

Abstract
A highly-conserved binding pocket on HIVgp41 is an important target for development of anti-viral inhibitors. Holden et al. (Bioorg. Med. Chem. Lett. 2012) recently reported 7 experimentally-verified leads identified through a computational screen to the gp41 pocket in conjunction with a new DOCK scoring method (termed FPS scoring) developed in our laboratory. The method employs molecular footprints based on per-residue van der Waals interactions, electrostatic interactions, or the sum. In this work, we critically examine the gp41 screening results, prioritized using different scoring methods, in terms of two main criteria: (1) ligand pose properties which include footprint and energy score decompositions, MW, number of rotatable bonds, ligand efficiency, formal charge, and volume overlap, and (2) ligand pose stability which includes footprint stability (changes in footprint overlap) and rmsd stability (changes in geometry). Relative to standard DOCK scoring, pose property analyses demonstrate how FPS scoring can be used to identify ligands that mimic a known reference (derived here from the native gp41 substrate), while pose stability analyses demonstrate how FPS scoring can be used to enrich for compounds with greater overall stability during molecular dynamics (MD) simulations. Compellingly, of the 115 compounds tested experimentally, the 7 active compounds, as a group, more closely mimic the footprints made by the reference and show greater MD stability compared to the inactive group. Extensive studies using 116 protein-ligand complexes as controls reveal that ligands in their crystallographic binding pose also maintain higher FPS scores and smaller rmsds than do accompanying decoys, confirming that native poses are indeed “stable” under the same conditions and that monitoring FPS variability during compound prioritization is likely to be beneficial. Overall, the results suggest the new scoring method will complement current virtual screening approaches for both the identification (FPS-ranking) and prioritization (FPS-stability) of target-compatible molecules in a quantitative and logical way.
Keywords: HIV, gp41, Protein-protein interactions, Docking, Virtual screening, DOCK, Footprint similarity, Scoring functions, Molecular dynamics
1. Introduction
The HIV glycoprotein-41 (gp41) represents an important and validated drug target for agents which inhibit key viral-host membrane fusion events required for infection and replication.1 Development of the first FDA-approved inhibitor in the membrane fusion class, a 36 a.a. C-helix peptide termed T20 (enfuvirtide, FUZEON),2 represents an important milestone in the treatment of HIV/AIDS. However, as a peptide-based drug, T20 is cost prohibitive (ca. USD $25,000 per person per year) for the majority of persons infected with HIV and it must be intravenously delivered. T20 efficacy is also negatively affected by gp41 resistance mutations3–8 that arise from continued use. Given these drawbacks, efforts to identify alternative inhibitors including modified C-peptides,9–11 D-peptides,12,13 and small organic molecules14–18 have been reported, using a variety of experimental and computational approaches. In particular, there has been an effort to develop compounds which target a highly-conserved hydrophobic pocket region on gp41 not exploited by T20, as described by Chan et al,19 which in principle could lead to drugs less affected by mutation. Although reported small molecule leads14–18 are presumed to interact within the gp41 pocket, concrete structural information on binding is lacking. Currently available crystal structures of gp41 have been limited to complexes containing C- or D-helix peptides. The fact that the gp41 pocket is highly solvent exposed presents additional challenges.
In terms of lead discovery, a practical dilemma for the computational chemist/biologist in both industry and academia is efficient prioritization and selection of virtual screening results derived from docking large ligand databases to the therapeutic target. The procedure requires careful application of a single or multiple scoring functions, used separately or in tandem, to filter the results down to manageably-sized sets for further study with the intent of identifying the most promising leads. Common methods for prioritization include use of physics-based, empirical, or pharmacophore scoring functions, among others, to rank-order the predicted ligand binding geometries (poses) for which some top-scoring fraction (i.e. 50–100 compounds) will be experimentally tested for activity. The well-known program DOCK20–22 employs a simple two-term scoring function consisting of intermolecular van der Waals and electrostatic interactions. Recently, our group developed a new method for DOCK termed footprint similarity (FPS) scoring23,24 which can be described as an energy-based pharmacophore-like approach. Defined as the per-residue breakdown of van der Waals and electrostatic interactions for a ligand with its target, footprints encode which groups of residues are most important for binding. Importantly, the overlap between two footprint patterns can be quantified using metrics such as Euclidian distance (d) or Pearson correlation (r, r2) which enables large databases to be rank-ordered in terms of similarity to a known reference. The FPS scoring method was developed through extensive validation and testing,23,24 using multiple experimental datasets, with regards to three key properties: pose reproduction, crossdocking, and enrichment. Subsequent application of the method led to the successful identification of experimentally verified drug-leads targeting both gp4118,25 and fatty acid binding protein.26
Our development of FPS scoring was motivated by the need to readily identify small organic molecules capable of mimicking specific footprint patterns (Figure 1b) made by key C-helix sidechains on gp41 that interact within the conserved hydrophobic pocket region formed at the interface of two N-helices (Figure 1a). We hypothesized that small molecules with sufficient footprint overlap (i.e. favorable FPS score) to key parts of native C-helix would also be capable of binding to the pocket and thereby inhibit viral replication. The identification of small molecules capable of mimicking specific protein-protein interactions has been described by Fry27,28 as molecular mimicry. FPS scoring provides a quantitative and logical way to computationally approach the problem. In general, compounds which interact with a binding environment in a similar manner as known drugs, substrates, cofactors, or as in the present case key side chains along a native protein-protein interface, are more likely to be inhibitors than molecules selected by other methods or randomly. Related approaches for scoring and pose selection have been reported by Deng et al29,30 using binary interaction fingerprints and Pfeffer et al31 using per-atom decomposition. As a visual example of bad vs good footprint overlap relative to a reference, Figure 2 shows two results from a virtual screen with, in this case, the overall worst (0.135 r) and best (1.975 r) FPSVDW+ES scores among the top 100,000/500,000 compounds docked to the gp41 target.
Figure 1.

Reference footprints constructed from a protein-protein interface on gp41. (a) Two views of the drug target site showing the N-helix hydrophobic binding pocket region in gray and four key sidechains from the native C-helix in green. (b) C34-derived van der Waals (VDW) and electrostatic (ES) reference footprints. Binding site residues involved in a salt-bridge (sb) with Asp121 and hydrogen bond (hb) with Trp120 on the reference are labeled in the electrostatic footprint and the pocket surface. Coordinates from PDB code 1AIK.32
Figure 2.
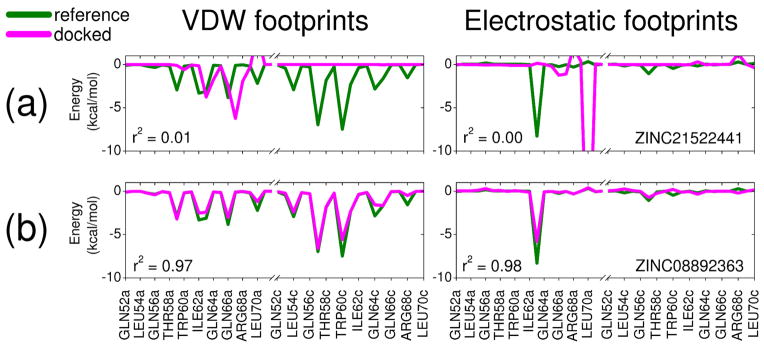
Footprint overlap comparisons for two molecules from the gp41 virtual screen showing (a) bad and (b) good FPSVDW+ES overlap with the C34-derived reference from Figure 1.
The primary goal of this study is to perform a critical retrospective analysis of the large-scale virtual screen to gp41 reported by Holden et al18 in which ca. 500,000 commercially available compounds were docked to the hydrophobic pocket, 115 were purchased, and 7 were identified as having favorable properties in three experimental assays (defined here as actives). Specifically, we compare results obtained using the standard DOCK energy score (DCEVDW+ES) with three footprint variants based on van der Waals interactions (FPSVDW), electrostatic interactions (FPSES), or the combination (FPSVDW+ES). The specific objectives are to quantify and compare how use of the new FPS method differs from the standard DOCK method in terms of two main criteria: (1) ligand pose properties and (2) ligand pose stability. Pose properties compared include DOCK and FPS score distributions, molecular weight, number of rotatable bonds, ligand efficiency, ligand formal charge, volume overlap, and footprint comparisons between active and inactive compounds which were tested experimentally. Pose stability comparisons are based on definitions which enumerate how a ligand changes relative to the initial docked pose in terms of footprint-stability (changes in footprint overlap) during a molecular dynamics (MD) simulation of the complex as well as rmsd-stability (changes in geometry). To verify that pose stability is an inherent property of experimentally-observed protein-ligand complexes under the same conditions, and therefore desirable, companion MD validation studies using crystallographic controls were also performed. Depending on the application, this computational approach that enables identification of favorably-scored ligands with high footprint overlap to a known reference, and which remain energetically and geometrically stable in their predicted binding pose, should be a useful tool to help researchers prioritize compounds for purchase.
2. Methods
2.1 Binding site setup for gp41
The docking setup for gp41 was constructed using coordinates from the trimeric coiled-coil construct reported by Chan et al32 (PDB code 1AIK) in which the three outer C-helix peptides (termed C34) were removed to expose the hydrophobic pocket formed at the interface of two of three inner N-helix peptides (termed N36). Following standard DOCK setup protocols33 a molecular surface of gp41 was computed (dms program),34 followed by sphere generation (sphgen program),35 and docking grid generation (grid program).36 The sphere set was augmented with heavy atom coordinates of the four key gp41 sidechains (Figure 1) and only one of the three symmetric pockets was targeted (N = 62 spheres total). The AMBER37 accessory program tleap was used to add hydrogen atoms and assign FF99SB38 partial charges to the protein, and the resultant structure was saved in mol2 format. Docking grids employed 6–9 Lennard-Jones exponents for intermolecular van der Waals energies, a ε = 4r distance-dependant dielectric to scale intermolecular Coulombic energies, a 0.325 Å grid spacing which extended 10 Å in all directions about the sphere set, and a 999 Å cutoff to ensure every grid point included all protein atoms.
2.2 Screening protocols
A library for virtual screening of ca. 500,000 molecules (N = 493,066) was derived from a subset of molecules taken from the ZINC39 database, using the supplied protonation states (pH = 7) and partial atomic charges (AMSOL), but filtered to remove compounds having a formal charge greater than ± 2 e−, or with more than 15 rotatable bonds. To optimize docking performance on the IBM BlueGene platform, the library was sorted in descending order (low to high) based on the number of ligand rotatable bonds so that at any given time during docking the compute nodes are receiving ligands of similar rigidity.40 Protocols for grid-based flexible docking were developed through extensive tests using a large validation test set (N = 780 systems) developed in our laboratory termed SB2010.33 Briefly, docking employed multi-anchor ligand scaffold orientation (max_orientations = 1000) followed by flexible growth (anchor and grow algorithm)41 with on-the-fly pruning and clustering (max_orients = 1000, clustering_cutoff = 100, conformer_score_cutoff = 100 kcal/mol). The calculations included a repulsive-only ligand internal energy term and each stage of growth employed 500 steps of simplex-based energy minimization to optimize poses with allowed step sizes of 1.0 Å for translational, 0.1π radians (18°) for rotational, and 10.0° for torsional degrees of freedom. Following docking to the grid, the top-scoring pose retained for each ligand was energy minimized in Cartesian space in order to compute residue-based footprints. The final minimizations employed a 6–12 Lennard-Jones intermolecular van der Waals term, a ε = 4r distance dependant dielectric intermolecular electrostatic term, and a repulsive-only ligand internal energy term. In addition, a 10 kcal/(mol Å2) harmonic tether was used to minimize root-mean-square-deviation (rmsd) differences between the original grid-based and coordinated-based docked poses. The final list of docked molecules was then rank-ordered using the DOCK Cartesian energy score termed DCEVDW+ES. All docking calculations employed DOCK6.4.42
2.3 Footprint rescoring and clustering
Per-residue footprints were computed for the top 20% (N = 100,000) Cartesian-minimized docked ligands and compared with that of the reference to yield van der Waals (FPSVDW), electrostatic (FPSES), and van der Waals plus electrostatic (FPSVDW+ES) footprint similarity scores. For this study, Pearson correlation coefficients (r and r2 values) were used to quantify similarity with values of 1.00 (FPSVDW or FPSES) or 2.00 (FPSVDW+ES) indicating perfect footprint overlap. Other methods, including threshold-based Pearson correlation, Euclidian distance, and a normalized version of Euclidian distance have also been explored.23 The gp41 reference was constructed from a single C34 peptide from PDB entry 1AIK32 truncated to include only residues 117–125 and modified so that sidechains were mutated to Ala with the exception of three key hydrophobic residues (Trp117, Trp120, Ile124) and one charged residue (Asp121) that interact with the known pocket region formed at the interface of two gp41 N-helices (Figure 1).19 The goal here was to focus the search for small molecules capable of mimicking the interaction patterns made by only this select group of residues while ignoring more distal residues. The mutated Ala construct yielded nearly identical footprint patterns to the truncated wildtype sequence as shown in Figure 3 (green line). For comparison, a difference plot employing only the four key residues (Trp117, Trp120, Ile124, Asp121) is also shown (Figure 3 blue line). The later case re-affirms that the overall signature, in this case, is primarily a function of the four specific pocket-binding residues. Standard ACE and NME capping groups were applied to the Ala construct and the crystallographic pose of the modified peptide was energy minimized with respect to the receptor coordinates following the same protocol as that described above for virtual screening. To increase chemotype diversity, the top 100,000 compounds were clustered using MACCS fingerprints43,44 as implemented in the program MOE45 using a Tanimoto coefficient of 0.75. Finally, rank-ordered lists based on the 500 top-scoring molecules as determined by each of the four methods (DCEVDW+ES, FPSVDW+ES, FPSVDW, FPSES) were generated. All footprint calculations employed a modified version of DOCK, now available in DOCK6.6 (http://dock.compbio.ucsf.edu).
Figure 3.

Differences in per-residue interactions between the Ala construct and wildtype gp41 sequence (green lines) for a 9 residue section of the native C-helix. For comparison, differences in energy using only the four key residues (Trp117, Trp120, Ile124, Asp121) and the 9-residue wildtype sequence are also shown (blue lines).
2.4 Molecular dynamics (MD) simulations
To evaluate ligand pose stability, molecular dynamics (MD) simulations were performed of the docked small molecules in complex with gp41. Stability was evaluated using both footprint similarity scores and rmsd. The accessory programs tleap and antechamber from the AMBER37 program suite were used to assemble, protonate, solvate (10 Å buffer), and assign force-field parameters for each complex consisting of FF99SB38 (protein), TIP3P46 (solvent), and GAFF47 (ligand). Ligands employed partial charges originally supplied with the ZINC database. A nine-step protocol (see Balius et al48 for details) was used to equilibrate each solvated structure prior to data collection. Briefly, a short steepest descent energy minimization followed by a short MD simulation was used to relax the added hydrogen atoms and water molecules. Additional short minimizations and MD simulations were performed with gradually decreasing restraints on non-hydrogen receptor and ligand atoms followed by an additional equilibration using a weak restraint only on the protein backbone. Production MD was conducted for 2 ns. Constant temperature (298.15 K) and pressure (1 bar) were maintained through use of Berendsen weak-coupling algorithms.49 Long range electrostatics were computed using the particle mesh Ewald (PME)50 with a real-space cut off of 8 Å. All MD simulations employed AMBER10.51
2.5 Control MD simulations
For stability comparisons with known ligand-binding geometries, 128 systems52 from 32 protein families (4 systems each) were selected from the SB2010 test set33 and MD simulations were performed using the same protocols described above with the exception that AM1-BCC53,54 partial charges were used for the ligands. Decoy poses were also simulated to evaluate stability in comparison with native (crystallographic) poses. The decoys were taken from ensembles generated by Mukherjee et al33 using the second lowest energy docked pose subject to the constraint the molecule had a different binding geometry (≥ 4Å rmsd) and poor footprint score (FPSVDW < 0.7 r or FPSES < 0.7 r) relative to the native pose. Overall, 116 out of the 128 systems had a companion decoy which was able to fulfill these conditions. For comparison purposes only the intersection of control and decoy sets (N = 116 systems) is presented in Results. In total, 744 explicit solvent MD simulations were performed in this study (116 × 2 based on the controls plus 128 × 4 based on the virtual screen).
3. Results and discussion
3.1 Ligand pose properties
3.1.1 Property comparisons
As illustrated in Figure 4, ligand pose properties obtained using the new FPS functions systematically differ from those obtained using the standard DOCK function. Specifically, FPS scoring, which decomposes the intermolecular score on a per-residue basis, can be used to selectively identify compounds with high footprint overlap to a reference query. In general, use of the standard scoring function DCEVDW+ES, consisting of a single numerical value representing the overall sum of intermolecular van der Waals plus electrostatic interactions, does not lead to as high of overlap. Results in this section include comparisons for molecules obtained from different subsets of the virtual screening pool as well as comparisons for the 115 compounds tested experimentally.
Figure 4.

Ligand pose properties based on different sets of 128 top-scoring molecules prioritized by one of four scoring schemes and then rescored with the other three functions: DOCK energy score (DCEVDW+ES, black), van der Waals footprint score plus electrostatic footprint score (FPSVDW+ES, orange), van der Waals footprint score (FPSVDW, green), and electrostatic footprint score (FPSES, blue). Histogrammed panels a-c show DOCK score distributions and individual energy components, panels d–f show different FPS score distributions, panels g–i show molecular weight, number of ligand rotatable bonds, and ligand efficiency, panels j–k show ligand formal charge, and volume overlap.
Figure 4 compares results obtained using FPS and DCE scoring in terms of their ligand pose properties for each of four different ensembles consisting 128 top-ranked ligands each. It is important to emphasize the ensembles in this figure each contain a different group of 128 molecules produced by re-ranking the top 100,000 molecules from the virtual screen (see Methods) using the DCEVDW+ES (black), FPSVDW+ES (orange), FPSVDW (green), or FPSES (blue) functions. Thus, the underlying properties computed from the four different ensembles will be different. Each ensemble is subsequently rescored (post-processed) with the other three scoring functions to yield the four colored distributions in each panel.
The individual ranking methods used to select different sets of 128 top-scoring molecules each produce an ensemble with properties that are physically consistent with the method employed. For example, molecules picked using the DCEVDW+ES function (Figure 4 black) have the most favorable energy scores in the DCEVDW+ES (Figure 4a black) histograms compared with the three footprint methods which show essentially the same distributions (Figure 4a orange, green, blue). Similarly, molecules chosen using FPSVDW+ES (Figure 4 orange), FPSVDW (Figure 4 green), or FPSES (Figure 4 blue) show larger peaks at higher Pearson coefficient values relative to the other ranking methods in their respective parent histograms (Figures 4d–f). Thus, on a case-by-case basis, large ensembles of molecules docked to a target can be subsequently mined to enrich for a particular energetic signature(s) which is not directly possible with the standard DCEVDW+ES function alone.
The DCEVDW+ES ensemble is strongly dominated by more favorable van der Waals components as illustrated by the left-shift of the black line to more favorable scores relative to other curves in Figure 4b. Here, stronger van der Waals (Figure 4b black) and DCEVDW+ES (Figure 4a black) energies for compounds selected using DCEVDW+ES are due to an increase in the average size of the molecules as shown by the higher molecular weights (Figure 4g black) and larger numbers of rotatable bonds (Figure 4h black). Size bias is a well-known problem in docking and strategies such as ligand efficiency55 have been devised in an attempt to address the fact that larger molecules generally have more favorable energy scores. Notably, use of FPS scoring generates a much wider range of molecular weights compared to DCEVDW+ES which appears to alleviate size bias (Figure 4g orange, green, blue). The relatively poor FPSVDW (Figure 4e black vs green) and FPSES (Figure 4f black vs blue) scores for the ensemble selected using DCEVDW+ES suggest, quantitatively, that a significant number of molecules identified using the standard DOCK function also interact with areas outside of the gp41 binding pocket. This is undesirable in this study as the goal is to mimic the interaction of the protein substrate. However, the fact that some DCEVDW+ES ranked compounds also have reasonably strong FPSVDW and FPSES Pearson coefficients (Figure 4e and 4f black, respectively) reveals a subset of compounds do have favorable consensus pose properties and thus would be good candidates for additional scrutiny. The prioritization protocol that led to the subset of 115 compounds which were purchased for experimental testing emphasized selection of ligands with favorable scores across multiple categories.18
The results in Figure 4 also reveal that different FPS ensembles have more favorable van der Waals energies (Figure 4b) when selected using FPSVDW (green) and more favorable electrostatic energies (Figure 4c) when using FPSES (blue). And, compounds selected using the combination FPSVDW+ES (Figure 4b,c orange) are roughly in-between those of FPSVDW or FPSES. Although the energetic magnitude for van der Waals and electrostatic components are independent of the quality of footprint correlations, the proclivity for FPSVDW to select molecules with good van der Waals energies and FPSES to select molecules with good electrostatic energies provides additional validation of the methodology.
Other properties of interest in Figure 4 include ligand efficiency (Figure 4i), ligand formal charge (Figure 4j), and percent volume overlap (Figure 4k) which is discussed further below. In terms of ligand efficiency (defined here as FPSVDW+ES score/ligand molecular weight), use of the FPSVDW+ES (Figure 4i orange) and FPSES (Figure 4i blue) functions leads to overall higher ligand efficiency, or compounds with more favorable interaction energies per molecule size. DCEVDW+ES and FPSVDW are substantially less efficient under these conditions. In terms of ligand formal charge, use of FPSVDW+ES (Figure 4j orange) and FPSES (Figure 4j blue) yields more molecules with a charge = −1 which is desirable given the known interaction involving the salt-bridge in the native gp41 system. In contrast, use of DCEVDW+ES and FPSVDW are more likely to enrich for compounds with formal charge = 0.
3.1.2 Envelope and energy comparisons
Figure 5 further illustrates differences between ensembles through visual comparison of the top 128 compounds in relation to the reference envelope (light green molecular surface) defined here by the four key sidechains from the native C34 peptide. Ensembles obtained using the three footprint functions (FPSVDW+ES, FPSVDW, FPSES) overlap structurally with the reference envelope more consistently than the DCEVDW+ES ensemble. Quantitatively, the correspondence can be expressed as a % volume overlap, shown in Figure 4k as histograms, computed between the four isolated reference sidechains and each docked pose. As expected, the averaged % volume overlap using DCEVDW+ES to choose compounds is lowest. The overall trend follows FPSVDW (45.1%) > FPSVDW+ES (44.0%) > FPSES (41.8%) > DCEVDW+ES (37.7%). Schiffer and coworkers have highlighted the importance of considering the “substrate envelope” when designing inhibitors against drug resistant HIV proteases.56 Use of footprints to enrich screening results towards that of a “reference envelope” is conceptually similar.
Figure 5.
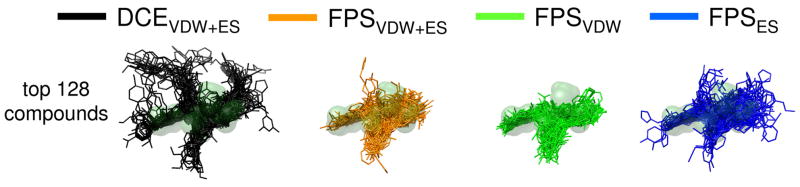
Visual overlays for the top 128 scoring compounds from each of the re-ranking methods in comparison with the reference envelope (light green surface).
In terms of energy, Figure 6 shows per-residue VDW and ES differences (Δ values) for the 20 top-scoring compounds from each ensemble relative to the reference. Consistent with the results presented earlier, use of the FPSVDW+ES function (Figure 6b) yields smaller per-residue differences for both energy components (ΔVDW = black, ΔES = red) compared to DCEVDW+ES indicating better molecular mimicry relative to the other methods. And, use of FPSVDW (Figure 6c) or FPSES (Figure 6d) functions alone each yield the smallest per-residue differences for their respective target function. Averaged absolute differences for the key residues (52a-70a, 52c-70c) shown in Figure 6 follow the trend DCEVDW+ES (0.47 kcal/mol) > FPSVDW (0.27 kcal/mol) = FPSES (0.27 kcal/mol) > FPSVDW+ES (0.25 kcal/mol). As expected, use of DCEVDW+ES (Figure 6a) leads to the largest variance in both energy terms indicating less effective mimicry, in this case by about double in magnitude relative to FPS (0.47 kcal/mol vs ~ 0.26 kcal/mol). Interestingly, only the DCEVDW+ES ensemble consistently yields dramatically more favorable ΔVDW deviations at specific residues (Figure 6a black lines below zero), a further indication of increased molecular size relative to the reference. However, as discussed in the next section, increased favorable interactions at individual receptor sites, in the present case, does not appear to be as important for activity as matching the overall pattern of the footprint comprised of a group of receptor residues.
Figure 6.
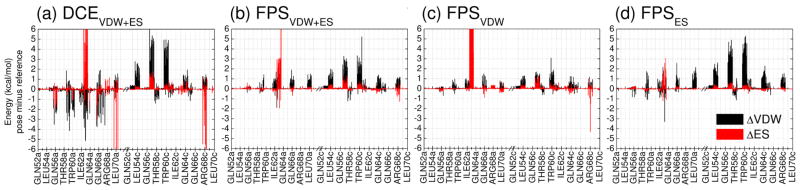
Per-residue energy differences (pose minus reference) for four ensembles comprised of the 20 top-scoring compounds derived from (a) DCEVDW+ES, (b) FPSVDW+ES, (c) FPSVDW, and (d) FPSES ranking methods. Steric packing differences (ΔVDW) are in black and the electrostatic differences (ΔES) are in red. Positive values indicate the reference makes stronger interactions at specific residues; negative values indicate the candidate molecule makes stronger interactions at specific residues. Energies in kcal/mol.
3.1.3 Footprint comparisons for experimentally tested compounds
As described in Holden et al,18 out of 115 compounds from the gp41 virtual screen tested experimentally, 24 showed affinity in a fluorescence-based binding assay and 7 were ultimately identified as having favorable characteristics (low Ki, low IC50, high CC50 values) based on three different types of experimental assays (binding, cell-cell fusion, cytotoxicity). Figure 7 shows the computational results in terms of the footprint patterns made by all the tested molecules when arranged into two groups: (i) the 7 compounds with favorable characteristics in all three experimental assays defined here as actives, and (ii) the remaining 108 compounds defined here as inactives. Strikingly, Figure 7 reveals how the smaller set of 7 active compounds (blue) more closely conform to the per-residue patterns of the reference (green) compared to the larger group of 108 inactives (red). However, it should also be emphasized that good correspondence between any individual candidate molecule and the reference does not necessarily imply activity as multiple inactives in Figure 7 also show good overlap. Nevertheless, the fact that all 7 active compounds show good overlap is notable, and suggests that footprints encode useful information. Overall, when faced with the daunting task of mining large libraries on the order of potentially millions of compounds, a prioritization method that includes mimicking known interaction patterns is a reasonable approach.
Figure 7.

Footprint similarity comparisons for 7 active compounds (blue) and 108 inactive compounds (red) relative to the reference (green).
It is important to emphasize the FPS method can be viewed as an attempt to capture which residues “as a group” facilitate ligand binding without a priori emphasizing which individual residue(s) are most important. Nonetheless, based on comparing the active vs inactive footprints in Figure 7 across this group of 115 compounds, specific interactions that appear to be important for activity include the need for: (1) favorable van der Waals interactions at positions Val59a, Lys63a, Gln66a, Leu57c, and Trp60c, (2) favorable electrostatic interactions at positions Lys63a, and Leu57c, (3) an absence of strong favorable electrostatic interactions at positions Gln56a, Gln66a, Arg68a, and Arg68c.
Finally, Table 1 shows the rank-ordered position for each of the 7 active compounds in four different lists obtained by re-scoring the top 100,000 compounds docked to gp41 with the different functions and examining only the top 500. Importantly, 5 out of the 7 actives were ranked within the top 62 in any of the ordered list and in all cases were ranked within the top 250. The lack of actives in the top 500 FPSVDW lists suggests including an electrostatic term is important. From an overall enrichment standpoint, three of the seven (SB-D10, SB-C01, and SB-H02) would not have been identified without FPS scoring. As was previously noted in Holden et al,18 the 115 compounds ultimately purchased for experimental evaluation was roughly evenly-split (21–33 compounds each) with regards to which criteria was used during selection with an emphasis placed on obtaining ligands with favorable scores across multiple categories. Thus, some compounds chosen based on DCE ranks also have reasonable FPS overlap. One compound in particular (SB-C09) was ranked well across three scoring functions (FPSVDW+ES = 3rd, DCEVDW+ES = 41st, and FPSES = 43rd). It should be emphasized that from a practical standpoint, choosing which compounds to purchase is not trivial. Beyond rank-ordered score, compound availability, chemical diversity, and visualization of the binding poses were among other criteria that were considered during selection. For the future, an ideal test to derive more complete enrichment statistics, if resources would allow, would be to purchase a reasonably large number (i.e. the top 100 compounds) from each rank-ordered list for experimental testing without consideration of rank in other scored lists.
Table 1.
Position in rank-order lists of up to 500 for the 7 active compounds.
| ZINC code | SB code | DCEVDW+ESa | FPSVDW+ESa | FPSVDWa | FPSESa |
|---|---|---|---|---|---|
| ZINC21093355 | SB-H11 | 24 | - | - | - |
| ZINC13898584 | SB-A05 | 40 | - | - | - |
| ZINC27552006 | SB-C09 | 41 | 3 | - | 43 |
| ZINC14010232 | SB-D04 | 61 | - | - | - |
| ZINC03659233 | SB-D10 | - | - | - | 148 |
| ZINC00304470 | SB-C01 | - | - | - | 247 |
| ZINC21537181 | SB-H02 | - | 46 | - | 450 |
Scoring functions used for ranking.
3.2 Ligand pose stability
3.2.1 Stability comparisons for experimentally tested compounds
The key utility of FPS scoring is to help identify and prioritize compounds from screens that mirror the interactions of a known substrate, inhibitor, or, as in this present case for gp41, significant protein sidechains. An alternative use is assessment of footprint stability with respect to the initial docked pose. Roughly analogous to rmsd (but computed in energetic rather than geometric space), footprint-stability provides a means to assess if key binding site interactions will persist during a molecular dynamics (MD) simulation of the complex while still allowing for the ligand to move in concert with the surrounding environment (i.e. sidechains and solvent). Here, footprint stability was specifically defined as maintaining an average FPSVDW+ES > 1.70 r relative to the initial docked pose, during an explicit solvent MD simulation of a docked complex with values being averaged over 2000 snapshots saved periodically (1 ps) during a 2 ns trajectory.
Although 2 ns is a relatively short simulation, under the present conditions employing a restrained protein backbone, the amount of ligand movement in terms of FPS or rmsd variability appears sufficient to establish whether any predicted pose can be categorized as stable. As examples, Figure 8 plots results for the subset of 115 compounds which were tested experimentally. Here, compounds which yield an average FPSVDW+ES score of 1.70 r or greater in Figure 8a or 8e are colored black, otherwise red. Under this criteria, and analogous to the results above which showed active compounds yielded a stronger footprint overlap with the reference compared to inactives (Figure 7), the 7 actives here also appear to be much more stable in terms of maintaining their FPS scores, especially for FPSVDW+ES and FPSES, during the MD trajectories (Figure 8a–c black vs 8e–g red). And as a group, no actives show large variability in terms of ligand rmsd (Figure 8d vs 8h), a general indication of binding stability, in contrast to multiple inactive trajectories. However, it should also be emphasized, again analogous to the FPS overlap discussion from above (Figure7), that good MD stability also does not necessarily indicate that a particular compound will be active as numerous inactives in Figure 8e-h do appear stable (black colored trajectories). Nevertheless, the fact that all of the 7 actives in Figure 8 maintain overall good stability is remarkable and suggests that use of MD in conjunction with FPS scoring to aid compound prioritization is a useful strategy.
Figure 8.

Ligand pose stability showing FPSVDW+ES (r), FPSVDW (r), FPSES (r), and rmsd (Å) values vs time (ps) relative to the initial docked position for 115 compounds from the gp41 virtual screen tested experimentally. For clarity, all values smoothed using adjacent averaging in blocks of 100. Compounds yielding an average FPSVDW+ES score of 1.70 r or greater in panels (a) and (e) are colored black across all panels, otherwise colored red.
3.2.2 Use of FPS to enrich for stability
We hypothesized that use of FPS scoring could lead to greater numbers of stable poses compared to the standard DCE function. To test this hypothesis on larger datasets, we retroactively examined the four groups of 128 top-ranked cluster heads based on different scoring functions used in the virtual screen (Figure 4). As above, we employed MD simulations of each complex to compute average FPSVDW+ES scores and rmsds relative to the initial docked poses, in addition to individual scores for FPSVDW and FPSES. For these experiments, a slightly more stringent criterion in terms of the magnitude of the FPS score was used. Table 2 quantifies the number of ligands in each group of 128 that maintained high average footprint scores (FPSVDW+ES > 1.80 r, FPSVDW > 0.90 r, or FPSES > 0.90 r) or low average rmsds (rmsd < 2.0 Å).
Table 2.
Number of ligands with high FPS scores and low rmsd values averaged from molecular dynamics simulations of docked complexes of gp41.
| Ranking Methoda | FPS-stability | rmsd-stability | ||
|---|---|---|---|---|
|
| ||||
| (a) | (b) | (c) | (d) | |
|
| ||||
| FPSVDW+ES N > 1.80 r |
FPSVDW N > 0.90 r |
FPSES N > 0.90 r |
rmsd N < 2 Å |
|
| DCEVDW+ES | 53/128 (41%) | 31/128 (24%) | 69/128 (54%) | 10/128 (8%) |
| FPSVDW+ES | 78/128 (61%) | 39/128 (30%) | 99/128 (77%) | 20/128 (16%) |
| FPSVDW | 12/128 (9%) | 53/128 (41%) | 18/128 (14%) | 16/128 (13%) |
| FPSES | 85/128 (66%) | 38/128 (30%) | 110/128 (86%) | 34/128 (27%) |
Scoring function used to identify 128 top-ranked cluster heads.
When using the FPSVDW+ES stability metric (Table 2a), compounds selected with FPSVDW+ES (61%) or FPSES (66%) functions do show enhanced stability relative to the DCEVDW+ES (41%) set. Mirroring this trend, when using the FPSES stability metric (Table 2c), use of FPSVDW+ES (77%) or FPSES (86%) functions to select compounds also yields more stable poses than DCEVDW+ES (54%). In contrast, use of the FPSVDW function yields few poses that are able to maintain good FPSVDW+ES (Table 2a, 9%) or FPSES stability (Table 2c, 14%). These results are consistent with the relatively featureless FPSVDW+ES and FPSES histograms for top-ranked compounds selected using the FPSVDW function (Figure 4d and 4f, green). Examination of the results in rmsd space (Table 2d) reveals a similar trend with functions containing electrostatic overlap terms, FPSVDW+ES (16%) and FPSES (27%), both yielding larger numbers of rmsd-stable compounds than DCEVDW+ES (8%). The previous observation that none of the 7 actives were in the rank-ordered lists of 500 derived using FPSVDW (Table 1) provides compelling support for including electrostatics. Overall, while additional systems need to be studied, the present studies suggest FPS scoring that includes the electrostatic term (FPSVDW+ES or FPSES) could be a tool to enrich for compounds that preferentially maintain their original docked interaction profiles relative to use of the DCEVDW+ES function. Importantly, monitoring of FPS emerges as a practical alternative to an rmsd-based measure of stability.
3.2.3 Control and decoy stability
To more thoroughly investigate if pose stability is a property inherent to known binding geometries (Figure 9), we examined the stability behavior of ligands taken from 32 unique protein-ligand families comprising 116 crystallographic complexes (termed controls) with accompanying decoys (see Methods describing decoy generation) under the same simulation conditions used to examine the gp41 complexes comprising the 115 experimentally tested compounds (Figure 8) and the four larger ensembles of 128 each (Table 2). As before, ligand FPS-stability and rmsd-stability were measured, in this case relative to either x-ray or decoy poses. Importantly, the control group of native x-ray poses (red histograms) more completely maintain their initial FPSVDW+ES (Figure 9a), FPSVDW (Figure 9b), and FPSES (Figure 9c) footprint patterns which yield higher r values relative to the corresponding decoy set (gray histograms). The controls also show greater rmsd-stability as indicated by lower rmsd values (Figure 9d). These tests help to confirm that pose stability is an inherent property of experimentally observed geometries, across a diverse group of protein-ligand families, and thus a potentially useful tool to aid ligand prioritization.
Figure 9.
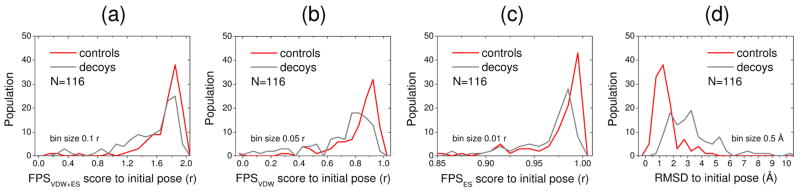
(a) FPSVDW+ES stability (r), (b) FPSVDW stability (r), (c) FPSES stability (r), and (d) rmsd stability (Å) based on MD simulations starting from known crystallographic controls (red)52 and associated decoys (gray).
3.2.4 Solvent accessible surface area (SASA)
While the percentage of decoys in Figure 9d that maintained a low rmsd (< 2.0 Å) was higher than might be expected (28%) for poses that are not observed experimentally, the percentage of favorably-scored compounds that maintained a low rmsd in the gp41 site was relatively small at 8–27% from simulations of the 4*128 rank-ordered lists (Table 2d) and 13–14% from simulations of the 115 compounds tested experimentally (Figure 8). An examination of solvent accessible surface area (SASA) shows that the crystallographic ligands (controls and decoys), on average, are significantly more buried (Figure 10 red) when bound to their respective targets versus the theoretical compounds docked into the gp41 site (Figure 10 colored lines). Taken together, the results suggest that use of rmsd as a metric to gauge pose stability could lead to false positives for highly buried ligands as well as false negatives for highly exposed ligands. In these instances, FPS definitions of pose stability provide a useful alternative.
Figure 10.

Ligand SASA of molecules from crystallographic complexes (red) and accompanying decoys (gray) vs molecules docked to gp41 prioritized using the standard DOCK energy score (DCEVDW+ES, black), van der Waals footprint score plus electrostatic footprint score (FPSVDW+ES, orange), van der Waals footprint score (FPSVDW, green), or electrostatic footprint score (FPSES, blue).
3.3 Ligand consensus
In a practical sense, the overall intersection between the ensemble derived using the standard DOCK DCEVDW+ES function with those of the three FPS methods is minimal at only 2–8 molecules (Table 3) across the four sets of 128 top-ranked compounds. Only the FPSVDW+ES and FPSES groups contain a significant number of molecules in common (N = 35) which is consistent with the observation that use of these two functions, in general, yields greater numbers of more stable poses (Table 2). In any event, it should be emphasized that footprint-based scoring provides alternative choices for ranking and was not necessarily designed to replace the standard DOCK scoring method. In fact, as the different ranking functions each produce different results (only 0% to 27% molecules are in common in Table 3), as emphasized earlier, it should be advantageous to look at top hits obtained using multiple scoring metrics. However, if the goal is to quickly identify molecules that mimic a known interaction pattern, then the FPS methods would have clear benefits.
Table 3.
Number of molecules in common across four sets of 128 top-ranked compounds using different rank-ordering methods.
| Methoda | DCEVDW+ES | FPSVDW+ES | FPSVDW | FPSES |
|---|---|---|---|---|
|
|
||||
| DCEVDW+ES | 128 | 8 | 2 | 6 |
| FPSVDW+ES | 128 | 8 | 35 | |
| FPSVDW | 128 | 0 | ||
| FPSES | 128 | |||
Scoring function used to identify top-ranked cluster heads (N = 128).
4. Conclusion
The goal of this work was to evaluate use of different scoring and ranking strategies to help prioritize compounds from large-scale computational virtual screening. The study compared the standard DOCK energy function (DCEVDW+ES) with three new footprint-based scoring functions (FPSVDW+ES, FPSVDW, and FPSES) developed in our laboratory.23,24 The experiments employed results from our prior virtual screen18 of ca. 500,000 publically available compounds docked to a hydrophobic pocket on the drug target HIVgp41 (Figures 3–8, Tables 1–3), as well as known crystallographic controls (Figures 9, 10). Ensembles of rank-ordered ligands were examined in terms of two main criteria: (1) ligand pose properties which included DCE and FPS score distributions, molecular weight, number of rotatable bonds, ligand efficiency, ligand formal charge, volume overlap, and footprint comparisons between experimentally tested active and inactive compounds, and (2) ligand pose stability which included footprint-stability (changes in footprint overlap) and rmsd-stability (changes in geometry).
Ligand pose property experiments demonstrate how FPS functions can be used to mine sets of compounds, from within a large pool of virtual screening results, which have properties that mimic a specific reference. In the case of gp41, the reference was a construct derived from the native peptide substrate sequence centered on four key sidechains that interact (Figure 3) at an important protein interface (Figure 1). Molecules with high FPS scores showed both good energetic (Figures 4, 6–7) and spatial (Figure 5) overlap with the reference. Importantly, the specific method used to prioritize the screening results each produces a set with properties physically consistent with the method employed (Figure 4). Molecules prioritized with FPSVDW+ES, FPSVDW, or FPSES have larger peaks at higher Pearson coefficient values in their respective parent histograms (Figure 4d–f) and smaller energy differences to the reference sidechains (Figures 6) compared to use of DCEVDW+ES. Notably, an examination of the 115 compounds tested experimentally reveals all 7 actives have high footprint overlap to the reference relative to the inactive group which shows much greater variability in terms of magnitude and position (Figure 7). Overall, the pose property studies suggest a consensus of scoring functions should be used when choosing compounds for experimental testing (Table 1).
Ligand pose stability experiments using MD simulations further demonstrate the utility of using FPS methods as an alternative to rmsd by allowing a ligand to move in concert with the surrounding environment and monitoring the extent with which binding site interactions can be maintained. Remarkably, the group of 7 actives all maintain high FPSVDW+ES and FPSES stability scores during the MD trajectories compared to the group of 108 inactive compounds for which multiple members show large variability (Figure 8). Tests to gauge pose stability across a much larger dataset (four groups of 128 compounds) show that use of FPSVDW+ES and FPSES functions to rank-order compounds yield more stable compounds relative to DCEVDW+ES or FPSVDW in all but one case (Table 2). Results for rmsd-stability follow the same trend. While additional studies on greater numbers of systems need to be performed, the present MD results suggest enhanced pose stability may be linked to favorable electrostatic compatibility better captured by electrostatic footprint patterns embodied in FPSVDW+ES and FPSES functions Consistent with this conclusion is the fact that 3 of the 7 experimentally-verified leads identified from among the 115 compounds tested for gp41 binding and activity were prioritized using FPSVDW+ES or FPSES (Table 3).18 Finally, concurrent control studies showed that known ligands in their crystallographic binding poses maintain higher FPS scores and smaller rmsds versus decoys (Figure 9). This confirms that native poses are indeed stable, and thus consideration of pose stability using FPS during compound prioritization is likely to be beneficial.
Overall, the results presented in this study suggest that footprint-based methods can play at least two useful roles in structure-based design: (a) footprints can aid identification of compounds biased to specific properties inherent to a known reference, and (b) they provide a useful alternative to rmsd for evaluating pose stability. Both uses complement current virtual screening approaches for the identification (FPS-ranking) and prioritization (FPS-stability) of target-compatible molecules in a quantitative and logical way.
Acknowledgments
Gratitude is expressed to Trent E. Balius and Sudipto Mukherjee for computational assistance and helpful discussions. This work was funded in part by the Stony Brook University Office of the Vice President for Research, the New York State Office of Science Technology and Academic Research, and NIH Grants R01GM083669 (to R.C.R), R01GM087998 (to M.G.), and F32GM105400 (to W.J.A). This research utilized resources at the New York Center for Computational Sciences at Stony Brook University/Brookhaven National Laboratory which is supported by the U.S. Department of Energy under Contract DE-AC02-98CH10886 and by the State of New York.
Abbreviations
- gp41
glycoprotein-41
- FPS
footprint similarity
- DCE
DOCK Cartesian energy
- VDW
van der Waals
- ES
electrostatic
- MD
molecular dynamics
- SASA
solvent accessible surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 2.Drugs@FDA. [last accessed June 27: 2013]; website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm.
- 3.Greenberg ML, Cammack N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother. 2004;54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 4.Mink M, Mosier SM, Janumpalli S, Davison D, Jin L, Melby T, Sista P, Erickson J, Lambert D, Stanfield-Oakley SA, Salgo M, Cammack N, Matthews T, Greenberg ML. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J Virol. 2005;79:12447–12454. doi: 10.1128/JVI.79.19.12447-12454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nameki D, Kodama E, Ikeuchi M, Mabuchi N, Otaka A, Tamamura H, Ohno M, Fujii N, Matsuoka M. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp4l and rev-responsive element functions. J Virol. 2005;79:764–770. doi: 10.1128/JVI.79.2.764-770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melby T, Sista P, DeMasi R, Kirkland T, Roberts N, Salgo M, Heilek-Snyder G, Cammack N, Matthews TJ, Greenberg ML. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Res Hum Retroviruses. 2006;22:375–385. doi: 10.1089/aid.2006.22.375. [DOI] [PubMed] [Google Scholar]
- 7.Davison DK, Medinas RJ, Mosier SM, Bowling TS, Delmedico MK, Dwyer JJ, Cammack N, Greenberg ML. New fusion inhibitor peptides, TRI-999 and TRI-1144, are potent inhibitors of enfuvirtide and T-1249 resistant isolates. XVI International AIDS Conference: Conference Reports for NATAP; 2006. Poster THPE0021. [Google Scholar]
- 8.Chinnadurai R, Rajan D, Munch J, Kirchhoff F. Human Immunodeficiency Virus Type 1 Variants Resistant to First- and Second-Version Fusion Inhibitors and Cytopathic in Ex Vivo Human Lymphoid Tissue. J Virol. 2007;81:6563–6572. doi: 10.1128/JVI.02546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci US A. 2001;98:11187–11192. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang SB, Lin K, Strick N, Neurath AR. Hiv-1 Inhibition by a Peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 11.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A Synthetic Peptide Inhibitor of Human Immunodeficiency Virus Replication: Correlation between Solution Structure and Viral Inhibition. Proc Natl Acad Sci US A. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 13.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci US A. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debnath AK, Radigan L, Jiang SB. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J Med Chem. 1999;42:3203–3209. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- 15.Jiang SB, Lu H, Liu SW, Zhao Q, He YX, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey G, Rits-Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc Natl Acad Sci US A. 2006;103:13938–13943. doi: 10.1073/pnas.0601036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai L, Gochin M. A Novel Fluorescence Intensity Screening Assay Identifies New Low Molecular Weight Inhibitors of the gp41 Coiled Coil Domain of HIV-1. Antimicrob Agents Chemother. 2007;51:2388–2395. doi: 10.1128/AAC.00150-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holden PM, Kaur H, Goyal R, Gochin M, Rizzo RC. Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorg Med Chem Lett. 2012;22:3011–3016. doi: 10.1016/j.bmcl.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci US A. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 21.Moustakas DT, Therese Lang PT, Pegg S, Pettersen E, Kuntz ID, Broojimans N, Rizzo RC. Development and Validation of a Modular, Extensible Docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 22.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balius TE, Mukherjee S, Rizzo RC. Implementation and Evaluation of a Docking-Rescoring Method using Molecular Footprint Comparisons. J Comput Chem. 2011;32:2273–2289. doi: 10.1002/jcc.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balius TE, Allen WJ, Mukherjee S, Rizzo RC. Grid-based Molecular Footprint Comparison Method for Docking and De Novo Design: Application to HIVgp41. J Comput Chem. 2013;34:1226–1240. doi: 10.1002/jcc.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen WJ, Rizzo RC. Computer-Aided Approaches for Targeting HIVgp41. Biology. 2012;1:311–338. doi: 10.3390/biology1020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG. Targeting Fatty Acid Binding Protein (FABP) Anandamide Transporters - A Novel Strategy for Development of Anti-Inflammatory and Anti-Nociceptive Drugs. PLoS One. 2012;7:e50968. doi: 10.1371/journal.pone.0050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fry DC. Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr Protein Pept Sci. 2008;9:240–247. doi: 10.2174/138920308784533989. [DOI] [PubMed] [Google Scholar]
- 28.Fry DC. Small-molecule inhibitors of protein-protein interactions: how to mimic a protein partner. Curr Pharm Des. 2012;18:4679–4684. doi: 10.2174/138161212802651634. [DOI] [PubMed] [Google Scholar]
- 29.Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem. 2004;47:337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 30.Brewerton SC. The use of protein-ligand interaction fingerprints in docking. Curr Opin Drug Discov Devel. 2008;11:356–364. [PubMed] [Google Scholar]
- 31.Pfeffer P, Neudert G, Klebe G. DrugScoreFP: profiling protein-ligand interactions using fingerprint simplicity paired with knowledge-based potential fields. Chem Cent J. 2008;2:S16. [Google Scholar]
- 32.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 33.Mukherjee S, Balius TE, Rizzo RC. Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J Chem Inf Model. 2010;50:1986–2000. doi: 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DMS, University of California at San Francisco Computer Graphics Laboratory. [last accessed June 27th 2013]; http://www.cgl.ucsf.edu/Overview/software.html.
- 35.DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J Med Chem. 1988;31:722–729. doi: 10.1021/jm00399a006. [DOI] [PubMed] [Google Scholar]
- 36.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 37.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Jr, MKM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct Funct Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters A, Lundberg ME, Lang PT, Sosa CP. IBM Red Paper. 2008. High Throughput Computing Validation for Drug Discovery Using the DOCK Program on a Massively Parallel System. REDP-441000. [Google Scholar]
- 41.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 42.DOCK Version 64. University of California at San Francisco; San Francisco, CA: 2011. [Google Scholar]
- 43.MDL Information Systems, Inc., 14600 Catalina Street, San Leandro, CA 94577.
- 44.Brown RD, Martin YC. Use of structure Activity data to compare structure-based clustering methods and descriptors for use in compound selection. J Chem Inf Comput Sci. 1996;36:572–584. [Google Scholar]
- 45.MOE Version 200810. Chemical Computing Group Inc; Montreal, Canada: [Google Scholar]
- 46.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 47.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 48.Balius TE, Rizzo RC. Quantitative Prediction of Fold Resistance for Inhibitors of EGFR. Biochemistry. 2009;48:8435–8448. doi: 10.1021/bi900729a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with Coupling to an External Bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 50.Darden T, York D, Pedersen L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 51.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER. 2010;10 [Google Scholar]
- 52.PDB codes used as controls include: acetylcholinesterase (1E66, 1EVE, 1GPN, 1H22), aspartate transcarbamoylase (1ACM, 1D09, 1Q95, 8ATC), beta trypsin (1BJU, 1BJV, 1GHZ, 1GI4), carbonic anhydrase (1BN1, 1BN3, 1BN4, 1BNN), carboxypeptidase a (1CBX, 1CPS, 1F57, 1HDQ), cel7 (1DY4, 1H46, 1Z3T, 1Z3V), cox (1EQG, 1EQH, 1HT5, 1HT8), estrogen receptor (1A52, 1ERE, 1GWQ, 1GWR), factor xa (1LPG, 1LPK, 1Z6E, 2J95), glucoamylase (1AGM, 1GAH, 1GAI, 1LF9), hiv protease (1A8K, 1CPI, 1HPV, 1HSH), hmg coa reductase (1HW9, 1HWI, 1HWJ, 1HWK), lysozyme (1HEW, 1LZB, 1LZC, 1LZY), mmp (1RMZ, 1ROS, 1Y93, 2OXW), neuraminidase (1A4G, 1A4Q, 1B9S, 1B9T), omp decarboxylase (1DBT, 1DQX, 1EIX, 1LOQ), phosphodiesterase (1OYN, 1XON, 1XOQ, 1XOR), phospholipase a2 (1HN4, 1KPM, 1KVO, 1OYF), hiv reverse transcriptase (1C1B, 1C1C, 1EP4, 1RTH), ribonuclease a (1JN4, 1W4O, 1W4P, 1W4Q), ribonuclease t1 (1BIR, 1RGK, 1RGL, 3BIR), scytalone dehydratase (3STD, 4STD, 5STD, 6STD), sialidase (1EUS, 1MZ6, 1N1T, 1N1V), streptavidin (1DF8, 1SRG, 1SWP, 1SWR), t4 lysozyme (181L, 182L, 1LI6, 2OU0), thermolysin (2TMN, 4TLN, 4TMN, 5TMN), thrombin (1K21, 1KTS, 1KTT, 1T4V), thymidylate synthase (1F4E, 1F4F, 1F4G, 1JMF), triosephosphate isomerase (2YPI, 4TIM, 6TIM, 7TIM), trypsin (1C5P, 1PPC, 1PPH, 1TPS), tryptophan synthase (1A5S, 1C9D, 1CW2, 1CX9), tyrosine phosphatase (1C83, 1C84, 1C86, 1C87).
- 53.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 54.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 55.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci US A. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MN, Nalivaika EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, Schiffer CA. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses. 2010;2:2509–2535. doi: 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]