

Abstract
We developed synthetic chemistry to access the marine alkaloid rigidins and over forty synthetic analogues based on the 7-deazaxanthine, 7-deazaadenine, 7-deazapurine and 7-deazahypoxanthine skeletons. Analogues based on the 7-deazahypoxanthine skeleton exhibited nanomolar potencies against cell lines representing cancers with dismal prognoses, tumor metastases and multidrug resistant cells. Studies aimed at elucidating the mode(s) of action of the 7-deazahypoxanthines in cancer cells revealed that they inhibited in vitro tubulin polymerization and disorganized microtubules in live HeLa cells. Experiments evaluating the effects of the 7-deazahypoxanthines on the binding of [3H]colchicine to tubulin identified the colchicine site on tubulin as the most likely target for these compounds in cancer cells. Because many microtubule-targeting compounds are successfully used to fight cancer in the clinic, we believe the new chemical class of antitubulin agents represented by the 7-deazahypoxanthine rigidin analogues have significant potential as new anticancer agents.
Introduction
In light of their fascinating novel structures, intriguing biological properties and the difficulty in obtaining large quantities from natural sources, marine pyrrole-derived alkaloids have attracted considerable attention from organic and medicinal chemists.1–8 At present, several marine natural products are in clinical trials, and one drug, trabectedin, was recently approved in Europe as the first ever marine-derived anticancer agent.9 In this context, the marine alkaloid rigidins A, B, C, D (Figure 1) and E isolated from the tunicate Eudistoma cf. rigida found near Okinawa and New Guinea have remained largely unexplored despite initial reports of promising biological activities.10–12
Figure 1.

Structures of rigidins A, B, C and D
These compounds were shown to exhibit cytotoxicity against murine leukemia L1210 cells,11 while rigidin A was also found to possess anticalmodulin activity.10 Further work in this area was hampered by the miniscule isolation yields of these natural products (0.0015 % for A, 0.00031 % for B, 0.00008 % for C, 0.00004 % for D, and 0.018 % for E based on dry weight). Up to 2011, the synthetic accomplishments in the rigidin area included three total syntheses of rigidin A13–16 and one of rigidin E.16 These approaches involved lengthy synthetic sequences and/or low overall yields and did not offer a solution to the natural product supply problem. More importantly, however, the lack of synthetic flexibility impeded their use for the preparation of analogues for structure-activity relationship (SAR)a studies. Because our research program aims to develop multicomponent reactions (MCR) to access “privileged medicinal scaffolds,”17–20 we became interested in the rigidins, which incorporate the purine-like pyrrolo[2,3-d]pyrimidine ring system, a common motif in natural products (e.g., tubercidin, toyocamycin, sangivamycin21) and medicinal agents with diverse bioactivities (e.g., anti-inflammatory,22,23 anticancer,24–29 antiviral,30,31 adenosine A1 and A3 receptor inhibition,32 adenosine kinase inhibition,33,34 and dihydrofolate reductase inhibition35).
In 2011, we reported in a communication format a general total synthesis of rigidins A, B, C and D, which involved only four steps from commercially available materials and was amenable to the production of synthetic rigidin analogues.36 This work has led to further developments and discoveries, which we describe here. More specifically, first, we detail the synthesis of first-generation synthetic analogues based on the 7-deazaxanthine skeleton. Second, we describe an anticancer evaluation of synthetically prepared rigidins A, B, C and D that, together with the first generation analogues, fail to display any significant effects on cancer cell proliferation. Third, we disclose novel MCRs leading to the preparation of the second-generation rigidin analogues based on the 7-deazaadenine, 7-deazapurine and 7-deazahypoxanthine (Figure 2) skeletons. Fourth, we show that the 7-deazahypoxanthines display potent antiproliferative activities. Fifth, we demonstrate that these compounds maintain their activity against tumor cells derived from cancers associated with dismal prognoses, metastatic tissues as well as multidrug resistant cells. Finally, we show that the anticancer properties of these hypoxanthine-like compounds are most likely caused by their pronounced effects on microtubule organization in cancer cells.
Figure 2.

Structures of pyrrolo[2,3-d]pyrimidines prepared in this work
Results and Discussion
Chemistry
The synthesis of natural rigidins A, B, C and D, as well as synthetic 7-deazaxanthine analogues, is based on the chemistry shown in Table 1. Sulfonamides 6–11, prepared by reacting commercially available aminoacetophenones with methanesulfonyl chloride, are utilized in a three-component reaction (3-CR) with various aromatic aldehydes 1–5 and cyanoacetamide in the presence of potassium carbonate in refluxing ethanol to give aminopyrroles 12–21 (Table 1). The synthesis of the 7-deazaxanthine framework is completed by carbonylation with oxalyl chloride in diglyme to give analogues 22a–j possessing diversely substituted aromatic moieties Ar1 and Ar2. Compounds 22a–d are further hydrogenolyzed to yield natural rigidins with overall yields of 61% for A, 58% for B, 60% for C and 53% for D from commercially available aminoacetophenones. This sequence constitutes the first synthesis of rigidins B, C and D, while rigidin A is accessed via a significantly improved method compared with the previously described routes.13–16 In addition, our reaction sequence leads to an expedient preparation of what we call the first-generation library of rigidin analogues based on the 7-deazaxanthine skeleton and possessing variable substituents in the aromatic moieties. Finally, in addition to these synthetic 7-deazaxanthines 22a–j, we prepared a dithia variant 22k, which is obtained by treating pyrrole 21 with carbon disulfide in pyridine (Figure 3).
Table 1.
MCR-based total general synthesis of rigidins A–D and their first-generation 7-deazaxanthine synthetic analogues 22a–j
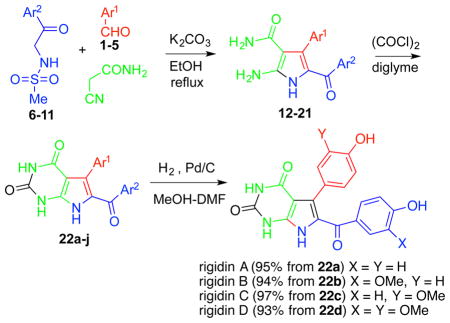
| ||||||
|---|---|---|---|---|---|---|
| aldehyde | methanesulfonylamide | pyrrole | 7-deazaxanthine | Ar1 | Ar2 | overall yield % |
| 1 | 6 | 12 | 22a |
|
|
64 |
| 1 | 7 | 13 | 22b |
|
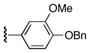
|
62 |
| 2 | 6 | 14 | 22c |
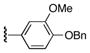
|
|
62 |
| 2 | 7 | 15 | 22d |
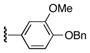
|
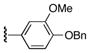
|
57 |
| 3 | 8 | 16 | 22e |
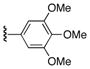
|
|
74 |
| 3 | 9 | 17 | 22f |
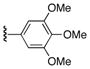
|
|
64 |
| 3 | 7 | 18 | 22g |
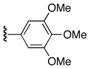
|
|
59 |
| 4 | 10 | 19 | 22h |
|
|
57 |
| 4 | 11 | 20 | 22i |
|
Ph | 62 |
| 5 | 11 | 21 | 22j |
|
Ph | 43 |
Figure 3.
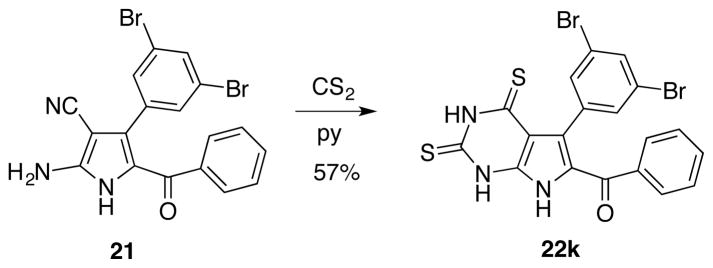
Preparation of an additional 7-deazapurine 22k
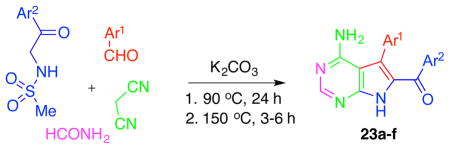
Our further studies of the above-described three-component reaction led to the discovery of its four-component (4-CR) variants. Thus, we found that if the reaction is conducted in formamide as a solvent and cyanoacetamide is replaced with malononitrile (Table 2) or cyanomethylketones (Table 3), the entire pyrrolo[2,3-d]pyrimidine skeleton is assembled in one step giving rise to 7-deazaadenines 23 (Table 2) or 7-deazapurines 24 (Table 3), respectively. The success of these 4-CRs also depends on an optimized temperature regime, in which the reaction is first kept at 90 °C to allow for a clean formation of intermediate aminopyrroles (similar to 12–21 in Table 1) and then at 150 °C to bring the fourth component formamide into the process, leading to closure of the pyrimidine portion of the molecule.
Table 2.
4-CR leading to the formation of the second-generation analogues based on the 7-deazaadenine skeleton.
| |||
|---|---|---|---|
| 7-deazaadenine | Ar1 | Ar2 | % yield |
| 23a |
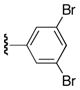
|
Ph | 58 |
| 23b |
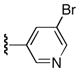
|
Ph | 66 |
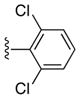
| 23c | Ph | Ph | 48 |
| 23d |
|
Ph | 85 |
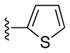
| 23e |
|
|
54 |
| 23f |
|
Ph | 72 |
Table 3.
4-CR leading to the formation of the second-generation analogues based on the 7-deazapurine skeleton.
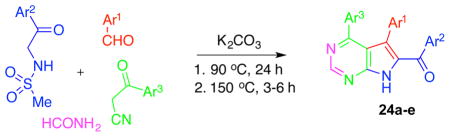
| ||||
|---|---|---|---|---|
| 6,7-dideazaadenine | Ar1 | Ar2 | Ar3 | % yield |
| 24a |
|
|
|
63 |
| 24b |
|
Ph | Ph | 30 |
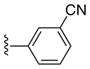
| 24c |
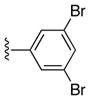
|
Ph |
|
33 |
| 24d |
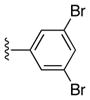
|
Ph | Ph | 39 |
| 24e |
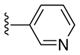
|
Ph | Ph | 22 |
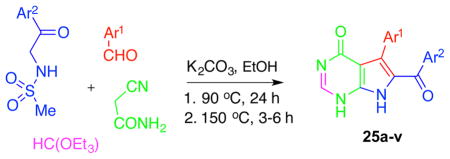
Additionally, we discovered a 4-CR leading to the formation of a 7-deazahypoxanthine skeleton 25 (Table 4). In this modification, the starting sulfonamides, aldehydes and cyanoacetamide are reacted in a 1:1 mixture of EtOH and triethyl orthoformate under a similar temperature regime, involving reflux at 90 °C followed by a gradual increase in temperature to 150 °C, with the concomitant slow evaporation of ethanol.
Table 4.
4-CR leading to the formation of the second-generation analogues based on the 7-deazahypoxanthine skeleton.
| |||
|---|---|---|---|
| 7-deazahypoxanthine | Ar1 | Ar2 | % yield |
| 25a | Ph | Ph | 71 |
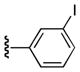
| 25b |
|
Ph | 68 |
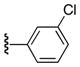
| 25c |
|
Ph | 61 |
| 25d |
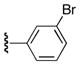
|
Ph | 54 |
| 25e |
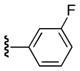
|
Ph | 53 |
| 25f |
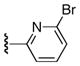
|
Ph | 60 |
| 25g |
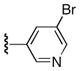
|
Ph | 88 |
| 25h |
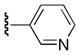
|
Ph | 61 |
| 25i |
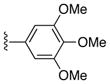
|
Ph | 64 |
| 25j |
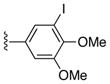
|
Ph | 37 |
| 25k |
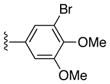
|
Ph | 51 |
| 25l |
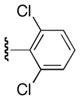
|
Ph | 74 |
| 25m |
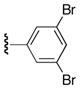
|
Ph | 77 |
| 25n |
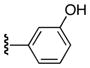
|
Ph | 26 |
| 25o |
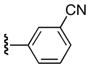
|
Ph | 47 |
| 25p |
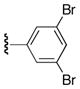
|
|
56 |
| 25q |
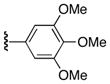
|
|
56 |
| 25r |
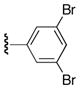
|
|
60 |
| 25s |
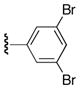
|
|
61 |
| 25t |
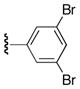
|
|
54 |
| 25u | Ph |
|
68 |
| 25v |
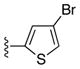
|
Ph | 71 |
The analogues accessed by these 4-CRs (Tables 2, 3 and 4) are generally obtained in good yields given the complexity of the newly devised reactions. The product pyrrolopyrimidines contain diversely substituted aromatic moieties, including alkoxy (e.g., 22a–i, 23e, 24a, 25i–k, 25q, etc), halogen (e.g., 23a,b, 25b–g, 25r–t, etc), cyano (e.g., 25o) and heterocycles (e.g., 23b, 25f–h, 25v, etc). Additionally, the hydroxy-substituted pyrrolopyrimidines can be obtained by the use of precursors containing benzyl-protected phenolic oxygens, with subsequent hydrogenolysis (similar to the synthesis of rigidins A–D from 22a–d in Table 1) or through another modification in the MCR chemistry involving the use of HCOOH as the fourth component (Table 5). Formic acid plays a dual role, providing the necessary carbon for pyrimidine ring-closure and promoting in situ debenzylation of the phenolic benzyl ethers, resulting in analogues 25w–z (Table 5). Interestingly, a search of the literature reveals no examples of metal-free utilization of formic acid for deprotection of benzyl ethers. To our knowledge the described chemistry represents the first examples of construction of the pyrrolo[2,3-d]pyrimidine skeleton in a one-step MCR process.
Table 5.
4-CR with concomitant debenzylation leading to the formation of hydroxyl-substituted 7-deazahypoxanthine analogues.
| |||
|---|---|---|---|
| 7-deazahypoxanthine | Ar1 | Ar2 | % yield |
| 25w | Ph | Ph | 88 |
| 25x |
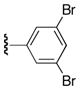
|
|
70 |
| 25y |
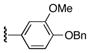
|
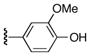
|
64 |
| 25z |
|
|
71 |
Pharmacology
(a) Antiproliferative Activities
The synthetically prepared natural products and their synthesized analogues were evaluated for antiproliferative activities using the HeLa cell line as a model for human cervical adenocarcinoma and MCF-7 cells as a model for breast adenocarcinoma. The cells were treated with each compound for 48 h, and cell viability was assessed using the MTT method37 (Table 6). The natural alkaloids and synthetic 7-deazaxanthine 22e–k analogues were inactive or weakly active, producing double-digit micromolar GI50 values in some instances (e.g., analogue 22i). Further, scaffold modifications, such as those resulting in 7-deazaadenine analogues 23 and 7-deazapurine compounds 24, led to the discovery of micromolar potencies associated with selected compounds (e.g., 23c and 24c). An unexpected and most gratifying observation was, however, the discovery of nanomolar antiproliferative activity in compounds based on the 7-deazahypoxanthine skeleton 25. Thus, analogues 25a, 25e, 25g, 25m and 25u all exhibited double-digit nanomolar potencies. A closer analysis of the SAR data indicates that the favored substitution pattern is the unsubstituted or para-substituted benzene ring at the sulfonamide-derived C-8 position (Ar2) and either the unsubstituted or meta-halogen-substituted benzene group at the aldehyde-derived C-7 position (Ar1). These structural features are found in the most potent analogues 25a–g, 25m, 25r–u. The replacement of the Ph group at C-7 (as in 25a) with the pyridine moiety (as in 25h) led to a drop in potency by a factor of 15. Together, these data indicate that the C-7 and, possibly, C-8 aromatic groups bind in a pocket where hydrophobic interactions predominate, and introduction of hydrophilic pyridine nitrogen (25h) or hydroxyl groups (25n, 25y,z) leads to an unfavorable loss of solvation energy.
Table 6.
Antiproliferative activities of the synthesized 7-deazapurines
| 7-deazapurine | cell viabilitya GI50, μM
|
|
|---|---|---|
| HeLa | MCF-7 | |
| rigidin A | > 100 | > 100 |
| rigidin B | > 100 | > 100 |
| rigidin C | > 100 | > 100 |
| rigidin D | > 100 | > 100 |
| 22e | > 100 | > 100 |
| 22f | > 100 | > 100 |
| 22g | > 100 | > 100 |
| 22h | > 100 | > 100 |
| 22i | 80 ± 10 | 14 ± 1 |
| 22j | 71 ± 12 | 47 ± 5 |
| 22k | > 100 | > 100 |
| 23a | > 100 | > 100 |
| 23b | > 100 | > 100 |
| 23c | 4.4 ± 0.4 | 3.9 ± 0.5 |
| 23d | 28 ± 4 | 15 ± 0 |
| 23e | > 100 | > 100 |
| 23f | > 100 | > 100 |
| 24a | 20 ± 1 | 8.0 ± 1 |
| 24b | 70 ± 15 | 75 ± 11 |
| 24c | 9.0 ± 1 | 7.0 ± 1 |
| 24d | 18 ± 10 | 10 ± 2 |
| 24e | 45 ± 9 | 51 ± 2 |
| 25a | 0.035 ± 0.007 | 0.040 ± 0.024 |
| 25b | 0.2 ± 0.1 | 0.150 ± 0.060 |
| 25c | 0.10 ± 0.07 | 0.06 ± 0.02 |
| 25d | 0.13 ± 0.05 | 0.06 ± 0.02 |
| 25e | 0.065 ± 0.010 | 0.070 ± 0.025 |
| 25f | 0.15 ± 0.05 | 0.135 ± 0.075 |
| 25g | 0.080 ± 0.025 | 0.087 ± 0.020 |
| 25h | 0.88 ± 0.06 | 1.26 ± 0.18 |
| 25i | 48 ± 8 | 33 ± 4 |
| 25j | 8.2 ± 1 | 40 ± 4 |
| 25k | 7.1 ± 1 | 10 ± 5 |
| 25l | 4.9 ± 0.3 | 2.2 ± 0.1 |
| 25m | 0.095 ± 0.040 | 0.095 ± 0.010 |
| 25n | 0.86 ± 0.06 | 1.09 ± 0.12 |
| 25o | 0.19 ± 0.01 | 0.25 ± 0.02 |
| 25p | 6.3 ± 1 | 0.80 ± 0.01 |
| 25q | >100 | 7.0 ± 1 |
| 25r | 0.30 ± 0.07 | 0.11 ± 0.035 |
| 25s | 0.40 ± 0.01 | 0.31 ± 0.01 |
| 25t | 2.2 ± 1 | 0.6 ± 0.1 |
| 25u | 0.045 ± 0.004 | 0.053 ± 0.005 |
| 25v | 43 ± 6 | 56 ± 2 |
| 25w | 0.4 ± 0.1 | 0.3 ± 0.07 |
| 25x | 0.30 ± 0.01 | 0.26 ± 0.05 |
| 25y | 5.0 ± 0.6 | 3.0 ± 0.6 |
| 25z | 4.0 ± 0.6 | 2.8 ± 0.8 |
Concentration required to reduce the viability of cells by 50% after a 48 h treatment with the indicated compounds relative to a DMSO control ± SD from two independent experiments, each performed in 4 replicates, as determined by the MTT assay.
(b) Cell Cycle Analysis
The structural similarities between the rigidin analogues and previously described podophyllotoxin analogues,19 as well as characteristic cellular morphological changes induced by the 7-deazahypoxanthines, led us to hypothesize that these compounds exert their antiproliferative properties through inhibition of tubulin dynamics, thus inducing mitiotic arrest in cancer cells. To test this hypothesis, cell cycle analysis was performed using the cell permeable DNA binding dye Vybrant Orange. In a normal population, cells are distributed among the three major phases of the cell cycle, G0/G1, in which there is one set of paired chromosomes per cell (2n), followed by the S or synthesis phase, in which DNA is synthesized in preparation for division, and finally the G2/M phase, in which the chromosomes of each cell have now been duplicated (4n). The DNA content can be measured by flow cytometry using fluorescent DNA selective probes such as Vybrant Orange that produce signals that are proportional to DNA mass per cell. Cell cycle analysis performed on HeLa cells using 25g and 25m with 0.1% dimethyl sulfoxide (DMSO) as a control revealed a pronounced cell cycle arrest in the G2/M phase (Figure 4), and this effect is characteristic of antimitotic agents disrupting microtubule assembly.38
Figure 4.
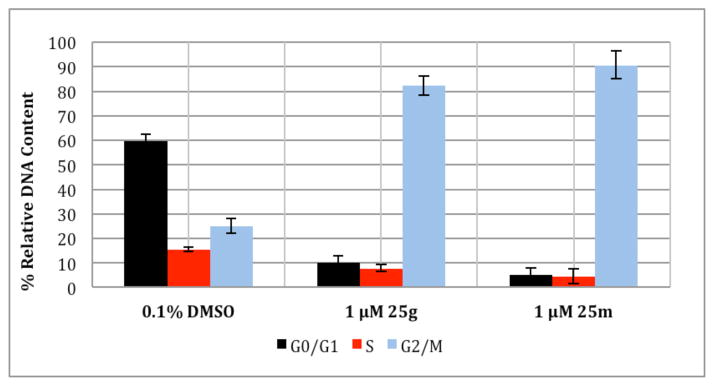
Flow cytometric cell cycle analysis using HeLa cells treated for 32 h with 25g or 25m. Error bars represent the standard error between triplicate experiments.
(c) Inhibition of tubulin assembly and colchicine binding to tubulin
To gain further insight into the possible antitubulin mechanism of action of the 7-deazahypoxanthines, the effects of these compounds on in vitro tubulin polymerization were assessed.39 In our initial experiment, polymerization was followed by fluorescence enhancement due to the incorporation of a fluorescent reporter, in this case 4′,6-diamidino-2-phenylindole (DAPI), into microtubules as polymerization occurs. The test compounds were compared to three different controls, a known microtubule stabilizer (taxol), a known microtubule destabilizer (colchicine) as well as a solvent control sample. Whereas taxol induced potent enhancement of microtubule formation relative to the effect of the DMSO control, 7-deazahypoxanthines 25a and 25m displayed potent inhibition of microtubule assembly in a manner similar to the known tubulin polymerization inhibitor colchicine (Figure 5).
Figure 5.

Effect of 25a and 25m on in vitro tubulin polymerization. Taxol (3 μM) promotes microtubule formation relative to 0.1% DMSO control. 25a (25 μM), 25m (25 μM) and colchicine (25 μM) completely suppress tubulin polymerization.
We next examined 25a and 25m in a quantitative turbidimetric assay,40 in comparison with combretastatin A-4, a well-described potent inhibitor of the reaction (Table 7).41 While 25a differed little from combretastatin A-4 in its inhibitory effect, 25m was distinctly less active. We also examined 25a and 25m for potential inhibitory effects on the binding of [3H]colchicine to tubulin (Table 7).42 We found 25a to be almost as active as combretastatin A-4, while 25m was less active, in accord with their relative activities in the assembly assay.
Table 7.
Inhibition of tubulin assembly and colchicine binding to β-tubulin
| compound | inhibition of tubulin assemblyb | inhibition of colchicine bindingc | |
|---|---|---|---|
|
| |||
| IC50 (μM) ± SD | % inhibition ± SD | ||
|
| |||
| 5 μM inhibitor | 1 μM inhibitor | ||
|
|
|||
| CA-4a | 0.96 ± 0.07 | 99 ± 0.4 | 89 ± 0.6 |
| 25a | 1.2 ± 0.1 | 88 ± 0.7 | 58 ± 4 |
| 25m | 1.6 ± 0.04 | 71 ± 0.5 | ND |
CA-4 = combretastatin A-4.
Inhibition of tubulin polymerization by selected compounds. Tubulin was at 10 μM.
% Inhibition of [3H]colchicine (5 μM) binding to tubulin (1 μM) by selected compounds.
There are three well-documented binding domains on tubulin, the first two of which include the pocket for taxanes and epothilones43,44 and the pocket for vinca alkaloids.45 FDA approved tubulin inhibitors binding to these two domains are all derivatives of complex natural products and are characterized by high bulkiness, hydrophobicity, and complicated structure. In addition, many tend to be easily recognized by P-glycoprotein (P-gp) and pumped out of cancer cells, which causes drug resistance.46 The third binding domain of tubulin is the pocket for colchicine and podophyllotoxin, small natural products with relatively simple structures. So far, there are no FDA approved anticancer drugs targeting this domain,47 although many colchicine site agents inhibit the growth of multidrug resistant (MDR) tumor cell lines and have reduced neurotoxicity and good oral bioavailability. These properties should be advantageous compared to the currently FDA approved tubulin inhibitors.48–49 The results shown in Table 7 indicate that the 7-deazahypoxanthines, as represented by compounds 25a and 25m, constitute a new class of colchicine site agents.
(d) Microtubule organization in cells
To evaluate the effects of 7-deazahypoxanthines on the microtubule cytoskeleton in cells, we cultured HeLa cells in the presence of compounds 25a and 25m at a range of concentrations and examined microtubule morphology (Figure 6). At concentrations between 1 and 10 nM, interphase microtubule organization (Figure 6, panels A–C and M–O) was indistinguishable from DMSO controls (not shown). Similarly, mitotic spindle morphology and spindle length was unaffected at concentrations at or below 10 nM for both analogues. However, there was a pronounced change in both interphase and mitotic microtubule organization at concentrations between 10 and 100 nM, where both compounds caused a complete collapse of mitotic microtubule organization (Figure 6, Panel J–L and V–X). This was also the concentration range that encompassed the antiproliferative GI50 values for both compounds (GI50 (25a) = 35 nM, GI50 (25m) = 95 nM, Table 6). Additionally, while both compounds dramatically affected the polymerization and organization of dynamic mitotic microtubules, 25a had a more drastic effect on interphase microtubule organization (Figure 6, panels D–F) in comparison with 25m (Figure 6, Panels P–R), suggesting that 25a was capable of affecting both dynamic and stable microtubule arrays. Together, these results lend further evidence indicating that tubulin-targeting is likely the main mechanism responsible for the antiproliferative effects of the 7-deazahypoxanthines.
Figure 6.
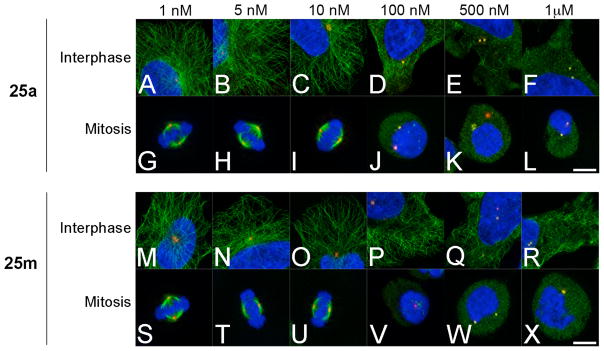
Microtubule organization in HeLa cells during interphase and mitosis. HeLa cells were treated for 3 h with compound 25a (panels A–L) and 25m (M–X) at a range of concentrations. Following drug treatment, cells were probed for microtubules (green), the centrosome marker pericentrin (red) and DNA (blue). Bar, 10 μm.
(e) Aqueous Solubility
Because low aqueous solubility is an intrinsic property of many natural and synthetic drug candidates and is usually associated with poor absorption and bioavailability,50,51 the water solubilities of the most potent nanomolar compounds 25a, 25e, 25g and 25m were determined with a UV/vis spectrophotometer at a wavelength of 335–336 nm. The solubility of compounds 25e, 25g and 25m were 65 μM, 30 μM and 40 μM, respectively, while under the same conditions 25a (10 μM) was less soluble. It appears that the meta-halogen substitution present in compounds 25e, 25g and 25m is beneficial for the enhanced water solubility and will be considered for further development of this series of compounds. As a whole, these numbers fall in the range of water solubilities of the other reported colchicine-site targeting anti-tubulin compounds.52
(f) Activity against MDR cells
Because it is known that colchicine site agents are often insensitive to the action of P-gp,53 we tested potent 7-deazahypoxanthines against MDR cells. The MDR uterine sarcoma cell line MES-SA/Dx5 was utilized for this experiment.54 This cell line was established from the parent uterine sarcoma MES-SA, grown in the presence of increasing concentrations of doxorubicin and is known to be resistant to a number of P-gp substrates. Both taxol and vinblastine displayed more than a thousand fold drop in potency when tested for antiproliferative activity against the MDR cell line as compared with the parent line (Table 8). In contrast, there was only a small variation in the sensitivities of the two cell lines towards 7-deazahypoxanthines 25a and 25m. Of note, the variation in sensitivity was slightly larger for the more lipophilic 25m than that for 25a, underscoring the idea that smaller, less lipophilic agents are poorer P-gp substrates.
Table 8.
Antiproliferative effect of selected compounds against MDR cells
| GI50
in vitro Values (nM)a
|
||
|---|---|---|
| MES-SA | MES-SA/Dx5 | |
|
| ||
| Taxol | 7 ± 1 | 9800 ± 283 |
| Vinblastine | 6 ± 1 | 5000 ± 1414 |
| 25m | 81 ± 6 | 394 ± 10 |
| 25a | 30 ± 4 | 70 ± 4 |
Concentration required to reduce the viability of cells by 50% after a 48 h treatment with the indicated compounds relative to a DMSO control ± SD from two independent experiments, each performed in 4 replicates, as determined by the MTT assay.
(g) Activity against cancer cell lines representing cancers with dismal prognoses or derived from tumor metastases
To study the antiproliferative effects of our potent 7-deazahypoxanthines 25 further, we investigated whether they retained their potency against a panel of cancer cell lines, representing cancers known to be associated with dismal prognoses (Table 9). These include glioma (U373 and Hs683), melanoma (SKMEL-28 and B16F10), non-small-cell lung (NSCLC, A549) and head-and-neck cancers (UM-SCC10A and UM-SCC22A). In addition, because more than 90% of cancer deaths are caused by tumor metastases, two head-and-neck cancer cell lines derived from lymph node metastases (UM-SCC10B and UM-SCC22B) were added to the panel. 7-Deazahypoxanthines 25a, 25g and 25m were found to retain their nanomolar potency against cells in this challenging in vitro panel (Table 9). Especially noteworthy are antiproliferative effects of 25a against human glioma (U373) and melanoma (SKMEL-28) cells, which had single digit nanomolar GI50 values.
Table 9.
Antiproliferative properties of potent 7-deazahypoxanthines 25 against cancer cell lines representing cancers with dismal prognoses or derived from tumor metastases
| compound | GI50 in vitro values (nM)a
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| glioma | melanoma | NSCLC | head and neck cancer | head and neck metastases | |||||
|
| |||||||||
| U373 | Hs683 | SKMEL-28 | B16F10 | A549 | UM-SCC10A | UM-SCC22A | UM-SCC10B | UM-SCC22B | |
| 25m | 30 | 20 | 20 | 50 | 30 | 90 | 110 | 100 | 20 |
| 25g | 30 | 20 | 20 | 40 | 20 | 90 | 120 | 130 | 20 |
| 25a | <10 | 10 | <10 | 20 | 10 | ND | ND | ND | ND |
Concentration required to reduce the viability of cells by 50% after a 48 h treatment with the indicated compounds relative to a DMSO control, as determined by the MTT assay
Conclusion
The research reported in this paper involves the utilization of the pyrrolo[2,3-d]pyrimidine-based natural alkaloid rigidins to discover novel anticancer agents. We described the development of novel synthetic chemistry allowing rapid access to various structural variants of this heterocyclic scaffold, which led to the generation of a library of diverse synthetic rigidin analogues. Although the natural products themselves were found to have little if any anticancer activity, synthetic analogues based on the 7-deazahypoxanthine skeleton were found to have promising double to single digit nanomolar antiproliferative potencies in human cancer cells. These compounds maintain potency in a panel of human cancer cells including those representing cancers with dismal prognoses as well as cells derived from metastatic lymph node tumors. All evidence obtained to date indicated that these rigidin analogues exert their antiproliferative action by inhibiting microtubule dynamics in cancer cells. Because many microtubule-targeting compounds are successfully used to fight cancer in the clinic, we believe the new chemical class of antitubulin agents represented by the 7-deazahypoxanthine rigidin analogues have significant potential as new anticancer agents.
Experimental Section
General Synthetic Methods
All reagents, solvents and catalysts were purchased from commercial sources (Acros Organics and Sigma-Aldrich) and used without purification. All reactions were performed in oven-dried flasks open to the atmosphere or under nitrogen and monitored by thin layer chromatography (TLC) on TLC precoated (250 μm) silica gel 60 F254 glass-backed plates (EMD Chemicals Inc.). Visualization was accomplished with UV light. Flash column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). 1H and 13C NMR spectra were recorded on Jeol 300 or Bruker 400 spectrometers. Chemical shifts (δ) are reported in ppm relative to the TMS internal standard. Abbreviations are as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). HRMS analyses were performed at the Mass Spectrometry Facility, University of New Mexico or using Waters Synapt G2 LCMS. The synthesized compounds are at least 95% pure according to UPLC/MS analysis.
General Procedure for the Synthesis of 7-Deazaxanthines (22a–j)
A required pyrrole36 (0.3478 mmol) was co-evaporated with 5 mL of toluene and dissolved in 3 mL of diglyme. Nitrogen gas was bubbled through the mixture for 5 min, and 0.20 mL (0.3825 mmol) of oxalyl chloride (2 M in CH2Cl2) was added dropwise while vigorously stirring the mixture. The mixture was refluxed for 10 h under nitrogen. After that time, the solvent was evaporated under reduced pressure, and 5 mL of 50% MeOH in CH2Cl2 was added to the solid in the flask. The precipitate was removed by filtration and rinsed sequentially with 2 mL of MeOH and 5 mL of diethyl ether. The mother liquor was chromatographed on silica gel using 30/1 to 30/5 CH2Cl2/MeOH gradient. The material from filtration and column chromatography was combined to give 7-deazaxanthines 22a–j.
6-[4-(Benzyloxy)benzoyl]-5-[4-(benzyloxy)phenyl]-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22a)
81% as a brown powder, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 5.02 (s, 2H), 5.06 (s, 2H), 6.69 (d, J = 8.8 Hz, 2H), 6.74 (d, J = 8.8 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 7.29–7.40 (m, 12H), 10.71 (bs, 1H), 11.31 (bs, 1H), 11.94 (bs, 1H). 13C NMR (DMSO-d6) δ: 69.8, 70.0, 98.9, 113.8, 114.3, 128.0, 128.1, 128.3, 128.5, 128.9, 129.0, 131.7, 132.8, 136.8, 137.6, 151.3, 158.1, 160.3, 161.2, 185.7. HRMS m/z (ESI+) calc’d for C33H26N3O5 (M+H+) 542.1721, found 542.1714.
6-[4-(Benzyloxy)-3-methoxybenzoyl]-5-[4-(benzyloxy)phenyl]-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22b)
74% as a brown powder, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.57 (s, 3H), 5.01 (s, 2H), 5.05 (s, 2H), 6.72 (s, 1H), 6.87–7.10 (m, 7H), 7.37 (m, 10H), 10.72 (bs, 1H), 11.27 (bs, 1H), 11.94 (bs, 1H). 13C NMR (DMSO-d6) δ: 56.3, 70.3, 71.2, 99.1, 114.0, 114.1, 123.5, 125.4, 125.6, 127.9, 128.0, 128.2, 128.3, 128.9, 131.9, 132.8, 137.4, 137.8, 142.0, 149.2, 150.9, 151.7, 158.5, 160.1, 185.7. HRMS m/z (ESI+) calc’d for C34H27N3NaO6 (M+Na+) 596.1798, found 596.1792.
6-[4-(Benzyloxy)benzoyl]-5-[4-(benzyloxy)-3-methoxyphenyl]-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22c)
76% as a yellow powder, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.60 (s, 3H), 4.97 (s, 2H), 5.03 (s, 2H), 6.66–6.74 (m, 5H), 7.34–7.38 (m, 12H), 10.70 (bs, 1H), 11.48 (bs, 1H), 11.96 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.9, 70.0, 70.6, 98.9, 113.1, 114.2, 116.2, 124.4, 125.5, 125.8, 128.1, 128.3, 128.5, 128.9, 129.0, 129.1, 131.2, 131.6, 137.0, 137.7, 151.3, 160.3, 161.5, 185.8. HRMS m/z (ESI+) calc’d for C34H27N3NaO6 (M+Na+) 596.1798, found 596.1804.
6-[4-(Benzyloxy)-3-methoxybenzoyl]-5-[4-(benzyloxy)-3-(methoxy)phenyl]-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22d)
72% as a yellow powder, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.52 (s, 3H), 3.55 (s, 3H), 4.98 (s, 2H), 5.04 (s, 2H), 6.74 (s, 1H), 6.78 (s, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.93 (d, J = 1.5 Hz, 1H), 7.10 (dd, J = 1.6 Hz, J = 8.3 Hz, 1H), 7.31–7.39 (m, 10H), 10.73 (bs, 1H), 11.27 (bs, 1H), 11.96 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.6, 55.7, 70.5, 70.7, 98.8, 112.7, 113.2, 123.3, 124.3, 125.5, 125.6, 128.0, 128.1, 128.2, 128.4, 128.8, 128.9, 131.4, 137.0, 137.6, 141.9, 147.7, 148.1, 148.3, 151.1, 151.2, 160.2, 185.7. HRMS m/z (ESI+) calc’d for C35H29N3NaO7 (M+Na+) 626.1903, found 626.1908.
6-(4-Fluorobenzoyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22e)
74%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.56 (s, 9H), 6.38 (s, 2H), 6.92 (m, 2H), 7.41 (m, 2H), 10.76 (bs, 1H), 11.32 (bs, 1H), 12.13 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.5, 60.3, 99.2, 109.2, 113.8, 114.9, 126.1, 127.6, 130.0, 131.5, 137.2, 151.1, 160.5, 156.3, 185.5. HRMS m/z (ESI+) calc’d for C22H18FNaN3O6 (M+Na+) 462.1077, found 462.1064.
6-(4-Bromobenzoyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22f)
64%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.50 (s, 9H), 6.33 (s, 2H), 7.23 (m, 4H). 13C NMR (DMSO-d6) δ: 56.2, 60.7, 99.4, 109.7, 125.1, 125.7, 127.5, 130.7, 130.8, 130.9, 137.7, 138.0, 142.4, 151.1, 151.9, 160.0, 185.6. HRMS m/z (ESI+) calc’d for C22H18BrNaN3O6 (M+Na+) 524.0256, found 524.0259.
6-(4-Benzyloxy-3-methoxybenzoyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22g)
59%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.52 (s, 12H), 4.98 (s, 2H), 6.46 (m, 2H), 6.86 (m, 2H), 7.04 (m, 1H), 7.35 (m, 5H), 10.72 (bs, 1H), 11.25 (bs, 1H), 11.99 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.5, 56.0, 70.4, 98.8, 109.6, 112.6, 112.7, 122.9, 125.8, 127.7, 128.0, 128.4, 128.9, 129.1, 131.2, 131.6, 137.0, 137.3, 148.1, 151.0, 151.7, 160.5, 185.8. HRMS m/z (ESI+) calc’d for C30H27N3NaO8 (M+Na+) 580.1696, found 580.1697.
6-(4-Methoxybenzoyl)-5-(4-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22h)
76% as a yellow powder, mp > 300 °C decomp.1H NMR (DMSO-d6) δ: 3.66 (s, 3H), 3.70 (s, 3H), 6.60 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 10.70 (bs, 1H), 11.32 (bs, 1H), 11.93 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.7, 55.9, 70.0, 99.1, 113.1, 113.6, 125.0, 125.7, 128.9, 130.9, 131.6, 141.9, 151.1, 159.1, 160.2, 162.5, 185.8. HRMS m/z (ESI+) calc’d for C21H18N3O5 (M+H+) 392.1246, found 392.1245.
6-Benzoyl-5-(4-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22i)
81% as a brown powder, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.63 (s, 3H), 6.57 (d, J = 8.8 Hz, 2H), 7.01–7.13 (m, 5H), 10.73 (bs, 1H), 11.30 (bs, 1H), 11.99 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.6, 99.4, 113.0, 124.7, 125.6, 128.1, 129.3, 130.1, 131.6, 132.8, 138.6, 142.3, 151.2, 159.1, 160.2, 186.9. HRMS m/z (ESI+) calc’d for C20H15N3NaO4 (M+Na+) 362.1141, found 362.1141.
6-Benzoyl-5-(3,5-dibromophenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (22j)
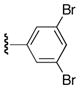
43%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ 7.14–7.35 (m, 8H), 10.8 (bs, 1H), 11.48 (bs, 1H), 12.37 (bs, 1H). 13C NMR (DMSO-d6) δ: 99.3, 121.2, 126.3, 126.7, 128.2, 129.0, 132.1, 133.2, 136.5, 138.4, 142.2, 151.2, 160.1, 186.6. HRMS m/z (ESI+) calc’d for C19H11Br2N3NaO3 (M+Na+) 511.9044, found 511.9052.
6-Benzoyl-5-(3,5-dibromophenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dithione (22k)
57%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.30–7.47 (m, 10H), 8.63 (bs, 1H). 13C NMR (DMSO-d6) δ: 112.3, 120.8, 126.6, 128.1, 129.0, 132.1, 132.4, 133.9, 136.3, 138.1, 171.6, 187.1, 191.5. HRMS m/z (ESI+) calc’d for C19H12Br2N3OS2 (M+H+) 519.8789, found 519.8788.
Synthesis of Rigidins A, B, C and D
A selected benzyl-protected 7-deazaxanthine 22a–d (0.0735 mmol) and Pd/C (20 mg, 10%) were suspended in DMF (0.3 mL) and MeOH (1 mL). The suspension was degassed three times by the freeze-pump-thaw method at −196 °C and then stirred under a hydrogen balloon for 3 h. After that time, the solvent was removed from the frozen sample by lyophilization, and the crude residue was dissolved in MeOH/CH2Cl2 and subjected to silica gel column chromatography (MeOH/CH2Cl2/AcOH=10/50/1) to afford pure alkaloids.
6-(4-Hydroxybenzoyl)-5-(4-hydroxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (Rigidin A)
94.7% as a yellow solid, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 6.46 (d, J = 8.5 Hz, 2H), 6.47 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 9.28 (bs, 1H), 10.02 (bs, 1H), 10.63 (bs, 1H), 11.32 (bs, 1H), 11.84 (bs, 1H). 13C NMR (CD3OD) δ: 98.5, 113.6, 114.1, 123.9, 128.5, 129.3, 131.5, 132.3, 156.0, 160.2, 160.8, 162.9, 185.4. HRMS m/z (ESI+) calc’d for C19H13N3NaO5 (M+Na+) 386.0753, found 386.0742.
6-(4-Hydroxy-3-methoxybenzoyl)-5-(4-hydroxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (Rigidin B)
95% as a brown solid, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.56 (s, 3H), 6.47 (d, J = 8.5 Hz, 2H), 6.57 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 6.99–7.01 (m, 3H), 9.30 (bs, 1H), 10.54 (bs, 1H), 11.99 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.7, 98.8, 113.7, 114.3, 115.0, 123.6, 124.2, 125.3, 128.8, 129.6, 133.0, 147.0, 150.7, 152.7, 156.9, 160.7, 185.8. HRMS m/z (ESI+) calc’d for C20H16N3O6 (M+H+) 394.1039, found 394.1032.
6-(4-Hydroxybenzoyl)-5-(4-hydroxy-3-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (Rigidin C)
96% as a dark yellow solid, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.58 (s, 3H), 6.53 (m, 3H), 6.69–6.76 (m, 2H), 7.33 (d, J = 8.5 Hz, 2H), 8.69 (bs, 1H), 9.66 (bs, 1H), 10.21 (bs, 1H). 13C NMR (DMSO-d6) δ: 56.2, 98.7, 114.8, 115.6, 116.4, 123.5, 125.5, 129.3, 131.7, 132.1, 135.4, 141.8, 147.1, 148.1, 151.1, 161.1, 185.7. HRMS m/z (ESI+) calc’d for C20H16N3O6 (M+H+) 394.1039, found 394.1028.
6-(4-Hydroxy-3-methoxybenzoyl)-5-(4-hydroxy-3-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (Rigidin D)
93% as a maroon solid, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.52 (s, 3H), 3.54 (s, 3H), 6.49 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 6.66 (dd, J = 1.7 Hz, J = 8.1 Hz, 1H), 6.73 (d, J = 1.6 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 7.05 (dd, J = 1.6 Hz, J = 8.2 Hz, 1H), 8.89 (bs, 1H), 9.65 (bs, 1H), 10.60 (bs, 1H), 11.83 (bs, 1H). 13C NMR (DMSO-d6) δ: 55.8, 55.8, 98.8, 113.7, 114.7, 116.5, 123.7, 124.0, 124.8, 125.3, 128.8, 129.7, 146.3, 146.6, 146.9, 150.8, 160.6, 185.8. HRMS m/z (ESI+) calc’d for C14H7N3NaO7 (M+Na+) 446.0964, found 446.0964.
General procedure for the synthesis of 4-amino-7(H)pyrrolo[2,3-d]pyrimidines (7-deazaadenines 23a–f)
To a solution of N-(2-oxo-2-arylethyl)methanesulfonamide (2 mmol), the selected aldehyde (2.6 mmol) and malononitrile (2.6 mmol) in formamide (2 mL) was added anhydrous granulated K2CO3 (1.2 mmol) in one portion. The mixture was purged with nitrogen for 5 min and then heated at 100 °C for 24 hours under the nitrogen atmosphere. The formation of the intermediate pyrrole was monitored by TLC. After that, the reaction temperature was increased to 150 °C, and the reaction mixture was heated for 3–6 h. The mixture was cooled to room temperature and poured into water (30 mL). The formed precipitate was collected by filtration and washed with EtOH (2 mL) and diethyl ether (2 mL) to give the desired 7-deazaadenine 23a–f.
4-Amino-6-benzoyl-5-(3,5-dibromophenyl)-7(H)pyrrolo[2,3-d]pyrimidine (23a)
58%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.23 (t, J = 7.41, 2H), 7.39 (m, 5H), 7.45 (d, J = 7.2 Hz, 2H), 8.24 (s, 1H),12.63 (s, 1H). 13C NMR (DMSO-d6) δ: 101.4, 119.7, 121.9, 127.5, 128.6, 129.2, 131.9, 132.2, 132.3, 137.1, 137.7, 150.9, 155.0, 158.8, 187.7. HRMS m/z (ESI+) calcd for C19H13Br2N4O (M+H+) 470.9456, found 470.9455.
4-Amino-6-benzoyl-5-(6-bromopyridin-2-yl)-7(H)pyrrolo[2,3-d]pyrimidine (23b)
66%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 6.37 (bs, 2H), 7.20 (m, 2H), 7.38 (m, 3H), 7.79 (s, 1H), 8.23 (s, 1H), 8.32 (s, 1H), 8.42 (s, 1H), 12.66 (s, 1H). 13C NMR (DMSO-d6) δ: 102.2, 118.0, 120.0, 128.3, 129.3, 130.1, 131.5, 132.4, 138.1, 140.5, 149.1, 149.2, 151.6, 155.5, 159.5, 188.1. HRMS m/z (ESI+) calcd for C18H13BrN5O (M+H+) 394.0303, found 394.0312.
4-Amino-6-benzoyl-5-phenyl-7(H)pyrrolo[2,3-d]pyrimidine (23c)
48%, mp 284 °C. 1H NMR (DMSO-d6) δ: 7.13 (m, 2H), 7.29 (m, 5H), 7.31 (m, 1H), 7.45 (d, J = 6.4 Hz, 2H), 7.97 (d, J = 8.8 Hz, 2H), 8.23 (s, 1H), 12.52 (s, 1H). 13C NMR (DMSO-d6) δ: 101.7, 122.5, 127.5, 127.6, 128.1, 128.9, 130.3, 131.7, 133.5, 137.5, 150.7, 154.9, 158.9, 162.9, 187.8. HRMS m/z (ESI+) calcd for C19H15N4O (M+H+) 315.1246, found 315.1238.
4-Amino-6-benzoyl-5-(thiophen-2-yl)-7(H)pyrrolo[2,3-d]pyrimidine (23d)
85%, mp 260 °C. 1H NMR (DMSO-d6) δ: 6.92 (m, 1H), 7.03 (s, 1H), 7.25 (t, J = 6.8 Hz, 2H), 7.4 (m, 3H), 7.58 (d, J = 7.4 Hz, 2H), 7.98 (d, J = 8.9 Hz, 1H), 8.24 (s, 1H), 12.65 (s, 1H). 13C NMR (DMSO-d6) δ: 102.0, 113.2, 127.5, 127.9, 128.2, 128.8, 129.5, 132.2, 133.6, 137.4, 150.6, 154.9, 158.8, 162.8, 187.7. HRMS m/z (ESI) calcd for C17H13N4OS (M+H+) 321.0810, found 321.0801.
4-Amino-6-(3-cyanobenzoyl)-5-(4-benzyloxyphenyl)-7(H)pyrrolo[2,3-d]pyrimidine (23e)
54%, mp 252 °C. 1H NMR (DMSO-d6) δ: 5.12 (s, 2H), 6.82 (d, J = 8.08 Hz, 2H), 7.4 (m, 7H), 7.47 (d, J = 7.3 Hz, 2H), 7.67 (d, J = 6.5 Hz, 2H), 8.24 (s, 1H), 12.60 (s, 1H). 13C NMR (DMSO-d6) δ: 69.5, 69.5, 109.8, 114.1, 118.8, 119.6, 127.6, 128.0, 128.5, 129.4, 130.1, 131.3, 131.6, 131.9, 136.4, 138.7, 150.9, 154.5, 158.6, 161.6, 186.3. HRMS m/z (ESI+) calcd for C27H20N5O2 (M+H+) 446.1617, found 446.1609.
4-Amino-6-benzoyl-5-(2,6-dichlorophenyl)-7(H)pyrrolo[2,3-d]pyrimidine (23f)
72%, mp 262 °C. 1H NMR (DMSO-d6) δ: 7.22 (m, 3H), 7.35 (m, 3H), 7.49 (d, J = 6.7 Hz, 2H), 8.22 (s, 1H), 12.69 (s, 1H). 13C NMR (DMSO-d6) δ: 102.5, 116.5, 128.1, 128.4, 128.8, 129.5, 131.4, 131.9, 132.4, 135.7, 138.3, 151.9, 154.3, 155.8, 159.4, 187.7. HRMS m/z (ESI+) calcd for C19H13Cl2N4O (M+H+) 383.0466, found 383.0474.
General procedure for the synthesis of 4-aryl-7(H)pyrrolo[2,3-d]pyrimidines (7-deazapurines 24a–e)
To a solution of the selected N-(2-oxo-2-arylethyl)methanesulfonamide (3 mmol), the selected arylcyanomethyl ketone (3.9 mmol), the selected aldehyde (3.9 mmol) in formamide (2 mL) was added anhydrous granulated K2CO3 (1.8 mmol). The mixture was purged with nitrogen and stirred overnight at 110 °C. The TLC monitoring of the reaction indicated the disappearance of the sulfonamide and the formation of the intermediate pyrrole (bright yellow spot on TLC). The temperature was then raised to 160 °C and the reaction was stirred for 3–6 h under nitrogen. After the pyrrole disappeared (TLC), the reaction mixture was cooled to room temperature and poured in a beaker with water (40 mL). The precipitate was collected by filtration and purified by column chromatography (CH2Cl2/EtOAc, 10:1) to yield the desired 7-deazapurine 24a–e.
6-(4-Methoxybenzoyl)-4-(4-methoxyphenyl)-5-(4-Methoxyphenyl)-7(H)pyrrolo[2,3-d]pyrimidine (24a)
63%, mp 224 °C. 1H NMR (DMSO-d6) δ: 3.64 (s, 3H), 3.78 (s, 3H), 3.83 (s, 3H), 6.65 (s, 1H), 6.84 (d, J = 7.9 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 7.9 Hz, 2H), 8.13 (d, J = 7.9 Hz, 2H), 8.80 (s, 1H); 13C NMR (DMSO-d6) δ: 54.9, 55.1, 55.3, 113.7, 114.1, 115.4, 125.8, 126.9, 127.8, 129.1, 131.2, 133.3, 134.8, 146.8, 157.0, 158.6, 161.2, 173.7; HRMS m/z (ESI+) calc’d for C28H24N3O4 (M+H+) 466.1767, found 466.1761.
(5-(4-Methoxyphenyl)-4-phenyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)(phenyl)methanone (24b)
30%, mp 208 °C. 1H NMR (DMSO-d6) δ: 3.56 (s, 3H), 6.37 (d, J = 6.8 Hz, 1H), 6.66 (d, J = 6.8 Hz, 1H), 7.06 (dd, J = 8.2 Hz, 2H), 7.23 (m, 5H), 7.39 (t, J = 7.4 Hz, 1H), 7.53 (dd, J = 7.4 Hz, 2H), 8.99 (s, 1H); 13.03 (s, 1H). 13C NMR (DMSO-d6) δ: 55.4, 113.1, 114.5, 120.8, 125.4, 127.5, 128.4, 129.1, 129.7, 129.8, 132.1, 133.1, 137.1, 137.3, 152.14, 153.5, 158.5, 162.3, 189.8. HRMS m/z (ESI+) calc’d for C26H19N3O2 (M+H+) 406.1556, found 406.1557.
(5-(3,5-Dibromophenyl)-4-(p-tolyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)(phenyl)methanone (24c)
33%, mp 250 °C. 1H NMR (DMSO-d6) δ: 2.27 (s, 3H), 6.90 (d, J = 1.7 Hz, 2H), 6.93 (d, J = 7.8 Hz, 1H), 7.10 (dd, J = 8.0 Hz, 2H), 7.25 (dd, J = 8.0 Hz, 2H), 7.37 (t, J = 1.7 Hz, 1H), 7.45 (t, J = 7.4 Hz, 1H), 7.55 (dd, J = 8.1 Hz, 2H), 9.01 (s, 1H); 13.25 (s, 1H). 13C NMR (DMSO-d6) δ: 20.9, 114.0, 118.0, 120.8, 127.8, 128.0, 128.9, 129.2, 131.1, 132.6, 132.9, 133.9, 136.6, 137.0, 138.9, 151.5, 151.5, 162.1, 188.8. HRMS m/z (ESI+) calc’d for C26H18Br2N3O (M+H+) 545.9817, found 545.9819.
(5-(3,5-Dibromophenyl)-4-phenyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)(phenyl)methanone (24d)
39%, mp 257 °C. 1H NMR (DMSO-d6) δ: 6.94 (d, J = 1.8 Hz, 2H), 7.13 (dd, J = 8.0 Hz, 2H), 7.25 (m, 5H), 7.35 (t, J = 1.7 Hz, 1H), 7.45 (t, J = 7.4 Hz, 1H), 7.55, (dd, J = 8.2 Hz, 2H), 9.03 (s, 1H); 13.29 (s, 1H). 13C NMR (DMSO-d6) δ: 114.4, 118.4, 121.3, 127.7, 128.4, 129.4, 129.6, 130.0, 131.7, 133.0, 133.3, 133.4, 137.1, 137.4, 152.0, 153.9, 162.4, 189.2. HRMS m/z (ESI+) calc’d for C25H16Br2N3O (M+H+) 531.9660, found 531.9653.
Phenyl(4-phenyl-5-(pyridin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)methanone (24e)
22%, mp 232 °C. 1H NMR (DMSO-d6) δ: 6.82 (m, 1H), 7.03 (dd, J = 8.0 Hz, 2H), 7.20 (m, 6H), 7.42 (t, J = 7.4 Hz, 1H), 7.55 (dd, J = 8.2 Hz, 2H), 7.96, (d, = 1.08 Hz, 1H), 8.10, (dd, J = 4.8, Hz, 2H), 9.04 (s, 1H); 13.28 (s, 1H). 13C NMR (DMSO-d6) δ: 114.3, 117.2, 121.9, 127.3, 128.1, 129.0, 129.1, 129.2, 129.5, 132.9, 133.2, 132.9, 133.2, 136.6, 136.7, 137.5, 147.3, 150.5, 151.8, 153.4, 162.0, 188.9. HRMS m/z (ESI+) calc’d for C24H17N4O (M+H+) 377.1402, found 377.1403.
General procedure for the synthesis of 1H-pyrrolo[2,3-d]pyrimidin-4(7H)-ones (7-deazahypoxanthines 25a–v)
To a solution of N-(2-oxo-2-arylethyl)methanesulfonamide (0.676 mmol), the selected aldehyde (0.879 mmol) and cyanoacetamide (0.072 g, 0.879 mmol) in a mixture of EtOH (2.5 mL) and HC(OEt)3 (2.5 mL) was added anhydrous granulated K2CO3 (0.052g, 0.372 mmol) in one portion. The mixture was purged with nitrogen for 5 min and then heated at 90 °C for 24 hours under the nitrogen atmosphere. The formation of the intermediate pyrrole was monitored by TLC. After that the reaction temperature was increased to 150 °C, and the reaction mixture was heated for 3–6 h. The mixture was cooled to room temperature, and the formed precipitate was collected by filtration and washed with EtOH (2 mL) and diethyl ether (2 mL) to give the desired 7-deazahypoxanthine 25a–v. An additional amount of the product was obtained by the evaporation of the mother liquor and purification of the residue by column chromatography with MeOH/CH2Cl2=1/40 to 1/20 gradient.
6-Benzoyl-5-phenyl-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25a)
71%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.04 (m, 3H), 7.15 (m, 4H), 7.33 (t, J = 6.8 Hz, 1H), 7.43 (d, J = 7.12 Hz, 2H), 8.02 (s, 1H), 12.01 (s, 1H), 12.79 (s, 1H). 13C NMR (DMSO-d6) δ: 106.3, 126.6, 126.7, 126.8, 127.5, 127.7, 129.0, 131.1, 131.8, 132.4, 137.4, 146.7, 149.3, 158.6, 187.7. HRMS m/z (ESI+) calcd for C19H14N3O2 (M+H+) 316.1086, found 316.1087.
6-Benzoyl-5-(3-iodophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25b)
68%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 6.87 (t, J = 7.52 Hz, 1H), 7.19 (t, J = 7.44 Hz, 2H), 7.25 (d, J = 7.52 Hz, 1H), 7.42 (m, 5H), 8.04 (s, 1H), 12.90 (s, 1H), 12.09 (s, 1H). 13C NMR (DMSO-d6) δ: 93.0, 106.3, 125.0, 127.8, 128.7, 128.8, 130.4, 131.9, 134.6, 135.2, 137.4, 139.5, 146.9, 149.4, 158.5, 187.6. HRMS m/z (ESI+) calcd for C19H13IN3O2 (M+H+) 443.0086, found 443.0080.
6-Benzoyl-5-(3-chlorophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25c)
61%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.04 (t, J = 7.48 Hz, 1H), 7.11 (d, J = 7.52 Hz, 2H), 7.19 (m, 3H), 7.38 (t, J = 6.2 Hz, 1H), 7.46 (d, J = 7.4 Hz, 2H), 8.04 (s, 1H), 12.05 (s, 1H), 13.00 (s, 1H). 13C NMR (DMSO-d6) δ: 106.4, 125.0, 126.5, 127.7, 128.5, 128.9, 129.7, 130.7, 131.5, 132.0, 134.6, 137.5, 146.8, 149.4, 158.5, 187.6. HRMS m/z (ESI+) calcd for C19H13ClN3O2 (M+H+) 350.0696, found 350.0694.
6-Benzoyl-5-(3-bromophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25d)
54%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.00 (t, J = 7.08 Hz, 1H), 7.19 (m, 3H), 7.25 (d, J = 7.76 Hz, 1H), 7.34 (s, 1H), 7.38 (t, J = 6.96 Hz, 1H), 7.45 (d, J = 7.32 Hz, 2H), 8.04 (s, 1H), 12.05 (s, 1H), 12.90 (s, 1H). 13C NMR (DMSO-d6) δ: 106.4, 120.1, 124.9, 127.7, 127.8, 128.8, 128.9, 129.4, 132.0, 130.1, 133.6, 134.8, 137.4, 146.9, 149.3, 158.5, 187.5. HRMS m/z (ESI+) calcd for C19H13BrN3O2 (M+H+) 394.0191, found 394.0183.
6-Benzoyl-5-(3-fluorophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25e)
53%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.08-6.88 (m, 2H), 7.19 (m, 1H), 7.36 (m, 1H), 7.46 (m, 2H), 7.61 (d, J = 7.2 Hz, 1H), 7.96 (d, J = 11.5 Hz, 1H), 8.01 (d, J = 7.2 Hz, 1H), 12.30 (s, 1H), 12.96 (s, 1H). 13C NMR (DMSO-d6) δ: 105.6, 114.1, 114.9, 115.1, 118.3, 124.5, 127.8, 129.0, 129.5, 131.2, 131.3, 132.5, 137.9, 159.8, 188.0. HRMS m/z (ESI+) calcd for C19H13FN3O2 (M+H+) 334.0992, found 395.0999.
6-Benzoyl-5-(6-bromopyridin-2-yl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25f)
60%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.19 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 7.2 Hz, 2H), 7.40 (t, J = 6.4 Hz, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.61 (t, J = 7.6 Hz, 1H), 8.03 (s, 1H), 8.09 (d, J = 8.0 Hz, 1H), 12.17 (s, 1H), 13.05 (s, 1H). 13C NMR (DMSO-d6) δ: 105.5, 122.1, 124.6, 125.4, 128.0, 128.5, 129.7, 132.2, 137.8, 138.7, 139.3, 146.8, 149.3, 152.7, 158.8, 188.5. HRMS m/z (ESI+) calcd for C18H12BrN4O2 (M+H+) 395.0144, found 395.0125.
6-Benzoyl-5-(5-bromopyridin-3-yl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25g)
88%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.21 (d, J = 7.2 Hz, 2H), 7.38 (t, J = 6.4 Hz, 1H), 7.46 (d, J = 6.4 Hz, 2H), 7.81 (s, 1H), 8.05 (s, 1H), 8.31 (s, 1H), 8.35 (s, 1H), 12.12 (s, 1H), 13.01 (s, 1H). 13C NMR (DMSO-d6) δ: 107.3, 119.0, 121.9, 128.4, 129.1, 129.6, 131.1, 132.6, 138.0, 140.8, 147.6, 148.3, 150.0, 150.2, 159.2, 187.7. HRMS m/z (ESI+) calcd for C18H11BrN4NaO2 (M+Na+) 416.9963, found 416.9965.
6-Benzoyl-5-(pyridin-3-yl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25h)
61%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.08 (m, 1H), 7.18 (t, J = 7.24 Hz, 2H), 7.36 (t, J = 6.88 Hz, 1H), 7.46 (d, J = 7.12 Hz, 2H), 7.60 (d, J = 7.72 Hz, 1H), 8.06 (s, 1H), 8.23 (m, 1H), 8.31 (s, 1H), 12.12 (s, 1H), 13.00 (s, 1H). 13C NMR (DMSO-d6) δ: 106.7, 121.9, 123.0, 127.8, 127.9, 128.6, 129.2, 132.0, 137.3, 138.0, 147.0, 147.3, 149.5, 150.8, 158.6, 187.3. HRMS m/z (ESI+) calcd for C18H13N4O2 (M+H+) 317.1039, found 317.1038.
6-Benzoyl-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25i)
64%, mp > 300 °C decomp., 1H NMR (DMSO-d6) δ: 3.32 (s, 3H), 3.54 (s, 3H), 3.56 (s, 3H), 6.46 (s, 2H), 7.16 (t, J = 8.0 Hz, 2H), 7.32 (t, J = 8.0 Hz, 1H), 7.44 (d, J = 7.7 Hz, 2H), 8.02 (m, 1H), 11.98 (s, 1H), 12.78 (s, 1H). 13C NMR (DMSO-d6) δ: 55.6, 59.7, 106.3, 109.4, 127.1, 127.5, 127.7, 128.8, 131.6, 137.8, 146.8, 149.4, 151.3, 158.5, 187.7. HRMS m/z (ESI+) calcd for C22H20N3O5 (M+H+) 406.1403, found 406.1390.
6-Benzoyl-5-(3-iodo-4,5-dimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25j)
37%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.58 (s, 6H), 6.82 (s, 1H), 7.07 (s, 1H), 7.16 (d, J = 7.6 Hz, 2H), 7.33 (t, 1H), 7.40 (d, J = 7.2 Hz, 2H), 8.02 (s, 1H), 12.00 (s, 1H), 12.85 (s, 1H). 13C NMR (DMSO-d6) δ: 56.1, 60.0, 91.3, 106.8, 117.2, 125.7, 128.1, 128.3, 129.2, 130.9, 132.2, 133.2, 138.2, 147.4, 147.6, 149.9, 150.9, 159.0, 188.1. HRMS m/z (ESI+) calcd for C21H17IN3O4 (M+H+) 503.0297, found 503.0280.
6-Benzoyl-5-(3-bromo-4,5-dimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25k)
51%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.59 (s, 6H), 6.77 (s, 1H), 6.95 (s, 1H), 7.16 (d, J = 7.2 Hz, 2H), 7.34 (t, 1H), 7.41 (d, J = 7.2 Hz, 2H), 8.04 (s, 1H), 12.01 (s, 1H), 12.87 (s, 1H). 13C NMR (DMSO-d6) δ: 56.3, 60.2, 106.9, 115.5, 116.3, 125.8, 127.4, 128.0, 128.2, 129.2, 130.2, 132.2, 138.2, 145.0, 147.5, 150.0, 152.0, 159.0, 188.0. HRMS m/z (ESI+) calcd for C21H17BrN3O4 (M+H+) 456.0382, found 456.0370.
6-Benzoyl-5-(2,6-dichlorophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25l)
74%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.12 (d, J = 6.8 Hz, 2H), 7.20 (m, 3H), 7.34 (m, 1H), 7.47 (d, J = 6.0 Hz, 2H), 8.02 (s, 1H), 12.05 (s, 1H), 13.01 (s, 1H). 13C NMR (DMSO-d6) δ: 107.6, 120.2, 127.4, 127.5, 127.9, 128.2, 130.0, 132.0, 132.2, 135.3, 137.8, 147.6, 150.0, 158.0, 187.1. HRMS m/z (ESI) calcd for C19H12Cl2N3O2 (M+H+) 384.0307, found 384.0299.
6-Benzoyl-5-(3,5-dibromophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25m)
77%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.23 (t, J = 7.24 Hz, 2H), 7.36 (s, 2H), 7.41 (t, J = 7.68 Hz, 1H), 7.46 (d, J = 7.68 Hz, 2H), 7.51 (s, 1H), 8.06 (s, 1H), 12.10 (s, 1H), 13.01 (s, 1H). 13C NMR (DMSO-d6) δ: 107.0, 107.1, 124.0, 128.3, 128.6, 129.3, 131.9, 132.6, 133.3, 137.0, 138.0, 147.6, 149.9, 159.0, 187.9. HRMS m/z (ESI+) calcd for C19H12Br2N3O2 (M+H+) 471.9296, found 471.9301.
6-Benzoyl-5-(3-hydroxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25n)
26%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 6.43 (s, 1H), 6.51 (s, 1H), 6.65 (s, 1H), 6.76 (s, 1H), 7.17 (s, 2H), 7.34 (s, 1H), 7.45 (s, 2H), 7.98 (s, 1H), 9.08 (s, 1H), 11.97 (s, 1H), 12.72 (s, 1H). 13C NMR (DMSO-d6) δ: 106.7, 114.2, 118.6, 122.6, 127.0, 128.0, 128.2, 129.4, 132.4, 134.1, 138.0, 147.1, 149.7, 156.5, 159.0, 188.4. HRMS m/z (ESI+) calcd for C19H14N3O3 (M+H+) 332.1035, found 332.1032.
3-(6-Benzoyl-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)benzonitrile (25o)
47%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.19 (t, J = 7.64 Hz, 1H), 7.26 (t, J = 7.2 Hz, 1H), 7.38 (t, J = 6.64 Hz, 1H), 7.44 (d, J = 6.0 Hz, 2H), 7.51 (d, J = 6.96 Hz, 2H), 7.59 (s, 1H), 8.06 (s, 1H), 12.11 (s, 1H), 12.99 (s, 1H). 13C NMR (DMSO-d6) δ: 106.5, 109.9, 118.6, 118.6, 127.8, 128.0, 129.0, 130.2, 131.9, 133.8, 134.5, 135.8, 137.4, 147.0, 149.4, 158.6, 187.3. HRMS m/z (ESI+) calcd for C20H13N4O2 (M+H+) 341.1039, found 341.1041.
5-(3,5-Dibromophenyl)-6-(4-fluorobenzoyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25p)
56%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.03 (t, J = 8.0 Hz, 2H), 7.35 (s, 2H), 7.52 (t, J = 6.4 Hz, 2H), 7.56 (s, 1H), 8.05 (s, 1H), 12.11 (s, 1H), 13.02 (s, 1H). 13C NMR (DMSO-d6) δ: 80.3, 107.1, 115.5, 121.3, 124.4, 128.7, 132.0, 133.5, 134.9, 137.1, 147.9, 150.3, 159.4, 163.2, 166.1, 186.4. HRMS m/z (ESI+) calcd for C19H11Br2FN3O2 (M+H+) 489.9202, found 489.9182.
6-(4-Methoxybenzoyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25q)
56%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 3.32 (s, 3H), 3.56 (s, 6H), 3.71 (s, 3H), 6.51 (s, 2H), 6.71 (d, J = 9.6 Hz, 2H), 7.46 (d, J = 9.6 Hz, 2H), 8.01 (s, 1H), 11.97 (s, 1H). 13C NMR (DMSO-d6) δ: 56.0, 56.3, 60.4, 106.5, 110.0, 113.6, 126.4, 128.4, 131.9, 136.6, 146.9, 149.8, 152.1, 159.3, 162.8, 187.0. HRMS m/z (ESI+) calcd for C23H22N3O6 (M+H+) 436.1509, found 436.1501.
5-(3,5-Dibromophenyl)-6-(4-methoxybenzoyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25r)
60%, mp > 300 °C decomp., 1H NMR (DMSO-d6) δ: 3.77 (s, 3H), 6.79 (d, J = 8.36 Hz, 2H), 7.39 (s, 2H), 7.50 (d, J = 8.36 Hz, 2H), 7.57 (s, 1H), 8.04 (s, 1H), 12.01 (s, 1H), 12.92 (s, 1H). 13C NMR (DMSO-d6) δ: 55.5, 106.1, 113.3, 120.8, 122.3, 128.5, 129.9, 131.2, 131.4, 132.7, 136.6, 146.7, 149.1, 158.5, 162.5, 186.1. HRMS m/z (ESI+) calcd for C20H13Br2N3NaO3 (M+Na+) 523.9221, found 523.9221.
6-(4-(Benzyloxy)benzoyl)-5-(3,5-dibromophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25s)
61%, mp 252 °C. 1H NMR (DMSO-d6) δ: 5.14 (s, 2H), 6.86 (d, J = 7.92 Hz, 2H), 7.36 (m, 3H), 7.41 (m, 4H), 7.56 (m, 3H), 8.04 (s, 1H), 12.15 (s, 1H), 12.95 (s, 1H). 13C NMR (DMSO-d6) δ: 69.4, 106.1, 106.2, 120.8, 122.3, 127.5, 127.9, 128.5, 130.0, 131.2, 131.4, 132.7, 136.5, 136.6, 146.7, 149.1, 158.5, 161.6, 186.0. HRMS m/z (ESI+) calcd for C26H17Br2N3NaO3 (M+Na+) 599.9534, found 599.9532.
6-(4-Bromobenzoyl)-5-(3,5-dibromophenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25t)
54%, mp 256 °C. 1H NMR (DMSO-d6) δ: 7.03 (d, J = 8.66 Hz, 2H), 7.36 (s, 2H), 7.52 (d, J = 8.06 Hz, 2H), 7.56 (s, 1H), 8.04 (s, 1H), 12.08 (s, 1H), 13.00 (s, 1H). 13C NMR (DMSO-d6) δ: 107.3, 114.9, 115.2, 121.2, 124.3, 131.4, 132.7, 132.9, 133.6, 135.4, 159.1, 163.2, 165.7, 186.1. HRMS m/z (ESI+) calcd for C19H11Br3N3O2 (M+H+) 549.8401, found 549.8395.
5-Benzoyl-6-(4-benzyloxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25u)
68%, mp 246 °C. 1H NMR (DMSO-d6) δ: 5.07 (s, 1H), 6.77 (d, J = 8.3 Hz, 2H), 7.06 (m, 3H), 7.20 (m, 2H), 7.38 (m, 4H), 7.45 (d, J = 8.3 Hz, 2H), 7.98 (d, J = 3.7 Hz, 1H), 11.97 (s, 1H), 12.71 (s, 1H). 13C NMR (DMSO-d6) δ: 69.7, 106.6, 114.5, 125.8, 127.0, 127.4, 128.1, 128.2, 128.4, 128.9, 130.5, 131.5, 132.0, 133.1, 136.9, 1 46.8, 149.5, 159.1, 161.9, 186.9. HRMS m/z (ESI+) calcd for C26H20N3O3 (M+H+) 422.1505, found 422.1502.
6-Benzoyl-5-(4-bromothiophen-2-yl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25v)
71%, mp 300 °C decomp., 1H NMR (DMSO-d6) δ: 6.95 (s, 1H), 7.33 (m, 2H), 7.48 (m, 2H), 7.58 (d, J = 6.9 Hz, 2H), 8.04 (s, 1H), 12.11 (s, 1H), 12.99 (s, 1H). 13C NMR (DMSO-d6) δ: 79.6, 79.6, 108.0, 117.0, 125.0, 128.5, 129.0, 129.3, 132.2, 133.0, 135.2, 137.9, 147.5, 149.8, 158.9, 187.9. HRMS m/z (ESI+) calcd for C17H10BrN3NaO2S (M+Na+) 421.9575, found 421.9568.
General procedure for the synthesis of 5-aryl-6-(4-hydroxybenzoyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-ones (7-deazahypoxanthines 25w–z)
To a solution of N-(2-oxo-2-arylethyl)methanesulfonamide (2 mmol), the selected aldehyde (2.6 mmol) and 2-cyanoacetamide (2.6 mmol) in ethanol (2 mL) was added anhydrous granulated K2CO3 (1.2 mmol) in one portion. The mixture was purged with nitrogen for 5 min and then refluxed for 24 hours under the nitrogen atmosphere. The formation of the intermediate pyrrole was monitored by TLC. After that, ethanol was evaporated under reduced pressure. To the dry residue, formic acid (4 mL) was added, and the mixture was refluxed for 24 hours. The formation of the 7-deazahypoxanthines 25w–z was monitored by TLC. The mixture was cooled to room temperature and purified by flash chromatography to give the desired product.
6-(4-Hydroxybenzoyl)-5-phenyl-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25w)
88%, mp > 300 °C decomp., 1H NMR (DMSO-d6) δ: 6.52 (d, J = 7.88 Hz, 2H), 7.11 (m, 3H), 7.22 (m, 2H), 7.40 (d, J = 7.4 Hz, 2H), 7.99 (s, 1H), 10.17 (br.s, 1H), 11.93 (s, 1H), 12.66 (s, 1H). 13C NMR (DMSO-d6) δ: 106.0, 114.6, 124.6, 126.5, 126.9, 127.9, 128.3, 131.0, 131.9, 132.7, 146.2, 148.9, 158.8, 161.5, 186.7. HRMS m/z (ESI+) calcd for C19H13KN3O3 (M+K+) 370.0594, found 370.0595.
5-(3,5-Dibromophenyl)-6-(4-hydroxybenzoyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25x)
70%, mp > 300 °C decomp. 1H NMR (DMSO-d6) δ: 7.42 (m, 4H), 7.57 (s, 1H), 7.61 (d, J = 7.8 Hz, 2H), 8.03 (s, 1H), 10.26 (s, 1H), 12.06 (s, 1H), 12.88 (s, 1H). 13C NMR (DMSO-d6) δ: 106.4, 115.2, 121.3, 122.0, 128.7, 129.1, 131.6, 132.3, 133.2, 137.2, 147.0, 149.4, 159.0, 162.2. HRMS m/z (ESI+) calcd for C19H12Br2N3O3 (M+H+) 489.9225, found 489.9220.
6-(4-Hydroxybenzoyl)-5-(4-hydroxy-3-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25y)
64%, mp > 300 °C decomp., 1H NMR (DMSO-d6) δ: 3.54 (s, 3H), 6.76-6.47 (m, 5H), 7.39 (d, J = 8.1 Hz, 2H), 8.81 (s, 1H), 7.95 (s, 1H), 10.12 (s, 1H), 11.88 (s, 1H), 12.52 (s, 1H). 13C NMR (DMSO-d6) δ: 121.9, 115.7, 115.1, 114.4, 128.8, 134.4, 141.3, 146.6, 149.9, 154.4, 157.1, 159.8, 162.5, 168.1, 187.6. HRMS m/z (ESI+) calcd for C20H15KN3O5 (M+K+) 400.0909, found 400.0917.
6-(4-Hydroxybenzoyl)-5-(4-hydroxyphenyl)-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (25z)
71%, mp > 300 °C decomp., 1H NMR (DMSO-d6) δ: 6.46 (d, J = 7.9 Hz, 2H), 6.51 (d, J = 7.9 Hz, 2H), 7.00 (d, J = 7.7 Hz, 2H), 7.37 (d, J = 7.7 Hz, 2H), 7.94 (s, 1H), 9.24 (s, 1H), 10.13 (s, 1H), 11.89 (s, 1H), 12.51 (s, 1H). 13C NMR (DMSO-d6) δ: 106.3, 114.3, 115.0, 123.8, 125.6, 127.8, 128.8, 129.0, 132.4, 132.7, 146.5, 149.3, 156.7, 159.2, 161.7, 186.0. HRMS m/z (ESI+) calcd for C19H14N3O4 (M+H+) 348.0984, found 348.0987.
Cell Culture
Human cancer cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), the European Collection of Cell Culture (ECACC, Salisbury, UK) and the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). The origin and histological type of each human cell line analyzed are as follows: glioma model lines were the Hs683 oligodendroglioma (ATCC code HTB-138) and the U373 glioblastoma (ECACC code 89081403) cell lines, melanoma model was the SKMEL-28 (ATCC code HTB-72) cell line, and carcinoma model was the A549 NSCLC (DSMZ code ACC107) cell line. Human cervical adenocarcinoma HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Human mammary carcinoma MCF-7 cells were cultured in DMEM supplemented with 10% FBS. Human uterine sarcoma MES-SA and MES-SA/Dx5 cells were cultured in RPMI-1640 medium supplemented with 10% FBS. The U373 cells were cultured in MEM culture medium (Lonza code 12-136F, Vervier, Belgium), while the Hs683, SKMEL-28 and A549 cells were cultured in RPMI culture medium (Lonza; code 12-115F) supplemented with 10% heat-inactivated FBS (Lonza, FBS South America code DE14-801F). Cell culture media were supplemented with 4 mM glutamine (Lonza code BE17-605E), 100 μg/mL gentamicin (Lonza code 17-5182), and penicillin-streptomycin (200 units/ml and 200 μg/ml) (Lonza code 17-602E). All cell lines were cultured in T25 flasks, maintained and grown at 37° C, 95% humidity, 5% CO2.
Human head and neck cancer cell lines UMSCC10A, 10B, 22A and 22B were provided by Dr. Thomas Carey of University of Michigan under the MTA agreement with the University of Colorado. UMSCC10A and 22A were originated from the primary tumors of two individual head and neck squamous cell carcinoma patients, and 10B and 22B were from their corresponding lymph node metastases. Cells were cultured in DMEM medium supplemented with 10% FBS, and antibiotics.
Antiproliferative Properties
To evaluate antiproliferative properties of the rigidins and rigidin analogues, the MTT assay was used. The HeLa, MCF-7, MES-SA, and MES-SA/Dx5 cell lines were assessed by trypsinizing each cell line and seeding 4 × 103 cells per well into microtiter plates. All compounds were dissolved in DMSO at a concentration of either 100 mM or 25 mM prior to cell treatment. The cells were grown for 24 h before treatment at concentrations ranging from 0.004 to 100 μM and incubated for 48 h in 200 μL media. 20 μL of MTT reagent in serum free medium (5 mg/mL) was added to each well and incubated further for 2 h. Media was removed, and the resulting formazan crystals were re-solubilized in 100 μL of DMSO. A490 was measured using a Thermomax Molecular Device plate reader. The experiments were performed in quadruplicate and repeated at least twice for each compound per cell line. Cells treated with 0.1% DMSO were used as a control, and phenyl arsine oxide (PAO) was used as a positive killing control.
For UMSCC-10A, 10B, 22A and 22B cells, the MTT assay was performed in a 24-well plate with 40000 cells in 500 uL media. The cells were grown for 24 h before treatment at concentrations ranging from 0.025 to 0.4 μM and incubated for additional 4 days. MTT was added for 3 h before harvest. A560 was measured using a BioTek plate reader. The experiments were performed in quadruplicate and repeated at least twice for each compound per cell line. Cells treated with 0.1% DMSO were used as a control.
Cell Cycle Analysis
To assess the effects of the analogues on the cell cycle, cell cycle analysis was performed by flow cytometry. 70–80 % confluent HeLa cells were trypsinized and seeded at an initial density of 2 × 105 cell per well into 6 well plates in DMEM supplemented with 10% FBS, 100 mg/L penicillin G and 100 mg/L streptomycin. The cells were allowed to adhere overnight before treatment with 25g or 25m at 1 μM. After 32 h, the cells were washed 3x with DMEM, trypsinized, pelleted, and re-suspended in 200 μL of culture medium containing 10 μM (2 μL/mL) Vybrant Orange dye. The samples in the labeling solution were transferred into Falcon tubes and incubated in a water bath for 30 min at 37 °C. The samples were then analyzed using a Becton Dickinson FACscan flow cytometer with CellQuest software. Cells treated with 0.1% DMSO were used as a control.
In Vitro Tubulin Polymerization Assay
To determine whether the compounds bind to tubulin, initial experiments were performed with the tubulin polymerization assay obtained from Cytoskeleton Inc. A 10x stock solution of each test compound (0.1% DMSO, taxol, colchicine, 25a and 25m) was prepared using molecular grade water. The tubulin reaction mix was prepared by mixing 243 μL of buffer 1 [80 mM PIPES sequisodium salt; 2.0 mM MgCl2; 0.5 mM ethylene glycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, pH 6.9, 10 μM DAPI], 112 μL tubulin glycerol buffer (80 mM PIPES sequisodium salt; 2.0 mM MgCl2; 0.5 mM ethylene glycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 60% v/v glycerol, pH 6.9), 1 mM GTP (final concentration) and 2 mg/mL tubulin protein (final concentration). The reaction mixture was kept on ice until needed, but it was used within an hour of preparation. The 10x test compounds were pipetted into the corresponding wells and warmed in the plate reader for 1 min, after which time they were diluted with the reaction mixture to their final 1x concentrations and placed in the fluorescent plate reader. The test compounds were incubated with the tubulin reaction mixture at 37 °C each in a separate experiment. The effect of each agent on tubulin polymerization was monitored in a temperature-controlled TECAN GENios Multifunction Fluorescence TRF, FI, FRET, Absorbance and Luminescence Microplate Reader for one hour, with readings acquired every 60 s.
Quantitative Effects on Tubulin Polymerization and on Colchicine Binding to Tubulin
To evaluate the quantitative effect of the compounds on tubulin assembly in vitro, varying concentrations of compounds were preincubated with 10 μM (1.0 mg/mL) bovine brain tubulin in 0.8 M monosodium glutamate (pH 6.6 in 2M stock solution) at 30 °C for 15 min and then cooled to 0 °C. After addition of 0.4 mM GTP, the mixtures were transferred to 0 °C cuvettes in recording spectrophotometers equipped with electronic temperature controller and rapidly (less than one minute) warmed to 30 °C. Tubulin assembly was followed turbidimetrically at 350 nm. The IC50 was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The methodology was described in detail previously.40 The capacity of the test compounds to inhibit colchicine binding to tubulin was measured as described.55 The reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine, and 5 or 1 μM test compound. Combretastatin A-4 was generously provided by Dr. G. A. Pettit, Arizona State University.
Morphological Analysis of Microtubule Organization in HeLa Cells
HeLa cells were cultured in EMEM (Lonza, Walkersville, MD) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), sodium pyruvate and sodium bicarbonate. For compound treatments, cells were treated for 3 h with either carrier (0.1% DMSO) or compounds solubilized in DMSO and prediluted in media prior to fixation by immersion in methanol at −20°C for 30 min. Cells were subsequently rehydrated in phosphate-buffered saline (1XPBS), and blocked by incubation in 3% bovine serum albumin (dissolved in 1XPBS) for one hour at room temperature. Cells were then incubated overnight at 4 °C with mouse anti-tubulin antibody (Sigma) and rabbit anti-pericentrin antibody (Abcam, Cambridge, MA) in blocking buffer. Primary antibodies were detected using Alexafluor-conjugated secondary antibodies (Molecular Probes), while DNA was detected using Hoechst 33342 (Invitrogen). All images were acquired using a Zeiss Axiovert 200M inverted microscope equipped with epifluorescence optics and an Apotome module (Carl Zeiss). Acquired images were then exported into eight bit tiff files, and figures were prepared using Adobe Photoshop software.
Water Solubility
A known mass of each molecule was dispersed in ultrapure deionized water (Millipore) at a concentration > 0.5 mg/mL. The supersaturated mixtures were sonicated for 1 minute in a bath sonicator and allowed to equilibrate for > 24 hours at room temperature. The supersaturated mixtures were then centrifuged for 10 minutes at 10,000 rpm (9,391 × g) in an Eppendorf microcentrifuge to remove the insoluble fraction of the drug. The supernatant was centrifuged further in a separate tube and the centrifugation process was repeated until no pellet was visible. The solution of the drug was then diluted in dimethyl sulfoxide (DMSO) at a 1:2 volume ratio of solution-to-DMSO. The concentration of the molecule in the water/DMSO solution was determined based on a concentration-absorbance calibration line created with solutions of the drug in a 1:2 water/DMSO co-solvent system. The peak absorbance of the drug (335–336 nm) was used to create the calibration. The water solubility of the drug was then determined by taking into account the dilution performed with the addition of DMSO.
Selection of Doxorubicin Resistant Cells
To assess the effects of the hypoxanthine-like rigidin analogues on MDR cells, the MES-SA/Dx5 cell line had to first be selected for. This was done according to Harker et al.52 The cells were split and allowed to adhere overnight. The next day cells were initially exposed to a DOX concentration of 100 nM, which represented the GI50 concentration. The cells were maintained at this DOX concentration until their growth rate reached that of the untreated cells. The DOX concentration was then increased in two-fold increments and following the same growth criteria at each concentration. A final DOX concentration of 500 nM was used as the stopping point. Each new DOX concentration required approximately 2 passages to reach the growth rate of the untreated cells.
Supplementary Material
Acknowledgments
This project was supported by grants from the National Institute of General Medical Sciences (P20GM103451), National Cancer Institute (CA-135579) and National Science Foundation (NSF award 0946998). AK is grateful to the Texas State University for start-up funding. TB thanks the Texas Emerging Technology fund for start-up support. Mary R. Reisenauer is thanked for performing the MTT assay with compounds 24b–e. LMYB and RK are research assistant and director of research, respectively, with the Fonds National de la Recherche Scientifique (FRS-FNRS, Belgium).
Abbreviations Used
- ATCC
American Type Culture Collection
- 3-CR
three-component reaction
- 4-CR
four-component reaction
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DSMZ
Deutsche Sammlung von Mikroorganismen and Zellkulturen
- ECACC
European Collection of Cell Culture
- EDTA
diaminoethanetetraacetic acid
- EtOH
ethanol
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FOG
Ficol Orange G
- HEPES
4-(2-hydroxyethyl)-1-piperazinethanesulfonic acid
- HHB
Heinz-HEPES buffer
- HRMS
high resolution mass spectrometry
- MCR
multicomponent reaction
- MDR
multidrug resistant
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PAO
phenyl arsine oxide
- P-gp
P-glycoprotein
- SAR
structure-activity relationship
- TLC
thin layer chromatography
- SD
standard deviation
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Urban S, Hickford SJH, Blunt JW, Munro MHG. Bioactive marine alkaloids. Curr Org Chem. 2000;4:765–807. [Google Scholar]
- 2.Hill RA. Marine natural products. Annu Rep Prog Chem Sect B. 2005;1001:124–136. [Google Scholar]
- 3.Simmons TL, Andrianasolo E, McPhail K, Flatt P, Gerwick WH. Marine natural products as anticancer drugs. Mol Cancer Ther. 2005;4:333–342. [PubMed] [Google Scholar]
- 4.Dembitsky VM, Gloriozova TA, Poroikov VV. Novel antitumor agents: marine sponge alkaloids, their synthetic analogs and derivatives. Mini-Rev Med Chem. 2005;5:319–336. doi: 10.2174/1389557053175362. [DOI] [PubMed] [Google Scholar]
- 5.Nakao Y, Fusetani N. Enzyme inhibitors from marine invertebrates. J Nat Prod. 2007;70:679–710. doi: 10.1021/np060600x. [DOI] [PubMed] [Google Scholar]
- 6.Sugumaran M, Robinson WE. Bioactive dehydrotyrosyl and dehydrodopyl compounds of marine origin. Mar Drugs. 2010;8:2906–2935. doi: 10.3390/md8122906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan H, Peng J, Hamann MT, Hu J-F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem Rev. 2008;108:264–287. doi: 10.1021/cr078199m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ploypradith M, Batsomboon P, Ruchirawat S, Ploypradith P. Cytotoxicities and structure-activity relationships of natural and unnatural lamellarins toward cancer cell lines. Chem Med Chem. 2009;4:457–465. doi: 10.1002/cmdc.200800339. [DOI] [PubMed] [Google Scholar]
- 9.Mabuchi S, Hisamatsu T, Kawase C, Hayashi M, Sawada K, Mimura K, Takahashi K, Takahashi T, Kurachi H, Kimura T. The activity of trabectedin as a single agent or in combination with everolimus for clear cell carcinoma of the ovary. Clin Cancer Res. 2011;17:4462–4473. doi: 10.1158/1078-0432.CCR-10-2987. [DOI] [PubMed] [Google Scholar]
- 10.Kabayashi J, Cheng J, Kikuchi Y, Ishibashi M, Yamamura S, Ohizumi Y, Ohta T, Nozoe S. Rigidin, a novel alkaloid with calmodulin antagonistic activity from the Okinawan marine tunicate Eudistma CF. Rigida Tetrahedron Lett. 1990;31:4617–4620. [Google Scholar]
- 11.Tsuda M, Nozawa K, Shimbo K, Kobayashi J. Rigidins B-D, new pyrrolopyrimidine alkaloids from a tunicate Cystodytes species. J Nat Prod. 2003;66:292–294. doi: 10.1021/np020393a. [DOI] [PubMed] [Google Scholar]
- 12.Davis RA, Christensen LV, Richardson AD, Moreira da Rocha R, Ireland CM. Rigidin E, a new pyrrolopyrimidine alkaloid from a Papua New Guinea tunicate Eudistoma species. Mar Drugs. 2003;1:27–33. [Google Scholar]
- 13.Edstrom ED, Wei Y. Synthesis of a novel pyrrolo[2,3-d]pyrimidine alkaloid, rigidin. J Org Chem. 1993;58:403–407. [Google Scholar]
- 14.Sakamoto T, Kondo Y, Sato S, Yamanaka H. Total synthesis of a marine alkaloid, rigidin. Tetrahedron Lett. 1994;35:2919–2920. [Google Scholar]
- 15.Sakamoto T, Kondo Y, Sato S, Yamanaka H. Condensed heteroaromatic ring systems. Part 24. Synthesis of rigidin, a pyrrolo[2,3-d]pyrimidine marine alkaloid. J Chem Soc, Perkin Trans I. 1996:459–464. [Google Scholar]
- 16.Gupton JT, Banner EJ, Scharf AB, Norwood BK, Kanters RPF, Dominey RN, Hempel JE, Kharlamova A, Bluhn-Chertudi I, Hickenboth CR, Little BA, Sartin MD, Coppock MB, Krumpe KE, Burnham BS, Holt H, Du KX, Keertikar KM, Diebes A, Ghassemi S, Sikorski JA. The application of vinylogous iminium salt derivatives to an efficient synthesis of the pyrrole containing alkaloids rigidin and rigidin E. Tetrahedron. 2006;62:8243–8255. [Google Scholar]
- 17.Magedov IV, Kireev AS, Jenkins AR, Evdokimov NM, Lima DT, Tongwa P, Altig J, Steelant WFA, Van slambrouck S, Antipin MY, Kornienko A. Structural simplification of bioactive natural products with multicomponent synthesis. 4. 4H-Pyrano-[2,3-b]naphthoquinones with anticancer activity. Bioorg Med Chem Lett. 2012;22:5195–5198. doi: 10.1016/j.bmcl.2012.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magedov IV, Kornienko A. Multicomponent reactions in alkaloid-based drug discovery. Chem Heterocycl Comp. 2012:38–43. doi: 10.1007/s10593-012-0965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magedov IV, Frolova L, Manpadi M, Bhoga UD, Tang H, Evdokimov NM, George O, Georgiou KH, Renner S, Getlic M, Kinnibrugh TL, Fernandes MA, Van slambrouck S, Steelant WFA, Shuster CB, Rogelj S, van Otterlo WAL, Kornienko A. Anticancer properties of an important drug lead podophyllotoxin can be efficiently mimicked by diverse heterocyclic scaffolds accessible via one-step synthesis. J Med Chem. 2011;54:4234–4246. doi: 10.1021/jm200410r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evdokimov NM, Van slambrouck S, Heffeter P, Tu L, Le Calve B, Lamoral-Theys D, Hooten CJ, Uglinskii PY, Rogelj S, Kiss R, Steelant WFA, Berger W, Yang JJ, Bologa CJ, Kornienko A, Magedov IV. Structural simplification of bioactive natural products with multicomponent synthesis. 3. Fused uracil-containing heterocycles as novel topoisomerase-targeting agents. J Med Chem. 2011;54:2012–2021. doi: 10.1021/jm1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta PK, Daunert S, Nassiri MR, Wotring LL, Drach JC, Townsend LB. Synthesis, cytotoxicity, and antiviral activity of some acyclic analogues of the pyrrolo[2,3-d]pyrimidine nucleoside antibiotics tubercidin, toyocamycin, and sangivamycin. J Med Chem. 1989;32:402–408. doi: 10.1021/jm00122a019. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed MS, Kamel R, Fatahala SS. Synthesis and biological evaluation of some thio-containing pyrrolo[2,3-d]pyrimidine derivatives for their anti-inflammatory and anti-microbial activities. Eur J Med Chem. 2010;45:2994–3004. doi: 10.1016/j.ejmech.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 23.Mohamed MS, Rashad AE, Adbel-Monem M. New anti-inflammatory agents. Z Naturforsch. 2007;62c:27–31. doi: 10.1515/znc-2007-1-205. [DOI] [PubMed] [Google Scholar]
- 24.Abou El Ella DA, Ghorab MM, Noaman E, Heiba HI, Khalil AI. Molecular modeling study and synthesis of novel pyrrolo[2, 3-d]pyrimidines and pyrrolotriazolopyrimidines of expected antitumor and radioprotective activities. Bioorg Med Chem. 2008;16:2391–2402. doi: 10.1016/j.bmc.2007.11.072. [DOI] [PubMed] [Google Scholar]
- 25.Willemann C, Grunert R, Bednarski PJ. Synthesis and cytotoxic activity of 5,6-heteroaromatically annulated pyridine-2,4-diamines. Bioorg Med Chem. 2009;17:4406–4419. doi: 10.1016/j.bmc.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Tangeda SJ, Garrlapati A. Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives and evaluation of their activities against human colon cancer cell lines. Eur J Med Chem. 2010;45:1453–1458. doi: 10.1016/j.ejmech.2009.12.050. [DOI] [PubMed] [Google Scholar]
- 27.Ghorab MM, Ragab FA, Heiba HI, Youssef HA, El-Gazzar MG. Synthesis of novel pyrrole and pyrrolo[2,3-d]pyrimidine derivatives bearing sulfonamide moiety for evaluation as anticancer and radiosensitizing agents. Bioorg Med Chem Lett. 2010;20:6316–6320. doi: 10.1016/j.bmcl.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Gangjee A, Zhao Y, Lin L, Raghavan S, Roberts EG, Risinger AL, Hamel E, Mooberry SL. Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicines site on tubulin and circumvent P-gp mediated resistance. J Med Chem. 2010;53:8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gangjee A, Kurup S, Ihnat MA, Thorpe JE, Shenoy SS. Synthesis and biological activity of N4-phenylsubstituted-6-(2,4-dichlorophenylmethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic and antitumor agents. Bioorg Med Chem. 2010;18:3575–3587. doi: 10.1016/j.bmc.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennet SM, Nguyen-Ba N, Ogilvie KK. Synthesis and antiviral activity of some acyclic and C-acyclic pyrrolo[2,3-d]pyrimidine nucleoside analogues. J Med Chem. 1990;33:2162–2173. doi: 10.1021/jm00170a019. [DOI] [PubMed] [Google Scholar]
- 31.Krawczyk SH, Nassiri MR, Kucera LS, Kern ER, Ptak RG, Wotring LL, Drach JC, Townsend LB. Synthesis and antiproliferative and antiviral activity of 2′-deoxy-2′-fluoroarabinofuranosyl analogs of the nucleoside antibiotics toyocamycin and sangivamycin. J Med Chem. 1995;38:4106–4114. doi: 10.1021/jm00020a026. [DOI] [PubMed] [Google Scholar]
- 32.Hess S, Muller CE, Frobenius W, Reith U, Klotz K-N, Eger K. 7-deazaadenunes bearing polar substituents: structure-activity relationships of new A1 and A3 adenosine receptor antagonists. J Med Chem. 2000;43:4636–4646. doi: 10.1021/jm000967d. [DOI] [PubMed] [Google Scholar]
- 33.Bookser BC, Ugarkar BG, Matelich MC, Lemus RH, Allan M, Tsuchiya M, Nakane M, Nagahisa A, Wiesner JB, Erion MD. Adenosine kinase inhibitors. 6. Synthesis, water solubility, and antinociceptive activity of 5-phenyl-7-(5-deoxy-b-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidines substituted at C4 with glycinamides and related compounds. J Med Chem. 2005;48:7808–7820. doi: 10.1021/jm050394a. [DOI] [PubMed] [Google Scholar]
- 34.Kim YA, Sharon A, Chu CK, Rais RH, Al Safarjalani ON, Naguib FNM, el Kouni MH. Structure-activity relationships of deaza-6-benzylthioinosine analogues as ligands of Toxoplasma gondii adenosine kinase. J Med Chem. 2008;51:3934–3945. doi: 10.1021/jm800201s. [DOI] [PubMed] [Google Scholar]
- 35.Gangjee A, Jain HD, Queener SF, Kisliuk RL. The effect of 5-alkyl modification on the biological activity of pyrrolo[2,3-d]pyrimidine containing classical and nonclassical antifolates as inhibitors of dihydrofolate reductase and as antitumor and/or antiopportunistic infection agents. J Med Chem. 2008;51:4589–4600. doi: 10.1021/jm800244v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frolova LV, Evdokimov NM, Hayden K, Malik I, Rogelj S, Kornienko A, Magedov IV. One-pot multicomponent synthesis of diversely substituted 2-aminopyrroles. A short general synthesis of rigidins A, B, C, and D. Org Lett. 2011;13:1118–1121. doi: 10.1021/ol103149b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosmann T. Rapid colorimetric assay for cellular growth and survival - application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.See for example: Kemnitzer W, Drewe J, Jiang S, Zhang H, Wang Y, Zhao J, Jia S, Herich J, Labreque D, Storer R, Meerovitch K, Bouffard D, Rej R, Denis R, Blais C, Lamothe S, Attardo G, Gourdeau H, Tseng B, Kasibhatla S, Cai SX. Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 1. Structure-activity relationships of the 4-aryl group. J Med Chem. 2004;47:6299–6310. doi: 10.1021/jm049640t.
- 39.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nature Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 40.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 41.Lin CM, Ho HH, Pettit GR, Hamel E. The antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 42.Tahir SK, Kovar SH, Ng RSC. Rapid colchicine competition-binding scintillation proximity assay using biotin-labeled tubulin. BioTechniques. 2000;29:156–160. doi: 10.2144/00291rr02. [DOI] [PubMed] [Google Scholar]
- 43.Löwe J, Li H, Downing KH, Nogales E. Refined structure of αβ-tubulin at 3.5 Å resolution. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 44.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 45.Gigant B, Wang C, Ravelli RBG, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435:519–522. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- 46.Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, Yang CP. Taxol: mechanisms of action and resistance. J Natl Cancer Inst Monogr. 1993;15:55–61. [PubMed] [Google Scholar]
- 47.Kuppens IE. Current state of the art of new tubulin inhibitors in the clinic. Curr Clin Pharmacol. 2006;1:57–70. doi: 10.2174/157488406775268200. [DOI] [PubMed] [Google Scholar]
- 48.Beckers T, Reissmann T, Schmidt M, Burger AM, Fiebig HH, Vanhoefer U, Pongratz H, Hufsky H, Hockemeyer J, Frieser M, Mahboobi S. 2-Aroylindoles, a novel class of potent, orally active small molecule tubulin inhibitors. Cancer Res. 2002;62:3113–3119. [PubMed] [Google Scholar]
- 49.Hande KR, Hagey A, Berlin J, Cai Y, Meek K, Kobayashi H, Lockhart AC, Medina D, Sosman J, Gordon GB, Rothenberg ML. The pharmacokinetics and safety of ABT-751, a novel, orally bioavailable sulfonamide antimitotic agent: results of a phase 1 study. Clin Cancer Res. 2006;12:2834–2840. doi: 10.1158/1078-0432.CCR-05-2159. [DOI] [PubMed] [Google Scholar]
- 50.Rupp C, Steckel H, Mueller BW. Solubilization of poorly water-soluble drugs by mixed micelles based on hydrogenated phosphatidylcholine. Int J Pharm. 2010;395:272–280. doi: 10.1016/j.ijpharm.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 51.Mueller CE. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodiversity. 2009;6:2071–2083. doi: 10.1002/cbdv.200900114. [DOI] [PubMed] [Google Scholar]
- 52.For a recent example see: Regina G, Bai R, Rensen WM, Di Cesare E, Coluccia A, Piscitelli F, Famiglini V, Reggio A, Nalli M, Pelliccia S, Da Pozzo E, Costa B, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Santoni A, Li J, Cona MM, Chen F, Ni Y, Brancale A, Dondio G, Vultaggio S, Varasi M, Mercurio C, Martini C, Hamel E, Lavia P, Novellino E, Silvestri R. Toward Highly Potent Cancer Agents by Modulating the C-2 Group of the Arylthioindole Class of Tubulin Polymerization Inhibitors. J Med Chem. 2013;56:123–149. doi: 10.1021/jm3013097.
- 53.See for example: Romagnoli R, Baraldi PG, Salvador MK, Preti D, Tabrizi MA, Brancale A, Fu X-H, Li J, Zhang S-Z, Hamel E, Bortolozzi R, Basso G, Viola G. Synthesis and evaluation of 1, 5-disubstituted tetrazoles as rigid analogues of combretastatin A-4 with potent antiproliferative and antitumor Activity. J Med Chem. 2012;55:475–488. doi: 10.1021/jm2013979.
- 54.Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Research. 1985;45:4091–4096. [PubMed] [Google Scholar]
- 55.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–67. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.