

Abstract
Histone deacetylases (HDACs) are important regulators of gene expression. Specific structural features and distinct regulative mechanisms rationalize the separation of the 18 different human HDACs into four classes. The class II comprises a heterogeneous group of nuclear and cytosolic HDACs involved in the regulation of several cellular functions, not just limited to transcriptional repression. In particular, HDAC4, 5, 7 and 9 belong to the subclass IIa and share many transcriptional partners, including members of the MEF2 family. Genetic studies in mice have disclosed the fundamental contribution of class IIa HDACs to specific developmental/differentiation pathways. In this review, we discuss about the recent literature, which hints a role of class IIa HDACs in the development, growth and aggressiveness of cancer cells.
Keywords: chromatin, differentiation, apoptosis, MEF2, epigenetic, HDAC4, HDAC5, HDAC7, HDAC9, cancer, RUNX2
Introduction
Chromatin organization plays a key role in the control of gene expression. In the nucleosome, DNA is wrapped around a core of positively charged proteins: the histones [1]. Post-translational modifications (PTM) of histones such as acetylation, phosphorylation, ubiquitination and methylation regulate chromatin architecture in response to environmental fluctuations. These PTMs, by locally influencing the accessibility to DNA of transcription factors, link extracellular signals to changes in gene expression. The final transcriptional landscape on a given enhancer/promoter is provided by the interplay between different PTMs of histones and epigenetic changes on DNA itself [2].
Acetylation at -amino groups of conserved lysine residues influences the activity of several proteins including histones, transcription factors and cytoskeletal elements. Lysines acetylation is controlled by the antagonistic engagement of two families of enzymes: the histone acetyltransferases (HATs) and the histone deacetylases (HDACs). On histones, lysines acetylation weakens the interaction of their positively charged tails with the negative backbone of DNA, thus augmenting chromatin accessibility. This loosening favours the recruitment of transcription factors at the promoter of specific genes. HDACs, on the opposite, by catalysing the removal of acetyl groups promote chromatin condensation and repress gene transcription [3].
The 18 human HDACs can be grouped into four different classes, based on sequence homologies to the yeast orthologues Rpd3, Hda1 and Sir2 (Fig. 1). Class I includes HDAC1, -2, -3 and -8, which show homologies with Rpd3. Class II comprises HDAC4, -5, -6, -7, -9, -10 and presents homology with Hda1. Class II HDACs is further subdivided into classes IIa and IIb. HDAC4, -5, -7 and -9 belong to class IIa whereas HDAC6 and 10 to class IIb. Class III groups Sirt1, 2, 3, 4, 5, 6 and 7, also known as silent information regulators (SIR) and are homologous to Sir2 in yeast. Finally, because HDAC11 shares sequence similarities with both classes I and II it forms, alone, the class IV [4–6]. Classes I, II and IV act with a zinc-dependent mechanism, whereas class III activity is NAD+ dependent and is not affected by the classical HDAC inhibitors. Classes I and II HDACs share homology in the catalytic motif, but diverge for the N-terminal region, which is extended only in class II [7].
Fig 1.
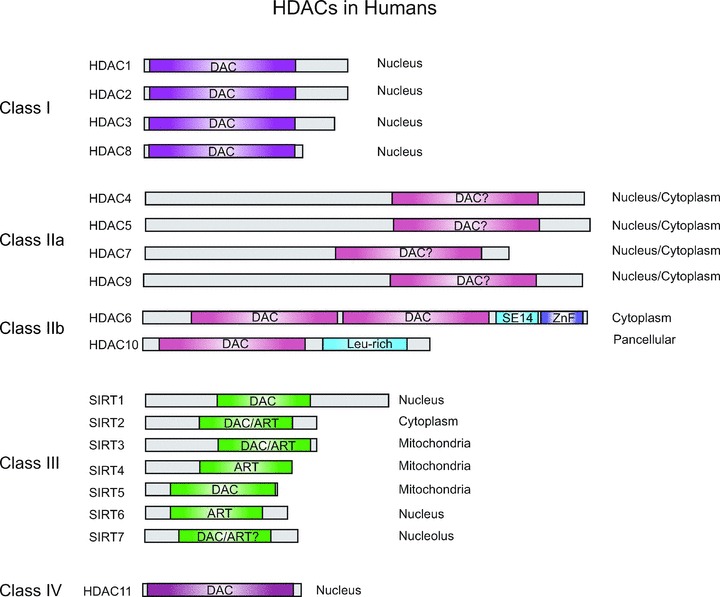
The histone deacetylase family. The histone deacetylase family is subdivided into different subfamilies according to homologies to yeast prototypes. Class I HDACs are mainly nuclear and display a zinc-dependent catalytic activity. Class II HDACs are localized both in the nucleus and in cytoplasm. The seven sirtuins in mammals are localized in different subcellular compartments including mitochondria. All sirtuins have a NAD+-dependent catalytic core domain that may act preferentially as a mono-ADP-ribosyl transferase (ART) and/or NAD+-dependent deacetylase (DAC). SIRT7 exhibits no DAC or ART activities [144]. The class IV HDACs includes HDAC11.
HDACs form high molecular weight complexes with different corepressors, which cooperate in silencing the transcription of several genes involved in various cancer-related functions, such as proliferation, migration, angiogenesis and differentiation. In addition to their chromatin-related functions, HDACs mediate also the deacetylation of a growing list of non-histone proteins, including transcription factors implicated in the control of cell growth, differentiation and apoptosis.
In this review, we will discuss about class IIa HDACs, of clues on their roles in the control of cell proliferation and of other tumour-related functions.
General concepts on class IIa HDACs
Regulation of subcellular localization
To negatively influence the transcriptional process class IIa HDACs need to be in the nucleus, where they associate in macromolecular complexes [8, 9]. An important strategy for restricting class IIa repressive activities is fulfilled by monitoring their subcellular localization. Class IIa HDACs carry both a nuclear localization signal (NLS) and a nuclear export signal (NES) that, once properly exposed, control the enzyme distribution in the two compartments [10, 11]. Inhibition of the Exportin 1 receptor (CRM-1) nuclear transporter by leptomycin B treatment determines the nuclear accumulation of class IIa HDACs, suggesting that these proteins shuttle continuously in and out the nucleus [12–14]. Several extracellular stimuli, through the activation of different kinases, are able to alter the balance between nuclear import and export of these repressors. Phosphorylation takes place on different serine residues, located in the amino-terminal region, which act as docking sites for 14-3-3 proteins binding [13–16]. Consequently, mutations of these residues into alanines relieve class IIa HDACs from kinases control, generating a super-repressive enzyme [12, 17, 18].
The interaction with 14-3-3 proteins influences class IIa subcellular localization through different mechanisms [19–21]. Binding of 14-3-3 proteins can mask the NLS sequence and thereby preventing the interaction with importin-α [13]. Alternatively, these interactions could promote a conformational change and the consequent exposition of the NES in the carboxy-terminal region that favours nuclear export [22].
In addition to the nuclear/cytoplasmic transport, further levels of regulations can influence class IIa activities. For example, HDAC5 mutants carrying an inactive NES sequence cannot exit the nuclei, but are unable to impact on muscle cell differentiation [11]. This result suggests that also the discharge of class IIa from their transcriptional partners could be an important step in the modulation of their repressive ability. On the same context, FRAP experiments have identified different nuclear pools of HDAC4. When HDAC4 is forced to localize in the nucleus, after a block of nuclear export or by mutations in the NES sequence, it binds chromatin for shorter times and in lower percentage, compared to a mutant incompetent for 14-3-3 proteins binding [21]. Hence, also within the nucleus, class IIa HDACs could be subjected to additional regulations. Within this scenario, the promotion of their cytoplasmic accumulation could serve as a mechanism to obtain maximal gene expression.
In response to specific extracellular signals different serine/threonine kinases regulate class IIa HDACs subcellular localization. The first family of protein kinases discovered to play this role was the calcium/calmodulin-dependent protein kinase family (CaMK) and in particular CaMK I and CaMK IV [17, 22–26]. In the muscle model, these kinases promote the nuclear export of class IIa HDACs, expression of MEF2 target genes and, myogenic differentiation. Although CaMK I and CaMK IV are promiscuous class IIa HDACs kinases, CaMK II manifests a specific activity against HDAC4 [27]. HDAC4 can subsequently integrate the export signal by physically associating with HDAC5.
PKD, MARK1 and MARK2, which belong to the same CaMK superfamily, are additional kinases involved in the regulation of class IIa HDACs shuttling [28, 29]. PKD is a downstream effector of the PKC pathway [30, 31]. It acts as an important regulator of class IIa HDACs during angiogenesis, in B and T cells and during muscle remodelling [32–38]. MARK1 and -2 are instead constitutively active enzymes, which target the first serine residue of the 14-3-3 binding site placed in the amino-terminal region [19].
Additional kinases add further complexity to the regulation of class IIa HDACs. The Salt inducible kinase 1 and Mirk/dyrk1B are class IIa kinases able to affect myogenic differentiation [39, 40]. This surplus suggests that multiple pathways control the function of these transcriptional repressors in a tight, quick and adequate manner.
Phosphorylation is a reversible PTM and phosphatases can rapidly and efficiently counteract a kinase’s impact. This general scheme holds true also in the case of class IIa HDACs. The phosphatase inhibitors calyculin A and okadaic acid cause the cytoplasmic accumulation of class IIa HDACs [13, 21, 41]. More detailed studies have discovered that the phosphatases targeting these transcriptional repressors are PP1β, MYPT1, as component of a multiprotein complex [42] and PP2A, which is required for nuclear import [21, 43].
Compared to the mechanisms controlling class IIa nuclear export, the nuclear import and retention pathways are less characterized. Initial discoveries pointed out that association of MEF2 transcription factors with class IIa HDACs promotes their accumulation in the nucleus [44]. Parathyroid hormone related peptide (PTHrP) and Forskolin, which activate protein kinase A (PKA), can promote class IIa HDACs nuclear retention [45]. An effect explained by the increased activity of PP2A. Moreover, PKA can also contribute to nuclear retention by phosphorylating a serine residue in the NLS and thus blocking the association with 14-3-3 proteins [46]. This effect seems to be restricted to HDAC5, because in other class IIa HDACs, such as HDAC7, the ‘RRXS’ motif is missing.
In cardiac myocytes another mechanism has been proposed to participate in the control of nuclear export. Two cysteine residues in the HDAC domain of HDAC4 are important for the formation of an intramolecular bond, which could mask the NES sequence [47]. The oxidation of the thiol groups or the mutation of one cysteine disrupts this regulation and favours the cytoplasmic localization even when Ser/Ala mutations are introduced in the 14-3-3 binding sites.
The control of class IIa activities is not limited to their subcellular localization. Additional PTMs can act on these HDACs. Ubiquitin-dependent degradation and caspase processing can influence class IIa HDACs behaviour. In muscle, degradation of class IIa plays a pivotal role during fibre type switching from fast and glycolytic to slow and oxidative [48]. Serum starvation can also elicit poly-ubiquitination and degradation of HDAC4. In this case, the kinase GSK3β seems to play an important role [49].
Apoptosis promotes cleavage of HDAC4 and HDAC7. In the case of HDAC4, the caspase-dependent cleavage generates a nuclear form of the enzyme with pro-apoptotic activity [50]. HDAC4 can be also sumoylated by RanBP2, at lysine 559 during the passage through the nuclear pore, but how this PTM influence HDAC4 repressive activity is presently unknown [51].
Finally, control of class IIa HDACs expression is an integrated process [52]. In particular, translation control is exerted by the action of specific microRNAs, and contributes to the tight regulation of muscle differentiation [53, 54].
Binding partners
To assert their repressive potential, class IIa HDACs must reside on chromatin. The interaction with selected transcription factors is the ‘conditio sine qua non’ for the chromatin recruitment of class IIa HDACs. The best-characterized class IIa partners are members of the MEF2 transcription factors family, which fine tune differentiation, cell growth and survival [17, 55–58]. Binding to MEF2 involves 12 well-conserved amino acids sited in the amino-terminal part of the deacetylase [10]. The HDAC–MEF2 interaction is dynamic and in differentiating myoblasts blocking the PI3K pathway enhances the stability of the MEF2–HDAC complex, maintaining chromatin in a repressive state [59]. Furthermore, this association can also trigger the sumoylation of MEF2s, which decreases the transcriptional potential [60].
MEF2 transcription factors are characterized by the presence of a MADS box DNA-binding domain, which is common to the serum response factor (SRF), another partner of class IIa HDACs [61]. SRF is required for cell differentiation in diverse contexts and class IIa HDACs are important regulators of its function [25, 53, 62]. Runx2 is another transcription factor modulated by the association with these repressors and, again, the interaction involves the amino-terminal region of the deacetylases [63]. Binding to Runx2 has multiple outcomes. It favours the deacetylation of target promoters and of the transcription factor, which is thus targeted for degradation [64]. Besides, in a sort of ‘full service’, HDAC4 is also capable of repressing transcription of RUNX2 gene [63].
MEF2, SRF and Runx2 are not the only transcriptional partners of class IIa HDACs. GATA [65, 66], Forkhead [67], Ying and Yang 1 [68, 69] Hypoxia-inducible factors (HIFs), and several others are proposed partners of class IIa HDACs. For many of these putative partners, validation of the interaction would be desirable at the level of endogenous proteins.
Class IIa HDACs can be recruited to chromatin also by the association with nuclear hormone receptors. Estrogen receptor can interact with HDAC4, HDAC5 and HDAC9 [70, 71] and this association requires the presence of the MEF2-binding sequence [71]. Also androgen receptor (AR) can make complexes with class IIa HDACs, specifically HDAC4 and HDAC7 [72, 73]. In particular, HDAC4 seems to negatively modulate AR promoting its sumoylation [73]. Furthermore, class IIa HDACs can combine with different nuclear hormone corepressors such as REA, ARR19 or RIP140, which negatively impact gene transcription [74–76].
The ability to interact with different partners, forming macromolecular structures is even important for the correct establishment of chromatin modifications. In fact, class IIa HDACs associate with HDAC3 in high molecular weight complexes, which manifest the deacetylating activity [9]. In this circumstance, the interaction involves the carboxy-terminal domain as a docking site for the recruitment of the class I HDAC (Fig. 2).
Fig 2.
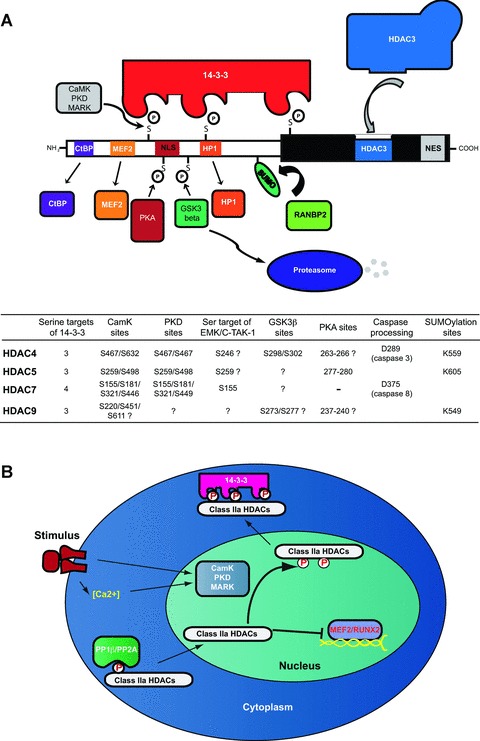
Class IIa HDACs domain organization and shuttling regulation. (A) The amino-terminal domain is subjected to different post-translational modifications such as phosphorylation, sumoylation and caspase cleavage. CaMK, PDK and MARK kinases phosphorylate several Ser residues in the amino-terminal domain thereby promoting the association with 14-3-3 proteins. Additional kinases such as PKA and GSKβ affect class IIa HDACs stimulating nuclear retention and protein degradation respectively. Different interactors associate with the N-terminal domain of class IIa HDACs such as CtBP, MEF2 or HP1, whereas the C-terminal domain can recruit HDAC3-NCoR/SMRT complex to promote Lys deacetylation. In the chart are reported aminoacidic residues involved in several post-translational processes including phosphorylation, sumoylation, caspase processing. (B) Extracellular stimuli that are able to activate CaMK, PDK and MARK kinases trigger the phosphorylation of class IIa HDACs and their association with 14-3-3 proteins, thereby promoting nuclear export. Conversely, the removal of the phosphate groups catalysed by PP1 or PP2A stimulates class IIa HDACs nuclear accumulation and repression of MEF2-dependent gene expression.
Finally, class IIa can interact with H3K9 methyltransferase SUV39H1 and HP1, structural components of heterochromatin that are essential for compact DNA packaging [26]. These additional players with nucleosomal remodelling properties point to the existence of complex molecular machineries devote to the orchestration of the epigenetic changes.
An additional peculiar feature of the amino-terminal region of class IIa HDACs is the presence of a glutamine rich domain (aa 62–129) [77]. This domain folds in straight α-helix that assemble in tetramers. It has been proposed that glutamine-rich domain could modulate dynamic protein–protein interactions thus possibly justifying the long list of class IIa HDACs interactors.
Catalytic activity
Although in vertebrates HDACs class IIa display catalytic sites similar to class I, surprisingly, they are inefficient enzymes and only a weak deacetylase activity can be measured in vitro on acetylated lysines [9, 78]. Classes I and IIa both present an active zinc ion in their catalytic pocket, which is surrounded by two His-Asp dyads [79]. The difference that accounts for the scant deacetylating activity of vertebrate class IIa HDACs lies in the substitution of the conserved ‘catalytic’ tyrosine with a histidine. This tyrosine acts as a transition-state stabilizer, a function that is not accomplished by the replaced histidine. In HDAC4, the mutation H976Y results in a gain of function and restores the catalytic activity comparable to that of class I HDACs [78].
Two studies have revealed the crystal structure of HDAC7 and HDAC4 catalytic domains. Both reported the presence of a class II specific additional zinc-binding domain, next to the active site, that coordinates four conserved amino acids and connects two segments of the protein. This site can conform differently according to the presence of inhibitors and seems to be involved in substrate specificity [80, 81].
Class IIa HDAC orthologues in model organisms
Class IIa HDAC orthologues have been described in Drosophlia melanogaster and Caenorhabditis elegans. These orthologues conserve the critical tyrosine in the HDAC domain and possess an elevated deacetylase activity [7, 82]. In C. elegans, the MEF2 orthologue MEF-2 and the class II HDAC protein HDA-4 control the expression of chemoreceptors in chemosensory neurons. Expression of chemoreceptors is under the control of integrated environmental signals such as food, pheromone and neuronal activity. Phosphorylation, by KIN-29/SIK (salt-inducible kinase) and 14-3-3 binding unlock HDA-4 repression [83]. Interestingly in C. elegans HAD-4 is not subjected to nuclear-cytoplasmic shuttling, probably because of the lack of a nuclear export signal [7, 83]. Although some peculiarities can be observed in C. elegans model, overall the principal mechanism keeping in check class IIa HDAC is conserved through the evolution.
Sequence analysis of the D. melanogaster class IIa orthologue, dHDAC4 confirms the presence of MEF-2 and 14-3-3 binding sites [7]. Experimental data indicate an involvement of dHDAC4 in the segmentation regulatory pathway and suggest that a complex transcriptional network is involved in the regulation of dHDAC4 expression [82].
HDAC4
HDAC4 and differentiation
HDAC4 expression is widespread in different tissues and organs, by contrast the generation of knock-out mice has testified an irreplaceable contribution of this repressor in the regulation of the ossification process. The lack of HDAC4 determines the appearance of numerous skeletal abnormalities because of premature endochondral ossification [63], which affected the development of mice and caused problems to the skull, the breathing and mobility. Initially, the phenotypic effect of HDAC4 mutant mice was explained by the dysregulation of Runx2 transcriptional activity. Runx2 is a central player in endochondral bone ossification, by promoting chondrocyte hypertrophy [84]. A careful examination of the role of MEF2 transcription factors in bone development, using both a conditional knock-out and transgenic mice bearing MEF2 mutant forms, discovered their requirement for the correct bone development. In particular, MEF2 family members promote the endochondral ossification regulating the hypertrophic gene program in chondrocytes [55]. Moreover hemizygous deletion of MEF2C rescued the phenotypic abnormalities seen in HDAC4 null mice demonstrating the need of a balance between HDAC4 and MEF2C for a correct ossification program. In adults, HDAC4 plays important role in the control of neuro-muscolar homeostasis. HDAC4 can coordinate the genetic response to denervation [85, 86]. Moreover, during reinnervation, HDAC4 is under the control miR-206 and this circuit can influence ALS progression [87].
HDAC4 and cancer
Several publications have underscored many roles played by HDAC4 in cancer cells. Some results point to a pro-proliferative role of HDAC4 whereas others sustain an anti-proliferative signalling of the deacetylase. For example, down-regulation of HDAC4 or inhibition of its function reduces cancer cell viability [88, 89], but HDAC4 mutations have been found in human breast carcinomas and melanomas [90, 91]. A possible explanation for these contradictory options could be found in the context-dependent status of the PTMs operating on HDAC4 and/or in the context dependent, multi-proteins complexes, recruiting HDAC4.
Initially, HDAC4 was described as a component of the p53 pathway that governs proliferation arrest [92]. Further studies have demonstrate additional connections between HDAC4 and p53, but unfortunately, a clear picture is not available at the moment. HDAC4 can be recruited to G2/M promoters upon DNA damage as the result of a complex formation with NF-Y and p53. On these sites HDAC4 could exert its repressive influence [93, 94]. HDAC4 was also reported to interact with p53BP and being involved in the DNA repair after ionizing radiation [88]. Once a role of HDAC4 in the regulation of DNA repair processes will be confirmed, developing specific ‘HDAC4 inhibitors’ capable of synergizing with DNA damaging agents to impact on cancer cells death, would be a promising option.
Among studies that support a proliferative function for HDAC4, we can include its contribution to the growth of epithelial cancer cells that do not express p53. In this context, down-regulation of HDAC4 causes mitotic blockage because of impairment of chromatids segregation and promotes apoptotic cell death [95]. Pro-growth functions are also suggested from its cellular subtype specific expression. HDAC4 is enriched in the proliferating zone of the colon crypts and is required for the negative modulation of the Cdk inhibitor p21 [96]. Its recruitment on p21 promoter does not involve the p53 binding sites but the Sp1/Sp3 sites. HDAC4 silencing inhibits cancer cell growth in vitro and in vivo [96, 97]. Deregulated expression of this enzyme was observed in hepatocellular carcinoma where its levels are increased because of the lack of two different miRNAs: miR-1 and miR-22 [98, 99]. Also in this context the down-regulation of HDAC4 reduces the growth rate [99]. miR-1 dysfunction and HDAC4 up-regulation, have been reported also in lung cancer cells [100].
As explained earlier, HDAC4 levels are also under the control of the ubiquitin–proteasome system. Growth factors can augment HDAC4 levels by suppressing its poly-ubiquitination. In this case, the molecular switch seems to be represented by GSK3β [49]. Not surprisingly, cancer cells have lost this regulation and HDAC4 levels are maintained elevated also in the absence of growth factors.
Surprisingly, whereas different mechanisms are described to affect HDAC4 expression in cancer cells little is known about the contribution of various oncogenic signalling pathways on its subcellular localization. Although preliminary studies suggested that oncogenic Ras could promote HDAC4 nuclear accumulation [101], how this impact on Ras-mediated transformation is unknown.
In addition, HDAC4 could contribute to cancer development independently from its chromatin attitude. This enzyme is able to associate with HIF1-α, a critical factor activated by hypoxia that plays an important role in tumour progression [102, 103]. The binding of HDAC4 protects HIF1-α from degradation and the use of HDACs inhibitors causes its acetylation and down-regulation [102]. Interestingly, other class IIa HDACs are able to bind HIF1-α and this binding seems to increase its transcriptional activity [104, 105]. Finally, HDAC4 is also a death substrate, cleaved by caspase-3 during apoptotic cell death [50]. The cleavage generates an amino-terminal fragment that can elicit mitochondrial apoptosis when ectopically expressed.
HDAC5
HDAC5 and differentiation
HDAC5 has been suggested to contribute in various cellular contexts to the regulation of differentiation-specific functions. Surprisingly, the generation of the knock-out mouse for HDAC5 did not revealed specific lethal abnormalities during development [56]. In adults, similarly to knock-out mice for HDAC9, the absence of HDAC5 favours the appearance of age-dependent, spontaneous, cardiac hypertrophy [56]. Interestingly, the absence of HDAC5 or HDAC9 protects female mice from myocardial infarction remodelling, possibly through the MEF2-dependent up-regulation of vascular endothelial growth factor (VEGF), which, in turns, promotes neo-angiogenesis [71].
Mice in which HDAC5 and HDAC9 have been contemporary removed were prone to perinatal death for cardiac defects. These defects are the consequence of abnormalities in growth and maturation of cardiomyocytes. It is not yet fully defined at the molecular level the origin of the proliferative/maturation deficit [56]. The contemporary loss of HDAC5 and HDAC9 had also enlightened their essential contribution in heart remodelling during pathological hypertrophic stimulation such as pressure overload or constitutive calcineurin activation [56].
Another context in which HDAC5 seems to carry out a regulatory function is the skeletal muscle. This repressor acts by blocking the differentiation of myoblasts to myotubes. The suppressive effect was the result of the inhibitory activity against MEF2 transcription factors [106]. Interestingly, the lack of a specific class IIa HDACs does not affect the development of the skeletal muscle suggesting, as in the heart, the possible redundant role for class IIa HDACs.
In addition, HDAC5 is also involved in controlling osteoblastic differentiation. In this case its action is to tether RUNX2, a critical transcription factor for osteoblast maturation [107]. In particular, upon TGF-β stimulation, HDAC5 can exert its function by forming a repressive complex with RUNX2 and SMAD3, which promotes RUNX2 degradation [64, 108]. Another piece of the complex puzzle controlling osteoblastic differentiation is provided by miR-2861, which targets HDAC5 and promotes its down-regulation at post-transcriptional level [109].
HDAC5 acts also as a central player in the regulation of the angiogenetic process. Silencing of HDAC5 can increase sprout length and endothelial migration, whereas the silencing of two other class IIa HDACs such as HDAC7 and HDAC9 promoted the opposite effect [110]. The anti-angiogenic behaviour of HDAC5 is independent from MEF2 and affects the secretion of FGF2 and Slit2. Moreover, VEGF stimulation of HUVEC cells promotes HDAC5 phosphorylation and nuclear export thereby stimulating MEF2 dependent transcription and in vitro angiogenesis [111].
HDAC5 and cancer
Although the contribution of HDAC5 to the control of angiogenesis is clearly a cancer-related process, there several other studies pointing out specific contributions of this deacetylase in cell growth and proliferation [112]. Initial observations based on overexpression studies described HDAC5 as a negative regulator of cell proliferation in different cancer cell lines [113]. Following this initial report it has also been found that HDAC5 repressed cyclin D3 promoter in fibroblast [114]. Conversely, HDAC5 could participate in p14 repression through the association with TBX3 in breast cancer cells, although detailed molecular data are lacking [115]. Recently, it was observed that HDAC5 plays an important role in neural stem cell proliferation [116, 117]. Specifically, the knock down of HDAC5 leads to increase expression of PTEN and p21 in a TLX-dependent manner and decreased cell proliferation. In pancreatic cancer, HDAC5 expression is indirectly stimulated by oxysterol binding protein-related protein (ORP) 5, a marker related to poor prognosis in this type of tumour [118]. Besides, the data suggest that HDAC5 can regulate PTEN expression in pancreatic cancer, although not in a TLX-dependent manner.
HDAC7
HDAC7 and vascular endothelium
HDAC7 is highly expressed in heart, lung, in CD4/CD8+ thymocytes, as well as in vascular endothelial cells during embryogenesis [14, 119]. Genetic studies have testified the important role of HDAC7 during early embryogenesis where it regulates angiogenesis and vascular cells homeostasis. Null mice for HDAC7 display embryonic lethality, principally because of defects in mutual adhesion of endothelial cells [57]. Both knock-out mice or mice bearing a form of HDAC7 mutated in the MEF-2 binding motif, die during embryogenesis with similar defects in endothelial cell–cell adhesion and consequent blood vessels damage [57]. Concomitantly, the matrix metalloproteinase 10 (MMP10) is up-regulated, whereas its inhibitor tissue inhibitor of metalloproteinase 1 (TIMP1) is down-regulated. These findings suggest that HDAC7 represses MMP10 expression in a MEF2-dependent manner. Within this scenario, loss of HDAC7 triggers MMP10 expression, leading to alterations in vascular homeostasis and blood vessel defects [57].
TNF-α can also modulate MMP10 expression in an HDAC7-dependent manner. Interaction between HDAC7 and promyelocytic leukaemia protein (PML) is mediated by TNF-α. This interaction influences the localization of HDAC7 to the nuclear bodies and the interaction with MEF2, releasing its repressive embrace and promoting MMP10 transcription [120].
Silencing of HDAC7 in HUVEC cells inhibits their outgrowth and the formation of capillary/tube-like structure, as well as their migration rate. The reduced motility is at least partially caused by altered levels of the growth factor PDGF-B and of its receptor PDGFR-β. On the other hand, decreased levels of HDAC7 do not seem to influence cell adhesion, proliferation or apoptosis in HUVEC cells [121].
HDAC7 can also influence other differentiation pathways. For example, HDAC7 is abundantly expressed in osteoblast-related cell lines, where it associates with Runx2 (an essential regulator of bone formation) thus repressing its transcriptional activity. This repressive influence can be regulated by BMP2 signalling [122, 123].
Is HDAC7 a negative regulator of cell proliferation?
A recent study has denoted that HDAC7 is critical for endothelial cells (EC) proliferation, through the modulation of β-catenin [124]. Overexpression of HDAC7 prevents the G1/S phase transition, and elevated HDAC7 retains β-catenin in the cytoplasm of EC by direct interaction, reducing the expression of its target genes and leading to decreased proliferation. VEGF stimulation induces proteasomal dependent degradation of HDAC7, thus allowing the nuclear localization of β-catenin and proliferation of EC. This work provides a new possible mechanism of gene expression control, exerted by the cytosolic pool of class IIa HDACs.
In breast cancer cells HDAC7 is necessary for estrogen-mediated repression of Reprimo (RPRM), a cell cycle inhibitor and tumour suppressor gene [125]. Silencing of HDAC7 in the breast cancer cell line MCF7 negatively influences 17-β-estradiol (E2)-mediated repression of RPRM, as well as that of other E2-repressed genes such as ENC1, NEDD9, OPG, CXCR4 and CERK. Upon treatment with E2, the estrogen receptor ER-α interacts with HDAC7 and mediates its recruitment to the RPRM promoter. The repressive activity of HDAC7 on RPRM is independent from its catalytic function. Also hormone binding to the AR induces stabilization and nuclear transfer of HDAC7. Nuclear HDAC7 represses AR-target genes transcription, and, here again, this function is mostly independent from the catalytic domain [72].
HDAC7 and cancer
The involvement of HDAC7 in the control of proliferation and angiogenesis is a smoking gun for its role in cancer. Not surprisingly, emerging evidence implicate an association of HDAC7 with cancer progression. High level of cytoplasmic HDAC7 was reported in pancreatic cancer patients [126]. Similarly, in children with acute lymphobastic leukaemia (ALL) high levels of HDAC7 and HDAC9 expression are associated with poor prognosis [127]. Interestingly, because in various cancer and normal cell lines HDAC7 expression was found to be specifically down-regulated by HDAC inhibitors (HDIs) [128, 129], tumours with high levels of HDAC7 could be more sensitive to HDIs treatment. For example, in cancer cells the repressive influence of HDAC7 is required for HIF-1–mediated cyclin D1 down-regulation [130]. In their study, the authors suggested that chemoresistance associated to HIF-1 expression might be dependent on the HDAC7-mediated repression on cyclin D1 expression [130]. In this model, cotreatment with HDIs and classical chemotherapeutics can overcome the drug-resistance phenotype.
It is important to note that HDAC7 is also implicated in the control of apoptosis in T cells, via the repression of Nur77 expression [119]. In cutaneous T cell lymphoma (CTCL), HDIs inhibit HDAC7 at multiple levels, including mRNA expression. HDAC7 inhibition permits Nur77 expression and its translocation to the mitochondria. Here, Nur77 triggers the intrinsic cell death pathway and synergizes with the Bcl-2/Bcl-xL antagonist ABT-737 to promote CTCL apoptosis [131].
During apoptosis HDAC7 is under the control of caspase-8. Processing by caspase-8 abolishes the transcription repression activity of HDAC7 thus, promoting Nur77 expression [132].
Very recently, using a trasposon-mediated mutagenesis approach in mice, HDAC7 has been identified as a cancer-related gene [133]. In conclusion, HDAC7 seems to contribute at several aspects of cancer, from its development up to drug resistance. Understanding the details of its fine regulation/dysregulation is pivotal to approach translation studies to develop new therapeutic approaches.
HDAC9
HDAC9 and differentiation
HDAC9 undergoes alternative splicing that gives origin to multiple isoforms, with putative distinct tissue-specific biological activities [134, 135]. MEF2-interacting transcription repressor/HDAC-related protein (MITR/HDRP) was the first form of HDAC9 to be characterized. This splice variant consists in the N-terminal part of the protein, devoid of the catalytic domain [134, 136]. HDAC9 transcripts are expressed at high levels in brain and skeletal muscle. HDAC9 and also MITR repress MEF2-mediated transcription, arguing again for deacetylase-independent activities. Interestingly, HDAC9 is also an MEF2 target, giving rise to a negative feedback loop that controls its expression [137]. Deletion of HDAC9 in mice leads to cardiac hypertrophy in advanced age and a concomitant exaggerated MEF2-dependent activation [18]. HDAC9 is also involved in the coordination of muscle homeostasis to electrical activity. Neural activity controls HDAC9 expression in muscle cells and HDAC9-null mice are supersensitive to changes in gene expression, as elicited by denervation [138]. More recent studies have unveiled a contribution of HDAC9 in the dendritic growth of cortical neurons [139]. Finally, HDAC9 seems important in the regulation of FOXP3, a transcription factor involved in Treg cells functions [140].
HDAC9 and cancer
qRT-PCR studies on HDACs in brain tumours have revealed a down-regulation of HDAC9 in glioblastoma, compared to low-grade astrocytoma and normal brain [141]. By contrast, high levels of HDAC5 and HDAC9 expression are associated with poor survival in medulloblastoma patients [112]. Both HDAC5 and 9 are highly expressed in the group with poorer prognosis, whereas the up-regulation of only one of the two genes gives an intermediate probability of survival. Silencing of HDAC5 or HDAC9 in medulloblastoma cell lines decreased cell growth and induced apoptosis after caspase activation [112]. High levels of mRNA expression for HDAC9 together with HDAC7 were observed also in bone marrow of children with acute lymphoblastic leukaemia, associated with bad tumour prognosis [127]. The high levels of expression and, more importantly, the role of HDAC9 in cancer cells survival is a strong impulse to investigate in more detail the relationships between HDAC9 and cancer growth. These studies, once confirmed, would fully justify additional efforts to identify specific HDAC9 antagonists.
It is not clear how HDAC9 could impact on cell growth and survival, whether or not the MEF2s are involved. Two recent reports, identifying non-histone proteins as HDAC9 substrates, open new possibilities, in addition to the MEF2 axis. HDAC9 can supervise metabolic response by regulating the acetylation status of USF-1, a transcription factor controlling the expression of fatty acid synthase (FAS). FAS is a key enzyme in the lipogeneic response to fasting and insulin signalling. Under fasting USF-1 is deacetylated by HDAC9 and the genetic program is repressed. USF-1 activation is elicited by its sequential phosphorylation/acetylation as mediated by DNA-PK and PCAF/HAT [142].
ATCD/TRIM29 (ataxia telangiectasia group D-complementing) is another, recently described non-histone substrate of HDAC9. HDAC9 can influence cell proliferation through the deacetylation of ATCD. In this model HDAC9 should act as an oncosuppressor by influencing the binding of ATCD to p53 and thus stimulating p21 expression [143].
The unclear point in both models concerns the extremely low enzymatic activity of class IIa HDACs in vertebrates, based on which we could assume that HDAC9 should recruit other HDACs to promote substrate deacetylation.
Conclusions
It is evident that the contribution of the class IIa HDACs to cancer development is complex. Different roles can be depicted in different tumour models. Paradoxically, there are examples of antagonistic contributions of the different deacetylases in the same cancer type or of the same deacetylase in different tumour models. Several cancer-related activities can be modulated by these repressors: proliferation, migration, motility, genomic stability, survival, apoptosis, angiogenesis and metabolism (Fig. 3). This plethora of biological activities further contributes to augment the landscape complexity. Can we solve and understand this complexity?
Fig 3.
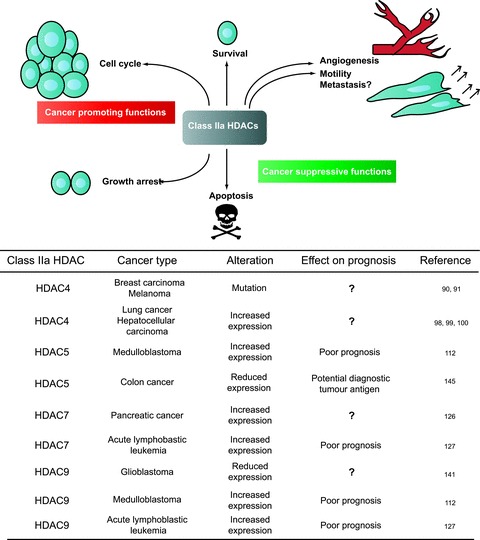
Schematic representation of the different influences exerted by class IIa on cancer-related cellular functions. Class IIa HDACs can participate in different cancer-related processes. According to the context they could behave as tumour promoter or tumour repressive players. In the chart are summarized data on class IIa HDACs status in human cancer.
Class IIa HDACs influence many of the earlier listed activities, as components of multiprotein complexes. Hence, the consequences of changes in class IIa HDACs (PTMs, expression levels and gene sequence) should always be considered respect to the status (mutations, expression levels and specific PTMs) of the other elements of these regulatory complexes. In this intricate scenario, the detailed definition of the contribution, in specific cancer types and in specific stages of cancer development, for each class IIa HDAC seems to be the only possible option that we have to unveil their role in tumourigenesis.
Acknowledgments
Work in Claudio Brancolini Lab is supported by AIRC (Associazione Italiana Ricerca sul Cancro IG-10437), MIUR and Regione FVG (AITT-LR25/07).
Conflict of interest
The authors declare no competing interests.
References
- 1.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 2.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 3.Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–40. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 4.de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25:2873–84. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischle W, Dequiedt F, Fillion M, et al. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem. 2001;276:35826–35. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- 9.Fischle W, Dequiedt F, Hendzel MJ, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 10.Wang AH, Yang XJ. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol Cell Biol. 2001;21:5992–6005. doi: 10.1128/MCB.21.17.5992-6005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21:6312–21. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miska EA, Karlsson C, Langley E, et al. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–40. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kao HY, Downes M, Ordentlich P, et al. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 15.Wang AH, Kruhlak MJ, Wu J, et al. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol Cell Biol. 2000;20:6904–12. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Randall WR, Schneider MF. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J Cell Biol. 2005;168:887–97. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinsey TA, Zhang CL, Lu J, et al. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–11. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang CL, McKinsey TA, Chang S, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–88. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dequiedt F, Martin M, Von Blume J, et al. New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol Cell Biol. 2006;26:7086–102. doi: 10.1128/MCB.00231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carnegie GK, Soughayer J, Smith FD, et al. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol Cell. 2008;32:169–79. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paroni G, Cernotta N, Dello Russo C, et al. PP2A regulates HDAC4 nuclear import. Mol Biol Cell. 2008;19:655–67. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97:14400–5. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao HY, Verdel A, Tsai CC, et al. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem. 2001;276:47496–507. doi: 10.1074/jbc.M107631200. [DOI] [PubMed] [Google Scholar]
- 24.Chawla S, Vanhoutte P, Arnold FJ, et al. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 2003;85:151–9. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- 25.Davis FJ, Gupta M, Camoretti-Mercado B, et al. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J Biol Chem. 2003;278:20047–58. doi: 10.1074/jbc.M209998200. [DOI] [PubMed] [Google Scholar]
- 26.Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22:7302–12. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Backs J, Backs T, Bezprozvannaya S, et al. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol. 2008;28:3437–45. doi: 10.1128/MCB.01611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 2003;4:111–7. doi: 10.1186/gb-2003-4-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73:667–77. doi: 10.1016/j.cardiores.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 30.Zugaza JL, Sinnett-Smith J, Van Lint J, et al. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 1996;15:6220–30. [PMC free article] [PubMed] [Google Scholar]
- 31.Zugaza JL, Waldron RT, Sinnett-Smith J, et al. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–60. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 32.Parra M, Kasler H, McKinsey TA, et al. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J Biol Chem. 2005;280:13762–70. doi: 10.1074/jbc.M413396200. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Li X, Parra M, et al. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A. 2008;105:7738–43. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dequiedt F, Van Lint J, Lecomte E, et al. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med. 2005;201:793–804. doi: 10.1084/jem.20042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matthews SA, Liu P, Spitaler M, et al. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol. 2006;26:1569–77. doi: 10.1128/MCB.26.4.1569-1577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–85. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fielitz J, Kim MS, Shelton JM, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A. 2008;105:3059–63. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim MS, Fielitz J, McAnally J, et al. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol. 2008;28:3600–9. doi: 10.1128/MCB.00189-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berdeaux R, Goebel N, Banaszynski L, et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. 2007;13:597–603. doi: 10.1038/nm1573. [DOI] [PubMed] [Google Scholar]
- 40.Deng X, Ewton DZ, Mercer SE, et al. Mirk/dyrk1B decreases the nuclear accumulation of class II histone deacetylases during skeletal muscle differentiation. J Biol Chem. 2005;280:4894–905. doi: 10.1074/jbc.M411894200. [DOI] [PubMed] [Google Scholar]
- 41.Sucharov CC, Langer S, Bristow M, et al. Shuttling of HDAC5 in H9C2 cells regulates YY1 function through CaMKIV/PKD and PP2A. Am J Physiol Cell Physiol. 2006;291:C1029–37. doi: 10.1152/ajpcell.00059.2006. [DOI] [PubMed] [Google Scholar]
- 42.Parra M, Mahmoudi T, Verdin E. Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev. 2007;21:638–43. doi: 10.1101/gad.1513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin M, Potente M, Janssens V, et al. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc Natl Acad Sci USA. 2008;105:4727–32. doi: 10.1073/pnas.0708455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borghi S, Molinari S, Razzini G, et al. The nuclear localization domain of the MEF2 family of transcription factors shows member-specific features and mediates the nuclear import of histone deacetylase 4. J Cell Sci. 2001;114:4477–83. doi: 10.1242/jcs.114.24.4477. [DOI] [PubMed] [Google Scholar]
- 45.Kozhemyakina E, Cohen T, Yao TP, et al. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol. 2009;29:5751–62. doi: 10.1128/MCB.00415-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha CH, Kim JY, Zhao J, et al. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2010;107:15467–72. doi: 10.1073/pnas.1000462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ago T, Liu T, Zhai P, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–93. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 48.Potthoff MJ, Wu H, Arnold MA, et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007;117:2459–67. doi: 10.1172/JCI31960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cernotta N, Clocchiatti A, Florean C, et al. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol Biol Cell. 2010;22:278–89. doi: 10.1091/mbc.E10-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paroni G, Mizzau M, Henderson C, et al. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell. 2004;15:2804–18. doi: 10.1091/mbc.E03-08-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirsh O, Seeler JS, Pichler A, et al. The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J. 2002;21:2682–91. doi: 10.1093/emboj/21.11.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu F, Pore N, Kim M, et al. Regulation of histone deacetylase 4 expression by the SP family of transcription factors. Mol Biol Cell. 2006;17:585–97. doi: 10.1091/mbc.E05-08-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–33. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Y, Ge Y, Drnevich J, et al. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J Cell Biol. 2010;189:1157–69. doi: 10.1083/jcb.200912093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnold MA, Kim Y, Czubryt MP, et al. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–89. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 56.Chang S, McKinsey TA, Zhang CL, et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–76. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang S, Young BD, Li S, et al. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–34. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 58.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–53. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serra C, Palacios D, Mozzetta C, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28:200–13. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–87. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Molkentin JD, Olson EN. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc Natl Acad Sci USA. 1996;93:9366–73. doi: 10.1073/pnas.93.18.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Margariti A, Xiao Q, Zampetaki A, et al. Splicing of HDAC7 modulates the SRF-myocardin complex during stem-cell differentiation towards smooth muscle cells. J Cell Sci. 2009;122:460–70. doi: 10.1242/jcs.034850. [DOI] [PubMed] [Google Scholar]
- 63.Vega RB, Matsuda K, Oh J, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–66. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 64.Jeon EJ, Lee KY, Choi NS, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. 2006;281:16502–11. doi: 10.1074/jbc.M512494200. [DOI] [PubMed] [Google Scholar]
- 65.Ozawa Y, Towatari M, Tsuzuki S, et al. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood. 2001;98:2116–23. doi: 10.1182/blood.v98.7.2116. [DOI] [PubMed] [Google Scholar]
- 66.Watamoto K, Towatari M, Ozawa Y, et al. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene. 2003;22:9176–84. doi: 10.1038/sj.onc.1206902. [DOI] [PubMed] [Google Scholar]
- 67.Liu R, Wang L, Chen G, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. 2009;69:2252–9. doi: 10.1158/0008-5472.CAN-08-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Feng Y, Xu L, et al. YY1 restrained cell senescence through repressing the transcription of p16. Biochim Biophys Acta. 2008;1783:1876–83. doi: 10.1016/j.bbamcr.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 69.Sucharov CC, Dockstader K, McKinsey TA. YY1 protects cardiac myocytes from pathologic hypertrophy by interacting with HDAC5. Mol Biol Cell. 2008;19:4141–53. doi: 10.1091/mbc.E07-12-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leong H, Sloan JR, Nash PD, et al. Recruitment of histone deacetylase 4 to the N-terminal region of estrogen receptor alpha. Mol Endocrinol. 2005;19:2930–42. doi: 10.1210/me.2005-0178. [DOI] [PubMed] [Google Scholar]
- 71.van Rooij E, Fielitz J, Sutherland LB, et al. Myocyte enhancer factor 2 and class II histone deacetylases control a gender-specific pathway of cardioprotection mediated by the estrogen receptor. Circ Res. 2010;106:155–65. doi: 10.1161/CIRCRESAHA.109.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karvonen U, Janne OA, Palvimo JJ. Androgen receptor regulates nuclear trafficking and nuclear domain residency of corepressor HDAC7 in a ligand-dependent fashion. Exp Cell Res. 2006;312:3165–83. doi: 10.1016/j.yexcr.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 73.Yang Y, Tse AK, Li P, et al. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene. 2011 doi: 10.1038/onc.2010.600. ; doi: 10.1038/onc.2010.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurtev V, Margueron R, Kroboth K, et al. Transcriptional regulation by the repressor of estrogen receptor activity via recruitment of histone deacetylases. J Biol Chem. 2004;279:24834–43. doi: 10.1074/jbc.M312300200. [DOI] [PubMed] [Google Scholar]
- 75.Jeong BC, Hong CY, Chattopadhyay S, et al. Androgen receptor corepressor-19 kDa (ARR19), a leucine-rich protein that represses the transcriptional activity of androgen receptor through recruitment of histone deacetylase. Mol Endocrinol. 2004;18:13–25. doi: 10.1210/me.2003-0065. [DOI] [PubMed] [Google Scholar]
- 76.Wei LN, Hu X. Receptor interacting protein 140 as a thyroid hormone-dependent, negative co-regulator for the induction of cellular retinoic acid binding protein I gene. Mol Cell Endocrinol. 2004;218:39–48. doi: 10.1016/j.mce.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 77.Guo L, Han A, Bates DL, et al. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc Natl Acad Sci USA. 2007;104:4297–302. doi: 10.1073/pnas.0608041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lahm A, Paolini C, Pallaoro M, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA. 2007;104:17335–40. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vanommeslaeghe K, De Proft F, Loverix S, et al. Theoretical study revealing the functioning of a novel combination of catalytic motifs in histone deacetylase. Bioorg Med Chem. 2005;13:3987–92. doi: 10.1016/j.bmc.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 80.Schuetz A, Min J, Allali-Hassani A, et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–63. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bottomley MJ, Lo Surdo P, Di Giovine P, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283:26694–704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeremski M, Stricker JR, Fischer D, et al. Histone deacetylase dHDAC4 is involved in segmentation of the Drosophila embryo and is regulated by gap and pair-rule genes. Genesis. 2003;35:31–8. doi: 10.1002/gene.10159. [DOI] [PubMed] [Google Scholar]
- 83.van der Linden AM, Nolan KM, Sengupta P. KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. EMBO J. 2007;26:358–70. doi: 10.1038/sj.emboj.7601479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stein GS, Lian JB, van Wijnen AJ, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23:4315–29. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 85.Tang H, Macpherson P, Marvin M, et al. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol Biol Cell. 2009;20:1120–31. doi: 10.1091/mbc.E08-07-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohen TJ, Waddell DS, Barrientos T, et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J Biol Chem. 2007;282:33752–9. doi: 10.1074/jbc.M706268200. [DOI] [PubMed] [Google Scholar]
- 87.Williams AH, Valdez G, Moresi V, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–54. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kao GD, McKenna WG, Guenther MG, et al. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–27. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Geng L, Cuneo KC, Fu A, et al. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- 90.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 91.Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–42. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- 92.Berns K, Hijmans EM, Mullenders J, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 93.Imbriano C, Gurtner A, Cocchiarella F, et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol. 2005;25:3737–51. doi: 10.1128/MCB.25.9.3737-3751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Basile V, Mantovani R, Imbriano C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J Biol Chem. 2006;281:2347–57. doi: 10.1074/jbc.M507712200. [DOI] [PubMed] [Google Scholar]
- 95.Cadot B, Brunetti M, Coppari S, et al. Loss of histone deacetylase 4 causes segregation defects during mitosis of p53-deficient human tumor cells. Cancer Res. 2009;69:6074–82. doi: 10.1158/0008-5472.CAN-08-2796. [DOI] [PubMed] [Google Scholar]
- 96.Wilson AJ, Byun DS, Nasser S, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mottet D, Pirotte S, Lamour V, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–56. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- 98.Datta J, Kutay H, Nasser MW, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68:5049–58. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Zhang J, Yang Y, Yang T, et al. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer. 2010;103:1215–20. doi: 10.1038/sj.bjc.6605895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nasser MW, Datta J, Nuovo G, et al. Down-regulation of micro-RNA-1 (miR-1) in lung cancer. Suppression of tumorigenic property of lung cancer cells and their sensitization to doxorubicin-induced apoptosis by miR-1. J Biol Chem. 2008;283:33394–405. doi: 10.1074/jbc.M804788200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Zhou X, Richon VM, Wang AH, et al. Histone deacetylase 4 associates with extracellular signal-regulated kinases 1 and 2, and its cellular localization is regulated by oncogenic Ras. Proc Natl Acad Sci U S A. 2000;97:14329–33. doi: 10.1073/pnas.250494697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 103.Bodily JM, Mehta KP, Laimins LA. Human Papillomavirus E7 enhances hypoxia-inducible factor 1 mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2010;73:1187–95. doi: 10.1158/0008-5472.CAN-10-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seo HW, Kim EJ, Na H, et al. Transcriptional activation of hypoxia-inducible factor-1alpha by HDAC4 and HDAC5 involves differential recruitment of p300 and FIH-1. FEBS Lett. 2009;583:55–60. doi: 10.1016/j.febslet.2008.11.044. [DOI] [PubMed] [Google Scholar]
- 105.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 106.Lu J, McKinsey TA, Nicol RL, et al. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–5. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 108.Kang JS, Alliston T, Delston R, et al. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24:2543–55. doi: 10.1038/sj.emboj.7600729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li H, Xie H, Liu W, et al. A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J Clin Invest. 2009;119:3666–77. doi: 10.1172/JCI39832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Urbich C, Rossig L, Kaluza D, et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–79. doi: 10.1182/blood-2009-01-196485. [DOI] [PubMed] [Google Scholar]
- 111.Ha CH, Wang W, Jhun BS, et al. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem. 2008;283:14590–9. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Milde T, Oehme I, Korshunov A, et al. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16:3240–52. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 113.Huang Y, Tan M, Gosink M, et al. Histone deacetylase 5 is not a p53 target gene, but its overexpression inhibits tumor cell growth and induces apoptosis. Cancer Res. 2002;62:2913–22. [PubMed] [Google Scholar]
- 114.Roy S, Shor AC, Bagui TK, et al. Histone deacetylase 5 represses the transcription of cyclin D3. J Cell Biochem. 2008;104:2143–54. doi: 10.1002/jcb.21771. [DOI] [PubMed] [Google Scholar]
- 115.Yarosh W, Barrientos T, Esmailpour T, et al. TBX3 is overexpressed in breast cancer and represses p14 ARF by interacting with histone deacetylases. Cancer Res. 2008;68:693–9. doi: 10.1158/0008-5472.CAN-07-5012. [DOI] [PubMed] [Google Scholar]
- 116.Sun G, Yu RT, Evans RM, et al. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc Natl Acad Sci U S A. 2007;104:15282–7. doi: 10.1073/pnas.0704089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sun G, Alzayady K, Stewart R, et al. Histone demethylase LSD1 regulates neural stem cell proliferation. Mol Cell Biol. 2010;30:1997–2005. doi: 10.1128/MCB.01116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ishikawa S, Nagai Y, Masuda T, et al. The role of oxysterol binding protein-related protein 5 in pancreatic cancer. Cancer Sci. 2010;101:898–905. doi: 10.1111/j.1349-7006.2009.01475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dequiedt F, Kasler H, Fischle W, et al. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 2003;18:687–98. doi: 10.1016/s1074-7613(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 120.Gao C, Cheng X, Lam M, et al. Signal-dependent regulation of transcription by histone deacetylase 7 involves recruitment to promyelocytic leukemia protein nuclear bodies. Mol Biol Cell. 2008;19:3020–7. doi: 10.1091/mbc.E07-11-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mottet D, Bellahcene A, Pirotte S, et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–46. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 122.Jensen ED, Gopalakrishnan R, Westendorf JJ. Bone morphogenic protein 2 activates protein kinase D to regulate histone deacetylase 7 localization and repression of Runx2. J Biol Chem. 2009;284:2225–34. doi: 10.1074/jbc.M800586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jensen ED, Schroeder TM, Bailey J, et al. Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J Bone Miner Res. 2008;23:361–72. doi: 10.1359/JBMR.071104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Margariti A, Zampetaki A, Xiao Q, et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ Res. 2010;106:1202–11. doi: 10.1161/CIRCRESAHA.109.213165. [DOI] [PubMed] [Google Scholar]
- 125.Malik S, Jiang S, Garee JP, et al. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol. 2010;30:399–412. doi: 10.1128/MCB.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–76. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 127.Moreno DA, Scrideli CA, Cortez MA, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150:665–73. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- 128.Dokmanovic M, Perez G, Xu W, et al. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther. 2007;6:2525–34. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- 129.Duong V, Bret C, Altucci L, et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol Cancer Res. 2008;6:1908–19. doi: 10.1158/1541-7786.MCR-08-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wen W, Ding J, Sun W, et al. Suppression of cyclin D1 by hypoxia-inducible factor-1 via direct mechanism inhibits the proliferation and 5-fluorouracil-induced apoptosis of A549 cells. Cancer Res. 2010;70:2010–9. doi: 10.1158/0008-5472.CAN-08-4910. [DOI] [PubMed] [Google Scholar]
- 131.Chen J, Fiskus W, Eaton K, et al. Cotreatment with BCL-2 antagonist sensitizes cutaneous T-cell lymphoma to lethal action of HDAC7-Nur77-based mechanism. Blood. 2009;113:4038–48. doi: 10.1182/blood-2008-08-176024. [DOI] [PubMed] [Google Scholar]
- 132.Scott FL, Fuchs GJ, Boyd SE, et al. Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J Biol Chem. 2008;283:19499–510. doi: 10.1074/jbc.M800331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rad R, Rad L, Wang W, et al. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science. 2010;330:1104–7. doi: 10.1126/science.1193004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou X, Marks PA, Rifkind RA, et al. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A. 2001;98:10572–7. doi: 10.1073/pnas.191375098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Petrie K, Guidez F, Howell L, et al. The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem. 2003;278:16059–72. doi: 10.1074/jbc.M212935200. [DOI] [PubMed] [Google Scholar]
- 136.Sparrow DB, Miska EA, Langley E, et al. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–98. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haberland M, Arnold MA, McAnally J, et al. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol. 2007;27:518–25. doi: 10.1128/MCB.01415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mejat A, Ramond F, Bassel-Duby R, et al. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci. 2005;8:313–21. doi: 10.1038/nn1408. [DOI] [PubMed] [Google Scholar]
- 139.Sugo N, Oshiro H, Takemura M, et al. Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci. 2010;31:1521–32. doi: 10.1111/j.1460-9568.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- 140.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 141.Lucio-Eterovic AK, Cortez MA, Valera ET, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer. 2008;8:243–52. doi: 10.1186/1471-2407-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wong RH, Chang I, Hudak CS, et al. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136:1056–72. doi: 10.1016/j.cell.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yuan Z, Peng L, Radhakrishnan R, et al. Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J Biol Chem. 2010;285:39329–38. doi: 10.1074/jbc.M110.179333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.North BJ, Marshall BL, Borra MT, et al. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–44. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 145.Scanlan MJ, Welt S, Gordon CM, et al. Cancer-related serological recognition of human colon cancer: identification of potential diagnostic and immunotherapeutic targets. Cancer Res. 2002;62:4041–7. [PubMed] [Google Scholar]