

Background: The activity of PTEN tumor suppressor is tightly controlled.
Results: SUMOylation of PTEN enhanced by PIASxα regulates PTEN activity.
Conclusion: PIASxα is a novel SUMO E3 ligase to promote SUMOylation of PTEN.
Significance: PIASxα-mediated SUMOylation of PTEN has a central role in tumor inhibition.
Keywords: PI 3-Kinase (PI3K), PTEN, Sumoylation, Tumor, Ubiquitination, PIASxα, SUMO E3 Ligase, Tumor Inhibition
Abstract
The tumor suppressor PTEN plays a critical role in the regulation of multiple cellular processes that include survival, cell cycle, proliferation, and apoptosis. PTEN is frequently mutated or deleted in various human cancer cells to promote tumorigenesis. PTEN is regulated by SUMOylation, but the SUMO E3 ligase involved in the SUMOylation of PTEN remains unclear. Here, we demonstrated that PIASxα is a SUMO E3 ligase for PTEN. PIASxα physically interacted with PTEN both in vitro and in vivo. Their interaction depended on the integrity of phosphatase and C2 domains of PTEN and the region of PIASxα comprising residues 134–347. PIASxα enhanced PTEN protein stability by reducing PTEN ubiquitination, whereas the mutation of PTEN SUMO1 conjugation sites neutralized the effect of PIASxα on PTEN protein half-life. Functionally, PIASxα, as a potential tumor suppressor, negatively regulated the PI3K-Akt pathway through stabilizing PTEN protein. Overexpression of PIASxα led to G0/G1 cell cycle arrest, thus triggering cell proliferation inhibition and tumor suppression, whereas PIASxα knockdown or deficiency in catalytic activity abolished the inhibition. Together our studies suggest that PIASxα is a novel SUMO E3 ligase for PTEN, and it positively regulates PTEN protein level in tumor suppression.
Introduction
PTEN3 (phosphatase and tensin homologue deleted on chromosome TEN) (1) is a well defined tumor suppressor that plays a critical role in multiple cellular processes, such as cell proliferation, apoptosis, cell cycle arrest, and genomic stability maintenance (2–7). PTEN is reduced in expression, deleted, or mutated with high frequency in various types of human cancers and is considered to function as a tumor suppressor (8, 9). The classic function of PTEN is to inhibit the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway through its lipid phosphatase activity. PTEN dephosphorylates phosphatidylinositol 3,4,5-triphosphate and converts it to phosphatidylinositol 4,5-bisphosphate to antagonize PI3K activity (10, 11). In addition to its phosphatase activity for lipid, PTEN is also found to function as a protein phosphatase (12).
It has been demonstrated that the PTEN level in cells is crucial for predicting tumor susceptibility (8, 9). Indeed, several mechanisms are involved in the exquisite regulation of PTEN protein level in vivo. Several transcription factors have been identified to bind directly to the specific sites of PTEN promoter and regulate PTEN transcription, such as transcription factor EGR1, which up-regulates PTEN, and NFκB, which negatively regulates PTEN (13–18). At the post-translational level, PTEN is regulated by manifold modifications including oxidation, acetylation, phosphorylation, and ubiquitination. Monoubiquitination and polyubiquitination serve to regulate PTEN nuclear import and its degradation, respectively (19–23). Recently, PTEN has been reported to be SUMOylated at both Lys-254 and Lys-266 sites. SUMO1 modification of Lys-266 is mainly responsible for PTEN association with the plasma membrane and the inhibition of PI3K-Akt signaling pathway (24). However, it remains unclear which SUMO E3 ligase is involved in the SUMOylation of PTEN.
SUMO (small ubiquitin-related modifier) is structurally similar to ubiquitin (25). So far three SUMO family members, SUMO1, SUMO2, and SUMO3, have been identified to exist in mammals (26, 27). These SUMO homologs conjugate to the lysine residue in target protein mostly by recognizing the consensus sequence ψKX(D/E) (ψ is a hydrophobic amino acid, and X is any amino acid) (28). SUMOylation, analogous to ubiquitination, is catalyzed by a set of enzymes: E1-activating enzyme (Aos1 and Uba2), E2-conjugating enzyme (Ubc9), and E3 ligase (29, 30). To date, three different types of proteins have been suggested to have SUMO E3 ligase activity: PIAS (protein inhibitor of activated STAT), RanBP2, and Pc2 (31–33). The PIAS proteins were initially described to inhibit DNA binding and transcriptional activation by STAT proteins. In mammals five PIAS family members were identified, including PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy (34–36). The PIAS proteins, similar to ubiquitin E3 ligases, contain a RING domain that is required for their SUMO E3 ligase activity. In addition, the PIAS proteins contain a SAP domain and SUMO binding domain required for noncovalent binding to SUMO. The various SUMO E3 ligases select different target proteins properly and promote their SUMOylation efficiently.
In this study we investigated the effect of PIAS proteins on the SUMO1 modification of PTEN and found an intricate post-translational mechanism involved in regulating tumorigenesis. We demonstrated that PIASxα is a novel SUMO E3 ligase for PTEN. Specifically, PIASxα promoted the SUMO1 modification of PTEN by physically interacting with PTEN both in vivo and in vitro. The interaction between PIASxα and PTEN was dependent on the integrity of the phosphatase and C2 domains of PTEN and the region of PIASxα comprising residues 134–347. Then we further assessed the regulation of SUMOylation on PTEN. The SUMOylation of PTEN enhanced by PIASxα increased PTEN protein stability by reducing its ubiquitination. Thus, PIASxα inhibited PI3K-Akt pathway by up-regulating PTEN at post-translational level and caused cell cycle arrest and proliferation inhibition. Overexpression of PIASxα in tumor cells even inhibited their tumorigenesis in nude mice, whereas PIASxα knockdown or deficiency in catalytic activity abolished the inhibition. Together, our data suggest that PIASxα functions as a positive regulator of PTEN through promoting its SUMO1 modification and highlight the importance of PIASxα/PTEN in tumor-suppressive functions.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, and RNA Interference
The human cell lines HEK293T, HeLa, U2OS, PC-3, PC-3M-2B4, H1299, A549, HCT116, and MDA-MB-231 cells were procured from ATCC and cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 unit/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. Cells were transiently transfected with plasmid using TurboFect in vitro Transfection Reagent (Fermentas) following the manufacturer's protocol. 48 h after transfection, cells were harvested and lysed to evaluate the transfection efficiency. PIASxα target siRNA sequence was 5′-AAG ATA CTA AGC CCA CAT TTG-3′. The lentivirus vector pLL3.7-shPTEN expresses shRNA that targets PTEN mRNA (5′-AAG ATC TTG ACC AAT GGC TAA-3′).
Real-time PCR
Total RNA was isolated using the RNApure High-purity Total RNA Rapid Extraction kit (BioTeke) following the manufacturer's protocol. Then the cDNA was synthesized using ReverAid First Strand cDNA Synthesis kit (Fermentas) followed by real-time PCR analysis with Maxima SYBR Green qPCR Master Mix (Fermentas). The DNA sequences of the human PIASxα primers are 5′-CTCATCAAGCCCACGAGTTTAG-3′ and 5′-CCAGGCAAAGTCTCAACTGAA-3′. These primers result in a product of 169 bp. The DNA sequences of the human PTEN primers are 5′-TTTGAAGACCATAACCCACCAC-3′ and 5′-ATTACACCAGTTCGTCCCTTTC-3′. These primers result in a 134-bp product. The DNA sequences of the human p27Kip1 primers are 5′-AACGTGCGAGTGTCTAACGG-3′ and 5′-CCCTCTAGGGGTTTGTGATTCT-3′, and the amplicon size is 209 bp. The human GAPDH primers are 5′-CCATGGAGAAGGCTGGGG-3′ and 5′-CAAAGTTGTCATGGATGACC-3′ with a 195-bp product (37). GAPDH is applied as an internal control for normalizing the real-time PCR results.
Western Blot and Antibodies
The whole cell lysates for Western blot analysis were prepared in radioimmune precipitation assay buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) containing Protease Inhibitor Mixture (Amresco). After the insoluble part of the lysates was cleared by centrifugation, protein concentrations were measured by the BCA Protein Assay kit (Pierce). 25 μg of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membrane. The primary antibodies used for immunoblotting analysis were against FLAG (F1804, Sigma), HA (MMS-101P, Covance), GST (IT003M, Macgene), His (IT005M, Santa Cruz), PIAS1 (sc-8152, Santa Cruz), PIAS3 (sc-46682, Santa Cruz), PIASxα (sc-30879, Santa Cruz), PIASxβ (sc-18245, Santa Cruz), PIASy (sc-30875, Santa Cruz), PTEN (sc-6817-R, Santa Cruz), Phospho-Akt (Ser473) (4060, Cell Signaling Technology), Akt (4685, Cell Signaling Technology), p27Kip1 (554, B&M Biotech Co., Ltd.), GAPDH (KM9002, Sungene), SUMO1 (sc-5308, Santa Cruz), and ubiquitin (D058-3, B&M Biotech Co., Ltd.). The secondary antibodies anti-mouse IgG antibody IRDye 800 conjugated (610-132-121) and DyLight 800 conjugated affinity-purified anti-rabbit IgG (611-145-002) were purchased from Rockland.
Immunoprecipitation
Cells for immunoprecipitation assay were lysed in immunoprecipitation lysis buffer (25 mm Tris-HCl pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 5% glycerol) containing Protease Inhibitor Mixture (Amresco). The whole cell lysates obtained by centrifugation were incubated with specified antibodies and protein A-Sepharose (GE Healthcare) overnight at 4 °C with constant rotation. The immunocomplexes were then washed with immunoprecipitation lysis buffer three times, boiled in SDS sample buffer, and subjected to SDS-PAGE followed by Western blot analysis.
GST Pulldown Assay
The control GST and GST-tagged proteins were expressed in Escherichia coli strain BL21 (DE3). Then the bacterial lysates were prepared in ice-cold binding buffer (PBS) by sonication and incubated with glutathione-Sepharose beads (GE Healthcare) overnight at 4 °C with rocking. After the incubation, His-tagged proteins were added to each tube for 4 h at 4 °C. The beads were washed with binding buffer three times and eluted with elution buffer (50 mm Tris-HCl, pH 8.0) containing 10 mm reduced glutathione. The elution was separated by SDS-PAGE, and the interactions were analyzed by Western blot with specified antibody.
Purification of Recombinant Proteins
The His-tagged recombinant protein expression vectors pET-SUMO1, pET-Aos1&Uba2 (38), pET-Ubc9, pET-PIAS1, pET-PIAS3, pET-PIASxα, pET-PIASxβ, pET-PIASy, pET-p53, pET-K386R, and pET-PTEN were constructed on the base of pET-28b (+) vector. The vectors were transformed into E. coli strain BL21 (DE3), and recombinant protein expression was induced by 0.1 mm isopropyl-β-d-thiogalactoside at 30 °C for 8 h. After sonication, the bacterial lysates were incubated with Ni2+-Sepharose beads (GE Healthcare) overnight at 4 °C with rotation. Then the beads were washed with binding buffer (20 mm sodium phosphate, 0.5 m NaCl, 40 mm imidazole, pH 7.4) 3 times, and the purified proteins were eluted using elution buffer (20 mm sodium phosphate, 0.5 m NaCl, 500 mm imidazole, pH 7.4). The purification efficiency and protein concentration were determined using silver staining as described previously (39). The purified proteins were stored at −20 °C in small aliquots for further experiment.
In Vitro SUMO1 Conjugation Assay
His-tagged PTEN, SUMO1, Aos1&Uba2 (SUMO E1-activating enzyme) and Ubc9 (SUMO E2-conjugating enzyme) expressed in E. coli BL21 (DE3) were purified by Ni2+-Sepharose beads (GE Healthcare). Each in vitro SUMOylation reaction mixture contained 125 ng of PTEN, 1 μg of SUMO1, 250 ng of Aos1&Uba2, 500 ng of Ubc9, and 8 μl of 5× reaction buffer (100 mm Hepes, pH 7.5, 25 mm MgCl2, 125 mm NaCl, 1 mm DTT, 2 mm ATP); H2O was added to make the final volume of 40 μl. The reaction mixture was incubated at 37 °C for 1.5 h and stopped by adding SDS sample buffer. The reaction mixture was separated by SDS-PAGE and immunoblotted with anti-PTEN antibody to detect the SUMO1 modification level of PTEN.
In Vivo SUMO1 Conjugation Assay
HeLa cells were transfected with either control plasmid or PIASxα plasmid. 48 h after transfection, cells were harvested and lysed in SDS buffer (5% SDS, 0.15 m Tris-HCl, pH 6.7, 30% glycerol) diluted 1:3 in radioimmune precipitation assay buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) containing 10 mm iodoacetamide, 20 mm N-ethylmaleimide, and Protease Inhibitor Mixture (Amresco). Lysates were sonicated briefly and cleared by centrifugation. Then the cell lysates were diluted in PBS, 0.5% Nonidet P-40 before incubation with anti-PTEN antibody and Protein A-Sepharose (GE Healthcare) overnight at 4 °C. The beads were collected, washed 3 times with ice-cold PBS, 0.5% Nonidet P-40, and the antigen-antibody complexes were recovered by boiling in SDS sample buffer. The samples were subjected to Western blot with anti-PTEN antibody or anti-SUMO1 antibody.
In Vivo Ubiquitination Assay
HeLa cells were transfected with various plasmids as indicated in individual experiments. 36 h after transfection, cells were treated with 10 μm MG132 for 6 h, and the whole cell lysates were prepared by immunoprecipitation lysis buffer containing Protease Inhibitor Mixture were subjected to immunoprecipitation with anti-PTEN antibody. The immunoprecipitated PTEN were released from the beads by boiling in SDS sample buffer. The analysis of ubiquitination was carried out by immunoblotting with anti-ubiquitin antibody.
Protein Half-life Assay
HeLa cells were transfected with 3 μg/dish PTEN plasmid and 3 μg/dish PIASxα plasmid or control plasmid. 36 h after transfection, 100 μg/ml cycloheximide was added to the dishes, and the cycloheximide treatment was terminated at 0, 3, 6, 9, and 12 h time points as indicated. The whole cell lysates were made, and protein concentration was determined. Subsequently, 25 μg of total protein from each sample was analyzed by immunoblotting with anti-PTEN antibody. Quantification of PTEN protein level was determined using TotalLab software, normalized to GAPDH.
Flow Cytometry Assay
The cells transfected with various plasmids as indicated in individual experiment were washed with PBS when the confluency was 70–80%. After digestion with 0.25% trypsin, cells were fixed with 70% ethanol overnight at 4 °C. The cells were resuspended in PBS buffer treated with 150 μg/ml RNase A (Sigma) at 37 °C for 30 min. Then the cells were stained with 25 μg/ml propidium iodide (PI) in the dark at 4 °C for 30 min. The cell cycle was measured using the FACScan flow cytometry system (BD Biosciences).
MTT (3-[4, 5-Dimethylthyazol-2-yl]-2,5-diphenyltetrazolium Bromide) Assay
HeLa and U2OS cells were stably transfected with either control vector or PIASxα vector. After puromycin selection, cells were seeded into 96-well plates at a density of 1000 cell/well. After culturing for 1, 2, 3, 4, 5, 6, 7, or 8 days, 20 μl of MTT solution (5 mg/ml) was added to each well followed by further incubation at 37 °C for 4 h. Medium was removed, and 150 μl of DMSO was added to each well to dissolve the formazan crystals. The absorbance at 490 nm was read using the microplate reader.
Soft Agar Colony Formation Assay
Single-cell suspensions of 1.5–3 × 104 cells were plated per 60-mm dish in 3 ml of DMEM containing 10% FBS and 0.35% agar on a layer of 5 ml of the same medium containing 0.7% agar. 1–2 weeks after culture, the colonies were stained with 0.05% crystal violet in PBS. Photographs were taken, and the number and size of colonies were determined by TotalLab software (40).
Tumorigenicity in Nude Mice
HeLa and U2OS cells were stably transfected with either control vector or PIASxα vector. After puromycin selection, 3 × 106 cells were suspended in 200 μl of PBS and subcutaneously injected into the left or right hind leg of 6-week-old female nude mice. 3–4 weeks after injection, the mice were killed, the tumors were weighed, and the size was measured. Each cell subline was evaluated in three different animals.
Statistical Analysis
The data are expressed as the mean ± S.E. (standard error of the mean) from an appropriate number of experiments as indicated in the figure legends. The statistical analysis was done by using Student's t test, and p < 0.01 or 0.05 was considered significant.
RESULTS
PTEN Is Modified by SUMO1 in Vitro
To determine whether PTEN is modified by SUMO1 in vitro, we established an in vitro system for SUMO1 modification as described previously (41). In this system the substrates were incubated with assay mix containing SUMO1, Aos1&Uba2 (E1-activating enzyme), and Ubc9 (E2-conjugating enzyme) in the presence of ATP. The inspection of p53 protein indicated the presence of a sequence (F-K386-T-E) that fits the conserved motif (ψKX(D/E)) of SUMO conjugation, and it has been reported that p53 is modified by SUMO1 at lysine 386 (42, 43), so we took wild-type p53 and its point mutant K386R (change lysine of 386 to arginine) as controls in SUMO1 modification in vitro system (Fig. 1B). First, we purified reaction reagents His-SUMO1, His-Aos1&Uba2, and His-Ubc9 with Ni2+-Sepharose beads. Western blot was carried out to analyze the protein purification efficiency (Fig. 1A). The substrates of His-p53 and His-K386R were also tagged with HA at C-terminal (Fig. 1C). The purified HA-tagged p53 or Lys-386 were incubated with SUMO1, Aos1&Uba2, and Ubc9 in the absence or presence of ATP followed by Western blot analysis with anti-HA antibody. As shown in Fig. 1D, in the absence of ATP, p53 was not modified by SUMO1 (p53 band was detected between 55 and 72 kDa), whereas in the presence of ATP, p53 was modified by SUMO1 (SUMO1-p53 band was detected between 72 and 95kDa). But either way, in the absence or presence of ATP, K386R was not modified by SUMO1. This result indicated that p53 was modified by SUMO1 at Lys-386 in vitro, which confirmed the previous result (42, 43). The established system can be used to test the SUMO1 modification in vitro.
FIGURE 1.

PTEN is modified by SUMO1 in vitro. A, purified proteins used for SUMOylation assay in vitro. His-tagged proteins were expressed in E. coli strain BL21 (DE3) and purified with Ni2+-Sepharose. The purified proteins were detected by Western blot with anti-His antibody. The protein ladder in the left lane was used to evaluate the molecular size of purified proteins. B, schematic representation of wild-type p53 and its point mutant K386R. p53 is modified by SUMO1 at lysine 386 site. C, the wild-type p53 and its mutant K386R were purified and subjected to Western blot. D, the SUMOylation assay in vitro was carried out to detect SUMO1 modification of p53 and K386R. The assay mix contained SUMO1, Aos1&Uba2 (E1-activating enzyme), Ubc9 (E2-conjugating enzyme), and ATP. p53 and K386R were used as the substrates. The reaction mix was detected by Western blot with anti-p53 antibody. E, schematic diagram of SUMO1 modification sites in PTEN. PTEN is modified by SUMO1 at lysine 254 and 266 sites. F, the wild-type PTEN was purified and subjected to Western blot. G, SUMO1 modification of PTEN in vitro relies on every component in the assay mix. The SUMOylation assay in vitro was performed with components added as indicated. The reaction mix was incubated and subjected to Western blot to detect the SUMO1 modification of PTEN. H, PTEN was incubated with assay mix containing SUMO1 or SUMO2. The reaction mix was subjected to Western blot with anti-PTEN antibody. I, PTEN or its point mutant K254R, K266R, or K254R/K266R were incubated with assay mix separately and then subjected to Western blot.
PTEN protein contains two sequences (IKVE, Lys at 254; LKKD, Lys at 266) that fit the conserved motif (ψKX(D/E)) of SUMO conjugation (Fig. 1E). It was reported that PTEN can be SUMOylated in cells and there are two SUMO1 modification sites, lysine 254 and lysine 266 (Fig. 1E) (24). Next, we tried to test whether PTEN can be SUMOylated in vitro with the established system. The purified HA-tagged PTEN (Fig. 1F) were incubated with SUMO1, Aos1&Uba2, and Ubc9 in the absence or presence of ATP, then immunoblotted against HA tag. As presented in Fig. 1G, the band detected between 72 and 95 kDa was SUMO1-modified PTEN in the presence of ATP. The covalent bonding of SUMO1 to PTEN was an ATP-consuming reaction dependent on the catalysis of Aos1&Uba2 and Ubc9. Taken all together, we confirmed that PTEN is definitely modified by SUMO1 in vitro with our established system. At the same time, we took SUMO2 as a control and assessed the SUMO2 modification of PTEN. The data indicated that only slight amount of SUMO2-modified PTEN, compared with SUMO1-modified PTEN, was detected (Fig. 1H). It has been reported that PTEN is heavily conjugated with SUMO1 at both Lys-254 and Lys-266 sites (24). Based on our observation and the report, our studies were focused on SUMO1 modification of PTEN. To confirm this result, we evaluated the SUMO1 modification of point mutants (K254R, K266R, and K254R/K266R) of PTEN. As shown in Fig. 1I, the double mutant K254R/K266R completely abolished SUMOylation, whereas the single mutants K254R or K266R greatly reduced SUMOylation in comparison with wild-type PTEN. However, we did not observe a shift in PTEN from 55 to 95 kDa, which is presumably conjugated with two molecules of SUMO1: (SUMO1)2-PTEN. The reason is that the SUMO1 protein contains more than 90 residues, and the requirement of space near the modified site is much larger than for other posttranslational modifications (for example, methylation, acetylation, phosphorylation), leading to the impossibility of dual SUMOylation of adjacent Lys-254 and Lys-266. Our data are consistent with the previous studies (24).
PIASxα Promotes SUMO1 Modification of PTEN in Vitro
In the SUMOylation process, the SUMO E3 ligases interact with both the E2-conjugating enzyme and the substrate, bringing the two together and thereby increasing the efficiency of SUMO conjugation (44, 45). Next, we asked if any known SUMO E3 ligase could enhance the SUMO1 modification of PTEN. We expressed His-tagged SUMO E3 ligase PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy in E. coli strain BL21 (DE3) and purified these proteins with Ni2+-Sepharose beads. Western blot analysis with anti-His antibody shown in Fig. 2A indicated the purification efficiency of the SUMO E3 ligases. In the SUMO1 modification in vitro system, different concentrations of PIAS3 were incubated with p53 in the presence of SUMO1, Aos1&Uba2, and Ubc9 as the positive control (46, 47). The result confirmed that the SUMO E3 ligase PIAS3 enhances SUMO1 modification of p53 in a dose-dependent manner (Fig. 2B).
FIGURE 2.

PIASxα stimulates SUMO1 modification of PTEN in vitro. A, the purified His-tagged recombinant proteins (SUMO E3 ligase) used for SUMOylation assay in vitro. His-tagged PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy were expressed in E. coli strain BL21 (DE3) and purified with Ni2+-Sepharose. The purified proteins were detected by Western blot against the His tag. Protein ladder in the left lane was used to evaluate the molecular size of purified proteins, and arrows denoted the different PIASs. B, PIAS3 promotes SUMOylation of p53 in vitro. PIAS3 and p53 were purified and incubated with the assay mix containing SUMO1, Aos1&Uba2 (E1-activating enzyme), and Ubc9 (E2-conjugating enzyme) either in the absence or presence of ATP. PIAS3 was added at concentrations of 0, 125, and 250 ng. C, the effect of PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy on SUMOylation of PTEN in vitro. Different PIASs were expressed and purified then incubated with PTEN in assay mix. The reaction products were separated by SDS-PAGE and immunoblotted with anti-PTEN antibody. The arrow denotes that a high level SUMOylation of PTEN was detected in assay mix containing PIASxα. D, PIASxα enhances SUMOylation of PTEN in a dose-dependent manner. Different concentrations of PIASxα were incubated with PTEN in the assay mix. Then the samples were analyzed by Western blot with anti-PTEN antibody. PIASxα was added at concentrations of 0, 50, 150, and 450 ng. E, PIASxα and its deletion mutant ΔRING were incubated with PTEN in assay mix and then subjected to Western blot.
To further find out which one of the PIAS proteins can stimulate PTEN SUMO1 modification in vitro, we performed the SUMO1 modification assay in the presence of PIASs. We incubated PTEN with PIAS1, PIAS3, PIASxα, PIASxβ, or PIASy in the presence of SUMO1, Aos1&Uba2, and Ubc9 and carried out Western blot to detect the SUMO1 modification of PTEN. Among all of the various PIAS proteins, PIASxα was able to facilitate the conjugation of SUMO1 to PTEN more significantly (Fig. 2C). To further confirm the SUMO1 modification assay result, we incubated PTEN with 0, 50, 150, or 450 ng of PIASxα in the presence of SUMO1, Aos1&Uba2, and Ubc9. Western blot results indicated that the SUMO E3 ligase PIASxα efficiently stimulated SUMO1 modification of PTEN in a dose-dependent manner in vitro (Fig. 2D). Previous studies indicated that the RING domain is indispensible to the catalytic activity of PIASxα (48, 49). Then we constructed the deletion mutant ΔRING to examine whether the catalytic activity of PIASxα is important for its role in SUMOylation. As presented in Fig. 2E, PIASxα deletion mutant ΔRING expectedly failed to stimulate SUMO1 modification of PTEN.
PIASxα Promotes SUMO1 Modification of PTEN in Vivo
Using the in vitro SUMO1 modification system described above, we have demonstrated that PTEN is modified by SUMO1 and PIASxα stimulates SUMO1 modification of PTEN in vitro. Next, we addressed the issue as to whether the same result could be shown with the SUMO1 modification assay in vivo. Before verifying this hypothesis, we first checked the endogenous protein level of PIASxα in several tumor cell lines, such as PC-3, PC-3M-2B4, H1299, A549, HCT116, MDA-MB-231, U2OS, and HeLa cells. The whole cell lysates were extracted and subjected to Western blot with anti-PIASxα antibody. As shown in Fig. 3A, left panel, the results indicated that the PIASxα was expressed at low levels in these tumor cell lines, whereas HeLa cells overexpressing PIASxα were used as a positive control. To confirm this result, we carried out real-time PCR to check the mRNA level of PIASxα in the various tumor cells (Fig. 3A, middle panel). We found that the mRNA level of PIASxα was also low in the tumor cells. Furthermore, we also evaluated the protein levels of PIAS1, PIAS3, PIASxβ and PIASy in these cancer cell lines by carrying out Western blot (Fig. 3A, right panel). The results showed that the protein levels were low. Together, our data indicated that the five members of PIAS family, as specific SUMO E3 ligases, were expressed at low levels in tumor cells.
FIGURE 3.

PIASxα promotes SUMO1 modification of PTEN in vivo. A, the whole cell lysates from several different tumor cell lines, PC-3, PC-3M-2B4, H1299, A549, HCT116, MDA-MB-231, U2OS, and HeLa, were subjected to Western blot with anti-PIASxα antibody to check the endogenous protein level of PIASxα. HeLa cells overexpressing PIASxα were used as positive control. The mRNA level of PIASxα in tumor cells was evaluated by real-time PCR analysis, normalized to the mRNA level of GAPDH (middle panel). The expression levels of PIAS1, PIAS3, PIASxβ, and PIASy in these cancer cell lines were evaluated by Western blot analysis as well (right panel). B, HeLa cells were transfected with either control vector or PIASxα. 48 h after transfection cells were harvested and lysed. The whole cell lysates were subjected to immunoprecipitation with anti-PTEN antibody. The immunoprecipitation (IP) products of PTEN were detected by Western blot (IB) with anti-PTEN antibody. Western blot results with short exposure (left panel) and long exposure (right panel) were shown. C, the same nitrocellulose membrane used above was stripped. After stripping, the membrane was detected with anti-SUMO1 antibody. Western blot results with short exposure (left panel) and long exposure (right panel) were shown in the same way. D, HeLa cells transfected with FLAG-SUMO1, HA-PTEN, and control vector or PIASxα were subjected to an in vivo SUMO1 conjugation assay described above (left panel). The cells in the right panel were transfected with FLAG-SUMO1, HA-PTEN, and control siRNA or PIASxα siRNA.
Next we took advantage of a HeLa cell line overexpressing PIASxα to perform a in vivo SUMO1 modification assay. HeLa cells were transfected with either control plasmid or PIASxα plasmid. 48 h after transfection, cells were lysed and subjected to immunoprecipitation with anti-PTEN antibody. The immunoprecipitated PTEN were released from the beads and separated by SDS-PAGE. Western blot analysis with anti-PTEN antibody (Fig. 3B) indicated that an additional band between 72 and 95 kDa was detected and enhanced by PIASxα overexpression. Judging from the molecular mass of PTEN (between 55 and 72 kDa), we considered the additional band to represent SUMO1-modified PTEN. To assess that this additional slower-migrating band above PTEN was SUMO1-PTEN, we had the same nitrocellulose membrane stripped and immunoblotted with anti-SUMO1 antibody. A Western blot result as shown in Fig. 3C confirmed that the additional slower-migrating band above PTEN was SUMO1-modified PTEN.
To further confirm our findings, we overexpressed or knocked down PIASxα by siRNA in HeLa cells transiently overexpressing FLAG-tagged SUMO1 and HA-tagged PTEN. As shown in Fig. 3D, PIASxα overexpression enhanced the conjugation of FLAG-SUMO1 to HA-PTEN, whereas PIASxα knockdown caused a decrease in the SUMO1 modification of PTEN.
PIASxα and PTEN Interact with Each Other Both in Vivo and in Vitro
It has been established that PIASxα enhances SUMO1 modification of PTEN as a SUMO E3 ligase in vitro and in vivo from the data described above, and it has been reported that PTEN associates with SUMO E2-conjugating enzyme Ubc9 in cells (50). With this background, we assumed that PIASxα and PTEN might interact with each other. To investigate this possibility, we first carried out the co-immunoprecipitation experiment. Cells were transfected with either control plasmid or FLAG-PIASxα plasmid, and the whole cell lysates were subjected to immunoprecipitation with anti-PIASxα antibody followed by Western blot analysis against PTEN. The result indicated that PIASxα interacted with PTEN in HeLa cells (Fig. 4A). The reciprocal co-immunoprecipitation analysis with anti-PTEN antibody was conducted, and the immunoprecipitated complex was immunoblotted against PIASxα. As shown in Fig. 4B, PTEN also interacted with PIASxα in HeLa cells.
FIGURE 4.

PIASxα and PTEN interact with each other both in vivo and in vitro. A, co-immunoprecipitation of PIASxα with PTEN from HeLa cells. Cells were transfected with FLAG-PIASxα. 48 h after transfection, cells were harvested and lysed. The whole cell lysates were immunoprecipitated with either control IgG or anti-PIASxα antibody, and co-immunoprecipitated (IP) PTEN was subsequently detected by Western blot (IB) with anti-PTEN antibody (top panel). The same samples were immunoblotted against PIASxα to determine co-immunoprecipitation efficiency (bottom panel). B, the interaction of PTEN with PIASxα in cells was further confirmed by reciprocal experiment analysis using co-immunoprecipitation as described above. C, GST-tagged PIASxα pulls down PTEN in vitro. A GST pulldown assay was carried out using immobilized control GST or GST-tagged PIASxα on glutathione-Sepharose followed by incubation with extracts prepared from E. coli strain BL21 (DE3) expressing His-PTEN. The interaction of PIASxα with PTEN was assessed by Western blot against the His tag (top panel). The same samples were immunoblotted with anti-GST antibody to evaluate GST pulldown efficiency (bottom panel). D, the interaction of PTEN with PIASxα in vitro was further confirmed by reciprocal experiment analysis. A GST pulldown assay was carried out as described above.
Next, we performed the GST pulldown assay to identify whether PTEN and PIASxα physically interact with each other directly in vitro. The purified protein GST or GST-PIASxα were incubated with the bacterially expressed PTEN in vitro and subjected to the GST pulldown assay. As shown in Fig. 4C, a Western blot result indicated that GST-PIASxα but not GST alone pulled down PTEN in vitro. Subsequently, we conducted the reciprocal GST pulldown assay to further test the interaction of PIASxα and PTEN in vitro. The assay confirmed that GST-PTEN also pulled down PIASxα in vitro (Fig. 4D). In summary, it can be concluded that PIASxα physically interacts with PTEN as a SUMO E3 ligase both in vivo and in vitro.
The Interaction between PIASxα and PTEN Depends on the Integrity of Phosphatase, C2 Domains of PTEN, and the Region of PIASxα Comprising Residues 134–347
The full-length PTEN has four domains that are critically important for its function, and they are phosphatase domain, C2 domain, CT domain, and PDZ binding domain. Based on the observation that PIASxα and PTEN interacted with each other directly, we further tried to define the structural requirements for their interaction. To test this possibility we carried out the GST pulldown assay with full-length PTEN and its various generated deletion mutants lacking different function domains (Fig. 5A). To map the PIASxα binding region on PTEN, we incubated GST, GST-PTEN, GST-M1 (1–187), GST-M2 (1–350), GST-M3 (1–400), GST-M4 (188–403), or GST-M5 (351–403) with bacterially expressed PIASxα in vitro followed by immunoblotting against PIASxα. As shown in Fig. 5B, the deletion mutants containing either phosphatase domain (M1) or C2 domain (M4) alone reduced the ability of interacting with PIASxα, whereas the double deletion mutant (M5) abolished the ability completely. The result indicated that the integrity of both the phosphatase domain and C2 domain is essential to the interaction of PTEN and PIASxα.
FIGURE 5.

The interaction between PIASxα and PTEN depends on phosphatase and C2 domains of PTEN but not on SAP, RING, and SUMO binding domains of PIASxα. A, schematic representation of N-terminal GST-tagged full-length PTEN (FL) along with its various deletion mutants (M1, M2, M3, M4, and M5). The full-length PTEN has four domains (phosphatase, C2, CT, and PDZ binding domains). B, a GST pulldown assay was carried out to determine the domain of PTEN essential for its interaction with PIASxα. The protein control GST, full-length PTEN, and its deletion mutants immobilized on glutathione-Sepharose were incubated with extracts prepared from E. coli stain BL21 (DE3) expressing His-tagged PIASxα. Then the interaction was assessed by a Western blot (IB) with anti-PIASxα antibody (top panel). GST pulldown efficiency was evaluated by immunoblotting with anti-GST antibody (bottom panel). C, schematic diagram of N-terminal GST-tagged full-length PIASxα (FL) and its generated deletion mutants (M1, M2, M3, M4, and M5). The known PIASxα domains (SAP, RING, and SUMO binding domains) are indicated in dark gray. D, the interactions of full-length PIASxα and its deletion mutants with PTEN were determined by GST pulldown assay. The experiment procedure was carried out as described above. PTEN was detected by Western blot with anti-PTEN antibody (top panel), and immunoblotting against GST-tag was to evaluate GST pulldown efficiency (bottom panel).
Next, we tried to delimit the regions of PIASxα responsible for its interaction with PTEN. According to the structural function domains of PIASxα, SAP domain, RING domain, and SUMO binding domain, we constructed GST-tagged full-length PIASxα and a series of its deletion mutants (Fig. 5C). For the GST pulldown assay, the purified proteins GST, GST-PIASxα, GST-M1 (134–572), GST-M2 (1–418), GST-M3 (1–347), GST-M4 (134–418), and GST-M5 (Δ134–347) were incubated with bacterially expressed PTEN in vitro. After the incubation, we carried out Western blot with anti-PTEN antibody. As shown in Fig. 5D, the deletion mutants of PIASxα, lacking the SAP domain, RING domain, or SUMO binding domain, were still able to pull down PTEN in vitro. We further delimited the structural requirement of PIASxα for its interaction with PTEN within a region comprising residues 134–347. Therefore, these observations strongly suggested that the interaction between PIASxα and PTEN depends on the integrity of phosphatase, C2 domains of PTEN, and the region of PIASxα comprising residues 134–347.
The SUMOylation of PTEN Enhanced by PIASxα Promotes PTEN Protein Stability by Reducing PTEN Ubiquitination
Similar to the post-translational modification by ubiquitin, the SUMO modification regulates protein degradation and stabilization. In some cases SUMOylation stabilizes protein by decreasing its ubiquitination. There has been evidence that SUMO can act as an antagonist of ubiquitin, as SUMO-modified IκBα and PCNA are resistant to proteasome-mediated degradation (51, 52). Because PIASxα interacts with PTEN and stimulates its SUMO1 modification, it is now a matter of interest to check whether PIASxα can enhance PTEN protein stability by reducing its ubiquitination. We performed in vivo ubiquitination assay in HeLa cells. We overexpressed control vector, FLAG-SUMO1, or FLAG-PIASxα in HeLa cells. 36 h after transfection, cells were treated with MG132 for 6 h and lysed. The whole cell lysates were directly subjected to Western blot with anti-ubiquitin antibody to evaluate the total ubiquitination level in cells. As shown in Fig. 6A, overexpression of SUMO1 or PIASxα had no effect on the total ubiquitination level in cells. To further determine PTEN ubiquitination level, the whole cell lysates were immunoprecipitated with anti-PTEN antibody and analyzed by immunoblotting against ubiquitin. As shown in Fig. 6B, PTEN ubiquitination level was reduced by overexpression of SUMO1 or PIASxα. The ubiquitination of PTEN regulates its proteasome-mediated degradation. To examine the rate of PTEN degradation, protein half-life assay was performed. As shown in Fig. 6C, PTEN protein level was detected at the indicated time points. The result showed that overexpression of PIASxα caused an increase in PTEN protein half-life (Fig. 6C, upper panels), whereas PIASxα knockdown led to diminished PTEN protein half-life (Fig. 6C, bottom panels).
FIGURE 6.

PIASxα enhances PTEN protein stability by reducing PTEN ubiquitination level. A, overexpression of SUMO1 and PIASxα has no effect on the global ubiquitination level in cells. HeLa cells were transfected with control vector, FLAG-SUMO1, or FLAG-PIASxα. 36 h post-transfection, cells were treated with MG132 (10 μm) for 6 h and harvested. The whole cell lysates were subjected to Western blot with anti-ubiquitin antibody. The protein expression was confirmed by immunoblotting using anti-FLAG antibody. B, SUMO1 and PIASxα reduce ubiquitination of PTEN. HeLa cells were transfected with control vector, SUMO1, or PIASxα. 36 h after transfection cells were treated with 10 μm MG132 for 6 h. Subsequently, the cells were harvested and lysed. The PTEN ubiquitination level was evaluated by immunoprecipitation (IP) with anti-PTEN antibody followed by anti-ubiquitin immunoblotting (IB). C, PIASxα overexpression increases PTEN stability (top panels). HeLa cells were transfected with either control vector or FLAG-tagged PIASxα. 36 h post-transfection, cells were treated with 100 μg/ml cycloheximide (CHX) and collected at the indicated time points. Then immunoblotting against FLAG, PTEN, and GAPDH was performed. Quantification of PTEN protein level was determined using TotalLab software normalized to GAPDH. PIASxα knockdown with siRNA technology in HeLa cells followed by Western blot analysis was carried out in the bottom panels. D, schematic diagram of wild-type PTEN and its generated point mutants (K254R, K266R, and K254R/K266R). All of them were fused with a HA tag at both the N and C termini. E, mutation at both Lys-254 and Lys-266 of PTEN increases PTEN ubiquitination level. HeLa cells were cotransfected with FLAG-PIASxα and HA-PTEN, HA-K254R, HA-K266R, or HA-K254R/K266R. Then the ubiquitination of wild-type PTEN and its generated point mutants was assessed by carrying out the experiment procedure described above. F, protein half-life assay of wild-type PTEN and its point mutant K254R/K266R. HeLa cells transiently expressing FLAG-tagged PIASxα were transfected with either HA-PTEN or HA-K254R/K266R. 36 post-transfection cells were subjected to protein half-life assay described above. Quantification of relative PTEN and K254R/K266R protein level was shown in the right panel. G, HeLa cells transfected with HA-PTEN, HA-K254R, HA-K266R, or HA-K254R/K266R were subjected to immunoprecipitation with anti-HA antibody followed by Western blot against Ser(P) (pSer) and Thr(P) (pThr; left panel). The SUMO1 modification of PTEN and its deletion mutant ΔC-terminal were assessed by SUMO1 conjugation assay (right panel).
It has been known that PTEN is modified by SUMO1 at both lysine 254 and lysine 266 sites in vivo (24), so we raised the question of whether the SUMOylation at Lys-254 and Lys-266 sites could interfere PTEN ubiquitination. Based on the wild-type PTEN overexpression plasmid, we constructed its point mutant plasmids K254R, K266R, and K254R/K266R with overlap PCR (Fig. 6D). The HA-tagged plasmid PTEN, K254R, K266R, or K254R/K266R were cotransfected with FLAG-tagged PIASxα overexpression plasmid into HeLa cells. 36 h after transfection cells were treated with MG132 for 6 h and lysed. The whole cell lysates were subjected to immunoprecipitation with anti-HA antibody followed by Western blot against ubiquitin. The result showed that the mutation of either Lys-254 or Lys-266 site did not increase PTEN ubiquitination level, but the mutation at both Lys-254 and Lys-266 sites significantly increased PTEN ubiquitination level (Fig. 6E). To further determine the effect of mutation on protein degradation, we also conducted a protein half-life assay in HeLa cells. As shown in Fig. 6F, the mutation of PTEN at both Lys-254 and Lys-266 sites caused a decrease in PTEN protein half-life. The phosphorylation of PTEN in CT domain, which is mainly phosphorylated on Ser and Thr, also has the function to stabilize PTEN (53). To further figure out the relationship between SUMOylation and phosphorylation of PTEN, we evaluated the phosphorylation level of wild type PTEN and its various point mutants without SUMO1-modified sites, respectively (Fig. 6G). The result indicated that SUMO1 modification of PTEN had no effect on its phosphorylation. In addition, we constructed a CT deletion mutant (deficiency in phosphorylation) and performed in vitro SUMO1 conjugation assay with the mutant. The data also indicated that phosphorylation deficiency of PTEN did not interfere with its SUMO1 modification. Together, we concluded that there is no functional interaction between phosphorylation of PTEN in the CT domain and SUMO1 modification of PTEN protein.
In sum, our data indicate that the SUMOylation of PTEN enhanced by PIASxα increases PTEN protein stability by reducing PTEN ubiquitination, and the integrity of SUMO1-modified sites (Lys-254/266) is required for sustaining PTEN ubiquitination level. We demonstrated that there is no functional relationship between phosphorylation of PTEN in CT domain and SUMO1 modification of PTEN.
PIASxα Inhibits PI3K-Akt Pathway by Up-regulating the PTEN Protein Level
PTEN acts as a potent tumor suppressor that negatively regulates the PI3K-Akt pathway. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate and converts it to phosphatidylinositol 3,4,5-triphosphate. In turn, phosphatidylinositol 3,4,5-triphosphate accumulation at the cellular membrane results in the recruitment of Akt, leading to Akt activation by phosphorylating Akt. PTEN, as a lipid phosphatase, inhibits the PI3K-Akt pathway by dephosphorylating phosphatidylinositol 3,4,5-triphosphate to phosphatidylinositol 4,5-bisphosphate. Therefore, PIASxα, being a SUMO E3 ligase and a positive regulator of PTEN, might inhibit the PI3K-Akt pathway by regulating the PTEN protein level. To test this possibility we assessed the effect of PIASxα overexpression and knockdown on the PI3K-Akt pathway. HeLa cells with PIASxα-overexpressing or knockdown were harvested, and the whole cell lysates were subjected to Western blot with antibodies as indicated in Fig. 7A. Consistent with our previous results, PIASxα overexpression caused an increase in endogenous PTEN. The phosphorylation of Akt at serine 473 site was decreased in PIASxα overexpression cells without any detectable change in the total Akt protein level. p27Kip1, as a target of the PI3K-Akt pathway and a cyclin-dependent kinase inhibitor, was found to be increased in PIASxα overexpression cells. And PIASxα knockdown by siRNA resulted in opposite effects. To further confirm that PIASxα caused an increase in PTEN and p27Kip1 protein levels but not mRNA levels, we conducted the real-time PCR to detect the specific mRNA levels. As shown in Fig. 7B, the results indicated that mRNA levels of PTEN and p27Kip1 were not increased in PIASxα overexpression cells.
FIGURE 7.

PIASxα inhibits PI3K-Akt signaling by up-regulating PTEN protein level. A, Western blot showing the inhibition of PI3K-Akt signaling by PIASxα. HeLa cells were transfected with either control vector or FLAG-PIASxα. 48 h after transfection the whole cell lysates were subjected to Western blot with anti-FLAG, anti-PTEN, anti-pAkt (Ser-473), and anti-p27Kip1 antibodies. Akt and GAPDH were detected by immunoblotting as controls (left panel). The cells transfected with either control siRNA or PIASxα siRNA were lysed and immunoblotted with the antibodies indicated above (right panel). B, real-time PCR analysis of PIASxα, PTEN, and p27Kip1 mRNA levels. HeLa cells were transfected with either control vector or PIASxα. 48 h post-transfection cells were harvested, and RNA was extracted. Then the cDNA was synthesized and subjected to real-time PCR to check the mRNA levels of PIASxα, PTEN, and p27Kip1, normalized to the mRNA level of GAPDH. Results are representative of three independent experiments, and values are the mean ± S.E. *, p < 0.01. C, flow cytometry analysis of HeLa and U2OS cells transfected with either control vector or PIASxα. 48 h after transfection cells were stained with PI, and DNA contents were measured by flow cytometry to evaluate the effect of PIASxα on cell cycle. The percentages of each cell type at the G0/G1, S, and G2/M phases are shown in panels as the mean ± S.E. *, p < 0.01; **, p < 0.05. The PIASxα and p27Kip1 protein levels were assessed by Western blot. D, PC-3 cells (PTEN-null) transfected with control or PIASxα plasmid were lysed and subjected to Western blot with antibodies indicated above. E, HeLa-shPTEN cell line (PTEN-null) was established by using lentivirus transfection and infection system. A Western blot was carried out to assess knockdown efficiency of PTEN (left panel). HeLa-Vector and HeLa-shPTEN cell lines transiently transfected with FLAG-PIASxα were subjected to Western blot (middle panels). The effect of PIASxα on the cell cycle in HeLa-shPTEN cells was evaluated by flow cytometry analysis (right panel).
Considering the significant role of p27Kip1 in the progression of cell cycle, as a cyclin-dependent kinase inhibitor, we performed a flow cytometry assay to determine the effect of PIASxα on the cell cycle in HeLa and U2OS cells. Cells transiently expressing PIASxα were stained with propidium iodide, and the distribution of cells in G0/G1, S, and G2/M phases of the cell cycle was measured by the FACScan flow cytometry system. The overexpression of PIASxα induced G0/G1 cell cycle arrest in HeLa and U2OS cells compared with the control cells (Fig. 7C).
To confirm the direct connection between PIASxα and PTEN, we applied a human prostate cancer cell line (PC-3) deficient for PTEN. As shown in Fig. 7D, we found that in PC-3 cells, which are PTEN null, PIASxα overexpression had no effect on PI3K-Akt pathway. This indicates that PIASxα inhibits PI3K-Akt pathway by definitely regulating PTEN protein level. Furthermore, we established the PTEN knockdown cell line with lentivirus vector pLL3.7-shPTEN and came to the same result that PIASxα was not able to decrease phosphorylation of Akt in HeLa-shPTEN cells (Fig. 7E, middle panels). The flow cytometry assay also indicated that PTEN played a critical role in PIASxα-mediated G0/G1 cell cycle arrest (Fig. 7E, right panel).
PIASxα Causes Cell Proliferation Inhibition and Tumor Suppression in a PTEN-dependent Manner
With the finding that PIASxα inhibits the PI3K-Akt pathway through positively regulating PTEN SUMOylation, we further tried to determine the effect of PIASxα on cell proliferation. First, MTT assay was carried out to determine the impact of PIASxα on cell growth in the four cell lines, such as HeLa, U2OS, PC-3, and HeLa-shPTEN cells. As shown in Fig. 8A, stable overexpression of PIASxα resulted in a decreased rate of cell proliferation in HeLa and U2OS cells, whereas there was no change in PC-3 and HeLa-shPTEN cells when compared with that in control cells. Second, soft agar colony formation assay was performed to measure the anchorage-independent growth of the tumor cells overexpressing PIASxα. As shown in Fig. 8B, both colony number and colony size were decreased in HeLa and U2OS cells stably expressing PIASxa compared with those in control cells, whereas no significant changes in PC-3 and HeLa-shPTEN cells were shown. Collectively, these results indicated that PIASxα, as an inhibitor of PI3K-Akt pathway, caused cell growth inhibition by regulating PTEN protein level. Therefore, PIASxα functions as a potential tumor suppressor in a PTEN-dependent manner. Furthermore, the tumor suppression potential of PIASxα was also supported by our tumorigenicity in nude mice experiments (Fig. 8C). Nude mice injected with HeLa or U2OS cells overexpressing PIASxα showed reduced tumor growth rate when compared with those injected with the cells transfected with control plasmid, whereas no significant change was found in nude mice injected with HeLa-shPTEN cells overexpressing PIASxα. Based on our findings that PIASxα deletion mutant ΔRING failed to stimulate SUMO1 modification of PTEN, we further examined whether the catalytic activity domain of PIASxα is important for its tumor suppressor activity. As shown in Fig. 8C, right panels, overexpression of ΔRING failed to suppress tumor growth in nude mice. Our data indicate that PIASxα, as a SUMO E3 ligase for PTEN, might be a potential tumor suppressor to prevent tumorigenesis.
FIGURE 8.
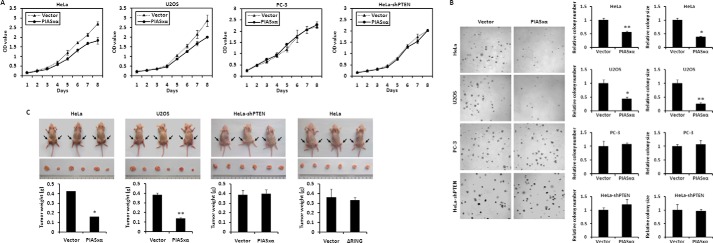
PIASxα causes cell proliferation and tumorigenesis inhibition in a PTEN-dependent manner. A, HeLa, U2OS, PC-3, and HeLa-shPTEN cells were stably transfected with either control vector or PIASxα. After puromycin selection, growth rates of cells were measured by MTT assay. OD, optical density. B, soft agar colony formation assay of HeLa, U2OS, PC-3, and HeLa-shPTEN cells stably transfected with control vector and PIASxα. Cells were cultured in soft agar for 1–2 weeks. The colonies were stained with 0.05% crystal violet, and the photographs of the stained colonies were taken. The number and size of colonies in three different microscope fields were determined by TotalLab software and are shown as the mean ± S.E. *, p < 0.01; **, p < 0.05. C, HeLa, U2OS, and HeLa-shPTEN cells stably transfected with control plasmid or PIASxα plasmid were subcutaneously injected into the left or right hind leg of three nude mice. Four weeks after injection the tumors were weighed, and size was measured. Data are shown as the mean ± S.E., n = 3. *, p < 0.01; **, p < 0.05). HeLa cells overexpressing PIASxα deletion mutant ΔRING were also subjected to the assay of tumorigenicity in nude mice.
DISCUSSION
The activity of the PTEN tumor suppressor protein is regulated by post-translational modifications, such as phosphorylation, acetylation, or ubiquitination. In addition, covalent attachment of the ubiquitin-like modifier SUMO appears to modulate PTEN activity. SUMOylation proceeds via an enzymatic pathway that is mechanistically analogous to ubiquitination but requires a different E1-activating enzyme and a SUMO-specific E2-conjugating enzyme. Here, we show that one member of the PIAS family, PIASxα, acts as specific E3 ligase that promotes SUMOylation of PTEN in vitro and in vivo.
PIASxα Is a SUMO E3 Ligase for PTEN
Our work is consistent with a model in which PIASxα regulates PI3K-Akt signaling pathway and cell proliferation by enhancing PTEN SUMOylation (see the model in Fig. 9). PIASxα is a SUMO E3 ligase for PTEN. It activates SUMOylation of PTEN. The SUMO1 modification of PTEN enhances its protein stability by protecting PTEN from ubiquitin modification and proteasome-mediated degradation. These conclusions are supported by the following experimental results. (i) PTEN is definitely modified by SUMO1 at Lys-254 and Lys-266 with our established system (Fig. 1). (ii) PIASxα stimulates SUMO1 modification of PTEN in a dose-dependent manner. The catalytic activity of PIASxα (RING domain) is indispensable for its role in SUMOylation (Figs. 2 and 3). (iii) PIASxα physically interacts with PTEN as a SUMO E3 ligase both in vivo and in vitro. The integrity of both the phosphatase domain and the C2 domain of PTEN and the region comprising residues 134–347 of PIASxα is essential to the interaction of PTEN and PIASxα. Actually, the physical interaction of PIASxα with PTEN is a prerequisite for promoting SUMO1 modification of PTEN (Figs. 4 and 5). (iv) PIASxα enhances PTEN protein stability by reducing PTEN ubiquitination, and the integrity of SUMO1-modified sites (Lys-254/266) is required for sustaining PTEN ubiquitination level. SUMO1 modification of PTEN has no effect on its phosphorylation in CT domain. There is no functional interaction between phosphorylation and SUMO1 modification of PTEN protein (Fig. 6). (v) PIASxα-mediating PTEN protein level results in the down-regulation of the PI3K-Akt pathway and, consequently, suppression of anchorage-independent cell proliferation and tumor growth in vivo (Figs. 7 and 8).
FIGURE 9.
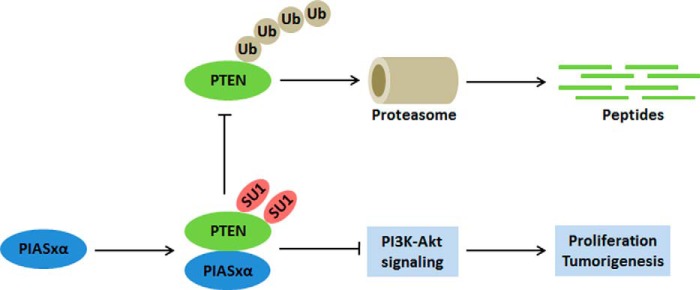
Model for the regulation of PTEN by PIASxα and its role in cell proliferation inhibition. In tumor cells such as HeLa and U2OS, PIASxα is expressed at low levels. A portion of PTEN is modified by ubiquitin. The polyubiquitination decreases PTEN protein level through ubiquitin-mediated proteasomal degradation. After overexpression of PIASxα in tumor cells, PIASxα interacts with PTEN and promotes its SUMO1 modification. The SUMOylation stabilizes PTEN by antagonizing its ubiquitination. The overexpression of PIASxα negatively regulates PI3K-Akt signaling pathway and inhibits tumor cell proliferation by up-regulating PTEN.
SUMOylation involves a three-enzyme cascade: a single E1-activating enzyme, a SUMO-specific E2-conjugating enzyme, and a substrate-specific E3 ligase. E3 ligase binds both the target protein and the E2 enzyme to facilitate SUMO conjugation. SUMOylation regulates several aspects of a target protein including protein stability, subnuclear localization, transcriptional activity, and protein-protein interactions. It has been reported that PTEN is SUMOylated at Lys-254/266 (24), but the molecular mechanisms underlying PTEN SUMOylation are unknown. In this study we demonstrate that PIASxα enhances SUMO1 modification of PTEN as a SUMO E3 ligase. PIASxα promotes PTEN protein stability by reducing PTEN ubiquitination, which indicates the existence of cross-talk between PTEN SUMOylation and ubiquitination. The integrity of SUMO1-modified sites (Lys-254/266) is also required for sustaining the PTEN ubiquitination level. These results suggest that the SUMOylation-ubiquitination interaction plays a critical role in the regulation of PTEN degradation and stability.
PTEN is heavily phosphorylated on Ser and Thr in the C-terminal region by a series of kinases, such as RhoA-associated kinase, glycogen synthase kinase 3β, and casein kinase 2, which regulates protein stability and function in cells. A reasonable question was raised of whether there is a relationship between phosphorylation and SUMO1 modification of PTEN. As shown in Fig. 6G, our data indicate that there is no functional interaction between phosphorylation and SUMO1 modification of PTEN.
PIASxα Is a Potential Tumor Inhibitor
The model in Fig. 9 also represents that PIASxα-enhanced PTEN SUMOylation inhibits PI3K-Akt signaling pathway and thus triggers G0/G1 cell cycle arrest, cell proliferation inhibition, and tumor suppression. Tumor suppressor PTEN participates in regulating multiple important cellular processes, such as cell cycle, cell proliferation, and tumorigenesis. Molecularly, PTEN acts as a tumor suppressor by negatively regulating PI3K-Akt signaling pathway. The overexpression of PIASxα caused an increase in PTEN protein level by stabilizing PTEN. Next we investigated whether PIASxα, a SUMO E3 ligase for PTEN, was involved in these cellular processes by regulating PTEN. Indeed, our findings indicated that PIASxα was able to negatively regulate PI3K-Akt signaling pathway through up-regulating PTEN protein level. Furthermore, we found that overexpression of PIASxα caused G0/G1 cell cycle arrest, cell proliferation, and tumor inhibition both in HeLa and U2OS cells (proficient for PTEN) but no influence in PC-3 and HeLa-shPTEN cells (deficient for PTEN). We also showed that catalytic activity of PIASxα (RING domain) is indispensable for its tumor suppression. The results indicate that PIASxα causes G0/G1 arrest, proliferation, and tumorigenesis inhibition in a PTEN-dependent manner.
All these data suggest that PIASxα, acting as a SUMO E3 ligase for PTEN, has the potential to function as a novel tumor suppressor by positively regulating PTEN. The PIASxα-PTEN interaction provides a new perspective on regulating of PTEN, which warrants future studies. The studies in this paper are the first steps toward understanding this circuitry, of which the PIASxα-mediated regulation of PTEN has a central role in tumor inhibition. Our studies underscore the need to elevate PIASxα level as part of therapeutic regiments to improve cancer prognosis.
Acknowledgments
We thank Dr. Amy Yee for providing the plasmids of the lentivirus infection system. We also thank Dr .Yali Dou for critical reading of the manuscript.
This work was supported by National Natural Science Foundation of China Grants 81170320 and 81230008 and Beijing Natural Science Foundation Grant 5122023.
- PTEN
- phosphatase and tensin homologue deleted on chromosome TEN
- PIAS
- protein inhibitor of activated STAT
- SUMO
- small ubiquitin-related modifier
- IB
- immunoblotted
- Ub
- ubiquitin
- PI
- propidium iodide
- MTT
- 3-[4,5-dimethylthyazol-2-yl]-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Steck P. A., Pershouse M. A., Jasser S. A., Yung W. K., Lin H., Ligon A. H., Langford L. A., Baumgard M. L., Hattier T., Davis T., Frye C., Hu R., Swedlund B., Teng D. H., Tavtigian S. V. (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 [DOI] [PubMed] [Google Scholar]
- 2. Stambolic V., Suzuki A., de la Pompa J. L., Brothers G. M., Mirtsos C., Sasaki T., Ruland J., Penninger J. M., Siderovski D. P., Mak T. W. (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 [DOI] [PubMed] [Google Scholar]
- 3. Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K. M. (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280, 1614–1617 [DOI] [PubMed] [Google Scholar]
- 4. Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P. P. (1998) Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 [DOI] [PubMed] [Google Scholar]
- 5. Sun H., Lesche R., Li D. M., Liliental J., Zhang H., Gao J., Gavrilova N., Mueller B., Liu X., Wu H. (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 6199–6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weng L. P., Brown J. L., Eng C. (2001) PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 10, 599–604 [DOI] [PubMed] [Google Scholar]
- 7. Shen W. H., Balajee A. S., Wang J., Wu H., Eng C., Pandolfi P. P., Yin Y. (2007) Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170 [DOI] [PubMed] [Google Scholar]
- 8. Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., Puc J., Miliaresis C., Rodgers L., McCombie R., Bigner S. H., Giovanella B. C., Ittmann M., Tycko B., Hibshoosh H., Wigler M. H., Parsons R. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 [DOI] [PubMed] [Google Scholar]
- 9. Teng D. H., Hu R., Lin H., Davis T., Iliev D., Frye C., Swedlund B., Hansen K. L., Vinson V. L., Gumpper K. L., Ellis L., El-Naggar A., Frazier M., Jasser S., Langford L. A., Lee J., Mills G. B., Pershouse M. A., Pollack R. E., Tornos C., Troncoso P., Yung W. K., Fujii G., Berson A., Steck P. A. (1997) MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 57, 5221–5225 [PubMed] [Google Scholar]
- 10. Maehama T., Taylor G. S., Dixon J. E. (2001) PTEN and myotubularin. Novel phosphoinositide phosphatases. Annu. Rev. Biochem. 70, 247–279 [DOI] [PubMed] [Google Scholar]
- 11. Luo J., Manning B. D., Cantley L. C. (2003) Targeting the PI3K-Akt pathway in human cancer. Rationale and promise. Cancer Cell 4, 257–262 [DOI] [PubMed] [Google Scholar]
- 12. Tamura M., Gu J., Takino T., Yamada K. M. (1999) Tumor suppressor PTEN inhibition of cell invasion, migration, and growth. Differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 59, 442–449 [PubMed] [Google Scholar]
- 13. Virolle T., Adamson E. D., Baron V., Birle D., Mercola D., Mustelin T., de Belle I. (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 3, 1124–1128 [DOI] [PubMed] [Google Scholar]
- 14. Patel L., Pass I., Coxon P., Downes C. P., Smith S. A., Macphee C. H. (2001) Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Curr. Biol. 11, 764–768 [DOI] [PubMed] [Google Scholar]
- 15. Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T. W. (2001) Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325 [DOI] [PubMed] [Google Scholar]
- 16. Shen Y. H., Zhang L., Gan Y., Wang X., Wang J., LeMaire S. A., Coselli J. S., Wang X. L. (2006) Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J. Biol. Chem. 281, 7727–7736 [DOI] [PubMed] [Google Scholar]
- 17. Xia D., Srinivas H., Ahn Y. H., Sethi G., Sheng X., Yung W. K., Xia Q., Chiao P. J., Kim H., Brown P. H., Wistuba I. I., Aggarwal B. B., Kurie J. M. (2007) Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NFκB-dependent pathway. J. Biol. Chem. 282, 3507–3519 [DOI] [PubMed] [Google Scholar]
- 18. Hettinger K., Vikhanskaya F., Poh M. K., Lee M. K., de Belle I., Zhang J. T., Reddy S. A., Sabapathy K. (2007) c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ. 14, 218–229 [DOI] [PubMed] [Google Scholar]
- 19. Trotman L. C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N. P., Carver B. S., Cordon-Cardo C., Erdjument-Bromage H., Tempst P., Chi S. G., Kim H. J., Misteli T., Jiang X., Pandolfi P. P. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X., Trotman L. C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument-Bromage H., Tempst P., Cordon-Cardo C., Pandolfi P. P., Jiang X. (2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Themsche C., Leblanc V., Parent S., Asselin E. (2009) X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 284, 20462–20466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maddika S., Kavela S., Rani N., Palicharla V. R., Pokorny J. L., Sarkaria J. N., Chen J. (2011) WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 13, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahmed S. F., Deb S., Paul I., Chatterjee A., Mandal T., Chatterjee U., Ghosh M. K. (2012) The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J. Biol. Chem. 287, 15996–16006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang J., Yan J., Zhang J., Zhu S., Wang Y., Shi T., Zhu C., Chen C., Liu X., Cheng J., Mustelin T., Feng G. S., Chen G., Yu J. (2012) SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 3, 911. [DOI] [PubMed] [Google Scholar]
- 25. Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J. (1998) Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 280, 275–286 [DOI] [PubMed] [Google Scholar]
- 26. Melchior F. (2000) SUMO–nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 16, 591–626 [DOI] [PubMed] [Google Scholar]
- 27. Müller S., Hoege C., Pyrowolakis G., Jentsch S. (2001) SUMO, ubiquitin's mysterious cousin. Nat. Rev. Mol. Cell Biol. 2, 202–210 [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez M. S., Dargemont C., Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]
- 29. Desterro J. M., Thomson J., Hay R. T. (1997) Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 417, 297–300 [DOI] [PubMed] [Google Scholar]
- 30. Gill G. (2003) Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr. Opin. Genet. Dev. 13, 108–113 [DOI] [PubMed] [Google Scholar]
- 31. Kahyo T., Nishida T., Yasuda H. (2001) Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol. Cell 8, 713–718 [DOI] [PubMed] [Google Scholar]
- 32. Pichler A., Gast A., Seeler J. S., Dejean A., Melchior F. (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120 [DOI] [PubMed] [Google Scholar]
- 33. Kagey M. H., Melhuish T. A., Wotton D. (2003) The polycomb protein Pc2 is a SUMO E3. Cell 113, 127–137 [DOI] [PubMed] [Google Scholar]
- 34. Sturm S., Koch M., White F. A. (2000) Cloning and analysis of a murine PIAS family member, PIASγ, in developing skin and neurons. J. Mol. Neurosci. 14, 107–121 [DOI] [PubMed] [Google Scholar]
- 35. Kotaja N., Aittomäki S., Silvennoinen O., Palvimo J. J., Jänne O. A. (2000) ARIP3 (androgen receptor-interacting protein 3) and other PIAS (protein inhibitor of activated STAT) proteins differ in their ability to modulate steroid receptor-dependent transcriptional activation. Mol. Endocrinol. 14, 1986–2000 [DOI] [PubMed] [Google Scholar]
- 36. Shuai K. (2000) Modulation of STAT signaling by STAT-interacting proteins. Oncogene 19, 2638–2644 [DOI] [PubMed] [Google Scholar]
- 37. Ramakrishnan S., Eppenberger U., Mueller H., Shinkai Y., Narayanan R. (1998) Expression profile of the putative catalytic subunit of the telomerase gene. Cancer Res. 58, 622–625 [PubMed] [Google Scholar]
- 38. Tatham M. H., Jaffray E., Vaughan O. A., Desterro J. M., Botting C. H., Naismith J. H., Hay R. T. (2001) Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276, 35368–35374 [DOI] [PubMed] [Google Scholar]
- 39. Yan J. X., Wait R., Berkelman T., Harry R. A., Westbrook J. A., Wheeler C. H., Dunn M. J. (2000) A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis 21, 3666–3672 [DOI] [PubMed] [Google Scholar]
- 40. Kakuguchi W., Kitamura T., Kuroshima T., Ishikawa M., Kitagawa Y., Totsuka Y., Shindoh M., Higashino F. (2010) HuR knockdown changes the oncogenic potential of oral cancer cells. Mol. Cancer Res. 8, 520–528 [DOI] [PubMed] [Google Scholar]
- 41. Sarge K. D., Park-Sarge O. K. (2009) Detection of proteins sumoylated in vivo and in vitro. Methods Mol. Biol. 590, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodriguez M. S., Desterro J. M., Lain S., Midgley C. A., Lane D. P., Hay R. T. (1999) SUMO-1 modification activates the transcriptional response of p53. EMBO J. 18, 6455–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gostissa M., Hengstermann A., Fogal V., Sandy P., Schwarz S. E., Scheffner M., Del Sal G. (1999) Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 18, 6462–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sapetschnig A., Rischitor G., Braun H., Doll A., Schergaut M., Melchior F., Suske G. (2002) Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO J. 21, 5206–5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mabb A. M., Wuerzberger-Davis S. M., Miyamoto S. (2006) PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 8, 986–993 [DOI] [PubMed] [Google Scholar]
- 46. Schmidt D., Müller S. (2003) PIAS/SUMO. New partners in transcriptional regulation. Cell. Mol. Life Sci. 60, 2561–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu S. Y., Chiang C. M. (2009) p53 sumoylation. Mechanistic insights from reconstitution studies. Epigenetics 4, 445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kotaja N., Vihinen M., Palvimo J. J., Jänne O. A. (2002) Androgen receptor-interacting protein 3 and other PIAS proteins cooperate with glucocorticoid receptor-interacting protein 1 in steroid receptor-dependent signaling. J. Biol. Chem. 277, 17781–17788 [DOI] [PubMed] [Google Scholar]
- 49. Rytinki M. M., Kaikkonen S., Pehkonen P., Jääskeläinen T., Palvimo J. J. (2009) PIAS proteins. Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 66, 3029–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waite K. A., Eng C. (2003) BMP2 exposure results in decreased PTEN protein degradation and increased PTEN levels. Hum. Mol. Genet. 12, 679–684 [PubMed] [Google Scholar]
- 51. Desterro J. M., Rodriguez M. S., Hay R. T. (1998) SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 2, 233–239 [DOI] [PubMed] [Google Scholar]
- 52. Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]
- 53. Tamguney T., Stokoe D. (2007) New insights into PTEN. J. Cell Sci. 120, 4071–4079 [DOI] [PubMed] [Google Scholar]