

Abstract
Development of conformationally restricted nucleotide building blocks continues to attract considerable interest due to their successful use within antisense, antigene and other gene-targeting strategies. Locked nucleic acid (LNA) and its diastereomer α-L-LNA are two interesting examples hereof. Oligonucleotides modified with these units display greatly increased affinity toward nucleic acid targets, improved binding specificity and enhanced enzymatic stability relative to unmodified strands. Here, we present the synthesis and biophysical characterization of oligodeoxyribonucleotides (ONs) modified with 2′-amino-α-L-LNA adenine monomers W–Z. The synthesis of target phosphoramidites 1–4 initiates from pentafuranose 5, which upon Vorbrüggen glycosylation, O2′-deacylation, O2′-activation and C2′-azide introduction yields nucleoside 8. A one-pot tandem Staudinger/intramolecular nucleophilic substitution converts 8 into 2′-amino-α-L-LNA adenine intermediate 9, which after a series of non-trivial protecting group manipulations affords key intermediate 15. Subsequent chemoselective N2′-functionalization and O3′-phosphitylation gives targets 1–4 in ~1–3% overall yield over eleven steps from 5. ONs modified with pyrene-functionalized 2′-amino-α-L-LNA adenine monomers X-Z display greatly increased affinity toward DNA targets (ΔTm/modification up to +14 °C). Results from absorption and fluorescence spectroscopy suggest that the duplex stabilization is a result of pyrene intercalation. These characteristics render N2′-pyrene-functionalized 2′-amino-α-L-LNA of considerable interest for DNA-targeting applications.
Introduction
Major efforts have been devoted over the past twenty years to develop conformationally restricted nucleotides. Oligonucleotides that are modified with these building blocks often display markedly increased affinity toward nucleic acid targets, improved discrimination of non-targets and greater resistance against enzymatic degradation relative to reference strands.1 Such oligonucleotides are accordingly widely used for nucleic acid targeting applications in molecular biology, biotechnology and medicinal chemistry.2 Locked Nucleic Acid (LNA) is amongst the most prominent members of this compound class due to its extraordinary affinity toward DNA and RNA complements (Figure 1); thus, increases in thermal denaturation temperatures (Tm’s) of up to +10 °C have been observed per modification.3–5 The diastereomeric α-L-LNA shares many characteristics with LNA, including very high affinity toward DNA/RNA targets, but is less well characterized due to more limited commercial availability (Fig. 1).6 The interesting properties of LNA, α-L-LNA and other conformationally restricted nucleotides, has spurred development of many analogues.1,7
Figure 1.

Structures of LNA, α-L-LNA and 2′-amino-α-L-LNA monomers studied herein. T = thymin-1-yl.
As part of our ongoing interest in LNA chemistry and diagnostic applications of pyrene-functionalized oligonucleotides,2c,8 we recently pursued the development of N2′-pyrene-functionalized 2′-amino-α-L-LNA thymine monomers (Fig. 1).9 Oligodeoxyribonucleotides (ONs) modified with these monomers display remarkable affinity toward complementary DNA as the pyrene moieties are preorganized to intercalate and engage in stacking with neighboring nucleobases upon duplex formation.9b Thus, increases in Tm’s of up to 20 °C per modification have been observed for short ONs modified with these building blocks. We have taken advantage of these characteristics and have developed probes for a variety of diagnostic applications. For example, ONs modified with two next-nearest neighbor incorporations of 2′-N-(pyren-1-yl)acetyl-2′-amino-α-L-LNA-T monomers are promising tools for detection of single nucleotide polymorphisms, i.e., the most prevalent type of genetic mutation in the human genome. These probes efficiently discriminate between complementary and single-base mismatched DNA/RNA targets through differences in pyrene excimer emission levels. In another example, the fluorescence of ONs modified with 2′-N-(pyren-1-yl)acetyl-2′-amino-α-L-LNA-T monomers was found to increase 16-fold upon hybridization with DNA targets featuring abasic sites, i.e., DNA lesions that result in genomic mutations and emergence of cancers if unrepaired.11 We have also utilized N2′-pyrene-functionalized 2′-amino-α-L-LNA-T monomers as the key activating components of the so-called Invader probes, which recognize mixed-sequence double-stranded DNA.12 Unfortunately, the synthesis of the N2′-pyrene-functionalized 2′-amino-α-L-LNA thymine phosphoramidites is challenging (~20 steps, <4% overall yield from diacetone-α-D-glucose), mainly due to unsuccessful attempts of introducing the necessary C2′-azido group at the nucleoside level without concomitant O2,O2′-anhydronucleoside formation.9a This forced us to introduce the azido group at the carbohydrate level, which resulted in a loss of anchimeric assistance from the O2-position during Vorbrüggen glycosylation and the formation of anomeric nucleoside mixtures. We hypothesized that these synthetic difficulties might be overcome with the corresponding adenine derivates, as formation of anhydronucleosides is unlikely. Easier access to N2′-pyrene-functionalized 2′-amino-α-L-LNA monomers is desirable in order to evaluate the diagnostic potential of these building blocks in greater detail.
In the present article, we report the synthesis and characterization of ONs modified with four different 2′-amino-α-L-LNA adenine monomers W–Z (Fig. 1). The ONs are characterized via thermal denaturation, UV-Vis and fluorescence experiments and shown to display extraordinary thermal affinity toward complementary DNA (ΔTm/modification up to 19.5 °C) and photophysical characteristics consistent with intercalative binding modes for the pyrene moieties.13
Results and Discussion
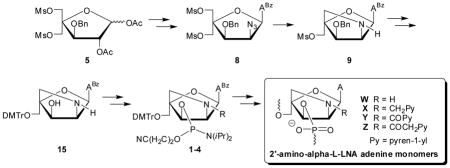
Retrosynthetic analysis of 2′-amino-α-L-LNA-A monomers
Inspired by our previously reported synthesis of the corresponding thymine monomers,9 we identified adenine derivative 15 as a suitable substrate for chemoselective N2′-functionalization and subsequent O3′-phosphitylation, which was expected to yield target 6-N-benzoyl-adenin-9-yl nucleosides 1–4 (Scheme 1). We surmised that key intermediate 15 could be obtained from nucleoside 9 via a series of protecting group manipulations. The O3′-benzyl group was selected due to i) its stability in acidic and basic reaction conditions, allowing it to be introduced at the beginning of the synthetic route, and ii) its ability to be removed under conditions that only have a minimal effect on the 6-N-benzoyl group of adenine.14 We expected to construct the 2-oxo-5-azabicyclo[2.2.1]heptane skeleton of 9 in a similar manner as originally reported for 2′-amino-β-D-LNA,15 i.e., via an one-pot tandem Staudinger/intramolecular nucleophilic substitution reaction, which revealed nucleoside 8 as an early target. In contrast to our synthesis of the corresponding thymine monomer where the C2′-azido group had to be introduced at the carbohydrate stage,9a we decided to introduce the 2′-azido group of 8 at the nucleoside stage, anticipating that selective O2′-deacylation of known nucleoside 66b and subsequent O2′-activation and introduction of the azido group would provide 8. Importantly, the anchimeric assistance provided by the O2-substituent during the Vorbrüggen glycosylation of 5 to give 6 prevents formation of anomeric mixtures, which is one of the drawbacks in the synthesis of 2′-amino-α-L-LNA-T monomers.9a
Scheme 1.

Retrosynthetic analysis for 2′-amino-α-L-LNA adenine monomers W–Z. PG = PN(iPr)2OCH2CH2CN; R = H/CH2Py/COPy/COCH2Py; py = pyren-1-yl; ABz = 6-N-benzoyl-adenin-9-yl.
Synthesis of key intermediate 15
Fully protected pentafuranose 5, which is obtained from diacetone-α-D-glucose in six steps and ~30% overall yield,6b,16 serves as the starting material for the synthesis of key intermediate 15 (Scheme 2). Glycosyl donor 5 was converted into alcohol 7 following protocols that deviated from the known route6b in the following manner: i) the use of trimethylsilyl triflate as Lewis acid and 1,2-dicholoroethane as reaction solvent during Vorbrüggen glycosylation of 5 affords 6 in higher yield and with easier workup than the original protocol involving tin(IV) chloride and acetonitrile (70% vs 57%, respectively), and ii) the use of the guanidinium nitrate/sodium methoxide reagent mixture17 results in slightly more efficient O2′-deacylation of 6 than the original protocol entailing dilute methanolic ammonia (88% vs 79% yield); however, the dilute reaction conditions of the former approach rendered it less practical for large scale reactions. Alcohol 7 was O2′-triflated and treated with sodium azide and 15-crown-5 in DMF at elevated temperatures to furnish azide 8 in excellent yield (89% over two steps). IR spectra of 8 verified the presence of the azide group (sharp band at 2115 cm−1). Nucleoside 8 was converted into the desired 2′-amino-α-L-LNA intermediate 9 via a one-pot tandem Staudinger/intramolecular nucleophilic substitution reaction using alkaline trimethylphosphine15 in a satisfying 85% yield.18 The 2-oxo-5-azabicyclo[2.2.1]heptane skeleton and stereochemical configuration of 9 was verified by NOE difference experiments on downstream products (vide infra).
Scheme 2.
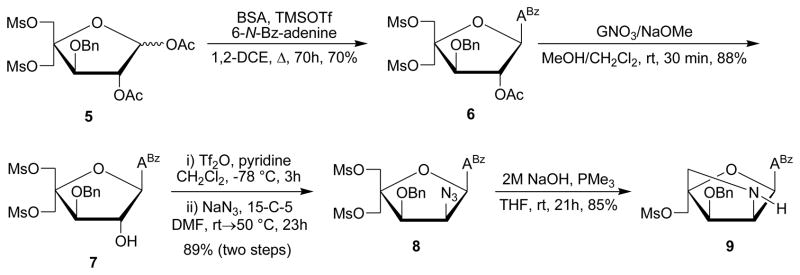
Synthesis of intermediate 9. BSA = N,O-bis(trimethylsilyl)acetamide; 1,2-DCE = 1,2-dichloroethane; ABz = 6-N-benzoyl-adenin-9-yl; GNO3 = guanidinium nitrate; 15-C-5: 15-crown-5.
The protecting group manipulations needed to convert nucleoside 9 into key intermediate 15 proved surprisingly challenging (Scheme 3). For example, O3′-debenzylation of bicyclic nucleoside 9 was unsuccessful using boron tricholoride or methanesulfonic acid in dichloromethane. Similarly, the corresponding N2′-Fmoc-protected nucleoside 10 - obtained by reacting 9 with 9′-fluorenylmethyl chloroformate under Schotten-Baumann conditions (results not shown) - failed to undergo O3′-debenzylation with BCl3 in dichloromethane or by standard hydrogenolysis. Additional attempted strategies are outlined in the Supporting information (Scheme S1). Instead, it proved necessary to protect the N2′-position of bicyclic nucleoside 9 as a trifluoroacetamide to give nucleoside 11 in 62% yield, which was then O3′-debenzylated with BCl3 in hexanes to give nucleoside 12 in 87% yield (Scheme 3). Protection of the O3′-position as a napthyl ether presents itself as an attractive alternative option as it can be cleaved using DDQ,7a but this option was not considered at the time of synthesis. Subsequent nucleophilic substitution of the C5′-mesylate group of 12 was attempted using potassium acetate and 18-crown-6, but this resulted in the formation of numerous side products according to analytical TLC and the approach was abandoned (Scheme 3). Instead, treatment of 12 with sodium benzoate and 15-crown-5 in hot DMF afforded O5′-benzoylated nucleoside 13 in 83% yield. Subsequent treatment of nucleoside 13 with aqueous sodium hydroxide in 1,4-dioxane effected the cleavage of both the O5′-benzoyl and N2′-trifluroacetamide protecting groups to afford polar amino alcohol 14 in 60% yield after column chromatography. The O5′-hydroxyl group of nucleoside 14 was subsequently protected as the 4,4′-dimethoxytrityl (DMTr) ether using standard conditions to afford key intermediate 15. Curiously, the reaction proceeded in no more than 38% yield as significant amounts of N2′,O5′-di-DMTr-protected nucleoside 16 were produced as well (30% yield). Efforts to optimize the yield of 15 were unsuccessful (e.g., slow addition of DMTr-Cl at low temperatures). However, it was possible to recycle nucleoside 16 into amino diol 14 using 3% dichloroacetic acid in a mixture of methanol and nitromethane19 in up to 96% yield. An alternative strategy toward 15, involving N2′-Fmoc protection of 14 and subsequent O5′-DMTr-protection and N2′-Fmoc deprotection, was dismissed due to inefficient O5′-DMTr protection (~40% yield, results not shown). Thus, the preferred route (5 →→ 9 → 11 →→ 15) affords intermediate 15 in ~5% overall yield from 5, not taking the recycling step 16 → 14 into account.
Scheme 3.

Synthesis of key intermediate 15. ABz = 6-N-benzoyl-adenin-9-yl; DMTr = 4,4′-dimethoxytrityl; FmocCl = 9′-fluorenylmethyl chloroformate.
Structural verification of 2′-amino-α-L-LNA configuration
In agreement with previously reported 1H NMR signals of other α-L-LNA nucleosides,6b,9 the 1H NMR signals of H1′, H2′ and H3′ of 2′-amino-α-L-LNA nucleosides appear as singlets or narrow doublets (J < 2 Hz),20 since the torsion angles described by H1′-C1′-C2′-H2′ and H2′-C2′-C3′-H3′ are fixed in +gauche and -gauche conformations, respectively. The structure of bicyclic nucleoside 14 was ascertained by NOE difference spectroscopy. NOE contacts between H1′/H2′ (7%), H1′/H3′ (5%) and H2′/H3′ (2%) suggest a cis relationship between these protons (Fig. 2). Since the stereochemical configuration at C3′ is defined by the choice of starting material and remains unchanged throughout synthesis, H1′ and H2′ must be pointing “down”, which confirms the nucleobase as pointing “up” and hence establishes the 2′-amino-α-L-LNA configuration. This is substantiated by signal enhancements between H5″A/H8′ (6%) indicating a cis relationship between the nucleobase and H5″ (H5″A is tentatively assigned as the H5″ closest to the nucleobase).
Figure 2.

Key NOE contacts in nucleoside 14.
Synthesis of phosphoramidite building blocks 1–4
Chemoselective N2′-functionalization of key intermediate 15 to give nucleosides 17–20 was realized: i) using 9′-fluorenylmethyl chloroformate (17: 51% yield; Schotten-Baumanm conditions could not be used due to the low solubility of 15 in dioxane/water), ii) via reductive amination using 1-pyrenecarboxaldehyde and sodium triacetoxyborohydride21 as the reducing agent (18: 68% yield), or iii) via EDC-mediated coupling of 1-pyrenecarboxylic acid or 1-pyreneacetic acid (19: 64% yield; 20: 79% yield) (Scheme 4). Subsequent phosphitylation using 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite afforded target compounds 1–4 in 46–71% yield after column chromatography and precipitation.
Scheme 4.

Synthesis of phosphoramidites 1–4. ABz = 6-N-benzoyl-adenin-9-yl; DIPEA = N,N′-diisopropylethylamine; DMTr = 4,4′-dimethoxytrityl; EDC·HCl = 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride; FmocCl = 9′-fluorenylmethyl chloroformate; NMI = N-methylimadazole; PCl = 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite; Py = pyren-1-yl.
Synthesis of modified ONs and experimental design
Phosphoramidites 1–4 were used in machine-assisted solid-phase DNA synthesis (0.2 μmol scale) to incorporate monomers W-Z into ONs using the following hand-coupling conditions (activator; coupling time; stepwise coupling yields): monomer W (pyridinium hydrochloride; 30 min; ~82%) and monomers X-Z (pyridinium hydrochloride; 15 min; ~95%). Following workup and HPLC purification, the composition and purity of all modified ONs was ascertained by MALDI MS analysis (Tables S1 and S2) and ion-pair reversed-phase HPLC, respectively. ONs containing a single incorporation in the 5′-GTG BTA TGC context are denoted W1, X1, Y1 and Z1, respectively. Similar conventions apply for ONs in the B2–B6 series (Table 1). Reference DNA and RNA strands are denoted D1/D2 and R1/R2, respectively. The following descriptive nomenclature is also used: N2′-PyMe (X-series), N2′-PyCO (Y-series) and N2′-PyAc (Z-series). We have previously used this 9-mer mixed-sequence context to study the hybridization properties of ONs modified with N2′-pyrene-functionalized 2′-amino-α-L-LNA thymine monomers, which facilitates direct comparison.9
Table 1.
Tm values of duplexes between B1–B6 and complementary DNA targets.a
| ON | Duplex | B = | ΔTm/°C
|
|||
|---|---|---|---|---|---|---|
| W | X | Y | Z | |||
| B1 | 5′-GTG BTA TGC | −0.5 | +5.0 | +11.0 | +6.0 | |
| D2 | 3′-CAC TAT ACG | |||||
|
| ||||||
| B2 | 5′-GTG ATB TGC | +0.5 | +7.0 | +14.0 | +7.5 | |
| D2 | 3′-CAC TAT ACG | |||||
|
| ||||||
| B3 | 5′-GTG BTB TGC | −1.0 | +5.0 | +13.5 | +16.0 | |
| D2 | 3′-CAC TAT ACG | |||||
|
| ||||||
| D1 | 5′-GTG ATA TGC | ±0.0 | +6.5 | +11.5 | +7.5 | |
| B4 | 3′-CAC TBT ACG | |||||
|
| ||||||
| D1 | 5′-GTG ATA TGC | ±0.0 | +5.5 | +12.0 | +6.0 | |
| B5 | 3′-CAC TAT BCG | |||||
|
| ||||||
| D1 | 5′-GTG ATA TGC | −2.0 | +5.0 | +13.5 | +14.5 | |
| B6 | 3′-CAC TBT BCG | |||||
ΔTm = change in Tm relative to unmodified reference duplex D1:D2 (Tm ≡ 27.5 °C); Tm’s determined as the maximum of the first derivative of melting curves (A260 vs T) recorded in medium salt buffer ([Na+] = 110 mM, [Cl−] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4)), using 1.0 μM of each strand. Tm’s are averages of at least two measurements within 1.0 °C; A/C/G/T = adenin-9-yl/cytosin-1-yl/guanin-9-yl/thymin-1-yl DNA monomers. For structures of monomers W–Z, see Figure 1. Data for the B1/B2/B4/B5-series (for monomers X/Y/Z) have previously been reported in reference 12b.
Thermal denaturation experiments – thermal affinity toward complementary DNA/RNA
The thermostability of duplexes between W/X/Y/Z-modified ONs and complementary DNA/RNA was evaluated by determining their thermal denaturation temperature (Tm) in medium salt buffer ([Na+] = 110 mM, pH 7.0). Changes in Tm’s of modified duplexes are discussed relative to Tm’s of unmodified reference duplexes.
ONs with one or two incorporations of 2′-amino-α-L-LNA adenine monomer W display very similar thermal affinity toward complementary DNA as unmodified reference ONs (ΔTm between −1.0 to +0.5 °C, Table 1). Thus, unlike the corresponding 2′-oxy-α-L-LNA adenine monomer,6b,22 the 2-oxo-5-azabicyclo[2.2.1]heptane skeleton of monomer W is not inherently beneficial for DNA duplex formation. We have made similar observations with ONs modified with the thymine counterpart of monomer W.9a This indicates that the unfunctionalized 2′-nitrogen of 2′-amino-α-L-LNA monomers either stabilizes the single-stranded ON or destabilizes the duplex, e.g., by perturbing the hydration spine in the major groove. In contrast, ONs modified with N2′-pyrene-functionalized 2′-amino-α-L-LNA adenine monomers X, Y, or Z, display greatly increased thermal affinity toward DNA targets (ΔTm/modification between +2.5 and +14 °C, Table 1). The observed Tm trends for singly modified ONs (Y > Z ≥ X) indicate that: i) monomers, in which the pyrene moiety is attached to the 2-oxo-5-azabicyclo[2.2.1]heptane skeleton via an alkanoyl linker, induce greater thermostabilization than corresponding monomers employing alkyl linkers (N2′-PyCO Y > N2′-PyMe X) and ii) monomers with short alkanoyl linkers result in more thermostable DNA duplexes than the corresponding monomers with longer alkanoyl linkers (N2′-PyCO Y > N2′-PyAc Z). We have observed similar structure-property relationships with the analogous thymine monomers, which – based on results from UV-Vis spectroscopy and molecular modeling – were explained by differential placement of the pyrene moiety for affinity-enhancing intercalation.9b Similar structural underpinnings are likely in effect for monomers X–Z (vide infra). However, the electron density of the pyrene moieties, which differs between the three monomers, may also be a contributing factor to the differential duplex stabilization.
An interesting trend is observed for the doubly modified ONs. Thus, incorporation of a second N2′-PyMe X or N2′-PyCO Y monomer does not result in additionally increased thermal affinity against DNA targets, whereas a second incorporation of the more flexible N2′-PyAc Z monomer does (compare ΔTm for the B1–B3 and B4–B6 series, Table 1). We hypothesize that the former observation is due to violation of the ‘nearest neighbor exclusion principle’, which states that free intercalators - at most - bind to every second base pair of a DNA duplex due to limits in local expandability of duplexes.23 Extrapolating the principle to tethered intercalators and assuming 3′-intercalative binding modes of the pyrene moieties of monomers X–Z (vide infra), the resulting duplexes involving B3 and B6 would feature a localized region with two intercalators in an area defined by four base pairs, which is at the saturation threshold. We speculate that the greater flexibility of the N2′-PyAc Z monomer, allows the modified DNA duplexes to structurally compensate for any stress induced by the high intercalator density.
Interestingly, the modified ONs display rather different thermal denaturation characteristics with RNA targets. Thus, ONs modified with 2′-amino-α-L-LNA adenine monomer W display significantly higher affinity against RNA than DNA targets (ΔTm/modification between +1.5 and +7.0 °C, Table 2). Further, X- and Y-modified ONs display markedly lower thermal affinity toward RNA than DNA targets, with N2′-PyMe X-modified DNA:RNA duplexes displaying similar thermostability as unmodified reference duplexes (ΔTm/modification between −0.5 and +2.5 °C, Table 2) and N2′-PyCO Y-modified duplexes being moderately stabilized (ΔTm/modification between +1.5 and +6.5 °C, Table 2). N2′-PyAc Z-modified ONs also display lower affinity toward RNA than DNA targets, but result in more thermostable heteroduplexes (ΔTm/modification between +4.0 and +8.0 °C, Table 2). These differences may again reflect the greater flexibility of monomer Z, which allows the pyrene moieties to adopt more favorable positions for affinity-enhancing intercalation (vide infra). DNA-selective hybridization (Table S3) - as seen for X/Y/Z-modified ONs and ONs modified with the thymine counterparts9b - is often observed for ONs modified with intercalating moieties,24 as intercalators favor the B-type helix geometry of DNA:DNA duplexes over the more compressed A/B-type helix geometry of DNA:RNA duplexes.25
Table 2.
T m values of duplexes between B1–B6 and complementary RNA targets.a
| ON | Duplex | B = | ΔTm/°C
|
|||
|---|---|---|---|---|---|---|
| W | X | Y | Z | |||
| B1 | 5′-GTG BTA TGC | +2.5 | −1.0 | +1.5 | +4.0 | |
| R2 | 3′-CAC UAU ACG | |||||
|
| ||||||
| B2 | 5′-GTG ATB TGC | +2.5 | +1.0 | +5.0 | +4.5 | |
| R2 | 3′-CAC UAU ACG | |||||
|
| ||||||
| B3 | 5′-GTG BTB TGC | +3.0 | −1.5 | +4.5 | +11.0 | |
| R2 | 3′-CAC UAU ACG | |||||
|
| ||||||
| R1 | 5′-GUG AUA UGC | +4.5 | −0.5 | +2.5 | +6.5 | |
| B4 | 3′-CAC TBT ACG | |||||
|
| ||||||
| R1 | 5′-GUG AUA UGC | +7.0 | +2.5 | +6.5 | +8.0 | |
| B5 | 3′-CAC TAT BCG | |||||
|
| ||||||
| R1 | 5′-GUG AUA UGC | +7.0 | ±0.0 | +7.0 | +14.5 | |
| B6 | 3′-CAC TBT BCG | |||||
ΔTm = change in Tm relative to unmodified reference duplexes D1:R2 (Tm ≡ 26.0 °C) and R1:D2 (Tm ≡ 24.5 °C); for conditions of thermal denaturation experiments, see Table 1.
Thermal denaturation studies – mismatch discrimination
The binding specificity of centrally modified ONs was determined against DNA/RNA strands with mismatched nucleotides opposite of the W–Z monomers. W4 displays improved discrimination of mismatched DNA targets relative to reference strand D2, with the AG-mismatch being discriminated particularly well (Table 3). X4/Y4/Z4 display surprisingly efficient discrimination of mismatched DNA targets - and AG mismatches in particular (Table 3) - considering the likely intercalative binding mode of the pyrene moieties, which is known to typically decrease base pairing fidelity.9b,26 However, ONs with X/Y/Z monomers positioned as next-nearest neighbors display poor discrimination of centrally mismatched DNA target (B3 series, Table S4), indicating that modification patterns have a major influence on binding specificity. Discrimination of RNA mismatches is generally less efficient with W4/X4/Y4 than with reference ONs, but improved with Z4 (Table 4).
Table 3.
Discrimination of mismatched DNA targets by B4-series and reference ONs.a
| ON | Sequence | B = | DNA: 5′-GTG ABA TGC
|
|||
|---|---|---|---|---|---|---|
|
Tm/°C
|
ΔTm/°C
|
|||||
| T | A | C | G | |||
| D2 | 3′-CAC TAT ACG | 27.5 | −17.0 | −15.5 | −9.0 | |
| W4 | 3′-CAC TWT ACG | 27.5 | −20.0 | −17.0 | −16.0 | |
| X4 | 3′-CAC TXT ACG | 34.0 | −16.5 | −7.5 | −17.0 | |
| Y4 | 3′-CAC TYT ACG | 39.0 | −21.0 | −12.0 | −17.0 | |
| Z4 | 3′-CAC TZT ACG | 35.0 | −21.5 | −14.0 | −14.5 | |
For conditions of thermal denaturation experiments, see Table 1. Tm’s of fully matched duplexes are shown in bold. ΔTm = change in Tm relative to fully matched DNA:DNA duplex.
Table 4.
Discrimination of mismatched RNA targets by B4-series and reference ONs.a
| ON | Sequence | B = | RNA: 5′-GUG ABA UGC
|
|||
|---|---|---|---|---|---|---|
|
Tm/°C
|
ΔTm/°C
|
|||||
| U | A | C | G | |||
| D2 | 3′-CAC TAT ACG | 24.5 | −15.0 | −15.0 | −11.0 | |
| W4 | 3′-CAC TWT ACG | 29.0 | −13.5 | −13.0 | −11.5 | |
| X4 | 3′-CAC TXT ACG | 24.0 | −10.5 | −8.0 | −10.0 | |
| Y4 | 3′-CAC TYT ACG | 27.0 | −10.5 | −11.0 | −8.5 | |
| Z4 | 3′-CAC TZT ACG | 31.0 | <−19.0 | −15.5 | −14.5 | |
For conditions of thermal denaturation experiments, see Table 1. Tm’s of fully matched duplexes are shown in bold. ΔTm = change in Tm relative to fully matched RNA:DNA duplex.
Optical spectroscopy
UV-Vis absorption and steady-state fluorescence emission spectra of X/Y/Z-modified ONs and the corresponding duplexes with DNA/RNA targets were recorded to gain additional insight into the binding modes of the pyrene moieties of monomers X/Y/Z. Bathochromic shifts of pyrene absorption maxima, which are indicative of strong interactions between pyrenes and nucleobases,27 were generally observed upon hybridization with complementary DNA/RNA (Δλmax = 0–6 nm, Table 5, Figs. 3, S2 and S3), with the most pronounced increases being observed for duplex formation of -modified ONs with DNA. These results are consistent with intercalative binding modes for the pyrene moieties of monomers X/Y/Z and in agreement with our previous observations with the corresponding thymine analogues.9b
Table 5.
Absorption maxima in the 300–400 nm region for ONs modified with N2′-pyrene functionalized 2′-amino-α-L-LNA adenine monomers X/Y/Z in the presence or absence of complementary DNA/RNA targets.a
| ON | Sequence | B= |
λmax[Δλmax] (nm)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X
|
Y
|
Z
|
|||||||||
| SSP | +DNA | +RNA | SSP | +DNA | +RNA | SSP | +DNA | +RNA | |||
| B1 | 5′-GTG BTA TGC | 348 | 350 [+2] | 350 [+2] | 349 | 352 [+3] | 351 [+2] | 348 | 350 [+2] | 351 [+3] | |
| B2 | 5′-GTG ATB TGC | 348 | 350 [+2] | 349 [+1] | 349 | 353 [+4] | 351 [+2] | 350 | 351 [+1] | 351 [+1] | |
| B3 | 5′-GTG BTB TGC | 347 | 349 [+2] | 348 [+1] | 349 | 351 [+2] | 351 [+2] | 347 | 350 [+3] | 351 [+4] | |
| B4 | 3′-CAC TBT ACG | 348 | 350 [+2] | 348 [±0] | 347 | 353 [+6] | 352 [+5] | 348 | 351 [+3] | 351 [+3] | |
| B5 | 3′-CAC TAT BCG | 348 | 350 [+2] | 350 [+2] | 350 | 351 [+1] | 350 [±0] | 348 | 351 [+3] | 351[+3] | |
| B6 | 3′-CAC TBT BCG | 348 | 350 [+2] | 348 [±0] | 348 | 352 [+4] | 353 [+5] | 348 | 351 [+3] | 351 [+3] | |
Δλmax = change in absorption maximum relative to single stranded probe (SSP). Measurements were performed at 5 °C except for single-stranded X/Y-modified probes, which were recorded at room temperature. Buffer conditions are as for thermal denaturation experiments.
Figure 3.
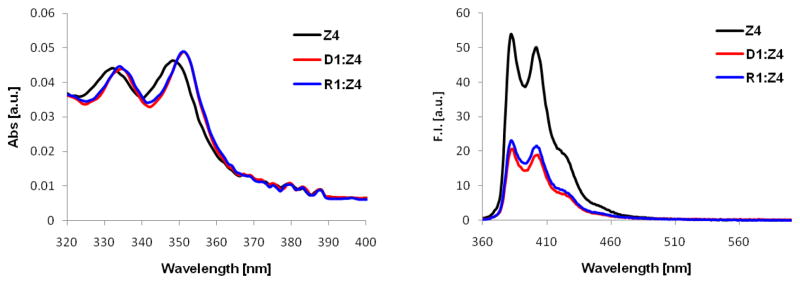
UV-vis absorption (left) and steady-state fluorescence emission (right) spectra of Z4 in the presence or absence of complementary DNA/RNA. Texp = 5 °C; each ON used at 1 μM concentration in Tm buffer; λex = 350 nm (fluorescence).
Steady-state fluorescence emission spectra were recorded at 5 °C using an excitation wavelength of λex = 350 nm. Hybridization of N2′-PyMe X-modified or N2′-PyAc Z-modified ONs with complementary DNA/RNA results in decreased emission (Figs. 3, S4 and S7), which is consistent with intercalation-induced quenching by flanking nucleobases.27,28 The resulting duplexes are only weakly fluorescent and exhibit defined vibronic bands at ~380 nm and ~400 nm along with a shoulder at 420–425 nm. On the other hand, a range of responses is observed upon hybridization of N2′-PyCO Y-modified ONs with complementary DNA/RNA, varying from ~50% reduction to ~3-fold enhancement of emission (Fig. S5). The resulting duplexes are strongly fluorescent (nearly two orders of magnitude more fluorescent than X-modified duplexes) with little or no vibronic fine structure (λem,max ~400 nm, broad band). The lack of a consistent response upon hybridization to DNA/RNA presumably is not due to different pyrene binding modes in the resulting duplexes but rather a reflection of different fluorescence intensities of the single-stranded probes, possibly due to different sequence-dependent rotational barriers around the Py-CO bond. Thus, singly modified DNA duplexes display similar emission intensities (Fig. S6), suggesting that the pyrene moieties are in similar microenvironments in the duplex which is consistent with an intercalative binding mode.
To further substantiate the proposed intercalative binding mode of N2′-PyCO monomer Y, we synthesized three additional Y-modified ONs in which the 3′-flanking nucleotide was varied systematically (Y7–Y9, Table 6). ONs with 3′-flanking purines display greater relative increases in thermal affinity against DNA targets than the corresponding ONs with 3′-flanking pyrimidines (compare ΔTm for Y7:D3 and Y9:D5 vs Y8:D4 and Y4:D1, Table 6). This is consistent with the proposed binding mode as 3′-intercalating pyrenes are expected to interact more strongly with large purines.
Table 6.
T m values for duplexes between Y4/Y7/Y8/Y9 and complementary DNA targets.a
| ON | Duplex | ΔTm/°C | reference Tm/°C |
|---|---|---|---|
|
|
|
|
|
| D3 | 5′-GTG TT ATGC | +17.0 | 27.5 |
| Y7 | 3′-CAC AY TACG | ||
|
|
|
|
|
| D4 | 5′-GTG GT ATGC | +10.0 | 33.0 |
| Y8 | 3′-CAC CY TACG | ||
|
|
|
|
|
| D5 | 5′-GTG CT ATGC | +19.5 | 24.0b |
| Y9 | 3′-CAC GY TACG | ||
|
|
|
|
|
| D1 | 5′-GTG AT ATGC | +11.5 | 27.5 |
| Y4 | 3′-CAC TY TACG | ||
ΔTm = change in Tm relative to unmodified reference duplex. For experimental conditions, see Table 1.
The low Tm of this reference duplex was confirmed by two independent operators using strands from three different commercial batches.
Steady-state fluorescence emission experiments provide further evidence for an intercalative binding mode of the pyrenes as the emission levels of these four DNA duplexes decrease in the anticipated order of nucleobase quenching efficiency (from least to most quenched: 3′-flanking A > T > C > G, Fig. 4).28b,29
Figure 4.
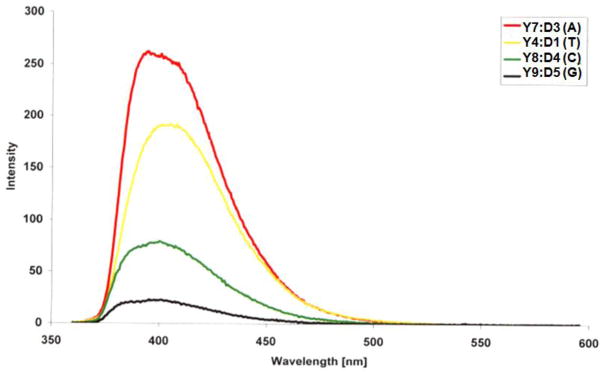
Fluorescence emission spectra of duplexes between Y4/Y7/Y8/Y9 and complementary DNA (nucleotide flanking the Y monomer on its 3′-side is listed in the parenthesis). Texp = 5 °C; each ON used at 0.15 μM concentration in Tm buffer; λex = 350 nm.
Conclusion
ONs modified with N2′-pyrene-functionalized 2′-amino-α-L-LNA adenine monomers X-Z display very high affinity toward DNA targets. The DNA-selective hybridization, together with hybridization-induced bathochromic shifts of pyrene absorption maxima and quenching of pyrene fluorescence, is indicative of intercalative binding modes for the pyrene moieties. ONs with such characteristics are likely to be of significant interest for applications in nucleic acid diagnostics and biotechnology.30 However, the synthesis of the corresponding phosphoramidites is very challenging due to non-trivial group manipulations (<1% overall yield from diacetone-α-D-glucose), which emphasizes the need for a more efficient synthetic route toward these building blocks or access to functional analogues of N2′-functionalized 2′-amino-αL-LNA monomers, which are easier to synthesize. In fact, we have already discovered the first examples of such analogues, i.e., N2′-pyrene-functionalized 2′-N-methyl-2′-amino-DNA and O2′-pyrene-functionalized RNA monomers,8a,12b and are exploring their use for applications in nucleic acid diagnostics.8b,8c
Experimental section
9-[2-O-Acetyl-3-O-benzyl-5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-α-L-threo-pentofuranosyl]-6-N-benzoyladenine (6)
6-N-benzoyladenine (28.6 g, 0.12 mol) and glycosyl donor 56b (40.6 g, 79.8 mmol) were co-evaporated with 1,2-dichloroethane (2×150 mL) and resuspended in anhydrous 1,2-dichloroethane (270 mL). To this was added N,O-bis(trimethylsilyl)acetamide (BSA, 49.2 mL, 0.20 mol) and the suspension was heated at reflux until turning homogenous. The solution was then cooled to rt, TMSOTf (43.1 mL, 0.24 mol) was slowly added, and the reaction mixture heated at reflux for 70h. The mixture was then cooled to rt and slowly poured into a solution of sat. aq. NaHCO3 and crushed ice (500 mL, 1:1, v/v). Additional crushed ice (~400 mL) was added and the mixture was stirred for 30 min. The resulting precipitate was removed via filtration and the filtrate was extracted with CH2Cl2 (1.5 L). The organic layer was washed with sat. aq. NaHCO3 (2×500 mL) and the combined aqueous layer back-extracted with CH2Cl2 (2×500 mL). The combined organic layer was evaporated to afford a crude residue, which was purified by silica gel column chromatography (0–10% i-PrOH in CH2Cl2, v/v) to provide nucleoside 6 (38.3 g, 70%) as a white foam. Rf = 0.4 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 712.1380 ([M+Na]+, C29H31N5O11S2·Na+, Calc. 712.1354). The observed 13C NMR data (75.5 MHz, CDCl3) are in good agreement with previously reported data for this compound.6b
9-[3-O-Benzyl-5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-α-L-threo-pentofuranosyl]-6-N-benzoyladenine (7)
Fully protected nucleoside 6 (4.66 g, 6.76 mmol) was dissolved in solution17 of guanidinium nitrate (4.91 g, 40.2 mmol) and NaOMe (0.24 g, 4.44 mmol) in MeOH:CH2Cl2 (450 mL, 9:1, v/v). The reaction mixture was stirred at rt for 30 min at which point sat. aq. NH4Cl (200 mL) was added. The resulting white precipitate was filtered off, washed with CH2Cl2, and the filtrate concentrated. The aqueous layer was extracted with CH2Cl2 (5×200 mL) and the combined organic layers were evaporated to near dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford alcohol 7 (3.84 g, 88%) as a white foam. Rf = 0.5 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 670.1234 ([M+Na]+, C27H29N5O10S2·Na+, Calc. 670.1248). The observed 13C NMR data (75.5 MHz, CDCl3) are in good agreement with previously reported data for this compound.6b
9-[2-C-Azido-3-O-benzyl-2-deoxy-4-C-methanesulfonyloxymethyl-5-O-methanesulfonyl-α-L-erythro-pentofuranosyl]-6-N-benzoyladenine (8)
Alcohol 7 (18.6 g, 28.7 mmol) was co-evaporated with anhydrous pyridine (50 mL) and dissolved in anhydrous CH2Cl2 (200 mL). The solution was cooled to −78 °C and anhydrous pyridine (7.3 mL, 90.6 mmol) was added, followed by dropwise addition of trifluoromethanesulfonyl anhydride (Tf2O, 9.90 mL, 58.9 mmol). The reaction mixture was allowed to warm to rt and stirred for 3h. At this point, crushed ice (50 mL) was added, and the layers were separated. The organic layer was washed with sat. aq. NaHCO3 (2×50 mL), evaporated to near dryness, and co-evaporated with abs. EtOH (2×50 mL) to afford the crude O2′-triflate as a lightly brown residue, which was used in the next step without further purification.
NaN3 (19.6 g, 0.30 mol) and 15-crown-5 (6.0 mL, 0.30 mol) were added to a solution of the crude O2′-triflate in anhydrous DMF (300 mL). The reaction mixture was first stirred at rt for 15h, then for an additional 8h at 50 °C. After cooling the mixture to rt, solids were filtered off and washed with EtOAc, and the combined organic layer was concentrated until near dryness. The concentrate was taken up in EtOAc (200 mL) and brine (200 mL), the layers were separated, and the aqueous layer extracted with EtOAc (4×100 mL). The combined organic layer was evaporated to near dryness and the resulting crude residue was purified by silica gel column chromatography (0–90% EtOAc in petroleum ether, v/v) to afford azide 8 (17.9 g, 89% over two steps) as a white solid material. Rf = 0.4 (EtOAc); IR (KBr): 2116 cm−1 (N3); MALDI-HRMS m/z 695.1295 ([M+Na]+, C27H28N8O9S2·Na+, Calc. 695.1313); 1H NMR (300 MHz, DMSO-d6) δ 11.30 (s, 1H, ex), 8.79 (s, 1H), 8.54 (s, 1H), 8.05 (d, 2H, J = 7.0 Hz), 7.30–7.67 (m, 8H), 6.74 (d, 1H, J = 4.4 Hz), 5.08–5.16 (m, 1H), 4.89 (d, 1H, J = 5.1 Hz), 4.74–4.83 (m, 2H), 4.69–4.72 (d, 1H, J = 11.4 Hz), 4.47–4.51 (d, 1H, J = 11.4 Hz), 4.42 (s, 2H), 3.28 (s, 3H), 3.24 (s, 3H); 13C NMR (75.5 MHz, DMSO-d6) δ 151.9, 150.4, 142.7, 136.9, 133.2, 132.5, 128.5, 128.0, 81.9, 81.7, 80.1, 73.5, 68.3, 61.9, 36.98, 36.94. The carbonyl group of the 6-N-benzoyl group was not visible.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-benzyloxy-1-methanesulfonyloxymethyl-2-oxa-5-azabicyclo[2.2.1]heptane (9)
Aqueous NaOH (2M, 38.9 mL, 77.8 mmol) and trimethylphosphine (1M in THF, 77.8 mL, 77.8 mmol) were added to an ice-cold solution of azido nucleoside 8 (35.2 g, 51.8 mmol) in THF (500 mL). The reaction mixture was allowed to warm up to rt and was stirred at this temperature for 21h. The mixture was then evaporated to near dryness and the resulting crude residue taken up in EtOAc (200 mL) and brine (200 mL). After separating the layers, the aqueous layer was extracted with MeOH:CH2Cl2 (3×200 mL, 2:8, v/v). The combined organic layer was evaporated to dryness, and the resulting crude purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to provide bicyclic nucleoside 9 (24.2 g, 85%) as a solid brown material. Rf = 0.3 (EtOAc); MALDI-HRMS m/z 573.1517 ([M+Na]+, C26H26N6O6S·Na+, Calc. 573.1527); 1H NMR31 (500 MHz, DMSO-d6) δ 11.17 (s, 1H, ex, NH), 8.77 (s, 1H, H8), 8.73 (s, 1H, H2), 8.06 (d, 2H, J = 7.0 Hz, Bz), 7.54–7.67 (m, 3H, Bz), 7.29–7.47 (m, 5H, Ph), 6.52 (d, 1H, J = 1.8 Hz, H1′), 4.72–4.76 (d, 1H, J = 11.7 Hz, CH2Ph), 4.62–4.67 (d, 1H, J = 11.7 Hz, CH2Ph), 4.57–4.60 (d, 1H, J = 11.7 Hz, H5′a), 4.49–4.53 (d, 1H, J = 11.7 Hz, H5′b), 4.45 (s, 1H, H3′), 3.93 (br s, 1H, H2′), 3.28–3.31 (m, 1H, H5″a, partial overlap with H2O), 3.22 (s, 3H, CH3SO2), 3.10–3.13 (d, 1H, J = 9.9 Hz, H5″b); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 152.1, 151.4 (C2), 150.0, 143.1 (C8), 137.8, 133.3, 132.3 (Bz), 128.4 (Ar), 128.2 (Ar), 127.62 (Ar), 127.60 (Ar), 125.1, 87.2, 84.3 (C1′), 80.4 (C3′), 71.0 (CH2Ph), 66.8 (C5′), 59.8 (C2′), 51.1 (C5″), 36.8 (CH3SO2). The 1H and 13C NMR data are in reasonable agreement with previously reported data from the patent literature.18 The 2-oxo-5-azabicyclo[2.2.1]heptane skeleton and stereochemistry of 9 was verified via NOE experiments on downstream product 14.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-benzyloxy-1-methanesulfonyloxymethyl-5-trifluoroacetyl-2-oxa-5-azabicyclo[2.2.1]heptane (11)
Bicyclic nucleoside 9 (8.68 g, 15.8 mmol) was co-evaporated with pyridine (2×20 mL) and dissolved in anhydrous CH2Cl2 (200 mL) and anhydrous pyridine (5.09 mL, 63 mmol). The solution was cooled to 0 °C and trifluoroacetic acid anhydride (4.45 mL, 31.5 mmol) was added. The reaction mixture was stirred for 2 h at 0 °C, at which point crushed ice (50 mL) was added. The layers were separated and the organic layer was washed with sat. aq. NaHCO3 (2×50 mL). The combined aqueous layer was back-extracted with CH2Cl2 (2×100 mL) and MeOH:CH2Cl2 (100 mL, 2:8, v/v), and the combined organic layer evaporated to near dryness. The resulting crude residue was sequentially co-evaporated with toluene (50 mL) and abs. EtOH:toluene (50 mL, 1:1, v/v) and purified by silica gel column chromatography (0–100% EtOAc in petroleum ether, v/v) to afford fully protected nucleoside 11 (6.27 g, 62%) as a white foam. Rf = 0.5 (10% MeOH:EtOAc, v/v). Physical data for mixture of rotamers (~4:6 by 1H NMR): MALDI-HRMS m/z 647.1541 ([M+H]+, C28H25F3N6O7S·H+, Calc. 647.1530); 1H NMR31,32 (500 MHz, DMSO-d6) δ 11.21 (s, 0.6H, ex, NHB), 11.19 (s, 0.4H, ex, NHA), 8.78 (s, 0.6H, H2B), 8.76 (s, 0.4H, H2A), 8.63 (s, 0.4H, H8A), 8.60 (s, 0.6H, H8B), 8.05 (d, 2H, J = 7.0 Hz, Bz-A+B), 7.52–7.67 (m, 3H, Bz-A+B), 7.31–7.41 (m, 5H, Ph-A/B), 6.83 (d, 0.6H, J = 1.1 Hz, H1′B), 6.80 (d, 0.4H, J = 1.1 Hz, H1′A), 5.26 (s, 0.4H, H2′A), 5.17 (s, 0.6H, H2′B), 4.84 (s, 0.6H, H3′B), 4.82 (s, 0.4H, H3′A), 4.62–4.79 (m, 4H, CH2Ph-A+B, H5′A+B), 4.53 (d, 0.4H, J = 10.6 Hz, H5″A), 4.35 (d, 0.6H, J = 12.1 Hz, H5″B), 4.07 (d, 0.4H, J = 10.6 Hz, H5″A), 3.91 (d, 0.6H, J = 12.1 Hz, H5″B), 3.28 (s, 3H, CH3SO2); 13C NMR (125 MHz, DMSO-d6) δ 165.49, 165.48, 155.3 (q, J = 37 Hz, COCF3), 155.0 (q, J = 37 Hz, COCF3), 151.8 (C2B), 151.65 (C2A), 151.59, 150.4, 150.3, 141.2 (C8B), 141.0 (C8A), 137.2, 137.0, 133.3, 132.4 (Bz), 128.43 (Bz), 128.40 (Ar), 128.39 (Ar), 128.38 (Ar), 128.34 (Ph), 127.91 (Ph), 127.88 (Ph), 127.53 (Ph), 127.51 (Ph), 125.5, 125.3, 115.5 (q, J = 288 Hz, CF3), 115.2 (q, J = 288 Hz, CF3), 86.16, 86.15, 84.9 (C1′A), 84.0 (C1′B), 79.2 (C3′B), 77.4 (C3′A), 71.6 (CH2Ph), 65.3 (C5′), 65.0 (C5′), 63.0 (C2′B), 61.4 (C2′A), 53.2 (C5″B), 53.1 (C5″A), 37.0 (CH3SO2); 19F NMR (376 MHz, DMSO-d6) δ −71.3 (CF3-B), −72.1 (CF3-A).
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-hydroxy-1-methanesulfonyloxymethyl-5-trifluoroacetyl-2-oxa-5-azabicyclo[2.2.1]heptane (12)
Fully protected nucleoside 11 (19.6 g, 30.4 mmol) was co-evaporated with 1,2-dichloroethane (3×100 mL) and dissolved in anhydrous CH2Cl2 (600 mL). The solution was cooled to −78 °C and BCl3 (1M solution in hexanes, 370 mL, 0.37 mol) was added. The reaction mixture was allowed to warm to rt and was stirred for 17h. The mixture was then cooled to 0 °C and crushed ice (800 mL) was slowly added. The layers were separated and the organic phase was washed with sat. aq. NaHCO3 (2×300 mL). The combined aqueous layer was back-extracted with EtOAc (4×500 mL) and the combined organic phase was evaporated to near dryness. The resulting residue was purified using silica gel column chromatography (0–20% MeOH in CH2Cl2, v/v) to afford alcohol 12 (14.7 g, 87%) as a white solid material. Rf = 0.3 (50% acetone in CH2Cl2, v/v). Physical data for mixture of rotamers (~4:6 by 1H NMR): MALDI-HRMS m/z 579.0871 ([M+Na]+, C21H19F3N6O7S·Na+, Calc. 579.0880); 1H NMR31,32 (500 MHz, DMSO-d6) δ 11.21 (s, 1H, ex, NHA+B), 8.76 (s, 0.6H, H2B), 8.75 (s, 0.4H, H2A), 8.60 (s, 0.4H, H8A), 8.56 (s, 0.6H, H8B), 8.04 (d, 2H, J = 7.7 Hz, BzA+B), 7.48–7.66 (m, 3H, BzA+B), 6.83 (d, 0.6H, J = 1.4 Hz, H1′B), 6.80 (d, 0.4H, J = 1.4 Hz, H1′A), 6.72 (d, 0.6H, ex, J = 4.1 Hz, 3′-OHB), 6.68 (d, 0.4H, ex, J = 4.1 Hz, 3′-OHA), 4.91 (br s, 0.4H, H2′A), 4.82 (d, 0.6H, J = 4.1 Hz, H3′B), 4.76 (d, 0.4H, J = 4.1 Hz, H3′A), 4.70 (br s, 0.6H, H2′B), 4.60–4.67 (m, 2H, H5′A+B, 4.46 (d, 0.4H, J = 10.3 Hz, H5″A), 4.26 (d, 0.6H, J = 11.7 Hz, H5″B), 4.03 (d, 0.6H, J = 10.3 Hz, H5″A), 3.86 (d, 0.5H, J = 11.7 Hz, H5″B), 3.29 (s, 3H, CH3SO2); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 155.3 (q, J = 36.6 Hz, COCF3), 155.1 (q, J = 36.6 Hz, COCF3), 151.7 (C2A/B), 151.6 (C2A/B), 150.3, 150.2, 141.2 (C8B), 141.0 (C8A), 132.4 (Bz), 128.44 (Bz), 128.42 (Bz), 128.39 (Bz), 128.38 (Bz), 125.5, 125.3, 115.5 (q, J = 288 Hz, CF3), 115.2 (q, J = 288 Hz, CF3), 87.0, 85.8, 84.8 (C1′A), 83.9 (C1′B), 72.5 (C3′B), 70.7 (C3′A), 65.7 (C5′), 65.4 (C5′), 65.3 (C2′B), 63.7 (C2′A), 52.8 (C5″B), 52.6 (C5″A), 37.0 (CH3SO2); 19F NMR (376 MHz, DMSO-d6) δ −71.1 (CF3-B), −72.1 (CF3-A).
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-benzoyloxymethyl-7-hydroxy-5-trifluoroacetyl-2-oxa-5-azabicyclo[2.2.1]heptane (13)
NaOBz (2.99 g, 20.8 mmol) and 15-crown-5 (2.07 mL, 10.4 mmol) were added to a solution of alcohol 12 (5.79 g, 10.4 mmol) in anhydrous DMF (100 mL). The reaction mixture was first stirred at 90 °C for 5h and then at rt for additional 18h. The mixture was concentrated to nearly dryness and taken up in EtOAc and brine. The layers were separated and the aqueous layer was extracted with EtOAc (4×200 mL). The combined organic layer was evaporated to near dryness, and the resulting residue purified by silica gel column chromatography (0–3.5% i-PrOH in CHCl3, v/v) to afford O5′-benzoylated nucleoside 13 (5.05 g, 83%) as a white foam. Rf = 0.4 (10% i-PrOH in CHCl3, v/v). Physical data for the mixture of rotamers (~4.5:5.5 by 1H NMR): MALDI-HRMS m/z 605.1337 ([M+Na]+, C27H21F3N6O6·Na+ Calc. 605.1367); 1H NMR31,32 (500 MHz, DMSO-d6) δ 11.21 (br s, 1H, ex, NHA+B), 8.69 (s, 1H H2A+B), 8.59 (s, 0.45H, H8A), 8.54 (s, 0.55H, H8B), 8.05–8.13 (m, 4H, BzA+B), 7.49–7.72 (m, 6H, BzA+B), 6.86 (d, 0.55H, J = 1.7 Hz, H1′B), 6.83 (d, 0.45H, J = 1.7 Hz, H1′A), 6.60–6.80 (br s, 1H, ex, 3′-OHA+B), 4.93 (s, 0.55 H, H3′B), 4.89–4.91 (m, 0.9H, H2′A, H3′A), 4.74–4.79 (m, 1.0H, H5′A+B), 4.68 (br s, 0.55H, H2′B), 4.58–4.64 (m, 1.0H, H5′A+B), 4.54 (d, 0.45H, J = 10.7 Hz, H5″A), 4.35 (d, 0.55H, J = 11.5 Hz, H5″B), 4.10 (d, 0.45H, J = 10.7 Hz, H5″A), 3.93 (d, 0.55H, J = 11.5 Hz, H5″B); 13C NMR (125 MHz, DMSO-d6) δ 166.5, 165.3, 155.1 (2q, J = 36 Hz, COCF3), 151.8 (C2A/B), 151.6 (C2A/B), 151.4, 140.6 (C8A/B), 140.4 (C8A/B), 134.5, 134.4, 133.6 (Bz), 131.9 (Bz), 129.55 (Bz), 129.54 (Bz), 129.18, 129.17, 128.7 (Bz), 128.4 (Bz), 128.2 (Bz), 125.5, 125.3, 115.5 (2q, CF3, J = 286 MHz, CF3), 87.4, 86.0, 84.5 (C1′A), 83.7 (C1′B), 72.9 (C3′B), 71.0 (C3′A), 65.3 (C2′B), 63.7 (C2′A), 60.7 (C5′), 60.2 (C5′), 53.0 (C5″B), 52.9 (C5″A) – minor impurities were observed at 121.3, 119.9 and 79.1 ppm; 19F NMR (376 MHz, DMSO-d6) δ −71.1 (CF3-B), −72.0 (CF3-A).
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-hydroxy-1-hydroxymethyl-2-oxa-5-azabicyclo[2.2.1]heptane (14)
From 13: Aqueous NaOH (2M, 17.5 mL, 0.35 mol) was added to an ice-cold solution of alcohol 13 (3.41 g, 5.85 mmol) in 1,4-dioxane and water (225 mL, 2:1, v/v). The reaction mixture was stirred at 0 °C for 2h, at which point sat. aq. NH4Cl (25 mL) was added. The solution was evaporated to dryness, and the resulting crude was adsorbed on silica gel and purified by silica gel column chromatography (0–20% MeOH in CH2Cl2, v/v) to afford amino alcohol 14 (1.33 g, 60%) as a white solid material. Rf = 0.4 (20% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 405.1294 ([M+Na]+, C18H18N6O4·Na+, Calc. 405.1282); 1H NMR (300 MHz, DMSO-d6) δ 11.17 (br s, 1H, ex, NH), 8.73 (s, 1H, ABz), 8.71 (s, 1H, ABz), 8.05 (d, 2H, J = 8.1 Hz, Bz), 7.49–7.67 (m, 3H, Bz), 6.44 (d, 1H, J = 1.9 Hz, H1′), 5.70 (d, 1H, ex, J = 4.0 Hz, 3′-OH), 4.82 (t, 1H, ex, J = 5.5 Hz, 5′-OH), 4.30 (d, 1H, J = 4.0 Hz, H3′), 3.69 (d, 2H, J = 5.5 Hz, H5′), 3.51 (s, 1H, H2′), 3.15–3.20 (d, 1H, J = 10.6 Hz, H5″), 2.94–3.01 (d, 1H, J = 10.6 Hz, H5″); 13C NMR (75.5 MHz, DMSO-d6) δ 165.5, 152.0, 151.2, 149.8, 143.2, 133.3, 132.3, 128.4, 125.2, 91.3, 83.9, 73.4, 62.2 (C5″), 58.3 (C2′), 50.6 (C5′).
From 16: Nucleoside 16 (1.00 g, 1.01 mmol) was dissolved in a solution of CHCl2COOH/MeOH/CH3NO2 (50 mL, 3:5:92, v/v/v) and stirred at 0 °C for 15 min. Sat. aq. NaHCO3 was carefully added to neutralize the solution and the mixture was evaporated to dryness and adsorbed on silica gel. The resulting residue was purified by silica gel column chromatography (0–20% MeOH in CH2Cl2, v/v - initially built including 1% Et3N) to afford amino diol 14 as a white solid material (0.37 g, 96%).
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2-oxa-5-azabicyclo[2.2.1]heptane (15)
Amino alcohol 14 (1.25 g, 3.27 mmol) was co-evaporated with anhydrous pyridine (30 mL) and redissolved in anhydrous pyridine (65 mL). The solution was cooled using a ice-salt mixture and DMTrCl (1.55 g, 4.58 mmol) was added in one portion (addition over several portions did not influence reaction outcome). The reaction mixture was warmed to rt and stirred for 23 h, at which point MeOH (10 mL) was added. The mixture was diluted with EtOAc (100 mL) and washed with sat. aq. NaHCO3 (2×15 mL). The combined aqueous layer was back-extracted with EtOAc (3×30 mL) and the combined organic phase was evaporated to dryness and subsequently co-evaporated with abs. EtOH:toluene (3×50 mL, 2:1, v/v). The resulting residue was purified by silica gel column chromatography (0–8% MeOH in CH2Cl2, v/v - initially built with 0.5% Et3N, v/v) and subsequently co-evaporated with abs. EtOH:toluene (2×50 mL, 1:1, v/v) to afford nucleoside 15 (0.85 g, 38%) as a white solid material, along with nucleoside 16 (0.97 g, 30%) as a yellow foam. Physical data for 15: Rf = 0.4 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 707.2609 ([M+Na]+, C39H36N6O6·Na+, Calc. 707.2589); 1H NMR31 (500 MHz, DMSO-d6) δ 11.16 (br s, 1H, ex, NH), 8.78 (s, 1H, H8), 8.73 (s, 1H, H2), 8.06 (d, 2H, J = 7 Hz, Bz), 7.63–7.67 (t, 1H, J = 7.7 Hz, Bz), 7.54–7.58 (d, 2H, J = 7.0 Hz, Bz), 7.19–7.44 (m, 9H, DMTr), 6.86–6.92 (m, 4H, DMTr), 6.54 (d, 1H, J = 1.9 Hz, H1′), 5.66 (d, 1H, ex, J = 4.8 Hz, 3′-OH), 4.40 (d, 1H, J = 4.8 Hz, H3′), 3.74 (s, 6H, CH3O), 3.50 (br s, 1H, H2′), 3.30–3.32 (m, 1H, H5′–overlap with H2O), 3.25–3.27 (d, 1H, J = 10.7 Hz, H5′), 3.19 (br s, 2H, H5″), 2.90 (br s, 1H, ex, 2′-NH); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 158.0, 152.2, 151.3 (C2), 149.9, 144.8, 143.3 (C8), 135.5, 135.4, 133.4, 132.3 (Ar), 129.71 (Ar), 129.68 (Ar), 128.45, 128.39 (Ar), 127.8 (Ar), 127.7 (Ar), 126.6 (Ar), 125.2, 113.2 (DMTr), 89.6, 85.1, 84.1 (C1′), 74.0 (C3′), 62.2 (C2′), 61.2 (C5′), 55.0 (CH3O), 51.2 (C5″).
Physical data for (1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-5-(4,4′-dimethoxytrityl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2-oxa-5-azabicyclo[2.2.1]heptane (16)
Rf = 0.5 (3% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 1009.3900 ([M+Na]+, C60H54N6O8·Na+, Calc. 1009.3895); 1H NMR31 (500 MHz, DMSO-d6) δ 11.33 (br s, 1H, ex, NH), 9.13 (s, 1H, H8), 8.77 (s, 1H, H2), 8.10 (d, 2H, J = 7.5 Hz, Bz), 7.64–7.67 (t, 1H, J = 7.5 Hz, Bz), 7.52–7.58 (t, 2H, J = 7.5 Hz, Bz), 6.78–7.41 (m, 22H, DMTr), 6.58 (d, 1H, J = 1.1 Hz, H1′), 6.53 (d, 2H, J = 9.1 Hz, DMTr), 6.43 (d, 2H, J = 9.1 Hz, DMTr), 4.26 (d, 1H, J = 5.0 Hz, H3′), 3.82 (d, 1H, J = 9.9 Hz, H5″), 3.75 (br s, 1H, H2′), 3.73 (s, 6H, CH3O), 3.65 (s, 3H, CH3O), 3.62 (s, 3H, CH3O), 3.55 (d, 1H, ex, J = 5.0 Hz, 3′-OH), 3.17–3.19 (d, 1H, J = 11.0 Hz, H5′), 3.09–3.11 (d, 1H, J = 11.0 Hz, H5′), 2.98 (d, 1H, J = 9.9 Hz, H5″); 13C NMR (125 MHz, DMSO-d6) δ 165.7, 158.0, 157.05, 156.96, 152.0, 151.6 (C2), 150.3, 145.2, 144.8, 142.9 (C8), 136.7, 136.2, 136.0, 135.4, 135.3, 133.3, 132.4 (Bz), 130.5 (DMTr), 130.2 (DMTr), 129.8 (DMTr), 129.7 (DMTr), 128.8 (DMTr), 128.44 (Bz), 128.39 (Bz), 127.73 (DMTr), 127.70 (DMTr), 127.1 (DMTr), 126.5 (DMTr), 126.3, 125.5 (DMTr), 123.8 (DMTr), 113.1 (DMTr), 112.5 (DMTr), 112.4 (DMTr), 88.4, 86.6 (C1′), 85.2, 74.3, 74.0 (C3′), 64.3 (C2′), 60.7 (C5′), 55.0 (C5″), 54.9 (CH3O), 54.8 (CH3O), 54.75 (CH3O), 54.73 (CH3O). A trace impurity of pyridine was identified in the 13C NMR spectrum at 149.5 ppm.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-5-(9′-fluorenylmethoxycarbonyl)-7-hydroxy-2-oxa-5-azabicyclo[2.2.1]heptane (17)
Amino alcohol 15 (200 mg, 0.29 mmol) was co-evaporated in anhydrous pyridine (2×2 mL) and re-dissolved in anhydrous pyridine (1.5 mL). The solution was cooled to 0 °C and 9′-fluorenylmethyl chloroformate (100 mg, 0.38 mmol) added hereto. The reaction mixture was warmed to rt and stirred for 6h, at which point it was diluted with EtOAc (30 mL) and washed with sat. aq. NaHCO3 (30 mL). The aqueous layer was back-extracted with EtOAc (25 mL) and the combined organic layer evaporated to near dryness. The resulting crude was co-evaporated with abs. EtOH:toluene (2 × 6 mL, 2:1, v/v) and purified by silica gel column chromatography (30–100% EtOAc in petroleum ether, v/v) to afford target nucleoside 17 (136 mg, 51%) as a white foam. Rf = 0.4 (EtOAc). Physical data for mixture of rotamers (~1:1.2 by 1H NMR): MALDI-HRMS m/z 929.3241 ([M + Na]+, C54H46N6O8·Na+ Calc. 929.3269); 1H NMR (300 MHz, DMSO-d6) δ 11.29 (br s, ex), 8.74 (s, 1H), 8.72 (s, 1.2H), 8.55 (s, 1.2H), 8.49 (s, 1H), 6.90–8.08 (m, 57.2H), 6.80 (d, 1.2H, J = 1.7 Hz), 6.73 (d, 1H, J = 1.7 Hz), 6.25 (d, 1.2H, ex, J = 4.4 Hz), 6.22 (d, 1H, ex, J = 4.4 Hz), 4.50 (br s, 1.2H), 4.45 (br s, 1H), 4.22 (d, 1H, J = 6.9 Hz), 4.15 (d, 1.2H, J = 6.9 Hz), 3.87 (d, 1H, J = 6.9 Hz), 3.83 (d, 1.2H, J = 6.9 Hz), 3.62–3.76 (m, 17.6H), 3.34–3.43 (m, 6.6H); 13C NMR (75.5 MHz, DMSO-d6) δ 165.66, 165.63, 158.1, 154.7, 154.6, 151.8, 151.5, 150.3, 150.2, 144.7, 143.7, 143.6, 143.5, 142.8, 141.3, 141.2, 140.6, 140.4, 140.3, 135.3, 135.2, 133.3, 132.4, 129.7, 128.8, 128.4, 127.9, 127.7, 127.6, 127.5, 127.2, 127.1, 126.9, 126.7, 125.4, 125.2, 125.1, 124.9, 124.6, 121.3, 119.98, 113.2, 88.9, 88.4, 85.4, 84.9, 84.7, 84.6, 72.5, 72.1, 66.8, 66.7, 63.9, 63.4, 60.6, 60.5, 55.0, 52.7, 52.6, 46.4, 45.9.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-5-(pyren-1-yl)methyl-2-oxa-5-azabicyclo[2.2.1]heptane (18)
Nucleoside 15 (225 mg, 0.33 mmol) was co-evaporated with anhydrous 1,2-dichloroethane (2×5 mL) and re-dissolved in anhydrous 1,2-dichloroethane (4 mL). 1-Pyrenecarboxaldehyde (115 mg, 0.49 mmol) and NaBH(OAc)3 (104 mg, 0.49 mmol) were added, and the resulting suspension was stirred at rt for 17 h, at which point sat. aq. NaHCO3 (30 mL) was added. The mixture was extracted with CH2Cl2 (3×15 mL) and the combined organic layers dried (Na2SO4) and evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford N2′-alkylated nucleoside 18 as a white foam (201 mg, 68%). Rf = 0.4 (EtOAc); MALDI-HRMS m/z 921.3326 ([M+Na]+, C56H46N6O6·Na+, Calc. 921.3371); 1H NMR31 (500 MHz, DMSO-d6) δ 11.18 (s, 1H, ex, NH), 8.53 (s, 1H, H2), 8.46 (s, 1H, H8), 7.97–8.24 (m, 8H, Ar), 7.60–7.85 (m, 6H, Ar), 7.41–7.44 (m, 2H, DMTr), 7.19–7.32 (m, 7H, DMTr), 6.85–6.92 (m, 4H, DMTr), 6.50 (d, 1H, J = 1.9 Hz, H1′), 6.14 (d, 1H, ex, J = 3.6 Hz, 3′-OH), 4.72–4.75 (d, 1H, J = 12.5 Hz, CH2Py), 4.60 (d, 1H, J = 3.6 Hz, H3′), 4.52–4.56 (d, 1H, J = 12.5 Hz, CH2Py), 3.73 (s, 6H, CH3O), 3.66 (s, 1H, H2′), 3.34–3.45 (m, 3H, 2× H5′, H5″), 3.17–3.20 (d, 1H, J = 10.3 Hz, H5″); 13C NMR (125 MHz, DMSO-d6) δ 165.6, 158.0, 151.7, 151.0 (C2), 149.9, 144.8, 142.8 (C8), 135.5, 135.4, 133.6, 133.2, 132.4 (Ar), 130.6, 130.2, 130.0, 129.71 (DMTr), 129.68 (DMTr), 128.7, 128.5 (Ar), 128.4 (Ar), 127.8 (DMTr), 127.7 (DMTr), 127.6 (Ar), 127.2 (Ar), 126.9 (Ar), 126.8 (Ar), 126.6 (DMTr), 125.9 (Ar), 125.5, 124.9 (Ar), 124.8 (Ar), 124.2 (Ar), 123.9, 123.7, 123.1 (Ar), 113.2 (DMTr), 90.3, 85.2, 84.8 (C1′), 75.4 (C3′), 66.3 (C2′), 61.3 (C5′), 59.2 (C5″), 58.2 (CH2Py), 55.0 (CH3O).
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-5-(pyren-1-yl)carbonyl-2-oxa-5-azabicyclo[2.2.1]heptane (19)
Amino alcohol 15 (0.41 g, 0.59 mmol) was co-evaporated with anhydrous 1,2-dichloroethane (2 × 10 mL), dissolved in anhydrous CH2Cl2 (11.9 mL), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochlorid (EDC·HCl, 226 mg, 1.19 mmol) and 1-pyrenecarboxylic acid (0.29 g, 1.19 mmol) added hereto. The reaction mixture was stirred for 45h at rt, at which point it was diluted with CH2Cl2 (50 mL) and washed with water (20 mL). The two phases were separated and the aqueous phase was back-extracted with CH2Cl2 (3 × 50 mL). The combined organic phase was evaporated to dryness and the resulting residue was purified by silica gel column chromatography (0–99% EtOAc and 1% pyridine in petroleum ether, v/v). The resulting product was co-evaporated with abs. EtOH:toluene (2 × 50 ml, 1:1, v/v) to afford nucleoside 19 (343 mg, 64%) as a yellow solid material. Physical data for the mixture of rotamers (~0.4:1 by 1H NMR): Rf: 0.5 (5% MeOH: CH2Cl2, v/v); MALDI-HRMS m/z 935.3138 ([M+Na]+, C56H44N6O7·Na+ Calc. 935.3164). 1H NMR31,32 (300 MHz, DMSO-d6) δ 11.21 (br s, 1.4H, ex, NH), 8.55 (s, 1H, ABz-B), 8.53 (s, 0.4H, ABz-A), 8.45 (s, 1H, ABz-B), 8.40 (s, 0.4H, ABz-A), 6.82–8.23 (m, ~37.8H, ArA+B), 6.50 (d, 1H, J = 2.2 Hz, H1′B), 6.40 (d, 0.4H, J = 1.8 Hz, H1′A), 6.15 (d, 1H, ex, J = 4.0 Hz, 3′-OHB), 6.07 (d, 0.4H, ex, J = 3.7 Hz, 3′-OHA), 4.42–4.77 (m, 4.2H, H3′A+B, H5′A+B), 3.55–3.75 (m, ~9.8H, CH3OA+B, H2′A+B), 3.15–3.44 (m, ~2.8H, H5″A+B); 13C NMR (75.5 MHz, DMSO-d6) δ 165.5, 157.9, 157.7, 151.7, 150.98, 149.8, 148.2, 144.7, 142.8, 140.1, 135.4, 135.3, 133.5, 133.1, 132.3, 130.5, 130.2, 129.9, 129.6, 128.8, 128.6, 128.4, 127.8, 127.6, 127.5, 127.3, 127.2, 126.9, 126.5, 126.3, 125.9, 125.5, 124.9, 124.8, 124.78, 124.2, 123.8, 123.7, 123.1, 113.1, 112.7, 92.0, 90.3, 85.1, 84.7, 79.8, 75.3, 74.7, 66.4, 61.2, 59.1, 58.6, 58.3, 58.2, 54.6.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-5-(pyren-1-yl)acetyl-2-oxa-5-azabicyclo[2.2.1]heptane (20)
Amino alcohol 15 (0.25 g, 0.37 mmol) was co-evaporated with anhydrous 1,2-dichloroethane (2×5 mL) and dissolved in anhydrous CH2Cl2 (10 mL). To this was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 108 mg, 0.55 mmol) and 1-pyreneacetic acid (145 mg, 0.55 mmol). The reaction mixture was stirred for 2.5h at rt, at which point it was diluted with CH2Cl2 (20 mL) and washed with water (2 ×10 mL). The aqueous layer was back-extracted with CH2Cl2 (10 mL) and the combined organic layer evaporated to dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 20 as a white solid (0.27 g, 79%). Physical data for the mixture of rotamers (~0.5:1.0 by 1H NMR): Rf = 0.5 (10% MeOH:CH2Cl2, v/v); MALDI-HRMS m/z 949.3321 ([M+Na]+, C57H46N6O7·Na+, Calc. 949.3320); 1H NMR31,32 (500 MHz, DMSO-d6) δ 11.29 (br s, 1.5H, ex, NHA+B), 8.97 (s, 1H, H8B), 8.73 (s, 0.5H, H2A), 8.71 (s, 1H, H2B), 8.55 (s, 0.5H, H8A), 6.91–8.32 (m, 40.5H, ArA+B), 6.83 (d, 0.5H, J = 1.7 Hz, H1′A), 6.80 (d, 1H, J = 1.4 Hz, H1′B), 6.32 (d, 0.5H, ex, J = 4.4 Hz, 3′-OHA), 6.19 (d, 1H, ex, J = 4.4 Hz, 3′-OHB), 5.07 (s, 0.5H, H2′A), 4.71–4.73 (m, 1.5H, H2′B, H3′A), 4.67 (d, 1H, J = 10.4 Hz, H5″B), 4.63 (d, 1H, J = 4.4 Hz, H3′B), 4.55–4.60 (d, 1H, J = 17.0 Hz, CH2Py-B), 4.36–4.40 (d, 1H, J = 17.0 Hz, CH2Py-B), 4.11 (d, 0.5H, J = 16.2 Hz, CH2Py-A), 3.96–4.03 (m, 1.5H, H5″A, H5″B), 3.66–3.77 (m, 9.5H, CH3O, H5″A), 3.34–3.48 (m, 3.5H, H5′A+B, CH2Py-A); 13C NMR (125 MHz, DMSO-d6) δ 169.9, 169.6, 165.6, 165.5, 158.1, 158.0, 152.0, 151.7 (ABz-A), 151.5, 151.4 (ABz-B), 150.6, 150.2, 144.71, 144.68, 141.7 (ABz-B), 141.5 (ABz-A), 135.4, 135.2, 133.54, 133.47, 132.5 (Ar), 130.70, 130.69, 130.28, 130.1, 129.91, 129.88, 129.82 (Ar), 129.79 (Ar), 129.77, 129.74 (Ar), 129.68, 129.62, 129.4, 129.2, 128.8, 128.7, 128.58 (Ar), 128.56 (Ar), 128.54 (Ar), 128.46 (Ar), 128.2 (Ar), 127.85 (Ar), 127.77 (Ar), 127.72 (Ar), 127.4 (Ar), 127.27 (Ar), 127.2 (Ar), 127.1 (Ar), 126.9, 126.8 (Ar), 126.68 (Ar), 126.2, 126.0 (Ar), 125.9 (Ar), 125.4, 125.3, 125.00 (Ar), 124.98 (Ar), 124.82 (Ar), 124.76 (Ar), 124.7 (Ar), 124.6 (Ar), 124.5 (Ar), 124.4 (Ar), 124.3 (Ar), 124.1, 123.89, 123.79, 123.76, 123.72, 123.2 (Ar), 113.2 (Ar), 89.0, 88.4, 85.5, 85.4, 84.73 (C1′A), 84.66 (C1′B), 72.9 (C3′A), 72.0 (C3′B), 64.5 (C2′A), 61.8 (C2′B), 60.9 (C5′B), 60.8 (C5′A), 55.0 (CH3O), 52.8 (C5″B), 52.3 (C5″A), 37.8 (CH2Py-B), 37.7 (CH2Py-A). Trace impurities of dichloromethane and 1-pyreneacetic acid were identified in the 13C NMR spectrum. The compound was used in the next step without further purification.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-5-(9′-fluorenylmethoxycarbonyl)-2-oxa-5-azabicyclo[2.2.1]heptane (1)
Nucleoside 17 (225 mg, 0.25 mmol) was co-evaporated with anhydrous 1,2-dichloroethane (2×4 mL) and re-dissolved in anhydrous CH2Cl2 (3.5 mL). Anhydrous N,N′-diisopropylethylamine (DIPEA, 195 μL, 1.12 mmol) and N-methylimidazole (NMI, 16 μL, 0.20 mmol) were added followed by dropwise addition of 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (111 μL, 0.50 mmol). The reaction mixture was stirred for 4h at rt, evaporated to dryness, and the resulting residue purified by silica gel column chromatography (0–2% MeOH in CH2Cl2, v/v, initially built in 0.5% Et3N) and subsequent precipitation from CH2Cl2/petroleum ether to afford target amidite 1 (126 mg, 46%) as a white foam. Physical data for mixture of rotamers: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1129.4356 ([M+Na]+, C63H63N8O9P·Na+, Calc. 1129.4348); 31P NMR (121 MHz, CDCl3) δ 150.32, 150.26, 150.1, 149.5.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-5-(pyren-1-yl)methyl-2-oxa-5-azabicyclo[2.2.1]heptane (2)
Nucleoside 18 (155 mg, 0.17 mmol) was co-evaporated with 1,2-dichloroethane (2×5 mL) and dissolved in a 20% N,N′-diisopropylethylamine solution in anhydrous CH2Cl2 (5.0 mL, v/v). To this was added 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (0.10 mL, 0.44 mmol) and the reaction mixture was stirred at rt for 17h whereupon the reaction mixture was quenched with abs EtOH (1 mL) and evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–90% EtOAc in petroleum ether, v/v) and precipitated from CH2Cl2/petroleum ether to afford amidite 2 as a white solid material (131 mg, 69%). Rf = 0.7 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1099.4642 ([M+H]+, C65H63N8O7P·H+, Calc.1099.4630); 31P NMR (121 MHz, CDCl3) δ 150.6, 148.4.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-5-(pyren-1-yl)carbonyl-2-oxa-5-azabicyclo[2.2.1]heptane (3)
Nucleoside 19 (0.30 g, 0.33 mmol) was co-evaporated with 1,2-dichloroethane (2×5 mL) and dissolved in 20% N,N′-diisopropylethylamine in anhydrous CH2Cl2 (3.4 mL, v/v). To this was added 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (0.11 mL, 0.49 mmol) and the reaction mixture was stirred for 22 h at rt, at which point abs. EtOH (2 mL) was added. The mixture was taken up in CH2Cl2 (50 mL) and washed with sat. aq. NaHCO3 (50 mL) and brine (50 mL). The combined aqueous layer was back-extracted with CH2Cl2 (2×20 ml) and the combined organic layer evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–99% EtOAc in petroleum ether containing 1% pyridine, v/v/v), co-evaporated with abs. EtOH:toluene (2×50 mL, 1:1, v/v) and precipitated from EtOAc/hexane to afford phosphoramidite 3 as a white foam (247 mg, 67%). Physical data for the mixture of rotamers: Rf = 0.5 (EtOAc); ESI-HRMS m/z 1113.4462 ([M+H]+, C65H61N8O8P·H+, Calc.1113.4422); 31P NMR (121 MHz, CDCl3) δ 152.3, 151.8, 150.2.
(1S,3R,4S,7R)-3-(6-N-Benzoyladenin-9-yl)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-5-(pyren-1-yl)acetyl-2-oxa-5-azabicyclo[2.2.1]heptane (4)
Nucleoside 20 (235 mg, 0.25 mmol) was co-evaporated with anhydrous 1,2-dichloroethane (2×5 mL) and re-dissolved in anhydrous CH2Cl2 (5 mL). Anhydrous N,N′-diisopropylethylamine (220 μL, 1.27 mmol) was added, followed by dropwise addition of 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (115 μL, 0.51 mmol). The reaction mixture was stirred at rt for 22 h, at which point abs. EtOH (1 mL) was added. The solution was evaporated to dryness and the resulting crude residue purified by silica gel column chromatography (0–2% MeOH in CH2Cl2, v/v) and precipitation from CH2Cl2/petroleum ether to afford target amidite 4 (203 mg, 71%) as a white foam. Physical data for the mixture of rotamers: Rf = 0.6 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 1149.4358 ([M+Na]+, C66H63N8O8P·Na+, Calc. 1149.4399); 31P NMR (121 MHz, CDCl3) δ 150.4, 150.31, 150.26, 148.1.
Synthesis of ONs
W/X/Y/Z-modified oligodeoxyribonucleotides were made on an automated DNA synthesizer using 0.2 μmol scale succinyl linked LCAA-CPG (long chain alkyl amine controlled pore glass) columns with a pore size of 500Å. Phosphoramidites 1–4 were incorporated into ONs using the following hand-coupling conditions (activator; coupling time; stepwise coupling yield): monomer W (pyridinium hydrochloride; 30 min; ~82%) and monomers X-Z (pyridinium hydrochloride; 15 min; ~95%). Standard protocols for incorporation of DNA phosphoramidites were used. Modified ONs were deprotected using 32% aq. NH3 (55 °C, 2–12 h) and purified (DMTr-ON) by ion-pair reversed-phase HPLC using either an ammonium formate – acetonitrile gradient or a triethylammonium acetate - acetonitrile gradient, which was followed by detritylation (80% aq. AcOH, 20 min) and precipitation (abs. EtOH or acetone, −18 °C, 12 h). The composition of the modified ONs was verified by MALDI-MS analysis (Tables S1 and S2) recorded either in positive or negative ion mode on a Quadrupole Time-Of-Flight Tandem Mass Spectrometer equipped with a MALDI source using 3-hydroxypicolinic acid as a matrix. Purity (>90%, unless stated otherwise) was verified either by the ion-pair reverse phase HPLC system running in analytical mode or ion-exchange HPLC using a Tris-Cl/EDTA - NaCl gradient.
Protocol - thermal denaturation studies
Concentrations of ONs were calculated using the following extinction coefficients (OD260/μmol): G, 12.0; A, 15.2; T, 8.4; U, 10.0; C, 7.1; pyrene, 22.4. ONs (each strand at 1.0 μM) were thoroughly mixed, denatured by heating and subsequent cooling to the starting temperature of the experiment. Quartz optical cells with a path length of 10.0 mm were used. Thermal denaturation temperatures (Tm’s/°C) were measured on a temperature-controlled UV-Vis spectrophotometer and determined as the maximum of the first derivative of the thermal denaturation curve (A260 vs. T) recorded in medium salt buffer (Tm buffer: 100 mM NaCl, 0.1 mM EDTA and pH 7.0 adjusted with 10 mM NaH2PO4/5 mM Na2HPO4). The temperature of the denaturation experiments ranged from at least 15 °C below Tm to 20 °C above Tm (although not below 3 °C). A temperature ramp of 0.5 °C/min or 1.0 °C/min was used in the experiments. Reported thermal denaturation temperatures are an average of two measurements within ±1.0 °C.
Protocol – UV-Vis absorption spectroscopy
UV-Vis spectra were recorded on a spectrophotometer at 5 °C (except for single-stranded X/Y-modified probes, which were recorded at room temperature) using each strand at 1 μM concentration in Tm buffer and quartz cells with 1 cm path lengths.
Protocol – fluorescence emission spectroscopy
Steady-state fluorescence spectra were recorded at 5 °C using an excitation wavelength of λex = 350 nm, each strand at 1 μM concentration in Tm buffer (except with Y-modified ONs where strands were used at 0.15 μM concentration), and quartz cells with 1 cm path lengths.
Supplementary Material
Acknowledgments
PJH appreciates financial support from Award Number R01 GM088697 from the National Institute of General Medical Sciences, National Institutes of Health, and from the Institute of Translational Health Sciences (ITHS) (supported by grants UL1 RR025014, KL2 RR025015, and TL1 RR025016 from the NIH National Center for Research Resources). JW appreciates financial support from The Danish National Research Foundation and the Danish Agency for Science Technology and Innovation. We thank Dr. T. Santhosh Kumar (Univ. Southern Denmark) for providing us with starting material and Dr. Lee Deobald (EBI Murdock Mass Spectrometry Center, Univ. Idaho) for mass spectrometric analyses.
Footnotes
Supporting information: General experimental section; additional synthetic strategies; NMR spectra for all new compounds; MS data for all modified ONs; representative thermal denaturation curves; additional Tm data; representative absorption and fluorescence emission spectra; evaluation of SNP-discriminatory potential. This material is available free of charge via the Internet at http://pubs.acs.org/.
References and notes
- 1.For reviews on conformationally restricted nucleotides, see e.g., Leumann CJ. Bioorg Med Chem. 2002;10:841–854. doi: 10.1016/s0968-0896(01)00348-0.Obika S, Abdur Rahman SM, Fujisaka A, Kawada Y, Baba T, Imanishi T. Heterocycles. 2010;81:1347–1392.Prakash TP. Chem Biodiv. 2011;8:1616–1641. doi: 10.1002/cbdv.201100081.Deleavey GF, Damha MJ. Chem Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011.Zhou C, Chattopadhyaya J. Chem Rev. 2012;112:3808–3832. doi: 10.1021/cr100306q.
- 2.(a) Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. Nucleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bennett CF, Swayze EE. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]; (c) Østergaard ME, Hrdlicka PJ. Chem Soc Rev. 2011;40:5771–5788. doi: 10.1039/c1cs15014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Watts JK, Corey DRJ. Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Matsui M, Corey DR. Drug Disc Today. 2012;17:443–450. doi: 10.1016/j.drudis.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. Chem Rev. 2013;113:6207–6233. doi: 10.1021/cr300362f. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Nielsen P, Koshkin AA, Wengel J. Chem Commun. 1998:455–456. [Google Scholar]
- 4.Obika S, Nanbu D, Hari Y, Andoh JI, Morio KI, Doi T, Imanishi T. Tetrahedron Lett. 1998;39:5401–5404. [Google Scholar]
- 5.Kaur H, Babu BR, Maiti S. Chem Rev. 2007;107:4672–4697. doi: 10.1021/cr050266u. [DOI] [PubMed] [Google Scholar]
- 6.(a) Rajwanshi VK, Håkansson AE, Dahl BM, Wengel J. Chem Commun. 1999:1395–1396. [Google Scholar]; (b) Sørensen MD, Kværnø L, Bryld T, Håkansson AE, Verbeure B, Gaubert G, Herdewijn P, Wengel J. J Am Chem Soc. 2002;124:2164–2176. doi: 10.1021/ja0168763. [DOI] [PubMed] [Google Scholar]
- 7.Recent examples include: Seth PP, Vasquez G, Allerson CA, Berdeja A, Gaus H, Kinberger GA, Prakash TP, Migawa MT, Bhat B, Swayze EE. J Org Chem. 2010;75:1569–1581. doi: 10.1021/jo902560f.Li Q, Yuan F, Zhou C, Plashkevych O, Chattopadhyaya J. J Org Chem. 2010;75:6122–6140. doi: 10.1021/jo100900v.Liu Y, Xu J, Karimiahmadabadi M, Zhou C, Chattopadhyaya J. J Org Chem. 2010;75:7112–7128. doi: 10.1021/jo101207d.Upadhayaya R, Deshpande SA, Li Q, Kardile RA, Sayyed AY, Kshirsagar EK, Salunke RV, Dixit SS, Zhou C, Foldesi A, Chattopadhyaya J. J Org Chem. 2011;76:4408–4431. doi: 10.1021/jo200073q.Shrestha AR, Hari Y, Yahara A, Osawa T, Obika S. J Org Chem. 2011;76:9891–9899. doi: 10.1021/jo201597e.Hanessian S, Schroeder BR, Giacometti RD, Merner BL, Østergaard ME, Swayze EE, Seth PP. Angew Chem, Int Ed. 2012;51:11242–11245. doi: 10.1002/anie.201203680.Haziri AI, Leumann CJ. J Org Chem. 2012;77:5861–5869. doi: 10.1021/jo300554w.Gerber AB, Leumann CJ. Chem Eur J. 2013;19:6990–7006. doi: 10.1002/chem.201300487.Morihiro K, Kodama T, Kentefu, Moai Y, Veedu RN, Obika S. Angew Chem Int Ed. 2013;52:5074–5078. doi: 10.1002/anie.201300555.Hari Y, Osawa T, Kotobuki Y, Yahara A, Shrestha AR, Obika S. Bioorg Med Chem. 2013;21:4405–4412. doi: 10.1016/j.bmc.2013.04.049.Hari Y, Morikawa T, Osawa T, Obika S. Org Lett. 2013;15:3702–3705. doi: 10.1021/ol401566r.Migawa MT, Prakash TP, Vasquez G, Seth PP, Swayze EE. Org Lett. 2013;15:4316–4319. doi: 10.1021/ol401730d.Hanessian S, Schroeder BR, Merner BL, Chen B, Swayze EE, Seth PP. J Org Chem. 2013;78:9051–9063. doi: 10.1021/jo401166q.Hanessian S, Wagger J, Merner BL, Giacometti RD, Østergaard ME, Swayze EE, Seth PP. J Org Chem. 2013;78:9064–9075. doi: 10.1021/jo401170y.
- 8.For recent examples, see: Karmakar S, Anderson BA, Rathje RL, Andersen S, Jensen T, Nielsen P, Hrdlicka PJ. J Org Chem. 2011;76:7119–7131. doi: 10.1021/jo201095p.Didion BA, Karmakar S, Guenther DC, Sau S, Verstegen JP, Hrdlicka PJ. ChemBioChem. 2013;14:1534–1538. doi: 10.1002/cbic.201300414.Denn B, Karmakar S, Guenther DC, Hrdlicka PJ. Chem Commun. 2013;49:9851–9853. doi: 10.1039/c3cc45705b.
- 9.(a) Kumar TS, Madsen AS, Wengel J, Hrdlicka PJ. J Org Chem. 2006;71:4188–4201. doi: 10.1021/jo060331f. [DOI] [PubMed] [Google Scholar]; (b) Kumar TS, Madsen AS, Østergaard ME, Sau SP, Wengel J, Hrdlicka PJ. J Org Chem. 2009;74:1070–1081. doi: 10.1021/jo802037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar TS, Wengel J, Hrdlicka PJ. ChemBioChem. 2007;8:1122–1125. doi: 10.1002/cbic.200700144. [DOI] [PubMed] [Google Scholar]
- 11.Kumar TS, Madsen AS, Østergaard ME, Wengel J, Hrdlicka PJ. J Org Chem. 2008;73:7060–7066. doi: 10.1021/jo800551j. [DOI] [PubMed] [Google Scholar]
- 12.(a) Sau SP, Kumar TS, Hrdlicka PJ. Org Biomol Chem. 2010;8:2028–2036. doi: 10.1039/b923465a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sau SP, Madsen AS, Podbevsek P, Andersen NK, Kumar TS, Andersen S, Rathje RL, Anderson BA, Guenther DC, Karmakar S, Kumar P, Plavec J, Wengel J, Hrdlicka PJ. J Org Chem. 2013;78:9560–9570. doi: 10.1021/jo4015936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.A conference proceeding outlining a portion of of this study has been published, see: Andersen NK, Wengel J, Hrdlicka PJ. Nucleosides Nucleotides Nucleic Acids. 2007;26:1415–1417. doi: 10.1080/15257770701539153.
- 14.Wenska M, Honcharenko D, Pathmasiri W, Chattopadhyaya J. Heterocycles. 2007;73:303–324. [Google Scholar]
- 15.Rosenbohm C, Christensen SM, Sørensen MD, Pedersen DS, Larsen LE, Wengel J, Koch T. Org Biomol Chem. 2003;1:655–663. doi: 10.1039/b208864a. [DOI] [PubMed] [Google Scholar]
- 16.Hrdlicka PJ, Andersen NK, Jepsen JS, Hansen FG, Haselmann KF, Nielsen C, Wengel J. Bioorg Med Chem. 2005;77:2597–2621. doi: 10.1016/j.bmc.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ellervik U, Magnusson G. Tetrahedron Lett. 1997;38:1627–1628. [Google Scholar]; (b) Rigoli JW, Østergaard ME, Canady KM, Guenther DC, Hrdlicka PJ. Tetrahedron Lett. 2009;50:1751–1753. [Google Scholar]
- 18.The conversion of 7 into 9 has been outlined in the patent literature, see: Sørensen MD, Wengel J, Koch T, Christensen SM, Rosenbohm C, Pedersen DS. 2003095467 A1. WO.
- 19.Takaku H, Morita K, Sumiuchi T. Chem Lett. 1983;11:1661–1664. [Google Scholar]
- 20.The signal for H3′ appears as a doublet in nucleosides with unmodified 3′-OH groups.
- 21.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 22.Hakansson AE, Wengel J. Bioorg Med Chem Lett. 2001;11:935–938. doi: 10.1016/s0960-894x(01)00110-x. [DOI] [PubMed] [Google Scholar]
- 23.Crothers DM. Biopolymers. 1968;6:575–584. doi: 10.1002/bip.1968.360060411. [DOI] [PubMed] [Google Scholar]
- 24.(a) Yamana K, Iwase R, Furutani S, Tsuchida H, Zako H, Yamaoka T, Murakami A. Nucleic Acids Res. 1999;27:2387–2392. doi: 10.1093/nar/27.11.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Christensen UB, Pedersen EB. Nucleic Acids Res. 2002;30:4918–4925. doi: 10.1093/nar/gkf624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bryld T, Højland T, Wengel J. Chem Commun. 2004:1064–1065. doi: 10.1039/b402414a. [DOI] [PubMed] [Google Scholar]
- 25.Marin V, Hansen HF, Koch TR, Armitage BA. J Biomol Struc Dyn. 2004;21:841–850. doi: 10.1080/07391102.2004.10506974. [DOI] [PubMed] [Google Scholar]
- 26.(a) Korshun VA, Stetsenko DA, Gait MJ. J Chem Soc, Perkin Trans 1. 2002:1092–1104. [Google Scholar]; (b) Dohno C, Saito I. ChemBioChem. 2005;6:1075–1081. doi: 10.1002/cbic.200400325. [DOI] [PubMed] [Google Scholar]
- 27.(a) Dougherty G, Pilbrow JR. Int J Biochem. 1984;16:1179–1192. doi: 10.1016/0020-711x(84)90215-5. [DOI] [PubMed] [Google Scholar]; (b) Nakamura M, Fukunaga Y, Sasa K, Ohtoshi Y, Kanaori K, Hayashi H, Nakano H, Yamana K. Nucleic Acids Res. 2005;33:5887–5895. doi: 10.1093/nar/gki889. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Asanuma H, Fujii T, Kato T, Kashida H. J Photochem Photobiol C. 2012;13:124–135. [Google Scholar]
- 28.(a) Okamoto A, Kanatani K, Saito I. J Am Chem Soc. 2004;126:4820–4827. doi: 10.1021/ja039625y. [DOI] [PubMed] [Google Scholar]; (b) Østergaard ME, Kumar P, Baral B, Guenther DC, Anderson BA, Ytreberg FM, Deobald L, Paszczynski AJ, Sharma PK, Hrdlicka PJ. Chem Eur J. 2011;17:3157–3165. doi: 10.1002/chem.201002109. [DOI] [PubMed] [Google Scholar]
- 29.Manoharan M, Tivel KL, Zhao M, Nafisi K, Netzel TL. J Phys Chem. 1995;99:17461–17472. [Google Scholar]; (b) Seo YJ, Ryu JH, Kim BH. Org Lett. 2005;7:4931–4933. doi: 10.1021/ol0518582. [DOI] [PubMed] [Google Scholar]
- 30.Persil O, Hud NV. Trends Biotechnol. 2007;25:433–436. doi: 10.1016/j.tibtech.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Assignments of 1H NMR signals (and of the corresponding 13C signals) of H5′, H5″ and CH2Ph (if present) are interchangeable.
- 32.The least predominant rotamer is denoted as ‘A’.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.