

Abstract
The human immunodeficiency virus type 1 (HIV-1) integrase (IN) protein plays an important role during the early stages of the retroviral life cycle and therefore is an attractive target for therapeutic intervention. We immunized rabbits with HIV-1 IN protein and developed a combinatorial single-chain variable fragment (scFv) library against IN. Five different scFv antibodies with high binding activity and specificity for IN were identified. These scFvs recognize the catalytic and C-terminal domains of IN and block the strand-transfer process. Cells expressing anti-IN–scFvs were highly resistant to HIV-1 replication due to an inhibition of the integration process itself. These results provide proof-of-concept that rabbit anti-IN–scFv intrabodies can be designed to block the early stages of HIV-1 replication without causing cellular toxicity. Therefore, these anti-IN–scFvs may be useful agents for “intracellular immunization”-based gene therapy strategies. Furthermore, because of their epitope binding characteristics, these scFvs can be used also as new tools to study the structure and function of HIV-1 IN protein.
Keywords: intracellular antibodies, single-chain variable fragment, HIV-1 integrase protein, HIV-1 neutralization, gene therapy
1. Introduction
The spread of human immunodeficiency virus type 1 (HIV-1) has been dramatic since the early 1980s, when the virus was discovered to be the causative agent of AIDS [1],[2]. Over the past two decades, researchers worldwide have sought to develop small-molecule inhibitors to target essential steps in the viral cycle [3]. Highly active antiretroviral therapy (HAART), one of the most widely used treatment regimens, employs a combination of therapeutic agents that target the viral reverse transcriptase (RT) and protease (PR) enzymes [3],[4]. In the developed world, access to HAART has led to significant reductions in the morbidity and mortality attributed to HIV/AIDS. However, the emergence of drug-resistant virus isolates is causing an increasingly detrimental impact on disease outcome [5],[6]. As a result, there is a pressing need to identify and develop new therapies that can be effective against virus isolates resistant to HAART [7],[8].
The HIV-1 integrase (IN) protein is currently the focus of an intense research effort to develop improved anti-HIV-1 drugs [9–11]. This enzyme catalyzes the integration of the HIV genome into the chromosome of the host cell, arguably the most insidious step in the infection process [12]. The HIV-1 IN consists of a 288 amino-acid (aa) protein (32 kDa) encoded at the 3′-end of the HIV pol gene and it contains three distinct domains: N-terminal, catalytic core, and the C-terminal [12–14]. The N-terminal domain (aa 1–49) is believed to be involved in protein multimerization and contains a histidine–histidine–cysteine–cysteine motif that coordinates zinc binding [13],[15–18]. The central or catalytic core domain (aa 50–212) contains a highly conserved triad of acidic residues D64, D116, and E152 (DDE motif) that are involved in catalysis [12],[19],[20]. Mutation of any of these acidic residues abolishes HIV-1 IN activity [13],[20]. The C-terminal domain (aa 213–288), the least conserved of the three protein domains, binds the viral DNA ends and might also contribute to the binding of chromosomal DNA during integration [20–23]. All three canonical IN structural domains may be involved in extensive protein–DNA and protein–protein interactions as similarly described for a retroviral IN from prototype foamy virus (PFV), a close relative of HIV-1 [24]. In this case, the structural data reveal that the retroviral intasome, the minimal functional complex involving viral DNA and IN, comprises an IN tetramer tightly associated with a pair of viral DNA ends [24].
During the past 10 years, several IN inhibitors have been developed to block the integration step [9–11],[25]. However, progress in the development of IN inhibitors has been slow; most of these compounds have not met the minimum standards required to be defined as lead molecules in the search for clinically useful applications or have been toxic in cell cultures [9–11],[25]. Recently, the first IN inhibitor, MK-0518 (ral-tegravir), received US Food and Drug Administration approval and entered clinical use [26]. Although a second compound, GS-9137 (JTK-303, elvitegravir) is in clinical trials [27], new agents and therapeutic approaches must continue to be identified and developed to block this crucial step in HIV-1 replication.
In recent years, gene therapy has been considered in treatment of HIV/AIDS, either as an alternative or as a complement to antiretroviral chemotherapy [28–30]. Intracellular antibodies (intrabodies) represent a new class of neutralizing molecules with potential use in gene therapy approaches [31–33]. An in-trabody is an antibody designed to be expressed intracellularly and directed to a particular subcellular compartment where it can exert its function [34–36]. Intrabodies can be expressed in several forms. The most commonly used format consists of a single-chain variable fragment (scFv) in which the variable domain of the heavy chain (VH) is connected to the light chain (VL) through a peptide linker. The specificity and affinity of the parent antibody are preserved in this process [34],[35]. The binding of an intrabody to its molecular target has the potential to block, suppress, alter, or even enhance the process mediated by that molecule [37–39].
A number of murine monoclonal antibodies (mAbs) directed against HIV-1 IN have been raised and characterized by several groups [40–48]. Some of these mAbs have been cloned into scFvs and tested as intrabodies [45],[46]. Although some of these block HIV-1 replication, they are all derived from murine immunoglobulin G molecules and were generated by hybridoma technology. The rabbit antibody repertoire has been used for decades in diagnostic applications in the form of polyclonal antibodies, and it offers an attractive alternative to the murine antibody repertoire for the generation of therapeutic mAbs. Rabbits, which belong to the order Lagomorpha (lagomorphs), are evolutionarily distant from mice and rats, which belong to the order Rodentia (rodents) [49]. As a result, epitopes that might be invisible to rodent mAbs and also human mAbs generated from transgenic mice with human immunoglobulin genes, can often be recognized by rabbit antibodies. Moreover, as was previously demonstrated, rabbit antibodies can be converted to humanized antibodies that retain both high specificity and affinity for the antigen [50],[51]. We have recently demonstrated that rabbit antibody fragments against viral proteins can be highly expressed in reducing environments of mammalian cells and efficiently block viral functions [52–54].
In the present study, we immunized rabbits with HIV-1 IN and generated a combinatorial scFv library against IN. We identified five different anti-IN–scFvs with high binding activity and specificity for IN. These scFvs bound to the catalytic and C-terminal domains of IN and inhibited the strand-transfer process without affecting the 3′-end processing reaction. Notably, cells expressing intrabodies were highly resistant to HIV-1 infection due to an inhibition of the integration process itself. These findings provide proof-of-principle that rabbit anti-IN intrabodies can be designed to block early stages of HIV-1 replication. Therefore, these intrabodies might be useful agents for “intra-cellular immunization” and will prove useful as tools to probe the structure and function of HIV-1 IN.
2. Materials and methods
2.1. Plasmids
The plasmid coding for HIV-1NL4-3 was obtained from the AIDS Research and Reference Reagent Program (Germantown, MD, USA). Plasmid pComb3X was derived from pComb3H [55]. The envelope plasmid pMD.G was kindly provided by Dr. Didier Trono. Plasmid pcDNA3.1/Zeo(+) and pBR322 were obtained from Invitrogen (Carlsbad, CA, USA). pMX-KRAB8FPBS2puro is a retroviral vector derived from pMX-puro, which comprises the 5′ long terminal repeat (LTR) and 3′ LTR regions, lacks the complete gag, env, and pol genes, and contains a puromycin-selectable marker [56]. The pMX-KRAB8FPBS2puro vector expresses a single bicistronic message for the translation of the 8FPBS2 zinc finger protein and additionally appends a hemagglutinin (HA) tag [57]. Plasmid pNde675 was previously described and is derived from pCR2.1 (Invitrogen) [58].
2.2. Antibodies and cell culture
The following antibodies were used: anti-M13–horseradish peroxidase (HRP) mAb (Roche Diagnostics GmbH, Basel, Switzerland), anti-HA–HRP mAb (Roche Diagnostics GmbH), rhodamine-conjugated anti-HA mAb (Roche Diagnostics GmbH). Anti-IN polyclonal antibody (#7375, #756, and #758) and anti-p24 mAb were from the AIDS Research and Reference Reagent Program. 293T cells (American Type Culture Collection), Gag-Pol 293 cells (Clontech, Palo Alto, CA, USA), and HeLa CD4 LTR-β-Gal cells (AIDS Research and Reference Reagent Program) were maintained in Dulbecco’s modified Eagle medium (DMEM) and Jurkat cells were maintained in Roswell Park Memorial Institute 1640 medium. Media were supplemented with 10% fetal calf serum (FCS), antibiotics (100 units/mL penicillin, and 100 μg/mL streptomycin), and 2 mM l-glutamine. Stable Jurkat clones expressing anti-IN–scFvs were maintained in the presence of puromycin (1 μg/mL). All cell cultures were maintained at 37 °C in 5% CO2. Tissue culture media and reagents were from Lonza (Basel, Switzerland).
2.3. Rabbit immunization
Two New Zealand white rabbits were treated with four subcutaneous injections containing 200 μg of purified HIV-1 IN protein in a 1-mL emulsion of adjuvant (Ribi Immunochem Research, Hamilton, MT, USA). The injections were administered at 2–3 week intervals. Antisera from the immune animals was analyzed for binding to HIV-1 IN protein by enzyme-linked immunosorbent assay (ELISA) using HRP-conjugated goat antirabbit Fc poly-clonal antibodies as secondary antibodies (Pierce, Rockford, IL, USA). Five days after the final boost, spleen and bone marrow from one leg were harvested and used for total RNA preparation with TRI-REAGENT (Invitrogen) according to the manufacturer’s protocol.
2.4. cDNA synthesis, antibody library construction, and phage display
First-strand cDNA was synthesized using the Superscript Pre-amplification System with oligo(dT) priming (Invitrogen). Specific oligonucleotide primers covering all known variable rabbit antibody family sequences were used to amplify VH and VL gene segments separately [55]. The purified variable region products were then assembled into scFv format by overlapping PCR. An 18 aa linker fragment (SSGGGGSGGGGGGSSRSS) was used to connect the VL and VH fragments. The final DNA fragments encoding a library of scFv antibody fragments (VL–linker–VH) were gel purified, digested with SfiI, and cloned into the appropriately cut phagemid vector pComb3X containing a suppressor stop codon and sequences encoding peptide tags for purification (6-His) and detection (HA). The recombinant phagemid was introduced into competent Escherichia coli ER2538 cells by electroporation. The phage library displaying the scFvs was panned against immobilized HIV-1 IN antigen using a solid-phase protocol [55]. One hundred and fifty scFv phage clones from the final output were randomly selected for ELISA to evaluate their binding activity against HIV-1 IN. HRP-conjugated anti-M13 mAb (Roche Diagnostics GmbH) was used for detection. All clones with a significant signal above background were further analyzed by DNA fingerprinting and sequencing.
2.5. Expression and purification of anti-IN antibody fragments
To express and purify selected anti-IN–scFvs, phagemid DNA was transformed into nonsuppressor E. coli strain TOP10F. A fresh colony of each clone was grown at 37 °C overnight in Super Broth (SB) medium containing 100 μg/mL of ampicillin. A 10 mL sample of cells was used to inoculate 1 L of SB medium containing 100 μg/mL of ampicillin. Cells were grown at 37 °C until A600 nm reached 0.9. scFv expression was induced by the addition of 0.5 mM isopropyl-1-thio-β-d-galactoside and growth was continued for 18 H. After induction, bacteria were harvested by centrifugation (4,000g, 4 °C, 15 Min) and resuspended in 50 mL equilibration buffer (20 mM NaH2PO4, 500 mM NaCl, 30 mM imidazole, and pH 7.4) supplemented with PR inhibitors (Roche Diagnostics GmbH). Cells were lysed by sonication. Centrifugation (14,000g, 4 °C, 30 Min) was used to remove cellular debris, and the supernatant was filtered through a 0.2-μm syringe filter. All chromatographic steps were performed at 4 °C. First, scFv extracts were purified by nickel chelate affinity chromatography using the C-terminal His tag. Bound proteins were eluted with a linear imidazole gradient from 0 to 300 mM imidazole in 20 mM NaH2PO4 and 0.5 M NaCl (pH 7.4). The appropriate fractions were pooled, dialyzed against 20 mM Tris (pH 8.0), 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and loaded onto a Mono Q anion exchange column (Amersham Biosciences, GE Healthcare, Fairfield, CA, USA), equilibrated in the same buffer. Elution from the anion exchange column was achieved with a linear gradient of 0 to 800 mM NaCl. Pooled fractions of all antibody fragments were dialyzed against phosphate-buffered saline (PBS). Protein purity was analyzed by nonreducing 15% SDS-PAGE. Protein quantification was determined using the classic Bradford reagent (Bio-Rad, Hercules, CA, USA).
2.6. In vitro binding studies of anti-IN antibody fragments
Relative binding of purified anti-IN antibody fragments was determined via ELISA after coating the wells with 200 ng of purified recombinant HIV-1 IN protein, HIV-1 PR, or bovine serum albumin (BSA) overnight at 4 °C in PBS. Wells were blocked for 1 H at 37 °C with 3% BSA in PBS. Purified anti-IN–scFvs were added to the wells and incubated for 1 H at 37 °C. After washing the wells with PBS, anti-HA–HRP mAb (Roche Diagnostics GmbH) was used for detection. Optical density at 405 nm was measured and assays were performed in triplicate. Specificity was demonstrated by Western blot analysis. Various amounts of HIV-1 IN and HIV-1 PR proteins were separated by SDS-PAGE and transferred to n-trocellulose membranes. After blocking, proteins were probed with each purified anti-IN–scFv clone (20 μg/mL) and then with anti-HA–HRP mAb (Roche Diagnostics GmbH) as a secondary antibody. Proteins were visualized using the ECL system (Amersham Biosciences, GE Healthcare). Antilatency-associated nuclear antigen (LANA) scFv (BM10) was used as a control antibody in both assays [54].
2.7. Mapping HIV-1 IN epitopes
To assess the IN-specific epitopes, purified anti-IN antibody fragments were tested for binding to a set of linear 20-mer peptides representing the entire HIV-1 IN sequence (AIDS Research and Reference Reagent Program). Briefly, Covalink ELISA plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated overnight with 5 μg of each peptide. After blocking with 3% BSA in PBS, purified anti-IN–scFvs were added to the wells and incubated 1 H. After washing the wells with low-ionic-strength buffer (150 mM NaCl, 50 mM Tris, pH 7.5, and 0.1% Triton X-100) or high-ionic-strength buffer (500 mM NaCl, 50 mM Tris, pH 7.5, and 1% Triton X-100), HRP-conjugated anti-HA mAb (Roche Diagnostics GmbH) was used for detection. All steps were performed at room temperature. To control for the quality of peptides and ELISA conditions, two polyclonal antibodies (AIDS Research and Reference Reagent Program) that specifically recognize residues 1–16 (Cat #756) and 276–288 in the IN protein (Cat #758) were used. Optical density at 405 nm was measured and assays were performed in triplicate. BM10 scFv was used as a control antibody.
2.8. DNA substrates for IN activity assays
The 3′-end processing substrate consisted of the terminal 21 bp from the U5 end of viral DNA and was prepared using the oligonucleotide RZ132 (5′-GTGTGGAAAATCTCTAGCAGT-3′) and AE118 (5′-ACTGCTAGAGATTTTCCACA-3′). RZ132 was treated with alkaline phosphatase and 5′-end labeled with [γ-32P] ATP by T4 polynucleotide kinase. The radiolabeled oligonucleotide was then annealed to AE118 and unincorporated ATP was removed by centrifugation through a quick-spin column (Millipore, Billerica, MA, USA). The precut DNA substrate for the strand-transfer assay was prepared by NdeI digestion of pNde675 plasmid, resulting in a 675-bp linear fragment flanked by 31 bp of HIV-1NL4-3 U5 LTR and U3 LTR sequences at each end. The precut DNA fragments were 5′-end labeled with [γ-32P] ATP by T4 polynucleotide kinase. The target DNA used in the strand-transfer integration assay was supercoiled pBR322 (Invitrogen).
2.9. IN activity assays
All IN activity assays were performed essentially as described previously with minor modifications [58],[59]. For the 3′-end processing assay the reaction mixtures (25 μL final volume) were assembled by incubating 200 nM IN on ice in 20 mM 3-(N-morpholino)propanesulfonic acid, pH 7.2, 0.1 mg/mL BSA, 7.5 mM MnCl2, 10% glycerol, and 10 mM 2-mercaptoethanol. These components were preincubated on ice for 15 Min with anti-IN–scFvs at various IN/scFv ratios. Then, 20 nM oligonucleotide substrate was added. Samples were incubated 1 H at 37°C and then reactions were quenched by adding 20 μL of sequencing gel loading dye (95% formamide, 10 mM EDTA, 0.03% bromophenol blue, and 0.03% xylene cyanol). A 2.5-μL aliquot of each reaction mixture was subjected to electrophoresis in a 20% denaturating polyacrylamide gel. Gels were dried, exposed to imaging plates, and visualized and quantified with a Fuji BAS-2500 bio-imaging analyzer (FujiFilm, Tokyo, Japan). For the strand-transfer assay, reaction mixtures (25 μL final volume) were assembled by incubating 80 nM IN on ice in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 12% dimethyl sulfoxide, 5 mM dithiothreitol, 10% polyethylene glycol-6000, 10 mM MgCl2, 20 mM ZnCl2, and 100 mM NaCl. These components were preincubated on ice for 15 Min with anti-IN–scFvs at various IN/scFv ratios. After addition of 10 nM DNA precut substrate, samples were further incubated on ice 30 Min. Then, 500 ng target plasmid DNA pBR322 was added. After a 30-Min preincubation on ice, the reaction was initiated by transferring to 37°C and incubation was continued for 90 Min. The reactions were stopped by addition of 0.1% SDS, 10 mM EDTA, and 5 μg of proteinase K. A 2.5-μL aliquot of each reaction mixture was subjected to electrophoresis in a 0.8% agarose gel in 1 × Tris–Borate–EDTA buffer. Gels were dried, exposed to imaging plates, and visualized and quantified with a Fuji BAS-2500 bio-imaging analyzer.
2.10. Expression of anti-IN antibody fragments in eukaryotic cells
After expression studies in E. coli TOP10F and analysis of binding activities, genes encoding anti-IN–scFvs were transferred into pcDNA3.1/Zeo+ (Invitrogen) by NotI and XhoI digestion. A methionine initiation codon was added into all scFvs by PCR. The sequence encoding the HA-tag sequence (YPYDVPDYA) was added at the C-terminus. The primers used for cloning in pcDNA3.1/Zeo+ were: Babe ScFv5 5′-GGCATGGGGGCCCAGGCGGCCCAGCTC-3′ and Babe ScFv3 5′-GCCACCACCCTCCTAAGAAGC-3′. Anti-IN–scFvs were also cloned into pcDNA3.1/Zeo+ with a nuclear localization signal (NLS). The NLS sequence was added at the N-terminal end of each fragment by using the primers Babe ScFv5NLS 5′-GGCATGGGGGCCCAGGCGGCCCAGCTC-3′ and Babe ScFv3 5′-GCCACCACCCTCCTAAGAAGC-3′. To analyze intrabody expression in eukaryotic cells, 293T cells (1–2 × 106) were transfected by a standard calcium phosphate method with 5 μg of plasmids encoding the scFv of interest. Efficiency of transfection is 70%–80%. Forty-eight hours posttransfection, 293T cells were washed with 5 mL of cold PBS. Cells were lysed in 1 mL 50 mM Tris (pH 8.0), 100 mM NaCl, 1% Nonidet P-40 containing PR inhibitors (Roche Diagnostics GmbH) for 60 Min on ice. The lysates were cleared by centrifugation for 30 Min at 14,000g, separated by 12% SDS-PAGE, and transferred to nitrocellulose membrane. Western blots were performed with HRP-conjugated anti-HA mAb (Roche Diagnostics GmbH).
2.11. Immunofluorescence staining
HeLa CD4 cells (0.5 × 106/well) transfected by calcium phosphate method with 4 μg of plasmid were fixed in PBS with 4% paraformaldehyde for 10 Min at room temperature, permeabilized with PBS plus 0.1% Triton X-100 for 5 Min, and washed with PBS plus 2% FCS before staining. For immunostaining of scFv, direct immunofluorescence was performed with rhodamine-conjugated anti-HA mAb (5 μg/mL) (Roche Diagnostics GmbH). Slides were mounted with DAPI Vectashield (Vector Labs, Burlingame, CA, USA) to stain nuclei, and cells were visualized using an Olympus IX-50 inverted microscope (Olympus Portugal, Lisbon, Portugal) equipped with Ludl Bio-Point filter wheels, and a 12-bit PCO (Kelheim, Germany) Sensicam QE cool CCD (Ludl Electronic Products, New York, NY, USA). Integrated control of the filter wheel and image acquisition was achieved by Image-Pro Plus 4.5 and Scope-Pro 3.1 (Media Cybernetics, Rockville, MD, USA). Settings for image acquisition (camera exposure time, filters and time interval) were determined by custom-made macros. Images were collected with Olympus 40 × or 100 × plan apo objectives (Numerical Aperture = 0.95 and 1.4, respectively).
2.12. One-step viral replication analysis
One-step viral replication was tested using HeLa CD4 LTR-β-Gal reporter (MAGI) cells, which contain a stably integrated LTR-β-galactosidase expression cassette that reports productive HIV-1 infection following Tat expression and trans-activation [60]. Briefly, HeLa CD4 LTR-β-Gal cells (1 × 106/well) were transfected by the calcium phosphate method with 4 μg of pcDNA3.1/Zeo(+) encoding scFv. Twenty-four hours after transfection, HIV-1NL4-3 was used to infect cells at a multiplicity of infection (MOI) of 1.0 for 5 H and enhanced by spinoculation. To produce HIV-1NL4-3 for infectivity assays 1 × 106 293T cells were transfected with 5 μg of plasmid encoding HIV-1NL4-3. After 48 H, lentiviral particles were collected and quantified by p24 ELISA. Cells were washed with prewarmed serum-free medium and cultured for 48 H. The ability of anti-IN–scFvs to inhibit early steps of replication was measured by quantification of β-galactosidase activity in cell lysates using a colorimetric assay based on the cleavage of chlorophenol red-β-d-galactopyranoside (CPRG). HeLa CD4 LTR-β-Gal cells were washed with PBS and lysed with lysis buffer (50 mM Tris, pH 8.0, 100 mM NaCl, and 1% Non-idet P-40), supplemented with PR inhibitors. After incubation for 30 Min on ice, CPRG reaction buffer (6 mM in lysis buffer) was added to the cell lysates and incubated for 2 H at 37 °C. The optical density at 570 nm was measured. BM10 was used as a control antibody.
2.13. Quantification of newly synthesized HIV-1 DNA
Real-time fluorescence-monitored PCR, using the LightCycler Instrument (Roche Diagnostics GmbH), was used to monitor, over a 24 H period postinfection, the synthesis of viral cDNA intermediates in HeLa-P4 cells. These cells were transfected with scFv and infected with HIV-1 in presence of 1 μM saquinavir, to limit viral replication to a single round. Equal amounts of DNase-treated virions (100 ng of p24) were used to infect 5 × 106 HeLa cells at 4 °C for 2 H. The cells were washed twice with PBS, and plated into 24-well plates containing complete DMEM pre-warmed to 37 °C and incubated at 37 °C. At different time points postinfection, equal aliquots of cells were collected and washed with PBS, and cellular DNA was extracted using the DNeasy tissue kit (Qiagen, Hilden, Germany). Equal amounts of cellular genomic DNA (determined spectrophotometrically at an optical density of 260 nm) were used to quantitate viral cDNA intermediates containing sequences for either R-U3 or U5-gag, which represents, respectively, minus strand-transfer DNA/early minus single-strand DNA and plus strand-transfer DNA. Two pairs of primers were used: R-U3 forward, 5′ CGGGACTGGGGAGTGGC-GAGC; R-U3 reverse, 5′ CAGAGTCACACAACAGACGGGC; U5-gag forward, 5′ TGTGTGCCCGTCTGTTGTGTGA; and U5-gag reverse, 5′ GAGTCCTGCGTCGAGAGAGCT [61]. All analyses were done in triplicate, with triplicate samples in each experiment. The statistical analyses employed column statistics and one-way analysis of variance. The lowest level of significance was set at P < 0.05.
2.14. Quantification of integrated viral DNA during HIV-1 Infection
Hela-P4 cells transfected with scFv and infected with HIV-1 were placed in the presence of 1 μM saquinavir, to limit viral replication to a single round, and harvested at 24 and 48 H postinfection. Samples were washed in PBS and treated with 500 units of DNase I (Roche Diagnostics GmbH) for 1 H at 37 °C, before DNA extraction using a QIAamp blood DNA minikit (Qiagen). The amount of integrated HIV-1 DNA was quantified by real-time PCR performed with the Light Cycler instrument (Roche Diagnostics GmbH) as described previously [62]. Each sample was analyzed in duplicate. Briefly, integrated DNA was quantified using an Alu-LTR-based nested PCR procedure. In a first round of PCR, integrated HIV-1 sequences were amplified with two outward facing Alu primers and a HIV-1 LTR-specific primer (L-M667) containing a λ phage-specific sequence at the 5′-end of the oligonucleotide. In a second round of PCR, we used specific primers for the λ sequence (λT) and the LTR region (AA5M) (Brussel and Sonigo [62]). To eliminate the signal due to the primer extension carried out by the L-M667 primer during the first round PCR, a control PCR assay was performed without Alu primers. The signal of the nested PCR obtained in the absence of Alu primers was subtracted from the integrated HIV-1 DNA signal. Copy numbers of total DNA two-LTR circles and integrated DNA were determined in reference to standard curves prepared by amplification of cloned DNA with matching sequences [62]. Results were normalized by the number of cells and the amount of cellular DNA quantified by PCR of the β-globin gene according to the manufacturer’s instructions (Roche Diagnostics GmbH).
2.15. Generation of stable anti-IN–scFv–Jurkat cell clones
pcDNA3.1-anti-IN–scFv plasmids were digested with SfiI, and the anti-IN–scFv genes were cloned between the two SfiI sites of pMX-KRAB8FPBS2puro, replacing the KRAB8FPBS2 zinc finger gene. pMX-anti-IN–scFv and pMD-G plasmids were cotransfected into the Gag-Pol-293 packaging cell line (Clontech) using the standard calcium phosphate method. After 48 H of incubation, culture supernatants were used for transduction of Jurkat cells. After 2–3 weeks of selection in puromycin-containing medium, stable anti-IN–scFv–Jurkat clones were isolated and analyzed for expression of anti-IN–scFv by Western blot with HRP-conjugated anti-HA mAb (Roche Diagnostics GmbH).
2.16. HIV-1 challenge of stable anti-IN–scFv–Jurkat cell clones
Anti-IN–scFv–Jurkat clones were infected at MOIs of 0.1–0.5 for 6 H at 37 °C, washed, and then cultured for up to 20 days. To monitor infection, aliquots were taken from the cultures at the indicated time points and p24 antigen in supernatant was determined using a HIV-1 p24 ELISA (Innovagen, Lund, Sweden). Cellular proliferation and viability of infected cells were analyzed using the tetrazolium salt WST-1 (Roche Diagnostics GmbH) according to the manufacturer’s protocol.
3. Results
3.1. Selection of specific anti-IN antibody fragments
Two rabbits from the New Zealand white strain were immunized and boosted four times with 200 μg of purified HIV-1 IN protein. ELISA of the rabbit sera from both animals showed that the immunization resulted in a strong immune response against HIV-1 IN (data not shown). Rabbits were euthanized and bone marrow and spleen were extracted for total RNA preparation and cDNA synthesis. To generate the chimeric scFv library, specific oligonucleotide primers covering all known variable rabbit antibody family sequences were used to amplify VH and VL gene segments [55]. The purified products of variable regions were assembled into scFv format by overlapping PCR as described in Materials and methods. The recombinant phagemid pComb3X was transformed into E. coli ER2538 cells to yield 8.3 × 107 individual clones. The phage library displaying the scFvs was then panned against immobilized HIV-1 IN protein using a solid-phase assay as previously described [55]. From the final phage-display panning, 150 scFv phage clones were randomly selected for ELISA and binding activity against HIV-1 IN was evaluated (data not shown). A total of 22 clones with higher-than-background signal were isolated and sequenced. Fourteen clones showed sequence variations in the frameworks and complementary determining regions (CDR) (data not shown) and were chosen for further expression and binding characterization.
3.2. Relative binding activity and specificity of anti-IN antibody fragments
The 14 clones were expressed in the periplasm of the nonsuppressor E. coli strain TOP10F. After induction, cells were lysed and the soluble fraction was subjected to immobilized metal affinity chromatography and anion exchange chromatography as described in Materials and methods. All scFvs were highly expressed in the form of soluble proteins and typical yields of pure scFv (>95%) were in the range of 1 ± 0.5 mg/L of bacterial culture. The relative binding activity and specificity of anti-IN–scFvs were assessed by ELISA and Western blotting. As shown in Fig. 1a, all anti-IN–scFv clones specifically bound to HIV-1 IN protein. In contrast, no binding to IN protein was observed with a scFv (clone BM10) that specifically recognizes the LANA1 from Kaposi’s sarcoma–associated herpesvirus [54]. Moreover, the anti-IN–scFvs did not bind to HIV-1 PR protein or BSA. To further test binding specificity of scFv to IN protein, four scFv clones with high binding activity to IN (clones 104, 135, 142, and 144) and another that gave a weaker signal (clone 7) were evaluated in immunoblotting assays. These five clones showed sequence variations in the CDR (Fig. S1, Supporting Information). HIV-1 IN and PR proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. After blocking, proteins were probed with purified anti-IN–scFvs and then with HRP conjugated anti-HA mAb as a secondary antibody. As a control antibody, we used the anti-LANA1 scFv (BM10). As shown in Fig. 1b, the anti-IN–scFvs specifically recognized the 32 kDa HIV-1 IN protein. Importantly, recombinant anti-IN–scFvs did not recognize HIV-1 PR and no bands were detected using the scFv BM10. Thus, ELISA and immunoblotting results showed that scFv clones 7, 104, 135, 142, and 144 specifically recognize the HIV-1 IN protein, and therefore, were chosen for further antibody characterization and functional studies.
Fig. 1.

Relative binding activities and specificity of anti-IN–scFvs. (a) The relative binding of anti-IN–scFvs to 200 ng of HIV-1 IN, PR, and BSA was evaluated by ELISA. The anti-LANA1 scFv (BM10) was used as a control antibody. Optical density at 405 nm was measured. Shown are average values of three independent experiments. (b) The indicated amounts of purified HIV-1 IN protein were separated by 15% SDS-PAGE and transferred to nitrocellulose membranes. For immunodetection, purified anti-IN–scFv clones 7, 104, 135, 142, and 144 were used (20 μg/mL). HRP-conjugated anti-HA mAb was used as a secondary antibody. HIV-1 PR at 800 nM was used to test anti-IN specificity and anti-LANA1 scFv (BM10) was used as a control antibody. Molecular weight is indicated in kDa.
3.3. Mapping HIV-1 IN epitopes
The five selected anti-IN–scFvs apparently recognized a continuous epitope of IN because they reacted with denatured IN on immunoblots (Fig. 1b) and ELISA (data not shown). To identify the epitopes recognized by the different antibodies, we used a set of 30 overlapping synthetic peptides (obtained from the AIDS Research and Reference Reagent Program) covering the entire IN molecule. Each peptide contained 20 residues, with the first 10 residues overlapping those found in the peptide immediately preceding it in the primary IN sequence. Interactions between the scFvs and the synthetic IN peptides were analyzed by ELISA as described in Materials and methods. To control the quality of peptides and ELISA conditions for epitope mapping, two polyclonal antibodies (AIDS Research and Reference Reagent Program) that specifically recognize residues 1–16 (Cat #756) and 276–288 in the IN protein (Cat #758) were used. Results showed that both polyclonal antibodies specifically recognize the expected peptides of HIV-1 IN (Table 1), thus confirming the quality of peptides and its binding to the ELISA plate. The background signal was low and no peptide binding was observed with anti-LANA1 scFv (clone BM10) but an unexpected pattern of peptide reactivity was observed for all five anti-IN–scFvs. As shown in Table 1, each anti-IN–scFv clone bound to peptide 4,338 (residues 126–145), peptide 4,343 (residues 176–195), and also to two downstream peptides 4,346 and 4,347 (residues 206–235). Because all anti-IN–scFvs bound to three regions separated in the linear sequence of IN (shown schematically in Fig. 2a), we were concerned that nonspecific peptide binding was occurring. The ELISA epitope mapping was repeated with a high-ionic-strength washing buffer (see Materials and methods). Under these conditions, a small decrease in the relative antigen-binding activity was observed (data not shown); nevertheless, the same peptide binding pattern was maintained for all anti-IN–scFv clones. Therefore, we sought to determine whether the three IN regions (residues 126–145, 176–195, and 206–235) were proximal in the folded IN structure forming a conformational epitope. Although the structure of full-length HIV-1 IN protein has not been determined, a crystal structure of the catalytic core and C-terminal region is available [63]. The structure suggests that the three epitope regions are very close and form a cavity (Fig. 2b). Therefore, to our knowledge, these scFvs constitute the first set of anti-IN antibodies that recognize and bind to epitope regions in both the catalytic and C-terminal domains of IN.
Table 1.
Epitope mapping of anti-IN–scFvs
| Peptide # | Amino acid position | Amino acid sequence | 7 | 104 | 135 | ScFv# 142 | 144 | BM10 | 756 | IN anti serum 758 |
|---|---|---|---|---|---|---|---|---|---|---|
| 4324 | 1–5 | EQVDKLVSAGIRKVLFLDGI | − | − | − | − | − | − | + | − |
| 4325 | 6–15 | IRKVLFLDGIDKAQDEHEKY | − | − | − | − | − | − | + | − |
| 4326 | 6–25 | IRKVLFLDGIDKAQDEHEKY | − | − | − | − | − | − | + | − |
| 4327 | 16–35 | HSNWRAAIASDFNLPPVVAKE | − | − | − | − | − | − | − | − |
| 4328 | 26–45 | FNLPPVVAKEIVASCDKCQL | − | − | − | − | − | − | − | − |
| 4329 | 36–55 | IVASCDKCQLKGEAMHGQVD | − | − | − | − | − | − | − | − |
| 4330 | 46–65 | KGEAMHGQVDCSPGIWQLDC | − | − | − | − | − | − | − | − |
| 4331 | 56–75 | CSPGIWQLDCTHLEGKYILV | − | − | − | − | − | − | − | − |
| 4332 | 66–85 | THLEGKVILVAVHVASGYIE | − | − | − | − | − | − | − | − |
| 4333 | 76–95 | AVHVASGYIEAEVIPAETGQ | − | − | − | − | − | − | − | − |
| 4334 | 86–105 | AEVIPAETGQETAYFLLKLA | − | − | − | − | − | − | − | − |
| 4335 | 96–115 | ETAYFLLKLAGRWPYKTIHT | − | − | − | − | − | − | − | − |
| 4336 | 106–125 | GRWPVKTIHTDNGSNFTGAT | − | − | − | − | − | − | − | − |
| 4337 | 116–135 | DNGSNFTGATVRAACWWAGI | − | − | − | − | − | − | − | − |
|
| ||||||||||
| 4339 | 136–155 | KQEFGIPYNPQSQGVVESMN | − | − | − | − | − | − | − | − |
| 4340 | 146–165 | QSQGYYESMNKELKKIIGQY | − | − | − | − | − | − | − | − |
| 4341 | 156–175 | KELKKIIGOY’RDQAEHLKTA | − | − | − | − | − | − | − | − |
| 4342 | 166–185 | RDQAEHLKTAYQMAYFIHNF | − | − | − | − | − | − | − | − |
|
| ||||||||||
| 4344 | 186–205 | KRKGGIGGYSAGERIVDIIA | − | − | − | − | − | − | − | − |
| 4345 | 196–215 | AGERIVDIIATDIOTKELQK | − | − | − | − | − | − | − | − |
|
| ||||||||||
| 4348 | 226–245 | YYRDSRXPLWKGPAKLLWKG | − | − | − | − | − | − | − | − |
| 4349 | 236–255 | KGPAKLLWKGEGAVVIQDNS | − | − | − | − | − | − | − | − |
| 4350 | 246–265 | EGAVVIQDNSDIKVVPRRKA | − | − | − | − | − | − | − | − |
| 4351 | 256–275 | DIKVVPRRKAKIIRDYGKQM | − | − | − | − | − | − | − | − |
| 4352 | 266–285 | KIIRDYGKQMAGDDCVASRQ | − | − | − | − | − | − | − | + |
| 4353 | 276–288 | AGDDCVASRQDED | − | − | − | − | − | − | − | + |
To perform epitope mapping, purified anti-IN–scFv clones 7, 104, 135, 142, and 144 were tested for binding to a set of 30 overlapping synthetic peptides covering the entire HIV-1 IN protein sequence. CovaLink ELISA plates (Thermo Fisher Scientific) were coated with 5 μg of each peptide and incubated overnight at 4 °C. The plates were then blocked with BSA and incubated with purified antibody fragments. After washing the wells with low-ionic-strength buffer or high-ionic-strength buffer (see Materials and methods), HRP-conjugated anti-HA mAb was used for detection. The same binding pattern was observed in both washing conditions. The anti-LANA1 scFv (BM10) was used as a control antibody. Integrase antiserum #756 and #758 were used to control for quality of peptides and ELISA conditions. Black boxes highlight the IN peptides bound by scFvs. The (+) symbol indicates binding and the (−) symbol means no binding.
Fig. 2.
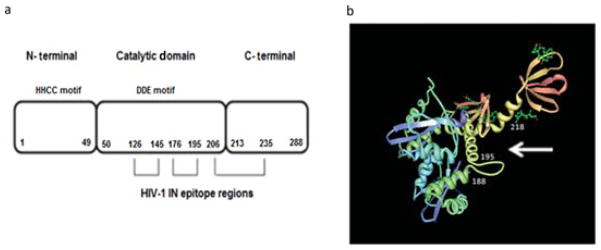
Schematic representation of HIV-1 IN and its interactions with anti-IN–scFvs. (a) Linear model of HIV-1 IN protein and the domains recognized by anti-IN–scFv clones 7, 104, 135, 142, and 144. The IN enzyme is composed of the N-terminal domain (NTD, aa 1–49), catalytic core domain (CCD, aa 50–212), and C-terminal domain (CTD, aa 213–288). The H and C residues within the NTD are conserved among retrotransposon integrase proteins. The D and E residues in the CCD form the DDE motif. All anti-IN–scFvs tested recognized and bound to residues 126–145 (peptide 4,338), residues 176–195 (peptide 4,343), and to the upstream residues 206–235 (peptides 4,346 and 4,347). (b) Model of the crystal structure of HIV-1 catalytic and C-terminal domains (PDB code 1EX4) [63] showing the epitopes recognized by the anti-IN–scFvs. The probable binding site of scFvs to HIV-1 IN is indicated by a white arrow.
3.4. Anti-IN antibody fragments strongly inhibit the strand-transfer reaction
Integration of retroviral DNA into the host chromosomal DNA involves two chemical steps. The newly synthesized blunt-ended viral DNA first undergoes 3′-end processing. In this reaction, two nucleotides are removed from each 3′-end. Next, the exposed hydroxyl groups attack a pair of phosphodiester bonds on opposite strands of the target DNA to complete the strand-transfer step. Both the 3′-end processing and DNA strand-transfer activities of HIV-1 IN can be recapitulated in vitro with DNA substrates that mimic the viral DNA ends. Therefore, to interrogate the effects of our scFvs on the in vitro activities of IN, both 3′-end processing and strand-transfer assays were performed in the presence of anti-IN–scFvs. As shown in Fig. 3a, none of anti-IN–scFvs tested interfered with 3′-end processing reaction. Cleavage of the 21-bp oligonucleotide substrate was not inhibited even when anti-IN–scFvs were added to IN at molar ratios of 4:1 (800 nM). To evaluate the effect of each anti-IN–scFv in the strand-transfer reaction, we used a preprocessed DNA substrate. As shown in Fig. 3b, scFvs 104, 135, 142, and 144 clearly inhibited the strand-transfer reaction as demonstrated by the decreases of half-site and full-site (concerted) integration of the 32P-labeled preprocessed DNA substrate into the target DNA (pBR322). These four antibody fragments inhibited more than 50% of the IN activity when present in the reaction at a twofold molar excess over IN (200 nM). At a higher concentration (400 nM), scFv-104 and scFv-142 inhibited the reactions almost completely. On the other hand, scFv-7 had a weaker inhibitory effect, and scFv anti-LANA1 (BM10) had no detectable effect on the strand-transfer activity of IN. The strand-transfer reaction was also performed with a substrate that was not precut (Fig. S2, Supporting Information) and similar inhibition results were obtained with the preprocessed and blunt DNA. Therefore, scFvs 104, 135, 142, and 144 selectively inhibited DNA strand transfer.
Fig. 3.

Effect of anti-IN–scFv binding on in vitro activities of HIV-1 IN. (a) IN (200 nM) was preincubated on ice with each scFv at various scFv/IN molar ratios. Radioactively labeled oligonucleotide substrate was added, and the reactions were allowed to proceed at 37 °C. Each reaction mixture was subjected to electrophoresis in a 20% denaturating polyacrylamide gel. The positions of the 21-bp oligonucleotide substrate (S) and the −2 cleavage product (P) are indicated. No inhibition of 3′-end processing was observed with any of the anti-IN–scFvs. (b) Purified scFv was preincubated with 80 nM IN at various scFv/IN molar ratios on ice. Radioactively labeled preprocessed DNA substrate and supercoiled target DNA (pBR322) were added and incubated as described in Materials and methods. Each reaction mixture was subjected to electrophoresis in 0.8% agarose gel. Half-site integration involves the insertion of one LTR end per target, and full-site integration involves the concerted insertion of two LTR ends per target. The positions of the concerted integration product, the half-site product, and products resulting from integration of viral DNA substrate into itself (Donor/Donor) are indicated. In both assays, anti-LANA1 scFv (BM10) was used as a control antibody. Gels were dried, exposed to imaging plates, and visualized and quantified with a Fuji BAS-2500 bio-imaging analyzer.
3.5. HIV-1 replication is inhibited by anti-IN–scFv intrabodies localized in cytoplasm and nucleus
After showing inhibition of HIV-1 IN activity in vitro, anti-IN–scFv genes with and without a NLS were transferred into pcDNA3.1/Zeo+. To evaluate the expression of anti-IN–scFv in eukaryotic cells, 293T cells were transfected by a standard calcium phosphate method with each scFv plasmid. The data in Fig. 4a demonstrate that all anti-IN–scFv clones were highly expressed. Cellular localization in HeLa cells was determined using an immunofluorescence assay. As shown in Fig. 4b, scFv-142 was expressed in the cytoplasm and scFv-142-NLS was found in the nucleus of eukaryotic cells. The same expression pattern was also observed for the other scFvs (data not shown). To evaluate whether the expression of anti-IN–scFvs in either the cytoplasm or nucleus inhibits HIV-1 replication, we performed a one-step viral replication assay in HeLa CD4 LTR-β-Gal cells. The results shown in Fig. 5 represent the percentage of viral replication events relative to the value obtained for HIV-1NL4-3 infection in control HeLa CD4 LTR-β-Gal cells (positive control, C +). Cytoplasmic or nuclear expression of anti-IN–scFv clones 104, 135, 142, and 144 resulted in more than 70% decrease in HIV-1 replication compared with the control experiments without scFv. On the other hand, scFv-7 expression was less effective (less than a 20% decrease in replication). Expression of control anti-LANA1 scFv (BM10) with or without the NLS did not inhibit HIV-1 replication. The biological activity of anti-IN–scFvs correlated with relative binding activities of scFvs and extent of strand-transfer inhibition.
Fig. 4.

Expression and localization of anti-IN intrabodies in eukaryotic cells. (a) Anti-IN intrabody expression vectors were transfected into 293T cells and after 48 H cells were lysed and cleared by centrifugation. Proteins were separated by 15% SDS-PAGE and visualized by probing with HRP-conjugated anti-HA mAb. Mock lysates of 293T cells were used as controls. Molecular weights are indicated in kDa. (b) Transfected HeLa cells expressing scFv-142 and scFv-142-NLS were stained with rhodamine-conjugated anti-HA mAb. ScFv is shown in red (rhodamine). Immunofluorescence microscopy was performed as described in Materials and methods using the appropriate excitation and emission filters.
Fig. 5.
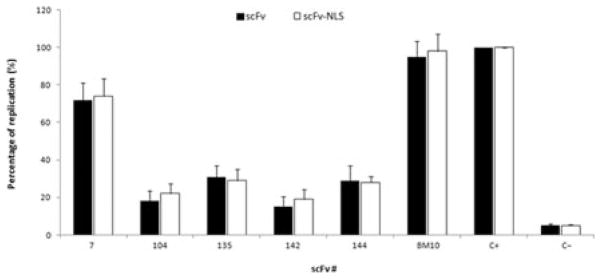
Neutralization of IN function in one-cycle replication assay. Values represent the percentages of one cycle replication in HeLa CD4 LTR-β-Gal cells relative to the value obtained for the wild-type virus. Cells were transfected with plasmids encoding anti-IN antibody fragments as described in Materials and methods. The ability of anti-IN antibody fragments to inhibit a single round of replication was measured by quantification of the β-galactosidase activity in cell lysates using a colorimetric assay based on the cleavage of CPRG by β-galactosidase as described in Materials and methods. Anti-LANA1 scFv (BM10) was used as a control antibody. C + indicates HIV-1NL4-3 infection of control HeLa CD4 LTR-β-Gal cells; C− indicates uninfected HeLa CD4 LTR-β-Gal cells. Optical density at 570 nm was measured and data represent mean ± SEM (n = 3).
3.6. Anti-IN–scFv intrabodies specifically inhibit HIV-1 DNA integration
To assess whether anti-IN–scFvs specifically inhibit the integration step during HIV replication and have an effect on the integration process itself, viral cDNA intermediates and provirus integration were measured at different times postinfection. Briefly, a real-time fluorescence-monitored PCR was first used to monitor over a 24 H period postinfection the synthesis of viral cDNA intermediates in Hela-P4 cells. These cells were transfected with scFv plasmids and infected with HIV-1 in the presence of 1 μM saquinavir, to limit viral replication to a single round. Specific primers were used to measure viral cDNA intermediates containing sequences for either R-U3 or U5-gag, which represent respectively the minus and plus strand-transfer DNA (see Material and methods). As shown in Figs. 6a and 6b, in the presence of azidothymidine (control), R-U3 and U5-gag intermediate levels were as expected low from 0 to 24 H. On the other hand, R-U3 and U5-gag intermediates peaked with all antibody constructs between 8 and 12 H and decreased over time. This pattern was similar to HIV-1 (positive control), which shows that expression of anti-IN intrabodie had no significant impact on viral cDNA intermediate synthesis. Then, to determine if anti-IN–scFvs were able to specifically block the integration step itself during HIV replication, the amount of integrated HIV-1 DNA was quantified by real-time PCR in postinfected scFv transfected Hela-P4 cells (see Material and methods). In the presence of scFv-BM10 and scFv-7, viral integration was not inhibited (Fig. 6c). In contrast, all other anti-IN constructs showed an effect between twofold and eightfold less provirus than HIV-1 wild-type. In particular, scFv 142 show the more pronounced effect compared with the other constructs. Therefore, combining these results with data of in vitro IN inhibition and neutralization of viral replication it is possible to consider scFv 104, 135, 142, and 144 as authentic IN inhibitors that owe their antiviral activity to inhibition of the integration process in HIV-infected cells.
Fig. 6.

Effect of scFv intrabodies on production of HIV-1 cDNA. (a and b) Cellular DNA was extracted at different time points from HeLa-P4 cells transfected with scFv expression plasmids and infected with HIV-1. Viral cDNA intermediates were monitored by real-time PCR, as described in Materials and methods. PCR primers used to detect the following DNA intermediates are indicated: R-U5 (minus strand strong stop) and U5-gag (DNA made after plus strand transfer). The results represent experiments performed three times. (c) Quantification of integrated viral cDNA during HIV-1 infection. HeLa P4 cells were transfected with scFv expressing plasmids and infected with HIV-1. At 24 and 48 H after infection cellular DNA was extracted and subjected to Q-PCR analysis to quantify integrated proviral DNA. Error bars represent variations between duplicate Q-PCR assays.
3.7. Jurkat cells stably expressing anti-IN intrabodies are protected from HIV-1 replication
The experiments described above examined the biological activity of anti-IN–scFvs in a transient assay under conditions where most virus transmission occurs by cell-to-cell spread. To determine whether anti-IN–scFv intrabodies inhibited IN function during several rounds of HIV-1 replication, we generated Jurkat cell lines stably expressing scFv intrabodies. We transduced Jurkat cells with retroviral vectors encoding anti-IN intrabodies and anti-LANA1 (BM10) was used as a control. As shown in Fig. 7a, Jurkat–scFv cell lines showed homogeneous and stable expression of intrabodies. To determine whether the intracellular expression of anti-IN–scFvs was able to prevent HIV-1 replication, Jurkat–scFv cell lines were challenged with the HIV-1NL4-3. Infectivity assays were performed with MOIs of 0.1–0.5 to mimic natural infections. The infected cell cultures were maintained for up to 20 days and infection was monitored at the indicated time points by p24 antigen ELISA. As shown in Fig. 7b, parental Jurkat cell lines supported vigorous replication of HIV-1 (positive control, C +) that peaked at approximately day 2 and up to day 12. In contrast, from days 2 to 18 postinfection, Jurkat–scFv-104, scFv-135, scFv-142, and scFv-144 cell lines showed approximately 60%–80% inhibition of HIV-1 p24 antigen production compared with the parental Jurkat cells. Anti-IN–scFv-7 demonstrated a modest inhibitory effect and control anti-LANA1 scFv (BM10) did not show an inhibitory effect on HIV-1 replication. In addition to neutralization of IN by anti-IN–scFv in infected cells, it is possible that nonspecific incorporation of scFv in HIV-1 particles might also affect virus replication by interfering with reverse transcription. This hypothesis was tested by endogenous RT assays in viral particles produced from scFv-expressing cells. The results showed that reverse transcription can be affected when anti-IN–scFv is present in the viral particle (Fig. S3, Supporting Information). Previous studies have shown that intracellular antibody expression has no effect on cell viability or proliferation [52–54]. Nevertheless, we quantified cell proliferation and cell viability of Jurkat–scFv cells. We observed that the kinetics of WST-1 metabolism in Jurkat–scFv cells infected with HIV-1 were similar to that of noninfected cell lines (data not shown). Therefore, the expression of anti-IN intrabodies did not result in cell toxicity.
Fig. 7.
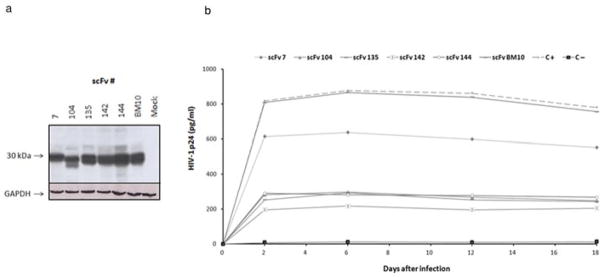
HIV-1 challenge of stable anti-IN–scFv–Jurkat cell clones. (a) Western blot of Jurkat cells stably expressing anti-IN–scFvs and anti-LANA1 scFv (clone BM10). Lysates of Jurkat cells not expressing scFv were used as negative controls (mock). Loading was controlled with anti-GAPDH antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). (b) Stable Jurkat cell clones expressing anti-IN–scFv intrabodies were infected with HIV-1NL4-3 at a MOI of 0.1–0.5. The cultures were maintained for up to 20 days. To monitor infection, aliquots were taken at the indicated time points and p24 levels were determined by ELISA. BM10 was used as a control antibody. C + indicates HIV-1NL4-3 infected cells; C− indicates uninfected cells. The data are representative of two independent experiments.
4. Discussion
Controlling HIV infection continues to be a major challenge worldwide. Although the drug cocktails used in HAART have markedly changed the profile of progression to AIDS in HIV-infected individuals, drug failure continues to occur as a consequence of viral resistance and complications arising from a lifelong regimen of chemotherapy. The development of new modes of inhibition is of paramount importance and has been the focus of recent research efforts. Gene therapy has captured the interest of a number of investigators as an attractive addition to conventional pharmacologic therapies because the alteration of the host cell could potentially confer permanent suppression of viral replication in infected individuals or could provide protection against viral infection. Nevertheless, the gene therapy community is still coming to grips with the reality that although a significant body of basic science and animal modeling has been compiled to indicate the possibility of gene transfer, an efficient and stable gene transfer has yet to be convincingly demonstrated. Thus, in this work we bring a new approach for sustained cell resistance to HIV-1 infection by developing rabbit-derived scFv intrabodies to the HIV-1 IN protein. The IN protein plays a crucial role in the early stages of HIV-1 infection by catalyzing the integration of viral cDNA into a host chromosome. Therefore, scFv-based strategies directed against IN may represent an effective approach to inhibit this crucial step of the viral replication cycle. It is conceivable that a strategy of gene therapy using intrabodies against IN may be applicable to modify hematopoietic stem/progenitor cells (HSPCs) to express antiviral proteins. This strategy may provide the immune system with enough HIV-resistant cells to vanquish the virus. Recently, evidence has suggested that the modified HSPCs have given rise to cells of multiple lineages (e.g., T cells, B cells, and macrophages) carrying the anti-HIV genes, albeit at very low numbers [64],[65].
In the present study, we immunized rabbits with HIV-1 IN and developed a combinatorial scFv phage library against IN. Although the generation of rabbit mAbs by hybridoma technology has also been reported [66], the phage display approach with its inherent linkage of phenotype and genotype provides ready access to antibody sequences and facilitates further in vitro optimizations, such as affinity maturation or humanization. Furthermore, combinatorial scFv libraries potentially allow display of the entire immunological record of an individual, allowing the detection and recovery of any antibody ever made. It was recently determined that combinatorial antibody libraries may be a very good mimic of antibodies existing in the organism [67]. The libraries can be created from immune individuals, biased for clones against the immunogen, or be as diverse as possible, producing so-called naive libraries from which theoretically antibodies to any antigen may be isolated. The technology is so robust that isolation of the type of antibodies searched for most often is successful. For each antigen panned, specific antibody clusters can be identified that explore multiple distinct epitopes across their respective target’s surface. This indicates that the display of antibody on phage in a scFv format does not significantly restrict the available diversity accessible to in vitro panning, resulting in antibodies that are specific and potentially different from those expressed naturally. Moreover, epitopes that are invisible in rodents or humans might be immunogenic in rabbits and, therefore, be easily selected by combinatorial rabbit-derived scFv libraries.
We were able to identify five scFv intrabodies that specifically recognize and bind HIV-1 IN protein. These scFvs bound to two epitope regions in the catalytic domain (aa 126–145 and 176–195) and to one upstream region in the C-terminal domain (aa 206–235) of IN protein. As recently published by Cherepanov’s group [24], the IN structure of PFV was determined to be bound to DNA, revealing the organization of the retroviral intasome. Using the described intasome structure, the scFv interacting epitopes of HIV-1 IN seem to be arranged in proximity to an exposed region of PFV IN. The scFvs described in this work differ substantially from previous anti-IN antibodies that have been raised [40–48]. With the combinatorial scFv library, we were able to identify a panel of new anti-IN–scFvs that can recognize epitopes not easily accessible by conventional antibody molecules, such as small pockets or canyons in the IN protein. Because our anti-IN intrabodies bind to three regions in the IN protein, we may speculate that the development of HIV-1 escape mutants should be minimized.
The anti-IN–scFvs inhibited the strand-transfer reaction catalyzed by IN in vitro, whereas no effect was observed on the 3′-end processing reaction in vitro. Thus, the region of IN recognized by the scFvs must be essential to the strand transfer but not to the 3′-end processing activity. The expression of anti-IN–scFvs in eukaryotic cells showed that our intrabodies were correctly folded and were efficiently delivered into the nucleus. We showed that HIV replication was strongly inhibited in cells transiently expressing anti-IN–scFv clones in either the cytoplasmic or nuclear compartments. These results, together with quantification of proviral integration, demonstrate that anti-IN–scFv specifically neutralize the IN activity before integration. Moreover, the stable expression of anti-IN intrabodies in Jurkat cells protects it from HIV-1 infection, indicating that scFvs are capable of blocking the IN activity during multiple rounds of infection. In addition, anti-IN–scFv might be non-specifically incorporated in viral particles during viral replication and interfere with reverse transcription. Therefore, the scFv strategy reported in this study can be used to block early stages of viral replication, due to the capacity of the scFvs to interfere with the establishment of a provirus.
In summary, our data show that combinatorial rabbit scFv phage libraries can be efficiently used to identify scFvs with novel epitope binding characteristics. Moreover, our data provide proof-of-principle that rabbit anti-IN intrabodies can be designed to block early stages of HIV-1 replication. Although we may envision the use of rabbit anti-IN intrabodies as agents for HIV-1 gene therapy, these antibodies can also be used to define specific functional properties of IN and as tools to better understand the structure HIV-1 IN.
Supplementary Material
Acknowledgments
We thank Dr. Zeger Debyser for providing the plasmid pCEP-IN. The envelope plasmid pMD.G was kindly provided by Dr. Didier Trono. HeLa CD4 LTR-β-Gal cells, plasmid HIV-1NL4-3 and HIV-1 IN peptides were obtained from the AIDS Research and Reference Reagent Program. This work was supported by grants from the Fundação para a Ciência e Tecnologia (PCTI/BIA-MIC/60038/2004). F.A.S. was supported with a doctoral fellowship from Fundação para a Ciência e Tecnologia (SFRH/BD/17039/2004). S.M. was supported with a postdoctoral fellowship from Fundação para a Ciência e Tecnologia (SFRH/BPD/21011/2004). R.C. was supported by the intramural research programs of the NIDDK of the National Institutes of Health and by the NIH AIDS Targeted Antiviral Program. C. B. was supported by a grant from the National Institutes of Health (R01GM065059).
Abbreviations
- aa
amino-acid
- BSA
bovine serum albumin
- CDR
complementary determining regions
- CPRG
chlorophenol red-beta-d-galactopyranoside
- E. coli
Escherichia coli
- ELISA
enzyme-linked immunosorbent assay
- HA
hemagglutinin
- HAART
highly active antiretroviral therapy
- HIV-1
human immunodeficiency virus type 1
- HRP
horseradish peroxidase
- IN
integrase
- IPTG
isopropyl-1-thio-β-d-galactoside
- mAbs
monoclonal antibodies
- MOI
multiplicity of infection
- NLS
nuclear localization signal
- PBS
phosphate-buffered saline
- PFV
prototype foamy virus
- PR
protease
- RT
reverse transcriptase
- scFv
single-chain variable fragment
- VH
heavy-chain variable domain
- VL
light-chain variable domain
Footnotes
Supporting Information is available in the online issue at wileyonlinelibrary.com.
The authors declare no conflict of interest.
Contributor Information
Carlos Barbas, III, Email: carlos@scripps.edu.
Joao Goncalves, Email: joao.goncalves@ff.ul.pt.
References
- 1.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret J, Gruest C, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozen-baum W, Montagnier L. Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B, White G, Foster P, Markham PD. Science. 1984;224:500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 3.Flexner F. Nat Rev Drug Discov. 2007;12:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]
- 4.Hughes A, Barber T, Nelson M. J Infect. 2008;57:1–10. doi: 10.1016/j.jinf.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Adamson CS, Freed EO. Drug Discov Today. 2008;13:424–432. doi: 10.1016/j.drudis.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortez KJ, Maldarelli F. Viruses. 2011;3:347–378. doi: 10.3390/v3040347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merry T, Astrautsova S. Biotechnol Appl Biochem. 2010;56:103–109. doi: 10.1042/BA20100010. [DOI] [PubMed] [Google Scholar]
- 8.Arts EJ, Hazuda DJ. Cold Spring Harb Perspect Med. 2012;2:a007161. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingale KB, Bhatia MS. Antivir Chem Chemother. 2011;17:95–105. doi: 10.3851/IMP1740. [DOI] [PubMed] [Google Scholar]
- 10.Pace P, Rowley M. Curr Opin Drug Discov Dev. 2008;11:471–479. [PubMed] [Google Scholar]
- 11.Semenova EA, Marchand C, Pommier Y. Adv Pharmacol. 2008;56:199–228. doi: 10.1016/S1054-3589(07)56007-2. [DOI] [PubMed] [Google Scholar]
- 12.Esposito D, Craigie R. Adv Virus Res. 1999;52:319–333. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 13.Engelman A, Bushman FD, Craigie R. EMBO J. 1993;12:3269–3275. doi: 10.1002/j.1460-2075.1993.tb05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Gent DC, Vink C, Groeneger AA, Plasterk RH. EMBO J. 1993;12:3261–3267. doi: 10.1002/j.1460-2075.1993.tb05995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng R, Jenkins TM, Craigie R. Proc Natl Acad Sci USA. 1996;93:13659–13664. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SP, Han MK. Biochemistry. 1996;35:3837–3844. doi: 10.1021/bi952056p. [DOI] [PubMed] [Google Scholar]
- 17.Burke CJ, Sanyal G, Bruner MW, Ryan JA, LaFemina RL, Robbins HL, Zeft AS, Middaugh CR, Cordingley MG. J Biol Chem. 1992;267:9639–9644. [PubMed] [Google Scholar]
- 18.Bushman FD, Engelman A, Palmer I, Wingfield P, Craigie R. Proc Natl Acad Sci USA. 1993;90:3428–3432. doi: 10.1073/pnas.90.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman A, Craigie R. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vink C, Lutzke RAP, Plasterk RHA. Nucleic Acids Res. 1994;22:4125–4131. doi: 10.1093/nar/22.20.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman A, Hickman AB, Craigie R. J Virol. 1994;68:5911–5917. doi: 10.1128/jvi.68.9.5911-5917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jenkins TM, Engelman A, Ghirlando R, Craigie R. J Biol Chem. 1996;271:7712–7718. doi: 10.1074/jbc.271.13.7712. [DOI] [PubMed] [Google Scholar]
- 24.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchand C, Maddali K, Métifiot M, Pommier Y. Curr Top Med Chem. 2009;9:1016–1037. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sayana S, Khanlou H. Expert Rev Anti Infect Ther. 2008;6:419–426. doi: 10.1586/14787210.6.4.419. [DOI] [PubMed] [Google Scholar]
- 27.Shimura K, Kodama EN. Antivir Chem Chemother. 2009;1920:79–85. doi: 10.3851/IMP1397. [DOI] [PubMed] [Google Scholar]
- 28.Strayer DS, Akkina R, Bunnell BA, Dropulic B, Planelles V, Pomerantz RJ, Rossi JJ, Zaia JA. Mol Ther. 2005;6:823–842. doi: 10.1016/j.ymthe.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Rossi JJ, June CH, Kohn DB. Nat Biotechnol. 2007;25:1444–1454. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bovolenta C, Porcellini S, Alberici L. Curr Pharm Biotechnol. 2012 doi: 10.2174/138920101405131111104009. Epub ahead of print http://www.ncbi.nlm.nih.gov/pubmed/22429132. Abstract. [DOI] [PubMed]
- 31.Lo AS, Zhu Q, Marasco WA. Handb Exp Pharmacol. 2008;181:343–373. doi: 10.1007/978-3-540-73259-4_15. [DOI] [PubMed] [Google Scholar]
- 32.Chen SY, Bagley J, Marasco WA. Hum Gene Ther. 1994;5:595–601. doi: 10.1089/hum.1994.5.5-595. [DOI] [PubMed] [Google Scholar]
- 33.Marasco WA. Immunotechnology. 1995;1:1–19. doi: 10.1016/1380-2933(95)00001-1. [DOI] [PubMed] [Google Scholar]
- 34.Cattaneo A, Biocca S. Intracellular Antibodies: Development and Applications. Springer-Verlag Incorporated; New York, NY: 1997. [Google Scholar]
- 35.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Pope SH, Riordan GS, Whitlow M. Science. 1988;242:423– 426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 36.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotný J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, Oppermann H. Proc Natl Acad Sci USA. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen PA. Methods Mol Biol. 2002;178:367–378. doi: 10.1385/1-59259-240-6:367. [DOI] [PubMed] [Google Scholar]
- 38.Erdag B, Balcioglu BK, Bahadir AO, Serhatli M, Kacar O, Bahar A, Seker UO, Akgun E, Ozkan A, Kilic T, Tamerler C, Baysal K. Biotechnol Appl Biochem. 2011;58:412–422. doi: 10.1002/bab.61. [DOI] [PubMed] [Google Scholar]
- 39.Lo AS, Zhu Q, Marasco WA. Handb Exp Pharmacol. 2008;181:343–373. doi: 10.1007/978-3-540-73259-4_15. [DOI] [PubMed] [Google Scholar]
- 40.Ramcharan J, Colleluori DM, Merkel G, Andrake MD, Skalka AM. Retrovirology. 2006;21:3–34. doi: 10.1186/1742-4690-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi J, Cheng H, Andrake MD, Dunbrack RL, Jr, Roder H, Skalka AM. J Biol Chem. 2003;277:12164–12174. doi: 10.1074/jbc.M105072200. [DOI] [PubMed] [Google Scholar]
- 42.Yi J, Skalka AM. Biopolymers. 2000;55:308–318. doi: 10.1002/1097-0282(2000)55:4<308::AID-BIP1004>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Yi J, Arthur JW, Dunbrack RL, Jr, Skalka AM. J Biol Chem. 2000;275:38739–38748. doi: 10.1074/jbc.M005499200. [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa T, Okui N, Kobayashi N, Sakuma R, Kitamura T, Kitamura Y. J Virol. 1999;73:4475–4480. doi: 10.1128/jvi.73.5.4475-4480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamura Y, Ishikawa T, Okui N, Kobayashi N, Kanda T, Shimada T, Miyake K, Yoshiike K. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:105–114. doi: 10.1097/00042560-199902010-00001. [DOI] [PubMed] [Google Scholar]
- 46.Levy-Mintz P, Duan L, Zhang H, Hu B, Dornadula G, Zhu M, Kulkosky J, Bizub-Bender D, Skalka AM, Pomerantz RJ. J Virol. 1996;70:8821–8832. doi: 10.1128/jvi.70.12.8821-8832.1996. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 47.Barsov EV, Huber WE, Marcotrigiano J, Clark PK, Clark AD, Arnold E, Hughes SH. J Virol. 1996;70:4484–4494. doi: 10.1128/jvi.70.7.4484-4494.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nilsen BM, Haugan IR, Berg K, Olsen L, Brown PO, Helland DE. J Virol. 1996;70:1580–1587. doi: 10.1128/jvi.70.3.1580-1587.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rader C. Methods Protocols. 2009;525:101–128. doi: 10.1007/978-1-59745-554-1_5. [DOI] [PubMed] [Google Scholar]
- 50.Steinberger P, Sutton JK, Rader C, Elia M, Barbas CF., III J Biol Chem. 2003;275:36073–36078. doi: 10.1074/jbc.M002765200. [DOI] [PubMed] [Google Scholar]
- 51.Rader C, Ritter G, Nathan S, Elia M, Gout I, Jungbluth AA, Cohen LS, Welt S, Old LJ, Barbas CF., III J Biol Chem. 2000;275:13668–13676. doi: 10.1074/jbc.275.18.13668. [DOI] [PubMed] [Google Scholar]
- 52.Aires da Silva F, Santa-Marta M, Freitas-Vieira A, Mascarenhas P, Barahona I, Moniz-Pereira J, Gabuzda D, Goncalves J. J Mol Biol. 2004;340:525–542. doi: 10.1016/j.jmb.2004.04.062. [DOI] [PubMed] [Google Scholar]
- 53.Goncalves J, Silva F, Freitas-Vieira A, Santa-Marta M, Malhó R, Yang X, Gabuzda D, Barbas C., 3rd J Biol Chem. 2002;277:32036–32045. doi: 10.1074/jbc.M201906200. [DOI] [PubMed] [Google Scholar]
- 54.Corte-Real S, Collins C, Aires da Silva F, Simas JP, Barbas CF, III, Chang Y, Moore P, Goncalves J. Blood. 2005;106:3797–3802. doi: 10.1182/blood-2005-04-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbas CF, III, Kang AS, Lerner RA, Benkovic SJ. Proc Natl Acad Sci USA. 1991;88:7978–7982. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, Kitamura T. Mol Cell Biol. 1998;18:3871–3879. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eberhardy SR, Goncalves J, Coelho S, Segal DJ, Berkhout B, Barbas CF., III J Virol. 2006;80:2873–2883. doi: 10.1128/JVI.80.6.2873-2883.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Craigie R, Hickman A, Engelman A. In: HIV: A Practical Approach. Karn J, editor. Vol. 2. IRL Press; Oxford, United Kingdom: 1995. pp. 53–71. [Google Scholar]
- 59.Li M, Craigie R. J Biol Chem. 2005;280:29334–29339. doi: 10.1074/jbc.M505367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kimpton J, Emerman M. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li XY, Guo F, Zhang L, Kleiman L, Cen S. J Biol Chem. 2007;282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 62.Brussel A, Sonigo P. J Virol. 2003;77:10119–10124. doi: 10.1128/JVI.77.18.10119-10124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Proc Natl Acad Sci USA. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Nat Biotechnol. 2010;8:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, Mi S, Yam P, Stinson S, Kalos M, Alvarnas J, Lacey SF, Yee JK, Li M, Couture L, Hsu D, Forman SJ, Rossi JJ, Zaia JA. Sci Transl Med. 2010;16:36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spieker-Polet H, Sethupathi P, Yam PC, Knight KL. Proc Natl Acad Sci USA. 1995;92:9348–9352. doi: 10.1073/pnas.92.20.9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GM, Cox D, Rajpal A, Pons J. Proc Natl Acad Sci USA. 2009;106:20216–20221. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.