz, 1H), 7.13 (t, J = 8.1 Hz, 1H), 5.70 (s, 1H), 4.64-4.52 (m, 3H), 4.19 (dd, J = 11.4, 1.7 Hz, 1H), 3.80 (s, 3H), 3.77-3.68 (m, 1H), 3.69-3.67 (m, 1H), 2.48 (s, 3H), 2.31-2.18 (m, 3H), 1.54-1.41 (m, 2H), 0.67 (s, 9H), 0.09 (s, 3H), 0.07 (s, 3H); LRMS-ES+ m/z (relative intensity) 599 (MH+, 75); HRMS-ES+ (C31H43N2O6SSi) calcd 599.2611 (MH+), found 599.2585.
To a solution of crude alcohol 48 and 2,6-lutidine (270 μL, 1.23 mmol) in CH2Cl2 (2.5 mL) was added chloromethyl(dimethyl)silyl triflate (289 μL, 2.47 mmol). The resulting mixture was stirred for 1.5 h at rt and then washed with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo to afford a residue, which was purified by flash chromatography on silica gel (20% EtOAc/hexanes) to give chloromethyl(dimethyl)silyl ether 49 (colorless gum, 32.9 mg, 76% from 47). 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 7.67-7.62 (m, 3H), 7.40-7.33 (m, 3H), 7.20-7.12 (m, 2H), 4.73 (dd, J = 10.1, 1.5 Hz, 1H), 4.45-4.39 (m, 2H), 3.89 (t, J = 10.1 Hz, 1H), 3.82 (s, 3H), 3.78-3.70 (m, 2H), 2.79 (d, J = 13.8 Hz, 1H), 2.74 (d, J = 13.8 Hz, 1H), 2.48 (s, 3H), 2.26-2.17 (m, 3H), 1.51-1.42 (m, 1H), 1.37-1.26 (m, 1H). 1.04 (s, 9H), 0.32-0.28 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 173.4, 144.1, 136.8, 133.4, 130.1, 128.7, 128.1, 127.0, 121.9, 119.81, 119.78, 111.8, 108.2, 76.2, 64.0, 60.8, 56.8, 52.9, 47.7, 46.4, 39.8, 31.5, 26.8, 24.2, 22.0, 21.5, 19.2, 14.6, 0.2, −0.1, −4.6, −5.0; LRMS-ES+ m/z (relative intensity) 705 (MH+, 50).
Methyl 11-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-(((iodomethyl)dimethylsilyl)oxy)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (50)
A sealed tube was charged with chloride 49 (77.4 mg, 0.110 mmol), acetone (5 mL) and then NaI (411 mg, 2.74 mmol). The vessel was sealed and the resulting mixture was heated at reflux and stirred for 15 h. After cooling to rt, the reaction mixture was diluted with ether and the organic phase was washed with water followed by aqueous Na2S2O3. The aqueous layers were extracted with ether and the combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue. This material was purified by flash chromatography on silica gel (20% EtOAc/hexanes) to give iodide 50 (colorless gum, 84.4 mg, 97%). 1H NMR (300 MHz, CDCl3) δ 9.17 (s, 1H), 7.69-7.64 (m, 3H), 7.40-7.47 (m, 3H), 7.21-7.11 (m, 2H), 4.73 (d, J = 9.9 Hz, 1H), 4.48-4.42 (m, 2H), 3.92 (t, J = 9.8 Hz, 1H), 3.82 (s, 3H), 3.78-3.70 (m, 2H), 2.48 (s, 3H), 2.28-2.16 (m, 3H), 2.03 (q, J = 8.7 Hz, 2H), 1.50-1.28 (m, 3H), 1.05 (s, 9H), 0.38-0.28 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 173.4, 144.1, 136.8, 133.4, 130.1, 128.7, 128.1, 127.0, 121.8, 119.8, 111.7, 108.2, 76.2, 64.0, 56.8, 52.8, 47.8, 46.4, 39.9, 39.8, 26.8, 24.2, 22.0, 19.2, 1.3, 0.7, −4.5, −4.9, −12.4; LRMS-ES+ m/z (relative intensity) 797 (MH+, 40).
Methyl 11-(((tert-Butyldimethylsilyl)oxy)methyl)-13,13-dimethyl-2-tosyl-1,2,3,4,4a,5,6,11-octahydro-11a,5-(epoxysilanomethano)pyrido[4,3-b]carbazole-5-carboxylate (51)
To a solution of indole iodo ester 50 (36.9 mg, 0.0463 mmol) at −78 °C was added KHMDS (0.1 M solution in THF, 1.39 mL, 0.139 mmol). The resulting mixture was stirred for 1 h at −78 °C, then transferred to a ice bath and stirred for an additional 15 min. The reaction mixture was diluted with aqueous NH4Cl, the organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue, which was purified by flash chromatography on silica gel (25% EtOAc/hexanes) to afford pentacycle 51 (white foam, 31.0 mg, 97%). 1H NMR (300 MHz, CDCl3) δ 9.00 (s, 1H), 8.06 (d, J = 7.08 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.39-7.34 (m, 3H), 7.21 (t, J = 7.7 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 4.50 (d, J = 10.7 Hz, 1H), 5.60 (dd, J = 10.4, 4.8 Hz, 1H), 4.17 (dd, J = 10.5, 5.1 Hz, 1H), 3.80-3.72 (m, 5H), 2.47 (s, 3H), 2.34-2.26 (m, 1H), 2.08 (d, J = 11.1 Hz, 1H), 1.91-1.86 (m, 1H), 1.53-1.41 (m, 3H), 1.06 (s, 9H), 0.97-0.85 (m, 2H), 0.29 (s, 3H), 0.27 (s, 3H), 0.10 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 176.1, 143.8, 137.8, 133.6, 132.1, 130.0, 128.4, 127.1, 122.4, 121.8, 119.6, 111.5, 110.4, 75.5, 63.8, 55.6, 52.8, 49.9, 46.9, 46.7, 40.5, 30.1, 28.0, 26.7, 25.5, 22.0, 19.1, 2.23, 2.16, −4.8, −4.9; LRMS-ES+ m/z (relative intensity) 669 (MH+, 30); HRMS-ES+ (C34H49N2O6SSi2) calcd 669.2850 (MH+), found 669.2857.
Methyl 7-(Hydroxymethyl)-1-oxo-4-tosyl-3,4,5,6,6a,7,8,12c-octahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7-carboxylate (55)
To a solution of indole β-lactone 36 (255 mg, 0.531 mmol) in THF (25 mL) cooled to −78 °C was added a solution of LiHMDS (1.0 M in THF, 1.60 mL, 1.60 mmol) dropwise with stirring. The resulting orange red solution was stirred at −78 °C for another 30 min. A solution of freshly distilled monomeric formaldehyde15 in THF (~0.5 M, 10.6 mL, 5.3 mmol) was added dropwise. The resulting brownish red solution was stirred at −78 °C for 5 min, then warmed to −40 °C and stirred for another 15 min. The reaction mixture was then quenched at −40 °C by addition of glacial acetic acid (0.10 mL, 1.6 mmol). The bright yellow solution was diluted with dichloromethane, quickly washed with ice cold water and dried over Na2SO4. After concentration of the solution, the residue was purified by column chromatography on silica gel (5% Et2O/CH2Cl2, then 50% EtOAc/hexanes) to afford α-hydroxymethyl ester 55 as an off-white solid (160 mg, 59%). FT-IR (film) 3403, 1824 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.43 (s, 1H, NH), 7.62–7.66 (m, 4H), 7.34–7.42 (m, 4H), 7.16–7.25 (m, 2H), 4.86 (s, 1H), 3.97–4.02 (m, 3H), 3.83 (s, 3H), 3.68 (app. d, J = 15.6 Hz, 1H), 2.74 (d, J = 15.7 Hz, 1H), 2.57 (dd, J = 6.4, 15.9 Hz, 1H), 2.46 (s, 3H), 2.26 (td, J = 4.2, 15.7 Hz, 1H), 2.13 (t, J = 8.2 Hz, 1H), 1.43–1.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.4, 167.1, 144.7, 136.6, 132.9, 131.6, 130.2, 128.0, 125.7, 123.2, 120.8, 118.6, 111.8, 101.0, 69.5, 55.4, 54.1, 53.1, 51.7, 45.3, 40.7, 26.8, 21.8; ESI MS (m/z): [M + H]+ 511.3; HRMS-ES (m/z): [M + H]+ calcd for C26H27N2O7S, 511.1539; found, 511.1538.
Methyl 11a-Hydroxy-12-oxo-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-5,11-(methanooxymethano)pyrido[4,3-b]carbazole-5-carboxylate (57)
To a solution of β-lactone 55 (200 mg, 0.39 mmol) in dichloromethane (2.6 mL) at rt was added triethylamine (10.6 mL, excess). The resulting slightly yellow solution was stirred at rt for 2 h, evaporated to dryness and the residue was purified by flash column chromatography on silica gel (5% Et2O/CH2Cl2, then 50% EtOAc/hexanes) to afford ε-lactone 57 as an off white solid (65 mg, 33%). The recovered β-lactone 55 was subjected to another translactonization under the same conditions. After 2 runs, 40 mg of β-lactone 55 was recovered and ε-lactone 57 was isolated by chromatography on silica gel (gradient 20–50% EtOAc/hexanes) as an off white foam (92 mg, BRSM, 58%). FT-IR (neat) 3407, 1725 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H, NH), 7.61 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 7.5 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.25 – 7.27 (m, 4H), 7.21 (t, J = 7.4 Hz, 1H), 7.15 (t, J = 7.5 Hz), 4.90 (d, J = 11.7 Hz, 1H), 4.37 (d, J = 11.7 Hz, 1H), 4.16 (s, 1H), 3.93 (s, 3H), 3.65 (br s, 1H), 3.58 (d, J = 13.5 Hz, 1H), 3.52–3.58 (m, 1H), 3.01 (m, 1H), 2.81 (dd, J = 3.8, 13.6 Hz, 1H), 2.59 (d, J = 13.6 Hz, 1H), 2.37 (s, 3H), 1.40 (m, 1H), 0.76 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 171.0, 167.8, 144.2, 135.9, 134.4, 132.0, 130.2, 127.4, 124.9, 122.8, 120.9, 118.0, 113.7, 112.1, 107.0, 76.0, 73.6, 53.7, 53.0, 52.8, 52.2, 50.6, 42.4, 23.8, 21.7; ESI MS (m/z): [M + H]+ 511.3; HRMS (m/z): [M + NH4]+ calcd for C26H30N3O7S 528.1804; found 528.1797.
Methyl 7-(((tert-Butyldimethylsilyl)oxy)methyl)-1-oxo-4-tosyl-3,4,5,6,6a,7,8,12c-octahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7-carboxylate (56)
To a solution of hydroxymethyl compound 55 (100 mg, 0.196 mmol) in CH2Cl2 (15 mL) at 0 °C was added 2,6-lutidine (226 μL, 1.95 mmol) and freshly distilled TBSOTf (225 μL, 0.98 mmol) with stirring. The colorless reaction mixture was stirred at 0 °C and monitored by TLC. Once the reaction was complete (~40 min), the solvent was removed in vacuo. The residue was purified by flash chromatography on silica gel (gradient 5–30% EtOAc/hexanes) to afford O-TBS ether 56 as a white foam (105 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 9.32 (br s, 1H, NH), 7.66-7.63 (m, 3H), 7.40 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 7.7 Hz, 2H), 7.23 (m, 1H), 7.19 (dd, J = 7.5, 7.6 Hz, 1H), 4.84 (s, 1H), 4.03 (d, J = 10.1 Hz, 1H), 4.00 (d, J = 12.4 Hz, 1H), 3.88 (d, J = 9.2 Hz, 1H), 3.81 (s, 3H), 3.70 (m, 1H), 2.74 (d, J = 11.6 Hz, 1H), 2.55 (dd, J = 4.7, 12.1 Hz, 1H), 2.46 (s, 3H), 2.27 (td, J = 3.0, 11.8 Hz,, 1H), 1.55-1.47 (m, 2H), 0.80 (s, 9H), −0.17 (s, 3H), −0.33 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 172.5, 167.4, 144.8, 136.4, 132.7, 132.4, 130.4, 128.2, 125.7, 123.1, 120.7, 118.7, 111.7, 100.4, 70.3, 55.8, 54.4, 53.0, 51.9, 45.5, 41.3, 27.1, 26.1, 25.9, 22.0, 18.4, −5.6; HRMS (m/z): [M + H]+ calcd for C32H41N2O7SSi, 625.2404; found 625.2390.
Methyl 11a-Hydroxy-5-(hydroxymethyl)-11-methyl-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (60)
To a stirred solution of aldehyde 58 (10 mg, 0.020 mmol) in CH2Cl2 (1.5 mL) at rt was added BF3·OEt2 (2.5 μL, 0.020 mmol) and 1,2-ethanedithiol (3.5 μL, 0.042 mmol). The reaction mixture was stirred at rt for 12 h, then diluted with CH2Cl2 and phosphate buffer (pH 7). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo to give a residue which was purified by column chromatography on silica gel (gradient 30–50% EtOAc/hexanes) to afford dithiane 59 as a white foam (10 mg, 87%) which was used directly in the next step. 1H NMR (400 MHz, CDCl3) δ 9.05 (br s, 1H), 7.68-7.60 (m, 3H), 7.44 (d, J = 7.8 Hz, 1H), 7.38 (d, J = 8.9 Hz, 2H), 7.23-7.12 (m, 2H), 5.87 (br s, 1H), 4.88 (s, 1H), 4.28 (d, J = 11.2 Hz, 1H), 4.17-4.02 (m, 3H), 3.87 (s, 3H), 3.70 (m, 1H), 3.05 (m, 1H), 2.78 (d, J = 11.7 Hz, 1H), 2.45 (s, 3H), 2.30 (m, 3H); LRMS-ES+ m/z (relative intensity) 589 (MH+).
To a slurry of freshly prepared W-2 Raney nickel (large excess) in ethanol was added a solution of dithiane 59 (12.0 mg, 0.020 mmol) in ethanol (5 mL). The reaction mixture was heated at 60 °C until TLC indicated consumption of the starting material (~45 min). The Raney nickel was filtered off through a pad of Celite, and the filtrate was evaporated to dryness in vacuo to give essentially pure methyl compound 60 as a white powder (7.5 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 8.64 (br s, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.35 (m, 3H), 7.20 (dd, J = 7.2, 7.2 Hz, 1H), 7.12 (dd, J = 7.2, 7.2 Hz, 1H), 4.11 (d, J = 11.2 Hz, 1H), 4.01 (d, J = 11.2 Hz, 1H), 3.93 (d, J = 10.6 Hz, 1H), 3.75 (s, 3H), 3.67 (q, J = 7.1 Hz, 1H), 2.45 (s, 3H), 2.27 (m, 1H), 2.13 (dd, J = 3.7, 12.5 Hz, 1H), 2.08 (d, J = 11.6 Hz, 1H), 1.56 (d, J = 7.0 Hz, 3H), 0.83 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 174.1, 137.9, 133.3, 130.2, 128.1, 122.5, 120.9, 120.0, 111.9, 78.8, 72.7, 53.0, 47.8, 46.1, 31.0, 30.1, 26.8, 22.0, 13.3.
Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-11-(hydroxymethyl)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (61)
To a solution of O-TBS β-lactone 56 (120 mg, 0.193 mmol) in THF at rt was added LiBH4 (22 mg, 1.0 mmol). The reaction mixture was stirred at rt for 12 h, diluted with aqueous NH4Cl and extracted with CH2Cl2. The extract was dried over Na2SO4 and the solvent was removed in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 2–5% Et2O/CH2Cl2) affording diol 61 as a white solid (90 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 8.76 (br s, 1H), 7.69-7.66 (m, 3H), 7.39 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 7.7 Hz, 2H), 7.22 (dd, J = 7.6, 7.8 Hz, 1H), 7.14 (dd, J = 7.1, 7.4 Hz, 1H), 4.87 (d, J = 12.2 Hz, 1H), 4.44 (d, J = 12.9 Hz, 1H), 4.23 (d, J = 8.7 Hz, 1H), 4.08 (d, J = 9.5 Hz, 2H), 3.71 (s, 3H), 3.67 (m, 1H), 2.45 (s, 3H), 2.25 (dd, J = 10.5, 10.8 Hz, 1H), 2.17 (d, J = 11.8 Hz, 1H), 1.92 (d, J = 12.2 Hz, 1H), 1.59 (m, 1H), 1.40 (m, 1H), 0.87 (s, 9H), −0.05 (s, 3H), −0.11 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 173.4, 144.3, 137.0, 135.0, 133.2, 130.2, 128.1, 125.8, 122.3, 120.2, 119.2, 111.9, 103.8, 74.5, 71.8, 61.0, 55.8, 54.9, 52.5, 47.6, 46.1, 37.4, 26.4, 26.2, 22.0, 18.5, −5.2, −5.5; HRMS (m/z): [M + H]+ calcd for C32H45N2O7SSi, 629.2717; found 629.2717.
Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-11-(iodomethyl)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (62)
To a solution of PPh3 (229 mg, 0.87 mmol) and I2 (222 mg, 0.87 mmol) in CH2Cl2 (9 mL) at rt was added imidazole (99 mg, 1.48 mmol). The resulting yellow suspension was stirred at rt for 10 min. A solution of diol 61 (122 mg, 0.195 mmol) in CH2Cl2 (15 mL) was added dropwise, and the bright yellow suspension was stirred at rt for 5 h. The mixture was diluted with CH2Cl2 and washed with 5% aqueous Na2S2O3. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, and the solvent was removed in vacuo. The residue was purified by flash column chromatography on silica gel (2% Et2O/CH2Cl2) to afford iodo alcohol 62 as a white foamy solid (133 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 8.80 (br s, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 7.7 Hz, 1H), 7.37-7.33 (m, 3H), 7.20 (dd, J = 7.1, 7.5 Hz, 1H), 7.14 (dd, J = 7.4, 7.4 Hz, 1H), 4.17-4.09 (m, 2H), 4.00-3.96 (m, 1H), 3.90-3.88 (m, 2H), 3.68 (s, 3H), 3.30 (s, 1H), 2.44 (s, 3H), 2.30 (m, 1H), 2.18 (m, 1H), 2.08 (dd, J = 3.1, 11.5 Hz, 1H), 1.63 (m, 1H), 1.43 (m, 1H), 0.84 (s, 9H), −0.08 (s, 3H), −0.10 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 172.9, 144.0, 136.5, 132.7, 132.3, 132.1, 129.9, 128.7, 127.9, 126.0, 122.0, 119.6, 111.4, 108.9, 72.9, 71.0, 54.8, 54.3, 52.3, 45.6, 36.4, 25.8, 21.6, 18.2, 4.8, 1.1, −5.6, −5.7; HRMS (m/z): [M + H]+ calcd for C32H44IN2O6SSi, 739.1734; found 739.1764.
Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-11-methyl-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (63)
10% Pd/C (158 mg) was suspended in a stirred solution of iodide 62 (29 mg, 0.039 mmol) in t-BuOH/EtOAc (1:1 v/v, 30 mL). The reaction mixture was evacuated and backfilled with H2 three times from a balloon and stirred under a H2 atmosphere at rt. After 3.5 h, another portion of 10% Pd/C (32 mg) was added and the reaction mixture was stirred under H2 at rt for another 12 h. The Pd/C was filtered off through a pad of Celite, and the solvent was removed in vacuo to afford a yellow foamy solid. This material was purified by flash column chromatography on silica gel (CH2Cl2, then 2% Et2O/CH2Cl2) to afford methyl compound 63 as a white solid (23 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 8.51 (br s, 1H), 7.72 (d, J = 7.9 Hz, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.34-7.32 (m, 3H), 7.17 (dd, J = 7.1, 7.6 Hz, 1H), 7.08 (dd, J = 7.2, 7.4 Hz, 1H), 4.13 (d, J = 9.4 Hz, 1H), 4.00 (d, J = 9.4 Hz, 1H), 3.96 (d, J = 11.8 Hz, 1H), 3.72 (s, 3H), 3.66 (q, J = 7.0 Hz, 1H), 3.09 (s, 1H), 2.45 (s, 3H), 2.26 (t, J = 10.4 Hz, 1H), 2.04 (d, J = 13.0 Hz, 1H), 2.00 (dd, J = 3.7, 12.6 Hz, 1H), 1.63 (qd, J = 4.4, 12.9 Hz, 1H), 1.57 (d, J = 6.9 Hz, 3H), 0.80 (s, 3H), −0.07 (s, 3H), −0.12 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 173.3, 143.9, 137.1, 133.0, 130.0, 129.9, 129.8, 127.9, 126.7, 121.7, 120.5, 119.1, 111.4, 111.2, 72.5, 70.7, 55.1, 53.4, 52.3, 48.2, 46.0, 30.5, 26.5, 25.8, 25.7, 21.7, 18.1, 12.8, −5.7, −5.8; HRMS (m/z): [M + H]+ calcd for C32H45N2O6SSi, 613.2768; found 613.2772.
Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-11-methyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (64)
Magnesium turnings (399 mg, excess) were added to a solution of sulfonamide 63 (21.6 mg, 0.035 mmol) in anhydrous methanol, and the mixture was sonicated at rt until all the magnesium turnings dissolved (~45 min). The reaction mixture was then poured into aqueous NH4Cl at 0 °C, and extracted with CHCl3. The organic extract was dried over Na2SO4, and evaporated to dryness in vacuo to give a white foamy solid. This material was purified by flash chromatography on silica gel (gradient 2–5% MeOH/CHCl3, then 1% NEt3 in 5% MeOH/CHCl3) to afford piperidine 64 as a white solid (15.6 mg, 96%). 1H NMR (400 MHz, CD3OD) δ 7.90 (br s, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.08 (dd, J = 7.2, 7.4 Hz, 1H), 6.98 (dd, J = 7.2, 7.3 Hz, 1H), 4.34 (d, J = 9.3 Hz, 1H), 4.20 (d, J = 9.4 Hz, 1H), 3.77 (s, 3H), 3.42 (q, J = 6.7 Hz, 1H), 3.28 (m, 1H), 3.03 (q, J = 7.2 Hz, 1H), 2.94 (br d, J = 12.6 Hz, 1H), 2.69 (m, 1H), 2.50 (d, J = 12.7 Hz, 1H), 2.29 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 0.83 (s, 9H), −0.02 (s, 3H), −0.10 (s, 3H); 13C NMR (75 MHz, CD3OD) δ 175.1, 138.6, 131.7, 127.9, 122.2, 120.8, 119.4, 112.7, 112.0, 79.6, 72.4, 71.6, 56.9, 54.0, 52.7, 47.8, 45.4, 32.1, 26.4, 19.2, 14.5, −5.4, −5.5; HRMS (m/z): [M + H]+ calcd for C25H39N2O4Si, 459.2674; found 459.2676.
Synthesis of (±)-Alstilobanine A (3)
A solution of O-TBS indole 64 (16.5 mg, 0.036 mmol) in CHCl3 (3.3 mL) at 0 °C was treated with a solution of hydrogen chloride in MeOH (1.25 M, 3.3 mL). The resultant colorless solution was stirred at rt for 2 h, and the volatiles were removed under high vacuum to afford (±)-alstilobanine A (3) hydrochloride salt as a white solid (12.2 mg, 100%). 1H NMR (400 MHz, CD3OD) δ 7.55 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.08 (dd, J = 7.4, 7.5 Hz, 1H), 6.98 (dd, J = 7.4, 7.5 Hz, 1H), 4.14 (d, J = 10.9 Hz, 1H), 4.04 (d, J = 10.8 H, 1H), 3.78 (s, 3H), 3.54 (d, J = 12.5 Hz, 1H), 3.26 (q, J = 7.1 Hz, 1H), 3.09 (m, 1H), 2.93 (m, 1H), 2.87 (d, J = 12.6 Hz, 1H), 2.52 (m, 1H), 2.06 (m, 1H), 1.87 (m, 1H), 1.43 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 174.9, 138.9, 130.9, 127.2, 122.5, 119.6, 119.5, 113.3, 112.2, 71.0, 68.7, 55.2, 53.0, 50.5, 42.4 (2C), 34.4, 22.6, 15.7; HRMS (m/z): [M + H]+ calcd for C19H25N2O4, 345.1809; found 345.1809.
Methyl 11a-Hydroxy-5,11-bis(hydroxymethyl)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (65)
To a solution of β-lactone alcohol 55 (10.0 mg, 0.020 mmol) in THF (1.7 mL) at rt was added dropwise a solution of DIBAL-H in toluene (1.5 M, 0.15 mL, 0.22 mmol). The reaction mixture was stirred at rt for 2 h, was then diluted with aqueous NH4Cl and 10% Rochelle’s salt and extracted with EtOAc. The organic layer was dried over Na2SO4 and evaporated in vacuo to give a brown residue which was purified by flash chromatography on silica gel (gradient 30–50% EtOAc/hexanes) to afford triol 65 as a slightly brown gum (4.9 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 8.94 (br s, 1H), 7.66 (d, J = 8.1 Hz, 3H), 7.41 (d, J = 8.1 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.22 (app t, J = 7.4 Hz, 1H), 7.14 (app t, J = 7.2 Hz, 1H), 5.11 (app br s, 1H), 4.78 (d, J = 12.9 Hz, 1H), 4.47 (t, J = 9.7 Hz, 1H), 4.19 (dd, J = 4.2, 11.1 Hz, 1H), 4.13-4.06 (m, 2H), 3.76 (s, 3H), 3.69 (m, 2H), 2.71 (m, 1H), 2.45 (s, 3H), 2.27 (m, 1H), 2.21 (d, J = 12.0 Hz, 1H), 2.10 (dd, J = 3.7, 12.2 Hz, 2H), 1.44 (m, 1H), 0.87 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 174.1, 144.3, 133.2, 130.2, 128.0, 122.6, 120.5, 119.3, 117.6, 112.1, 111.2, 74.4, 71.3, 70.8, 61.2, 55.1, 55.0, 52.9, 45.8, 30.1, 26.5, 22.0; LRMS-ES+ m/z (relative intensity) 515 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C26H31N2O7S, 515.1852; found 515.1851.
Methyl 11-(Hydroxymethyl)-2-tosyl-1,2,3,4,4a,5,6,11-octahydro-11a,5-(epoxymethano)pyrido[4,3-b]carbazole-5-carboxylate (67) and Methyl 7-(Hydroxymethyl)-4-tosyl-3,4,5,6,6a,7,8,12c-octahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7-carboxylate (68)
To a solution of PPh3 (8.5 mg, 0.032 mmol) and triol 65 (12.0 mg, 0.024 mmol) in THF (5 mL) at rt was added dropwise DIAD (5 μL, 0.016 mmol). The resulting orange solution was stirred at rt for 1 h. The volatiles were then removed in vacuo to give an orange residue which was purified by flash chromatography on silica gel (gradient 20–30% EtOAc/hexanes) to afford tetrahydrofuranyl ether 67 as the major product (white foam, 6.1 mg, 51%). 1H NMR (400 MHz, CDCl3) δ 9.04 (br s, 1H), 7.69 (d, J = 7.7 Hz, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.36 (m, 1H), 7.18 (dd, J = 7.1, 7.8 Hz, 1H), 7.13 (dd, J = 7.1, 7.8 Hz, 1H), 4.97 (d, J = 4.0 Hz, 1H), 4.73 (d, J = 10.8 Hz, 1H), 4.38 (d, J = 11.7 Hz, 1H), 4.21-4.18 (m, 3H), 3.97 (d, J = 9.9 Hz, 1H), 3.86 (s, 3H), 3.72 (d, J = 5.1 Hz, 1H), 2.45 (s, 3H), 2.41 (m, 1H), 2.29 (d, J = 12.0 Hz, 1H), 1.69-1.54 (m, 1H), 1.49 (dd, J = 4.3, 12.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 171.4, 144.0, 136.1, 134.3, 132.8, 130.0, 127.5, 126.6, 122.1, 120.2, 119.6, 113.9, 111.7, 106.9, 106.1, 83.1, 81.3, 62.1, 54.7, 54.0, 53.2, 51.8, 45.7, 39.4, 29.9, 22.8, 21.8; LRMS-ES+ m/z (relative intensity) 497 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C26H29N2O6S, 497.1746; found 497.1729.
Oxetane 68 (white solid, 2.8 mg, 23%). 1H NMR (400 MHz, CDCl3) δ 9.24 (br s, 1H), 7.69-7.23 (m, 8H), 5.13 (t, J = 6.1 Hz, 1H), 4.42 (m, 1H), 4.31 (m, 1H), 4.12 (m, 1H), 4.04 (dd, J = 3.5, 6.4 Hz, 1H), 3.96 (dd, J = 3.3, 5.5 Hz, 1H), 3.83 (s, 3H), 3.60 (m, 1H), 2.45 (s, 3H), 2.39 (d, J = 11.4 Hz, 1H), 2.23-2.00 (m, 2H), 1.39 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 173.7, 144.2, 136.8, 133.2, 132.7, 132.4, 131.7, 130.3, 128.7, 128.0, 126.1, 122.7, 120.0, 116.1, 110.4, 85.1, 75.3, 71.7, 55.7, 54.2, 52.9, 45.6, 43.0, 33.8, 26.0, 21.8; HRMS (m/z): [M + H]+ calcd for C26H29N2O6S, 497.1746; found 497.1752.
Oxetane 68 could be formed as the major product using the following procedure: to a solution of PPh3 (4.3 mg, 0.016 mmol) and triol 65 (6.5 mg, 0.013 mmol) in benzene at 0 °C was added dropwise a solution of DEAD in toluene (40 wt %, 6.5 μL, 0.014 mmol). The reaction mixture was stirred at 0 °C for 5 min, and then heated at reflux for 1 h. The mixture was cooled to rt and concentrated in vacuo to give a residue which was purified by preparative TLC (30% EtOAc/hexanes, plate developed 4 times) to afford oxetane 68 as a white solid (4.4 mg, 70%), together with small amounts of cyclopropane 66 (<0.5 mg, <8%) and tetrahydrofuranyl ether 67 (<0.5 mg, <8%).
Methyl 3-Tosyl-1,1a,2,3,4,5,5a,6-octahydro-1b,6-(epoxymethano)cyclopropa[d]pyrido[4,3-b]carbazole-6-carboxylate (66)
To a solution of PPh3 (21 mg, 0.080 mmol) and triol 65 (8.4 mg, 0.016 mmol) in THF (1.5 mL) at 0 °C was added dropwise DIAD (16 μL, 0.050 mmol). The resulting orange solution was stirred at 0 °C for 10 min, then allowed to warm to rt and stirred for 5 h. The volatiles were removed in vacuo to give an orange residue which was purified by flash chromatography on silica gel (gradient 20–30% EtOAc/hexanes) to afford cyclopropane 66 as a white foam (6.6 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.8 Hz, 1H), 7.68 (m, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.47 (m, 3H), 4.25 (d, J = 9.2 Hz, 1H), 4.21 (d, J = 10.2 Hz, 1H), 4.08 (d, J = 9.5 Hz, 1H), 3.86 (s, 3H), 3.81 (m, 1H), 3.17 (dd, J = 5.6, 8.5 Hz, 1H), 2.64 (t, J = 5.4 Hz, 1H), 2.47 (d, J = 10.5 Hz, 1H), 2.44 (s, 3H), 2.21 (m, 1H), 2.15 (dd, J = 3.2, 12.7 Hz, 1H), 2.06 (m, 1H), 1.93 (m, 1H), 0.95 (m, 1H), 0.77 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 170.2, 132.4, 132.3, 132.2, 130.1, 128.8, 128.7, 127.7, 127.2, 125.2, 121.8, 119.0, 116.6, 112.8, 107.9, 106.3, 78.9, 72.8, 60.5, 58.6, 53.5, 53.0, 46.0, 42.2, 24.3, 22.0, 21.8; LRMS-ES+ m/z (relative intensity) 479 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C26H27N2O5S, 479.1641; found 479.1631.
Methyl 6-(((tert-Butyldimethylsilyl)oxy)methyl)-1b-hydroxy-3-tosyl-1a,1b,2,3,4,5,5a,6-octahydro-1H-cyclopropa[d]pyrido[4,3-b]carbazole-6-carboxylate (69)
To a stirred solution of AgOTf (2.6 mg, 0.010 mmol) in CH2Cl2 (0.8 mL) at rt was added dropwise a solution of iodide 62 (4.0 mg, 0.0054 mmol) in CH2Cl2 (0.2 mL). After 15 min, the colorless solution became a yellow suspension. The reaction mixture was then filtered through a pad of Celite. The filtrate was concentrated in vacuo to give cyclopropane 69 as a white solid (3.0 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 8.1 Hz, 2H), 7.36 (dd, J = 7.5, 7.6 Hz, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.23 (m, 1H), 7.06 (d, J = 7.4 Hz, 1H), 4.16 (d, J = 9.7 Hz, 1H), 3.99 (d, J = 9.9 Hz, 1H), 3.52 (m, 2H), 3.48 (br s, 1H), 2.83 (s, 3H), 2.72-2.70 (m, 2H), 2.62 (t, J = 7.9 Hz, 1H), 2.37 (s, 3H), 2.30 (m, 1H), 1.77 (dd, J = 4.8, 8.0 Hz, 1H), 0.82 (m, 1H), 0.75 (s, 9H), −0.05 (s, 3H), −0.15 (s, 3H); LRMS-ES+ m/z (relative intensity) 611 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C32H42N2O6SSi, 611.2611; found 611.2602.
Methyl 1b-Hydroxy-3-tosyl-1a,1b,2,3,4,5,5a,7-octahydro-1H-cyclopropa[d]pyrido[4,3-b]carbazole-6-carboxylate (70)
To a stirred solution of OTBS iodomethyl indole 62 (28 mg, 0.038 mmol) in CH2Cl2 (5.6 mL) at rt was added a solution of TBAF in THF (1.0 M, 71 μL, 0.071 mmol). The reaction mixture was stirred at rt for 1.5 h, diluted with aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (gradient 20–50% EtOAc/hexanes) to afford cyclopropane indoline 70 as a white solid (17 mg, 97%). 1H NMR (400 MHz, CDCl3) δ 9.57 (br s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.27 (m, 2H), 7.17 (dd, J = 7.5, 7.6 Hz, 1H), 6.93 (dd, J = 7.5, 7.6 Hz, 1H), 6.85 (d, J = 7.7 Hz, 2H), 3.70 (s, 3H), 3.59 (d, J = 11.4 Hz, 1H), 3.47 (d, J = 11.9 Hz, 1H), 3.18 (s, 1H), 2.98 (d, J = 14.5 HZ, 1H), 2.56 (br s, 1H), 2.40 (s, 3H), 2.37-2.32 (m, 2H), 2.23-2.13 (m, 2H), 2.06 (m, 1H), 1.40 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 169.2, 162.5, 144.2, 143.9, 133.6, 131.0, 130.1, 127.7, 127.6, 121.2, 118.7, 116.7, 114.0, 111.5, 109.4, 85.9, 69.4, 51.7, 50.8, 42.7, 34.4, 32.6, 32.5, 29.8, 23.4, 21.7, 20.6; LRMS-ES+ m/z (relative intensity) 467 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C25H27N2O5S, 467.1641; found 467.1671.
8-Benzyl 7-Methyl 7-(((tert-Butyldimethylsilyl)oxy)methyl)-1-oxo-4-tosyl-3,4,5,6,6a,7-hexahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7,8(12cH)-dicarboxylate (71)
To a solution of NH indole-β-lactone 56 (28 mg, 0.045 mmol) in THF (2.5 mL) cooled to −78 °C was added a solution of KHMDS in toluene (0.5 M, 0.14 mL, 0.070 mmol). After the resulting yellow solution was stirred at −78 °C for 30 min, CbzCl (neat, 51 μL, 0.36 mmol) was added dropwise. The reaction mixture was allowed to warm to rt, stirred for another 5 h, and then diluted with CH2Cl2. The organic phase was washed with aqueous NH4Cl, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CH2Cl2) to afford the N-Cbz-β-lactone 71 as a colorless gum (31 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 7.98-7.95 (m, 1H), 7.66 (m, 3H), 7.48 (m, 2H), 7.42-7.29 (m, 6H), 5.46 (d, J = 12.0 Hz, 1H), 5.35 (d, J = 12.0 Hz, 1H), 4.71 (m, 2H), 4.41 (d, J = 10.1 Hz, 1H), 3.97 (d, J = 10.1 Hz, 1H), 3.82 (m, 1H), 3.47 (m, 1H), 3.39 (s, 3H), 2.95 (m, 2H), 2.46 (s, 3H), 2.43 (m, 1H), 1.64-1.59 (m, 1H), 0.73 (s, 9H), −0.04 (s, 3H), −0.11 (s, 3H).
6-Benzyl 5-Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11a-hydroxy-11-(hydroxymethyl)-2-tosyl-3,4,4a,5,11,11a-hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (72)
To a solution of N-Cbz-indole-β-lactone 71 (10.0 mg, 0.013 mmol) in THF (2.0 mL) at rt was added solid LiBH4 (2.0 mg, 0.091 mmol). The reaction mixture was stirred at rt for 3 h, diluted with aqueous NH4Cl, and extracted with CH2Cl2. The organic layer was dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (gradient 2–5% Et2O/CH2Cl2) to afford diol 72 as a white solid (6.4 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 8.02 (br m, 1H), 7.87 (m, 1H), 7.65 (m, 2H), 7.46 (m, 2H), 7.40-7.30 (m, 7H), 5.46 (d, J = 13.2 Hz, 1H), 5.37 (d, J = 12.0 Hz, 1H), 4.71 (s, 1H), 4.43 (m, 1H), 4.35 (m, 1H) 4.22 (m, 2H), 3.78 (m, 1H), 3.56 (s, 1H), 3.36 (s, 3H), 2.44 (s, 3H), 2.12 (m, 2H), 0.65 (s, 9H), −0.03 (s, 3H), −0.20 (s, 3H); LRMS-ES+ m/z (relative intensity) 763 (MH+, 100); HRMS (m/z): [M + H]+ calcd for C40H51N2O9SSi, 763.3085; found 763.3121.
6-Benzyl 5-Methyl 11-(Bromomethyl)-11a-hydroxy-5-(hydroxymethyl)-2-tosyl-3,4,4a,5,11,11a-hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (73)
To a solution of PPh3 (39 mg, 0.15 mmol) and CBr4 (45 mg, 0.14 mmol) in CH2Cl2 at rt was added dropwise a solution of N-Cbz diol 72 (41 mg, 0.054 mmol) in CH2Cl2 (8.2 mL). The resulting cloudy reaction mixture was stirred at rt for 20 h. The mixture was evaporated in vacuo to give a residue which was purified by preparative TLC on silica gel (5% Et2O/CH2Cl2) to afford the bromo alcohol 73 as a white solid (10 mg, 26%). 1H NMR (400 MHz, CDCl3) δ 8.05 (m, 1H), 7.67 (m, 3H), 7.48 (d, J = 7.6 Hz, 2H), 7.43-7.30 (m, 7H), 5.48 (d, J = 12.0 Hz, 1H), 5.35 (d, J = 12.0 Hz, 1H), 4.41 (br s, 2H), 4.22-4.11 (m, 2H), 4.03 (d, J = 6.6 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.66 (m, 1H), 3.33 (s, 3H), 2.45 (s, 3H), 2.28-2.14 (m, 2H), 1.49 (m, 1H), 1.00-0.86 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.0, 144.4, 135.0, 133.0, 130.2, 129.4, 129.3, 129.2, 128.4, 128.2, 125.7, 123.8, 120.2, 116.4, 112.3, 110.0, 70.9, 69.5, 54.9, 52.0, 31.2, 27.3, 22.0; HRMS (m/z): [M + H]+ calcd for C34H36N2O8SBr, 711.1376; found 711.1417.
8-Benzyl 7-Methyl 7-(Hydroxymethyl)-4-tosyl-3,4,5,6,6a,7-hexahydro-1H-oxeto[2,3-c]pyrido[4,3-b]carbazole-7,8(12cH)-dicarboxylate (74)
To a solution of bromide 73 (1.6 mg, 0.0023 mmol) in CH2Cl2 (0.8 mL) was added activated 4Å molecular sieves (5.2 mg), silica gel (5.2 mg), solid NaHCO3 (3.0 mg, 0.036 mmol) and solid AgOTf (12.2 mg, 0.047 mmol). The reaction mixture was stirred at rt for 2 h, then diluted with CH2Cl2 and filtered through a pad of Celite. The filtrate was concentrated in vacuo to afford oxetane 74 as a white solid (1.2 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 10.8 Hz, 1H), 7.67 (d, J = 7.2 Hz, 2H), 7.55-7.15 (m, 10H), 5.51 (d, J = 14.4 Hz, 1H), 5.37 (d, J = 14.4 Hz, 1H), 5.13 (m, 1H), 4.52 (d, J = 11.0 Hz, 1H), 4.43 (d, J = 11.0 Hz, 1H), 4.31 (t, J = 7.2 Hz, 1H), 3.98 (m, 1H), 3.86 (m, 2H), 3.66 (m, 1H), 3.37 (s, 3H), 2.46 (s, 3H), 2.35 (t, J = 7.2 Hz, 1H), 2.17-2.07 (m, 2H).
For characterization, N-Cbz-indole oxetane 74 (1.2 mg) and glacial HOAc (1.0 μL) were dissolved in EtOAc (1.0 mL) and hydrogenated with 10% Pd/C (5 mg) under H2 from a balloon at rt for 1.5 h. The reaction mixture was then filtered through a pad of Celite and the filtrate was evaporated to dryness in vacuo to give a white solid (0.8 mg, 85%), which was identical to oxetane 68.
Methyl 5-(((tert-Butyldimethylsilyl)oxy)methyl)-11-formyl-11a-hydroxy-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (75)
Freshly prepared IBX37 (46 mg, 0.16 mmol) was added to a solution of diol 61 (97 mg, 0.16 mmol) in dry DMSO (5.3 mL) and the mixture was stirred at rt for 20 h. The reaction mixture was diluted with water and extracted with ether. The organic phase was washed with aqueous NaHCO3, brine, and dried over MgSO4. The solvent was removed in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 20–40% EtOAc/hexanes) to afford aldehyde 75 as a white solid (91 mg, 94%). 1H NMR (300 MHz, CDCl3) δ 10.26 (s, 1H), 8.74 (br s, 1H, NH), 7.68 (d, J = 8.0 Hz, 2H), 7.42-7.32 (m, 4H), 7.21 (dd, J = 7.5, 7.5 Hz, 1H), 7.11 (dd, J = 7.5, 7.5 Hz, 1H), 4.70 (br s, 1H), 4.42 (s, 1H), 4.20 (d, J = 9.5 Hz, 1H), 4.13-4.08 (m, 1H), 4.02 (d, J = 9.5 Hz, 1H), 3.74 (s, 3H), 3.70 (m, 1H), 2.44 (s, 3H), 2.29 (m, 1H), 2.23 (m, 1H), 2.00 (dd, J = 3.8, 11.5 Hz, 1H), 1.54 (dd, J = 4.2, 12.8 Hz, 1H), 1.43 (m, 1H), 0.79 (s, 9H), −0.09 (s, 3H), −0.14 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 203.6, 172.8, 144.0, 136.9, 133.0, 131.4, 129.9, 127.9, 125.8, 122.4, 120.0, 119.4, 111.5, 103.7, 72.4, 70.5, 55.2, 55.1, 52.6, 48.9, 48.5, 45.7, 26.4, 25.7, 21.7, 18.1, −5.7, −5.8; HRMS (m/z): [M + H]+ calcd for C32H43N2O7SSi, 627.2560; found 627.2542.
Methyl 11-Formyl-11a-hydroxy-5-(hydroxymethyl)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (58)
Method A
To a solution of ε-lactone 57 (5.5 mg, 0.011 mmol) in CH2Cl2 (0.2 mL) at −78 °C was added BF3·OEt2 (2.7 μL, 0.022 mmol), followed by dropwise addition of a DIBAL-H solution in toluene (1.5 M, 14 μL, 0.022 mmol). The reaction mixture was stirred at −78 °C for 1 h, and then quenched by addition of 10% Rochelle’s salt and Na2HPO4/KH2PO4 buffer (pH 7). The mixture was extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4. The solvent was removed in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 30–50% EtOAc/hexanes) to afford aldehyde 58 as an off-white foam (1.2 mg, 22%). 1H NMR (400 MHz, CDCl3) δ 10.3 (s, 1H), 8.94 (s, 1H), 7.67 (m, 3H), 7.41 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.24 (m, 1H), 7.14 (dd, J = 7.4, 7.5 Hz, 1H), 4.51 (s, 1H), 4.19 (d, J = 11.1 Hz, 1H), 4.15-4.00 (m, 3H), 3.78 (s, 3H), 3.70 (m, 2H), 2.46 (s, 3H), 2.26 (m, 2H), 2.11 (td, J = 4.2, 11.4 Hz, 1H), 1.00-0.86 (m, 2H); 13C NMR δ (75 MHz, CDCl3) δ 203.1, 173.4, 144.5, 137.4, 133.2, 130.8, 130.3, 128.2, 126.4, 123.1, 120.5, 120.0, 112.1, 105.2, 72.5, 69.4, 55.5, 55.2, 53.2, 49.6, 48.7, 45.9, 26.8, 22.0; HRMS (m/z): [M + H]+ calcd for C26H29N2O7S, 513.1695; found 513.1696.
Method B
OTBS aldehyde 75 (233 mg, 0.36 mmol) was dissolved in a solution of HCl in EtOAc (1.0 M, 5 mL) and the mixture was stirred at rt for 1 h. The volatiles were removed in vacuo to afford hydroxy aldehyde 58 as a red solid (183 mg, 100%), which was used in the next step without further purification.
Methyl 11-(bis(Ethylthio)methyl)-11a-hydroxy-5-(hydroxymethyl)-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido[4,3-b]carbazole-5-carboxylate (76)
To a solution of hydroxymethyl aldehyde 58 (16 mg, 0.032 mmol) and ethanethiol (69 μL, 0.96 mmol) in CH2Cl2 (2.5 mL) at −50 °C was added a solution of TiCl4 in CH2Cl2 (1.0 M, 0.10 mL, 0.10 mmol). The resulting dark orange-brown mixture was stirred while allowed to warm slowly to rt over 15 h. The reaction mixture was then diluted with CH2Cl2, treated with aqueous NaHCO3, and extracted with CH2Cl2. The organic layers were washed with aqueous NH4Cl and brine, and dried over Na2SO4. The solvent was removed in vacuo and the residue was purified by flash chromatography on silica gel (gradient 10–50% EtOAc/hexanes) to afford dithioacetal 76 as a pink solid (18 mg, 94%). 1H NMR (300 MHz, CDCl3) δ 9.09 (br s, 1H), 8.15 (d, J = 6.3 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.36-7.33 (m, 3H), 7.22-7.12 (m, 2H), 4.70 (s, 1H), 4.19 (s, 3H), 4.13 (m, 1H), 3.63 (br m, 3H), 2.91 (m, 1H), 2.73 (q, J = 7.2 Hz, 4H), 2.44 (s, 3H), 2.23-2.12 (m, 3H), 1.31 (t, J = 7.2 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 174.2, 144.5, 137.1, 132.7, 132.2, 130.2, 128.5, 127.0, 122.6, 122.0, 119.9, 111.8, 109.2, 77.7, 70.1, 56.1, 54.5, 52.9, 52.5, 28.7, 27.4, 22.0, 14.7 (2C); HRMS (m/z): [M + Na]+ calcd for C30H38N2O6S3Na, 641.1790; found 641.1792.
Methyl 12-(Ethylthio)-11a-hydroxy-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-5,11-(methanooxymethano)pyrido[4,3-b]carbazole-5-carboxylate (77) and Methyl 12-(Ethylthio)-11a-hydroxy-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-5,11-(methanooxymethano)pyrido[4,3-b]carbazole-5-carboxylate (78)
To a solution of dithioacetal 76 (130 mg, 0.21 mmol) in nitromethane (10 mL) at rt was added a mixture of 4Å MS and flame dried silica gel (1:1 wt. mixture, 390 mg), followed by solid NaHCO3 (79 mg, 0.94 mmol). The mixture was stirred at rt for 40 min and then was treated dropwise with a solution of anhydrous AgClO4 (131 mg, 0.63 mmol) in nitromethane (1.7 mL). The resulting brown suspension was stirred at rt until TLC indicated completion of the reaction (~30 min). The mixture was diluted with CH2Cl2 and EtOAc, filtered through a pad of Celite, and the filtrate was evaporated to dryness in vacuo to give a yellow powder. This powder was stirred with 10 mL of CH2Cl2 at rt for 10 min and filtered to give the major C18 diastereomer 77 as a white solid (77 mg, 66%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (br s, 1H), 7.44 (d, J = 7.5 Hz, 3H), 7.37 (d, J = 7.6 Hz, 1H), 7.17 (d, J = 7.9 Hz, 2H), 7.04 (app t, J = 7.0 Hz, 1H), 6.98 (app t, J = 7.4 Hz, 1H), 5.49 (d, J = 3.1 Hz, 1H), 5.26 (br s, 1H), 4.07 (d, J = 11.4 Hz, 1H), 3.89 (d, J = 11.4 Hz, 1H), 3.78 (s, 3H), 3.30 (d, J = 12.2 Hz, 2H), 3.22 (d, J = 3.1 Hz, 1H), 3.07 (t, J = 12.7 Hz, 1H), 2.46-2.36 (m, 3H), 2.30 (s, 3H), 2.37 (d, J = 12.5 Hz, 1H), 1.51 (d, J = 11.0 Hz, 1H), 1.10 (t, J = 7.4 Hz, 3H), 0.57 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.2, 143.6, 136.6, 136.3, 135.8, 130.2, 127.6, 126.9, 121.2, 119.9, 118.1, 113.1, 111.7, 110.2, 82.4, 77.5, 68.3, 54.9, 53.5, 51.9, 48.3, 47.5, 42.2, 25.7, 25.4, 21.8, 16.0.
The filtrate was concentrated to give a residue which was purified by flash chromatography on silica gel (gradient 20–50% EtOAc/hexanes) to afford the minor C18 diastereomer 78 as an off-white solid (23 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 9.48 (s, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.20 (d, J = 8.0 Hz, 2H), 7.11–7.16 (m, 2H), 5.58 (d, J = 4.4 Hz, 1H), 4.44 (d, J = 11.3 Hz, 1H), 4.02 (d, J = 11.3 Hz, 1H), 3.85 (s, 3H), 3.59 (d, J = 13.5 Hz, 1H), 3.30–3.36 (m, 2H), 3.28 (d, J = 4.5 Hz, 1H), 2.65 (d, J = 13.5 Hz, 1H), 2.62 (dd, J = 4.6, 13.0 Hz, 1H), 2.41–2.48 (m, 2H), 2.37 (s, 3H), 1.52 (m, 1H), 1.16 (t, J = 7.4 Hz, 3H), 0.97 (dd, J = 5.7, 13.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 172.5, 143.3, 136.0, 135.5, 135.2, 129.6, 127.5, 126.6, 121.9, 120.4, 117.9, 115.6, 112.8, 111.8, 109.9, 83.5, 77.9, 66.9, 54.6, 53.0, 50.5, 49.8, 46.8, 41.9, 25.4, 25.1, 21.6, 15,3; HRMS (m/z): [M + H]+ calcd for C28H33N2O6S2, 557.1775; found 557.1780.
Methyl 4-Tosyl-3,4,5,6,6a,7,8,12c-octahydro-1H-1,7-(epoxymethano)oxeto[2,3-c]pyrido[4,3-b]carbazole-7-carboxylate (79)
The major hemithioacetal 77 (1.8 mg, 0.0032 mmol) was heated with a slurry of freshly prepared W-2 Raney nickel in ethanol (~0.5 mL, excess) at 60 °C for 1 h. The reaction mixture was diluted with CH2Cl2 and filtered through a pad of Celite. The filtrate was concentrated to give oxetane acetal 79 as a colorless gum (1.0 mg, 63%). 1H NMR (360 MHz, CDCl3) δ 9.49 (br s, 1H), 7.63 (d, J = 7.2 Hz, 2H), 7.44 (d, J = 7.0 Hz, 1H), 7.39 (d, J = 7.2 Hz, 1H), 7.17 (m, 3H), 6.80 (m, 1H), 4.36 (m, 1H), 4.17 (d, J = 11.0 Hz, 1H), 4.03 (m, 2H), 3.85 (s, 3H), 3.58 (m, 2H), 3.32 (m, 2H), 3.06 (app s, 1H), 2.68 (d, J = 12.0 Hz, 1H), 2.60 (m, 1H), 2.38 (s, 3H), 1.44 (m, 2H); HRMS (m/z): [M + H]+ calcd for C26H27N2O6S, 495.1590, found 495.1572.
Methyl 11a-Hydroxy-2-tosyl-2,3,4,4a,5,6,11,11a-octahydro-1H-5,11-(methanooxymethano)pyrido[4,3-b]carbazole-5-carboxylate (80)
To a solution of major hemithioacetal 77 (13.6 mg, 0.0245 mmol) in acetone (5.0 mL) was added 10% Pd/C (75 mg) and triethylsilane (0.7 mL, excess). The reaction mixture was heated at reflux. Additional Pd/C and triethylsilane were added every 2 h until TLC indicated completion of the reaction (6 additions of 10% Pd/C and Et3SiH were required in total). The reaction mixture was then filtered through a pad of Celite and the filtrate was concentrated in vacuo to give a residue which was purified by flash column chromatography on silica gel (gradient 5–10% EtOAc/hexanes, then 30% EtOAc/hexanes, then 5% Et2O/CH2Cl2, then 50% EtOAc/hexanes) to afford oxepane 80 as a white foamy solid (11.4 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 9.49 (s, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.11–7.17 (m, 2H), 4.38 (dd, J = 3.7, 11.4 Hz, 1H), 4.17 (d, J = 11.4 Hz, 1H), 3.84 (s, 3H), 3.83 (d, J = 11.4 Hz, 1H), 3.58 (d, J = 13.4 Hz, 1H), 3.54 (dd, J = 1.8, 11.3 Hz, 1H), 3.25–3.37 (m, 2H), 3.06 (t, J = 1.8 Hz, 1H), 2.67 (d, J = 13.3 Hz, 1H), 2.60 (dd, J = 4.5, 13.1 Hz, 1H), 2.36 (s, 3H). 1.48 (m, 1H), 0.95 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 173.0, 143.7, 135.8, 135.6, 135.4, 130.0, 127.8, 125.5, 122.0, 120.3, 117.9, 114.4, 112.5, 112.0, 74.2, 68.0, 55.4, 53.2, 52.7, 51.6, 42.4, 42.3, 25.5, 21.9, 18.4; HRMS (m/z): [M + H]+ calcd for C26H29N2O6S, 497.1746; found 497.1753. The minor hemithioacetal 78 could be converted to oxepane 80 under the same reaction conditions in essentially identical yield.
Synthesis of (±)-Alstilobanine E (2)
Magnesium turnings (451 mg, excess) were added to a solution of sulfonamide-protected oxepane 80 (20 mg, 0.040 mmol) in anhydrous methanol and the mixture was sonicated at rt until all the magnesium had dissolved (~45 min). The reaction mixture was then poured into aqueous NH4Cl at 0 °C, and extracted with CHCl3. The organic extract was dried over Na2SO4, and evaporated to dryness in vacuo. The residue was purified by flash chromatography on silica gel (gradient 1–5% MeOH/CH2Cl2, then 1% NEt3 in 5% MeOH/CH2Cl2, then 1% NEt3 in 10% MeOH/CH2Cl2) to afford racemic alstilobanine E (2) as an off-white solid (13 mg, 95%). 1H NMR (400 MHz, CD3OD, 1 μL of TFA added) δ 7.90 (s, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.43 (d, J = 9.0 Hz, 1H), 7.10 (dd, J = 7.0, 7.4 Hz, 1H), 7.03 (dd, J = 7.0, 7.3 Hz, 1H), 4.31 (dd, J = 3.0, 11.4 Hz, 1H), 4.14 (d, J = 11.4 Hz, 1H), 3.90 (s, 3H), 3.84 (d, J = 11.4 Hz, 1H), 3.57 (dd, J = 3.0, 11.6 Hz, 1H), 3.24 (m, 1H), 3.20 (m, 1H), 2.96 (d, J = 13.0 Hz, 1H), 2.74 (dd, J = 5.2, 13.0 Hz, 1H), 2.61 (d, J = 13.1 Hz, 1H), 1.69–1.74 (m, 1H), 1.19 (dddd, J = 5.9, 13.1, 13.3, 13.9 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 172.9, 137.8, 135.3, 126.4, 122.4, 120.6, 118.0, 113.0, 112.5, 75.6, 75.4, 67.2, 56.1, 53.2, 51.5, 49.0, 43.9, 39.8, 23.3; HRMS (m/z): [M + H]+ calcd for C19H23N2O4, 343.1652; found 343.1658.
Synthesis of (±)-Angustilodine (1)
Alstilobanine E (2) (10.4 mg, 0.0304 mmol) and paraformaldehyde (6.4 mg, ~7 equiv.) were stirred in methanol (3.0 mL) and treated with glacial acetic acid (7.5 μL) at rt. The resulting white suspension was stirred at rt for another 30 min, and solid NaBH4 was added every 60 min (4.0 mg, 0.105 mmol in each portion, 3 h total) until TLC (1% NEt3 in 5% MeOH/CH2Cl2) indicated completion of the reaction. The reaction mixture was treated with aqueous NH4Cl, and was extracted with CHCl3. The combined organic layers were washed with aqueous NaHCO3, brine, and dried over Na2SO4. The solvent was removed in vacuo to give a residue which was purified by flash chromatography on silica gel (gradient 1–5% MeOH/CH2Cl2, then 1% NEt3 in 5% MeOH/CH2Cl2) to afford racemic angustilodine (1) as an off-white gum (8.8 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 9.55 (s, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.17 (app t, J = 8.0 Hz, 1H), 7.12 (app t, J = 7.6 Hz, 1H), 4.39 (dd, J = 2.1, 11.3 Hz, 1H), 4.12 (d, J = 11.4 Hz, 1H), 3.86 (d, J = 11.4 Hz, 1H), 3.84 (s, 3H), 3.55 (dd, J = 3.3, 11.3 Hz, 1H), 3.06 (app t, J = 3.0 Hz, 1H), 3.03 (m, 1H), 2.66 (dd, J = 4.4, 13.1 Hz, 1H), 2.36 (d, J = 11.9 Hz, 1H), 2.31 (d, J = 11.3 Hz, 1H), 2.28 (m, 1H), 2.26 (s, 3H), 1.32 (m, 1H), 0.98 (m, 1H); 13C NMR (212.5 MHz, CDCl3) δ 173.3, 135.5, 135.0, 125.6, 121.2, 119.7, 117.4, 111.7, 111.3, 76.5, 72.9, 66.2, 61.0, 55.2, 52.6, 52.3, 49.9, 45.4, 41.9, 24.8; HRMS (m/z): [M + H]+ calcd for C20H25N2O4, 357.1809, found 357.1806.
Supplementary Material
Acknowledgments
The authors are grateful to the National Institutes of Health (GM-087733) and the National Science Foundation (CHE-1105653) for financial support of this research. Dr. H. Yennawar (Penn State Small Molecule X-Ray Cystallographic Facility) is thanked for the X-ray crystal structure determination.
Footnotes
Notes
The authors declare no competing financial interest.
Proton and carbon NMR spectra of new compounds. Included also are X-ray data for compound 30. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.For reviews of monoterpene indole alkaloids see inter alia: Cordell GA. Introduction to Alkaloids: A Biogenetic Approach. Wiley-Interscience; New York: 1981. Pelletier SW. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 1 Wiley; New York: 1983. Bisset NG. In: In Indoles and Biogenetically Related Alkaloids. Phillipson JD, Zenk MH, editors. Academic Press; London: 1980. Herbert RB. In The Chemistry of Heterocyclic Compounds. In: Saxton JE, editor. Indoles Part 4, The Monoterpenoid Indole Alkaloids. Wiley; New York: 1983. Atta-ur-Rahman A Basha. Biosynthesis of Indole Alkaloids. Clarendon Press; Oxford: 1983. Kutchan TM. Phytochemistry. 1993;32:493. doi: 10.1016/s0031-9422(00)95128-8.Ishikura M, Yamada K, Abe T. Nat Prod Rep. 2010;27:1630. doi: 10.1039/c005345g.Ishikura M, Abe T, Choshi T, Hibino S. Nat Prod Rep. 2013;30:694. doi: 10.1039/c3np20118j.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 3. Wiley; Chichester: 2009. p. 369.
- 2.Kam TS, Choo YM. Helv Chim Acta. 2004;87:366. [Google Scholar]
- 3.Koyama K, Hirasawa Y, Zaima K, Hoe TC, Chan KL, Morita H. Bioorg Med Chem. 2008;16:6483. doi: 10.1016/j.bmc.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 4.For a preliminary account of portions of this work see: Feng Y, Majireck MM, Weinreb SM. Angew Chem Int Ed. 2012;51:12846. doi: 10.1002/anie.201207949.
- 5.Taken in part from: Majireck MM. PhD Thesis. The Pennsylvania State University; 2011.
- 6.(a) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (b) Henry-Riyad H, Lee C, Purohit VC, Romo D. Org Lett. 2006;8:4363. doi: 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]; (c) Ma G, Nguyen H, Romo D. Org Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Purohit VC, Matla AS, Romo D. J Am Chem Soc. 2008;130:10478. doi: 10.1021/ja803579z. [DOI] [PubMed] [Google Scholar]; (e) Nguyen H, Ma G, Romo D. Chem Commun. 2010;46:4803. doi: 10.1039/c0cc00607f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Morris KA, Arendt KM, Oh SH, Romo D. Org Lett. 2010;12:3764. doi: 10.1021/ol101388h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nguyen H, Ma G, Gladysheva T, Fremgen T, Romo D. J Org Chem. 2011;76:2. doi: 10.1021/jo101638r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Leverett CA, Purohit VC, Romo D. Angew Chem Int Ed. 2010;49:9479. doi: 10.1002/anie.201004671. [DOI] [PubMed] [Google Scholar]; (i) Leverett CA, Purohit VC, Johnson AG, Davis RB, Tantillo DJ, Romo D. J Am Chem Soc. 2012;134:13348. doi: 10.1021/ja303414a. [DOI] [PubMed] [Google Scholar]; (j) Liu G, Shirley ME, Romo D. J Org Chem. 2012;77:2496. doi: 10.1021/jo202252y. [DOI] [PubMed] [Google Scholar]
- 7.For reviews of nitrosoalkenes see: Gilchrist TL. Chem Soc Rev. 1983;12:53.Lyapkalo IM, Ioffe SL. Russ Chem Rev. 1998;67:467.
- 8.(a) Korboukh I, Kumar P, Weinreb SM. J Am Chem Soc. 2007;129:10342. doi: 10.1021/ja074108r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li P, Majireck MM, Witek JA, Weinreb SM. Tetrahedron Lett. 2010;51:2032. doi: 10.1016/j.tetlet.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kumar P, Li P, Korboukh I, Wang TL, Yennawar H, Weinreb SM. J Org Chem. 2011;76:2094. doi: 10.1021/jo1024392. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sengupta R, Weinreb SM. Synthesis. 2012;44:2933. [Google Scholar]
- 9.(a) Denmark SE, Dappen MS. J Org Chem. 1984;49:798. [Google Scholar]; (b) Denmark SE, Dappen MS, Sternberg JA. J Org Chem. 1984;49:4741. [Google Scholar]; (c) Denmark SE, Dappen MS, Sear NL, Jacobs RT. J Am Chem Soc. 1990;112:3466. [Google Scholar]
- 10.Mahboobi S, Bernauer K. Helv Chim Acta. 1988;71:2034. [Google Scholar]
- 11.Chauhan PS, Majireck MM, Weinreb SM. Heterocycles. 2012;84:577. [Google Scholar]
- 12.Wildsmith E, Timms GH. Tetrahedron Lett. 1971;12:195. [Google Scholar]
- 13.For related acylations, see: McKew JC, Foley MA, Thakker P, Behnke ML, Lovering FE, Sum FW, Tam S, Wu K, Shen MWH, Zhang W, Gonzalez M, Liu S, Mahadevan A, Sard H, Khor SP, Clark JD. J Med Chem. 2006;49:135. doi: 10.1021/jm0507882.Raucher S, Klein P. J Org Chem. 1986;51:123.
- 14.Hlasta DJ, Luttinger D, Perrone MH, Silbernagel MJ, Ward SJ, Haubrich DR. J Med Chem. 1987;30:1555. doi: 10.1021/jm00392a005. [DOI] [PubMed] [Google Scholar]
- 15.DePuy CH, Ponder BW. J Am Chem Soc. 1959;81:4629. [Google Scholar]
- 16.Van den Broek LAGM, Breuer ML, Liskamp RMJ, Ottenheijm HCJ. J Org Chem. 1987;52:1511. [Google Scholar]
- 17.Schlosser M, Jenny T, Guggisberg Y. Synlett. 1990;11:704. [Google Scholar]
- 18.Cf, Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Some compounds were not fully characterized for unsuccessful routes.
- 20.For preparation of chloromethyldimethylsilyl triflate see: Bassindale AR, Borbaruah M, Glynn SJ, Parker DJ, Taylor PG. J Chem Soc, Perkin Trans. 1999;2:2099.
- 21.For reviews on the Tamao-Fleming oxidation see: Tamao K. Proc Jpn Acad, Ser B. 2008;84:123. doi: 10.2183/pjab.84.123.Mullins RJ, Jolley SL, Knapp AR. In: In Name Reactions for Functional Group Transformations. Li JJ, Corey EJ, editors. Wiley; Hoboken: 2007. Fleming I. Chemtracts: Org Chem. 1996;9:1.Jones GR, Landais Y. Tetrahedron. 1996;52:7599.Tamao K. Adv Silicon Chem. 1996;3:1.Schinzer D. In: In Organic Synthesis Highlights II. Waldmann H, editor. New York: 1995. Colvin EW. In: In Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 7 Pergamon Press; Oxford: 1991.
- 22.Huscroft IT, Carlson EJ, Chicchi GG, Kurtz MM, London C, Raubo P, Wheeldon A, Kulagowski JJ. Bioorg Med Chem Lett. 2006;16:2008. doi: 10.1016/j.bmcl.2005.12.069. [DOI] [PubMed] [Google Scholar]
- 23.Nyasse B, Grehn L, Ragnarsson U. Chem Commun. 1997:1017. [Google Scholar]
- 24.We are grateful to Professor H. Morita (Hoshi University) for providing copies of the proton and carbon NMR spectra of the TFA salt of natural alstilobanine A. We also thank Professor T.-S. Kam (University of Malaya) for copies of the spectra of angustilodine.
- 25.For reviews of the Mitsunobu reaction see: Hughes DL. Org React. 1992;42:335.Lawrence S. PharmaChem. 2002;1:12.Dembinski R. Eur J Org Chem. 2004:2763.
- 26.For formation of a tetrahydrofuran by a Mitsunobu reaction see: Pranger T, Rodriguez MS, Suarez E. J Org Chem. 2003;68:4422. doi: 10.1021/jo034129d.
- 27.For oxetane formation by a Mitsunobu reaction see: Popsavin V, Benedekovic G, Sreco B, Popsavin M, Francuz J, Kojic V, Bogdanovic G. Org Lett. 2007;9:4235. doi: 10.1021/ol701734s.Popsavin V, Benedekovic G, Sreco B, Francuz J, Popsavin M, Kojic V, Bogdanovic G, Divjakovic V. Tetrahedron. 2009;65:10596.
- 28.Cf, Johansen JE, Christie BD, Rapoport H. J Org Chem. 1981;46:4914. [Google Scholar]
- 29.Cf, Mihailovic ML, Gojkovic S, Konstantinovic S. Tetrahedron. 1973;29:3675. [Google Scholar]
- 30.Gerfaud T, Xie C, Neuville L, Zhu J. Angew Chem Int Ed. 2011;50:3954. doi: 10.1002/anie.201100257. [DOI] [PubMed] [Google Scholar]
- 31.(a) Kumar V, Dev S. Tetrahedron Lett. 1983;24:1289. [Google Scholar]; (b) Bulman-Page PC, Roberts RA, Paquette LA. Tetrahedron Lett. 1983;24:3555. [Google Scholar]
- 32.Nicolaou KC, Prasad CVC, Hwang CK, Duggan ME, Veale CA. J Am Chem Soc. 1989;111:5321. [Google Scholar]
- 33.Blom P, Ruttens B, Van Hoof S, Hubrecht I, Van der Eycken J, Sas B, Vanhemel J, Vandenkerckhove J. J Org Chem. 2005;70:10109. doi: 10.1021/jo051021k. [DOI] [PubMed] [Google Scholar]
- 34.Fukuyama T, Lin SC, Li L. J Am Chem Soc. 1990;112:7050. [Google Scholar]
- 35.Schonafinger K, Yasenchak CM, Vollman A, Ong HH. J Heterocycl Chem. 1988;25:535. [Google Scholar]
- 36.The R value for this structure was 0.1145. This high R value can be attributed to water molecules in the crystal lattice but the quality of the structure is sufficient to establish the stereochemical features of 30.
- 37.Boeckman RK, Jr, Shao P, Mullins JJ. Org Synth. 2000;77:141. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.