

Abstract
Polyprenols, a type of universal glycan lipid carrier, play important roles for glycan bio-assembly in wide variety of living systems. Chemical synthesis of natural polyisoprenols such as undecaprenol and dolichols, but especially their homologs, could serves as a powerful molecular tool to dissect and define the functions of enzymes involved in glycan biosynthesis. In this paper, we report an efficient and reliable method to construct this type of hydrophoic molecule through a base-mediated iterative coupling approach using a key bifunctional (Z, Z)-diisoprenyl building block. The ligation with N-acetyl-D-glactosamine (GalNAc) with a set of the synthesized lipid analogs forming polyprenol pyrophosphate linked GalNAc (GalNAc-PP-lipid) conjugates is also demonstrated.
Keywords: Polyprenol, Enzyme substrate specificity, Lipopolysaccharide
Introduction
Polyprenol phosphates, including undecaprenyl-phosphate in bacteria and dolichol-phosphate in archaea and eukaryotes, serve as specific membrane-bound glycan carriers throughout glycan bio-assembly.[1] These carriers, in the form of polyprenol pyrophosphate linked glycans (glycan-pp-lipid), subsequently act as donors for specific enzymes which translocate the glycan or glycopeptide moiety en-block onto macromolecules. This glycan transfer forms such important cellular structures as N-linked glycoproteins in archaea and eukaryotes[2] as well as peptidoglycan and lipopolysaccharide (LPS) in bacteria.[3] In the case of LPS, which contributes significantly to bacterium-host interactions, resistance to serum-mediated killing, and regulation of the host immune-response,[4] the polyprenol undecaprenol acts as the lipid carrier (Figure 1). Specifically, undecaprenol supports generation of the O-polysaccharide repeating unit structure and its subsequent polymerization by the polymerase Wzy before the glycan is transferred through the action of WaaL to complete LPS biosynthesis (Figure 1). In an effort to more fully understand this biosynthetic pathway, we have recently reconstituted the LPS biosynthetic pathway in vitro. As part of this work, we have developed a series of polyprenols that allows for a comprehensive evaluation of the most relevant structural elements of these lipids including: 1) length, 2) olefin stereochemistry, and 3) saturation of the α-isoprene unit. However, given that the majority of these polyprenols are not readily available commercially, synthetic methods had to be employed to construct this lipid library.
Figure 1.

Schematic illustration of E. coli O86 O-polysaccharide biosynthesis and its ligation with Lipid A core by O-antigens ligase WaaL forming lipopolysaccharide (LPS).
Luckily, advances in the methodology for production of polyprenols have attracted tremendous attention in the past decades. The first examples, which appeared in the early 1980s, detailed the synthesis of (all-E)-polyprenols.[5] However, a method for the stereoselective formation of trisubstituted Z-olefins was needed to gain access to the aforementioned undecaprenol and analogs thereof that contain multiple (Z)-isoprenyl units. The group of Still[6] and Sato et al.[7] respectively reported such an approach, in which various α-alkoxyketones were shown to be capable of yielding (Z)-olefins with high selectivity via Wittig olefination with unstablized ylides. The method later led to a very useful synthetic protocol which utilizes a bifunctional (Z, Z)-diene building block to elongate the lipid chain via transformation of its two terminal functional groups into two respective ligation-amenable coupling partners.[8] While additional methods have been reported for the solution-phase 29–31 and solid-phase synthesis32 of polyprenols, we found that slight modifications to the Sato protocol [7] provided a very robust and efficient method for obtaining (Z)-olefin containing polyprenols. Accordingly, reported herein is our application of the Sato approach to the development of a comprehensive polyprenol library which enables the detailed characterization of lipid substrate specificity for polyprenol-based bacterial systems. The subsequent conjugation of these lipids with N-acetyl-D-galactosamine (GalNAc) to form polyisoprenol pyrophosphate-linked GalNAc derivatives (GalNAc-PP-lipid) is also reported. While we focused on the application of this library towards understanding LPS biosynthesis, it should also be noted that this library can be readily extended to characterize the specificity of other systems such as the oligosaccharyltransferase PglL that is involved in bacterial O-linked protein glycosylation.[9]
Results and Discussion
To examine the polyprenol substrate specificity of Wzy and WaaL, a small library was envisioned (Figure 2). The first three polyprenols, undecaprenol, heptaprenol and pentaprenol, were selected to evaluate the effect of length on polymerization. As can be seen, the number of isoprene units decreases from 11 to 7 to 5 along this series. However, it was also noted that as the length decreases in this series, the number of (Z)-olefins also decreases. Given that these olefins are adjacent to the hydroxy terminus and thus in close proximity to the bond breaking event during Wzy-mediated polymerization, it is reasonable to believe that they may serve as an essential recognition element. A derivative of pentaprenol, (all-Z)-pentaprenol was therefore proposed in which the number of Z-olefins is increased to four with the total number of isoprene units remaining constant. The hypothesis for this design was that if Z-olefins are an important recognition element, then the degree of polymerization should increase from pentaprenol to (Z)-pentaprenol-based substrates. In contrast, if double bond geometry is irrelevant, then no increase in activity should be observed. The (all-E)-polyprenol solanesol was also proposed to support the results of the (all-Z)-pentaprenol experiment. Finally, while bacteria utilize isoprene-based polyprenols in which each isoprene unit is unsaturated, eukaryotes instead utilize a polyprenol class generally referred to as dolichols.[10] Dolichols are almost structurally identical to bacterial polyprenols with the exception of the α-isoprene unit which is saturated. Another derivative of pentaprenol, monosaturated (MS)-pentaprenol, was thus proposed to consider the importance of having an unsaturated α-isoprene unit.
Figure 2.

Structures of undecaprenol and associated analogs.
In order to construct this library, the bifunctional building block I, which contained two (Z)-isoprene units, was envisioned as the key intermediate (Scheme 1). I was constructed such that orthogonal hydroxyl protecting groups at the two termini would allow for selective functional group transformations and subsequent lipid backbone elongation. As illustrated in Scheme 1, coupling of two such modified diene-building blocks II and III through nucleophilic substitution provides access to a Z-isoprene rich polyprenol backbone (IV) in a highly convergent fashion. Coupling of this fragment to the readily accessible isoprenyl fragment VI completes assembly of the polyprenol upon final deprotection.
Scheme 1.

Retrosynthetic analysis for preparation of polyisoprenoid lipids.
Following this line of design, synthesis of heptaprenol, pentaprenol, and (Z)-pentaprenol commenced from the commercially available alcohol nerol (Scheme 2). Benzylation of the hydroxyl group, followed by regioselective epoxidation of the terminal olefin using N-bromosuccinimide (NBS) under basic conditions afforded epoxide 3. Oxidative cleavage of 3 using HIO4 and NaIO4 in aqueous THF then furnished the crude aldehyde intermediate. Reduction of the crude aldehyde by NaBH4 afforded the alcohol 4 in 62% yield over three steps. Conversion of mono-protected diol 4 to the corresponding iodide followed by treatment with PPh3 afforded phosphonium salt 5 (88%, two steps). Wittig olefination of 5 with tetrahydropyranyl (THP) protected α-hydroxyacetone 6 gave the desired (Z, Z)-diene building block 7 in 82% yield with over 90% stereoselectivity for the Z-isomer as determined by HPLC analysis. The pure Z-isomer was subsequently separated by column chromatography.
Scheme 2.

Synthesis of key bifunctional building block 7.
With building block 7 assembled, the two termini were subsequently modified to afford unique coupling partners for elongation of the (Z)-olefin backbone. The benzyl protecting group was first removed using dissolving metal reduction to afford 10, after which mesylation followed by nucleophilic substitution with LiCl yielded the alkyl halide 11. Further nucleophilic displacement with the sodium salt of p-toluenesulfinic acid provided access to allylic sulfone 12 in 66% yield over two steps. Alternatively, the tetrahydropyranyl (THP) protecting group of 7 was removed under acidic conditions, giving alcohol 8 in 98% yield. This alcohol was then converted into the corresponding alkyl chloride 9 using the same mesylation/nucleophilic substitution procedure described above (Scheme 3).
Scheme 3.
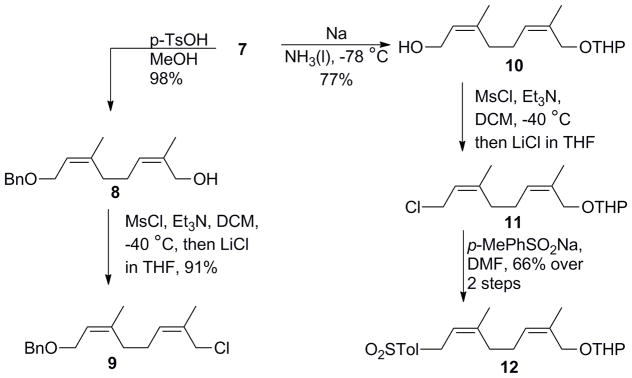
Derivatization of bifunctional intermediate 7 into allylic halide 9 and isoprenoid sulfone 12.
Having prepared the key synthons 12 and 9, these building blocks were connected to generate (all-Z)-tetraene 13. Specifically, monolithiation of allylic sulfone 12, followed by dropwise addition of isoprenoid chloride 9 afforded the (all-Z)-tetra-isoprenyl intermediate 13. The terminal THP group was then removed under acidic conditions to afford the polyprenoid alcohol 14 in 95% yield over two steps (Scheme 4). Utilizing the same mesylation/substitution approach as described above, alkyl halide 15 was successfully obtained from the corresponding alcohol. A terminal tri-olefin fragment was then coupled to 15 using allylic sulfone 16 which itself can be readily accessed via p-tolunesulfinate substitution of commercially available farnesyl bromide. Without further purification, the ligated product 17 was directly subjected to Li-mediated reduction to execute the debenzylation and concurrent desulfonation, affording heptaprenol 18 in 32% overall yield over two steps. Small amounts of regioisomers (<5%) were formed as byproducts due to the conjugative reduction, but could be easily separated through a sequence of acetylation, silver-nitrate based silica gel column chromatography, and deacetylation manipulations.
Scheme 4.

Synthesis of heptaprenol 18.
Having successfully prepared isoprenoid halides 9 and 15 as part of the heptaprenol synthesis, pentaprenol and (all-Z)-pentaprenol were readily accessed through coupling with farnesyl sulfone 16 and allyl sulfone 27, respectively, followed by debenzylation/desulfonylation (Table 1). As in the case of heptaprenol, reductive deprotection of (all-Z)-pentaprenol yielded small amounts of regioisomers (<5%) that were readily removed through acetylation, silver-nitrate based silica gel column chromatography, and deacetylation. Monosaturated pentaprenol was generated in a similar fashion through use farnesyl sulfone 16 and the isoprene halide 26 which was prepared through the same sequence outlined for 9 using commercially available citronellol rather than nerol (Scheme 5).
Table 1.
Synthesis of other polyisoprenols via conjugation, deprotection and desulfonation.
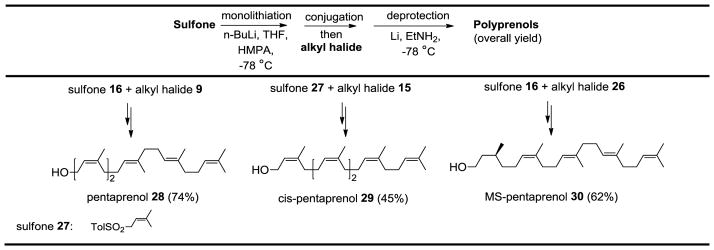
|
Scheme 5.

Synthesis of MS building block 26 via similar synthetic protocol as for 9.
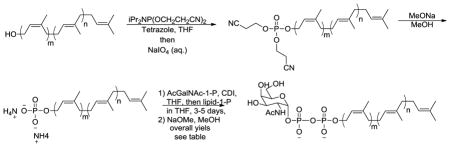
With the desired polyprenol library in hand, we then set out to elaborate these lipids into the corresponding GalNAc-PP-polyprenol substrates for eventual characterization of Wzy and WaaL substrate specificity. The general route utilized to prepare lipid-1-P involved phosphorylation of the isoprenoid alcohols with dicyanoethoxy N,N-diisopropylphosphoramidite in THF in the presence of tetrazole to afford the corresponding phophite. In situ oxidation with NaIO4 then gave the phosphate esters. The dicynoethoxy groups were lastly removed under basic conditions to give the phosphate salts. The lipid-1-P could be quickly purified through DEAE anion exchange column chromatography. The products were directly used in subsequent step for pyrophosphate formation. The corresponding GalNAc-1-P was prepared following literature protocols in similar fashion as for lipid-1-P synthesis except for using dibenzyl N,N-diethylphosphoramidite as a phosphorylating reagent (Table 2).[11]
Table 2.
Preparation of lipid-1-P and the conjugation with GalNAc-1-P forming GalNAc-PP-lipid.
| |||
|---|---|---|---|
| Lipid | Geometry | Structure R: GalNAc | GalNAc-PP-lipids (overall yield)* |
| undecaprenol | 7 Z, 3 E |
 31 |
59% |
| heptaprenol | 4 Z, 2 E |
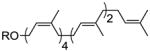 32 |
36% |
| pentaprenol | 2 Z, 2 E |
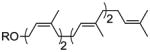 33 |
49% |
| (all-Z)-pentaprenol | 4 Z, 0 E |
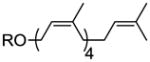 34 |
20% |
| solanesol | 0 Z, 8 E |
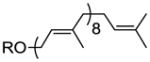 35 |
50% |
| MS-pentaprenol | 1 Z, 2 E |
36 |
26% |
overall yield is based on the polyprenols.
CDI activation of GalNAc-1-P at room temperature for 30 minutes led to formation of the corresponding phosphorimidazolidate intermediate, which, after methanol decomposition of excess CDI, was directly treated with lipid-1-P at room temperature for 3–5 days to furnish the coupled products. The undecaprenol and solanesol lipids are commercially available and thus directly used in the conjugation synthesis. After preparative C18 reverse phase column chromatographic purification, the product was then deacetylated under Źemplén deacetylation conditions affording the final GalNAc-PP-lipids derivatives with the overall yield for each lipid analog indicated in Table 2.
As we reported recently in related biochemical studies,[12] we used this set of GalNAc-PP-lipid derivatives as starting materials for the in vitro assembly of the E. coli O86 O-antigen repeating unit (RU) by means of sequential enzymatic glycosylation.[13] The well-characterized E. coli O86 O-antigen glycosyltransferases (WbnH, WbnJ, WbnK and WbnI) exhibit highly relaxed substrate specificity towards the lipid moiety, thus allowing for efficient assembly of RU-PP-lipid analogs. We were then able to utilize these RU-PP-lipid analogs to gain insight into the lipid substrate specificity of Wzy and WaaL. Interestingly, as was reported in these publications, Wzy and WaaL were observed to exhibit remarkably different lipid specificities when tested with the chemoenzymatic synthesized RU-PP-lipid analogs as chemical probes.
Conclusions
In summary, we have developed a polyprenol library which allows for the detailed characterization of enzyme lipid substrate specificity. The derivatives provide a valuable chemical tool in probing molecular details of the complicated E. coli LPS biosynthesis. We believe the method described in this paper will find its broader application in biological investigation of other related biological systems.
Experimental Section
General
All solvents were dried with a solvent-purification system from Innovative Technology, Inc. All reactions were performed in oven-dried glassware. All materials were obtained from commercial sources and used as received unless otherwise noted. Analytical TLC was carried out on silica gel 60 F254 aluminum-backed plates (E. Merck). EMD™ Slica Gel 60 (particle size: 0.040–0.063 mm, 230–400 mesh ASTM) was used for normal-phase flash column chromatography. EMD™ Slica Gel 60 RP-18 (particle size: 0.040–0.063 mm, 230–400 mesh ASTM) was utilized for reverse-phase purification of GalNAc-PP-lipids. 1H, 13C and 31P NMR spectra were recorded at the indicated field strengths using Bruker NMR instrument. High-resolution mass spectra were obtained on a Bruker micrOTOF instrument.
(3,7-Dimethyl-octa-2,6-dienyloxymethyl)-benzene (2)
To a stirred solution of nerol 1 (30.00 g, 194.49 mmol) in dry THF at 0 °C was added NaH (4.67 g, 194.49 mmol) in separate portions slowly under argon atmosphere. The reaction mixture was stirred at 0 °C for 30 min before benzyl bromide (23.10 mL, 194.49 mmol) and NaI (2.92 g, 19.45 mmol) was added sequentially. The resulting mixture was heated at reflux for 5 h, cooled down to rt and quenched with aq 1.0 M HCl. The reaction solvent was removed under reduced pressure and the residue was diluted with a hexane/EtOAc mixture (10:1, v/v, 500 mL). The organic layer was washed with water, aq satd NaHCO3, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 5.0%) to afford 2 as a colorless oil (43.80 g, 92%). 1H NMR (500 MHz, CDCl3): δ 7.38-7.25 (m, 5H), 5.41 (dt, J = 6.9 Hz, J = 1.8 Hz, 1H), 5.10-5.04 (m, 1H), 4.49 (s, 2H), 4.00 (d, J = 6.9 Hz, 2H), 2.07-2.02 (m, 4H), 1.75 (s, 3H), 1.66 (s, 3H), 1.57 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 140.8, 138.8, 132.1, 128.5, 128.0, 127.7, 124.1, 122.1, 72.3, 66.7, 32.5, 26.9, 25.9, 23.7, 17.8. HRESIMS calcd for C17H24ONa [M+Na]+: 267.1725, found 267.1780.
6-Benzyloxy-4-methylhex-4-en-1-ol (4)
To a stirred solution of 2 (43.80 g, 179.20 mmol) in dioxane (250 mL) and H2O (30 mL) at 0 °C were added N-Bromosuccinimide (31.89 g, 179.20 mmol) in three portions. The reaction mixture was stirred for 5 h at rt before a solution of KOH (10.05 g, 179.20 mmol) in MeOH (60 mL) was dropwise added. The stirring was continued for additional 3 h at rt, and then the reaction mixture was diluted with water (150 mL) and extracted with Et2O (3 × 100 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated. The crude expoxide 3 was utilized directly in the next step. Specifically, NaIO4 (19.16 g, 89.60 mmol) and periodic acid (49.01 g, 215.0 mmol) were added into a solution of 3 in THF (250 mL) and water (50 mL) at rt. The reaction mixture was stirred for 30 min at rt and then diluted with satd Na2SO4 (100 mL). The reaction mixture was then extracted with EtOAc (3 × 200 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated to give crude aldehyde which was used directly in the next reduction step. Specifically, to a stirred solution of the aldehyde (39.12 g, 179.20 mmol) in methanol (250 mL) at 0 °C was added NaBH4 (5.43 g, 143.40 mmol) was in three portions. The reaction mixture was stirred for 30 min 0 °C, after which the reaction solvent was removed. The residue was dissolved in EtOAc (200 mL) and the organic solution was washed with water, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 15% to 25%) to afford 4 as a colorless oil (24.48 g, 62% over 3 steps). 1H NMR (500 MHz, CDCl3): δ 7.33-7.25 (m, 5H), 5.49 (t, J = 7.2 Hz, 1H), 4.50 (s, 2H), 3.97 (d, J = 7.2 Hz, 2H), 3.55 (t, J = 6.5 Hz, 2H), 2.29 (bs, 1H), 2.17 (t, J = 7.3 Hz, 2H), 1.73 (s, 3H), 1.61–1.66 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 141.8, 138.3, 128.6, 128.2, 127.9, 122.0, 72.6, 66.0, 61.5, 30.3, 28.0, 23.3. HRESIMS calcd for C14H20O2Na [M+Na]+: 243.1361, found 243.1390.
(6-Benzyloxy-4-methylhex-4-enyl)-triphenylphosphonium iodide (5)
A mixture of 4 (13.00 g, 59.01 mmol), PPh3 (20.12 g, 76.71 mmol), imidazole (7.23 g, 106.2 mmol) and iodine (17.97 g, 70.81 mmol) in benzene (300 mL) was stirred at rt for 20 min, then the reaction solvent was removed under reduced pressure. The residue was diluted with ether (200 mL) and the organic solution was washed with water, 20% aq Citric Acid, satd NaHCO3, brine, dried over Na2SO4, filtered and concentrated. The residue was suspended in hexanes with vigorous stirring and filtered through a short bed of silica gel. The filtrate was concentrated, affording crude iodide intermediate which was directly used in the next step. Specifically, PPh3 (21.70 g, 82.68 mmol) was added to a solution of above prepared crude iodide (19.50 g, 59.05 mmol) in benzene (100 mL). The mixture was heated at reflux for 20 h with stirring, then cooled down to rt before the reaction solvent was removed under reduced pressure. The resultant white powder triturated with Et2O to remove residual PPh3 by filtration and the solid was collected and dried to afford the product 5 as a white powder (30.04 g, 88% yield over two steps), which is directly used without characterization.
1-(Tetrahydropyran-2-yloxy)-propan-2-one (6)
A mixture of dihydropyran (24.7 mL, 136.6 mmol), hydroxyacetone (9.35 mL, 270.40 mmol) and pyridinium p-toluenesulfonate (0.34 g, 1.35 mmol) was stirred at rt for 2.5 h, after which it was diluted with hexanes and subjected to flash column chromatography on silica gel (EtOAc in hexanes, 0 to 6%) to afford 6 as a colorless oil (17.20 g, 80% yield). 1H NMR (500 MHz, CDCl3): δ 4.58 (t, J = 3.2 Hz, 1H), 4.18 (d, J = 17.3 Hz, 1H), 4.05 (d, J = 17.3 Hz, 1H), 3.80-3.74 (m, 1H), 3.48-3.42 (m, 1H), 2.11 (s, 3H), 1.90-1.44 (m, 6H); 13C NMR (125 MHz, CDCl3): δ 206.8, 98.9, 72.4, 62.5, 30.4, 26.6, 25.4, 19.3. HRESIMS calcd for C8H14O3Na [M+Na]+: 181.0841, found 181.0905.
2-(8-Benzyloxy-2,6-dimethyl-octa-2,6-dienyloxy)-tetrahydropyran (7)
To a stirred solution of 5 (14.89 g, 25.13 mmol) in THF (100 mL) at −78 °C was added a solution of n-BuLi (13.35 mL, 21.36 mmol, 1.6 M) in hexane dropwise and the reaction was stirred for 30 min at such temperature. After a solution of 6 (4.93 g, 31.16 mmol) in THF (20 mL) was added, and the reaction mixture was then stirred overnight, during which time the reaction temperature was allowed to warm up to rt. The reaction was quenched with aq satd NH4Cl and the reaction solvent was removed under reduced pressure. The residue was diluted with EtOAc (100 mL) and the organic solution was washed with water, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 0% to 5%) to afford 7 as a colorless oil (7.12 g, 82%).1H NMR (500 MHz, CDCl3): δ 7.33-7.25 (m, 5H), 5.40 (t, J = 6.8 Hz, 1H), 5.30 (t, J = 7.1 Hz, 1H), 4.55 (t, J = 3.6 Hz, 1H), 4.47 (s, 2H), 4.12-4.01 (m, 2H), 3.98 (d, J = 6.9 Hz, 2H), 3.88-3.82 (m, 1H), 3.51-3.45 (m, 1H), 2.16-2.03 (m, 4H), 1.86-1.77 (m, 1H), 1.74 (s, 3H), 1.73 (s, 3H), 1.72-1.46 (m, 5H); 13C NMR (125 MHz, CDCl3): δ 140.3, 138.8, 132.6, 128.9, 128.6, 128.0, 127.7, 122.4, 97.7, 72.3, 66.6, 65.5, 62.3, 32.6, 30.9, 26.5, 25.7, 23.7, 22.0, 19.7. HRESIMS calcd for C22H32O3Na [M+Na]+: 367.2249, found 367.2289.
8-Benzyloxy-2,6-dimethyl-octa-2,6-dien-1-ol (8)
p-toluenesulfonic acid monohydrate (0.35 g, 1.84 mmol) was added to a stirred solution of 7 (1.80 g, 5.23 mmol) in methanol (50 mL) and the reaction mixture was stirred at rt overnight. The reaction solvent was then removed under reduced pressure, affording a residue which was dissolved in CH2Cl2 (75 mL). The organic solution was washed with water, satd NaHCO3, brine, dried over Na2SO4 and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 5% to 30%) to afford 8 as a colorless oil (1.33 g, 98% yield). 1H NMR (500 MHz, CDCl3): δ 7.36-7.30 (m, 4H), 7.29-7.25 (m, 1H), 5.43 (t, J = 6.9 Hz, 1H), 5.23 (t, J = 7.1 Hz, 1H), 4.48 (s, 2H), 4.02 (s, 2H), 3.95 (d, J = 6.9 Hz, 2H), 2.14-2.04 (m, 4H), 1.75 (s, 3H), 1.74 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 140.5, 138.6, 135.4, 128.6, 128.0, 127.8, 127.6, 122.4, 72.4, 66.5, 61.6, 32.5, 26.3, 23.8, 21.5. HRESIMS calcd for C17H24O2Na [M+Na]+: 283.1674, found 283.1652.
(8-Chloro-3,7-dimethyl-octa-2,6-dienyloxymethyl)-benzene (9)
To a stirred solution of 8 (1.34 g, 5.14 mmol) in CH2Cl2 (20 mL) and THF (10 mL) at −40 °C was added triethylamine (0.93 mL, 6.68 mmol). The reaction mixture was stirred for 5 min before mesyl chloride (0.48 mL, 6.17 mmol) was added. The stirring was continued for 30 min at such temperature and then a solution of LiCl (2.18 g, 51.40 mmol) in THF (35 mL) was added. The mixture was stirred for 3 h, during which time the reaction temperature was allowed to warm up to 0 °C. The reaction solvent was removed and the residue was diluted with EtOAc (100 mL). The organic solution was washed with water, satd NaHCO3, brine, dried over NaSO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 1% to 5%) to afford 9 as a colorless oil (1.29 g, 90%). 1H NMR (400 MHz, CDCl3): δ 7.43-7.25 (m, 5H), 5.44 (t, J = 8.4 Hz, 1H), 5.33 (t, J = 7.3 Hz, 1H), 4.49 (s, 2H), 4.00 (s, 2H), 3.98 (d, J = 7.0 Hz, 2H), 2.19-2.06 (m, 4H), 1.79 (s, 3H), 1.75 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 140.0, 138.7, 132.1, 130.6, 128.6, 128.0, 127.7, 122.7, 72.4, 66.5, 43.7, 32.2, 26.7, 23.6, 21.8. HRESIMS calcd for C17H23ClONa [M+Na]+: 301.1335, found 301.1378.
3,7-Dimethyl-8-(tetrahydro-pyran-2-yloxy)-octa-2,6-dien-1-ol (10)
NH3 (g) (100 mL) was condensed in a reaction vessel at −78 °C. Na (2.00 g, 87.09 mmol) was added and the reaction was stirred for 1 h at such low temperature. A solution of 7 (3.00 g, 8.709 mmol) in THF (10 mL) was then added dropwise. The reaction was stirred for 5 h at −78 °C before it was quenched with CH3OH (50 mL). NH3 (l) was allowed to evaporated overnight, after which the remaining solvent was removed under reduced pressure. The residue was diluted CH2Cl2 (100 mL). The organic solution was washed with water, brine, dried over NaSO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 10% to 50%) to afford 10 as a colorless oil (1.70 g, 77%). 1H NMR (400 MHz, CDCl3): 5.42 (t, J = 7.1 Hz, 1H), 5.34 (t, J = 7.0 Hz, 1H), 4.57 (t, J = 3.4 Hz, 1H), 4.12-3.98 (m, 4H), 3.90-3.78 (m, 1H), 3.54-3.44 (m, 1H), 2.20-2.00 (m, 4H), 1.75 (s, 3H), 1.72 (s, 3H), 1.88-1.46 (m, 6H); 13C NMR (100 MHz, CDCl3): 139.5, 132.8, 129.1, 125.0, 97.5, 65.4, 62.2, 59.1, 32.3, 30.8, 26.6, 25.7, 23.8, 22.0, 19.5. HRESIMS calcd for C15H26O3Na [M+Na]+: 277.1780, found 277.1768.
2-[2,6-Dimethyl-8-(toluene-4-sulfonyl)-octa-2,6-dienyloxy]-tetrahydropyran (12)
To a stirred solution of 10 (1.70 g, 6.68 mmol) in mixed solvent of CH2Cl2 (20 mL) and THF (45 mL) at −40 °C was added triethylamine (1.21 mL, 8.68 mmol). The reaction was stirred for 5 min before mesyl chloride (0.62 mL, 8.01 mmol) was added. The stirring was continued for 30 min at such temperature. Then a solution of LiCl (3.10 g, 73.03 mmol) in THF (10 mL) was added and the mixture was stirred for 3 h, during which time the reaction temperature was allowed to warm up to 0 °C. The reaction solvent was removed under reduced pressure, affording a residue which was diluted with 10% EtOAc in hexanes (100 mL). The organic solution was washed with water, satd NaHCO3, brine, dried over NaSO4, filtered and concentrated. The crude product was used directly in the next step. Specifically, crude 11 was dissolved in DMF (40 mL) and sodium p-toluenesulfinate (4.50 g, 8.01 mmol) was then added in one portion, and the reaction was stirred for 3 h at rt. The reaction solvent was removed under reduced pressure and the residue was diluted with CH2Cl2 (100 mL). This organic solution was washed with water, brine, dried over NaSO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 10% to 15%) to afford 12 as colorless oil (1.73 g, 66% over two steps). 1H NMR (400 MHz, CDCl3): 7.72 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 5.23-5.14 (m, 2H), 4.53 (t, J = 3.4 Hz, 1H), 4.04-3.96 (m, 2H), 3.88-3.80 (m, 1H), 3.76 (d, J = 8.1 Hz, 2H), 3.53-3.44 (m, 1H), 2.42 (s, 3H), 2.00-1.90 (m, 2H), 1.84-1.74 (m, 3H), 1.703 (s, 3H), 1.701 (s, 3H), 1.60-1.46 (m, 6H); 13C NMR (100 MHz, CDCl3): 145.6, 144.6, 136.2, 133.0, 129.8, 128.7, 128.3, 111.5, 97.5, 65.3, 62.3, 56.1, 32.3, 30.9, 25.9, 25.7, 23.7, 21.9, 21.8, 19.7. HRESIMS calcd for C22H32O4SNa [M+Na]+: 415.1919, found 415.1947.
16-Benzyloxy-2,6,10,14-tetramethyl-8-(toluene-4-sulfonyl)-hexadeca-2,6,10,14-tetraen-1-ol (14)
To a stirred solution of 12 (0.30 g, 0.76 mmol) in dry THF (6 mL) and HMPA (2 mL) was added n-BuLi (0.48 mL, 0.76 mmol) dropwise at −78 °C. After the reaction was stirred at such temperature for 10 min, a solution of compound 9 (0.18 g, 0.64 mmol) in THF (6 mL) was then added dropwise over 1 h via an automated injector. The reaction mixture was then stirred overnight, during which time the reaction temperature was allowed to warm up to rt slowly. The reaction was then quenched with 1% aq acetic acid. The mixture was extracted with diethyl ether (3 × 100 mL), and the combined organic layer was dried over NaSO4, filtered and concentrated. The crude intermediate 13 was utilized directly in the next step. Specifically, to a stirred solution of crude 13 (0.25 g, 0.39 mmol) in methanol (15 mL) was added p-toluenesulfonic acid monohydrate (0.10 g, 0.55 mmol). The reaction was stirred at rt overnight. The reaction solvent was removed under reduced pressure, the residue was diluted with CH2Cl2 (30 mL). The organic layer was washed with satd NaHCO3, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography (EtOAc in hexanes, 7% to 15%) to afford 14 as a colorless oil (0.21 g, 95% yield). 1H NMR (500 MHz, CDCl3): δ 7.68 (d, J = 8.2 Hz, 2H), 7.31-7.25 (m, 7H), 5.39 (t, J = 6.8 Hz, 1H), 5.13 (t, J = 6.0 Hz, 1H), 5.09 (t, J = 7.1 Hz, 1H), 4.92 (d, J = 10.4 Hz, 1H), 4.47 (s, 2H), 4.05-3.95 (m, 4H), 3.82 (td, J = 10.8 Hz, J = 2.8 Hz, 1H), 2.63 (dd, J = 13.2 Hz, J = 2.4 Hz, 1H), 2.41 (s, 3H), 2.04-1.93 (m, 4H), 1.92-1.57 (m, 5H), 1.73 (s, 3H), 1.69 (s, 3H), 1.65 (s, 3H), 1.56 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 145.1, 144.7, 140.2, 138.7, 135.4, 135.1, 130.7, 129.6, 129.4, 128.5, 128.3, 127.7, 127.3, 122.4, 117.9, 72.3, 66.5, 63.6, 61.2, 32.6, 32.3, 30.8, 26.7, 25.9, 23.9, 23.6, 23.6, 21.8, 21.5. HRESIMS calcd for C34H46O4SNa [M+Na]+: 573.3015, found 573.3087.
1-(16-Benzyloxy-1-chloro-2,6,10,14-tetramethyl-hexadeca-2,6,10,14-tetraene-8-sulfonyl)-4-methylbenzene (15)
To a stirred solution of 14 (0.21 g, 0.37 mmol) in a mixed solvent of CH2Cl2 (6 mL) and THF (3 mL) at −40 °C was added triethylamine (0.07 mL, 0.49 mmol) under nitrogen atmosphere. The reaction was stirred for 5 min at such temperature before mesyl chloride (0.04 mL, 0.45 mmol) was added. The stirring was continued for 30 min at such temperature. Then a solution of LiCl (0.20 g, 4.72 mmol) in THF (10 mL) was added and the reaction mixture was stirred for 3 h, during which time the reaction temperature was allowed to warm up to 0 °C. The reaction solvent was then removed and the residue was diluted with 10% EtOAc in hexanes (40 mL). The organic solution was washed with water, saturated NaHCO3, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography (EtOAc in hexanes, 5% to 11%) to afford 15 as a colorless oil (0.18 g, 85%). 1H NMR (400 MHz, CDCl3): 7.70 (d, J = 8.3 Hz, 2H), 7.34-7.25 (m, 7H), 5.39 (td, J = 6.8 Hz, J = 1.0 Hz, 1H), 5.21-5.10 (m, 2H), 4.97 (d, J = 10.5 Hz, 1H), 4.47 (s, 3H), 3.99-3.91 (m, 4H), 3.82 (td, J = 10.9 Hz, J = 2.8 Hz, 1H), 2.64 (dd, J = 13.2 Hz, J = 2.4 Hz, 1H), 2.41 (s, 3H), 2.03-1.94 (m, 4H), 1.93-1.58 (m, 6H), 1.76 (s, 3H), 1.69 (s, 3H), 1.66 (s, 3H), 1.57 (s, 3H); 13C NMR (100 MHz, CDCl3): 144.7, 144.5, 140.1, 138.7, 135.2, 132.3, 130.7, 130.2, 129.6, 129.4, 128.5, 128.3, 128.0, 127.7, 122.4, 118.3, 72.3, 66.5, 63.6, 43.6, 32.3, 32.1, 30.7, 26.7, 26.1, 23.9, 23.6, 23.6, 21.8, 21.8. HRESIMS calcd for C34H45O3ClSNa [M+Na]+: 591.2676, found 591.2605.
1-Methyl-4-(3,7,11-trimethyl-dodeca-2,6,10-triene-1-sulfonyl)-benzene (16)
To a stirred solution of farnesyl bromide (1.92 g, 6.74 mmol) in DMF (20 mL) was added sodium p-toluenesulfinate (1.44 g, 8.09 mmol) in one portion, and the reaction was stirred for 3 h at rt. The reaction solvent was removed under high vacuum and the residue was diluted with CH2Cl2 (100 mL). This organic solution was washed with water, brine, dried over Na2SO4, filtered and concentrated. The crude product was subjected to flash column chromatography on silica gel (EtOAc in hexanes, 5% to 12%) to afford 16 as a yellow oil (2.21 g, 91% yield). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 5.15 (t, J = 7.9 Hz, 1H), 5.09-4.99 (m, 2H), 3.76 (d, J = 7.9 Hz, 2H), 2.41 (s, 3H), 2.08-1.90 (m, 8H), 1.65 (s, 3H), 1.57 (s, 3H), 1.55 (s, 3H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 146.4, 144.6, 136.1, 136.0, 131.6, 129.7, 128.7, 124.4, 123.6, 110.7, 56.3, 39.9, 26.9, 26.4, 25.9, 21.8, 17.9, 16.4, 16.2. HRESIMS calcd for C22H32O2SNa [M+Na]+: 383.2021, found 383.2065.
Representative procedure for preparation of polyisoprenol 18. 3,7,11,15,19,23,27-Heptamethyl-octacosa-2,6,10,14,18,22,26-heptaen-1-ol (18)
To a stirred solution of 16 (0.47 g, 1.29 mmol) in freshly distilled THF (6 mL) and HMPA (2 mL) at −78 °C was added n-BuLi (0.806 mL, 1.290 mmol) dropwise and the mixture was stirred for 10 min at such temperature. A solution of alkyl chloride 15 (300 mg, 1.08 mmol) in THF (6 mL) was then added dropwise over 1 h via an automated injector. The mixture was then stirred overnight, during which time the reaction temperature was allowed to warm up to rt. The reaction was quenched by addition of 1% aq acetic acid. The mixture was then extracted with diethyl ether (3 × 50 mL), and the combined organic layer was dried over NaSO4, filtered and concentrated. The crude product 17 was directly used in next step without purification.
Ethylamine (25 mL) was condensed in a reaction flask under nitrogen atomosphere at −78 °C. Lithium metal (114 mg, 16.41 mmol) was then added, after which the mixture was stirred for 1 h to afford a dark blue solution. The solution of crude 17 in anhydrous THF (5 mL) was added dropwise over 1 min. The reaction was then immediately quenched with aq NH4Cl solution and then brought to rt. Following evaporation of ethylamine, the residue was diluted with EtOAc (50 mL). The organic solution was washed with water, brine, dried over NaSO4, filtered and concentrated. The crude product was subjected to flash column chromatography (EtOAc in hexanes, 2% to 5%) to afford Heptaprenol 18 as a colorless oil (0.09 g, 35%). 1H NMR (500 MHz, CDCl3): δ 5.42 (td, J = 7.1 Hz, J = 1.2 Hz, 1H), 5.17-5.04 (m, 6H), 4.07 (d, J = 7.1 Hz, 2H), 2.09-2.00 (m, 20H), 1.99-1.93 (m, 4H), 1.73 (s, 3H), 1.67 (s, 12H), 1.59 (s, 3H), 1.58 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 140.1, 136.3, 135.7, 135.6, 135.4, 135.2, 131.5, 125.2, 125.1, 124.8, 124.7, 124.6, 124.5, 124.4, 59.3, 40.0, 40.0, 32.5 (2), 32.4, 32.2, 27.0, 26.9, 26.9, 26.7, 26.6, 26.6, 25.9, 23.66, 23.67, 23.7, 23.6, 17.9, 16.2. HRESIMS calcd for C35H58ONa [M+Na]+: 517.4385, found 517.4376.
(3,7-Dimethyl-oct-6-enyloxymethyl)-benzene (20)
To a stirred solution of 19 (10.05 g, 63.99 mmol) in THF (50 mL) was added NaH (1.54 g, 63.99 mmol) at 0 °C. The reaction mixture was then stirred at such temperature for 30 min, after which benzyl bromide (10.94 g, 63.99 mmol) and sodium iodide (0.96 g, 6.40 mmol) were then added. The reaction mixture was then heated at reflux for 5 h, cooled down to rt, carefully quenched with aq 1.0 M HCl. The reaction solvent was then removed under reduced pressure, and the residue was diluted with a mixed hexane/EtOAc (10/1, v/v, 100 mL). The organic layer was washed with water, brine, dried over Na2SO4 and concentrated. The residue was subjected to flash column chromatography (EtOAc in hexanes, 2.5%) to afford 20 as a colorless oil (12.84 g, 81%). 1H NMR (500 MHz, CDCl3): δ 7.33 (d, J = 4.4 Hz, 4H), 7.28-7.23 (m, 1H), 5.09 (t, J = 7.1 Hz, 1H), 4.49 (s, 2H), 3.54-3.44 (m, 2H), 2.04-1.89 (m, 2H), 1.72-1.52 (m, 2H), 1.67 (s, 3H), 1.59 (s, 3H), 1.47-1.39 (m, 1H), 1.37-1.29 (m, 1H), 1.20-1.10 (m, 1H), 0.80 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 139.0, 131.3, 128.5, 127.8, 127.7, 125.1, 73.1, 69.0, 37.4, 37.0, 29.8, 25.9, 25.7, 19.8, 17.8. HRESIMS calcd for C17H26ONa [M+Na]+: 269.1881, found 269.1867.
6-Benzyloxy-4-methylhexan-1-ol (22)
To a stirred solution of 20 (7.53 g, 30.55 mmol) in dioxane (75 mL) and H2O (10 mL) was added N-Bromosuccinimide (5.44 g, 30.55 mmol) in three portions at 0 °C. The reaction mixture was stirred for 5 h at rt. A solution of KOH (1.72 g, 30.55 mmol) in MeOH (10 mL) was added dropwise into the mixture. After stirring for 3 h at rt, the reaction mixture was diluted with water (100 mL) and extracted with Et2O (3 × 50 mL). The organic layer was dried over Na2SO4 and concentrated. The crude epoxide 21 was then dissolved in THF (75 mL) and water (15 mL). NaIO4 (3.27 g, 15.27 mmol) and periodic acid (8.36 g, 36.66 mmol) were then added into the mixture. The reaction was stirred for 30 min at rt and subsequently diluted with saturated Na2SO4 (aq.) (60 mL). The reaction mixture was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over Na2SO4 and concentrated. The crude aldehyde was utilized directly in the next step. NaBH4 (0.92 g, 24.44 mmol) was added in three portions into the solution of crude aldehyde in methanol (10 mL) at 0 °C. The reaction was stirred at the same temperature for 30 min, then the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (100 mL) and washed with water, brine, dried over Na2SO4 and filtered. The filtrate was concentrated, affording a residue which was subjected to flash column chromatography (EtOAc in hexanes, 15% to 20%) to afford 22 as a colorless oil (2.17 g, 32% over 3 steps). 1H NMR (500 MHz, CDCl3): δ 7.32 (d, J = 4.6 Hz, 1H), 7.29-7.25 (m, 1H), 4.48 (s, 2H), 3.55 (t, J = 6.7 Hz, 2H), 3.53-3.45 (m, 2H), 2.06 (bs, 1H), 1.70-1.32 (m, 6H), 1.20-1.12 (m, 1H), 0.88 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 138.7, 128.5, 127.8, 127.6, 73.0, 68.7, 63.1, 36.8, 33.1, 30.2, 29.8, 19.7. HRESIMS calcd for C14H22O2Na [M+Na]+: 245.1517, found 245.1515.
(6-Benzyloxy-4-methylhexyl)-triphenylphosphonium iodide (23)
To a stirred solution of 22 (1.50 g, 6.75 mmol), PPh3 (2.30 g, 8.77 mmol), imidazole (0.83 g, 12.14 mmol) in benzene (30 mL) was added iodine (2.06 g, 8.10 mmol) at rt. The mixture was stirred for 30 minutes at rt, then the reaction solvent was removed under reduced pressure. The residue was diluted with ether (40 mL) and washed with water, 20% Citric Acid, saturated aq NaHCO3, brine and concentrated. The residue was then suspended in hexanes with vigorous stirring followed by filtration through a short bed of silica gel. The filtrate was concentrated under reduced pressure, affording crude idode intermediate (2.23 g, 6.75 mmol) which was then dissolved in benzene (10 mL), followed by addition of PPh3 (2.48 g, 9.45 mmol). The reaction mixture was then heated at reflux for 20 h, cooled down to rt. The reaction solvent was removed under reduced pressure, yielding a white powder, which was then triturated with ether (20 mL) to remove excess PPh3 via filtration. The collect solid was dried to afford 23 as white powder (1.80 g, 44% yield over two steps), which was used directly in next step without characterization.
2-(8-Benzyloxy-2,6-dimethyloct-2-enyloxy)-tetrohydropyran (24)
To a stirred solution of 23 (0.85 g, 1.43 mmol) in THF (10 mL) was added n-BuLi (0.76 mL, 1.22 mmol) dropwise at −78° C under argon atmosphere. After stirring at such temperature for 30 minutes, a solution of 6 (0.27 g, 1.72 mmol) in THF (3 mL) was added dropwise, and the reaction mixture was allowed to warm to room temperature overnight. The reaction was quenched with saturated NH4Cl and then was extracted with EtOAc (30 mL) for three times. The organic layer was dried over Na2SO4 and concentrated. The residue was subjected to flash column chromatography (EtOAc in hexanes, 1% to 10%) to afford 24 as a colorless oil (0.30 g, 61%).1H NMR (500 MHz, CDCl3): δ 7.36-7.30 (m, 4H), 7.28-7.25 (m, 1H), 5.33 (t, J = 7.2 Hz, 1H), 4.56 (t, J = 3.6 Hz, 1H), 4.49-4.47 (m, 2H), 4.10-4.05 (m, 2H), 3.90-3.82 (m, 1H), 3.52-3.44 (m, 3H), 2.13-1.96 (m, 2H), 1.88-1.77 (m, 1H), 1.74 (3H), 1.72-1.48 (m, 7H), 1.44-1.29 (m, 2H), 1.20-1.11 (m, 1H), 0.86 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 138.9, 131.8, 130.0, 128.5, 127.8, 127.7, 97.8, 73.1, 68.9, 65.6, 62.4, 37.6, 36.9, 30.9, 29.8, 25.7, 25.4, 22.0, 19.8, 19.7. HRESIMS calcd for C22H34O3Na [M+Na]+: 369.2406, found 369.2418.
8-Benzyloxy-2,6-dimethyl-oct-2-en-1-ol (25)
To a stirred solution of 24 (0.30 g, 0.87 mmol) in methanol (10 mL) was added p-toluenesulfonic acid monohydrate (0.20 g, 1.04 mmol). The reaction was stirred at rt overnight. The solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2 (30 mL). The organic layer was washed with saturated NaHCO3, brine, dried over anhydrous Na2SO4 and concentrated. The crude product was subjected to flash column chromatography (EtOAc in hexanes, 5% to 12%) to afford 25 as a colorless oil (0.22 g, 96%). 1H NMR (500 MHz, CDCl3): δ 7.35-7.29 (m, 4H), 7.28-7.24 (m, 1H), 5.24 (t, J = 7.3 Hz, 1H), 4.49-4.47 (m, 2H), 4.14 (d, J = 11.8 Hz, 1H), 4.03 (d, J = 11.8 Hz, 1H), 3.52-3.42 (m, 2H), 2.16-2.06 (m, 1H), 2.03-1.94 (m, 1H), 1.77 (s, 3H), 1.72-1.54 (m, 3H), 1.38-1.27 (m, 2H), 1.24-1.14 (m, 1H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 138.7, 134.5, 128.7, 128.6, 127.9, 127.7, 73.1, 68.5, 61.7, 37.4, 36.5, 29.2, 25.1, 21.5, 19.8. HRESIMS calcd for C17H26O2Na [M+Na]+: 285.1830, found 285.1843.
(8-Bromo-3,7-dimethyl-oct-6-enyloxymethyl)-benzene (26)
To a stirred solution of 25 (0.22 g, 0.83 mmol) in CH2Cl2 (15 mL) was added triethylamine (0.28 mL, 2.00 mmol) at −40 °C, followed by dropwise addition of mesyl chloride (0.08 mL, 0.10 mmol). The reaction mixture was stirred at such temperature for 30 minutes, a solution of LiBr (0.36 g, 4.16 mmol) in THF (2.8 mL) was then added, and the reaction temperature was allowed to warm to 0 °C over 3 h. The solvent was removed under reduced pressure, and the residue was diluted with EtOAc (30 mL). The organic layer was washed with saturated NaHCO3 and dried over Na2SO4. The crude product was subjected to flash column chromatography (EtOAc in hexanes, 0% to 2.5%) to afford 26 as a pale yellow oil (0.27 g, 99%). 1H NMR (500 MHz, CDCl3): δ 7.37-7.30 (m, 4H), 7.29-7.25 (m, 4H), 5.36 (td, J = 1.4 Hz, J = 7.4 Hz, 1H), 4.49-4.47 (m, 2H), 3.96 (s, 2H), 3.55-3.45 (m, 2H), 2.14-1.98 (m, 2H), 1.81 (s, 3H), 1.73-1.55 (m, 2H), 1.47-1.35 (m, 2H), 1.26-1.17 (m, 1H), 0.891 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 138.8, 132.1, 131.5, 128.5, 127.8, 127.7, 73.1, 68.8, 36.8, 36.8, 32.6, 29.7, 25.7, 22.1, 19.7. HRESIMS calcd for C17H25BrONa [M+Na]+: 347.0986, found 347.0956.
1-Methyl-4-(3-methylbut-2-ene-1-sulfonyl)-benzene (27)
To a stirred solution of 3,3-Dimethylallyl bromide (2.00 g, 13.49 mmol) in DMF (20 mL) was Sodium p-toluenesulfinate (2.87 g, 16.10 mmol) at rt. The reaction mixture was stirred for 3 h at rt, and then reaction solvent was removed under high vacuum. The residue was diluted with CH2Cl2 (100 mL). The organic solution was washed with water, brine, dried over Na2SO4, filtered and concentrated. The residue was subjected to flash column chromatography (EtOAc in hexanes, 10% to 15%) to afford 27 as a white solid (2.73 g, 91%). 1H NMR (500 MHz, CDCl3): δ 7.71 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 5.15 (t, J = 7.9 Hz, 1H), 3.74 (d, J = 7.9 Hz, 2H), 2.42 (s, 3H), 1.69 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 144.6, 142.9, 136.2, 129.8, 128.7, 110.8, 56.5, 26.0, 21.8, 18.0. HRESIMS calcd for C12H16O2SNa [M+Na]+: 247.0769, found 247.0723.
3,7,11,15,19-Pentamethyl-icosa-2,6,10,14,18-pentaen-1-ol (28)
Polyprenol 28 was prepared according to the general procedure as for compound 18 using sulfone 16 and alkyl halide 9 as starting materials. The product was obtained in 74% overall yield (145 mg, from 152 mg of 9) after flash column chromatographic purification (EtOAc in hexanes, 5%-10%). 1H NMR (500 MHz, CDCl3): δ 5.42 (t, J = 7.1 Hz, 1H), 5.16-5.04 (m, 4H), 4.07 (d, J = 7.1 Hz, 2H), 2.12-2.01 (m, 12H), 2.00-1.92 (m, 4H), 1.72 (s, 3H), 1.67 (s, 3H), 1.66 (s, 3H), 1.59 (s, 3H), 1.58 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 140.0, 136.4, 135.5, 135.2, 131.5, 124.7, 124.7, 124.6, 124.4, 124.2, 59.2, 39.9 (2), 32.4, 32.2, 27.0, 26.8, 26.8, 26.5, 25.9, 23.6, 23.6, 17.9, 16.2. HRESIMS calcd for C25H42ONa [M+Na]+: 381.3133, found 381.3137.
3,7,11,15,19-Pentamethyl-icosa-2,6,10,14,18-pentaen-1-ol (29)
Polyprenol 29 was prepared according to the general procedure as for compound 18 using sulfone 27 and alkyl halide 15 as starting materials. The product was obtained in 45% overall yield (98 mg, from 345.70 mg of 15) after flash column chromatographic purification (EtOAc in hexanes, 5%–10%). 1H NMR (500 MHz, CDCl3): δ 5.43 (t, J = 7.1 Hz, 1H), 5.17-5.02 (m, 4H), 4.07 (d, J = 6.4 Hz, 2H), 2.13-1.92 (m, 16H), 1.73 (s, 3H), 1.67 (s, 12H), 1.59 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 140.1, 136.3, 135.6, 135.6, 131.8, 125.2, 125.1, 124.8, 124.7, 124.5, 59.3, 32.5, 32.4, 32.2, 29.9, 26.9, 26.6, 26.6, 26.5, 23.7, 23.7, 23.6, 23.6, 23.6, 17.9. HRESIMS calcd for C25H42ONa [M+Na]+: 381.3133, found 381.3142.
3,7,11,15,19-Pentamethyl-icosa-6,10,14,18-tetraen-1-ol (30)
Polyprenol 30 was prepared according to the general procedure as for compound 18 using sulfone 16 and alkyl halide 26 as starting materials. The product was obtained in 62% overall yield (185 mg, from 268 mg of 26) after flash column chromatographic purification (EtOAc in hexanes, 5%–7%). 1H NMR (500 MHz, CDCl3): δ 5.15-5.05 (m, 4H), 3.72-3.60 (m, 2H), 2.10-2.01 (m, 8H), 2.00-1.90 (m, 6H), 1.663 (s, 3H), 1.661 (s, 3H), 1.62-1.52 (m, 10H), 1.41-1.27 (m, 2H), 1.24-1.12 (m, 2H), 0.89 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 135.4, 135.3, 135.2, 131.5, 125.6, 124.6, 124.5, 124.4, 61.4, 40.2, 39.9, 37.7, 32.2, 29.5, 27.0, 26.9, 26.8, 25.9, 25.5, 23.6, 19.7, 17.9, 16.22, 16.20. HRESIMS calcd for C25H44ONa [M+Na]+: 383.3290, found 383.3287.
General procedure for preparation of Polyprenol-phosphates
To a stirred solution of polyprenol (0.15 mmol) in THF (5 mL) was added bis-(2-cyanoethyl)-N,N-di-isopropyl phosphoramidite (50 mg, 0.18 mmol) at 0 °C, followed by Tetrazole (42 mg, 0.59 mmol). The reaction mixture was stirred at 0 °C for 10 min, after which it was warmed to rt and stirred for an additional 1 h. The reaction was cooled down to 0 °C again, pyridine (0.04 mL, 0.53 mmol) and NaIO4 (65 mg, 0.30 mmol) were added sequentially. After stirring for 15 min at 0 °C, the reaction was warmed to rt and stirred for an additional 1 h. The reaction mixture was then diluted with EtOAc (30 mL) and washed with water, aq 5% Na2SO3 and brine. The organic layer was dried over Na2SO4 and concentrated. The residue, after evacuated under high vacuum for several hours, was dissolved in anhydrous methanol (10 mL) and catalytic amount of NaOMe in CH3OH solution was added. The reaction mixture was stirred at rt for 24 hours and then neutralized by Amberlyst-15 ion exchange resin until pH around 7 and filtered. The filtration was concentrated, and the crude product was then purified on DEAE ion exchange column (pre-washed with 100% methanol, 50% and 25% methanol in CHCl3 sequentially, volume ratio) as following: after loading of crude product in methanol onto column, the column was washed first with 25% methanol in CHCl3 in excess amount to remove any impurities, then followed by collection of product when eluted with 30 mM NH4HCO3 in methanol. The product containing eluent was then concentrated to dryness and directly used in next step for GalNAc-PP-lipid preparation.
General procedure of preparation of GalNAc-PP-Polyprenol
To a stirred solution of diisopropylethylammonium salt of GalNAc-1-PO42− (23 mg, 0.034 mmol) in THF (3 mL) was added N,N′-carbonyldiimidazole (CDI) (23 mg, 0.14 mmol) at rt under argon atmosphere. After stirring at rt for 2h, dry methanol (42 μL) was added into the mixture and the stirring was kept for an additional 0.5 h at rt. The reaction solvent was then removed under reduced pressure, affording a residue which, after evacuation under high vacuum for 30 minutes, was dissolved in anhydrous THF (3 mL). The resultant THF solution was added to a solution of lipid phosphate (0.026 mmol) in THF (2 mL) under argon atmosphere. After stirring at rt for 3 days, the reaction mixture was then concentrated, the residue was purified by flash chromatography on a short bed of reverse phase C18 silica gel column eluting first with water, followed by 50% isopropanol in water (product is collect in this portion), removal of organic solvent under reduced pressure followed by lyophylization afforded crude AcGalNAc-PP-lipid.
The crude product was treated with a 0.1% NaOCH3 in CH3OH solution (3 mL). The reaction was stirred at rt for 40 min and then neutralized by Amberlyst-15 ion exchange resin until pH around 7. The resin was removed by filtration and the filtrate was concentrated, the residue was purified by C-18 reverse phase column chromatography using a gradient of A from 0% to 50% with 5% increment each time (solvent A: isopropanol (lipids: und, solan) or CH3CN (Lipids: hept, pent, (all-Z)-pent, MS-pent); Solvent B = 1.7% NH4HCO3 aqueous solution). The test tubes containing product (detected by MS) was pooled and then lyophilized to afford GalNAc-PP-lipid as a white powder. The reaction yield was based on lipid substrate over 4 steps.
GalNAc-PP-undecaprenyl (31)
GalNAc-PP-und was prepared according to the general procedure. However, the two purification steps were reversed as the final product proved too insoluble to obtain suitable spectral data. The crude product was thus purified via C-18 reverse phase column chromatography to afford peracetylated GalNAc-PP-und as a white solid (20 mg, 59%) from 23 mg of undeca-1-P. 1H NMR (500 MHz, CD3OD): δ 5.60 (dd, J = 3.2 Hz, J = 7.3 Hz, 1H), 5.44-5.41 (m, 2H), 5.21 (dd, J = 3.2 Hz, J = 11.3 Hz, 1H), 5.14-5.05 (m, 10H), 4.58 (t, J = 6.9 Hz, 1H), 4.54-4.45 (m, 3H), 4.21 (dd, J = 8.1 Hz, J = 10.9 Hz, 1H), 4.04 (dd, J = 5.8 Hz, J = 10.8 Hz, 1H), 2.10 (s, 3H), 2.08-2.00 (m, 34H), 1.98 (s, 3H), 1.97 (s, 3H), 1.96-1.92 (m, 6H), 1.89 (s, 3H), 1.71 (s, 3H), 1.66-1.62 (m, 21H), 1.58 (s, 3H), 1.57 (s, 9H); 13C NMR (125 MHz, CD3OD): δ 174.4, 172.3, 172.2, 171.9, 140.6, 136.5, 136.4, 136.3, 136.3, 136.1, 135.9, 132.1, 126.3, 126.3, 126.3, 126.0, 125.6, 125.6, 125.6, 123.6, 123.6, 96.5, 70.4, 68.8, 68.7, 63.8, 63.8, 62.3, 41.0, 41.0, 40.9, 34.4, 33.4, 33.4, 33.4, 33.2, 33.0, 30.8, 30.8, 30.7, 30.6, 30.5, 30.5, 30.4, 28.0, 27.8, 27.8, 27.8, 27.7, 27.7, 27.7, 26.1, 24.0, 23.9, 23.8, 23.0, 20.8, 20.8, 20.7, 17.9, 16.3, 14.5, 14.0; 31P NMR (162 MHz, CD3OD): δ −9.7, −12.6; HRESIMS calcd for C69H110NO15P2 [M-H]−: 1254.7356; found, 1254.7361. Final deprotection with 0.1% NaOCH3 in CH3OH, neutralization with Amberlyst-15 ion exchange resin and filtration though a short bed of C-18 reverse phase silica gel afforded GalNAc-PP-und (18 mg, 99%). HRESIMS calcd for C63H104NO12P2 [M-H]−:1128.7039; found, 1128.7045.
GalNAc-PP-heptaprenyl (32)
GalNAc-PP-Hept was prepared as described above. Following the deprotection step, the crude product was purified via C-18 reverse phase column chromatography to afford GalNAc-PP-hept as a white solid (22 mg, 36%) from 41 mg of Heptaprenyl-1-P. 1H NMR (500 MHz, D2O): δ 5.61 (dd, J = 2.7 Hz, J = 5.9 Hz, 1H), 5.50 (t, J = 5.5 Hz, 1H), 5.24-5.08 (m, 6H), 4.51 (t, J = 5.7 Hz, 2H), 4.29 (d, J = 10.8 Hz, 1H), 4.23 (t, J = 5.5 Hz, 1H), 4.07 (d, J = 1.4 Hz, 1H), 4.00 (dd, J = 2.2 Hz, J = 10.7 Hz, 1H), 3.88-3.75 (m, 2H), 2.20-1.96 (m, 27H), 1.79 (s, 3H), 1.72 (s, 3H), 1.70 (s, 3H), 1.68 (s, 3H), 1.62 (s, 3H), 1.60 (s, 9H); 13C NMR (125 MHz, D2O): δ 174.7, 141.3, 135.42, 135.40, 134.8, 134.7, 125.2, 125.1, 124.9, 124.41, 124.35, 94.8, 72.2, 68.6, 67.7, 62.8, 61.3, 49.9, 49.8, 32.10, 32.06, 31.84, 26.6, 26.4, 26.2, 25.5, 25.4, 23.22, 23.18, 23.04, 23.0, 22.3, 17.3, 15.7; 31P NMR (162 MHz, D2O): δ −9.7, −11.4; HRESIMS calcd. for C43H72NO12P2 [M-H]−: 856.4535 found, 856.4543.
GalNAc-PP-pentaprenyl (33)
GalNAc-PP-pent was prepared according to the general procedure. Following the deprotection step, the crude product was purified via C-18 reverse phase column chromatography to afford GalNAc-PP-pent as a white solid (22 mg, 49%) from 27 mg of pentaprenyl-1-P. 1H NMR (500 MHz, CD3OD): δ 5.67 (dd, J = 3.1 Hz, J = 6.8 Hz, 1H), 5.56 (t, J = 6.6 Hz, 1H), 5.30-5.16 (m, 4H), 4.58 (t, J = 6.5 Hz, 2H), 4.36 (d, J = 10.9 Hz, 1H), 4.30 (t, J = 6.0 Hz, 1H), 4.14 (d, J = 3.0 Hz, 1H), 4.07 (dd, J = 2.9 Hz, J = 10.9 Hz, 1H), 3.94-3.84 (m, 2H), 2.30-2.23 (m, 2H), 2.22-2.11 (m, 13H), 2.10-2.02 (m, 4H), 1.85 (s, 3H), 1.80 (s, 3H), 1.75 (s, 3H), 1.71 (s, 3H), 1.68 (s, 6H); 13C NMR (125 MHz, D2O): δ 174.7, 160.3, 141.2, 135.8, 134.9, 134.4, 130.6, 124.6, 124.5, 124.3, 121.2, 94.7, 72.2, 68.7, 67.7, 62.7, 61.3, 49.9, 48.9, 39.7, 39.7, 32.0, 31.9, 26.7, 26.6, 26.1, 25.4, 23.1, 22.9, 22.3, 17.4, 15.8, 15.7; 31P NMR (162 MHz, D2O): δ −10.4, −13.1; HRESIMS calcd for C33H56NO12P2 [M-H]−: 720.3283; found, 720.3276.
GalNAc-PP-(all-Z)-pentaprenyl (34)
GalNAc-PP-(all-Z)-pent was prepared according to the general procedure. Following the deprotection step, the crude product was purified via C-18 reverse phase column chromatography to afford GalNAc-PP-(all-Z)-Pent as a white solid (15 mg, 20%) from 46 mg of (all-Z)-pentaprenyl-1-P. 1H NMR (500 MHz, D2O): δ 5.62 (dd, J = 3.0 Hz, J = 6.5 Hz, 1H), 5.51 (t, J = 6.0 Hz, 1H), 5.24-5.12 (m, 4H), 4.53 (t, J = 6.2 Hz, 2H), 4.30 (d, J = 11.1 Hz, 1H), 4.25 (t, J = 5.9 Hz, 1H), 4.09 (d, J = 2.4 Hz, 1H), 4.01 (dd, J = 2.6 Hz, J = 10.9 Hz, 1H), 3.88-3.78 (m, 2H), 2.22-2.00 (m, 19H), 1.79 (s, 3H), 1.73 (s, 3H), 1.72-1.68 (m, 9H), 1.63 (s, 3H); 13C NMR (125 MHz, D2O): δ 174.7, 141.3, 135.4, 135.4, 134.8, 134.7, 125.2, 125.1, 124.9, 124.4, 124.4, 94.8, 72.2, 68.6, 67.7, 62.8, 61.3, 49.8, 32.1, 32.1, 31.8, 26.6, 26.4, 26.2, 25.5, 25.4, 23.2, 23.2, 23.0, 23.0, 22.3, 17.3, 15.7; 31P NMR (162 MHz, D2O): δ −10.1, −12.3; HRESIMS calcd for C33H56NO12P2 [M-H]−: 720.3283; found, 720.3265.
GalNAc-PP-solanesyl (35)
GalNAc-PP-solan was prepared according to the general procedure. However, the two purification steps were reversed as the final product proved too insoluble to obtain suitable spectral data. The crude product was thus purified via C-18 reverse phase column chromatography to afford peracetylated GalNAc-PP-solan as a white solid (35 mg, 50%) from 44 mg of solanesyl-1-P. 1H NMR (500 MHz, CD3OD): δ 5.61 (dd, J = 3.3 Hz, J = 7.3 Hz, 1H), 5.43 (t, J = 6.6 Hz, 2H), 5.22 (dd, J = 3.1 Hz, J = 11.3 Hz, 1H), 5.14-5.04 (m, 8H), 4.59 (t, J = 7.0 Hz, 1H), 4.53 (t, J = 6.3 Hz, 3H), 4.21 (dd, J = 8.2 Hz, J = 10.8 Hz, 1H), 4.04 (dd, J = 5.8 Hz, J = 10.8 Hz, 1H), 2.11 (s, 3H), 2.10-2.02 (m, 18H), 1.99 (s, 3H), 1.99-1.94 (m, 17H), 1.90 (s, 3H), 1.69 (s, 3H), 1.65 (s, 3H), 1.59 (s, 3H), 1.58 (s, 21H); 13C NMR (125 MHz, CD3OD): δ 174.5, 172.4, 172.2, 171.9, 140.8, 136.3, 135.9, 135.9, 135.9, 135.8, 132.1, 125.7, 125.7, 125.6, 125.4, 122.7, 122.6, 96.5, 70.6, 68.7, 64.0, 64.0, 62.3, 41.0, 41.0, 41.0, 40.9, 40.8, 34.5, 33.2, 30.9, 30.8, 30.7, 30.6, 30.5, 28.0, 27.9, 27.8, 27.7, 27.7, 27.7, 26.0, 24.0, 23.8, 23.0, 20.8, 20.7, 20.7, 17.9, 16.8, 16.2, 15.7, 14.5; 31P NMR (162 MHz, CD3OD): δ −9.6, −12.5; HRESIMS calcd for C59H94NO15P2, [M-H]−:1118.6104; found, 1118.6112. Final deprotection with 0.1% NaOCH3 in CH3OH and filtration though a short bed of C-18 reverse phase silica gel afforded GalNAc-PP-solan (31 mg, 99%). HRESIMS calcd for C53H88NO12P2 [M-H]−: 992.5787, found 992.5780.
GalNAc-PP-MS-pentaprenyl (36)
GalNAc-PP-MS-pent was prepared according to the general procedure. Following the deprotection step, the crude product was purified via C-18 reverse phase column chromatography to afford GalNAc-PP-MS-pent as a white solid (27 mg, 26%) from 54 mg of MS-pentaprenyl-1-P. 1H NMR (500 MHz, D2O): δ 5.55 (dd, J = 3.3 Hz, J = 7.1 Hz, 1H), 5.22-5.05 (m, 4H), 4.26 (dt, J = 2.9 Hz, J = 11.0 Hz, 1H), 4.19 (dd, J = 5.2 Hz, J = 7.1 Hz, 1H), 4.06-3.93 (m, 4H), 3.84-3.72 (m, 2H), 2.11-2.02 (m, 11H), 2.01-1.92 (m, 6H), 1.69 (s, 3H), 1.66 (s, 3H) 1.61 (s, 3H), 1.58 (s, 6H), 1.54-1.24 (m, 4H), 1.22-1.10 (m, 1H), 0.92 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, D2O): δ 174.7, 135.0, 134.8, 134.5, 130.8, 125.7, 124.4, 124.2, 94.7, 72.1, 68.6, 67.7, 64.9, 61.3, 49.9, 49.8, 39.6, 37.3, 37.1, 31.8, 29.0, 26.6, 26.6, 26.5, 25.4, 24.9, 23.2, 22.3, 18.8, 18.8, 17.4, 15.7, 15.7; 31P NMR (162 MHz, D2O): δ −9.9, −11.8; HRESI MS calcd for C33H58NO12P2 [M-H]−: 722.3440; found, 722.3437.
Supplementary Material
Acknowledgments
We thank NIH R01 (GM085267) for financial support of this work.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.xxxxxxxxx.
Supporting Information (see footnote on the first page of this article): Copies of the 1H NMR and 13C NMR spectra of all new compounds.
References
- 1.a) Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]; b) Hartley MD, Imperiali B. Arch Biochem Biophys. 2012;517:83–97. doi: 10.1016/j.abb.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Holstein SA, Hohl RJ. Lipids. 2004;39:293–309. doi: 10.1007/s11745-004-1233-3. [DOI] [PubMed] [Google Scholar]; d) Hug I, Feldman MF. Glycobiology. 2011;21:138–151. doi: 10.1093/glycob/cwq148. [DOI] [PubMed] [Google Scholar]
- 2.a) Yurist-Doutsch S, Chaban B, VanDyke DJ, Jarrell KF, Eichler J. Mol Microbiol. 2008;68:1079–1084. doi: 10.1111/j.1365-2958.2008.06224.x. [DOI] [PubMed] [Google Scholar]; b) Jones MB, Rosenberg JN, Betenbaugh MJ, Krag SS. Biochim Biophys Acta. 2009;1790:485–494. doi: 10.1016/j.bbagen.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Roth J, Zuber C, Park S, Jang I, Lee Y, Kysela KG, Le Fourn V, Santimaria R, Guhl B, Cho JW. Mol Cells. 2010;30:497–506. doi: 10.1007/s10059-010-0159-z. [DOI] [PubMed] [Google Scholar]; d) Schiller B, Hykollari A, Yan S, Paschinger K, Wilson IB. Biol Chem. 2012;393:661–673. doi: 10.1515/hsz-2012-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yan A, Lennarz WJ. J Bio Chem. 2005;280:3121–3124. doi: 10.1074/jbc.R400036200. [DOI] [PubMed] [Google Scholar]; f) Kornfeld R, Kornfeld S. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 3.a) Lovering AL, Safadi SS, Strynadka NCJ. Annu Rev Biochem. 2012;81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]; b) Osborn MJ. Annu Rev Biochem. 1969;38:501–538. doi: 10.1146/annurev.bi.38.070169.002441. [DOI] [PubMed] [Google Scholar]
- 4.Esko JD, Doering TL, Raetz CRH. In: Essentials of Glycobiology. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. The Consortium of Glycobiology Editors; La Jolla, California, Cold Spring Harbor NY: 2009. [Google Scholar]
- 5.a) Altman LJ, Ash L, Kowerski RC, Epstein WW, Larsen BR, Rilling HC, Muscio F, Gregonis DE. J Am Chem Soc. 1972;94:3257–3259. doi: 10.1021/ja00764a073. [DOI] [PubMed] [Google Scholar]; b) Sato K, Inoue S, Onishi A, Uchida N, Minowa N. J Chem Soc, Perkin Trans 1. 1981;0:761–769. [Google Scholar]; c) Altman LJ, Ash L, Marson S. Synthesis. 1974;1974:129–131. [Google Scholar]; Naruta dY. J Org Chem. 1980;45:4097–4104. [Google Scholar]
- 6.a) Sreekumar C, Darst KP, Still WC. J Org Chem. 1980;45:4260–4262. [Google Scholar]; b) Sato K, Miyamoto O, Inoue S, Kobayashi T, Furusawa F. Chem Lett. 1981:1711–1714. [Google Scholar]
- 7.Sato K, Miyamoto O, Inoue S, Kobayashi T, Furusawa F. Chem Lett. 1981:1711–1714. [Google Scholar]
- 8.Sato K, Miyamoto O, Inoue S, Furusawa F, Matsuhashi Y. Chem Lett. 1983:725–728. [Google Scholar]
- 9.a) Faridmoayer A, Fentabil MA, Haurat MF, Yi W, Woodward R, Wang PG, Feldman MF. J Bio Chem. 2008;283:34596–34604. doi: 10.1074/jbc.M807113200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Faridmoayer A, Fentabil MA, Mills DC, Klassen JS, Feldman MF. J Bacteriol. 2007;189:8088–8098. doi: 10.1128/JB.01318-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gebhart C, Ielmini MV, Reiz B, Price NL, Aas FE, Koomey M, Feldman MF. Glycobiology. 2012;22:962–974. doi: 10.1093/glycob/cws059. [DOI] [PubMed] [Google Scholar]; d) Musumeci MA, Hug I, Scott NE, Ielmini MV, Foster LJ, Wang PG, Feldman MF. J Bio Chem. 2013;288:10578–10587. doi: 10.1074/jbc.M112.432815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Lehle L, Strahl S, Tanner W. Angew Chem Int Ed. 2006;45:6802–6818. doi: 10.1002/anie.200601645. [DOI] [PubMed] [Google Scholar]; b) Parodi AJ. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]; c) Skorupinska-Tudek K, Wojcik J, Swiezewska E. Chem Rec. 2008;8:33–45. doi: 10.1002/tcr.20137. [DOI] [PubMed] [Google Scholar]; d) Welti M. Glycoconj J. 2013;30:51–56. doi: 10.1007/s10719-012-9417-y. [DOI] [PubMed] [Google Scholar]
- 11.Sim MM, Kondo H, Wong CH. J Am Chem Soc. 1993;115:2260–2267. [Google Scholar]
- 12.a) Woodward R, Yi W, Li L, Zhao G, Eguchi H, Sridhar PR, Guo H, Song JK, Motari E, Cai L, Kelleher P, Liu X, Han W, Zhang W, Ding Y, Li M, Wang PG. Nat Chem Bio. 2010;6:418–423. doi: 10.1038/nchembio.351. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Han W, Wu B, Li L, Zhao G, Woodward R, Pettit N, Cai L, Thon V, Wang PG. J Bio Chem. 2012;287:5357–5365. doi: 10.1074/jbc.M111.308486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi W, Shao J, Zhu L, Li M, Singh M, Lu Y, Lin S, Li H, Ryu K, Shen J, Guo H, Yao Q, Bush CA, Wang PG. J Am Chem Soc. 2005;127:2040–2041. doi: 10.1021/ja045021y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.