

Abstract
The chemistry of several iron(III) porphyrinates containing silylthiolate ligands is described. The complexes are prepared by protonolysis reactions of silanethiols with the iron(III) precursors, [Fe(OMe)(TPP)] and [Fe(OH)(H2O)(TMP)] (TPP = dianion of meso-tetraphenylporphine; TMP = dianion of meso-tetramesitylporphine). Each of the compounds has been fully characterized in solution and the solid state. The stability of the silylthiolate complexes versus other iron(III) porphyrinate complexes containing sulfur-based ligands allows for an examination of their reactivity with several biologically relevant small molecules including H2S, NO, and 1-methylimidazole. Electrochemically, the silylthiolate complexes display a quasi-reversible one-electron oxidation event at potentials higher than that observed for an analogous arylthiolate complex. The behavior of these complexes versus other sulfur-ligated iron(III) porphyrinates is discussed.
Introduction
The role of thiolate ligands in modulating the reactivity of biologically relevant transition metal ions has been well documented for a number of different ligand environments.1 For heme-iron, the presence of cysteinate ligands in enzymes such as cytochrome P450 has been shown to modulate the reactivity of the metal cofactor in ways unique to that of other axially coordinated ligands.2–8 In addition to thiols, the simplest of sulfhydryl containing species, hydrogen sulfide, has become of prominent interest due to its role in a variety of physiological processes in higher organisms.9,10 Roles for H2S in biology have now been demonstrated to include vasodilation,11 signal transduction,12 and protection against oxidative stress.13 In addition, the potential for H2S to serve as a source of elemental sulfur in the assembly of various cofactors makes the chemistry of this small molecule particularly intriguing. As a result, a great deal of recent work has centered on developing new platforms for detection of H2S in vivo.14–19
The role of H2S in biology begs the question of its molecular mechanisms of action. In this respect, parallels with the chemistry of other small signaling molecules such as NO and CO are notable.20,21 Unlike these small molecules, however, the fundamental coordination chemistry of hydrogen sulfide at biologically relevant transition metal scaffolds has not been explored in detail.22 This fact likely stems from a number of difficulties associated with H2S including its toxicity, its acidity, its reducing potential, and its propensity for forming intractable metal sulfides. In this vein, the porphyrin framework might be envisioned to provide a convenient platform for exploration of H2S chemistry since its resistance to protonolysis coupled with its ability to constrain reactivity to mutually trans axial sites is beneficial in overcoming several of the challenges mentioned above. Furthermore, H2S been demonstrated to effect a variety of processes in heme proteins making basic understanding of this molecule in such environments important.23–27
Previous examinations of H2S chemistry with metal porphyrinates have provided some key preliminary observations. With biological systems, the reaction of H2S with hemoglobin and myoglobin to form “sulfhemes” has been known for over a century. These green reaction products originally observed by Hoppe-Seyler in 1866 were later shown to result predominantly from modification of the periphery of the porphyrin cofactor.28–30 Bona fide binding of H2S and HS− to heme iron has also been reported,31 most notably for the clam hemoglobin HbI from Lucina pectinata,32–36. Despite this precedent, biological examples of hydrogen sulfide coordination remain scarce.
With synthetic porphyrin systems, work by Scheidt has demonstrated formation of hydrosulfide adducts of both iron(III)37 and iron(II).38 In related chemistry, Holm has reported a similar iron(III) hydrosulfide species as a transient intermediate in the reaction of [Fe(OEP)]2-μ-O (OEP = dianion of octaethylporphine) with H2S.39 [Fe(SH)(OEP)] was proposed to be high-spin in line with other five-coordinate iron(III) thiolates containing porphyrinate ligands based on its NMR features.40 More recently, H2S binding to iron(II) was reported by Collman for a synthetic cyctochrome c model complex incorporating a picket fence type porphyrin ligand.41,42
In all these previous reports, the difficulty in preparing and isolating H2S/HS− species of iron porphyrinates has precluded detailed examinations of their reactivity and spectroscopy. Even in ruthenium chemistry, where binding of H2S is well established,43–49 examples involving porphyrinate ligands are unknown. James has reported several thiol adducts of [RuII(TMP)] (TMP = meso-tetramesitylporphine), although analogous reactions with H2S did not afford well-defined species.50,51
We have been examining the coordination chemistry of H2S with transition metals in biologically relevant ligand scaffolds in order to elucidate the fundamental chemistry of such species relevant to the physiological action of hydrogen sulfide in biology. In this contribution, we describe our investigations of iron(III) porphyrinates with H2S and silanethiols (R3SiSH). The resulting silylthiolate complexes provide an alternative to hydrocarbyl-thiolates and appear to provide a stable platform for examining the chemistry of sulfur-bound iron(III) porphyrinates.
Results and Discussion
Preparation and characterization of iron(III) porphyrinates
Previous reports by Holm and Scheidt suggested the existence of the elusive iron(III) hydrosulfide complex, [Fe(SH)(P)] (P = porphyrinate ligand), although complete characterization and an examination of the reactivity of these species is lacking.37,39 We therefore first examined the preparation of an analogous hydrosulfide iron(III) porphyrinate, [Fe(SH)(TPP)], (TPP = dianion of meso-tetraphenylporphine). Both protonolysis reactions of the oxo-bridged complex [FeIII(TPP)]2-μ-O with H2S and salt metathesis reactions of [FeCl(TPP)] with NaSH or (Et4N)SH were examined in an attempt to prepare this species (Scheme 1). In each case, the iron(II) porphyrinate [FeII(TPP)], was the only metal-containing species recovered from the reaction, consistent with previous observations by Holm.39 For reactions employing hydrosulfide salts, we failed to observe formation of the previously reported hydrosulfide adduct of iron(II) under the conditions examined (1 – 3 equiv. of HS−).38
Scheme 1.
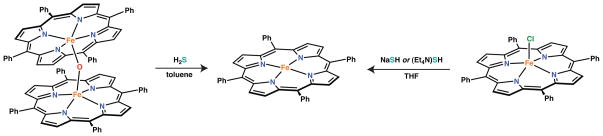
Replacement of TPP with the bulkier TMP ligand was examined in hopes of providing added stability to a putative hydrosulfide complex. Moreover, the TMP ligand allows for isolation of the terminal hydroxide complex, [Fe(OH)(H2O)(TMP)], which we envisioned as a superior precursor than the oxo-bridged complex for protonolysis reactions employing sulfhydryls.52 UV-vis spectroscopic observation of the reaction of [Fe(OH)(H2O)(TMP] with excess H2S at room temperature did lead to formation of a new iron(III) complex (Figure 1), which we tentatively assign as the desired [Fe(SH)(TMP)] complex based on the position of the Soret absorbance and the Q-bands.52 Repeating this reaction at the higher concentrations necessary for product isolation, however, resulted in formation of iron(II) as observed with the TPP ligand.
Figure 1.

Electronic absorption spectra of 10 μM [Fe(OH)(TMP)] in toluene before (blue) and after (red) addition of excess H2S at 23 °C. Inset displays a magnification of the Q-band region.
Based on the results obtained with H2S and HS−, we surmised that the propensity for reduction could be slowed by using an electronically similar ligand with greater steric bulk at sulfur. Select examples of five-coordinate iron(III) porphyrinates bearing thiolate ligands are known, all structurally characterized examples of which contain aryl thiolates.40,53–62. Believing the hydrocarbon substituents of these thiolates to be poor models for the proton of the HS− ligand, we sought to examine the use of silylthiolates. The use of these ligands was also inspired in part by the analogy drawn between protons and trialkylsilyl groups in organic chemistry,63–65 and the noted stabilizing effect of silicon atoms alpha to anionic centers.66,67 Furthermore, these ligands have been used as a source of sulfur equivalents previously,39,68 offering the potential for deprotection to afford a hydrosulfide species.
To install the silylthiolate we chose to examine protonolysis reactions akin to those described above for H2S, reasoning that avoidance of thiolate salts would be beneficial in preventing reduction to iron(II). Furthermore, such reactions are likely more relevant to potential biological chemistry of sulfhydryls with iron heme centers containing Brønsted basic ligands such as hydroxide. We therefore examined reactions of the methoxide derivative, [Fe(OMe)(TPP)], with HSSiiPr3 (hereafter HSTIPS) as shown in Scheme 2. This species was chosen because the corresponding iron(III) hydroxide complex is not stable for the TPP ligand.69 Reaction of HSTIPS with [Fe(OMe)(TPP)] proceeded rapidly in benzene or toluene affording the iron(III) silylthiolate complex as a red solid in high yield. All preparations of [Fe(STIPS)(TPP)] also produced 5 – 10% of [FeII(TPP)] due to competing reduction. Similar reactions with PhSH and tBuSH also afforded the desired thiolate complexes as judged by 1H NMR spectroscopy, however, in these instances a larger degree of reduction was observed precluding isolation of these species (see Supporting Information).
Scheme 2.
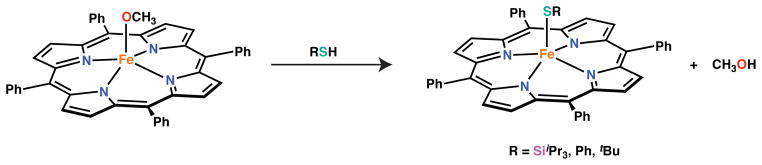
The 1H NMR features of [Fe(STIPS)(TPP)] are similar to those observed for other silylthiolate complexes of meso-substituted porphyrinates discussed below, and are consistent with other high-spin iron(III) complexes such as [FeCl(TPP)].70 The pyrrolic protons of [Fe(STIPS)(TPP)] appear as a broad peak near 80 ppm in benzene-d6. For all silylthiolate complexes investigated, this pyrrolic resonance is found 4 – 5 ppm downfield of that for the corresponding hydrocarbyl-thiolate complexes (c.f. 76 ppm for [Fe(StBu)(TPP)]). The meta hydrogen atoms of the phenyl substituents appear as two singlets at 12.31 and 11.00 ppm, the former of which overlaps a broad resonance at 12.7 ppm that is most likely due to the isopropyl groups of the STIPS ligand (see SI).
The electronic absorption spectrum of [Fe(STIPS)(TPP)] in toluene displays a Soret feature centered at 24,000 cm−1 (417 nm), which appears to overlap several higher energy bands (see SI). The appearance of this Soret feature may be due to the presence of ~5% [FeII(TPP)], however, similar overlapping Soret features were observed for other silylthiolate species containing the TMP ligand (vida infra). The spectrum of [Fe(STIPS)(TPP)] also contains several less intense bands in the Q region consistent with a five-coordinate iron(III) porphyrinate. These bands also resemble those observed in the reaction of [Fe(OH)(H2O)(TMP)] with H2S described above, further supporting the assignment of the species in Figure 1 as the hydrosulfide iron(III) complex.
The solid-state structure of [Fe(STIPS)(TPP)] is depicted in Figure 2. The iron atom resides 0.561 Å out of the best-fit plane containing the porphyrin atoms and the Fe-S bond vector shows a deviation from normal originating from the N4 centroid as observed in other five-coordinate thiolate complexes.61 The Fe-S bond distance of 2.269 Å is significantly below the range encountered for other structurally characterized [FeIII(SR)(P)] species (c.f. 2.32 – 2.36 Å),71,72 highlighting the difference between the silylthiolate ligand and those based on hydrocarbyl-substituted thiols. Despite the significant steric hindrance afforded by the SiiPr3 group, the Fe-S-Si bond angle remains substantially bent at 115.78°.
Figure 2.

Thermal ellipsoid (50%) drawing of [Fe(STIPS)(TPP)]. Hydrogen atoms and co-crystallized benzene molecule omitted for clarity. Selected bond distances (Å) and angles (deg): Fe(1)-S(1) = 2.269(1); Fe(1)-N(1) = 2.082(2); Fe(1)-N(2) = 2.074(2); Fe(1)-N(3) = 2.088(2); Fe(1)-N(4) = 2.087(2); S(1)-Si(1) = 2.152(1); Fe(1)-S(1)-Si(1) = 115.78 (4).
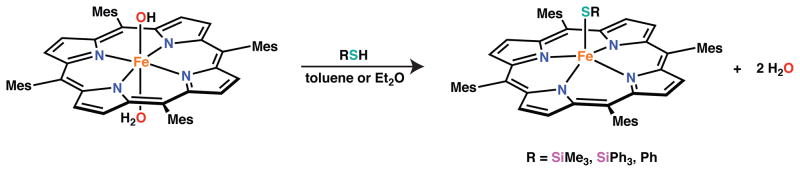
Protonolysis reactions with the bulkier, more electron-rich TMP ligand were next examined with the intent of further stabilizing a trivalent silylthiolate complex and preventing reduction to iron(II). Reaction of [Fe(OH)(OH2)(TMP)] with the silanethiols, HSTIPS and HSSiPh3 afforded the corresponding thiolate species in good yield (Scheme 3). In contrast to similar reactions with TPP supported complexes, no reduction to iron(II) was observed with the TMP ligand. The benzenethiolate complex, [Fe(SPh)(TMP)] was also prepared by this route in sufficient purity to allow for isolation. Reactions with tBuSH afforded the tert-butylthiolate complex as judged by 1H NMR spectroscopy. As with the TPP ligand, however, substantial reduction to [FeII(TMP)] was found to accompany the protonolysis reaction. Such a result is not surprising given the pronounced basicity of tBuS− with respect to PhS− and the silylthiolates. The 1H NMR spectroscopic features of [Fe(STIPS)(TMP)] and [Fe(SSiPh3)(TMP)] are similar to those of [Fe(STIPS)(TPP)] discussed above (see also SI). Furthermore, the absorption spectra for all TMP complexes also resemble those of [Fe(STIPS)(TPP)] demonstrating a Soret feature that is complicated by the coincidence of other high energy absorptions (see SI).
Scheme 3.
The solid-state structures of [Fe(STIPS)(TMP)] and [Fe(SSiPh3)(TMP)] are displayed in Figure 3 and show many similarities to the structure of [Fe(STIPS)(TPP)]. For the TMP complexes, the increased steric interactions between the silylthiolate ligand and the porphyrin meso substituents force the iron atom even farther out of the porphyrin plane (c.f. 0.645 Å for [Fe(STIPS)(TMP)]). This effect is also observed [Fe(SSiPh3)(TMP)], however the metric parameters of this complex should be viewed cautiously as the structure refinement was problematic (R1 = 9.0%). Crystals of [Fe(SPh)(TMP)] were also subjected to X-ray diffraction, however, the structure could not be refined satisfactorily (see SI). The diffraction data did establish the connectivity of the molecule unambiguously displaying a similar positional disorder of the Fe-SPh moiety as found in the published structure of [Fe(SPh)(TPP)].54
Figure 3.

Thermal ellipsoid drawings of [Fe(STIPS)(TMP)] (50%) and [Fe(SSiPh3)(TMP)] (35%). Hydrogen atoms and co-crystallized solvent molecules omitted for clarity. Selected bond distances (Å) and angles (deg) for [Fe(STIPS)(TMP)]: Fe(1)-S(1) = 2.273(1); Fe(1)-N(1) = 2.078(2); Fe(1)-N(2) = 2.080(2); Fe(1)-N(3) = 2.081(2); Fe(1)-N(4) = 2.081(2); S(1)-Si(1) = 2.134(1); Fe(1)-S(1)-Si(1) = 123.00(4); for [Fe(SSiPh3)(TMP)]: Fe(1)-S(1) 2.243(3); Fe(1)-N(1) = 2.062(5); Fe(1)-N(2) = 2.073(5); Fe(1)-N(3) = 2.090(6); Fe(1)-N(4) = 2.080(5); S(1)-Si(1) = 2.040(3); Fe(1)-S(1)-Si(1) = 127.1(2).
To further establish the electronic structure of the silylthiolate complexes, EPR spectra of [Fe(STIPS)(TMP)] and [Fe(SSiPh3)(TMP)] were recorded at 77 K in 2-MeTHF. Previous work has established that five-coordinate thiolate complexes of iron(III) porphyrinates adopt high-spin ground states displaying axial EPR signals with g⊥ ≈ 6.40,62,73 Such a result was confirmed for both [Fe(STIPS)(TMP)] and [Fe(SSiPh3)(TMP)] in 2-MeTHF at 77 K (see SI). In contrast, the EPR spectrum of [Fe(SPh)(TMP)] in 2-MeTHF at 4 K displayed three signals. One of these signals corresponds to high-spin iron(III) and matches that observed for the silylthiolate complexes. The other two signals are consistent with a low-spin iron(III) thiolate.74,75 Upon warming to 62 K, the high-spin signal decreases in intensity suggesting that the appearance of the low-spin signals is not due to an impurity (see SI) but to an equilibrium between different spin states, possibly involving solvent coordination.76 The two low-spin signals display very similar g values and likely result from slightly different conformations of the complex in the frozen glass.
Electrochemistry
The electrochemistry of the silylthiolate complexes was next examined to understand their redox properties and help establish whether the propensity for reduction of iron(III) porphyrinates in reactions employing H2S and other thiols is predominantly thermodynamic (high potential) or kinetic in origin. The CV for [Fe(STIPS)(TPP)] is displayed in Figure 4. A quasi-reversible one-electron reduction is found at a potential of E½ = −1.15 V (vs Fc/Fc+), suggesting that reduction to iron(II) is not very facile. The cyclic voltammograms of [Fe(STIPS)(TMP)] and [Fe(SSiPh3)(TMP)] demonstrate cathodic events at similar potentials, although in each case these events show a greatly attenuated return wave (see SI).
Figure 4.

Cyclic voltammogram of 2 mM [Fe(STIPS)(TPP)] in CH2Cl2 at a Pt disk electrode. Scan rate is 50 mV/s and the supporting electrolyte is 0.1 M Bu4NPF6.
In the CV for all silylthiolate complexes examined, a quasi-reversible anode event is observed near +0.45 V. In the case of the Ph3SiS− complex, this event becomes nearly reversible at faster scan rates (see SI). The potentials found for these anode events are similar to those assigned as porphyrin-based oxidations (P/P+·) with other iron porphyrinates (c.f. +0.61 V for [FeCl(TMP)]), however, they do not display the same degree of reversibility.77 A possible explanation for this electrochemical behavior is that the oxidized iron(III) thiolate complexes contain considerable thiyl radical character. For comparison, the oxidation of free −STIPS (as its Et3NH+ salt) under identical CV conditions occurs at a potential of +0.76 V (see SI). Partial dissociation of the thiyl radical on the electrochemical timescale would lead to formation of disulfide and iron(III) accounting for the observed quasi-reversibility. Consistent with this proposal is the observation of peaks assignable to the iron(II)/iron(III) couple of [Fe(TPP)] in the return wave near −0.15 V (see SI). Such an explanation is also consistent with the proposal by Green that oxygenated cyctochrome P450 intermediates display sulfur localized ligand radical character.78 Figure 5 shows the region of the differential pulse voltammogram for all four thiolate species highlighting this first anodic event. Of interest is the fact that this oxidation event occurs at the lowest potential for the benzenethiolate complex despite the lower pKa of PhSH versus trialkylsilanethiols.79 This event is also completely irreversible for the benzenethiolate complex (see SI), again underscoring the unique features of the silylthiolates versus thiolates containing hydrocarbyl substituents.
Figure 5.

DPV of iron(III) thiolate complexes in CH2Cl2 at a Pt disk electrode highlighting the first observed oxidation event. Supporting electrolyte: 0.1 M Bu4NPF6.
In addition to the first quasi-reversible anode event discussed above, higher potential oxidation processes are observed for each of the thiolate complexes out to +1.3 V (see SI). While we cannot assign these processes definitively at this time, the first of these high potential events coincides with the second of two reversible one-electron oxidation events displayed by other iron porphyrinate complexes.80 In the case of [Fe(STIPS)(TPP)], this event is clearly resolved and nearly reversible (E½ = +0.92 in Figure 4). In contrast to other iron porphyrinates, however, each of the thiolate complexes also displays an intense peak above +1.0 V that appears to correspond to a multi-electron redox process (E½ = +1.15 V in Figure 4). This multi-electron event is not unique to the silylthiolate complexes and is also observed with the benzenethiolate species (see SI).
Reaction chemistry
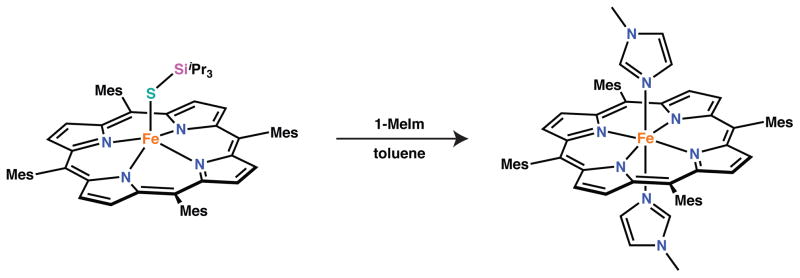
To probe the chemical behavior of the silylthiolate complexes, several reactions with biologically relevant molecules were investigated using the prototypical complex, [Fe(STIPS)(TMP)]. Treatment of the five-coordinate complex with 1-methylimidazole (1-MeIm) did not afford a six-coordinate thiolate species as judged by 1H NMR spectroscopy, but rather led to eventual reduction of iron(III) and isolation of [Fe(1-MeIm)2(TMP)] (Scheme 4; see also SI).81 Similar reduction was not observed in weakly coordinating solvents such as THF and 2-MeTHF (vida supra). This fact suggests that only strong Lewis bases are capable of binding to the silylthiolate complex, and that such coordination results in reduction to iron(II).
Scheme 4.
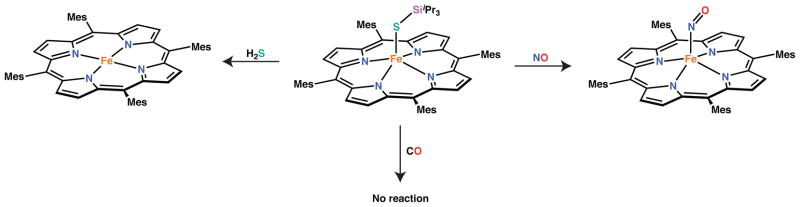
Exposure of the iron(III) thiolate complexes to ambient atmosphere resulted in little to no decomposition as judged by NMR spectroscopy. Furthermore, the preparation of each of the thiolate complexes shown in Scheme 3 could be conducted in the presence of air with no decrease in reaction yield demonstrating their relative stability towards oxygen. Reaction of [Fe(STIPS)(TMP)] with other small molecules, however, was found to lead to different outcomes (Scheme 5). Introduction of 1 atm of CO (g) to a benzene-d6 solution of [Fe(STIPS)(TMP)] resulted in no reaction after 24 hrs as judged by NMR spectroscopy. In contrast, introduction of 1 atm of NO (g) under identical conditions resulted in immediate formation of [Fe(NO)(TMP)] as judged by 1H NMR, IR, and UV-vis spectroscopy (see SI).82,83 The organic byproduct of this reaction is not known at this time, but is presumably either the disulfide or the nitrosothiol, TIPSSNO. We favor the former based on similar findings by Richter-Addo.61 The final gaseous small molecule examined was H2S. Treatment of [Fe(STIPS)(TMP)] with 1 atm of H2S in benzene-d6 lead to immediate reduction and formation of [FeII(TMP)] in similar fashion to reactions starting from [Fe(OH)(H2O)(TMP)].
Scheme 5.
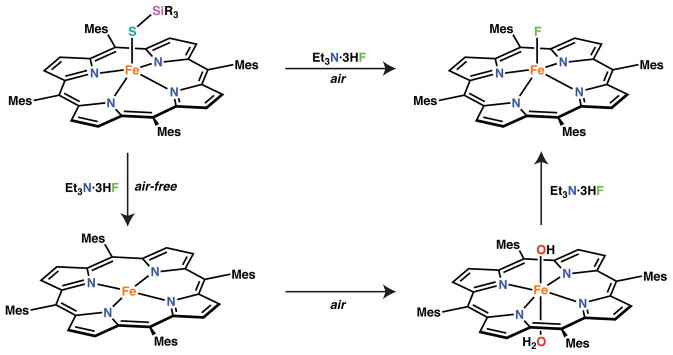
In addition to biologically relevant small molecules, we also examined the reactivity of the silylthiolate complex with a fluoride source, Et3N·3HF, anticipating that S-Si bond cleavage might represent another route to an iron(III) hydrosulfide complex. In the absence of oxygen, reaction of Et3N·3HF with [Fe(STIPS)(TMP)] in benzene-d6 afforded [FeII(TMP)] as judged by 1H NMR spectroscopy. We could not identify a signal in the 19F NMR spectrum of the reaction mixture corresponding to TIPSF and therefore do not know at this time whether the iron(II) porphyrinate is formed via the intermediacy of the desired iron(III) hydrosulfide, or via a different pathway. Under ambient conditions, the iron(III) fluoride complex, [FeF(TMP)] can be isolated from the reaction mixture. Control experiments with [Fe(OH)(H2O)(TMP)] and Et3N·3HF demonstrated formation of the same iron(III) fluoride complex consistent with previous work.84 Thus, formation of [FeF(TMP)] in the presence of oxygen likely follows the reaction sequence shown in Scheme 6.
Scheme 6.
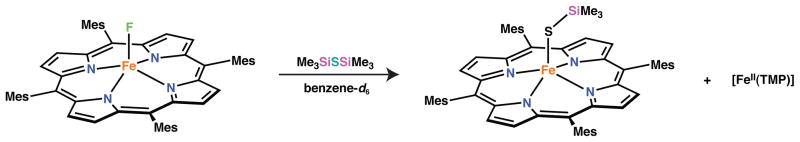
In order to assess the ability of [FeF(TMP)] to serve as a precursor to Fe-S bonds, the reaction shown in Scheme 7 was investigated. Upon treatment of [FeF(TMP)] with (Me3S)2Si in benzene-d6, a new iron(III) complex with meta-aryl resonances at 14.59 and 12.99 ppm was observed to form (see SI). We assign this new species as [Fe(STMS)(TMP)] based on the similarity of its NMR features to those compounds discussed above. Furthermore, the appearance of a resonance at −174 ppm in the 19F NMR spectrum of the reaction mixture suggests that S-Si bond scission is occurring with concomitant formation of TMSF. Substantial [FeII(TPP)] was also observed to form under these conditions demonstrating that the protonolysis route is a superior means of preparing silylthiolate ligated iron(III) porphyrinates.
Scheme 7.
Conclusions
We have reported the synthesis and characterization of several iron(III) porphyrinates containing silylthiolate ligands. The complexes are prepared by protonolysis reactions with the corresponding O-bound species representing a plausible reaction scenario for the interaction of sulfhydryls and iron(III) hemes in a biological setting. The presence of the silyl group on sulfur appears to give rise to notable differences in the structure and chemistry of these species compared with traditional iron(III) porphyrinates containing hydrocarbyl-thiolate ligands. The silylthiolate species are all high-spin, and display five-coordinate geometries in the solid-state with short Fe-S distances. Electrochemical measurements are consistent with a quasi-reversible initial oxidation event for each of the silylthiolate complexes, which contrasts with the completely irreversible event found for a related benzenethiolate complex. Finally, reaction pathways of the silylthiolate complexes with biologically relevant small molecules appear to be dominated by reduction to iron(II), including formation of a ferrous nitrosyl upon reaction with NO.
Experimental
General Comments
Unless otherwise noted, all manipulations were performed under an atmosphere of nitrogen gas using standard Schlenk technique or in a Vacuum Atmospheres glovebox under an atmosphere of purified nitrogen. Tetrahydrofuran, diethyl ether, methylene chloride, pentane, and toluene were purified by sparging with argon and passage through two columns packed with 4 Å molecular sieves. Benzene, benzene-d6, and 2-methyltetrahydrofuan were dried over sodium then vacuum-distilled. All solvents were stored in the glovebox over 4 Å molecular sieves prior to use. 1H NMR spectra were recorded in benzene-d6 on a Varian INOVA spectrometer operating at 500 MHz and referenced to the residual C6D5H peak of the solvent (δ 7.16 ppm vs TMS). UV-vis spectra were recorded at ambient temperature in dichloromethane on a Cary-60 spectrophotometer in air-tight Teflon-capped quartz cells. Cyclic voltammetry measurements were performed in dichloromethane in a single compartment cell under a nitrogen atmosphere (in the glovebox) at 25 °C using a CH Instruments 620D electrochemical workstation. A 3-electrode set-up was employed comprising a 1 mm diameter Pt disk working electrode, platinum wire auxiliary electrode, and Ag quasi-reference electrode. Triply recrystallized Bu4NPF6 was used as the supporting electrolyte. All electrochemical data were referenced internally to the ferrocene/ferrocenium couple at 0.00 V. EPR measurements were recorded in 4mm quartz tubes on a Bruker E500 EPR spectrometer operating at the X-band at a modulation frequency of 100 kHz and modulation amplitude of 10 G. Low temperature measurements were made in frozen 2-MeTHF glasses at temperatures between 4 and 77 K with temperature control maintained by a helium (4 – 65 K) or nitrogen (77K) flow cryostat (ESR900, Oxford Instruments, Inc.). Elemental analyses were performed by Atlantic Microlab, Inc. of Norcross, GA.
Materials
Metalloporphyrins [FeCl(TPP)], [Fe(OCH3)(TPP)], [FeIII(TPP)]-μ-O, [FeCl(TMP)], and [Fe(OH)(H2O)(TMP)] were prepared by literature procedures or slight modifications thereof.70,85–87 Et3N·3HF, iPr3SiSH, Ph3SiSH, PhSH, and TMS2S were purchased from Aldrich and used as received. NaSH was prepared by the method of Scheidt employing Na (s) and H2S (g) in THF.38 H2S gas was purchased from Praxair and used as received. For experiments employing gaseous H2S, the gas stream was delivered to the reaction vessel via a syringe needle interfaced to stainless steel tubing leading from the regulator. NO gas was purified by bubbling through 10 M NaOH (aq) then collected from the headspace of the solution.
X-ray Data Collection and Structure Solution Refinement
Crystals suitable for X-ray diffraction were mounted in Paratone oil onto a glass fiber and frozen under a nitrogen cold stream. The data were collected at 98(2) K using a Rigaku AFC12/Saturn 724 CCD fitted with Mo Kα radiation (λ = 0.71073 Å). Data collection and unit cell refinement were performed using Crystal Clear software.88 Data processing and absorption correction, giving minimum and maximum transmission factors, were accomplished with Crystal Clear and ABSCOR,89 respectively. All structures were solved by direct methods and refined on F2 using full-matrix, least-squares techniques with SHELXL-97.90,91 All non-hydrogen atoms were refined with anisotropic displacement parameters. All carbon bound hydrogen atom positions were determined by geometry and refined by a riding model.
[Fe(SSiiPr3)(TPP)]
To 50.0 mg (71.5 μmol) of [Fe(OCH3)(TPP)] in 2 mL of toluene was added 12.5 μL (65.6 μmol) of iPr3SiSH. The solution color immediately changed from brown to red and was allowed to stir at ambient temperature for 60 min. All volatiles were removed in vacuo and the resulting residue was washed with pentane to afford 60.0 mg (98%) of a purple microcrystalline solid. NMR spectroscopy of bulk material showed the presence of methanol and ~5% [FeII(TPP)]. Crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a saturated benzene solution at room temperature. 1H NMR: δ 79.9 (v br s, pyr-H), 12.6 (v br s, TIPS), 12.31 (s, m-ArH), 11.00 (s, m-ArH), 7.9 (v br s, o-ArH), 6.05 (s, p-ArH), 4.8 (v br s, o-ArH). UV-Vis λmax, cm−1 (ε, M−1cm−1): Soret 24000 (87000), Q-bands 19600 (13000), 17600, 15500, 14600. Anal. Calcd for C53H49FeN4SSi·CH3OH: C, 72.87; H, 6.00; N, 6.30. Found: C, 73.05; H, 5.78; N, 6.25.
[Fe(SSiiPr3)(TMP)]
To 50.3 mg (57.6 μmol) of [Fe(OH)(H2O)(TMP)] in 3 mL of toluene dissolved was added 13 μL (68.2 μmol) of iPr3SiSH. The solution color immediately changed from brown-yellow to red and was allowed to stir at ambient temperature for 60 min. All volatiles were removed in vacuo and the remaining residue was dissolved in 5 mL of pentane. The pentane solution was then evaporated to afford 56.6 mg (89%) of a dark purple microcrystalline solid. NMR spectroscopy of the bulk material from multiple syntheses showed the presence of pentane. Crystals suitable for X-ray diffraction were grown by slow cooling of a saturated pentane solution at −30 °C. 1H NMR: δ 77.9 (v br s, pyr-H), 14.31 (s, m-ArH), 12.92 (s, m-ArH), 11.7 (v br s, TIPS), 7.0 (v br s, o-CH3), 3.69 (s, p-CH3), 2.5 (v br s, o-CH3). UV-Vis λmax, cm−1 (ε, M−1cm−1): Soret 23200 (62000), Q-bands 19500 (11000), 17500, 15200, 14500. Anal. Calcd for C65H73FeN4SSi·C5H12: C, 76.54; H, 7.80; N, 5.10. Found: C, 75.77; H, 7.69; N, 4.49.
[Fe(SSiPh3)(TMP)]
To 50.2 mg (57.6 μmol) of [Fe(OH)(H2O)(TMP)] in 9 mL of toluene dissolved was added 16.8 mg (57.4 μmol) of Ph3SiSH. The solution color immediately changed from brown-yellow to red and was allowed to stir at ambient temperature for 60 min. All volatiles were removed in vacuo and the remaining residue was washed with pentane to afford 63.7 mg (98%) of a dark purple microcrystalline solid. Crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a saturated dichloromethane solution of the complex. 1H NMR: δ 78.4 (v br s, pyr-H), 15.09 (s, m-ArH), 14.6 (br s, SiPh3), 13.56 (s, m-ArH), 7.0 (v br s, o-CH3), 3.78 (s, p-CH3), 3.2 (br, SiPh3), 2.9 (v br s, o-CH3). UV-Vis λmax, cm−1 (ε, M−1cm−1): Soret 23900 (92000), Q-bands 19400 (17000), 17300, 14900, 14200. Anal. Calcd for C74H67FeN4SSi: C, 78.77; H, 5.99; N, 4.97. Found: C, 78.69; H, 6.19; N, 4.60.
[Fe(SPh)(TMP)]
To 50.3 mg (57.6 μmol) of [Fe(OH)(H2O)(TMP)] in 3 mL of toluene was added 5.7 μL (51.7 μmol) of benzenethiol. The solution colored changed from brown-yellow to red and was allowed to stir at ambient temperature for 60 min. All volatiles were removed in vacuo and the remaining residue was dissolved in 5 mL of pentane. The pentane solution was then evaporated to afford 55.7 mg (97%) of a dark purple microcrystalline solid. NMR spectroscopy of bulk material from multiple syntheses showed the presence of variable amounts of water. 1H NMR: δ 73.9 (v br s, pyr-H), 70.4 (v br s, SPh), 13.97 (s, m-ArH), 12.63 (m-ArH), 6.3 (v br s, o-CH3), 3.75 (s, p-CH3), 3.2 (br s, SPh), 3.0 (v br s, o-CH3). UV-Vis λmax, cm−1 (ε, M−1cm−1): Soret 24300 (98000), Q-bands 19500 (17000), 17500, 14700, 14300. Anal Calcd for C62H57FeN4S·(H2O)1.5: C, 76.53; H, 6.21; N, 5.76. Found: C, 76.51; H, 6.93, N, 4.81.
Procedure for NMR scale reactions with [Fe(STIPS)(TMP)]
To a septum-capped NMR tube containing a ~10 mM solution of [Fe(STIPS)(TMP)] in benzene-d6 was introduced an excess of gaseous reagent (H2S, NO, or CO). In the case of reactions employing 1-MeIm, Et3N·3HF, or (Me3Si)2S, a stoichiometric amount of the corresponding reagent was added as a neat liquid via microsyringe. Solutions were immediately subjected to 1H NMR spectroscopy. For the reaction with NO (g), the solution was evaporated to dryness and the remaining solid was used for IR spectroscopy (KBr). Crystals of [Fe(1-MeIm)2(TMP)] were grown by layering of a benzene solution of the complex with pentane at room temperature.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Welch Foundation (AX-1772 to ZJT). JDC is supported through the MBRS/RISE program (GM060655). The authors also acknowledge Mr. Salvador Echeveste, Jr. for experimental assistance through the support of the ACS Project SEED program.
Footnotes
Supporting Information. Additional spectra, thermal ellipsoid drawing for [Fe(1-MeIm)2(TMP)], crystallographic data and refinement parameters for all complexes and the corresponding CIF files. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Dawson JH, Holm RH, Trudell JR, Barth G, Linder RE, Bunnenberg E, Djerassi C, Tang SC. J Am Chem Soc. 1976;98:3707–3709. doi: 10.1021/ja00428a054. [DOI] [PubMed] [Google Scholar]
- 3.Dawson JH, Sono M. Chem Rev. 1987;87:1255–1276. [Google Scholar]
- 4.Ogliaro F, de Visser SP, Shaik S. J Inorg Biochem. 2002;91:554–567. doi: 10.1016/s0162-0134(02)00437-3. [DOI] [PubMed] [Google Scholar]
- 5.Meunier B, de Visser SlP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 6.Shaik S, Kumar D, de Visser SlP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 7.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2009;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 8.de Visser SP, Nam W. Handbook of Porphyrin Science. Vol. 10. World Scientific Publishing Co. Pte. Ltd; 2010. pp. 85–139. [Google Scholar]
- 9.Li L, Rose P, Moore PK. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 10.Czyzewski BK, Wang DN. Nature. 2012;483:494–497. doi: 10.1038/nature10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson KR. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shatalin K, Shatalina E, Mironov A, Nudler E. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 14.Lippert AR, New EJ, Chang CJ. J Am Chem Soc. 2011;133:10078–10080. doi: 10.1021/ja203661j. [DOI] [PubMed] [Google Scholar]
- 15.Qian Y, Karpus J, Kabil O, Zhang SY, Zhu HL, Banerjee R, Zhao J, He C. Nat Commun. 2011;2:495. doi: 10.1038/ncomms1506. [DOI] [PubMed] [Google Scholar]
- 16.Chan J, Dodani SC, Chang CJ. Nat Chem. 2012;4:973–984. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galardon E, Roger T, Deschamps P, Roussel P, Tomas A, Artaud I. Inorg Chem. 2012;51:10068–10070. doi: 10.1021/ic300952d. [DOI] [PubMed] [Google Scholar]
- 18.Montoya LA, Pluth MD. Chem Commun. 2012;48:4767–4769. doi: 10.1039/c2cc30730h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin VS, Chang CJ. Curr Opin Chem Biol. 2012;16:595–601. doi: 10.1016/j.cbpa.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R. FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 21.Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M. Antioxid Redox Signal. 2010;13:157–192. doi: 10.1089/ars.2009.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.James BR. Pure Appl Chem. 1997;69:2213–2220. [Google Scholar]
- 23.Nicholls P, Kim JK. Biochim Biophys Acta, Bioenerg. 1981;637:312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- 24.Nicholls P, Kim JK. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 25.Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Biochem J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackstone E, Morrison M, Roth MB. Science. 2005;308:518–518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 27.Pietri R, Roman-Morales E, Lopez-Garriga J. Antiox Redox Signal. 2011;15:393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson EA. Biochim Biophys Acta, Protein Struct. 1970;207:30–40. doi: 10.1016/0005-2795(70)90134-0. [DOI] [PubMed] [Google Scholar]
- 29.Berzofsky JA, Peisach J, Blumberg WE. J Biol Chem. 1971;246:3367–3377. [PubMed] [Google Scholar]
- 30.Berzofsky JA, Peisach J, Horecker BL. J Biol Chem. 1972;247:3783–3791. [PubMed] [Google Scholar]
- 31.Strianese M, De Martino F, Pellecchia C, Ruggiero G, D’Auria S. Protein Pept Lett. 2011;18:282–286. doi: 10.2174/092986611794578378. [DOI] [PubMed] [Google Scholar]
- 32.Kraus DW, Wittenberg JB. J Biol Chem. 1990;265:16043–16053. [PubMed] [Google Scholar]
- 33.Kraus DW, Wittenberg JB, Lu JF, Peisach J. J Biol Chem. 1990;265:16054–16059. [PubMed] [Google Scholar]
- 34.Cerda-Colón JF, Silfa E, López-Garriga J. J Am Chem Soc. 1998;120:9312–9317. [Google Scholar]
- 35.Pietri R, Lewis A, León RG, Casabona G, Kiger L, Yeh SR, Fernandez-Alberti S, Marden MC, Cadilla CL, López-Garriga J. Biochemistry. 2009;48:4881–4894. doi: 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Román-Morales E, Pietri R, Ramos-Santana B, Vinogradov SN, Lewis-Ballester A, López-Garriga J. Biochem Biophys Res Commun. 2010;400:489–492. doi: 10.1016/j.bbrc.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.English DR, Hendrickson DN, Suslick KS, Eigenbrot CW, Scheidt WR. J Am Chem Soc. 1984;106:7258–7259. [Google Scholar]
- 38.Pavlik JW, Noll BC, Oliver AG, Schulz CE, Scheidt WR. Inorg Chem. 2010;49:1017–1026. doi: 10.1021/ic901853p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai L, Holm RH. J Am Chem Soc. 1994;116:7177–7188. [Google Scholar]
- 40.Tang SC, Koch S, Papaefthymiou GC, Foner S, Frankel RB, Ibers JA, Holm RH. J Am Chem Soc. 1976;98:2414–2434. doi: 10.1021/ja00425a008. [DOI] [PubMed] [Google Scholar]
- 41.Collman JP, Ghosh S, Dey A, Decréau RA. Proc Natl Acad Sci USA. 2009;106:22090–22095. doi: 10.1073/pnas.0904082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collman JP, Ghosh S. Inorg Chem. 2010;49:5798–5810. doi: 10.1021/ic100472p. [DOI] [PubMed] [Google Scholar]
- 43.Kuehn CC, Taube H. J Am Chem Soc. 1976;98:689–702. [Google Scholar]
- 44.Sellman D, Lechner P, Knoch F, Moll M. Angew Chem Int Ed. 1991;30:552–553. [Google Scholar]
- 45.Ma ESF, Rettig SJ, James BR. Chem Commun. 1999:2463–2464. [Google Scholar]
- 46.Jessop PG, Lee CL, Rastar G, James BR, Lock CJL, Faggiani R. Inorg Chem. 2002;31:4601–4605. [Google Scholar]
- 47.Chatwin SL, Diggle RA, Jazzar RFR, Macgregor SA, Mahon MF, Whittlesey MK. Inorg Chem. 2003;42:7695–7697. doi: 10.1021/ic030241i. [DOI] [PubMed] [Google Scholar]
- 48.Ma ESF, Rettig SJ, Patrick BO, James BR. Inorg Chem. 2012;51:5427–5434. doi: 10.1021/ic3004118. [DOI] [PubMed] [Google Scholar]
- 49.Galardon E, Daguet H, Deschamps P, Roussel P, Tomas A, Artaud I. Dalton Trans. 2013;42:2817–2821. doi: 10.1039/c2dt31758c. [DOI] [PubMed] [Google Scholar]
- 50.Rebouças JS, Patrick BO, James BR. J Am Chem Soc. 2012;134:3555–3570. doi: 10.1021/ja211226e. [DOI] [PubMed] [Google Scholar]
- 51.Rebouças JS, James BR. Inorg Chem. 2013;52:1084–1098. doi: 10.1021/ic302401m. [DOI] [PubMed] [Google Scholar]
- 52.Brewer C. J Chem Soc, Chem Commun. 1990:344–346. [Google Scholar]
- 53.Koch S, Tang SC, Holm RH, Frankel RB, Ibers JA. J Am Chem Soc. 1975;97:916–918. doi: 10.1021/ja00837a052. [DOI] [PubMed] [Google Scholar]
- 54.Collman JP, Sorrell TN, Hodgson KO, Kulshrestha AK, Strouse CE. J Am Chem Soc. 1977;99:5180–5181. doi: 10.1021/ja00457a049. [DOI] [PubMed] [Google Scholar]
- 55.Miller KM, Strouse CE. Acta Crystallogr, Sect C. 1984;40:1324–1327. [Google Scholar]
- 56.Nasri H, Haller KJ, Wang Y, Huynh Boi H, Scheidt WR. Inorg Chem. 1992;31:3459–3467. [Google Scholar]
- 57.Ueyama N, Nishikawa N, Yamada Y, Okamura TA, Nakamura A. Inorg Chim Acta. 1998;283:91–97. [Google Scholar]
- 58.Ueyama N, Nishikawa N, Yamada Y, Okamura TA, Oka S, Sakurai H, Nakamura A. Inorg Chem. 1998;37:2415–2421. [Google Scholar]
- 59.Sun WY, Zhang L, Gu Q. J Electroanal Chem. 1999;469:84–87. [Google Scholar]
- 60.Tani F, Matsu-ura M, Nakayama S, Naruta Y. Coord Chem Rev. 2002;226:219–226. [Google Scholar]
- 61.Xu N, Powell DR, Cheng L, Richter-Addo GB. Chem Commun. 2006;0:2030–2032. doi: 10.1039/b602611g. [DOI] [PubMed] [Google Scholar]
- 62.Das PK, Chatterjee S, Samanta S, Dey A. Inorg Chem. 2012;51:10704–10714. doi: 10.1021/ic3016035. [DOI] [PubMed] [Google Scholar]
- 63.Fleming I. Chem Soc Rev. 1981;10:83–111. [Google Scholar]
- 64.Hwu JR, Wetzel JM. J Org Chem. 1985;50:3946–3948. [Google Scholar]
- 65.Hwu JR, Wang N. Chem Rev. 1989;89:1599–1615. [Google Scholar]
- 66.Brinkman EA, Berger S, Brauman JI. J Am Chem Soc. 1994;116:8304–8310. [Google Scholar]
- 67.Hwu JR, Wong FF, Huang JJ, Tsay SC. J Org Chem. 1997;62:4097–4104. [Google Scholar]
- 68.Kabytaev KZ, Everett TA, Safronov AV, Sevryugina YV, Jalisatgi SS, Hawthorne MF. Eur J Inorg Chem. 2013;2013:2488–2491. [Google Scholar]
- 69.Evans DR, Reed CA. J Am Chem Soc. 2000;122:4660–4667. [Google Scholar]
- 70.Cheng RJ, Latos-Grazynski L, Balch AL. Inorg Chem. 1982;21:2412–2418. [Google Scholar]
- 71.Scheidt WR, Reed CA. Chem Rev. 1981;81:543–555. [Google Scholar]
- 72.A short Fe-S distance of 2.177(1) Å was reported for a low-spin six-coordinate iron(III) alkyl thiolate complex. See Suzuki NHT, Urano Y, Kikuchi K, Uekusa H, Ohashi Y, Uchida T, Kitagawa T, Nagano T. J Am Chem Soc. 1999;121:11571–11572.
- 73.Franke A, Hessenauer-Ilicheva N, Meyer D, Stochel G, Woggon WD, van Eldik R. J Am Chem Soc. 2006;128:13611–13624. doi: 10.1021/ja060650o. [DOI] [PubMed] [Google Scholar]
- 74.Byrn MP, Strouse CE. J Am Chem Soc. 1981;103:2633–2635. [Google Scholar]
- 75.Higuchi T, Uzu S, Hirobe M. J Am Chem Soc. 1990;112:7051–7053. [Google Scholar]
- 76.Huang YP, Kassner RJ. J Am Chem Soc. 1979;101:5807–5810. [Google Scholar]
- 77.Jones DH, Hinman AS. Can J Chem. 2000;78:1318–1324. [Google Scholar]
- 78.Green MT. J Am Chem Soc. 1999;121:7939–7940. [Google Scholar]
- 79.Salinger RM, West R. J Organomet Chem. 1968;11:631–633. [Google Scholar]
- 80.Swistak C, Mu XH, Kadish KM. Inorg Chem. 1987;26:4360–4366. [Google Scholar]
- 81.Shirazi A, Barbush M, Ghosh S, Dixon DW. Inorg Chem. 1985;24:2495–2502. [Google Scholar]
- 82.Wyllie GRA, Scheidt WR. Chem Rev. 2002;102:1067–1090. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 83.Franke A, Roncaroli F, van Eldik R. Eur J Inorg Chem. 2007:773–798. [Google Scholar]
- 84.Anzai K, Hatano K, Lee YJ, Scheidt WR. Inorg Chem. 1981;20:2337–2339. [Google Scholar]
- 85.Adler AD, Longo FR, Kampas F, Kim J. J Inorg Nucl Chem. 1970;32:2443–2445. [Google Scholar]
- 86.Otsuka T, Ohya T, Sato M. Inorg Chem. 1984;23:1777–1779. [Google Scholar]
- 87.Fleischer EB, Srivastava TS. J Am Chem Soc. 1969;91:2403–2405. [Google Scholar]
- 88.Crystal Clear. Rigaku/MSC Inc., Rigaku Corporation; The Woodlands, TX: 2005. [Google Scholar]
- 89.ABSCOR. Higashi; Rigaku Corporation; Tokyo, Japan: 1995. [Google Scholar]
- 90.Sheldrick GM. SHELXTL97: Program for Refinement of Crystal Structures. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 91.Sheldrick GM. Acta Crystallogr, Sect A. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.