

Abstract
Soluble guanylate cyclase (sGC) is a heterodimeric heme protein and the primary nitric oxide receptor. NO binding stimulates cyclase activity, leading to regulation of cardiovascular physiology and making sGC an attractive target for drug discovery. YC-1 and related compounds stimulate sGC both independently and synergistically with NO and CO binding; however, where the compounds bind and how they work remains unknown. Using linked-equilibria binding measurements, surface plasmon resonance, and domain truncations in Manduca sexta and bovine sGC, we demonstrate that YC-1 binds near or directly to the heme-containing domain of the beta subunit. In the absence of CO, YC-1 binds with Kd = 9–21 μM, depending on construct. In the presence of CO, these values decrease to 0.6–1.1 μM. Pfizer compound 25 bound ~10-fold weaker than YC-1 in the absence of CO whereas compound BAY 41–2272 bound particularly tightly in the presence of CO (Kd = 30–90 nM). Additionally, we found that CO binding is much weaker to heterodimeric sGC proteins (Kd = 50–100 μM) than to the isolated heme domain (Kd = 0.2 μM for Manduca beta H-NOX/PAS). YC-1 greatly enhanced CO binding to heterodimeric sGC, as expected (Kd = ~1 μM). These data indicate the alpha subunit induces a heme pocket conformation with lower affinity for CO and NO. YC-1 family compounds bind near the heme domain, overcoming the alpha subunit effect and inducing a heme pocket conformation with high affinity. We propose this high-affinity conformation is required for the full-length protein to achieve high catalytic activity.
Nitric oxide (NO) regulates a phenomenal array of physiological processes, including blood pressure homeostasis, wound healing, memory formation, sexual response and the fighting of infectious disease.1 Impairment in NO signaling can lead to hypertension and atherosclerosis, and contribute to heart attack and stroke.2,3 NO is produced by a class of enzymes called nitric oxide synthases (NOSs) through the oxidation of L-arginine to L-citrulline.4,5 The primary receptor for NO is soluble guanylyl/guanylate cyclase (sGC), a heterodimeric heme protein of ~150 kDa that responds to NO binding to heme through enhanced cyclase activity, producing cGMP and a signaling cascade. Treatment of cardiovascular disease by stimulating the nitric oxide pathway has long been a treatment goal, beginning over 150 years ago with administration of amyl nitrite6 and nitroglycerin7 to relieve symptoms of angina pectoris, although the mode of action of these compounds (release of NO) was not discovered until many years later. More recently, sGC, the NO receptor, has been heavily targeted for drug discovery.
sGC is composed of two homologous subunits, α and β. Multiple isoforms of each subunit have been identified; however, the most common isoform is the α1/β1 heterodimer (reviewed in8). Each sGC subunit consists of four domains, an N-terminal Heme-Nitric Oxide Oxygen (H-NOX) domain9 (also called a SONO domain10), a central Per-Arnt-Sim (PAS) domain,11 a coiled-coil domain and a C-terminal catalytic cyclase domain.12 There is a single heme moiety in the heterodimer, associated with the β1 H-NOX domain. The equivalent domain in the α1 subunit has lost the ability to bind heme but appears to have retained an overall H-NOX-like fold and is therefore commonly referred to as the α1 H-NOX domain. During signaling, NO binding to heme in the β1-subunit leads to the formation of a pentacoordinated Fe-NO complex with proximal histidine bond breakage.13–15 The structural change due to this event is transferred to the cyclase domain, which in turn enhances cGMP production. How this structural change is translated to increased catalytic activity is poorly understood. Moreover, elusive structural details for sGC have hampered the understanding of allosteric regulation in the protein. Structures of individual sGC domains such as the β1 coiled-coil homodimer,16 the α1 PAS domain17 and the structure of the α1/β1 heterodimeric cyclase domain18 have recently been determined. Insight for H-NOX and PAS domain comes from the structures of prokaryotic homologous proteins.10,19–21 Yet an understanding of how these domains are arranged in the functional NO sensor remains unknown.
Small molecule stimulators of sGC have been discovered, opening new doors for drug discovery in the treatment of cardiovascular diseases.22 The first of these is compound YC-1, a benzylindazole derivative that inhibits platelet activation through stimulating sGC.23 YC-1 stimulates sGC two- to four-fold in the absence of NO but acts synergistically with CO or NO to achieve several hundred fold activation.24,25 Binding of YC-1 can also overcome inhibitory phosphorylation of sGC.26 Compound BAY 63–2521 (riociguat), a YC-1 derivative, has just completed phase III clinical trials27–30 and approved by the FDA for treatment of pulmonary hypertension (as Adempas). These compounds stimulate sGC activity in an NO independent and heme dependent manner, but how they bind to sGC and how they stimulate catalytic activity is unknown. Studies aimed at determining the binding site for YC-1-family compounds have suggested the pseudosymmetric site in the cyclase domain,31,32 the α1 H-NOX domain33,34 and the β1 H-NOX domain.35–38 A second class of compounds that function through replacing the sGC heme, which can be lost upon oxidation, have also been developed.22 Prominent among these are compounds BAY 58–2667 (cinaciguat),39 and HMR1766 (ataciguat).40
We developed sGC from the tobacco hornworm/hawkmoth (Manduca sexta) for biophysical and biochemical characterization, and to help with uncovering the mechanism underlying YC-1 stimulation.33,41–43 Manduca sexta sGC (Ms sGC) is highly homologous to its mammalian counterparts and responds well to YC-1-family compounds. Expression of N-terminal heterodimeric constructs lacking the α1/β1 cyclase domains (Ms sGC-NT constructs) leads to proteins that retain YC-1 binding. YC-1 binding to Ms sGC leads to enhanced CO and NO binding33 and to the trapping of CO in the heme pocket after laser photolysis, leading to rebinding with heme before escape from the protein (geminate recombination).41 Ms sGC-NT is an elongated molecule with a central parallel coiled-coil domain, based on chemical cross-linking, mass spectrometry and small angle X-ray scattering (SAXS) studies.43 In this model, the coiled-coil domain acts as an organizing center for the PAS, H-NOX and, presumably, cyclase domains. Here, we demonstrate that the alpha subunit serves to keep the beta subunit heme domain in a conformation with reduced affinity for CO, and that YC-1 binds directly to the beta subunit, inducing a high-affinity heme domain conformation.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were obtained from Sigma-Aldrich, restriction enzymes from Fermentas, and purification columns from GE Healthcare unless otherwise indicated. Pfizer compound 25 targeted to sGC (PF-25) was kindly provided by Dr. Lee Roberts of Pfizer Inc.44 DEA/NO was kindly provided by Dr. Katrina Miranda (University of Arizona).
sGC Protein Expression Vectors
Construct Ms sGC CT1 (α1 residues 272–699 and β1 residues 199–600) was obtained by PCR amplification from a full-length Ms sGC pETDuet1 construct.33 Forward primer 5′-ggatccgaccaaagtgacagattt-3′ and reverse primer 5′-gcggccgcctaagttggttcttct-3′ were used for the α1 subunit and the PCR product was cloned into the pETDuet1 vector using the restriction sites BamHI and NotI. Similarly, the Ms sGC CT1 β1-fragment was obtained by PCR amplification from the Ms sGC full-length pETDuet1 construct using primers 5′-catatgacgttgtctcttgaacca-3′ and 5′-gatatcttaatggatcttcctggt-3′ and the PCR product was cloned into the same pETDuet1 vector using the restriction sites NdeI and EcoRV. The final construct had a His6 purification tag fused to the N-terminus of the α1-subunit. Stop codons were inserted at α1 Asn 451 and β1 Thr 381 using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA), leading to constructs containing just the PAS and coiled coil domains (α1 272–450, β1 199–380).
Possible boundaries for stable PAS domain expression were surveyed using the Ms sGC α1 PAS-CC-Cyclase (residues 272–699) and β1 PAS-CC-Cyclase (residues 199–600) cloned into a single plasmid (pETDuet-1, Novagen) or cloned individually into the pETDuet-1 (α1) or pET28a+ (β1) plasmids. Domain boundaries were examined through introduction of stop codons, using the QuikChange mutagenesis kit. Ms sGC β1 PAS construct (residues 199–319) in pET28a+ was obtained by inserting a stop codon at position 320. The Ms sGC α1 PAS domain, spanning residues 279–425, was cloned into the pETHSUL vector, kindly provided by the Loll laboratory.45 A ligation independent cloning (LIC) approach was undertaken as described,45 using forward primer 5′-agattggtggcatcggcgtggctagcttctgc-3′, and reverse primer 5′-gaggagagtttagacttaaccatcctgagccctagcc-3′ (LIC overhang residues are underlined). The vector was made ready for ligation using the direct digestion method with BseRI (New England Biolabs). A stop codon was introduced at position 405 to yield wild-type construct Ms sGC-P25α, spanning residues 279–404. A triple cysteine-to-alanine mutant (C285A/C352A/C374A, Ms sGC-P35α) was produced to assist in crystallization.17 All mutations were introduced using the QuikChange lightning site-directed mutagenesis kit. Vector pSUPER, containing a dual-tagged catalytic domain of SUMO Hydrolase (dtUD1) fused to N-terminus SUMO, was also kindly provided by the Loll laboratory.45 Ms sGC β1(1–380), containing the H-NOX and PAS domains and most of the CC domain, was amplified by PCR and subcloned into the pGEM-T vector. The fragment was then cut with NcoI and NotI restriction enzymes and inserted into the pET28c vector, yielding a C-terminal His6 tag.
A single step insertion methodology46 was used for insertion of the BirA recognition sequence (Avi-tag, GLNDIFEAQKIEWHE) at the C-terminus of the Ms sGC-NT21 β1 subunit (residue 380, reference 43) and Ms sGC β1(1–380) using forward primers: 5′-ggaattggaaaaacagaagggtggcggtctgaacgacatcttcgaggctcaaaaaatagagtggcacgagtaggacaggcttctttactca gtg-3′ and 5′-ggaattggaaaaacagaagggtggcggtctgaacgacatcttcgaggctcaaaaaatagagtggcacgaggcggccgcactcgagcac caccac-3′ and a common reverse primer: 5′-cttctgtttttccaattccagctctcggaatgtttgttgaag-3′. The Avi-tags with two N-terminal glycine linker residues are underlined. Similarly, a C-terminal Avi-tag was added to α1 PAS domain construct Ms sGC-P25α using forward primer: 5′-gactcttcatatccgatataggtggcggtctgaacgacatcttcgaggctcaaaaaatagagtggcacgagtgacttcatgatgcgacgaga g-3′ and reverse primer: 5′-tatatcggatatgaagagtccccttccagtcagaccttcgag-3′. E. coli biotin protein ligase BirA in vector pGEX-4T-1, with an N-terminal GST-tag linked to a thrombin cleavage site and a C-terminal His6-tag, was kindly provided by Dr. Michael Kuhns (University of Arizona). The thrombin cleavage site was changed to a TEV cleavage site using forward primer: 5′-ccatcctccaaaatcgggcgaaaacttgtatttccagggatccaaggataacaccg-3′ and reverse primer: 5′-cggtgttatccttggatccctggaaatacaagttttcgcccgattttggaggatgg-3′ (TEV cleavage site underlined). Additionally, a stop codon was inserted in front of His6-tag.
Expression and Purification of Ms sGC PAS Domains
Ms sGC α1 PAS with an N-terminal His-tagged SUMO fusion was expressed in E. coli strain BL21 (DE3) pLysS. Cells were grown at 37 °C to an OD600 of 0.6 before induction with 0.5 mM isopropyl 1-thio-β-D-thiogalactopyranoside (IPTG) and growth continued at 20 °C; cells were harvested after 16 h. Purification steps were performed at 4 °C. Cell pellets were resuspended in lysis buffer (50 mM sodium phosphate, pH 7.4, 300 mM NaCl, 20 mM imidazole, 0.1 mg/ml DNase I, 2 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, and 1 μg/mL aprotinin, leupeptin and pepstatin), disrupted using a French press cell (1000 psi), clarified by ultracentrifugation (45Ti rotor, 40,000 rpm for 30 min), supplemented with 10% glycerol (w/v) and 5 mM β-mercaptoethanol, and loaded onto a 5 mL Ni-NTA column previously equilibrated with binding buffer (50 mM sodium phosphate pH 7.4, 300 mM NaCl, 20 mM imidazole, 5 mM β-mercaptoethanol). The column was washed with binding buffer until baseline was reached and bound protein was eluted using an imidazole gradient ranging from 20–300 mM over 100 mL (20 bed volumes) by mixing binding buffer and elution buffer (binding buffer supplemented with 500 mM imidazole). Cleavage of the N-terminal His-tagged SUMO domain was achieved by adding 1 mg of purified SUMO hydrolase (dtUD1) to the pooled PAS-containing fractions followed by overnight dialysis at 4 °C against two changes of dialysis buffer (50 mM sodium phosphate pH 7.4, 300 mM NaCl, 5% Glycerol (w/v), 10 mM β-mercaptoethanol). The dialyzed product was again loaded onto the Ni-NTA column to remove the His-tagged SUMO and SUMO hydrolase proteins, followed by concentration to ~3 mL and further purification over an S-200 size exclusion column previously equilibrated with equilibration buffer (50 mM Tris-HCl pH 7.4, 200 mM NaCl, 5% Glycerol (w/v), 5 mM dithiothreitol). The final material was concentrated to 10–15 mg/ml using a Vivaspin concentrator (Sartorius Stedim Biotech) and stored at −80°C. A final yield 2–3 mg of highly pure protein was obtained per liter of cell culture.
Ms sGC β1 PAS was expressed in E. coli strain Rosetta pLysS. Cells were grown at 37 °C to an OD600 of 1.0 and were induced with 0.2 mM IPTG after which they were grown with slow shaking (90 rpm) at 18 °C for 18 h before harvesting. Cell lysate was obtained as described for the α1 PAS domain, and the protein was purified using Ni-NTA followed by S-200 size exclusion chromatography. A yield 30–40 mg was obtained per liter of cell culture.
SUMO hydrolase (dtUD1) was expressed in strain BL21 (DE3) pLysS. Cells were grown at 37 °C until OD600 reached 0.6 and were induced with 0.5 mM IPTG and grown at 30 °C for 6 h before harvesting. Purification was performed using Ni-NTA column chromatography as described.45
BirA was expressed in strain BL21 (DE3) pLysS. Cells were grown at 37 °C to an OD600 1.0 and expression induced with 0.5 mM IPTG, followed by growth at 16 °C for 20 h before harvesting. Cells were lysed by sonication at 4 °C in lysis buffer, clarified by ultracentrifugation and supernatant loaded onto a GSTrap FF column previously equilibrated with 50 mM sodium phosphate buffer, pH 7.4, containing 300 mM NaCl. Bound GST-tagged BirA was eluted with the above buffer supplemented with 20 mM glutathione. Fractions were incubated for 24 h with 5 μM His6-tagged TEV-protease and the mixture loaded onto a GSTrap FF column in tandem with a Ni-NTA column. Flow through was collected and the protein concentrated and stored in −80 °C.
TEV protease with N-terminal polyhistidine, C-terminal polyarginine and mutation S219V was prepared from a previously described pRK793 vector.47 TEV was expressed in E. coli strain BL21 (DE3) pLysS. Cells were grown to an OD600 of 0.6 at 37 °C, induced with 1 mM IPTG and grown at 30 °C for 6 h before harvesting. Cell pellets were resuspended in binding buffer (50 mM sodium phosphate, pH 7.4, 300 mM NaCl, 20 mM imidazole), disrupted using a French press cell (1000 psi) and clarified by ultracentrifugation (45Ti rotor, 40,000 rpm for 30 min). The supernatant was supplemented with 10% glycerol (w/v) and 5 mM β-mercaptoethanol before loading onto a 5 mL Ni-NTA column previously equilibrated with the binding buffer (50 mM sodium phosphate pH 7.4, 300 mM NaCl, 20 mM imidazole, 5 mM β-mercaptoethanol). The column was washed with the binding buffer until baseline was reached and bound protein was eluted using an imidazole gradient ranging from 20–300 mM over 100 mL (20 bed volumes) by mixing binding buffer and elution buffer (binding buffer supplemented with 500 mM imidazole). Fractions containing TEV protease were pooled, buffer exchanged with final storage buffer (50 mM sodium phosphate, pH 7.4, 300 mM NaCl, 25% Glycerol, 1 mM EDTA and 1 mM DTT), concentrated to a final concentration of ~4 mg/mL and frozen in −80 °C.
Expression and Purification of Heme Containing Manduca and Bovine sGC Proteins
Ms sGC-NT13, Ms sGC-NT19 and Ms sGC-NT21 were expressed in E. coli and purified using Ni-NTA, StrepTactin (Ms sGC-NT19) and size exclusion chromatography, as previously described.33,43 Ms sGC β1(1–380) was expressed in E. coli strain Rosetta pLysS. Cells were grown at 37 °C to an OD600 of 1.0 and cooled on ice before induction. Culture was induced with 0.5 mM IPTG and supplemented with 25 μM δ-aminolevulinic acid and grown at 30 °C for 6 h before harvesting. Cell pellets were resuspended in lysis buffer B (50 mM Tris.HCl, pH 8.5, 10 mM NaCl, 0.1 mg/mL DNase I, 2 mM MgCl2, 1 mM PMSF, 1 mM Benzamidine, and 1 μg/mL aprotinin, leupeptin and pepstatin and 1 mM dithionite), disrupted by French pressure cell and clarified by ultracentrifugation. The supernatant was supplemented with 10% glycerol (w/v), 5 mM β-mercaptoethanol and ~100 fold excess of dithionite (~1 mM, assuming 2 mg of protein per litre of the cell culture). The sample was loaded onto a DEAE anion exchange column or Q-FF sepharose column previously equilibrated with buffer A (20 mM Tris.HCl, pH 8.5, 1 mM EDTA and 5 mM β-mercaptoethanol), the column washed with buffer A and protein eluted with a 0–300 mM NaCl elution gradient (200 mL) using buffer A (0 mM NaCl) and buffer B (buffer A + 500 mM NaCl). Colored fractions were pooled and loaded onto the Ni-NTA column and eluted with 30 mM EDTA in a single step elution. Fractions were supplemented with fresh dithionite and tris(2-carboxyethyl)phosphine (TCEP), concentrated and were further purified by running the sample through a size exclusion S200 column previously equilibrated with gel filtration buffer (50 mM potassium phosphate buffer, pH 7.4, 100 mM KCl, 2 mM EDTA, 5% glycerol, 1 mM TCEP), where it ran as a monomer. The purified protein was supplemented with dithionite, concentrated to 5–10 mg/mL and stored at −80 °C.
For the bovine sGC β1 H-NOX and β1 H-NOX-PAS, we used a systematic site-directed mutagenesis approach to determine the appropriate C-terminal ends for optimal expression and solubility of the proteins (described elsewherea). We used the untagged β1 1–385 (residues 1–385) construct cloned into the pET30b plasmid (kind gift of Judith Burstyn) and introduced stop codons at various positions using the QuikChange mutagenesis kit. Bt sGC β1 H-NOX (residues 1–197) and sGC β1 H-NOX-PAS construct (residues 1–359) in pET30b were obtained by inserting a stop codon at position 198 and 360, respectively. These constructs displayed the highest levels of expression and solubility in E. coli cells. Purification of both constructs was performed as follows. Each construct was expressed in E. coli strain BL21(DE3)pRIPL. Cells were grown at 37 °C to an OD600 of 1.0 and cooled on ice before induction. Culture was induced with 0.4 mM IPTG and supplemented with 450 μM δ-aminolevulinic acid and ferric citrate and grown at 20 °C for 24 h before harvesting. Cell pellets were resuspended in lysis buffer (20 mM Tris.HCl pH 7.5, 50 mM NaCl, DTT, 0.5 mg/mL lysozyme, 300 U benzonase (SIGMA), and one tablet of EDTA-free protease inhibitor cocktail tablet/50mL (Roche)), disrupted by sonication and clarified by ultracentrifugation. We used 10 mM DTT to keep the β1 H-NOX protein reduced, and 1 mM DTT for the β1 H-NOX-PAS construct. The clarified lysate was loaded onto a Q-FF sepharose column previously equilibrated with buffer A (20 mM Tris.HCl pH 7.5, 50 mM NaCl, and DTT) and the protein was eluted with a 0.05–1 M NaCl elution gradient (buffer B = buffer A + 1 M NaCl). Colored fractions were pooled, dialyzed into buffer A, and loaded onto an S75 size-exclusion column equilibrated in buffer A. Colored fractions were pooled and loaded onto a second QFF column pre-equilibrated in buffer C (20 mM Tris.HCl pH 8.5, 50 mM NaCl, TCEP). The protein was eluted with a 0.05–1M NaCl gradient (buffer D = buffer C + 1M NaCl). The colored fractions were pooled, dialyzed into buffer C (10 mM and 1 mM TCEP for β1 H-NOX and β1 H-NOX-PAS, respectively). The purified protein was concentrated to ~10 mg/mL and stored at −80 °C.
Determination of Dissociation Constants for CO
CO dissociation constants were measured by titrating CO from a saturated solution into sGC protein and monitoring the appearance of the CO-bound Soret absorption band, as described previously.33,43 The Ms sGC β1(1–380) and Bt sGC β1(1–197) samples were prepared in Ar-purged buffer supplemented with excess dithionite. CO binding experiments were performed in a 10 cm pathlength cuvette for Ms sGC-β1(1–380) and Ms sGC-NT21 using a Cary 50 spectrophotometer (Varian) with a modified sample holder. Binding data in the presence and absence of 50 μM YC-1 was plotted using a single site saturation ligand binding model in SigmaPlot (SPSS, Inc., Chicago, IL).
To extract linked equilibria binding behavior, CO binding assays were performed for Ms sGC-NT21 (10 cm cuvette) and Ms sGC-NT13 (1 cm cuvette) at various YC-1 concentrations. The stimulation of CO binding in the presence of YC-1 was described with a cooperative 2-site model with 4 states: free protein, protein bound to CO only, protein bound to YC-1 only, and protein bound to both CO and YC-1. Independent binding of CO and YC-1 are described with the association constants KaCO and KaYC-1, with an assumed cooperativity constant Kint representing coupling between the two binding processes. This model is described with a binding polynomial of the form:
| Eq. 1 |
in which each term represents the statistical weight for one of the four states. The fraction of CO sites occupied is given by the ratio of the weights for states with CO bound to all four states:
| Eq. 2 |
Estimates for the model parameters KaCO, KaYC-1 and Kint were obtained from a global fit of θ to the normalized absorbance changes at wavelengths 423 nm and 433 nm, using MATLAB (The MathWorks, Inc., USA). The product KaYC-1 Kint indicates the association constant for YC1 binding to the CO-bound complex, and so its inverse represents the dissociation constant KdYC-1′. Similarly, KdCO′ may be estimated from the inverse of the product KaCO Kint.
To directly measure YC-1 family compounds binding to Ms sGC-NT-CO, an ~2 nm shift in the Soret band maxima was monitored as a function of compound concentration. Compound was titrated into a 1 or 10 cm cuvette containing a CO saturated protein solution. Kd for ligand binding in the presence of CO was calculated by plotting the Soret shift difference with respect to the increasing concentrations of the ligand and fitting to a single site saturation ligand binding model in SigmaPlot.
Fluorescence Anisotropy
Fluorescence anisotropy was measured using a JASCO J-815 CD fluorescence spectrometer equipped with an anisotropy attachment (JASCO, Maryland, US). Average anisotropy was measured at 20 °C for 60 seconds using an excitation wavelength of 325 nm. Anisotropy was calculated using total fluorescence above 380 nm, which was measured 90° incident to the excitation beam. Initial anisotropy was measured for a 2 μM YC-1 solution in 10 mM Tris.HCl, pH 7.5, by titrating Ms sGC α1 P25α or β1 PAS or lysozyme, and the sample was mixed thoroughly for 30 seconds before anisotropy measurements were taken. Data were fit using a one site-total binding model implemented in GraphPad (GraphPad Software, Inc., USA).
Surface Plasmon Resonance Binding Experiments
In vitro biotinylation of Avi-tagged Ms sGC-NT21, Ms sGC α1 PAS and Ms sGC β1(1–380) were performed using E. coli BirA biotin ligase. A reaction mixture containing 30–40 μM Avi-tagged protein, 1–2 μM purified BirA, 0.5 mM biotin, 10 mM magnesium acetate and 10 mM ATP was incubated at 4 °C for 6 h and loaded onto Ni-NTA column (Ms sGC-NT21) or Superdex-75 analytical gel filtration column (Ms sGC α1 PAS and Ms sGC β1(1–380)) to remove excess reaction components. Biotinylation was confirmed by monoclonal anti-biotin antibody (Sigma-Aldrich) western blot. All SPR studies were performed on a Biacore T100 instrument at 20 °C. Both reference and sample CM5 sensor chip surfaces (GE Healthcare) were prepared by amine coupling methodology following the procedures in the Biacore T100 instrument manual. The chip surfaces were first activated by using a mixture of N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC). NeutrAvidin (Pierce), 100 μg/ml in 10 mM NaOAc pH 4.5, was then immobilized to 10,000 response units by flowing it over the activated surfaces. Running buffer was 10 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.05% Tween 20. Any remaining active esters were blocked with ethanolamine and the immobilized NeutrAvidin chip surface was washed three times with a 30 second pulse of 10 mM HCl. Biotinylated Ms sGC proteins were captured onto the NeutrAvidin- coated chip surfaces by injecting 25 μM protein at a flow rate of 10 μL/min until ~10,000 response units were achieved. Running buffer was 50 mM potassium phosphate buffer, pH 7.4, 100 mM KCl, 2 mM EDTA, 1 mM TCEP. The surfaces were washed with running buffer for 2 h at a flow rate 100 μL/min until a stable response was obtained, indicating no further dissociation of the biotinylated proteins. Each chip has four flow cells allowing simultaneous measurements on one reference and three active surfaces. The Biacore T100 MIX function was used to mix DEA/NO or NaOH alone (0.5 mM DEA/NO stock in 10 mM NaOH, 1% DMSO, contained in an Ar-purged sealed Biacore vial) with varied concentrations of PF-25 in 50 mM potassium phosphate buffer, pH 7.4, 100 mM KCl, 2 mM EDTA, 1 mM TCEP containing 1% DMSO to achieve a final concentration of 25 μM DEA/NO. Various concentrations of PF-25 containing DEA/NO or NaOH were injected over the surface at 25 μL/min with 115 s association and 240 s dissociation times. Running buffer was 50 mM potassium phosphate buffer, pH 7.4, 100 mM KCl, 2 mM EDTA, 1 mM TCEP, 1% DMSO. Solvent correction curves were used to compensate for any mismatch between the sample buffer and the running buffer. Data were analyzed with Biacore T100 evaluation software to obtain the offset corrected response (R, measured in response units RU) and the expected maximum response (Rmax) based on the response from the immobilized protein (Rimmob) and the relative molecular weights of ligand and analyte:
| Eq. 3 |
For measurements where binding was very weak (-NO), the dissociation constant was obtained from a single site saturation ligand binding model with Rmax constrained, while for tighter binding (+NO), Rmax was allowed to be fit. Fitting was with SigmaPlot:
| Eq. 4 |
RESULTS
The sGC Alpha Chain Inhibits CO Capture By Heme in the Beta Chain
To begin understanding how NO and CO affinity for sGC heme is modulated, we examined CO binding to a variety of sGC proteins lacking specific domains but retaining heme. Measurements were made in the presence or absence of YC-1 or related compounds. We focused on CO binding due to its weaker affinity for ferrous heme as compared with NO, allowing for the measurement of equilibrium dissociation constants, and for its clear response to YC-1 binding. Measurements were made with Ms sGC-NT constructs (Fig. 1), taking advantage of their high stability and high yields from bacterial expression.33,41–43 Ms sGC-NT constructs are heterodimeric proteins lacking the C-terminal cyclase domains while retaining YC-1 binding. We previously demonstrated that YC-1 binding leads to tighter CO and NO binding and to a geminate recombination phase upon CO photolysis.33,41 Here, we extend these studies to include proteins completely lacking the α1 chain and, where needed, using a cuvette with a 10 cm pathlength, allowing for more precise measurement of the tighter binding constants that occur in the presence of YC-1 (Fig. 2, Table 1).
Figure 1.
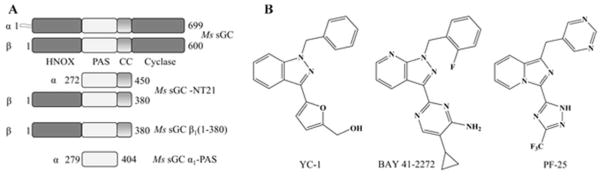
sGC constructs and ligand structures. (A) Schematic representation of the heterodimeric Ms sGC domains, expression constructs and their boundaries. Constructs used in the present study but not shown in the diagram are Ms sGC-NT13 (α1 49–450, β1 1–380), Bt sGC-β1 (1–197) and Bt sGC β1(1–359). (B) Structures of YC-1, BAY 41-2272 and PF-25.
Figure 2.
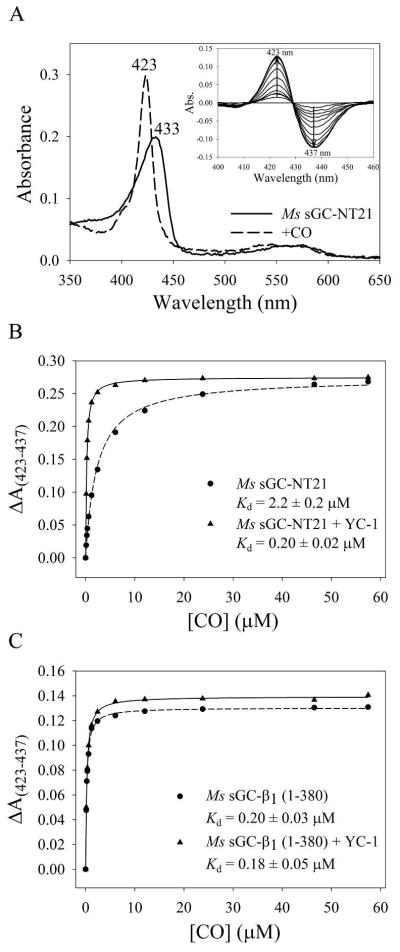
CO saturation binding analysis. (A) Absorption spectra of purified Ms sGC-NT21 before and after CO saturation. The A433/A280 ratio for the unliganded protein is ~1.8, consistent with high purity and full heme incorporation. Inset: difference spectra for Ms sGC-NT21 upon CO titration. (B) CO saturation binding curve for Ms sGC-NT21 ± YC-1, which displays a 10-fold tightening of the CO-dissociation constant upon YC-1 binding. (C) CO saturation binding curve for Ms sGC-β1 (1–380) ± YC-1, which displays little change in CO binding affinity upon YC-1 binding. Titrations were performed in a 10 cm cuvette at room temperature with 0.1 μM protein in buffer containing 50 mM potassium phosphate, pH 7.4, 100 mM KCl, 5% glycerol, and 50 μM YC-1. The data were corrected for dilution upon addition of CO-saturated buffer and were fitted to a single-site saturation model to obtain the CO dissociation constants.
TABLE 1.
CO Dissociation Constants for sGC Proteins
| Protein | α1 | β1 | KdCO (μM) | KdCO′ (μM, +Ligand) | Ref. | |
|---|---|---|---|---|---|---|
| Bt sGC (full length) | 1–691 | 1–619 | 127 | ~26 | YC-1 | 51 |
| 97 | 76 | |||||
| Hs sGC (full length) | 1–690 | 1–619 | 260 | 77 | ||
| Ms sGC-NT2 | 49–471 | 1–401 | 77 ± 7 | 1.7 ± 0.1 | YC-1 | 33 |
| 90 ± 9 | 1.0 ± 0.1 | YC-1 | 43 | |||
| Ms sGC-NT13 | 49–450 | 1–380 | 2.8 ± 0.4 | YC-1 | ||
| 53 ± 4 | 2.9 ± 0.2 | PF-25 | This work | |||
| 0.25 ± 0.02a | BAY | |||||
| Ms sGC-NT19 | 49–450 | 1–380 | 50 ± 3 | 0.8 ± 0.1 | YC-1 | 43 |
| Ms sGC-NT21 | 272–450 | 1–380 | 0.20 ± 0.02a | YC-1 | ||
| 2.2 ± 0.2a | 0.24 ± 0.01a | PF-25 | This work | |||
| 0.07 ± 0.01a | BAY | |||||
| Ms sGC β1(1–380) | absent | 1–380 | 0.20 ± 0.03a | 0.18 ± 0.05a | YC-1 | This work |
| Bt sGC β1(1–197) | absent | 1–197 | 1.6 ± 0.2b | 1.2 ± 0.2b | YC-1 | This work |
| Bt sGC β1(1–359) | absent | 1–359 | 15 ± 4 | 10 ± 3 | YC-1 | This work |
Titration binding data were measured using gas-tight syringes and 1 or 10 cm cuvettes fitted with rubber septum. Protein concentration was 1 μM unless otherwise indicated. Where included for measuring KdCO′, the YC-1 and PF-25 concentrations were 50 μM, and BAY 41-2272 concentrations were 2.5 μM (Ms sGC-NT21) or 10 μM (Ms sGC-NT13). The values listed are the mean and standard deviation of at least three independent measurements.
Measured in a 10 cm cuvette, using 0.1 μM protein.
Measured in a 1 cm cuvette, using 0.5 μM protein.
The Ms sGC coiled-coil likely ends at α1 Pro 460 and β1 Pro 390.16,42,43 We trimmed the C-terminal end of Ms sGC-NT2 by 21 residues to remove a portion of the linker between the coiled-coil and cyclase domains, as well as a small portion of the coiled-coil, yielding Ms sGC-NT13 and Ms sGC-NT19, which are identical except for addition of a Strep purification tag to Ms sGC-NT19. Both proteins display a small increase in CO binding affinity in the absence of YC-1, but no significant change in CO affinity in the presence of YC-1 (Table 1). Values for KdCO obtained with these proteins varied from 50–90 μM in the absence of YC-1, to 0.8–2.8 μM in the presence of YC-1 (referred to as KdCO′). Removal of the α1 H-NOX domain (Ms sGC-NT21) led to considerable tightening of CO binding (KdCO = 2.2 μM and KdCO′ = 0.2 μM, Table 1). These measurements were made in a 10 cm cuvette, allowing for protein concentrations as low as 50 nM to be used and minimizing depletion of free CO through heme binding. Thus, Ms sGC-NT21 binds CO 20–40 fold tighter than Ms sGC proteins containing the α1 H-NOX domain and still responds to YC-1, displaying a 10-fold increase in CO binding affinity when YC-1 is present. These data are consistent with previous studies indicating YC-1-family compounds stimulate NO-dependent catalysis in sGC proteins lacking the first 259 residues of α1.48–50
To further narrow the YC-1 binding location, we examined an Ms sGC protein lacking the entire α1 chain. This protein binds CO with high affinity (~0.2 μM) in the presence or absence of YC-1 (Fig. 2, Table 1). Thus, YC-1 appears to have no effect on CO binding affinity in the absence of the alpha chain and, importantly, CO binding is as tight in the absence of the alpha chain as it is in the presence of YC-1 for any of the heterodimeric Ms sGC proteins, displaying a value for KdCO of ~200 nM. We conclude that both the α1 PAS and H-NOX domains lower CO affinity for the sGC heme. Binding of YC-1 appears to relieve this restraint.
We examined the bovine β1 subunit to extend our results to a mammalian sGC. The bovine protein displays the same overall domain structure as Ms sGC and has an overall sequence identity of 60% with the β1 subunit and 39% with the α1 subunit. We produced two Bt sGC forms, one containing just the H-NOX domain and one containing the H-NOX and PAS domain. As with Ms sGC β1, neither Bt sGC β1 construct displayed YC-1 sensitivity (Table 1). Binding of CO was less tight to the bovine protein, however, and differed between the H-NOX and H-NOX-PAS containing proteins, with the shorter construct having the greatest affinity (~1 μM) and the longer construct binding with about 10-fold less affinity. Homodimer formation in Bt sGC β1(1–359) may contribute to the lower CO affinity (Ms sGC β1 (1–380) behaves as a monomer).
YC-1 Binding Affinity to Ms sGC
The data in Table 1 indicate the YC-1 binding site is within the Ms sGC-NT21 construct and, furthermore, that YC-1 enhancement of CO binding requires an intact α1 PAS domain. Measurement of YC-1 binding affinity for sGC, which is needed to clarify where on the protein binding takes place, is frustrated by the poor solubility of YC-1 in aqueous solutions and by the tendency for YC-1 to bind non-specifically to proteins. We therefore employed a multidimensional binding assay to extract the YC-1 dissociation constant through analysis of the linked equilibria between CO binding and YC-1 binding (Fig. 3, Table 2). For a system displaying linked equilibria, binding of either ligand, in this case CO or YC-1, will affect the binding of the other. We therefore measured CO binding affinity as a function of YC-1 concentration for Ms sGC-NT21 (10 cm cuvette) and Ms sGC-NT13 (1 cm cuvette). Linked equilibria analyses yielded KdYC-1 values of 9.3 μM and 21 μM, respectively, for the two proteins in the absence of NO and CO (Table 2). Cooperativity factors of 14 and 19 (Table 3), reflecting the influence of one ligand on binding of the other, were also derived from these data, from which the dissociation constant for YC-1 binding to the CO saturated protein (KdYC-1′) can be derived. These values were 0.7 μM and 1.1 μM, respectively, for Ms sGC-NT21 and Ms sGC-NT13 (Table 2).
Figure 3.
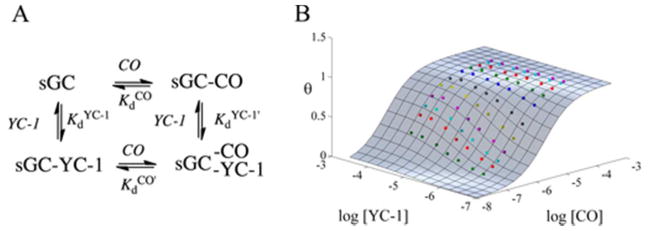
Ligand binding and linked equilibria in sGC. (A) Linked equilibria diagram showing four different states for CO and YC-1 binding to sGC. (B) Global fitting of the fraction of CO sites occupied (θ)to the normalized ΔA(423–437) for Ms sGC-NT21 (10 cm cuvette) using MATLAB. The surface represents the extent of CO binding as a function of CO and YC-1 concentration. Colored points indicate measured ΔA(423–437). Titrations were performed in a 10 cm cuvette at room temperature with 0.1 μM protein in buffer containing 50 mM potassium phosphate, pH 7.4, 100 mM KCl, 5% glycerol, and YC-1 concentrations ranging from 0–50 μM.
TABLE 2.
YC-1, PF-25 and BAY 41-2272 Dissociation Constants for sGC Proteins (μM).
| −NO/CO
|
+CO
|
+NO
|
|||||
|---|---|---|---|---|---|---|---|
| Protein | KdYC-1 | KdPF-25 | KdBAY | KdYC-1′ | KdPF-25′ | KdBAY′ | KdPF-25′ |
| Ms sGC-NT13 | 21 ± 5a | 155 ± 11a | 17 ± 3b | 1.1 ± 0.3a | 3.0 ± 0.3a | 0.08 ± 0.01e | |
| 0.8 ± 0.1d | 2.8 ± 0.2d | ||||||
| Ms sGC-NT21 | 9.3 ± 0.8a | 73 ± 21a | 2.0 ± 0.5a | 0.67 ± 0.06a | 3.8 ± 1.4a | 0.03 ± 0.01a | 11 ± 2c |
| 153 ± 5c | 0.6 ± 0.1e | 0.9 ± 0.3d | 0.09 ± 0.02e | ||||
| Ms sGC β1(1–380) | 92 ± 5c | 7 ± 1c | |||||
| Ms sGC α1 PAS | N.B.c | N.B.c | |||||
| Bt sGC (full length) | 52f | ~8h | |||||
| ~20g | |||||||
From global fitting of multidimensional titration data (see text). For Ms sGC-NT13, titration was in a 1 cm cuvette with 1 μM protein. For Ms sGC-NT21, titration was in a 10 cm cuvette with 0.1 μM protein. Values are the mean and standard deviation of three independent measurements. Values for KdYC-1′ were obtained using the cooperativity factors shown in Table 3.
Estimated assuming linked equilibria: (KdCO/KdCO′) KdBAY′.
From SPR (see text). Values in the absence of NO were fitted with calculated Rmax and those in the presence were fitted with a floating Rmax. Errors are from the fitting. N.B.: No binding.
From fitting of Soret shift (1 cm cuvette). Values are the mean and standard deviation of 3–5 independent measurements. For YC-1, 1 μM protein was used. For PF-25, both 1 and 0.5 μM protein was used.
From fitting of Soret shift (1 and 10 cm cuvette). Values are the mean and standard deviation of 3–10 independent measurements. For YC-1 (10 measurements), measurements with protein concentrations of 0.1 (10 cm cuvette), and 0.5 and 1.0 μM (1 cm cuvette) were included. For BAY 41-2272 (3 measurements), protein concentrations of 0.05 and 0.1 μM were included (10 cm cuvette).
From reference 32, measured in the presence of Mn2+.
From reference 24, EC50 value for stimulating bovine sGC in the absence of NO or CO.
From reference 51, measured in the presence of GTP.
TABLE 3.
Cooperativity Factors for sGC Proteins.
|
Kint (CO)a
|
KdCO/KdCO′
|
KdPF-25/KdPF-25′ (NO)
|
|||||
|---|---|---|---|---|---|---|---|
| Protein | YC-1 | PF-25 | BAY | YC-1 | PF-25 | BAY | PF-25 |
| Ms sGC-NT13 | 19 ± 2 | 53 ± 3 | N.M.b | 19 ± 3 | 18 ± 2 | 212 ± 23 | |
| Ms sGC-NT21 | 13.8 ± 0.3 | 19 ± 4 | 58 ± 7 | 11 ± 1 | 9 ± 1 | 31 ± 5 | 14 ± 3 |
| Ms sGC β1(1–380) | 13 ± 2 | ||||||
From global fitting.
Could not be readily measured by global fitting due to the large cooperativity factor, requiring both 1 and 10 cm cuvettes be used.
We also directly measured YC-1 binding to CO-saturated heterodimeric Ms sGC proteins by monitoring the ~2 nm blue-shift in the Soret absorption band that occurs upon YC-1 binding.41,51 Monitoring of this shift while titrating in YC-1 allowed for estimating compound affinity (Table 2). We examined binding of YC-1 to Ms sGC-NT21-CO and Ms sGC-NT13-CO in 10 cm and 1 cm cuvettes, respectively, which yielded values nearly identical to those obtained from the linked equilibria analysis (Table 2). Likewise, estimating cooperativity factors by linked equilibria analysis or by the ratio of KdCO and KdCO′ yielded similar values (Table 3), indicating good internal consistency between the two approaches. A shift in Soret band maxima was not observed for Ms sGC β1 (1–380).
Relative YC-1, BAY 41–2272 and PF-25 Binding Affinities
We examined binding by compounds BAY 41–2272, which is active at lower concentrations than YC-1,22 and Pfizer compound 25 (PF-25), a recently described compound with greater aqueous solubility.44 PF-25 behaves similarly to other YC-1 family compounds, stimulating human sGC in the presence of NO (EC20 = 80 nM) and relaxing pre-constricted aortic rings (IC50 = 60 nM). Both compounds are derived from YC-1 (Fig. 1), differing mainly through substitution of the YC-1 furan ring. Affinity measurements were made using linked equilibria and/or Soret band shift. Interestingly, differences were observed among the compounds for binding to both the unliganded and CO-liganded proteins, and for their associated cooperativity factors. As with CO, YC-1 family compounds also bind tighter to Ms sGC-NT21 than to Ms sGC-NT13, indicating that the α1 H-NOX domain not only interferes with CO binding but also interferes with YC-1 family compound binding (Table 2). The effect of H-NOX removal on stimulator binding is greater for BAY 41–2272 (~9-fold) than for YC-1 or PF-25 (~2-fold).
Binding of stimulator compounds is much tighter to the CO complexes than to the unliganded proteins. The greatest enhancement is seen for BAY 41–2272, which binds to the CO complex with Kd = 30–90 nM, whereas YC-1 binds with Kd = ~1 μM and PF-25 with Kd = ~3 μM (Table 2). PF-25 binds particularly poorly to the unliganded proteins (70–150 μM).
Measuring PF-25 Binding Using Surface Plasmon Resonance
We hypothesized that YC-1-family compounds bind to the α1 PAS domain since (i) PAS domains commonly bind small molecules in their capacity as signaling proteins;11 (ii) the α1 PAS domain is required for observing a YC-1-dependent enhancement in CO affinity (Table 1); and (iii) addition of YC-1 did not lead to a shift in the Soret absorption band for any of the proteins lacking the alpha chain. To directly address whether YC-1 binding is to the α1 PAS domain, we used surface plasmon resonance (SPR), allowing for binding measurements that were not dependent on heme spectra. To accomplish this, we specifically biotinylated Ms sGC NT21, α1 PAS and β1(1–380) at their C-termini, using biotin ligase (BirA) and sGC proteins modified to contain the BirA recognition sequence.52 The biotinylated proteins were captured in the SPR instrument on a NeutrAvidin-coated sensor chip and analyte binding examined. Unfortunately, neither YC-1 nor BAY 41–2272 was suitable for SPR binding measurements because of their poor solubility in aqueous buffer. Introducing these compounds required a greater percentage of DMSO than was well tolerated by the protein over the course of the measurements, which take hours to complete. We therefore turned to PF-25, a compound better suited to SPR measurements due to its greater aqueous solubility.
Binding of PF-25 to Ms sGC NT21 was clearly observed in the SPR instrument (Fig. 5). Binding was rapid, as expected from our CO titration experiments, making kon measurements unreliable. Release was also rapid and difficult to quantify. Ligand affinity was therefore estimated through saturation binding, leading to KdPF-25 = 153 μM, a value similar to that observed through analysis by linked equilibria. Unexpectedly, binding to Ms sGC β1(1–380) was also observed (KdPF-25 = 92 μM). In contrast, binding to Ms sGC α1 PAS was not observed. Thus, these data indicated PF-25 binds to the β1 chain between residues 1–380.
Figure 5.

PF-25 binding to Ms sGC constructs examined by surface plasmon resonance. Biotinylated Ms sGC-NT21, Ms sGC α1 PAS and Ms sGC β1(1–380) were captured on an SPR chip containing immobilized NeutrAvidin. Solutions of PF-25 ± DEA/NO were injected into the chip and response change recorded. The injected samples contained 50 mM potassium phosphate buffer, pH 7.4, 100 mM KCl, 2 mM EDTA, 1 mM TCEP, 1% DMSO, PF-25 (0–142.5 μM) and, where included, 25 μM DEA/NO. Shown are representative sensograms for PF-25 binding in the presence (a–c) and absence (d–f) of DEA/NO. Three trials are shown for each PF-25 concentration. Binding data were analyzed with the Biacore T-100 evaluation software. Values for Kd were obtained using a single-site saturation model (SigmaPlot) by plotting Response (RU) with respect to PF-25 concentration using floating Rmax (g) or calculated Rmax (h).
Measuring binding of PF-25 to the CO-saturated protein was not possible because the SPR instrument has an in-line degasser to prevent bubble formation. We therefore examined binding after addition of NO to the heme containing constructs, which could be saturated with small amounts of NO released in situ. Unlike with CO, only nanomolar concentrations of NO were required for these measurements due to the high affinity of NO for sGC heme (picomolar to nanomolar) and the small amount of protein captured on the chip surface (picomoles). NO binding to heme enhanced PF-25 binding to Ms sGC NT21 as expected (KdPF-25′ = 11 μM), but, in contrast to CO, also enhanced PF-25 binding to Ms sGC β1(1–380) (Fig. 5, Table 2), yielding KdPF-25′ = 7 μM. Surprisingly, PF-25 also bound slightly weaker to Ms sGC-NT21-NO than to Ms sGC-NT21-CO. The reason for these differences in binding is not yet clear but is presumably due to differences in the conformations of the NO and CO complexes.
YC-1 Binding to Ms sGC PAS Domains is Not Observed by Fluorescence Anisotropy
We examined YC-1 binding to PAS domains in solution using fluorescence anisotropy. Various proteins were titrated into a YC-1 containing solution and changes in YC-1-dependent fluorescence anisotropy were monitored. Weak binding (>100 μM) was indicated for Ms sGC α1 PAS, but not for lysozyme. However, the data were extremely noisy, non-saturable and slow to equilibrate, suggesting the observed interaction was non-specific. Likewise, a weak signal was also seen for the β1 PAS domain, although less substantial, and also likely to be due to non-specificity. Binding to heme-containing Ms sGC proteins could not be measured due to signal quenching by the heme. Although inconclusive, these data indicate the individual sGC PAS domains do not contain the YC-1 binding site, consistent with our SPR data.
DISCUSSION
YC-1 family compounds show great promise for the treatment of cardiovascular disease through their stimulation of sGC, yet their mechanism of action remains unknown. Likewise, the mechanism by which NO binding to sGC heme stimulates cyclase activity is unclear. Here, we show that (i) the α1 PAS domain inhibits CO – and presumably NO – binding to the β1 heme domain; (ii) binding of YC-1 overcomes this inhibition; (iii) binding of YC-1 and CO or NO to heterodimeric sGC display linked equilibria, with binding of one enhancing binding of the other; (iv) monomeric β1 sGC displays high CO affinity and loss of CO/YC-1 linked equilibria; and (v) YC-1 binding is to the β1 chain, most likely in the heme domain. A model emerges from our study in which the α1 H-NOX and PAS domains act to inhibit the β1 H-NOX domain, which, in turn, acts to inhibit the cyclase domain. Activation of cyclase can occur through the relieving of either inhibitory contact; maximal activity occurs when both inhibitory contacts are removed. In what follows, we expand upon these findings.
YC-1 Binding to or Near the sGC Heme Domain
Discovery of the YC-1 binding site has been challenging due to the difficulty in working with sGC protein. Suggestions in the literature for where YC-1 binds invoke nearly all sGC domains, including the α1 H-NOX domain,33,34,53 the catalytic domain,31,32,54 and the heme domain.35–38,55,56 Here, using SPR, we demonstrate that binding is to the N-terminal portion of the β1 chain. Since binding could not be detected to the β1 PAS domain by fluorescence anisotropy measurements, binding is most likely to the heme-containing β1 H-NOX domain. Additionally, a recent report notes that a human sGC construct lacking both H-NOX domains is not stimulated by BAY-41–2272, while full-length sGC is.57
Binding of YC-1 leads to an ~2 nm shift in the Soret absorption band for the CO complex,38,41,55 which we used to measure the YC-1 dissociation constants to be 0.8 and 0.6 μM, respectively, for Ms sGC-NT13-CO and Ms sGC NT21-CO (Fig. 4, Table 2). Others have used this shift in absorbance,38,55 or a shift in the Raman heme spectrum upon addition of YC-1 or BAY 41–2272,35–37,56,58 to argue that YC-1 family compounds bind to the heme domain. However, the high concentrations of YC-1 used in those studies (generally 200 μM) raises the concern that the spectral changes may have arisen from non-specific binding. Here, we demonstrate that only nanomolar quantities of YC-1 are required for inducing the shift in the Ms sGC-NT-CO Soret absorption band, consistent with a specific binding event.
Figure 4.

YC-1 binding to Ms sGC-NT-CO. Representative curve for PF-25 binding to Ms sGC-NT21-CO. Ms sGC-NT-CO complexes were prepared in either 1 cm (1 μM protein, shown) or 10 cm (0.1 μM protein, BAY 41-2272) septa-capped cuvettes containing CO-saturated buffer (50 mM potassium phosphate, pH 7.4, 100 mM KCl, 5% glycerol). YC-1 family compounds (1 mM or 15 mM in ethanol) were titrated into the cuvettes and the shift in the Soret band monitored (ΔA(416–427)). The data were fitted to a single-site saturation model to obtain the dissociation constants for compound binding to the Ms sGC-NT-CO complex. Inset: difference spectra upon compound titration. Ethanol or DMSO alone, or other unrelated ring-containing compounds, do not generate a shift in the Soret band (data not shown).
YC-1 readily enhances CO binding in Ms sGC-NT proteins (Table 1; reference 33), which lack the cyclase domain, but does not stimulate the isolated cyclase domains.59 YC-1 also readily enhances CO binding to Ms sGC-NT21 (Table 1; reference 43), which does not contain the α1 H-NOX domain, ruling out the α1 H-NOX domain as containing the binding site. A similar conclusion was reached for truncated versions of mammalian sGC proteins.48–50 These data, taken together with our SPR and spectral data, provide a compelling argument for heme-domain binding of YC-1 family compounds.
Results using the bovine protein are qualitatively the same as for the Manduca protein, but differ in the dissociation constants for CO binding. Bt sGC β1(1–197) binds CO ~8-fold less tightly than Ms sGC β1(1–380) (Table 1), while binding by Bt sGC β1(1–359) is weaker yet, ~9-fold weaker than binding by Bt sGC β1(1–197). Neither bovine protein displays a significant response to YC-1. The difference in CO affinity between Bt sGC β1(1–197) and Bt sGC β1(1–359) may result from stabilization of the lower affinity form of the H-NOX domain upon homodimer formation in the larger protein, or from interactions between the H-NOX and PAS domains.
YC-1 Binding Relieves Inhibition of the β1 Heme Domain by the α1 H-NOX and PAS Domains
Perhaps our most unexpected finding is that the affinity of CO for heme in the β1 H-NOX domain is as high in the absence of other domains as it is in the presence of YC-1 (Table 1). We showed previously that YC-1 binding leads to tighter CO binding in Ms sGC-NT constructs33 and that binding leads to the development of a geminate recombination phase upon CO photolysis.41 Negrerie and co-workers recently showed that the isolated human H-NOX domain (β1 1–190) also displays a geminate rebinding phase whereas the full-length protein does not.60 This tighter binding and trapped CO correlates with increased cyclase activity for full-length sGC proteins.25,61 We now show that both the α1 PAS and α1 H-NOX domains impair CO binding (Table 1). Removal of the α1 H-NOX domain enhances CO binding by 24-fold, and complete removal of the α1 chain enhances CO binding by 265-fold (Table 1). CO binding to Ms sGC β1(1–380) in the absence of YC-1 is as tight (KdCO = 0.20 μM) as to any of the Ms sGC-NT proteins in the presence of YC-1. Cooperativity in CO binding to any of the constructs missing the α1 chain is now largely absent (Kint ~1) and YC-1 binding does not appreciably enhance CO binding (Table 1). In addition to tightly binding CO, Ms sGC β1(1–380) presumably also binds tightly to YC-1 and BAY 41–2272.
The situation with NO binding is similar to that for CO, but apparently differs with respect to cooperativity. Unlike with CO, NO binding leads to proximal histidine cleavage and a five-coordinate Fe-NO complex. NO release is multiphasic. We previously showed that YC-1 binding to Ms sGC-NT-NO eliminated the faster of two NO release rates, yielding a protein with higher NO affinity, much as occurs with CO.33 However, unlike with CO, the data in Table 2 indicate that binding of NO enhances binding of PF-25 to Ms sGC β1(1–380) by ~10-fold, indicating that cooperativity occurs for NO with this protein but not for CO. The reason for this difference is unclear but may have to do with the difference in heme domain conformation for the five-coordinate and six-coordinate Ms sGC proteins.
We recently determined the crystal structure of Ms sGC α1 PAS17 and a molecular model for domain packing based on homology modeling, chemical cross-linking and SAXS analysis.43 These data indicate a direct contact between one face of the Fα helix in Ms sGC α1 PAS, involving residues Glu 340 and Lys 343, and the subdomain containing proximal His 105 that is proposed to be involved in signal transduction62–64 (Fig. 6). This contact in the intact protein may serve to stabilize the H-NOX domain in a low affinity conformation. A pocket in this subdomain identified in our homology model may be the YC-1 binding site and provide a means for counteracting the effects of α1 PAS inhibition. Our cross-linking data also indicate direct contact between the α1 and β1 H-NOX domains (Fig. 6). This contact is farther from the heme and may serve to enhance the α1 PAS/β1 H-NOX interaction.
Figure 6.

Contact residues between α1 H-NOX, α1 PAS and β1 H-NOX domains. Shown is the crystal structure for the α1 PAS domain (PDB entry 4GJ4, ref 17) and homology models for the α1 and β1 H-NOX domains. Cross-links between the two domains identified by mass spectrometry are shown,43 as are the heme and proximal histidine in the β1 H-NOX domain. Figure prepared using PyMOL (W. L. DeLano, http://www.pymol.org).
Binding Affinity of YC-1 Family Compounds
Just as YC-1 binding enhances CO binding to heterodimeric sGC, CO binding enhances YC-1 binding, highlighting both the allosteric nature of sGC and the linked equilibria between CO and YC-1 binding events. Binding to Ms sGC-NT13 in the absence of CO (Fig. 3, Table 2) varies from ~20 μM for YC-1 and BAY 41–2272, to 155 μM for PF-25. Binding to Ms sGC-NT21 is slightly tighter than to Ms sGC-NT13, particularly for BAY 41–2272, indicating that the α1 H-NOX domain inhibits not only CO binding, but also binding by stimulator compounds.
Binding of YC-1 family compounds to Ms sGC-NT-CO complexes is 10–200 fold tighter than to Ms sGC-NT (Fig. 4, Tables 2 and 3). BAY 41–2272 binds particularly tightly to the CO complexes, displaying dissociation constants of 30–90 nM. Thus, the enhanced stimulation of sGC by BAY 41–2272 as compared to other YC-1 family compounds appears to be due to especially tight binding to the active conformation.
Binding of PF-25 to Ms sGC-NT21-NO, examined using SPR, was ~2-fold weaker than to Ms sGC β1(1–380)-NO, consistent with the unliganded and CO-bound binding studies (Table 2), and consistent with a model in which the α1 PAS domain inhibits binding of CO, NO or YC-1 family compounds. Interestingly, PF-25 binding to Ms sGC-NT21-NO is 3–12 fold weaker than to Ms sGC-NT21-CO.
The YC-1 dissociation constants we measure for the unliganded and truncated Ms sGC proteins are similar to those reported for binding and stimulating the full-length protein. Binding to full-length bovine lung sGC was monitored using equilibrium dialysis, yielding Kd = 52 μM when measured in the presence of Mn2+,32 and by the effective concentration needed to achieve 50% maximal stimulation of unliganded bovine sGC, yielding EC50 ~20 μM.24 These values compare favorably with stimulator binding to Ms sGC-NT13 (KdYC-1 = 21 μM). Additionally, similar concentrations are needed for BAY 41–2272 to bind Ms sGC-NT21-CO (KdBAY′ = 0.09 μM) as for BAY 41–2272 to stimulate sGC in pre-constricted aortic rings (EC50 = 0.3 μM, reference 34). This agreement between binding affinity for Ms sGC-NT and the concentration needed to stimulate activity in full-length protein provides confidence that our measurements reflect the functionally important binding events.
A Model for sGC Regulation
The model that emerges from our data is one in which the low energy state for the heme pocket differs between the isolated β1 H-NOX domain and heterodimeric sGC. In the isolated domain, the heme pocket traps CO and NO, leading to high affinity for these ligands. In heterodimeric sGC, the heme pocket changes conformation such that NO and CO can more readily escape and binding affinity is reduced. Binding of YC-1 family compounds to heterodimeric sGC alters or severs the connection between the heme domain and α1 subunit, returning the heme domain to its high-affinity conformation. A prediction of this model is that CO/NO binding will induce the high-affinity heme domain conformation, leading to tighter binding by YC-1 family compounds. Linked equilibria were in fact observed: YC-1 family compounds bind 10–200 fold more tightly in the presence of CO or NO than in their absence (Table 2).
A second prediction of this model is that the low-affinity H-NOX conformation inhibits cyclase activity, while the high-affinity conformation relieves this inhibition or is stimulatory. In this way, binding of either YC-1 or CO/NO leads to cyclase activation, and binding of both yields full sGC activation. Support for this model comes from studies of sGC mutant β1H105C, in which the heme proximal histidine is mutated to cysteine, leading to heme-free sGC and a presumably high-affinity conformation for the H-NOX domain. This protein displays high basal catalytic activity and can still be stimulated by YC-1.65,66 Additional support for the model comes from studies of the rat cyclase domain alone and after addition of the H-NOX domain in trans, which leads to inhibition of cyclase activity67 and protection of cyclase from hydrogen/deuterium exchange.64 Although inhibition was insensitive to NO, these data support a model in which the H-NOX domain directly binds to and inhibits the cyclase domain.67 That the high-affinity heme domain conformation may in fact stimulate cyclase activity rather than simply remove an inhibitory contact is suggested by experiments with sGC lacking both H-NOX domains, which displays only basal catalytic activity.57 Interestingly, this protein is also insensitive to BAY 41–2272.
A third prediction of this model is that linked equilibria should exist between nucleotide binding to the cyclase domain and ligand binding to the heme domain. That such a link exists has been previously described.68–70 In the presence of nucleotide, NO release is slowed,69 full stimulation by NO enhanced68 and desensitization delayed.70
How the β1 H-NOX domain switches from low to high CO and NO affinity is unknown. One recently proposed possibility is that the proximal heme pocket is strained in the low-affinity form, leading to heme distortion and inherently poor CO and NO binding affinity. Upon bond breakage, CO and NO escape rather than rebind to heme.38,60,71,72 In the high-affinity state, proximal strain is relieved by moving of the proximal histidine and heme iron into the plane of the porphyrin ring, yielding greater CO/NO affinity and higher capture rate.72 This model is reminiscent of the “relaxed” and “tense” states described for hemoglobin.
A second possibility is for the heme domain to adopt “open” and “closed” conformations, in which the closed conformation hinders CO/NO escape and favors capture by heme. This is the strategy employed by Rhodnius prolixus nitrophorin 4, an NO transport protein. In nitrophorin 4, two loops collapse into the heme pocket at low pH, generating a closed conformation, increased geminate recombination and higher affinity for NO.73–75 Distinguishing between these models awaits high-resolution structure determinations.
Figure 7.

Model for sGC regulation. Shown is a proposed model for allosteric regulation in sGC in which the β1 H-NOX is in equilibria between high- and low-affinity conformations. YC-1, NO, CO and the absence of α1 chain H-NOX and PAS domains all shift the equilibria toward high affinity, while the α1 chain H-NOX and PAS domains shift the equilibria towards low affinity. The low-affinity conformer is inhibitory toward the cyclase domains, while the high-affinity conformer is non-inhibitory or possibly stimulatory.
Acknowledgments
Funding Sources: This work was supported in part by grants from the National Institutes of Health (HL062969 to WRM and U54 CA143924 to WRM and MJG), and the American Heart Association (11PRE7610113 to RP, and 10SDG2600345 to EDG).
We thank Alex Hailey, and Scott Ogley for help with evaluating expression of PAS domains containing differing C-termini. We are grateful to Jacquie Brailey for help with protein expression and to Dr. Patrick Loll for providing us with his SUMO-fusion ligation independent cloning vector. We thank Dr. Lee Roberts for Pfizer compound PF-25 and Dr. Katrina Miranda for DEA/NO. SPR data were acquired by the Arizona Proteomics Consortium supported by NIEHS grant ES06694 to the Southwest Environmental Health Sciences Center, NIH/NCI grant CA023074 to the Arizona Cancer Center and by the BIO5 Institute of the University of Arizona. The Biacore T100 biosensor was provided though generous support of the Prescott Friends of the Sarver Heart Center with leadership gifts from Jim and Linda Lee, Ron and Laura James, and Swayze and Kathy McCraine.
ABBREVIATIONS
- sGC
soluble guanylyl cyclase
- Ms sGC
Manduca sexta sGC
- Bt sGC
Bos taurus (bovine) sGC
- DEA/NO
2-(N,N-Diethylamino)-diazenolate-2-oxide
- CC
coiled coil
- PAS domain
Per-ARNT-Sim domain
- H-NOX domain
heme-nitric oxide/oxygen binding domain
- SPR
surface plasmon resonance
- SAXS
small angle X-ray scattering
Footnotes
ADDITIONAL NOTE
Judy Hines, Leida Rassouli-Taylor, Judith Burstyn, and Elsa Garcin, Raman studies of bovine soluble guanylate cyclase, in preparation.
References
- 1.Ignarro LJ, editor. Nitric Oxide Biology and Pathobiology. 2. Academic Press; San Diego: 2010. [Google Scholar]
- 2.Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens (Greenwich) 2008;10:304–310. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coggins MP, Bloch KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol. 2007;27:1877–1885. doi: 10.1161/ATVBAHA.107.142943. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Poulos TL. Structure-function studies on nitric oxide synthases. J Inorg Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Stuehr DJ, Tejero J, Haque MM. Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunton TL. Use of Nitrite of Amyl in Angina Pectoris. Lancet. 1867;90:97–98. [Google Scholar]
- 7.Murrell W. Nitro-glycerine as a remedy for angina pectoris. Lancet. 1879;113:113–115. [Google Scholar]
- 8.Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- 9.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 11.Moglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Ruoho AE, Rao VD, Hurley JH. Catalytic mechanism of the adenylyl and guanylyl cyclases: Modeling and mutational analysis. Proc Natl Acad Sci USA. 1997;94:13414–13419. doi: 10.1073/pnas.94.25.13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dierks EA, Hu S, Vogel KM, Yu AE, Spiro TG, Burstyn JN. Demonstration of the role of scission of the proximal histidine-iron bond in the activation of soluble guanylyl cyclase though metalloporphyrin substitution studies. J Am Chem Soc. 1997;119:7316–7323. [Google Scholar]
- 14.Stone JR, Marletta MA. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry. 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 15.Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, Böhme E, Schultz G, Koesling D. Mutation of His-105 in the β1 subunit yields a nitric oxide- insensitive form of soluble guanylate cyclase. Proc Natl Acad Sci USA. 1994;91:2592–2596. doi: 10.1073/pnas.91.7.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma X, Beuve A, van den Akker F. Crystal structure of the signaling helix coiled-coil domain of the beta1 subunit of the soluble guanylyl cyclase. BMC structural biology. 2010;10:2. doi: 10.1186/1472-6807-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purohit R, Weichsel A, Montfort WR. Crystal structure of the Alpha subunit PAS domain from soluble guanylyl cyclase. Protein Sci. 2013;22:1439–1444. doi: 10.1002/pro.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allerston CK, von Delft F, Gileadi O. Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS One. 2013;8:e57644. doi: 10.1371/journal.pone.0057644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma X, Sayed N, Baskaran P, Beuve A, van den Akker F. PAS-mediated Dimerization of Soluble Guanylyl Cyclase Revealed by Signal Transduction Histidine Kinase Domain Crystal Structure. J Biol Chem. 2008;283:1167–1178. doi: 10.1074/jbc.M706218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- 24.Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 25.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: Implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 26.Ramanathan S, Mazzalupo S, Boitano S, Montfort WR. Thrombospondin-1 and angiotensin II inhibit soluble guanylyl cyclase through an increase in intracellular calcium concentration. Biochemistry. 2011;50:7787–7799. doi: 10.1021/bi201060c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belik J. Riociguat, an oral soluble guanylate cyclase stimulator for the treatment of pulmonary hypertension. Curr Opin Investig Drugs. 2009;10:971–979. [PubMed] [Google Scholar]
- 28.Mittendorf J, Weigand S, Alonso-Alija C, Bischoff E, Feurer A, Gerisch M, Kern A, Knorr A, Lang D, Muenter K, Radtke M, Schirok H, Schlemmer KH, Stahl E, Straub A, Wunder F, Stasch JP. Discovery of riociguat (BAY 63–2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. ChemMedChem. 2009;4:853–865. doi: 10.1002/cmdc.200900014. [DOI] [PubMed] [Google Scholar]
- 29.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 30.Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, Mayer E, Simonneau G, Wilkins MR, Fritsch A, Neuser D, Weimann G, Wang C. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–329. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- 31.Lamothe M, Chang FJ, Balashova N, Shirokov R, Beuve A. Functional characterization of nitric oxide and YC-1 activation of soluble guanylyl cyclase: structural implication for the YC-1 binding site? Biochemistry. 2004;43:3039–3048. doi: 10.1021/bi0360051. [DOI] [PubMed] [Google Scholar]
- 32.Yazawa S, Tsuchiya H, Hori H, Makino R. Functional characterization of two nucleotide-binding sites in soluble guanylate cyclase. J Biol Chem. 2006;281:21763–21770. doi: 10.1074/jbc.M508983200. [DOI] [PubMed] [Google Scholar]
- 33.Hu X, Murata LB, Weichsel A, Brailey JL, Roberts SA, Nighorn A, Montfort WR. Allostery in recombinant soluble guanylyl cyclase from Manduca sexta. J Biol Chem. 2008;283:20968–20977. doi: 10.1074/jbc.M801501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Gerzer R, Minuth T, Perzborn E, Pleiss U, Schröder H, Schroeder W, Stahl E, Steinke W, Straub A, Schramm M. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 35.Ibrahim M, Derbyshire ER, Marletta MA, Spiro TG. Probing soluble guanylate cyclase activation by CO and YC-1 using resonance Raman spectroscopy. Biochemistry. 2010;49:3815–3823. doi: 10.1021/bi902214j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ibrahim M, Derbyshire ER, Soldatova AV, Marletta MA, Spiro TG. Soluble guanylate cyclase is activated differently by excess NO and by YC-1: resonance Raman spectroscopic evidence. Biochemistry. 2010;49:4864–4871. doi: 10.1021/bi100506j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin E, Czarnecki K, Jayaraman V, Murad F, Kincaid J. Resonance Raman and infrared spectroscopic studies of high-output forms of human soluble guanylyl cyclase. J Am Chem Soc. 2005;127:4625–4631. doi: 10.1021/ja0440912. [DOI] [PubMed] [Google Scholar]
- 38.Yoo BK, Lamarre I, Rappaport F, Nioche P, Raman CS, Martin JL, Negrerie M. Picosecond to second dynamics reveals a structural transition in Clostridium botulinum NO-sensor triggered by the activator BAY-41-2272. ACS Chem Biol. 2012;7:2046–2054. doi: 10.1021/cb3003539. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt PM, Schramm M, Schroder H, Wunder F, Stasch JP. Identification of residues crucially involved in the binding of the heme moiety of soluble guanylate cyclase. J Biol Chem. 2004;279:3025–3032. doi: 10.1074/jbc.M310141200. [DOI] [PubMed] [Google Scholar]
- 40.Schindler U, Strobel H, Schonafinger K, Linz W, Lohn M, Martorana PA, Rutten H, Schindler PW, Busch AE, Sohn M, Topfer A, Pistorius A, Jannek C, Mulsch A. Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol. 2006;69:1260–1268. doi: 10.1124/mol.105.018747. [DOI] [PubMed] [Google Scholar]
- 41.Hu X, Feng C, Hazzard JT, Tollin G, Montfort WR. Binding of YC-1 or BAY 41-2272 to Soluble Guanylyl Cyclase Induces a Geminate Phase in CO Photolysis. J Am Chem Soc. 2008;130:15748–15749. doi: 10.1021/ja804103y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fritz BG, Hu X, Brailey JL, Berry RE, Walker FA, Montfort WR. Oxidation and loss of heme in soluble guanylyl cyclase from Manduca sexta. Biochemistry. 2011;50:5813–5815. doi: 10.1021/bi200794c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fritz BG, Roberts SA, Ahmed A, Breci L, Li W, Weichsel A, Brailey JL, Wysocki VH, Tama F, Montfort WR. Molecular Model of a Soluble Guanylyl Cyclase Fragment Determined by Small-Angle X-ray Scattering and Chemical Cross-Linking. Biochemistry. 2013;52:1568–1582. doi: 10.1021/bi301570m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts LR, Bradley PA, Bunnage ME, England KS, Fairman D, Fobian YM, Fox DN, Gymer GE, Heasley SE, Molette J, Smith GL, Schmidt MA, Tones MA, Dack KN. Acidic triazoles as soluble guanylate cyclase stimulators. Bioorg Med Chem Lett. 2011;21:6515–6518. doi: 10.1016/j.bmcl.2011.08.071. [DOI] [PubMed] [Google Scholar]
- 45.Weeks SD, Drinker M, Loll PJ. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr Purif. 2007;53:40–50. doi: 10.1016/j.pep.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 48.Koglin M, Behrends S. A functional domain of the alpha1 subunit of soluble guanylyl cyclase is necessary for activation of the enzyme by nitric oxide and YC-1 but is not involved in heme binding. J Biol Chem. 2003;278:12590–12597. doi: 10.1074/jbc.M212740200. [DOI] [PubMed] [Google Scholar]
- 49.Kraehling JR, Busker M, Haase T, Haase N, Koglin M, Linnenbaum M, Behrends S. The amino-terminus of nitric oxide sensitive guanylyl cyclase alpha(1) does not affect dimerization but influences subcellular localization. PLoS One. 2011;6:e25772. doi: 10.1371/journal.pone.0025772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharina IG, Jelen F, Bogatenkova EP, Thomas A, Martin E, Murad F. Alpha1 soluble guanylyl cyclase (sGC) splice forms as potential regulators of human sGC activity. J Biol Chem. 2008;283:15104–15113. doi: 10.1074/jbc.M710269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kharitonov VG, Sharma VS, Magde D, Koesling D. Kinetics and equilibria of soluble guanylate cyclase ligation by CO: Effect of YC-1. Biochemistry. 1999;38:10699–10706. doi: 10.1021/bi990277f. [DOI] [PubMed] [Google Scholar]
- 52.Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 53.Pal B, Kitagawa T. Binding of YC-1/BAY 41-2272 to soluble guanylate cyclase: A new perspective to the mechanism of activation. Biochem Biophys Res Commun. 2010;397:375–379. doi: 10.1016/j.bbrc.2010.05.122. [DOI] [PubMed] [Google Scholar]
- 54.Makino R, Yazawa S, Hori H, Shiro Y. Interactions of soluble guanylate cyclase with a P-site inhibitor: effects of gaseous heme ligands, azide, and allosteric activators on the binding of 2′-deoxy-3′-GMP. Biochemistry. 2012;51:9277–9289. doi: 10.1021/bi3004044. [DOI] [PubMed] [Google Scholar]
- 55.Sharma VS, Magde D. Activation of soluble guanylate cyclase by carbon monoxide and nitric oxide: A mechanistic model. Methods. 1999;19:494–505. doi: 10.1006/meth.1999.0892. [DOI] [PubMed] [Google Scholar]
- 56.Li Z, Pal B, Takenaka S, Tsuyama S, Kitagawa T. Resonance Raman evidence for the presence of two heme pocket conformations with varied activities in CO-bound bovine soluble guanylate cyclase and their conversion. Biochemistry. 2005;44:939–946. doi: 10.1021/bi0489208. [DOI] [PubMed] [Google Scholar]
- 57.Sharina I, Sobolevsky M, Doursout MF, Gryko D, Martin E. Cobinamides are novel coactivators of nitric oxide receptor that target soluble guanylyl cyclase catalytic domain. J Pharmacol Exp Ther. 2012;340:723–732. doi: 10.1124/jpet.111.186957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denninger JW, Schelvis JPM, Brandish PE, Zhao Y, Babcock GT, Marletta MA. Interaction of soluble guanylate cyclase with YC-1: Kinetic and resonance Raman studies. Biochemistry. 2000;39:4191–4198. doi: 10.1021/bi992332q. [DOI] [PubMed] [Google Scholar]
- 59.Derbyshire ER, Fernhoff NB, Deng S, Marletta MA. Nucleotide regulation of soluble guanylate cyclase substrate specificity. Biochemistry. 2009;48:7519–7524. doi: 10.1021/bi900696x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoo BK, Lamarre I, Martin JL, Negrerie M. Quaternary structure controls ligand dynamics in soluble guanylate cyclase. J Biol Chem. 2012;287:6851–6859. doi: 10.1074/jbc.M111.299297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friebe A, Koesling D. Mechanism of YC-1-induced activation of soluble guanylate cyclase. Mol Pharmacol. 1998;53:123–127. doi: 10.1124/mol.53.1.123. [DOI] [PubMed] [Google Scholar]
- 62.Baskaran P, Heckler EJ, van den Akker F, Beuve A. Identification of residues in the heme domain of soluble guanylyl cyclase that are important for basal and stimulated catalytic activity. PLoS One. 2011;6:e26976. doi: 10.1371/journal.pone.0026976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baskaran P, Heckler EJ, van den Akker F, Beuve A. Aspartate 102 in the heme domain of soluble guanylyl cyclase has a key role in NO activation. Biochemistry. 2011;50:4291–4297. doi: 10.1021/bi2004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Underbakke ES, Iavarone AT, Marletta MA. Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc Natl Acad Sci USA. 2013;110:6777–6782. doi: 10.1073/pnas.1301934110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin E, Lee YC, Murad F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc Natl Acad Sci USA. 2001;98:12938–12942. doi: 10.1073/pnas.231486198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin E, Sharina I, Kots A, Murad F. A constitutively activated mutant of human soluble guanylyl cyclase (sGC): implication for the mechanism of sGC activation. Proc Natl Acad Sci USA. 2003;100:9208–9213. doi: 10.1073/pnas.1633590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry. 2005;44:4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 68.Russwurm M, Koesling D. NO activation of guanylyl cyclase. Embo J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cary SP, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc Natl Acad Sci USA. 2005;102:13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai AL, Berka V, Sharina I, Martin E. Dynamic ligand exchange in soluble guanylyl cyclase (sGC): implications for sGC regulation and desensitization. J Biol Chem. 2011;286:43182–43192. doi: 10.1074/jbc.M111.290304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsai AL, Martin E, Berka V, Olson JS. How do heme-protein sensors exclude oxygen? Lessons learned from cytochrome c′, Nostoc puntiforme heme nitric oxide/oxygen-binding domain, and soluble guanylyl cyclase. Antioxid Redox Signal. 2012;17:1246–1263. doi: 10.1089/ars.2012.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai AL, Berka V, Martin E, Olson JS. A “sliding scale rule” for selectivity among NO, CO, and O(2) by heme protein sensors. Biochemistry. 2012;51:172–186. doi: 10.1021/bi2015629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weichsel A, Andersen JF, Roberts SA, Montfort WR. Reversible nitric oxide binding to nitrophorin 4 from Rhodnius prolixus involves complete distal pocket burial. Nat Struct Biol. 2000;7:551–554. doi: 10.1038/76769. [DOI] [PubMed] [Google Scholar]
- 74.Kondrashov DA, Montfort WR. Nonequilibrium dynamics simulations of nitric oxide release: comparative study of nitrophorin and myoglobin. J Phys Chem B. 2007;111:9244–9252. doi: 10.1021/jp071136n. [DOI] [PubMed] [Google Scholar]
- 75.Benabbas A, Ye X, Kubo M, Zhang Z, Maes EM, Montfort WR, Champion PM. Ultrafast dynamics of diatomic ligand binding to nitrophorin 4. J Am Chem Soc. 2010;132:2811–2820. doi: 10.1021/ja910005b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stone JR, Marletta MA. The ferrous heme of soluble guanylate cyclase: formation of hexacoordinate complexes with carbon monoxide and nitrosomethane. Biochemistry. 1995;34:16397–16403. doi: 10.1021/bi00050a021. [DOI] [PubMed] [Google Scholar]
- 77.Martin E, Berka V, Bogatenkova E, Murad F, Tsai AL. Ligand selectivity of soluble guanylyl cyclase: effect of the hydrogen-bonding tyrosine in the distal heme pocket on binding of oxygen, nitric oxide, and carbon monoxide. J Biol Chem. 2006;281:27836–27845. doi: 10.1074/jbc.M601078200. [DOI] [PubMed] [Google Scholar]