

Abstract
The Agrobacterium tumefaciens VirB/D4 type IV secretion system (T4SS) translocates DNA and protein substrates across the bacterial cell envelope. Six presumptive channel subunits of this T4SS (VirD4, VirB11, VirB6, VirB8, VirB2, and VirB9) form close contacts with the VirD2-T-strand transfer intermediate during export, as shown recently by a novel transfer DNA immunoprecipitation (TrIP) assay. Here, we characterize the contribution of the hydrophobic channel component VirB6 to substrate translocation. Results of reporter protein fusion and cysteine accessibility studies support a model for VirB6 as a polytopic membrane protein with a periplasmic N terminus, five transmembrane segments, and a cytoplasmic C terminus. TrIP studies aimed at characterizing the effects of VirB6 insertion and deletion mutations on substrate translocation identified several VirB6 functional domains: (i) a central region composed of a large periplasmic loop (P2) (residues 84 to 165) mediates the interaction of VirB6 with the exiting T-strand; (ii) a multi-membrane-spanning region carboxyl-terminal to loop P2 (residues 165 to 245) is required for substrate transfer from VirB6 to the bitopic membrane subunit VirB8; and (iii) the two terminal regions (residues 1 to 64 and 245 to 290) are required for substrate transfer to the periplasmic and outer membrane-associated VirB2 and VirB9 subunits. Our findings support a model whereby the periplasmic loop P2 comprises a portion of the secretion channel and distinct domains of VirB6 participate in channel subunit interactions required for substrate passage to the cell exterior.
Keywords: conjugation, pathogenesis, secretion, DNA translocation, membrane protein topology
Introduction
The type IV secretion systems (T4SS) translocate DNA and proteins across the cell envelopes of bacteria.1 One large T4SS subfamily, the conjugation systems, deliver DNA substrates to target cells by a mechanism dependent on formation of a mating junction between donor and recipient cells.2 Conjugal DNA transfer is achieved through the coordinated actions of three distinct protein subcomplexes.3 First, the DNA transfer and replication (Dtr) proteins initiate transfer by binding at the origin-of-transfer (oriT) sequence of a mobile DNA element, forming the relaxosome. The catalytic subunit of the relaxosome, the relaxase, nicks the strand destined for transfer (T-strand) and then remains covalently bound to the 5′ end of the T-strand, generating the relaxase-T-strand transfer intermediate.4 Next, a homomultimer of an integral membrane ATP-binding protein termed the coupling protein (T4CP) recruits the DNA substrate to the secretory apparatus, probably through recognition of C-terminal secretion signals carried by the relaxase and other relaxosome subunits.5–8 Finally, the T4CP functions together with a trans-envelope secretion channel composed of the mating pair formation (Mpf) proteins to deliver the DNA transfer intermediate to the cell exterior.9 The processing and recruitment reactions are biochemically well characterized for several conjugation systems, and recent structure–function studies have been aided by available crystal structures for the TrwC10,11 and TraI12,13 relaxases of plasmids R388 and F, respectively, and TrwB T4CP14 of plasmid R388. Until recently, however, there has been very little understanding of the secretion channel architecture or mechanism of action.
The Agrobacterium tumefaciens VirB/D4 T4SS is an archetypal conjugation system used for delivery of oncogenic transfer DNA (T-DNA) and effector proteins to susceptible plant cells.15 In A. tumefaciens, the VirD2 relaxase and auxiliary Dtr proteins initiate T-DNA processing by binding the oriT-like T-DNA border repeat sequences. The VirD2-T-strand transfer intermediate, released from the tumor-inducing (Ti) plasmid, is recruited by the VirD4 T4CP to the VirB/D4 T4SS.16 To define the route by which the VirD2-T-strand exits the cell, we recently developed a three-stage in vivo formaldehyde crosslinking/immunoprecipitation/PCR amplification assay, termed transfer DNA immunoprecipitation (TrIP). This assay identifies close contacts between a translocating DNA substrate and channel subunits of the secretory apparatus.16 Results of TrIP studies of wild-type and T4SS mutant strains showed that the VirD2-T-strand exits the cell via the following temporal pathway: (i) the VirD4 T4CP recruits the DNA substrate to the T4SS; (ii) the T4CP delivers the substrate to the VirB11 homohexameric ATPase17 situated at the cytoplasmic face of the inner membrane;18 (iii) VirB11 mediates substrate transfer to the integral inner membrane channel components VirB6 and VirB8; and (iii) these inner membrane proteins deliver the substrate to the channel components VirB2 and VirB9 localized in the periplasm and outer membrane. The remaining VirB proteins do not form close contacts with the DNA substrate, but nevertheless are required for substrate passage and thus likely function as structural scaffolds or modulators of channel activity.16
These findings now permit detailed structure–function studies to better understand how each of the secretory components contributes to substrate transfer. In the present study, we characterized the contribution of the VirB6 channel subunit to substrate transfer. VirB6 is highly hydrophobic and predicted by several different computer methods to span the inner membrane multiple times (Figure 1(A)). We applied a combination of reporter protein fusion and cysteine accessibility approaches to develop a topology model for VirB6. Next, we characterized the effects of insertion and deletion mutations on the capacity of VirB6 to form close contacts with the T-DNA substrate and to deliver the substrate to other presumptive channel subunits, as monitored with the TrIP assay. Our results define a temporal order for formation of substrate close contacts with the VirB6 and VirB8 inner membrane channel subunits. Furthermore, we show that an extracytoplasmic domain mediates the interaction of VirB6 with the DNA substrate, whereas domains elsewhere in the protein mediate substrate passage from VirB6 through the portion of the secretion channel projecting through the periplasm to the cell exterior.
Figure 1.

Topology analyses of the VirB6 subunit of the A. tumefaciens VirB/D4 type IV secretion system. (A) The computer programs listed at left predict the transmembrane segments (TMS) shown as filled rectangles with residue numbers relative to the N terminus above and the orientation of the TMS across the membrane below (I, inside; O, outside). Filled triangles identify TMS supported by results of reporter protein fusion studies. (B) Reporter protein activity levels of virB6 null mutant strain PC1006 expressing 5′ fragments of virB6 translationally fused to lacZ (above the x-axis) or phoA (below); numbers identify the virB6 codon immediately 5′ to the fusion junction site. Reporter protein activities represent the average of at least three experiments with standard deviations indicated. (C) Steady-state levels of the fusion proteins in AS-induced cells. Total membrane proteins (5 μg) from strains producing the fusion proteins identified by the junction site residue number were subjected to SDS-PAGE and Western blot analysis with anti-PhoA (above) or anti-β-Gal (below) antibodies.
Results
VirB6 topology mapping by fusion to reporter proteins
Recent studies indicate that the topology of a membrane protein can be predicted reliably when several topology prediction methods show a consensus.19 Five widely used methods predicted five transmembrane segments (TMS) for VirB6, although several of these methods also predicted one or more additional TMS (Figure 1(A)). We tested these predictions, first by fusing VirB6 N-terminal fragments to periplasmically active alkaline phosphatase (PhoA) or cytoplasmically-active β-galactosidase (β-Gal).20 As shown in Figure 1(B), A. tumefaciens strains producing complementary sets of fusion proteins displayed a fairly consistent pattern of reporter protein activities. For example, strains producing fusion proteins with junction sites between residues 85 and 146, and 235 and 238 exhibited intermediate to high PhoA and low β-Gal activities, suggestive of a periplasmic location of the junction sites. Conversely, strains producing fusion proteins with junction sites between 29 and 70, 167 and 201, and 260 and 290 displayed high β-Gal and low PhoA activities, suggestive of a cytoplasmic location. Strains producing chimeric proteins with PhoA or β-Gal fused at codon 150 displayed intermediate reporter protein activities, preventing a conclusion regarding the subcellular location of this junction site. All fusion proteins except for VirB6-β-Gal derivatives with junction sites at putative periplasmic residues 85, 229, and 235 accumulated at comparable levels suggestive of stable protein production (Figure 1(C)).
Results of these reporter protein fusion studies provide support for four of the predicted TMS, two oriented in-to-out (i-o) across the membrane spanning residues 63 to 83 and 201 to 221, and two oriented out-to-in (o-i) most probably spanning residues 167 to 187 and 224 to 244 (Figure 1(A)). The data do not firmly establish the locations of the two o-i TMS, possibly because both are located in regions of long, contiguous stretches of hydrophobicity. Insertion of a reporter protein within such stretches might result in a non-native topology and, consequently, aberrant reporter protein activity. However, below we supply additional evidence for these four TMS assignments, plus one more TMS that spans residues 34 to 55.
A cytoplasmic C terminus and localization to the cell poles
The accuracy of membrane prediction methods increases further when experimental data are available about the location of the C terminus of a protein.21,22 The above findings suggest that the C terminus of VirB6 is located in the cytoplasm. We also fused the full-length protein to another cytoplasmic marker, the green fluorescent protein (GFP). GFP-tagged cytoplasmic proteins or domains generally are stable and fluorescent, whereas the corresponding fusions to periplasmic proteins or domains generally are unstable and non-fluorescent.22 The A. tumefaciens cells were highly fluorescent as shown by microscopy (Figure 2(A)), and synthesized a stable form of VirB6-GFP, as shown by immunostaining (Figure 2(B)), providing further support for a cytoplasmic C terminus.
Figure 2.
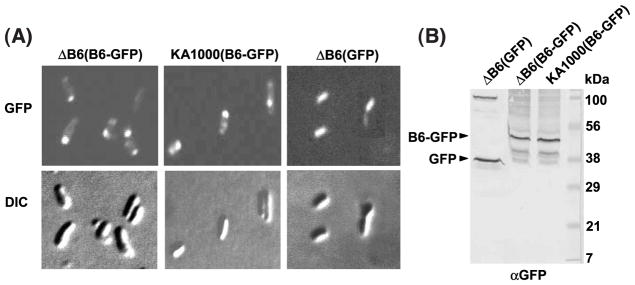
VirB6 topology and cellular location studies with GFP. (A) VirB6 fused at its C terminus to cytoplasmically active GFP. PC1006 deleted of virB6 (ΔB6) and Ti-plasmidless KA1000 cells expressing PvirB-virB6-GFP from pSJB6GFP (B6-GFP) display polar fluorescence; PC1006 cells (ΔB6) expressing PvirB-GFP from pZDB88 (GFP) are uniformly fluorescent. Top panels: GFP fluorescence monitored by fluorescence microscopy; lower panels: corresponding images by DIC microscopy. (B) Production of VirB6-GFP and GFP in A. tumefaciens. Membrane proteins (5 μg) from strains examined by microscopy were subjected to SDS-PAGE and Western blot analysis with anti-GFP antibodies (αGFP). Positions of stably-produced VirB6-GFP and GFP are indicated at the left; molecular mass markers are indicated (in kDa) at right.
Very intriguingly, the VirB6-GFP producing cells displayed fluorescence at the poles, whereas, cells producing only GFP were uniformly fluorescent (Figure 2(A)). Other studies have shown that the virB-encoded T pilus23 and the VirD4 T4CP24 are polar localized and, further, the VirD4 T4CP recruits the VirE2 protein substrate to the poles.25 These observations support a general model that the VirB/D4 T4SS assembles at the A. tumefaciens cell poles. To test whether VirB6 localization is dependent on synthesis of a T4SS subunit or another product of the Ti plasmid, we examined the fluorescence pattern of strain KA1000(pSJB6GFP), a Ti-plasmidless strain engineered to express PvirB-virB6-GFP upon induction with acetosyringone (AS) (see Materials and Methods). This strain produced a stable form of VirB6-GFP (Figure 2(B)) and displayed polar fluorescence (Figure 2(A)), suggesting that VirB6 targeting to the poles is mediated through contacts with chromosomally encoded factors. VirB6 polar localization might therefore represent an early and possible nucleating event in the VirB/D4 T4SS biogenesis pathway.
Topology mapping by cysteine accessibility
To further evaluate the topology of active forms of full-length VirB6, we assayed for accessibility in whole cells of single Cys residues introduced along the length of VirB6 to sulfhydryl reagents.26 Alleles for the ten Cys substitution mutant proteins under study fully complemented a virB6 null mutation in T-DNA transfer assays (data not shown) and accumulated at wild-type levels (Figure 3(A)), indicating that the Cys mutations did not grossly perturb the structure of VirB6 or contacts with other machine subunits. Whole cells were treated with 3-(N-maleimidylpropionyl) biocytin (MPB; a biotinylated sulfhydryl-specific maleimide probe), which passes readily through the pores of the outer membrane but only inefficiently through the inner membrane.27,28 Additionally, to verify that the MPB-labeled cysteine residues were exposed to the outer surface of the inner membrane, whole cells were pretreated with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; a highly hydrophilic non-biotinylated maleimide derivative) to block such residues prior to treatment with MBP.29 Initial studies showed that conditions described previously concerning the level of MBP and incubation temperature,30 labeled the periplasmic proteins VirB1031 and PhoA,32 but not cytoplasmic β-Gal (data not shown). Finally, to assay for solvent-accessibility of the introduced Cys residues, we assayed for labeling upon treatment of vesicles with MPB (see Materials and Methods).
Figure 3.

Accessibility of introduced cysteine residues. Immunodetection of native VirB6 and Cys substitution mutant proteins. Lanes 1, strain PC1006(pSJB964) producing native VirB6 from PvirB-virB6; lanes 2 (—), strain PC1006 (ΔvirB6); and lanes 3 through 12, PC1006 producing the VirB6 Cys substitution mutants listed. Membrane proteins (5 μg) were subjected to SDS-PAGE and Western blot analysis with anti-VirB6 antibodies (αB6). MPB labeling of VirB6 Cys substitution mutants. Whole cells: PC1006 (ΔvirB6) strains treated with MPB without (above) or with (below) pretreatment with AMS. Native and mutant forms of VirB6 were isolated by immunoprecipitation and analyzed for MPB labeling as described in Materials and Methods. Vesicles: solvent-accessible Cys residues, as shown by treatment of A. tumefaciens vesicles with MBP. The treatment of whole cells or of vesicles with MPB did not label the unique Cys42 residue of the native protein (B6). Note that MPB labeled a non-VirB6 species migrating slightly faster (whole cells) or slower (vesicles) than VirB6 in the protein gels; these species were labeled in strains producing and lacking VirB6, e.g. ΔvirB6, lane 2.
The computer algorithms predict that the unique Cys residue of native VirB6 at residue 42 is buried in a TMS (Figure 1(A)). However, the reporter protein fusion studies were inconclusive in providing evidence for this TMS (Figure 1(B)), possibly due to disruption of the N-terminal transmembrane topology accompanying fusions at residues 29 through 70. Treatments of whole cells or of vesicles with MPB, even at a high concentration (1 mM), failed to label Cys42 (Figure 3(B), first lane), supporting a prediction that this residue is sequestered in the lipid bilayer. To further evaluate whether an o-i TMS spans residues 33 to 54, we assayed for MPB accessibility of two Cys substitution mutations, I12C and D25C, in whole cells. As shown in Figure 3(B), both Cys residues were biotinylated and, further, biotinylation was completely blocked by pre-treatment with AMS. These findings place the N terminus of VirB6 in the periplasm and support the assignment of TMS1 as spanning residues 33 to 54; we designate the periplasmic N terminus as segment P1 in the proposed VirB6 topology model (Figure 4). Treatment of vesicles with MPB labeled the D25C substitution but not I12C (Figure 3(B)). However, this latter residue is located in a hydrophobic segment predicted by two computer algorithms to span the membrane (Figure 1(A)). The labeling of I12C upon treatment of whole cells with MPB argues against this prediction, and thus it seems more likely that I12C is sequestered in a solvent-inaccessible secondary structure that forms during preparation of A. tumefaciens vesicles.
Figure 4.

A membrane topology model for the VirB6 channel subunit. The model is derived from predictions by five widely used computer algorithms and results of β-Gal, PhoA, and GFP reporter protein fusion and Cys accessibility studies. Gray circles, Cys substitution mutations labeled by treatment of whole cells with MPB; open circles, Cys substitution mutations labeled only by treatment of vesicles with MPB; Open square, Cys42 of the native protein was not labeled by treatments of whole cells or vesicles with MPB. The experimental findings provide support for the five transmembrane segments (TMS) shown. The N-terminal segment and internal loops in the periplasm (P1,2,3) and the internal loops and C-terminal segment in the cytoplasm (C1,2,3) are shown.
Eight other Cys substitution mutations displayed MPB accessibility patterns that were consistent with results of the reporter protein fusion studies. Two substitution mutations, Q116C and S133C, located in the central hydrophilic region and a more C-terminal substitution, T224C, were biotinylated upon treatment of whole cells with MPB and labeling was blocked completely by pretreatment with AMS (Figure 3(B)). Both the reporter protein fusion and Cys accessibility data therefore support the assignments of periplasmic loops P2 and P3 in the topology model (Figure 4). Other substitution mutations were not labeled upon treatment of whole cells, suggestive of cytoplasmic locations. Three of these Cys substitutions were clearly solvent-accessible, as shown by abundant labeling upon treatment of vesicles with MPB (Figure 3(B)). These were R62C and T66C in the proposed cytoplasmic loop C1, and L263C in the C-terminal cytoplasmic segment C3 (Figure 4). Two other substitutions (D188C, Q291C) were less accessible to MPB upon treatment of vesicles; labeling was detectable only with a high concentration (1 mM) of MPB. The low level of accessibility of D188C located at the beginning of cytoplasmic loop C2 might be due to shielding by TMS3, which spans residues 166 to 187 (Figures 3 and 4). Additionally, the C terminus of VirB6 might adopt a secondary structure that buries Q291C. Regardless, results of the reporter protein fusion studies strongly support a proposal that the C terminus of VirB6 is in the cytoplasm (Figure 4).
Substrate transfer blocks imposed by VirB6 insertion mutations
With the TrIP assay, we showed that null mutations of virB6 and virB8 did not affect in vivo formaldehyde crosslinking of the T-strand substrate to the VirD4 T4CP or the VirB11 ATPase. However, the virB6 mutation blocked formation of the substrate–VirB8 crosslink, the virB8 mutation blocked formation of the substrate–VirB6 crosslink, and both mutations blocked formation of substrate contacts with VirB2 and VirB9. On the basis of these findings, we postulated that VirB6 and VirB8 function coordinately to mediate substrate transfer from VirD4 and VirB11 located at the cytoplasmic face of the inner membrane to the periplasmic and outer membrane-associated channel subunits VirB2 and VirB9.16 To further define the contribution of VirB6 to the dynamics of substrate transfer through the VirB/D4 T4SS, we characterized the effects of insertion and deletion mutations on steps of the postulated translocation pathway, beginning with substrate transfer from VirB11 to VirB6.
We first investigated the effects of synthesizing a set of VirB6 insertion mutants on the translocation pathway. We have reported that strains producing each of nine mutant proteins bearing a four-residue (i4) insertion fail to deliver the T-strand to plant cells, whereas a strain producing the A290.i4 mutant protein displays essentially wild-type function.33 As expected, this latter strain also formed a wild-type pattern of T-strand close contacts with VirD4 and the VirB channel subunits, as monitored by TrIP (Figure 5(B)). Also as expected, strains producing each of the nine remaining i4 mutant proteins, or producing one of five i31 mutant derivatives generated by transposon mutagenesis displayed substrate transfer blocks. All i4 and i31 mutant proteins accumulated at comparable levels (data not shown),33 suggesting that the transfer blocks cannot be attributed to destabilizing effects of the mutations.
Figure 5.

VirB6 domain structure defined by transfer DNA immunoprecipitation (TrIP) studies of PC1006 (ΔvirB6) producing VirB6 insertion mutants. (A) Postulated VirB6 domain structure. The broad line at the top identifies regions of VirB6 required for formation of T-strand close contacts with VirB6 (dark shading), VirB8 (light shading), and VirB2 and VirB9 (gray shading), no affect on substrate transfer (white shading). Numbers below the broad line and the vertical lines identify domain boundaries postulated on the basis of TrIP results and the VirB6 topology model, shown as a wavy line below; C, cytoplasm; IM, inner membrane; P, periplasm. The numbers along the VirB6 topology model identify the junction site residue for the i4 or i31 insertion mutations and the N and C termini of the 295 residue protein. VirB6 insertion mutations were assigned as classes I, II, or III on the basis of observed effects on substrate transfer through the VirB/D4 secretion channel, as shown by TrIP. (B) TrIP studies of T-strand interactions with VirB proteins from strains producing the VirB6 insertion mutants. Strains: WT, A348; ΔB6, PC1006; PvirB-B6, PC1006 producing native VirB6; PC1006 strains producing the VirB6.i4 or VirB6.i31 (marked by asterisk) mutants. Strains were treated in vivo with formaldehyde prior to lysis. S, supernatant after immunoprecipitation with the anti-VirB antibodies listed at left; IP, immunoprecipitate. The T-DNA substrate (T-DNA) and the Ti control fragment (Ct) were detected in the S and IP fractions by PCR amplification and gel electrophoresis. The WT strain and PC1006 producing native VirB6 or the A290.i4 mutant showed no substrate transfer arrest; PC1006 and PC1006 producing class I mutant proteins were blocked at the VirB11 to VirB6 transfer step; PC1006 producing class II mutants were blocked at the VirB6 to VirB8 transfer step, and PC1006 producing class III mutants were blocked at the VirB8 to VirB2 and VirB9 transfer step. Vertical lines separate the mutant classes and horizontal lines identify the substrate transfer arrest points.
Remarkably, strains producing mutant proteins with insertions mapping within potential VirB6 subdomains showed substrate transfer blocks at distinct steps of the translocation pathway. For example, mutations located within the central periplasmic loop P2 (Q85.i4, T124.i4, Q150.i4, Q150.i31) did not affect substrate recruitment by VirD4 or transfer to VirB11, but instead arrested transfer from VirB11 to VirB6 and, as expected, the remaining channel subunits (Figure 5(B)). These mutations, designated as class I mutations, were located at various positions throughout loop P2 (Figure 5(A)), suggesting that this entire loop might form a secondary structure required for the interaction of VirB6 with substrate during translocation.
Additional mutations arrested substrate transfer following formation of the VirB6 close contact. For example, strains producing mutations designated as class II (L175.i4, L201.i4, M229.i31, T234.i31, F235.i4) failed to deliver the T-strand to VirB8, VirB2, and VirB9 (Figure 5(B)). Thus, while VirB6 and VirB8 probably function coordinately to direct substrate transfer across the inner membrane,16 the identification of class II mutations defines a temporal order to substrate contacts with these inner membrane channel components; the T-strand interacts first with VirB6, then with VirB8. The class II mutations mapped to a region adjacent to loop P2, whose predicted topology encompasses TMS3 to TMS5, inclusive of the intervening loops C2 and P3 (Figure 5(A)).
Mutations designated as class III did not affect substrate transfer from VirD4 through VirB8, but blocked formation of substrate close contacts with VirB2 and VirB9 (Figure 5(B)). Interestingly, the class III mutations mapped to the two terminal regions of VirB6. Four mutations (E29.i4, T44.i31, I53.i31, and D60.i4) mapped to the N-terminal periplasmic segment P1, TMS1, and the small cytoplasmic loop C1. One mutation (I260.i4) mapped within the cytoplasmic C terminus. As noted above, the L290.i4 mutation near the C terminus of VirB6 did not affect substrate translocation.
Domains mediating substrate transfer identified by deletion mapping
The above studies suggest VirB6 can be dissected into four functional domains: (i) a central periplasmic region (loop P2) necessary for formation of a close contact with the VirD2-T-strand; (ii) an adjacent multi-membrane-spanning region (TMS3 through TMS5) required for substrate transfer from VirB6 to VirB8; and (iii) two terminal regions (segment P1, TMS1, loop C1; segment C3; Figure 4) required for substrate transfer from VirB8 to VirB2 and VirB9. To gain further evidence for the functional importance of these domains and to better define the boundaries, we characterized the effects of various deletion mutations on substrate transfer.
As shown in Figure 6, only the deletions extending into loop P2 abolished formation of substrate close contacts with VirB6. In fact, one deletion mutant (Δ1-85) is missing TMS2 and only one or two residues of loop P2, whereas a second (Δ158-295) is missing TMS3 and only about seven residues of loop P2. These findings suggest that, in addition to loop P2, the TMS flanking loop P2 contribute to formation of the VirB6–substrate contact. However, note that one deletion mutant (Δ160-221) is missing about five residues of loop P2 and TMS3, yet still formed a substrate close contact (Figure 6(B)). Interestingly, this mutant protein is also missing loop C2 and TMS4, such that TMS5 might functionally substitute for TMS3 (Figure 6(B); see Figure 4). If this is the case, the precise sequence composition of the flanking TMS might not be important for substrate transfer to VirB6, only that two TMS flank periplasmic loop P2; conceivably, these TMS function mainly to tether loop P2 to the membrane. Consistent with such a proposal, the L175.i4 mutation in TMS3 does not affect formation of the VirB6–T-strand close contact (Figure 5). We attempted to test whether a fragment of VirB6 spanning only residues 55 to 195 interacts with substrate by TrIP, but this fragment was unstable (data not shown). Thus, further studies are needed to ascertain whether the loop P2 domain with its flanking TMS is sufficient for substrate transfer from VirB11 to VirB6.
Figure 6.

VirB6 domain structure defined by TrIP studies of PC1006 (ΔvirB6) strains producing VirB6 deletion mutants. (A) VirB6 domain structure depicted as in Figure 5. The numbers along the VirB6 topology model identify the start or end-points of the deletion mutations. VirB6 deletion mutations are shown schematically, the top line represents the full-length (295 residues) VirB6 protein. Mutations were assigned as classes I, II, or III on the basis of observed effects on substrate transfer through the VirB/D4 secretion channel as shown by TrIP. The Δ160-221 mutation lacking TMS3 does not affect formation of the VirB6 substrate contact, possibly because TMS5 (denoted by the horizontal broken line) substitutes for TMS3. The Figure reflects the proposed requirement for the flanking TMS for loop P2 function. (B) TrIP studies of T-strand interactions with VirB proteins from strains producing the VirB6 deletion mutant proteins. Strains: WT, A348; ΔB6, PC1006; PvirB-B6, PC1006 producing native VirB6; PC1006 producing the VirB6 deletion derivatives indicated. S, supernatant after immunoprecipitation with the anti-VirB antibodies listed at left; IP, immunoprecipitate; T-DNA, T-DNA substrate; Ct, Ti control fragment. Deletion mutations extending into the postulated domains blocked the VirB-substrate contact listed at the top of A. Class I mutations arrested substrate transfer from VirB11 to VirB6; class II; from VirB6 to VirB8; class III from VirB8 to VirB2 and VirB9. Vertical lines separate the mutant classes and horizontal lines identify the substrate transfer arrest points.
Deletions projecting into the multi-membrane-spanning region between residues 166 to 245 abolished substrate transfer from VirB6 to VirB8 (Figure 6(B)). These findings are compatible with TrIP studies of the insertion mutants, pointing to the importance of this region for this transfer step. By contrast, the Δ261-290 mutation, as with the I260.i4 mutation, did not affect substrate transfer from VirB6 to VirB8, providing further evidence that the cytoplasmic C terminus of VirB6 does not contribute to this transfer step.
Finally, deletions of N-terminal (Δ1-30, Δ30-60) or C-terminal (Δ261-290) residues did not affect substrate transfer from VirD4 through VirB8, but abolished transfer to VirB2 and VirB9 (Figure 6(B)). These findings show that N and C-terminal residues are completely dispensable for substrate trafficking to and across the inner membrane. Interestingly, these termini are located on opposite faces of the inner membrane (Figure 4), yet both appear to mediate the delivery of substrate through a periplasmic portion of the secretion channel.
Discussion
Members of the VirB6 protein family are essential channel subunits of many type IV secretion systems of Gram-negative bacteria.1 Over 75 VirB6-like proteins have been identified, all of which are known or postulated components of T4SS.2,34 Despite overall low sequence similarities, VirB6-like proteins are highly hydrophobic with multiple predicted membrane-spanning segments.2,34 Previous studies showed that several VirB proteins are rapidly degraded in virB6 null mutants, implicating VirB6 as important for assembly of a stable secretory apparatus.33,35,36 With the TrIP assay (this work),16 we now have shown unequivocally that VirB6 participates directly in substrate trafficking. The contribution of VirB6 to machine biogenesis and to substrate transfer underscore the importance of detailed topology and structure–function studies of this T4SS subunit.
A VirB6 topology model
Five algorithms showed a consensus in predicting five TMS and a large central periplasmic loop, although nearly all methods also predicted additional TMS (Figure 1(A)). Results of reporter protein fusion and Cys accessibility studies support the predictions for four of the consensus TMS (TMS 1 to 4), but suggest that TMS5 is located between residues 224 and 244 as opposed to residues 250 to 275. The data favoring this TMS5 assignment include (i) high β-Gal and low PhoA activities of strains producing reporter protein fusions with junctions at residue 260; these activities are more consistent with a TMS spanning residues 224 to 244 (Figure 1), and (ii) labeling of the L263C substitution mutation upon treatment of vesicles with MPB, suggesting that this residue is not buried in the lipid bilayer (Figure 3).
In concert with the predictions by most algorithms for additional TMS, there was no consensus regarding the cellular locations of the termini of VirB6. At this point, we cannot completely exclude the possibility of additional TMS. However, our data strongly support assignments of the N terminus to the periplasm and the C terminus to the cytoplasm. Such a topology dictates that VirB6 possesses an odd number of TMS, and it is unlikely our studies have missed two TMS. Additionally, we note that the model depicted in Figure 4 fits very well with an average hydropathy profile of 14 VirB6 close homologs developed in a recent phylogenetic analysis.34 All five peaks of hydrophobicity identified in that analysis align very well with the five experimentally confirmed TMS, whereas central and C-terminal stretches of hydrophilic residues correspond to the periplasmic loop P2 and the cytoplasmic C terminus. Finally, our findings are compatible with results of an early PhoA fusion study that assigned residues 103, 113, and 138 to the periplasm; these residues are within the domain here designated as loop P2.37 Our topology model is the first for a polytopic channel subunit of a type IV secretion system. As with all early models, we acknowledge that further studies will probably refine the model and might supply information about contributions of other T4SS to the overall VirB6 architecture or possible structural changes accompanying substrate translocation.
Structure–function relationships
On the basis of the topology model, one can envision several possible VirB6 domains, five TMS that might form an inner membrane channel as well as several extramembranous loops that might form specific contacts with other T4SS subunits required for machine assembly and function. To characterize the domain structure of VirB6, we used the recently described TrIP assay to monitor effects of insertion and deletion mutations on substrate transfer across the cell envelope. As mentioned above, this assay is based on formation of in vivo formaldehyde crosslinks between channel subunits and the DNA substrate, most probably as the substrate threads through the secretion channel.16 Remarkably, we discovered that insertions or deletions within a given region of VirB6 exerted similar effects on the formation of close contacts between channel subunits and the translocating VirD2-T-strand. Although boundaries are not defined precisely, our findings support the assignments of four functional domains, one that mediates formation of substrate contacts with VirB6, a second mediating substrate transfer to the bitopic channel subunit VirB8, and two required for substrate transfer to the periplasmic/OM-associated channel subunits VirB2 and VirB9 (Figures 5 and 6).
All mutations within or projecting into loop P2 abolished formation of the VirB6 interaction with the T-strand substrate. The insertion mutations affecting T-strand transfer to VirB6 were at various positions of loop P2, suggesting that this region might adopt a specific secondary structure required for the substrate interaction. The TMS flanking loop P2 are necessary for formation of VirB6 close substrate contacts. However, the finding that the L175.i4 mutation does not abolish the VirB6–T-strand contact, coupled with evidence that TMS5 can functionally substitute for TMS3, suggests that the presence of these TMS but not their precise sequence compositions contribute to the substrate contact. These TMS might be important for the structural integrity of loop P2, although a more direct role in formation of an inner membrane channel cannot be excluded. Below, we present possible structures for loop P2 and its flanking TMS in the context of two mechanistic models. One depicts VirB6 as a channel subunit required for substrate passage across the inner membrane, whereas the second depicts the dominant structure of importance as a “vestibule” in the periplasm that corresponds to the substrate docking and entry point into the secretion channel in the periplasm.
In agreement with the present findings, i4 mutations in loop P2 abolish T-strand delivery to plant cells. Interestingly, however, strains producing the Q150.i4 (class I; Figure 5) as well as the L201.i4 (class II) mutant proteins are still capable of mobilizing transfer of the VirE2 effector protein to plant cells as well as the IncQ plasmid to agro-bacterial recipients.33 We suggested previously that these mutations might unveil a substrate discrimination mechanism. However, it is possible that these are partial function mutations in which case the apparent substrate discrimination phenotype might actually reflect differences in detection thresholds of assays used to monitor substrate transfer. A closer examination of the latter possibility was afforded with a quantitative version of the TrIP assay.16 With this assay, as expected, most strains producing VirB6.i4 mutant proteins displayed complete T-strand transfer arrest at the step of the pathway identified with the non-quantitative assay (Figure 5); in each of these cases, the amount of T-strand associated with channel subunits positioned downstream of the transfer arrest point was <0.5% of that associated with upstream channel subunits. The notable exceptions were the two strains producing the Q150.i4 and L201.i4 mutations, whereby the relative amounts of T-strand bound to VirB6, VirB8, VirB2 and VirB9 approximated 10% of levels found associated with VirD4 and VirB11 (data not shown). We conclude that the Q150.i4 and L201.i4 mutations indeed are partial function mutations that permit low-level transfer of all substrates, including the T-strand, through the secretion channel. These findings illustrate the robustness of the TrIP assay for monitoring substrate transfer, as well as the exquisite sensitivity of the quantitative assay for defining the magnitude of the effect a transfer arrest mutation exerts on substrate passage.
The identification of the class II mutations, those arresting substrate transfer from VirB6 to VirB8, establishes a temporal order to the translocation pathway that could not be detected by TrIP studies of the non-polar virB null mutations. The VirB6 domain responsible for this transfer step is next to loop P2 and consists minimally of three TMS and intervening loops. This transmembrane configuration, coupled with the bitopic topology of VirB8, suggests a model whereby the TMS of both proteins interact to form the secretion channel. Curiously, however, efforts to identify such a VirB6–VirB8 complex have been unsuccessful by use of IP or GST-pulldown assays33 or a yeast two-hybrid screen.38 The absence of a dihybrid interaction in yeast is not surprising in view of the overall hydrophobic character of VirB6. Moreover, the detergent treatments used to date, e.g. Triton X-100, dodecylmaltoside, and octylglucoside,33 for isolating membrane protein complexes from A. tumefaciens cells might have dissociated a VirB6–VirB8 complex. VirB6 might interact weakly with VirB8 or only upon substrate engagement. Regardless of the reasons, the results of the TrIP studies supply compelling evidence that VirB6 and VirB8 subunits interact functionally to mediate substrate transfer and, furthermore, the VirD2–T-strand complex forms a close contact first with VirB6 then with VirB8 in the export pathway.
There is accumulating evidence that VirB6 and VirB8 contribute to the biogenesis of this T4SS. VirB8 interactions with VirB9 and VirB10 were suggested with the yeast two-hybrid screen.38,39 Additionally, VirB8 production appears to nucleate assembly of a VirB8, VirB9, VirB10 complex in A. tumefaciens.40 VirB8 was shown to interact with the VirB1 putative transglycosylase.38 VirB8 might therefore contribute to machine biogenesis, first, by recruiting the transglycosylase to punch holes in the peptidoglycan, then by recruiting the structural subunits VirB9 and VirB10 to the site of assembly.38 Adding to this biogenesis pathway, we showed here that VirB6 localizes to the cell poles (Figure 2(A)), the probable site for assembly of the virB/virD4-encoded secretion channel and T pilus.23–25,41 VirB6 localization was independent of other channel subunits, suggesting that VirB6 positioning at the cell poles represents a very early step in this assembly pathway. VirB6 and VirB8 thus appear to coordinate their activities at two stages; during early machine biogenesis as polar positioning/nucleation factors, and later in a functional secretory apparatus for conveying substrate from VirD4 and VirB11 to the VirB2 and VirB9 channel subunits. Our recent studies of VirB10 (to be reported elsewhere) further suggest that this bitopic membrane protein plays an important role in biogenesis of this secretion system as a TonB-like energy coupler by sensing ATP-binding or hydrolysis activities of VirD4 and VirB11 and, in turn, forming a stable bridge between inner and outer membrane subcomplexes of the secretory apparatus.
Intact VirB6 termini were required for substrate transfer to VirB2 and VirB9 (Figures 5 and 6). These termini reside on opposite faces of the inner membrane (Figure 4), whereas VirB2 and VirB9 localize predominantly in the periplasm and outer membrane.42–45 The terminal domains might exert their effects on substrate transfer to these channel subunits through direct or indirect partner protein interactions. In support of direct protein–protein contacts, we have immunoprecipitated presumptive complexes of VirB6, VirB7, and VirB9 from extracts of wild-type cells as well as a strain producing only VirB6 through VirB10 among the VirB proteins.33 Of further interest, we showed that the synthesis of several VirB6.i4 mutant proteins is correlated with altered migration of higher-order VirB7 and VirB9 complexes in non-reducing protein gels.33 Mutants with i4 insertions in the terminal domains (E29.i4 and D60.i4; I260.i4 and A290.i4) induced formation of a similar pattern of VirB9 higher-order complexes, whereas mutants with insertions elsewhere in the protein induced a different pattern of VirB9 complexes.33 How these i4 mutations affect VirB9 oligomerization is unknown, but the combination of our earlier findings and the present TrIP studies suggest that the VirB6 terminal domains probably form subunit contacts required for machine assembly at the inner membrane–periplasm interface.
Channel subunit versus substrate entry portal?
A central outstanding question is how the VirD2–T-strand crosses the inner membrane. We envision two models to describe this process (Figure 7). According to the channel model, VirB6, VirB8 and perhaps other integral membrane VirB proteins form the translocation channel. Thus, VirD4 T4CP recruits the substrate and delivers it to VirB11; in turn, VirB11 utilizes the energy of ATP hydrolysis to “pump” the substrate through the channel. A contribution of VirB6 to channel formation is compatible with a demonstrated requirement for DNA transfer to the periplasmic proteins VirB2 and VirB9 (this study),16 a VirB6 stabilizing effect on other VirB proteins in vivo33,35,36 and the identification of higher-order complexes composed of VirB6 and other VirB proteins.33,46 Such an activity is reminiscent of known or postulated roles of polytopic subunits of dedicated protein translocation pathways in bacteria as well as eukaryotic cells.47,48
Figure 7.

Models for VirB6 function in mediating DNA transfer through the VirB/D4 T4SS. In the “channel” model, the VirD4 T4CP recruits the DNA substrate and delivers it to the VirB11 ATPase. VirB11 directs substrate transfer through an inner membrane channel composed of the five TMS of VirB6 (shaded) and the TMS of bitopic VirB8 (vertical dashes). Periplasmic loop P2 of VirB6 folds into the channel, possibly dynamically to facilitate DNA passage. In the “vestibule” model, VirD4 recruits and delivers substrate to the VirB11 chaperone for unfolding the VirD2 relaxase bound to the 5′ end of the T-strand. VirB11 delivers the translocation-competent substrate back to VirD4 for delivery across the membrane. At the periplasm-membrane interface, loop P2 of VirB6 forms a vestibule through which the substrate passes to gain entry into the secretion channel. In both models, VirB6 coordinates further progression through the periplasm via a channel composed of VirB8, VirB2 and VirB9. The models show only the six channel subunits identified by TrIP to form close contacts with the DNA substrate.16
Curiously, however, our TrIP studies identified loop P2 and not the TMS or cytoplasmic loops as necessary for the VirB6–substrate close contact. Loop P2 might thus form part of the channel architecture, embedding into the channel either stably or dynamically as a function of substrate transfer. Such a mechanism is reminiscent of the periplasmic loop of SecG, which is thought to invert across the membrane during SecA-mediated translocation via the general secretory pathway.49 Alternatively, it is noteworthy that loop P2 possesses one or more segments of contiguous hydrophobicity (95 to 115 by TopPredII; 144 to 164 by TopPredII, MEMSATII, PHDhtm) (Figure 1(A)). Our experimental findings do not support the TMS predictions, but these residues might still align with the membrane to coordinate substrate passage through the channel. By analogy, the second periplasmic loop of TatC possesses contiguous hydrophobic segments that do not span the inner membrane, and it is postulated that these residues align with the membrane in order to facilitate the transfer of folded proteins by a pushing or pulling mechanism.50
The alternative vestibule model postulates that the bulk of VirB6 functions as a structural scaffold, establishing subunit–subunit contacts to build the T4SS, and that only loop P2 participates directly in DNA transfer (Figure 7). In this model, the substrate is recruited to the T4SS by the VirD4 T4CP and delivered to the VirB11 chaperone for ATP-dependent unfolding of the VirD2 relaxase. VirB11 then delivers the translocation-competent VirD2-T-strand back to the T4CP translocase for substrate transfer across the inner membrane. At the periplasmic face of the inner membrane, the substrate gains access to the virB-encoded secretion channel through a vestibule formed by loop P2 of VirB6.
This model is compatible with the finding that loop P2, but not other regions of VirB6, is required for the substrate contact. Additionally, recent genetic, structural and biochemical findings strongly support a proposal that T4CPs function as translocases. For example, TrwB (R388) T4CP adopts a three-dimensional ball–stem structure, with the stem protruding across the inner membrane, reminiscent of F1-ATPase.51 The central cavity of the hexamer is ~20 Å in diameter, large enough to accommodate DNA and unfolded polypeptide substrates. T4CPs also have been shown to bind DNA and ATP,9,52,53 and these proteins exhibit sequence similarities with two known DNA translocases, Bacillus subtilis sporulation protein SpoIIIE and Escherichia coli cell division protein FtsK.14,54 Finally, this model might explain why small amounts of VirD2 relaxase and the VirE2 protein substrate were detected in the periplasmic fraction recovered by osmotic shock.55 Following translocation, the substrate could gain temporary access to the periplasm before entering the VirB6 portal.
In summary, VirB6 is an inner membrane component of the VirB/D4 T4SS with five experimentally-validated TMS and four functional domains. The central periplasmic loop P2 is required for the VirB6–substrate interaction, and might function as a domain of the inner membrane channel or a vestibule for substrate entry in the periplasm. An adjacent multi-membrane-spanning domain mediates substrate transfer to bitopic VirB8, and the two terminal domains residing on opposite faces of the membrane stimulate transfer from VirB8 to the periplasmic/OM-associated channel subunits VirB2 and VirB9. Further structure–function studies of these domains will generate a detailed mechanistic understanding of how this fascinating polytopic protein coordinates passage of a DNA substrate at several steps of a temporally ordered export pathway.
Materials and Methods
Bacterial strains and growth conditions
A. tumefaciens strain A348 served as the wild-type strain, the isogenic derivative PC1006 is a non-polar virB6 null mutant and strain A136 lacks the Ti plasmid.35 Strain KA1000 is strain A136 expressing the two-component regulatory system virA/virG on an IncP plasmid; the construction of this strain will be described elsewhere. Plasmids used in these studies are described below. The conditions and media for growth of A. tumefaciens and E. coli and for vir gene induction with 200 μM acetosyringone (AS) have been described.33 Plasmids were maintained in A. tumefaciens and E. coli by addition of carbenicillin (100 μg/ml) or kanamycin (100 μg/ml).
Constructs for topology studies with reporter proteins
In-frame fusions between N-terminal fragments of VirB6 and cytoplasmically active β-galactosidase (β-Gal) or periplasmically active alkaline phosphatase (PhoA)20 were constructed. Plasmid pXZ61 expressing Plac-virB6 was mutagenized with the TnlacZ/in or TnphoA/in transposons,55 as described.56 Plasmids from colonies displaying β-Gal or PhoA activities on histochemical plates were analyzed for insertions in the virB6 gene by restriction digest analysis and sequencing of fusion junctions. Plasmids pXZ605, pXZ601, pXZ602, pXZ603, and pXZ604 carry in-frame insertions at codons 44, 53, 150, 229, and 234, respectively, of virB6. However, multiple isolates bearing transposon insertions into only these few sites were recovered, prompting the construction of additional in-frame lacZ insertions by cloning. A ′lacZ gene lacking its first eight codons was isolated as a ~3.2 kb NotI-KpnI restriction fragment from pTAD25058 and inserted into each of ten similarly digested pSJ4xxx plasmids.33 As described below, the pSJ4xxx series of plasmids each carry a unique NotI restriction site inserted at ~30 codon intervals along the length of virB6. The resulting plasmid series, pSJ2xxx, express N-terminal fragments of virB6 fused in-frame to ′lacZ. In pSJ4xxx, pSJ2xxx, and other plasmid series constructed in this laboratory, xxx represents the number of the codon corresponding to the junction site of a chimeric gene of interest.57
We constructed a set of ten hybrid proteins in which ′phoA lacking both its signal sequence and translational stop codon was inserted in-frame at intervals of every ~30 codons of virB6. First, a BglII-SalI-TEV-NotI-′phoA-NotI-SalI-BglII gene cassette composed of codons for the TEV protease recognition site (ENLYFQS) and signal-sequenceless ′phoA was generated by PCR amplification with the transposon TnTAP59 as a template and the upstream (5′-ATACACAAGTTTGTCGACAGATCTGAAAACCTGTAC-3′) and downstream (5′-CGAATTGTAACCGTCGACAGATCTGCGGCCGCCTTTCAGCCCCAG-3′) primers; SalI (GTCGAC) and BglII (AGATCT) sites are in bold. Next, we introduced this cassette as a 1.4 kb BglII fragment into each of ten BamHI sites located at ~30 codon intervals along the length of virB6 in the previously-described pSJ6xxx plasmid series.33 The resulting plasmid series, designated pSJ8xxx, produce VirB6′-TEV-′PhoA-′VirB6 sandwich fusions. In our initial studies, strains producing fusion proteins with junction sites at residues 85 and 229 exhibited high PhoA activities, whereas all other strains displayed very low PhoA activities and the fusion proteins were not detected by immunostaining. In principle, the sandwich fusion approach offers a means of analyzing the topology of a full-length protein, but low reporter activities that often accompany such constructions60,61 can complicate assignments of domain topologies. For this reason, we did not analyze these sandwich fusions further, and instead used a combination of VirB6′ truncation reporter fusion and complementary cysteine accessibility approaches. However, the pSJ8xxx plasmids were digested by NotI digestion and religation to release the ′phoA fragment, generating the pSJ4xxx plasmids that were used as described above to construct the VirB6′-βgal fusion proteins.
We fused N-terminal fragments of VirB6 to signal-sequenceless ′phoA bearing a stop codon. The ′phoA cassette as a 2.2 kb BamHI-KpnI fragment from pUI115661 was inserted into each of ten similarly digested pSJ6xxx plasmids resulting in the pSJ3xxx plasmids. Additional VirB6′-PhoA hybrid proteins were constructed by introduction of ′phoA cassettes from pUI1156, pUI1158, pUI1160 (for the three reading frames62) into unique restriction enzyme sites in virB6. Plasmid pSJ964 was digested with the enzyme in parantheses and KpnI, which is located downstream of virB6: codon 70 (BstEII), 146 (StyI made blunt-ended with Klenow), 167 (EcoRI), 238 (SacII made blunt-ended with phage T4 DNA polymerase). Next, restriction fragments carrying ′phoA from the appropriate pUI plasmid (in parentheses) were ligated to these linearized plasmids: EcoRV-KpnI fragment (pUI1156) to the BstEII-KpnI fragment, EcoRV-KpnI fragment (pUI1160) to the StyI (blunted)-KpnI fragment, EcoR1-KpnI fragment (pUI1160) to the EcoRI-KpnI fragment, and EcoRV-KpnI fragment (pUI1158) to the SacII-KpnI fragment. The resulting plasmids are named pXZ671 (fusion junction at codon 70), pXZ673 (146), pXZ674 (167), and pXZ675 (238).
To construct a chimeric gene producing full-length VirB6 fused to the green fluorescent protein (GFP), we first introduced an XbaI restriction site immediately upstream of the virB6 stop codon by PCR using pXZ61 as a template and the following oligonucleotide primers: 5′-CAGGTTGAACGGGGTCTAGAGATCTCCATGGCTTAC-3′ and 5′-GCGTCTAGACCCCGTTCAACCTGAGC-3′ (XbaI site in bold). We next introduced the GFP gene as a 0.73 kb XbaI-SalI fragment from pXZ6257 into similarly digested pXZ6157 to yield pXZ176 expressing Plac-virB6-GFP. Next, a 1.3 kb NdeI fragment of pXZ176 carrying virB6 and the N-terminal 220 bp of GFP was introduced into similarly digested pZD8863 to yield pSJB6NGFP expressing virB6 and the 5′ end of GFP from PvirB. Finally, an 800 bp NcoI-KpnI fragment of pXZ176 carrying the 3′ half of GFP was cloned into similarly digested pSJB6NGFP to yield pSJB6GFP expressing PvirB-virB6-GFP.
Construction of cysteine substitution mutations
We introduced ten cysteine substitution mutations in virB6 with the PCR QuikChange® site-directed mutagenesis protocol (Strategene) with plasmid pSJ964 as a template and the complementary pairs of oligonucleotides listed in Table 1. Plasmids were isolated from E. coli DH5α and the substitution mutations were confirmed by restriction analysis (each substitution introduced an SphI or a PstI site) and then by sequencing. The plasmids, designated pKVD50-pKVD59, were ligated to the broad host range plasmid pXZ151 for introduction into the non-polar virB6 null mutant PC1006.
Table 1.
Oligonucleotide primers used to construct VirB6 cysteine substitution mutations
| Mutation | 5′ primer | 3′ primer |
|---|---|---|
| I12C | 5′-GCCGTTTACGGCATGCCATACGATCTTCGATG-3′ | 5′-AAGATCGTATGGCATGCCGTAAACGG-3′ |
| D25C | 5′-CTTCACGACAGGCCTCTGCAGGATGCTTGAG-3′ | 5′-GTCTCAAGCATCCTGCAGAGGCCTGTC-3′ |
| R62C | 5′-GAAGTCGATACCTGCAGCGGTATCACTCG-3′ | 5′-GTGATACCGCTGCAGGTATCGACTTC-3′ |
| T66C | 5′-CCCGAGCGGTATCTGCAGGGTGATCACGGTC-3′ | 5′-GTGATCACCCTGCAGATACCGCTGCG-3′ |
| Q116C | 5′-GACTGTTCCGGCATGCTTGGATACAATTTTCG-3′ | 5′-AATTGTATCCAAGCATGCCGGAACAGT-3′ |
| S133C | 5′-CAGAAAATCGCATGCGAAATCGGTCCG-3′ | 5′-CGGACCGATTTCGCATGCGATTTTCTG-3′ |
| D188C | 5′-GGATATATTTTCTGCAGGACGCGGGACATC-3′ | 5′-GATGTCCCGCGTCCTGCAGAAAATATATC-3′ |
| T224C | 5′-CCTAACCGAAGCATGCGCGCTCACCCTC-3′ | 5′-GAGGGTGAGCGCGCATGCTTCGGTTAGG-3′ |
| L263C | 5′-CGCTCATTGTCGCATGCCCGGCAATCGCCGGC-3′ | 5′-GGCGATTGCCGGGCATGCGACAATGAGC-3′ |
| Q291C | 5′-ACCGTCGCTTCGCATGCGTTGAACGGGGCTAG-3′ | 5′-GCCCCGTTCAACGCATGCGAAGCGACG-3′ |
Restriction sites SphI (GCATGC) and PstI (CTGCAG) in bold; Cys codon (TGC) underlined.
Construction of virB6 insertion, truncation, and internal deletion mutations
The pSJ6xxx series plasmids producing VirB6 derivatives bearing four-residue insertion (i4) mutations have been described.33 The ten insertion mutations were identified by the VirB6 residue adjacent to the insertion site; however, for the seven mutations positioned C-terminal to the Q85.i4 mutation, the insertion sites were labeled incorrectly. The correct insertion sites are shifted by about ten residues, e.g. the former T114.i4 mutation33 is now T124.i4, Q140.i4 is Q150.i4, and so on. We constructed a set of five mutant proteins bearing an i31 peptide insertion. Plasmids pXZ601-pXZ605 obtained by TnlacZ/in transposon mutagenesis (see above) were digested with BamHI and religated to remove the lacZ gene and transposon sequence. The resulting plasmids named pXZ621-pXZ625 carry the 31-peptide insertion (i31) SDSYTQVASWTEPFPFSIQGDPRSDQETVXX, with X indicating residues determined by the insertion target site.
Plasmids pXZ693 and pXZ694 synthesize N-terminal fragments of VirB6, designated Δ158-295 and Δ234-295, respectively. These plasmids were obtained by TnlacZ/in mutagenesis in which the transposon had inserted out-of-frame at codons 158 and 234, respectively, in both cases resulting in a stop codon near the 5′ end of the transposon sequence. The following internal deletions were constructed by cloning using fragments of virB6 obtained from the pSJ5xxx plasmid series. Plasmid pKVD61 expressing virB6Δ1-30 (ATG initiation at codon 30) was constructed by inserting an NcoI-XhoI fragment bearing codons 30–295 from plasmid pSJ5030 into similarly digested pPJC914KS+NcoI. Likewise, plasmid pKVD62 expressing virB6′Δ30-60 was constructed by inserting an NcoI-XhoI fragment bearing codons 60–295 from plasmid pSJ5061 into similarly digested pSJ5030. Plasmid pKVD64 expressing virB6Δ116-133 was constructed by inserting an NdeI-SphI fragment bearing codons 1–116 from pKVD51 into similarly digested pKVD52. Likewise, plasmid pKVD63 expressing virB6′Δ263-291 was constructed by inserting an NdeI-SphI fragment bearing codons 1–263 from pKVD58 into similarly digested pKVD59. Plasmid pKVD65 expressing virB6Δ160-221 was made by site-directed mutagenesis using the complementary mutagenic primers 5′-CGGCACGCTCTGGTCTGAAGCGACTGCGCTC-3′ and 5′-GGTGAGCGCAGTCGCTTCAGACCAGAGCGTGCCGTAAAG-3′ with pSJ964 as template. Finally, pKVD66 expressing an internal fragment of virB6 (codons 55–195) was constructed by PCR amplification of the fragment from the template pSJ964 using the forward primer 5′-CAGGGTATTTTACATATGCGCGGCGAAGTCGATAC-3′ and the reverse primer 5′-GATAAGTTGCTCGAGTCATTACGCTGCGATGTCCCG-3′. The fragment was introduced as an NdeI-XhoI fragment into similarly digested pSJ964.
Reporter protein activities and GFP detection by fluorescence microscopy
Alkaline phosphatase (AS) and β-galactosidase assays were performed according to the methods of Manoil20 and Miller.64 Reporter protein activities in PhoA or Miller units represent the average of at least three experiments with standard deviations indicated. AS-induced cells were examined by differential interference microscopy (DIC), and for GFP production by fluorescence microscopy. Each experiment was replicated at least three times. Images of cells were acquired with an Olympus BX60 microscope equipped with a 100× oil immersion phase-contrast objective as described.63
Chemical labeling of cysteine residues
Cysteine accessibility experiments were carried out as described by Bogdanov65 and Wang.66 A 75 ml culture of A. tumefaciens strain PC1006 producing cysteine-substituted VirB6 derivatives was induced for vir gene expression by growth in ABIM to an A600 of 0.5. Cells were harvested, resuspended in buffer A (100 mM Hepes (pH 7.5), 250 mM sucrose, 25 mM MgCl2, 0.1 mM KCl), and distributed into two centrifuge tubes to a final A600 of 12 in 500 μl of buffer A. To one cell suspension, 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; Molecular Probes) was added to a final concentration of 5 mM and cells were incubated for 30 minutes at 25 °C. AMS was removed by suspending cells in 5 ml of buffer A and centrifuging twice, and the washed cell pellet was suspended in 500 μl of buffer A. To the AMS-pretreated cells and the second cell suspension, 3-(N-maleimidylpropionyl) biocytin (MPB; Molecular Probes) was added to a final concentration of 100 μM (from a 20 mM stock freshly dissolved in DMSO) and the cells were incubated for five minutes at 25 °C. The final concentration of DMSO in the reaction mixture did not exceed 0.5% (v/v). β-Mercaptoethanol (20 mM final concentration) was added to quench biotinylation, and cells were washed twice in buffer A containing 20 mM β-mercaptoethanol and suspended in 200 μl of TES (10 mM Tris (pH 7.5), 5 mM EDTA, 2% (w/v) SDS).
Immunoprecipitation and detection of labeled Cys residues
Labeled cells suspended in TES were vortex mixed vigorously for 30 minutes at 37 °C and then diluted with 250 μl of buffer B (150 mM Tris (pH 8.0), 0.5 M sucrose, 10 mM EDTA). Lysozyme (1 mg/ml final concentration) was added and the samples were incubated on ice for one hour, then vortex mixed for 15 minutes at 37 °C. Twenty microliters of Triton X-100 and 30 μl of EDTA-free protease inhibitor cocktail (Pierce Biochemicals; one tablet dissolved in 500 μl of buffer B) were added and the samples were vortex mixed for ten minutes at 25 °C, then incubated with gentle rocking for three hours at 4 °C. Samples were diluted with 900 μl of buffer B, and cell debris was removed by centrifugation at 14,000g for 15 minutes. Protein A-Sepharose CL4B (Pharmacia) (30 μl bed volume) was incubated with the supernatant for 60 minutes at room temperature and centrifuged at 5000g to remove protein A-Sepharose and non-specifically bound proteins. Anti-VirB6 antibodies coupled to protein A-Sepharose were added to the supernatant and incubated overnight at 4 °C. The beads were washed twice in buffer B with 1% Triton X-100 for ten minutes, once in buffer B with 0.1% Triton X-100 for ten minutes, once in buffer B for five minutes, and resuspended in 30 μl of 2× Laemmli’s buffer. Samples were incubated at 37 °C for one hour with shaking, centrifuged, and the solubilized proteins were subjected to SDS-PAGE and transfered to nitrocellulose membranes (0.45 μm pore size). Membranes were incubated overnight in blocking buffer (PBS, 0.1% Tween 20, 5% (w/v) bovine serum albumin (BSA)) and then for four hours in the presence of avidin HRP (Pierce; 1: 10,000 dilution of 2 mg/ml stock solution). Blots were washed three times in buffer C (PBS, 0.1% Tween 20, 0.5% BSA) and biotinylated proteins were analyzed by chemiluminescence according to the manufacturer’s (Amersham Pharmacia) instructions.
Preparation of membrane vesicles and labeling with MPB
Cells from a 500 ml Agrobacterium culture grown to an A600 of 0.5 in ABIM were lysed by a single pass through a French pressure cell operated at 560 kg/cm2 (8000 psi; 1 psi ≈6.9 kPa). Cell debris was removed by centifugation at 20,000g for ten minutes, and the supernatant was centrifuged at 100,000g for one hour to recover vesicles. This method yielded predominantly inverted vesicles but a fraction was right-side-out, as shown by protease susceptibility of membrane proteins exposed on the cytoplasmic (e.g. VirB11) and periplasmic (e.g. VirB10) face of the membrane (data not shown). Vesicles served as controls to test for MPB accessibility of VirB6 Cys substitution mutations on either side of the inner membrane, thus the ratio of inverted versus right-side-out vesicles was not quantified. Vesicles were suspended in buffer A and total protein concentration was determined. Samples of vesicles containing equivalent amounts of protein in 200 μl of buffer A were labeled with MPB (20 μM final concentration) as described above. The reaction was quenched with β-mercaptoethanol (20 mM final concentration), the vesicles were solubilized, and VirB6 was recovered by immunoprecipitation as described above.
Transfer immunoprecipitation (TrIP) assay
The transfer immunoprecipitation (TrIP) assay was performed as described.16 Briefly, 6 ml of cells induced for vir gene expression was suspended in 20 mM sodium phosphate buffer (pH 6.8) with formaldehyde (FA; 0.1% (v/v) final concentration). Cells were incubated for 20 minutes at 18 °C with shaking, FA was added in 0.2% increments to reach a final concentration of 1% over 15 minutes, and the cells were incubated for 40 minutes at room temperature without shaking. Cells were pelleted, resuspended in 200 μl of TES buffer (50 mM Tris–HCl (pH 6.8), 2 mM EDTA, 1% β-mercaptoethanol, 1% SDS) and then incubated for 30 minutes at 37 °C with shaking. Next, 900 μl of NP1 buffer (150 mM Tris–HCl (pH 8.0), 0.5 M sucrose, 10 mM EDTA) supplemented with 1 mg/ml of lysozyme was added, the mixture was incubated for one to two hours on ice and then for 30 minutes at 37 °C with shaking. Triton X-100 was added to a 4% final concentration and the mixture was incubated for 15 minutes at room temperature on a wheel. A 5× solution of protease inhibitors cocktail, EDTA-free (Complete, Boehringer–Mannheim) in 25 mM MgCl2 was added and the mixture was incubated with rocking for 15 minutes at 37 °C and then for two to three hours at 4 °C on a wheel. Then, 3.2 ml of NP1 buffer was added and the insoluble material was removed by centrifugation for 15 minutes at 14,000g. Proteins of interest were immunoprecipitated as described above. For detection of DNA, 0.05 of the immunoprecipitates or 0.00001 of the soluble fraction was analyzed by PCR amplification using 50 pmol of each of four primers per reaction. Primers for amplification of the T-DNA region corresponding to gene 7 of the Tl-DNA were 5′-GGGCGATTATGGCATCCAGAAAGCC-3′ and 5′-GTCGGCGGCCCACTTGGCACACAG-3′. Primers for the non-transferred ophDC locus (a region of the Ti plasmid ~25 kb from the transferred T-DNA) were 5′-CCTGCGGATGTCAGGGCTCTCGT-3′ and 5-CTGTCCGTGCTTGCCAATCCCCG-3′. PCR products were separated by electrophoresis through 1.2% (w/v) agarose gels and visualized by staining with ethidium bromide. A quantitative version of the TrIP assay was carried out as described.16
Acknowledgments
We thank Xue-Rong Zhou and Krishnamohan Atmakuri for strain constructions. We thank Krishnamohan Atmakuri, Mikhail Bogdanov, and William Dowhan for helpful discussions, and Mikhail Bogdanov for guidance and supplies for the Cys accessibility studies. We thank William Margolin and members of the Margolin laboratory for GFP antibodies and use of their fluorescence microscopy facility. This work was supported by NIH grant GM48746.
Abbreviations used
- T4SS
type IV secretion systems
- Dtr
DNA transfer and replication
- oriT
the origin-of-transfer
- T-strand
the strand destined for transfer
- Mpf
mating pair formation
- T-DNA
transfer DNA
- TrIP
transfer DNA immunoprecipitation
- TMS
transmembrane segments
- PhoA
alkaline phosphatase
- β-Gal
β-galactosidase
- GFP
green fluorescent protein
- AS
acetosyringone
- MPB
3-(N-maleimidylpropionyl) biocytin
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
References
- 1.Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nature Rev Microbiol. 2003;1:137–150. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawley TD, Klimke WA, Gubbins MJ, Frost LS. F factor conjugation is a true type IV secretion system. FEMS Microbiol Letters. 2003;224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- 3.Lessl M, Lanka E. Common mechanisms in bacterial conjugation and Ti-mediated T-DNA transfer to plant cells. Cell. 1994;77:321–324. doi: 10.1016/0092-8674(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 4.Byrd DR, Matson SW. Nicking by transesterification: the reaction catalysed by a relaxase. Mol Microbiol. 1997;25:1011–1022. doi: 10.1046/j.1365-2958.1997.5241885.x. [DOI] [PubMed] [Google Scholar]
- 5.Llosa M, Gomis-Ruth FX, Coll M, de la Cruz F. Bacterial conjugation: a two-step mechanism for DNA transport. Mol Microbiol. 2002;45:1–8. doi: 10.1046/j.1365-2958.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 6.Gomis-Ruth FX, de la Cruz F, Coll M. Structure and role of coupling proteins in conjugal DNA transfer. Res Microbiol. 2002;153:199–204. doi: 10.1016/s0923-2508(02)01313-x. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton CM, Lee H, Li PL, Cook DM, Piper KR, von Bodman SB, et al. TraG from RP4 and TraG and VirD4 from Ti plasmids confer relaxosome specificity to the conjugal transfer system of pTiC58. J Bacteriol. 2000;182:1541–1548. doi: 10.1128/jb.182.6.1541-1548.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szpirer CY, Faelen M, Couturier M. Interaction between the RP4 coupling protein TraG and the pBHR1 mobilization protein Mob. Mol Microbiol. 2000;37:1283–1292. doi: 10.1046/j.1365-2958.2000.02077.x. [DOI] [PubMed] [Google Scholar]
- 9.Schroder G, Krause S, Zechner EL, Traxler B, Yeo HJ, Lurz R, et al. TraG-like proteins of DNA transfer systems and of the Helicobacter pylori type IV secretion system: inner membrane gate for exported substrates? J Bacteriol. 2002;184:2767–2779. doi: 10.1128/JB.184.10.2767-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arrondo JL, Echabe I, Iloro I, Hernando MA, de la Cruz F, Goni FM. A bacterial TrwC relaxase domain contains a thermally stable alpha-helical core. J Bacteriol. 2003;185:4226–4232. doi: 10.1128/JB.185.14.4226-4232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guasch A, Lucas M, Moncalian G, Cabezas M, Perez-Luque R, Gomis-Ruth FX, et al. Recognition and processing of the origin of transfer DNA by conjugative relaxase TrwC. Nature Struct Biol. 2003;10:1002–1010. doi: 10.1038/nsb1017. [DOI] [PubMed] [Google Scholar]
- 12.Larkin C, Datta S, Nezami A, Dohm JA, Schildbach JF. Crystallization and preliminary X-ray characterization of the relaxase domain of F factor TraI. Acta Crystallog sect D. 2003;59:1514–1516. doi: 10.1107/s0907444903012964. [DOI] [PubMed] [Google Scholar]
- 13.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure (Camb) 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Gomis-Ruth FX, Coll M. Structure of TrwB, a gatekeeper in bacterial conjugation. Int J Biochem Cell Biol. 2001;33:839–843. doi: 10.1016/s1357-2725(01)00060-7. [DOI] [PubMed] [Google Scholar]
- 15.Zhu J, Oger PM, Schrammeijer B, Hooykaas PJ, Farrand SK, Winans SC. The bases of crown gall tumorigenesis. J Bacteriol. 2000;182:3885–3895. doi: 10.1128/jb.182.14.3885-3895.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cascales E, Christie PJ. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science. 2004;304:1170–1173. doi: 10.1126/science.1095211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, et al. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J. 2003;22:1969–1980. doi: 10.1093/emboj/cdg223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rashkova S, Spudich GM, Christie PJ. Mutational analysis of the Agrobacterium tumefaciens VirB11 ATPase: identification of functional domains and evidence for multimerization. J Bacteriol. 1997;179:583–589. doi: 10.1128/jb.179.3.583-591.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson J, Persson B, von Heijne G. Consensus predictions of membrane protein topology. FEBS Letters. 2000;486:267–269. doi: 10.1016/s0014-5793(00)02321-8. [DOI] [PubMed] [Google Scholar]
- 20.Manoil C. Analysis of membrane protein topology using alkaline phosphatase and β-galactosidase gene fusions. Methods Cell Biol. 1991;34:61–75. doi: 10.1016/s0091-679x(08)61676-3. [DOI] [PubMed] [Google Scholar]
- 21.Melen K, Krogh A, von Heijne G. Reliability measures for membrane protein topology prediction algorithms. J Mol Biol. 2003;327:735–744. doi: 10.1016/s0022-2836(03)00182-7. [DOI] [PubMed] [Google Scholar]
- 22.Drew D, Sjostrand D, Nilsson J, Urbig T, Chin CN, de Gier JW, von Heijne G. Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc Natl Acad Sci USA. 2002;99:2690–2695. doi: 10.1073/pnas.052018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai EM, Chesnokova O, Banta LM, Kado CI. Genetic and environmental factors affecting T-pilin export and T-pilus biogenesis in relation to flagellation of Agrobacterium tumefaciens. J Bacteriol. 2000;182:3705–3716. doi: 10.1128/jb.182.13.3705-3716.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar RB, Das A. Polar location and functional domains of the Agrobacterium tumefaciens DNA transfer protein VirD4. Mol Microbiol. 2002;43:1523–1532. doi: 10.1046/j.1365-2958.2002.02829.x. [DOI] [PubMed] [Google Scholar]
- 25.Atmakuri K, Ding Z, Christie PJ. VirE2, a type IV secretion substrate, interacts with the VirD4 transfer protein at cell poles of Agrobacterium tumefaciens. Mol Microbiol. 2003;49:1699–1713. doi: 10.1046/j.1365-2958.2003.03669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seal RP, Leighton BH, Amara SG. Transmembrane topology mapping using biotin-containing sulfhydryl reagents. Methods Enzymol. 1998;296:318–331. doi: 10.1016/s0076-6879(98)96024-4. [DOI] [PubMed] [Google Scholar]
- 27.Loo TW, Clarke DM. Membrane topology of a cysteine-less mutant of human P-glycoprotein. J Biol Chem. 1995;270:843–848. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- 28.Wada T, Long JC, Zhang D, Vik SB. A novel labeling approach supports the five-transmembrane model of subunit α of the Escherichia coli ATP synthase. J Biol Chem. 1999;274:17353–17357. doi: 10.1074/jbc.274.24.17353. [DOI] [PubMed] [Google Scholar]
- 29.Long JC, Wang S, Vik SB. Membrane topology of subunit α of the F1F0 ATP synthase as determined by labeling of unique cysteine residues. J Biol Chem. 1998;273:16235–16240. doi: 10.1074/jbc.273.26.16235. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Bogdanov M, Pi J, Pittard AJ, Dowhan W. Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition. J Biol Chem. 2003;278:50128–50135. doi: 10.1074/jbc.M309840200. [DOI] [PubMed] [Google Scholar]
- 31.Beaupré CE, Bohne J, Dale EM, Binns AN. Interactions between VirB9 and VirB10 membrane proteins involved in movement of DNA from Agrobacterium tumefaciens into plant cells. J Bacteriol. 1997;179:78–89. doi: 10.1128/jb.179.1.78-89.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Geest M, Lolkema JS. Transmembrane segment (TMS) VIII of the Na(+)/Citrate transporter CitS requires downstream TMS IX for insertion in the Escherichia coli membrane. J Biol Chem. 1999;274:29705–29711. doi: 10.1074/jbc.274.42.29705. [DOI] [PubMed] [Google Scholar]
- 33.Jakubowski SJ, Krishnamoorthy V, Christie PJ. Agrobacterium tumefaciens VirB6 protein participates in formation of VirB7 and VirB9 complexes required for type IV secretion. J Bacteriol. 2003;185:2867–2878. doi: 10.1128/JB.185.9.2867-2878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao TB, Saier MH., Jr Conjugal type IV macromolecular transfer systems of Gram-negative bacteria: organismal distribution, structural constraints and evolutionary conclusions. Microbiology. 2001;147:3201–3214. doi: 10.1099/00221287-147-12-3201. [DOI] [PubMed] [Google Scholar]
- 35.Berger BR, Christie PJ. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol. 1994;176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hapfelmeier S, Domke N, Zambryski PC, Baron C. VirB6 is required for stabilization of VirB5 and VirB3 and formation of VirB7 homodimers in Agrobacterium tumefaciens. J Bacteriol. 2000;182:4505–4511. doi: 10.1128/jb.182.16.4505-4511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das A, Xie YH. Construction of transposon Tn3phoA: its application in defining the membrane topology of the Agrobacterium tumefaciens DNA transfer proteins. Mol Microbiol. 1998;27:405–414. doi: 10.1046/j.1365-2958.1998.00688.x. [DOI] [PubMed] [Google Scholar]
- 38.Ward DV, Draper O, Zupan JR, Zambryski PC. Peptide linkage mapping of the Agrobacterium tumefaciens vir-encoded type IV secretion system reveals protein subassemblies. Proc Natl Acad Sci USA. 2002;99:11493–11500. doi: 10.1073/pnas.172390299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das A, Xie YH. The Agrobacterium T-DNA transport pore proteins VirB8, VirB9, and VirB10 interact with one another. J Bacteriol. 2000;182:758–763. doi: 10.1128/jb.182.3.758-763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar RB, Xie YH, Das A. Subcellular localization of the Agrobacterium tumefaciens T-DNA transport pore proteins: VirB8 is essential for the assembly of the transport pore. Mol Microbiol. 2000;36:608–617. doi: 10.1046/j.1365-2958.2000.01876.x. [DOI] [PubMed] [Google Scholar]
- 41.Ding Z, Atmakuri K, Christie PJ. The outs and ins of bacterial type IV secretion substrates. Trends Microbiol. 2003;11:527–535. doi: 10.1016/j.tim.2003.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thorstenson Y, Kuldau G, Zambryski P. Subcellular localization of seven VirB proteins of Agrobacterium tumefaciens: implications for the formation of a T-DNA transport structure. J Bacteriol. 1993;175:5233–5241. doi: 10.1128/jb.175.16.5233-5241.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirasu K, Kado C. Membrane location of the Ti plasmid VirB proteins involved in the biosynthesis of a pilin-like conjugative structure on Agrobacterium tumefaciens. FEMS Microbiol Letters. 1993;111:287–294. doi: 10.1111/j.1574-6968.1993.tb06400.x. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez D, Dang TAT, Spudich GM, Zhou XR, Berger BR, Christie PJ. The Agrobacterium tumefaciens virB7 gene product, a proposed component of the T-complex transport apparatus, is a membrane-associated lipoprotein exposed at the periplasmic surface. J Bacteriol. 1996;178:3156–3167. doi: 10.1128/jb.178.11.3156-3167.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sagulenko V, Sagulenko E, Jakubowski S, Spudich E, Christie PJ. VirB7 lipoprotein is exocellular and associates with the Agrobacterium tumefaciens T pilus. J Bacteriol. 2001;183:3642–3651. doi: 10.1128/JB.183.12.3642-3651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krall L, Wiedemann U, Unsin G, Weiss S, Domke N, Baron C. Detergent extraction identifies different VirB protein subassemblies of the type IV secretion machinery in the membranes of Agrobacterium tumefaciens. Proc Natl Acad Sci USA. 2002;99:11405–11410. doi: 10.1073/pnas.172390699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saier MH., Jr Tracing pathways of transport protein evolution. Mol Microbiol. 2003;48:1145–1156. doi: 10.1046/j.1365-2958.2003.03499.x. [DOI] [PubMed] [Google Scholar]
- 48.Cao TB, Saier MH., Jr The general protein secretory pathway: phylogenetic analyses leading to evolutionary conclusions. Biochim Biophys Acta. 2003;1609:115–125. doi: 10.1016/s0005-2736(02)00662-4. [DOI] [PubMed] [Google Scholar]
- 49.Nishiyama K, Suzuki T, Tokuda H. Inversion of the membrane topology of SecG coupled with SecA-dependent preprotein translocation. Cell. 1996;85:71–81. doi: 10.1016/s0092-8674(00)81083-1. [DOI] [PubMed] [Google Scholar]
- 50.Gouffi K, Santini CL, Wu LF. Topology determination and functional analysis of the Escherichia coli TatC protein. FEBS Letters. 2002;525:65–70. doi: 10.1016/s0014-5793(02)03069-7. [DOI] [PubMed] [Google Scholar]
- 51.Gomis-Ruth FX, Moncalian G, Perez-Luque R, Gonzalez A, Cabezon E, de la Cruz F, Coll M. The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature. 2001;409:637–641. doi: 10.1038/35054586. [DOI] [PubMed] [Google Scholar]
- 52.Hormaeche I, Alkorta I, Moro F, Valpuesta JM, Goni FM, de La Cruz F. Purification and properties of TrwB, a hexameric, ATP-binding integral membrane protein essential for R388 plasmid conjugation. J Biol Chem. 2002;277:46456–46462. doi: 10.1074/jbc.M207250200. [DOI] [PubMed] [Google Scholar]
- 53.Schroder G, Lanka E. TraG-like proteins of type IV secretion systems: functional dissection of the multiple activities of TraG (RP4) and TrwB (R388) J Bacteriol. 2003;185:4371–4381. doi: 10.1128/JB.185.15.4371-4381.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Errington J, Bath J, Wu LJ. DNA transport in bacteria. Nature Rev Mol Cell Biol. 2001;2:538–545. doi: 10.1038/35080005. [DOI] [PubMed] [Google Scholar]
- 55.Pantoja M, Chen L, Chen Y, Nester EW. Agrobacterium type IV secretion is a two-step process in which export substrates associate with the virulence protein VirJ in the periplasm. Mol Microbiol. 2002;45:1325–1335. doi: 10.1046/j.1365-2958.2002.03098.x. [DOI] [PubMed] [Google Scholar]
- 56.Manoil C, Bailey J. A simple screen for permissive sites in proteins: analysis of Escherichia coli lac permease. J Mol Biol. 1997;267:250–263. doi: 10.1006/jmbi.1996.0881. [DOI] [PubMed] [Google Scholar]
- 57.Zhou XR, Christie PJ. Mutagenesis of Agrobacterium VirE2 single-stranded DNA-binding protein identifies regions required for self-association and interaction with VirE1 and a permissive site for hybrid protein construction. J Bacteriol. 1999;181:4342–4352. doi: 10.1128/jb.181.14.4342-4352.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dang TAT, Christie PJ. The VirB4 ATPase of Agrobacterium tumefaciens is a cytoplasmic membrane protein exposed at the periplasmic surface. J Bacteriol. 1997;179:453–462. doi: 10.1128/jb.179.2.453-462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehrmann M, Bolek P, Mondigler M, Boyd D, Lange R. TnTIN and TnTAP: mini-transposons for site-specific proteolysis in vivo. Proc Natl Acad Sci USA. 1997;94:13111–13115. doi: 10.1073/pnas.94.24.13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blank TE, Donnenberg MS. Novel topology of BfpE, a cytoplasmic membrane protein required for type IV fimbrial biogenesis in enteropathogenic Escherichia coli. J Bacteriol. 2001;183:4435–4450. doi: 10.1128/JB.183.15.4435-4450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexeyev MF, Winkler HH. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J Mol Biol. 1999;285:1503–1513. doi: 10.1006/jmbi.1998.2412. [DOI] [PubMed] [Google Scholar]
- 62.Ouchane S, Kaplan S. Topological analysis of the membrane-localized redox-responsive sensor kinase PrrB from Rhodobacter sphaeroides 2.4.1. J Biol Chem. 1999;274:17290–17296. doi: 10.1074/jbc.274.24.17290. [DOI] [PubMed] [Google Scholar]
- 63.Ding Z, Zhao Z, Jakubowski SJ, Krishnamohan A, Margolin W, Christie PJ. A novel cytology-based, two-hybrid screen for bacteria applied to protein–protein interaction studies of a type IV secretion system. J Bacteriol. 2002;184:5572–5582. doi: 10.1128/JB.184.20.5572-5582.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 65.Bogdanov M, Heacock PN, Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002;21:2107–2116. doi: 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Bogdanov M, Dowhan W. Topology of polytopic membrane protein subdomains is dictated by membrane phospholipid composition. EMBO J. 2002;21:5673–5681. doi: 10.1093/emboj/cdf571. [DOI] [PMC free article] [PubMed] [Google Scholar]