

Abstract
Antiplatelet agents, sarpogrelate (SAR), a 5-HT2A receptor antagonist, and cilostazol (CIL), a phosphodiesterase III (PDE-III) inhibitor, are used for the treatment of peripheral vascular disease. We tested whether these agents affect cardiac function and subcellular remodelling in congestive heart failure (CHF) induced by myocardial infarction (MI). Three weeks after MI, rats were treated daily with 5 mg/kg SAR or CIL as well as vehicle for 5 weeks. Sham-operated animals served as controls. At end of the treatment period, haemodynamic measurements were performed and the left ventricle was processed for the determination of sarcoplasmic reticulum (SR) Ca2+-uptake and -release activities, and expression of SR Ca2+-pump, phospholamban and ryanodine receptors, as well as myofibrillar ATPase activities, expression of α- and β-myosin heavy chain (MHC) isoforms, and phosphorylation of phospholamban and cardiac troponin-I (c Tn-I). Marked haemodynamic changes in the MI-induced CHF were associated with depressions in SR Ca +-uptake and -release activities as well as in protein content and gene expression for SR proteins. Furthermore, myofibrillar Ca2+-stimulated ATPase activity, as well as protein content and gene expression for α-MHC were decreased whereas those for β-MHC were increased in the failing heart. Also, phosphorylation levels of phospholamban and cTn-I were reduced in failing hearts. The MI-associated changes in cardiac function, SR and myofibillar activities, as well as SR and myofibrillar protein and gene expression were attenuated by treatment with SAR or CIL. The results suggest that SAR and CIL improve cardiac function by ameliorating subcellular remodelling in the failing heart and indicate the potential therapy of CHF with antiplatelet agents.
Keywords: SR function, myofibrillar proteins, cardiac function, heart failure, gene expression
Introduction
By virtue of its antiplatelet action, sarpogrelate (SAR), a serotonin antagonist (5-HT2A receptor antagonist), is indicated for the therapy of peripheral vascular disease [1, 2]. Not only does this agent inhibit 5-HT induced increase in intracellular Ca2+[Ca2+]i and cell proliferation in vascular smooth muscle cells [3, 4], it has also been reported to produce similar effects in the coronary artery smooth muscle cells [5]. Furthermore, SAR has been shown to attenuate arrhythmias and heart dysfunction due to acute myocar-dial infarction (MI) in rats [6] and improve the recovery of cardiac performance in ischaemic reperfused hearts by reducing changes in the high energy phosphate stores and myocardial ultrastructure [7]. Since prolonged activation of 5-HT2A receptors and associated signal transduction mechanisms have been observed to produce cardiac hypertrophy and congestive heart failure (CHF) [8], it is possible that SAR may exert some beneficial effects in CHF by attenuating changes in cardiac performance. This study was therefore undertaken to determine if the animals with CHF showed improvement in heart function upon treatment with SAR. In view of the critical role played by subcellular remodelling in the development of CHF [9, 10], alterations in biochemical activities and molecular composition of sarcoplasmic reticulum (SR) and myofibrils in the failing heart were examined with or without SAR treatment. To test if other antiplatelet agents exert beneficial effects similar to those for SAR in CHF, cilostazol (CIL), a phos-phodiesterase-III (PDE-III) inhibitor [11], which has similar properties as SAR in producing antiplatelet effects and vasodilation [11], was used in this study for the purpose of comparison. It should be mentioned that both SAR and CIL are novel antiplatelet agents, which exert vasodilatory effects and are used for the treatment of complications associated with peripheral thromboem-bolism as well as prevention of coronary restenosis [1, 2, 11–13].
Materials and methods
Experimental model and study design
All experimental protocols were approved by the Animal Care Committee of the University of Manitoba following guidelines established by the Canadian Institutes of Health Research. These guidelines conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996). MI was induced in male Sprague–awley rats (175–200 g) by occlusion of the left coronary artery as described earlier [14–16]. Briefly, the heart in anaesthetized animals was exposed and the left coronary artery was ligated at about 2 mm from its origin at the aorta. Sham-operated rats underwent the same procedure except coronary ligation. All rats received standard care, kept at 12 hrs day/night cycle and fed rat chow and water ad libitum. Three weeks after the operation, echocardiographic measurements were performed (SONOS 5500, Agilent Technologies Inc., Andover, MA, USA) for baseline values [17]. Since myocardial infarct is fully healed at about 3 weeks after the coronary occlusion, this time-point was chosen for starting drug treatment. Sham-operated rats (left ventricular (LV) ejection fraction (>80%) were assigned to control, the surviving coronary-ligated rats with ejection fraction (<50% were randomized to vehicle-treated infarcted (MI), SAR-treated infarcted (MI+SAR) and CIL-treated infarcted (MI+CIL) groups. Both SAR and CIL were given daily at a dose of 5 mg/kg for 5 weeks via a gastric tube starting at 3 weeks after the induction of MI; control animals received vehicle alone. The selected doses of drugs are considered to be within the safe and effective therapeutic range [1, 6, 11].
Haemodynamic studies
Haemodynamic measurements were carried out at 8 weeks after surgery as described previously [14–16]. Briefly, animals were anaesthetized with an intraperitoneal injection of a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg). The right carotid artery was exposed and cannulated with a microtip pressure transducer (SPR-249, Millar Instruments, Houston, TX, USA); mean arterial pressure (MAP) was determined and then the catheter was advanced to the LV cavity. Measurements of LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), heart rate, rate of pressure development (+dP/dt) and rate of pressure decay (−dP/dt) in addition to MAP were performed using AcqKnowledge software (3.0.3 MP100, BIOPAC System Inc., Goleta, CA, USA). After removing the hearts, the ventricles and scar tissue were separated and weighed. The lungs and the liver were also weighed for wet and dry weights.
Determination of myofibrillar Mg2+-ATPase and Ca2+-stimulated ATPase activities
The myofibrillar fraction was isolated as described earlier [14] and suspended in a final solution containing 100 mM KCl, 20 mM Tris-HCl (pH 7.0). Mg2+-ATPase activity was measured at 30°C in a medium containing (in mM) 20 imidazole (pH 7.0), 2 MgCl2, 2 Na2ATP, 10 NaN3, 1.6 ethylene glycoltetra acetic acid (EGTA) and 50 KCl [14]. Total ATPase activity was determined in the same medium except that EGTA was replaced by 10 μM of free Ca2+. Ca2+-stimulated ATPase activity was taken as the difference between values obtained for total and Mg +-ATPase activities. All reactions were terminated at 5 min. by the addition of 12% trichloroacetic acid. The samples were centrifuged at 1000 g and the phosphate was determined in the supernatant by colorimetric method.
Analysis of cardiac myosin heavy chain isoforms
Cardiac MHC isoforms were determined under denaturing conditions; both α-and β-MHCs were separated on a 4% polyacrylamide gel upon loading 4μg protein/well as described previously [14, 18]. The electrophoresis was carried out at a constant 220 V for 3.5 hrs with cooling between 9°C and 13°C. The gels were stained with coomassie brilliant blue R250 for 2 hrs and were destained with acetic acid and methanol. The relative amount of isoforms was estimated by GS-800 imaging densitometer (Quantity One 4.4.0 Software, Bio-Rad Laboratories, Mississauga, Canada).
Determination of SR Ca2+-uptake and -release activities
SR vesicles were isolated as described previously [15, 19]. For Ca2+-uptake assay [15], the reaction mixture contained (in mM) 50 Tris-maleate (pH 6.8), 5 NaN3, 5 ATP, 5 MgCl2, 120 KCl, 5 potassium oxalate, 0.1 EGTA, 0.1 45CaCl2 (20 mCi/l), and 25 μM ruthenium red. The reaction was initiated by adding 20 μg of SR vesicles at 37°C and terminated at 1 min. by filtration. The filters were then washed, dried and counted in a β scintillation counter. The Ca2+-release activity was measured as described previously [15]. The SR fraction was incubated in a medium containing (in mM) 100 KCl, 5 MgCl2, 5 NaN3, 20 Tris-HCl (pH 6.8) and 5 potassium oxalate, along with 10 μM 45CaCl2 (20 mCi/l) and 5 mM ATP for a period of 45 min. Ca2+-release was initiated by the addition of 1 mM EGTA and 1 mM Ca2+; the reaction was terminated at 15 sec by Millipore filtration. The filters were then counted in a β scintillation counter (Beckman Coulter Canada Inc., Mississauga, Canada). The Ca2+-induced Ca2+-release was inhibited by 95–100% upon incubating SR vesicles with 20 μM ryanodine.
Western blot analysis
The relative protein content of SR Ca2+-cycling proteins, Ca2+-pump ATPase (SERCA2a), Ca 2+-release channel or ryanodine receptor (RyR) as well as phospholamban (PLB) and its phosphorylated form, Ser-16 PLB, were determined by Western blot analysis as described previously [15, 19]. SR samples (20 |xg) were separated by SDS-polyacrylamide gel electrophoresis on a 4–20% gradient gel for RyR, 10% for SERCA2a and 15% for PLB and Ser-16 PLB and transferred to polyvinylidene difluoride membranes. Membranes were probed with a monoclonal anti-RyR antibody (1:1000) and monoclonal anti-SERCA2a antibody (1:1400) (Affinity Bioreagents, Golden CO, USA), as well as monoclonal anti-PLB polyclonal antibody (1:1000) (Upstate Inc., Charlottesville, VA, USA) and polyclonal anti-Ser-16 PLB antibody (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Following incubation with the appropriate peroxidase labelled secondary antibodies, Amersham ECL kit (GE Healthcare UK Ltd., Little Chalfont, Buckinghamshire, UK) was used for detection. Values for untreated and treated groups were expressed as % of the control group in each case. For the measurement of cardiac troponin I (cTnI) and phospho-rylated cTnI, membranes were incubated with monoclonal anti-cTnI (1:1000; Cell Signaling Technology, Inc.) and anti-phosphorylated cTnI specific for the phosphorylated form Ser22/Ser23 (1:1000; Cell Signaling Technology Inc.) antibodies, respectively (20). These membranes were then incubated with biotinylated anti-rabbit IgG (1:3000, Amersham) for 40 min. and finally with streptavidin conjugated horseradish peroxidase (1:3000, Amersham) for 40 min. The level of phosphorylated cTnI was expressed as % of total cTnI content. Gels were stained with coomassie blue after blotting, and blots were stained with Ponceau S solution to ensure uniform protein loading in all groups. The bands were analysed by the model GS-800 imaging densitometer (Bio-Rad Laboratories, Mississauga, Ontario, Canada) with the image analysis software (version 1.0). The values were expressed as a percentage of sham control values.
Northern blot analysis
Total RNA was isolated from the viable LV of sham control and infarcted rats with or without drug treatment by the acid guanidinium thiocyanate-phenol-chloroform method [15, 16, 18, 19]. Briefly, frozen samples of viable LV were homogenized with a Polytron homogenizer (model PT3000) at 12,000 rpm in the presence of 1.5 ml of TRIzol Reagent (1 ml/100 mg tissue; (GIBCO-BRL Life Technologies, Grand Island, NY, USA). The mixture was centrifuged at 12,000 g (model J2-HS, Beckman Coulter Canada Inc., Mississauga, Canada) for 10 min at 4°C. The supernatant was incubated with chloroform (0.3 ml/sample) for 5 min. at room temperature and then centrifuged at 12,000 g for 15 min. at 4°C. The RNA containing upper aqueous phase was kept at -20°C for 4 hrs after addition of 0.75 ml of iso-propyl alcohol. Upon centrifugation at 17,500 g for 10 min. at 4° C, RNA pellets were washed two times in 75% ethanol and vacuum dried by speed vac (model SC110, Savant Instruments, Farmingdale, NY, USA). The final RNA pellet was re-suspended in diethyl pyrocarbonate (DEPC)-treated water and stored at -70°C. The RNA concentration was calculated from the absorbance at 260 and 280 nm with SPECTRAmax PLUS (Molecular Devices, Sunnyvale, CA, USA). Total RNA (20 μg) was denatured at 65° C for 10 min and size fractionated on a 1.2% agarose gel containing 1.2 M formaldehyde. The blotted samples were transferred onto positively charged nytron nylon membranes (Schleicher & Schuell, Keene, NH, USA) UV cross-linked, and hybridized to randomly primed cDNA.
Statistical analysis
All values are presented as mean ±S.E.M. Statistical differences between the mean values were evaluated by one-way ANOVA followed by Duncan's new multiple test. The differences were considered significant at a P-value <0.05.
Results
General characteristics and mortality
Out of 117 rats, which underwent the operative procedure, 19 were sham-operated rats. None of the shams died during the study period. In the after surgical 3 week period, 35 coronary-ligated rats died, which corresponds to about 36% mortality. Total of 63 rats entered the treatment period. During the treatment period, the mortality for MI rats was 6/21, for MI+SAR rats was 3/21 and for MI+CIL rats was 9/21. As shown in Table 1, the surviving MI, MI+SAR and MI+CIL animals showed decreased body weight compared to sham controls. MI rats had clinical signs of CHF such as lung congestion as reflected by the increased lung wet/dry weight ratio, ventricular chamber enlargement, right ventricular (RV) hypertrophy and pulmonary oedema. They also had a scar with well-defined borders. Treatment with SAR and CIL prevented the increase in ventricular weight, RV weight, ventricular/body weight ratio and lung wet/dry weight ratio. However, no changes in scar weight or liver wet/dry weight ratio were observed among the different treated groups.
1.
General characteristics of sham, MI, MI+SAR and MI+CIL animals 8 weeks after surgery.
| SHAM (n = 19) | Ml (n = 15) | MI+SAR (n = 18) | MI+CIL (n = 12) | |
|---|---|---|---|---|
| Body weight (g) | 599 ± 14 | 521 ± 9 * | 527 ± 11 * | 523 ± 10 * |
| Ventricular weight (g) | 1.37 ± 0.04 | 1.58 ± 0.04 * | 1.41 ± 0.04 # | 1.43 ± 0.04 # |
| Ventricular weight/Body weight ratio (mg:g) | 2.38 ± 0.03 | 2.99 ± 0.08 * | 2.55 ± 0.05 # | 2.57 ± 0.07 # |
| RV (g) | 0.31 ± 0.02 | 0.48 ± 0.04 * | 0.36 ± 0.04 # | 0.37 ± 0.04 # |
| Scar weight (g) | — | 0.16 ± 0.01 | 0.15 ± 0.01 | 0.16 ± 0 |
| Lungs wet/dry ratio | 4.53 ± 0.06 | 5.11 ± 0.10 * | 4.95 ± 0.08 # | 4.95 ± 0.11 # |
| Liver wet/dry ratio | 3.18 ± 0.02 | 3.31 ± 0.06 | 3.30 ± 0.05 | 3.27 ± 0.03 |
Values are means ± SEM. MI, myocardial infarction; SAR, sarpogrelate; CIL, cilostazol; LV, left ventricle; RV, right ventricle. *P<0.05, significantly different from sham. #P<0.05, significantly different from MI.
Haemodynamic studies
Haemodynamic parameters obtained from different experimental groups 8 weeks after surgery are shown in Table 2. Infarcted animals exhibited significantly higher heart rate and developed LV dysfunction as reflected by decreased LVSP, +dP/dt, −dP/dt and MAP as well as elevated LVEDP compared with control animals. Heart rate and LVEDP were reduced, whereas the −dP/dt, (dP/dt, LVSP and MAP were increased towards sham levels upon treating MI animals with SAR or CIL (Table 2).
2.
Haemodynamic parameters of sham, MI, MI+SAR and MI+CIL animals 8 weeks after surgery.
| SHAM (n = 19) | MI (n = 15) | MI+SAR (n = 18) | MI+CIL (n = 12) | |
|---|---|---|---|---|
| HR (beats/min.) | 310 ± 2.1 | 365 ± 5.5 * | 348 ± 3.2 # | 342 ± 4.1 # |
| MAP (mm Hg) | 113 ± 3.2 | 72 ± 1.2 * | 112 ± 4.1 # | 102 ± 3.3 # |
| LVSP (mm Hg) | 130 ± 4.6 | 88 ± 2.9 * | 119 ± 4.4 # | 108 ± 3.5 # |
| LVEDP (mm Hg) | 8.1 ± 0.42 | 19.8 ± 0.68 * | 10.2 ± 0.54 # | 11.1 ± 0.5 # |
| +dP/dt (mm Hg/sec) | 8186 ± 290 | 3306 ± 118 * | 6172 ± 216 # | 6280 ± 247 # |
| −dP/dt (mm Hg/sec) | 6738 ± 561 | 2740 ± 105 * | 4301 ± 220 # | 4529 ± 295 # |
Values are means ±S.E.M. MI, myocardial infarction; SAR, sarpogrelate; CIL, cilostazol; HR, heart rate; MAP, mean arterial pressure; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; +dP/dt, peak rate of pressure development; −dP/dt, peak rate of pressure decay. *P<0.05, significantly different from sham. #P<0.05, significantly different from MI.
SR Ca2+-uptake and Ca2+-release activities, protein content and gene expression
Ca2+-uptake and -release activities were determined in SR vesicles obtained from the viable LV tissue of different groups (Fig. 1A and B). Both SR Ca2+-uptake and -release activities were lower in the MI group compared with the sham control group, but these were improved significantly upon SAR or CIL treatment. To study the effects of SAR and CIL on the expression of SR proteins, isolated SR samples were assayed by western immunoblotting. As shown in Figure 2 and Figure 3A, SERCA2a, RyR and PLB protein expression as well as the phosphorylated level of PLB were reduced by 71%, 62%, 23% and 63% in the failing LV compared to shams, respectively. SAR and CIL treatment effectively tended to normalize the MI-associated decrease in protein content. Northern blot analysis revealed that mRNA levels for SERCA2a, RyR and PLB proteins were decreased in the vehicle-treated infarcted hearts by 60%, 45% and 35% compared to controls, respectively (Fig. 4). This decrease in LV mRNA levels was largely prevented by treatments with both SAR and CIL.
1.
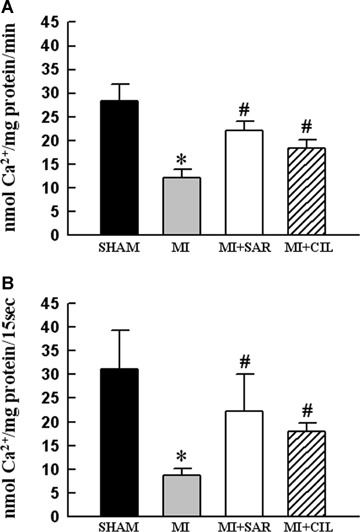
Sarcoplasmic reticulum (SR) Ca2+-uptake (A) and Ca2+-release (B) activities in sham, vehicle-treated (MI), sarpogrelate-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. MI, myocardial infarction. Values are means ± S.E.M. of 6–9 animals for each group. *P < 0.05 versus sham; #P < 0.05 versus MI.
2.
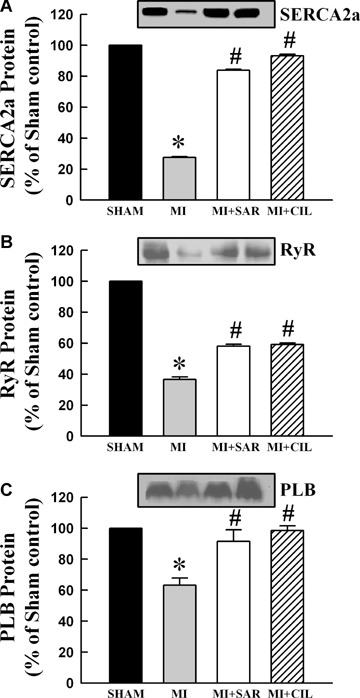
Representative western blots and densitometric analysis of protein content for sarcoplasmic reticulum SERCA2a (A), RyR (B) and PLB (C) proteins in sham, vehicle-treated (MI), sarpogrelate-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. SERCA2a: sarcoplasmic reticulum Ca2+-ATPase; RyR: ryanodine receptor. Values are means ± S.E.M. of 6–9 animals for each group. *P < 0.05 versus sham; #P < 0.05 versus MI.
3.
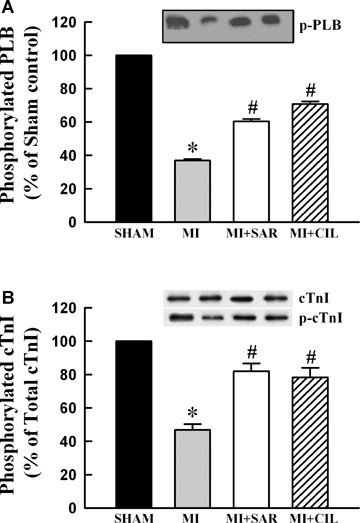
Representative western blots and densitometric analysis of protein contents for sarcoplasmic reticulum phosphorylated PLB (A) and phosphorylated cTnI (B) in sham, vehicle-treated (MI), sarpogrelate-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. PLB: phospholamban; cTnI: cardiac troponin I. Values are means ± SEM of 6–9 animals for each group. *P < 0.05 versus sham; # P < 0.05 versus MI.
4.
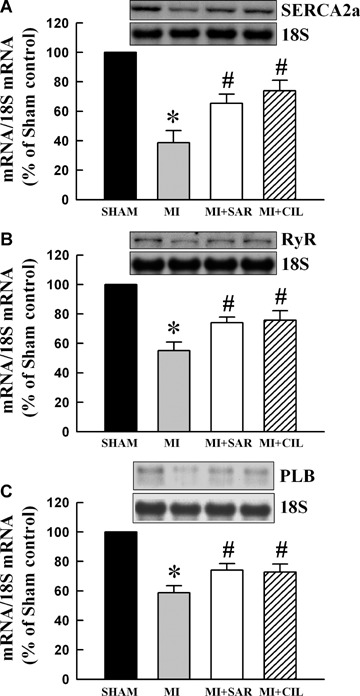
Representative northern blots and densitometric quantification of mRNA abundance for sarcoplasmic reticulum SERCA2 (A), RyR (B) and PLB (C) in sham, vehicle-treated (MI), sarpogrelate-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. SERCA2a: sarcoplasmic reticulum Ca2+-ATPase; RyR: ryanodine receptor; PLB: phospholamban. Values are means ± S.E.M. of 6–10 animals for each group. *P < 0.05 versussham; #P < 0.05 versus MI.
Myofibrillar ATPase activities and myosin heavy chain isoforms
The data in Figure 5 show that myofibrils isolated from the viable LV of MI hearts exhibited a lower Ca2+-stimulated ATPase activity compared with sham-operated rats. Treatment with SAR or CIL normalized the Ca +-stimulated ATPase activity comparable to sham values. Myofibrillar Mg2+-ATPase activity did not show any significant difference among various groups. As shown in Figure 6A, viable LV tissue of MI hearts showed a marked reduction in β-MHC isoform (from 72.2% to 44.0% of total MHC) and an increase in β-MHC isoform (from 27.84% to 55.98% of total MHC). At the same time, α-MHC mRNA level was decreased by 70% (Fig. 6B) and that for α-MHC mRNA was increased by 88% (Fig. 6C) in LV of the infarcted rats. Therapy with SAR and CIL significantly attenuated the increase in β-MHC as well as the decrease in α-MHC due to MI at both protein and mRNA levels (Fig. 6). In order to show if the phosphoryla-tion of cTnI is altered in the experimental animals, protein content for both unphosphorylated and phosphorylated forms of cTnI were determined. The data in Figure 3B indicate that protein content for phosphorylated cTnI decreased to 46.8% level in the MI group whereas SAR and CIL treatments restored these levels to 81.9% and 78.3% when compared with sham controls, respectively.
5.
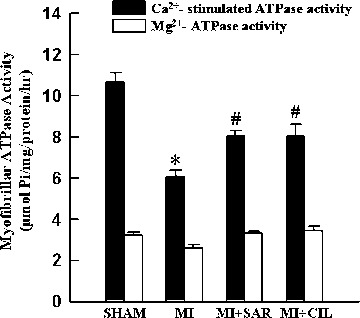
Myofibrillar Ca2+-stimulated and Mg2+-ATPase activities in sham, vehicle-treated (MI), sarpogrelate-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. MI: myocardial infarction; Values are means ± S.E.M. of 6–9 animals for each group. *P < 0.05 versus sham; #P < 0.05 versus MI.
6.
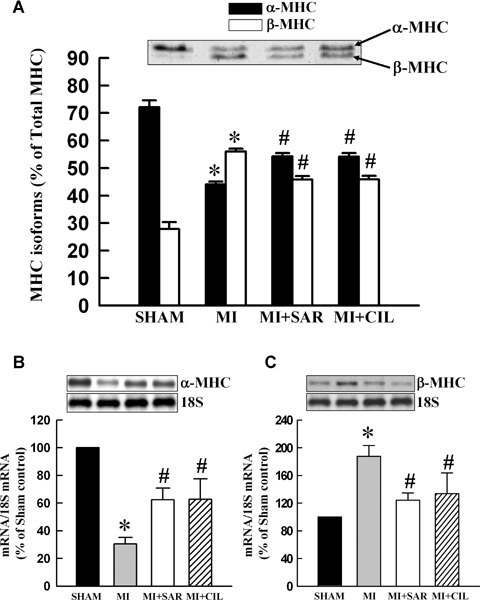
Coomassie blue-stained gels showing α- and β-MHC protein expression and their corresponding densitometric analysis (A) and representative northern blots and densitometric quantification of mRNA abundance for α-MHC (B) and β-MHC (C) in sham, vehicle-treated (MI), sarpogre-late-treated (MI+SAR) and cilostazol-treated (MI+CIL) rats at 8 weeks after surgery. MI: myocardial infarction; MHC: myosin heavy chain. Values are means ± S.E.M. of 6–9 animals for each group. *P < 0.05 versus sham; #P < 0.05 versus MI.
Discussion
In the present study, we showed for the first time that antiplatelet agents, SAR and CIL, attenuated remodelling of subcellular organelles such as SR and myofibrils as well as improved LV function in a rat model of CHF secondary to MI. These findings suggest that antiplatelet agents may occupy a unique niche temporally located between efforts to reduce platelet deposition and mitigating cardiac remodelling in CHF.
Antiplatelet agents attenuate structural ventricular remodelling and improve cardiac function
SAR has been shown to improve cardiac function and LV pressures in ischaemia/reperfusion injury in isolated hearts [17] as well as in acute MI in rats [6]. Although no such beneficial effects of CIL in heart failure have been reported previously, other PDE inhibitors are known to improve cardiac function and exert car-dioprotective action in the failing heart [11, 21, 22]. Now, we have observed that both SAR and CIL attenuated cardiac diastolic and systolic dysfunction in CHF due to MI. Such changes were associated with reduced alterations in +dP/dt and −dP/dt as well as attenuation in cardiac hypertrophy and pulmonary congestion in CHF animals upon treatment with both agents. SAR and CIL treatment also depressed MAP and reduced cardiac preload in the infarcted animals as illustrated by the reduction of LVEDP. These haemodynamic observations indicate that both SAR and CIL improve LV filling, enhance contraction and relaxation, prevent the deterioration of global LV function as well as limit cardiac hypertrophy in the failing hearts. Although the effects of SAR and CIL treatment seem to be comparable in qualitative terms, some differences in the magnitude of their effects on certain measured parameters in this study may be due to differences in the mechanisms of action of these two agents [2, 11]. In particular, it was observed that mortality in CIL-treated MI rats was higher and that in SAR-treated group was lower in comparison to the vehicle-treated MI animals. This increase in mortality in CIL-treated MI rats may be obvious because CIL treatment would increase the cyclic adenosine monophosphate (cAMP) level in the failing heart due to its inhibitory effect on PDE and thus, produce toxic effects on myocardial cells including arrhythmias [23]. In fact, various PDE inhibitors have been shown to cause arrhythmias and increase mortality in patients with CHF [23, 24].
Modulation of SR remodelling by antiplatelet agents in CHF
Remodelling of the SR and disturbance in the intracellular Ca2+-homeostasis have been considered as the main reasons for the inability of failing heart to generate contractile force [10, 16, 25, 26]. It is pointed out that alterations in Ca2+-handling have been reported to occur in cardiomyocytes from failing hearts due to MI [16, 27]. Furthermore, previous studies from our laboratory have shown depressed SR Ca2+-uptake, SR Ca2+-release as well as SR protein and gene expression in failing hearts due to MI [15, 19, 28, 29]. Similarly, in this study, CHF was associated with a depression in SR function as well as a down-regulation of SERCA2a, and RyR proteins which may result in Ca2+-handling abnormalities in cardiomyocytes and impaired muscle relaxation and contraction. Although PLB protein level, which inhibits SERCA2a activity in SR [30] was reduced in the infarcted hearts, it should be noted that the decrease in SERCA2a level was more than that in the level of PLB. Thus, the increase in PLB/SERCA2a protein ratio would enhance inhibition of SERCA2a by PLB in MI hearts. In this con-text, both SAR and CIL attenuated the depression in SR Ca2+-uptake, SR protein content and gene expression for SERCA2a and PLB in addition to improving the PLB/SERCA2a ratio which may contribute to the improvement of cardiac contraction and relaxation in the failing heart. Nonetheless, results reported in this study regarding the effects of both SAR and CIL on SR function as well as SR protein and gene expression suggest attenuation of SR remodelling in the failing hearts by some antiplatelet agents.
Increasing the Ca2+ -uptake into the SR either through stimulation of the SERCA2a [29] or by increasing the phosphorylation level of PLB [31] has been proposed as an attractive approach to improve diastolic and systolic function and thus represent a potential therapeutic strategy to restore the disturbed intracellular Ca2+-handling in CHF [32]. SAR and CIL treatment also improved SR regulation by recovering PLB phosphorylation, which was decreased in the failing hearts and contributed towards the inhibition of SERCA2a function. Of the several protein kinases, which mediate the phosphorylation of PLB, the cAMP-dependent protein kinase A (PKA) appears to be an important mediator of the enhanced cardiac contractility [33]. It should also be pointed out that in agreement with previous studies [34, 35], we found that failing hearts after MI have lower cTnI phosphorylation levels compared to healthy hearts. This down-regulation of cTnI phosphory-lation reduces the Ca2+ affinity of troponin C, decreases Ca2+-sen-sitivity of the contractile machinery and leads to enhanced relaxation [36]. Treatment with SAR or CIL restored MI-induced decrease in cTnI phosphorylation without affecting total cTnI content. Thus, it appears that the beneficial effects of SAR and CIL on contractile function partly rely on their ability to restore not only the PLB phosphorylation level but also the cTnI phosphorylation level in the failing heart.
Modulation of myofibrillar remodelling by antiplatelet agents in CHF
Consistent with our previous observations [14, 18], we found that the myofibrils isolated from the failing heart exhibited significantly depressed Ca2+-stimulated ATPase activity when compared to sham controls. The decrease in myofibrillar Ca2+-stimulated ATPase activity was accompanied by decreased expression of α-MHC isoform and increased expression of β-MHC isoform at both protein and mRNA levels, indicating a fetal gene re-expression like program and remodelling of myofibrils in the hypertrophic failing hearts. These alterations in myofibrils have been suggested to partly explain defects in cardiac contraction and relaxation of the failing heart due to MI [4, 18]. This point is further substantiated by our observations that improvement in cardiac function in the failing heart was associated with attenuation of myofibrillar remodelling upon treatment of infarcted animals with SAR or CIL. Interestingly, while a partial restoration of the α-MHC mRNA resulted in a partial restoration of the α-MHC protein level upon treating the failing hearts with SAR and CIL, near complete normalization of the β-MHC mRNA only produced a partial restoration of the β-MHC protein level. This indicates that the β-MHC expression is under a complex post-transcriptional regulation as suggested by Haddad et al. [37]. It is pointed out that myofibrillar ATPase activity is vastly determined by the ratio of the expressed MHC isoforms; α-MHC has a high ATPase activity and produces high cross-bridge force with less economy of energy consumption [38]. Since treatment with SAR or CIL was found to attenuate the depression in myofibrillar Ca2+-stimulated ATPase activity by increasing α-MHC, the improvement in cardiac function in the failing heart by these agents may be related to their effect on the cross bridge formation and energy consumption processes. Thus, in addition to preventing changes in the phosphorylation of cTnI, both SAR and CIL attenuate myofibrillar remodelling.
SAR and CIL as antiplatelet agents in CHF
Recently Nebigil et al.[8] have reported that the activation of 5HT2 receptors in the heart activates G-protein coupled phospholipase C (PLC), which initiates a rapid formation of IP3 and diacylglycerol (DAG), and increases intracellular Ca2+-levels leading to the development of intracellular Ca2+-overload and cardiac dysfunction. Thus, 5HT2A-receptor blockade by SAR may prevent the activation of the 5HT2A/PLC pathway, inhibit the excess release of Ca2+ from the internal stores and produce beneficial effects in CHF. On the other hand, CIL would increase the level of cAMP by inhibiting PDE-III within platelets and cardiomyocytes and thus can be seen to attenuate platelet aggregation [11] and increase cardiac contractility, respectively. Since the reduced phosphorylation of PLB in the failing heart would largely depress the Ca2+-dependent activity of SERCA2a, the increase in cAMP due to inhibition of PDE by CIL could contribute to an increase in the phosphorylation state of PLB and the improved intracellular Ca2+-handling. It should be pointed out that the potency of the PDE-III inhibitors as inotropic agents in CHF is considered to correlate with their potency to inhibit the SR membrane-bound PDE-III [39]. In addition, both SAR and CIL improved the depressed levels of cTnI which will increase the contractile function in the failing heart. Thus, this study has shown that antiplatelet agents, such as SAR and CIL improve cardiac performance in post-MI CHF, which beneficial effects are associated with attenuation of changes in biochemical activities of both SR and myofibrils as a consequence of their action on gene expression for subcellular organelle proteins. These results may open an entirely new perspective in the therapy of CHF involving antiplatelet agents.
Since treatment with both SAR and CIL decreased heart weight and heart weight/body weight ratio in the infarcted animals, it appears that these agents reduce cardiac hypertrophy due to their antihypertrophic action in animals with heart failure. Blockade of 5-HT receptors by various agents including SAR has also been reported to decrease cardiac hypertrophy in heart failure [40, 41]. Although this is a first study which has shown antihypertrophic effect of CIL, other PDE-III inhibitors, such as amrinone and milri-none have been reported to reduce cardiac hypertrophy in the failing heart [42, 43, 44]. While the exact mechanisms of the antihy-pertrophic action of both SAR and CIL in the failing heart remain to be established, afterload reduction as a consequence of decreased peripheral resistance in heart failure by these agents may play an important role in this regard. Nonetheless, on the basis of an association of antihypertrophic action of both SAR and CIL with improved cardiac function of the infarcted animals, it can be argued that the improvement in cardiac performance by these agents may be due to a reduction in the extent of cardiac hypertrophy. However, such a view is in contrast to the general concept that cardiac hypertrophy is beneficial in compensating cardiac function due to the loss of viable myocardium in the infarcted heart [45]. On the other hand, cardiac hypertrophy over a prolonged period is known to exert deleterious action on heart function as a consequence of sub-cellular remodelling under different pathophysiological conditions [9, 10] and the beneficial effect of SAR and CIL on cardiac function may be due to attenuation of subcellular remodelling. Accordingly, the significance of changes in heart weight in terms of cardiac performance during the development of heart failure as well as due to the treatment with agents such as SAR and CIL may depend upon the stage of cardiac hypertrophy. Furthermore, in view of the results presented in this study, it is proposed that the improvement of cardiac function by antiplatelet therapy may be related to the attenuation of subcellular remodelling in the failing heart.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR). JB was a fellow of the TACTICS program, in association with CIHR and Heart and Stroke Foundation of Canada. We wish to thank Donald Chapman for his excellent technical assistance during Northern blotting procedures.
References
- 1.Doggrell SA. Sarpogrelate: cardiovascular and renal clinical potential. Expert Opin Investig Drugs. 2004;13:865–74. doi: 10.1517/13543784.13.7.865. [DOI] [PubMed] [Google Scholar]
- 2.Saini HK, Takeda N, Goyal RK, Kumamoto H, Arneja AS, Dhalla NS. Therapeutic potentials of sarpogrelate in cardiovascular disease. Cardiovasc Drug Rev. 2004;22:27–54. doi: 10.1111/j.1527-3466.2004.tb00130.x. [DOI] [PubMed] [Google Scholar]
- 3.Sharma SK, Zahradka P, Chapman D, Kumamoto H, Takeda N, Dhalla NS. Inhibition of serotonin-induced vascular smooth muscle cell proliferation by sar-pogrelate. J Pharmacol Exp Therap. 1999;290:1475–81. [PubMed] [Google Scholar]
- 4.Saini HK, Sharma SK, Zahradka P, Kumamoto H, Takeda N, Dhalla NS. Attenuation of the serotonin-induced increase in intracellular calcium in rat aortic smooth muscle cells by sarpogrelate. Can J Physiol Pharmacol. 2003;81:1056–63. doi: 10.1139/y03-108. [DOI] [PubMed] [Google Scholar]
- 5.Sharma SK, Del Rizzo DF, Zahradka P, Bhangu SK, Werner JP, Kumamoto H, Takeda N, Dhalla NS. Sarpogrelate inhibits serotonin-induced proliferation of porcine coronary artery smooth muscle cells. Implications for long-term graft patency. Ann Thorac Surg. 2001;71:1856–64. doi: 10.1016/s0003-4975(01)02599-1. [DOI] [PubMed] [Google Scholar]
- 6.Brasil D, Temsah RM, Kumar K, Kumamoto H, Takeda N, Dhalla NS. Blockade of 5-HT(2A) receptors by sarpogrelate protects the heart against myocardial infarction in rats. J Cardiovasc Pharmacol Ther. 2002;7:53–9. doi: 10.1177/107424840200700i108. [DOI] [PubMed] [Google Scholar]
- 7.Temsah RM, Kumamoto H, Takeda N, Dhalla NS. Sarpogrelate diminishes changes in energy stores and ultrastruc-ture of the ischemic-reperfused rat heart. Can J Physiol Pharmacol. 2001;79:761–7. [PubMed] [Google Scholar]
- 8.Nebigil CG, Maroteaux L. Functional consequence of serotonin/5-HT2B receptor signaling in heart: role of mitochondria in transition between hypertrophy and heart failure? Circulation. 2003;108:902–8. doi: 10.1161/01.CIR.0000081520.25714.D9. [DOI] [PubMed] [Google Scholar]
- 9.Dhalla NS, Shao Q, Panagia V. Remodeling of cardiac membranes during the development of congestive heart failure. Heart Fail Rev. 1998;2:261–72. [Google Scholar]
- 10.Dhalla NS, Dent MR, Tappia PS, Sethi R, Barta J, Goyal RK. Subcellular remodeling as a viable target for the treatment of congestive heart failure. J Cardiovasc Pharmacol Therapeut. 2006;11:31–45. doi: 10.1177/107424840601100103. [DOI] [PubMed] [Google Scholar]
- 11.Sorkin EM, Markham A. Cilostazol. Drugs Aging. 1999;14:63–71. doi: 10.2165/00002512-199914010-00005. [DOI] [PubMed] [Google Scholar]
- 12.Fujita M, Mizuno K, Ho M, Tsukahara R, Miyamoto A, Miki O, Ishii K, Miwa K. Sarpogrelate treatment reduces restenosis after coronary stenting. Am Heart J. 2003;145:E16. doi: 10.1067/mhj.2003.176. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchikane E, Fukuhara A, Kobayashi T, Kirino M, Yamasaki K, Kobayashi T, Izumi M, Otsuji S, Tateyama H, Sakurai M, Awata N. Impact of cilostazol on restenosis after percutaneous coronary balloon angioplasty. Circulation. 1999;100:21–6. doi: 10.1161/01.cir.100.1.21. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Liu X, Ren B, Rupp H, Takeda N, Dhalla NS. Modification of myosin gene expression by imidapril in failing heart due to myocardial infarction. J Mol Cell Cardiol. 2002;34:847–57. doi: 10.1006/jmcc.2002.2023. [DOI] [PubMed] [Google Scholar]
- 15.Shao Q, Ren B, Saini HK, Netticadan T, Takeda N, Dhalla NS. Sarcoplasmic retic-ulum Ca2+-transport and gene expression in congestive heart failure are modified by imidapril treatment. Am J Physiol Heart Circ Physiol. 2005;288:H1674–82. doi: 10.1152/ajpheart.00945.2004. [DOI] [PubMed] [Google Scholar]
- 16.Saini HK, Shao Q, Musat S, Takeda N, Tappia PS, Dhalla NS. Imidapril treatment improves the attenuated inotropic and intracellular calcium responses to ATP in heart failure due to myocardial infarction. Br J Pharmacol. 2005;144:202–11. doi: 10.1038/sj.bjp.0705867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recommendations for quanti-tation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–67. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Guo X, Dhalla NS. Modification of myosin protein and gene expression in failing hearts due to myocardial infarction by enalapril or losartan. Biochim Biophys Acta. 2004;1690:177–84. doi: 10.1016/j.bbadis.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Guo X, Chapman D, Dhalla NS. Partial prevention of changes in SR gene expression in congestive heart failure due to myocardial infarction by enalapril or losar-tan. Mol Cell Biochem. 2003;254:163–72. doi: 10.1023/a:1027321130997. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Takeda N, Dhalla NS. Troponin I phosphorylation in heart homogenate from diabetic rat. Biochim Biophys Acta. 1996;1316:78–84. doi: 10.1016/0925-4439(96)00007-5. [DOI] [PubMed] [Google Scholar]
- 21.Wood MA, Hess ML. Long term oral therapy of congestive heart failure with phos-phodiesterase inhibitors. Am J Med Sci. 1989;297:105–13. doi: 10.1097/00000441-198902000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Sanada S, Kitakaze M, Papst PJ, Asanuma H, Node K, Takashima S, Asakura M, Ogita H, Liao Y, Sakata Y, Ogai A, Fukushima T, Yamada J, Shinozaki Y, Kuzuya T, Mori H, Terada N, Hori M. Cardioprotective effect afforded by transient exposure to phosphodiesterase III inhibitors: the role of protein kinase A and p38 mitogen-activated protein kinase. Circulation. 2001;104:705–10. doi: 10.1161/hc3201.092216. [DOI] [PubMed] [Google Scholar]
- 23.Barta J, Sanganalmath SK, Sethi R, Kumamoto H, Takeda N, Edes I, Dhalla NS. Antithrombotic agents sarpogrelate and cilostazol differentially affect ventricular arrhythmias and survival in rats. Exp Clin Cardiol. 2006;11:246. (Abstract) [Google Scholar]
- 24.Nony P, Boissel JP, Lievre M, Leizorovicz A, Haugh MC, Fareh S, De Breyne B. Evaluation of the effect of phosphodi-esterase inhibitors on mortality in chronic heart failure patients. J Clin Pharmacol. 1994;46:191–6. doi: 10.1007/BF00192547. [DOI] [PubMed] [Google Scholar]
- 25.Pieske B, Maier LS, Bers DM, Hasenfuss G. Ca2+ handling and sarcoplasmic reticu-lum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res. 1999;85:38–46. doi: 10.1161/01.res.85.1.38. [DOI] [PubMed] [Google Scholar]
- 26.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–64. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang XE, Musch TI, Zellis R, Cheung JY. Effects of impaired Ca2+ homeostasis on contraction in post infarction myocytes. J Appl Physiol. 1999;86:943–50. doi: 10.1152/jappl.1999.86.3.943. [DOI] [PubMed] [Google Scholar]
- 28.Afzal N, Dhalla NS. Differential changes in left and right ventricular SR Ca2+ transport in congestive heart failure. Am J Physiol. 1992;262:H868–74. doi: 10.1152/ajpheart.1992.262.3.H868. [DOI] [PubMed] [Google Scholar]
- 29.Netticadan T, Temsah RM, Kawabata K, Dhalla NS. Sarcoplasmic reticulum Ca2+/Calmodulin-dependent protein kinase is altered in heart failure. Circ Res. 2000;86:596–605. doi: 10.1161/01.res.86.5.596. [DOI] [PubMed] [Google Scholar]
- 30.Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, Date MO, Chrast J, Matsuzaki M, Peterson KL, Chien KR, Ross J., Jr Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004;113:727–36. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Jr, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sar-coplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–22. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 32.Hajjar RJ, Del Monte F, Matsui T, Rosenzweig A. Prospects for gene therapy for heart failure. Circ Res. 2000;86:616–21. doi: 10.1161/01.res.86.6.616. [DOI] [PubMed] [Google Scholar]
- 33.Kuschel M, Karczewski P, Hempel P, Schlegel WP, Krause EG, Bartel S. Ser16 prevails over Thr17 phospholamban phos-phorylation in the beta-adrenergic regulation of cardiac relaxation. Am J Physiol. 1999;276:H1625–33. doi: 10.1152/ajpheart.1999.276.5.H1625. [DOI] [PubMed] [Google Scholar]
- 34.Van Der Velden J, Papp Z, Zaremba R, Boontje NM, De Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 35.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 36.Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–35. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 37.Haddad F, Bodell PW, Qin AX, Giger JM, Baldwin KM. Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J Biol Chem. 2003;278:37132–8. doi: 10.1074/jbc.M305911200. [DOI] [PubMed] [Google Scholar]
- 38.VanBuren P, Harris DE, Alpert NR, Warshaw DM. Cardiac V1 and V3 myosins differ in their hydrolytic and mechanical activities in vitro. Circ Res. 1995;77:439–44. doi: 10.1161/01.res.77.2.439. [DOI] [PubMed] [Google Scholar]
- 39.Weishaar RE, Kobylarz-Singer DC, Steffen RP, Kaplan HR. Subclasses of cyclic AMP-specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ Res. 1987;61:539–47. doi: 10.1161/01.res.61.4.539. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda K, Tojo K, Tokudome G, Hosoya T, Harada M, Nakao K. The effects of sar-pogrelate on cardiomyocyte hypertrophy. Life Sci. 2000;67:2991–6. doi: 10.1016/s0024-3205(00)00879-1. [DOI] [PubMed] [Google Scholar]
- 41.Brattelid T, Qvigstad E, Birkeland JA, Swift F, Bekkevold SV, Krobert KA, Sejersted OM, Skomedal T, Osnes JB, Levy FO, Sjaastad I. Serotonin responsiveness through 5-HT(2A) and 5-HT(4) receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J Mol Cell Cardiol. 2007;43:767–79. doi: 10.1016/j.yjmcc.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Sweet CS, Ludden CT, Stabilito II, Emmert SE, Heyse JF. Beneficial effects of milrinone and enalapril on long-term survival of rats with healed myocardial infarction. Eur J Pharmacol. 1988;147:29–37. doi: 10.1016/0014-2999(88)90630-9. [DOI] [PubMed] [Google Scholar]
- 43.Osadchii O, Norton G, Woodiwiss A. Inotropic responses to phosphodi-esterase inhibitors in cardiac hypertrophy in rats. Eur J Pharmacol. 2005;514:201–8. doi: 10.1016/j.ejphar.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 44.Movsesian MA. PDE3 inhibition in dilated cardiomyopathy: reasons to reconsider. J Card Fail. 2003;9:475–80. doi: 10.1016/s1071-9164(03)00135-0. [DOI] [PubMed] [Google Scholar]
- 45.Dhalla NS, Afzal A, Beamish RE, Naimark B, Takeda N, Nagano M. Pathophysiology of cardiac dysfunction in congestive heart failure. Can J Cardiol. 1993;9:873–87. [PubMed] [Google Scholar]