

Abstract
Obesity-associated insulin resistance is accompanied by an alteration in the Th1/Th2 balance in adipose tissue. T-bet (Tbx21) is an immune cell transcription factor originally described as the master regulator of Th1 cell development, although is now recognized to have a role in both the adaptive and innate immune systems. T-bet also directs T-cell homing to pro-inflammatory sites by the regulation of CXCR3 expression. T-bet−/− mice have increased visceral adiposity but are more insulin-sensitive, exhibiting reduced immune cell content and cytokine secretion specifically in the visceral fat depot, perhaps due to altered T-cell trafficking. Studies of T-bet deficiency on Rag2- and IFN-γ-deficient backgrounds indicate the importance of CD4+ T cells and IFN-γ in this model. This favorable metabolic phenotype, uncoupling adiposity from insulin resistance, is present in young lean mice yet persists with age and increasing obesity. We suggest a novel role for T-bet in metabolic regulation.
Keywords: IFN-γ, T-bet, Th1, adipose, diabetes, immune, immunometabolism, inflammation, insulin resistance, metabolism, obesity
The close association between nutrient excess and alterations in the cellular and molecular mediators of inflammation and immunity is being increasingly appreciated. This is particularly evident in the development of obesity and insulin resistance. Insulin resistance is a major pathological feature of obesity, type 2 diabetes, and cardiovascular disease, causes of considerable morbidity and mortality worldwide. Multiple factors are thought to contribute to systemic insulin resistance, and work over the past decade has highlighted the role of chronic inflammation in visceral adipose tissue as a key component and promoter of this condition.1-3 Most early studies examined the role of adipose tissue macrophages in obesity-associated insulin resistance, but there is mounting evidence for a key role of the adaptive immune system, particularly T cells, in this process.4-7 We have recently described a novel role for the immune cell transcription factor, T-bet (Tbx21) in immunometabolism.8 Mice deficient for T-bet display greater visceral fat but are paradoxically more insulin-sensitive (Fig. 1). Here, we discuss the unusual metabolic phenotype of the T-bet-deficient mouse and how the absence of T-bet might uncouple obesity and insulin resistance as part of an emerging role for T-bet in metabolic physiology and pathophysiology.
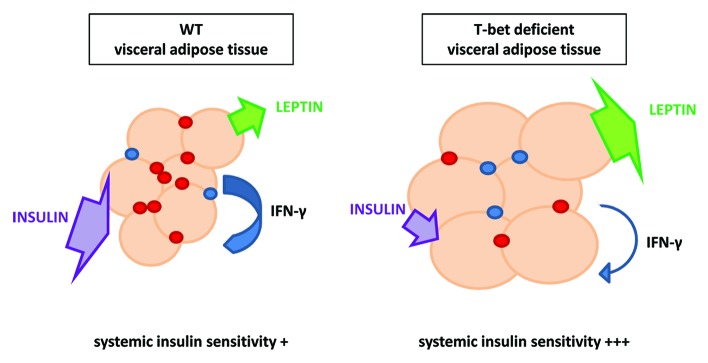
Figure 1. T-bet deficiency uncouples visceral adiposity from insulin resistance. T-bet-deficient mice have fewer immune cells and altered cytokine secretion in visceral fat compared with wild-type mice both in young chow-fed mice and following high fat diet feeding. Despite an increase in visceral fat mass with higher circulating leptin, T-bet−/− mice displayed enhanced systemic insulin sensitivity.
The immune and endocrine systems are functionally and anatomically integrated. We were the first to report that the adipocyte-derived hormone leptin, a key regulator of food intake and energy homeostasis, is also an important immune-modulatory cytokine linking nutritional status with immune function.9,10 Adipose tissue is now recognized to be complex; it comprises not only mature adipocytes, but also preadipocytes, endothelial cells, and immune cells in the stromal vascular fraction. Humoral mediators (adipokines) released from adipose tissue affect whole body energy homeostasis. Immune cells are present in normal lean adipose tissue prior to the onset of obesity, but increasing adiposity is accompanied by significant changes in the immune cell populations within fat. This alteration of immune cell populations in obesity is thought to lead to the adverse metabolic complications of obesity through the liberation of pro-inflammatory cytokines, such as interleukin (IL)-1 (IL-1), IL-6, and tumor necrosis factor-α (TNF-α).2,3,11
Alteration in the balance between different T-cell populations within visceral adipose tissue in particular appears to be an early event in obesity. Defined largely by their secreted cytokine profile, CD4+ T cells lineages include T helper (Th) 1, Th2, and Th17, and regulatory T cell (Treg) subsets.12 It is now over a decade since we first reported that the adipocyte-derived hormone leptin, which is increased in obesity, is able to influence T-cell development and survival10 and modulate the T-cell immune response by enhancing T-cell proliferation and inducing the production of pro-inflammatory cytokines such as interferon-γ (IFN-γ).9,13 Indeed, we found that leptin was able to alter the balance between Th1 and Th2 immune responses skewing toward a Th1 phenotype and suppressing Th2 responses.9,13 As with other chronic inflammatory conditions like atherosclerosis, the Th1/Th2 balance is now recognized to be altered in obesity.14,15 Obesity is thought to skew T cells toward a Th1 or pro-inflammatory phenotype in fat and this is associated with the development of insulin resistance. In obesity, the T cell population in adipose tissue changes: there is a significant increase in pro-inflammatory Th1 cell numbers but also a reduction in the proportion of Foxp3+ Tregs.5,6,15 The observed recruitment to, and expansion of, adipose tissue Th1 cells and decline in Tregs has been reported in both obese mice and humans.16 Interestingly, as well as promoting this Th1 bias,9 leptin has more recently been found to have a negative effect on the proliferative capacity of Tregs.17 IFN-γ, the signature pro-inflammatory cytokine secreted by Th1 cells, is a strong activator of macrophages. In contrast, Foxp3+ Tregs are thought to exert anti-inflammatory effects in adipose tissue.5,6 IL-10 is an anti-inflammatory cytokine produced by Tregs and can protect adipocytes from the negative effects on insulin signaling induced by TNF-α.18 Thus in obesity, there are alterations in adipose tissue immune cell populations, which regulate the inflammatory environment; this impacts on energy balance and glucose homeostasis.
Several recent studies have reported that T cells influence glucose homeostasis in mice with diet induced obesity.5-7,15 T-bet (Tbx21) is a member of the T-box transcription factor family member and regulates the differentiation and function of immune cells.19-21 Expression of T-bet is almost exclusively restricted to the immune system, although its expression in the olfactory bulb has been reported its functional role in this location is unclear.22,23 T-bet is best known, and was originally described, for its role in the development of Th1 cells, where it is directly responsible for the transactivation of the signature Th1 cytokine, IFN-γ.19 Given the recognition of altered Th1/Th2 balance in obesity-associated insulin resistance, it was therefore not surprising that we found T-bet-deficient mice were more insulin-sensitive than wild-type (WT) control mice.8 We also observed, however, than T-bet deficiency was associated with a modest increase in visceral adiposity. As increased visceral adipose tissue mass is usually associated with worsening insulin sensitivity rather than improved insulin sensitivity;24 this finding was unusual. Furthermore, the dissociated phenotype of increased visceral adiposity and better insulin sensitivity was not restricted to mice following high fat diet feeding, but was also present in mice as young as 8 weeks old, suggesting a role for T-bet in metabolic physiology.
So why are T-bet-deficient mice fatter than WT mice? The answer to this seemingly simple question is not so clear. We found that despite being heavier, the food intake of T-bet−/− mice was not greater and was, in fact, cumulatively slightly less than that observed in wild-type mice. Energy expenditure was, however, significantly less in T-bet-deficient mice and this appeared to be in part due to reduced physical activity. The mechanism for this intriguing observation remains unknown, but is unlikely to be due to a direct central effect, given T-bet’s near exclusive immune-restricted expression. Increased adiposity has been reported without adverse metabolic effects in the context of transgenic overexpression of adiponectin in the obese, leptin-deficient ob/ob mouse.25 It is possible that T-bet may interact with peroxisome proliferator-activated receptor gamma (PPAR-γ) signaling pathways. PPAR-γ is a key molecular mediator of both adipogenesis and insulin sensitivity.26 PPAR-γ agonists, such as pioglitazone, which are used as insulin sensitizing agents in the treatment of type 2 diabetes, also increase adiponectin levels25,27,28 and are associated with a concomitant increase in fat mass. These models however, differ from T-bet deficiency as their improved insulin sensitivity is accompanied by the expansion of the subcutaneous adipose depot but a reduction in mass of the visceral adipose tissue depot, rather than a significant increase in visceral adipose tissue mass as observed in the T-bet-deficient mouse. Interestingly, PPAR-γ expressed in visceral adipose tissue Tregs has recently been found to contribute substantially to the insulin sensitizing effect of pioglitazone as well as the upregulation of several genes involved in lipid metabolism.29 The role of PPAR-γ and its downstream signaling pathways in the adipose and metabolic phenotype of T-bet deficiency therefore warrants further investigation.
Recent years has seen the role of T-bet in the regulation and function of immune cells broaden from one where it was seen primarily as the master regulator of Th1 cell development, to one which involves multiple cells types of both the adaptive and innate immune systems.30-33 Indeed, a wide range of immune cell types from both the adaptive and innate immune system have been implicated in obesity associated adipose tissue inflammation and insulin resistance.5,6,34-36 The potential role of T-bet in the adaptive and innate immune system in this process was dissected by studies in Rag2-deficient mice which lack an adaptive immune system (B, T, and NKT cells). The similar body weight, fat distribution, and glucose homeostasis phenotype we found for T-bet-deficient mice on a Rag2-deficient background (Rag2−/− × T-bet−/−) and Rag2-deficient mice suggested, however, that it is T-bet expression in the adaptive immune system rather than in the innate immune system that is of prime importance to the favorable metabolic phenotype observed T-bet deficiency. Indeed, adoptive transfer of T-bet-deficient CD4+ T cells to a Rag2-deficient host resulted in a modest improvement of insulin sensitivity.
In fat, resident T cells have been found to be highly polarized to a Th1 phenotype with high expression of both T-bet and IFN-γ.6 The role of the signature Th1 cytokine, IFN-γ in the favorable metabolic phenotype of T-bet−/− mice was addressed by studies in IFN-γ-deficient mice. Deficiency of IFN-γ has been reported to improve glucose homeostasis in obesity, although there have been conflicting reports on its effect on adiposity.37-39 Production of IFN-γ, unlike T-bet, is not immune restricted and the relationship between the two is not a simple one: IFN-γ is both upstream and downstream of T-bet and not all effects of IFN-γ are T-bet-dependent and vice versa.20,40 In fact, the phenotypes of T-bet and IFN-γ deficiency are immunologically distinct.41,42 However, as we found that loss of T-bet on an IFN-γ-deficient background did not confer any additional metabolic benefit, it is likely that the improved glucose homeostasis observed in T-bet deficiency is IFN-γ-dependent. We also demonstrated that mice lacking T-bet on a Rag2-deficient background had significantly diminished IFN-γ levels (likely due to the marked T-bet-dependent reduction in NK cells) yet exhibited similar metabolic phenotype to Rag2-deficient mice that still expressed T-bet in cells of the innate immune system.8 These data are consistent with the emerging importance of adaptive immunity in immunometabolism and, together with the studies in IFN-γ-deficient mice, suggest that IFN-γ from the innate immune system is less important than that originating from cells of the adaptive immune system in the metabolic phenotype of these models.
T-bet−/− mice displayed a reduction of the immune cell content in the visceral fat depot specifically. In contrast, immune cell populations were similar in the subcutaneous fat depot as well as in muscle (unpublished observations). The only exception to these depot specific differences in immune cell content was the expected significant reduction of the natural killer (NK) cell population; NK cells are developmentally dependent on T-bet33,43 and were found to be lower in all tissues examined. The visceral depot specific reduction in immune cells was also seen in young mice, prior to the development of obesity, again indicating a physiological role for T-bet in fat distribution (Fig. 1). The proportion of Foxp3+ Tregs were, however, increased in this depot, consistent with recent reports of a potential role for this cell type in insulin sensitivity.6,29 Furthermore, adoptive transfer of CD4+ T-bet-deficient T cells to young Rag2-deficient mice resulted in an enhanced insulin-sensitive phenotype with fewer visceral adipose tissue T cells, suggesting a key role of T-bet-deficient CD4+ T cells in glucose homeostasis. Interestingly, T-bet specifies a transcriptional program that imprints the homing of T cells to pro-inflammatory sites. We have previously shown that the chemokine receptor CXCR3 is a direct transcriptional target of T-bet.44,45 Subtle changes in the balance of immune cell subtypes in adipose tissue may depend, in part, by differential expression of chemokines and their receptors. We did observe differences in molecules responsible for T cell trafficking in T-bet-deficient mice, such as CXCR3 and its ligands compared with wild-type mice. These and alterations in other trafficking molecules might account for the reduced immune cell numbers we observed in the visceral adipose tissue depot of T-bet−/− mice. It is likely that the subsequent alteration in the immune-inflammatory environment in adipose tissue then impacts on systemic insulin sensitivity. Whether T-bet deficiency improves whole body insulin sensitivity primarily through alterations in adipose tissue immune cell content and cytokine secretion remains to be determined. Alterations in leptin, and other adipocytokines and soluble mediators, such as free fatty acids, have broader effects on metabolism by affecting other insulin-sensitive tissues such as muscle, the liver, and the brain. Whether T-bet deficiency has a direct role in these organs, which may then also contribute to systemic insulin sensitivity, remains to be determined.
The nature and origin of the signals that trigger the pro-inflammatory response in visceral adipose tissue, however, remains unclear. Naïve T-cell activation and proliferation requires cognate recognition of antigen through a peptide/MHC (major histocompatability complex) II complex. Our original report that leptin able to enhance IFN-γ production from T cells has recently been confirmed9,13,46 and this has been found to induce the expression of MHC II on macrophages and adipocytes.37,46 The expression of MHC II on adipocytes is an early response to obesity. High fat diet-fed mice deficient for MHC II on adipocytes have reduced adipose tissue inflammation and are more insulin-sensitive.46 We found significant quantitative and qualitative differences between the immune cell and cytokine secretion profile of the visceral adipose tissue depot and the subcutaneous depot in T-bet-deficient mice. Depot differences in adipokine expression are well recognized47 and are likely to play a role in the increased risk of metabolic syndrome associated with enhanced visceral adiposity.48 Adipocytes express functional pattern recognition receptors suggesting their ability to respond to bacterial and viral antigens.49 This may be of relevance to the visceral adipose tissue depot in particular as it is in close proximity to the gut and the intestinal microbiota. Pre-adipocytes offer the first line of defense for translocation of antigens by exhibiting both antimicrobial and phagocytic activity. Recent reports suggest that this depot may be in a state of “immune activation” with increased expression of MHC class II on macrophages and, given the observation of a restricted T-cell receptor repertoire, that the accumulation of T cells in this location may be antigen-driven.5,6 Differential expression of chemokine receptors plays a key part in the process of T-cell migration to inflammatory sites, and as stated above, T-bet is known to regulate molecules that are critical to homing of T cells to inflamed sites.8,45 Whether any potential inciting antigens are gut-derived microbial, or intrinsic adipocyte, in origin remains to seen.
Deficiency of T-bet in the immune system therefore appears uncouple visceral adiposity from insulin resistance (Fig. 1). This effect appears to be dependent particularly on CD4+ T cells and the Th1 signature cytokine IFN-γ and adds to the emerging body of data suggesting a key role of the adaptive immune system in obesity-associated insulin resistance. However, T cells are already present fat depots prior to the onset of obesity. We have found that deficiency of T-bet is able to further enhance insulin sensitivity even in the lean state. To our knowledge, this is the first description of an immune-restricted molecule directly influencing metabolic physiology. A challenge now in the emerging field of immunometabolism is identification of the downstream molecular interactions through which T-bet is able to impact on adiposity and insulin action and the potential molecular drivers that seem to target this process to the visceral adipose tissue depot in particular. Targeting the T-bet axis may one day offer a novel therapeutic strategy in the treatment of conditions associated with insulin resistance, such as obesity and Type 2 diabetes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
J.K.H. and G.M.L. are funded by the Medical Research Council (MR/K002996/1 to J.K.H.; G0802068 to G.M.L.). G.M.L. is also funded by the Wellcome Trust (WT091009) and supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/26220
References
- 1.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 2.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, Fischer-Posovszky P, Barth TF, Dragun D, Skurk T, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1304–10. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 5.Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–9. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun. 2009;384:482–5. doi: 10.1016/j.bbrc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Stolarczyk E, Vong CT, Perucha E, Jackson I, Cawthorne MA, Wargent ET, Powell N, Canavan JB, Lord GM, Howard JK. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metab. 2013;17:520–33. doi: 10.1016/j.cmet.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 10.Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, Lechler RI, Bloom SR. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. 1999;104:1051–9. doi: 10.1172/JCI6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–74. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 12.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–69. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lord GM, Matarese G, Howard JK, Bloom SR, Lechler RI. Leptin inhibits the anti-CD3-driven proliferation of peripheral blood T cells but enhances the production of proinflammatory cytokines. J Leukoc Biol. 2002;72:330–8. [PubMed] [Google Scholar]
- 14.Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.RES.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 15.Lumeng CN, Maillard I, Saltiel AR. T-ing up inflammation in fat. Nat Med. 2009;15:846–7. doi: 10.1038/nm0809-846. [DOI] [PubMed] [Google Scholar]
- 16.Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, Perry K, Hazey J, Kampfrath T, Kollengode M, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One. 2011;6:e16376. doi: 10.1371/journal.pone.0016376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Rosa V, Procaccini C, Calì G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, Matarese G. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26:241–55. doi: 10.1016/j.immuni.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/S0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 20.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–42. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 21.Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, Lord GM, Jenner RG. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. 2012;3:1268. doi: 10.1038/ncomms2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faedo A, Ficara F, Ghiani M, Aiuti A, Rubenstein JL, Bulfone A. Developmental expression of the T-box transcription factor T-bet/Tbx21 during mouse embryogenesis. Mech Dev. 2002;116:157–60. doi: 10.1016/S0925-4773(02)00114-4. [DOI] [PubMed] [Google Scholar]
- 23.Mitsui S, Igarashi KM, Mori K, Yoshihara Y. Genetic visualization of the secondary olfactory pathway in Tbx21 transgenic mice. Neural Syst Circuits. 2011;1:5. doi: 10.1186/2042-1001-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–20. doi: 10.1016/j.cmet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–37. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 27.Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, Yano W, Ogata H, Tokuyama K, Takamoto I, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–55. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 28.Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–60. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 29.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–53. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, Lord GM, Glickman JN, Glimcher LH. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A. 2003;100:7749–54. doi: 10.1073/pnas.1332767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–94. doi: 10.1016/S1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 35.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–7. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, Andoh Y, Fujii S, Iwabuchi K, Onoé K, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2010;30:193–9. doi: 10.1161/ATVBAHA.109.198614. [DOI] [PubMed] [Google Scholar]
- 37.Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103:467–76. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong N, Fam BC, Cempako GR, Steinberg GR, Walder K, Kay TW, Proietto J, Andrikopoulos S. Deficiency in interferon-gamma results in reduced body weight and better glucose tolerance in mice. Endocrinology. 2011;152:3690–9. doi: 10.1210/en.2011-0288. [DOI] [PubMed] [Google Scholar]
- 39.O’Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, Marks DL. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism. 2012;61:1152–61. doi: 10.1016/j.metabol.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lametschwandtner G, Biedermann T, Schwärzler C, Günther C, Kund J, Fassl S, Hinteregger S, Carballido-Perrig N, Szabo SJ, Glimcher LH, et al. Sustained T-bet expression confers polarized human TH2 cells with TH1-like cytokine production and migratory capacities. J Allergy Clin Immunol. 2004;113:987–94. doi: 10.1016/j.jaci.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 42.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soderquest K, Powell N, Luci C, van Rooijen N, Hidalgo A, Geissmann F, Walzer T, Lord GM, Martín-Fontecha A. Monocytes control natural killer cell differentiation to effector phenotypes. Blood. 2011;117:4511–8. doi: 10.1182/blood-2010-10-312264. [DOI] [PubMed] [Google Scholar]
- 44.Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, Lord GM. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A. 2009;106:17876–81. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–9. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng T, Lyon CJ, Minze LJ, Lin J, Zou J, Liu JZ, Ren Y, Yin Z, Hamilton DJ, Reardon PR, et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 2013;17:411–22. doi: 10.1016/j.cmet.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes. 2007;56:1010–3. doi: 10.2337/db06-1656. [DOI] [PubMed] [Google Scholar]
- 49.Bès-Houtmann S, Roche R, Hoareau L, Gonthier MP, Festy F, Caillens H, Gasque P, Lefebvre d’Hellencourt C, Cesari M. Presence of functional TLR2 and TLR4 on human adipocytes. Histochem Cell Biol. 2007;127:131–7. doi: 10.1007/s00418-006-0230-1. [DOI] [PubMed] [Google Scholar]