

Abstract
Limited lifespan and senescence are quantitative traits, controlled by many interacting genes with individually small and environmentally plastic effects, complicating genetic analysis. We performed genome wide analysis of gene expression for two Drosophila melanogaster lines selected for postponed senescence and one control, unselected line to identify candidate genes affecting lifespan as well as variation in lifespan. We obtained gene expression profiles for young flies of all lines, all lines at the time only 10% of the control lines survived, and the time at which 10% of the selected lines survived. Transcriptional responses to aging involved 19% of the genome. The transcriptional signature of aging involved the down-regulation of genes affecting proteolysis, metabolism, oxidative phosphorylation, and mitochrondrial function; and the up-regulation of genes affecting protein synthesis, immunity, defense responses, and the detoxification of xenobiotic substances. The transcriptional signature of postponed senescence involved the up-regulation of proteases and phosphatases and genes affecting detoxification of xenobiotics; and the down-regulation of genes affecting immunity, defense responses, metabolism and muscle function. Functional tests of 17 mutations confirmed 12 novel genes affecting Drosophila lifespan. Identification of genes affecting longevity by analysis of gene expression changes in lines selected for postponed senescence thus complements alternative genetic approaches.
Keywords: aging, postponed senescence, gene expression, candidate genes, artificial selection
1. Introduction
Aging and senescence – the progressive deterioration of survivorship and fertility with age – are near universal phenomena among eukaryotes. Aging is associated with many complex phenotypes with variation attributable to quantitative trait loci (QTLs) with individually small effects whose expression is sensitive to the environment. Much progress has been made toward understanding the genetic and molecular basis of longevity by analysis of mutations in model organisms (Guarente and Kenyon, 2000). However, there is growing evidence that large numbers of genes may affect lifespan (Zou et al., 2000; Pletcher et al., 2002; Curtis et al., 2007); therefore, understanding the genetic architecture underlying this complex suite of traits requires the ability to study whole genomes.
Model organisms with increased longevity due to environmental manipulations or selection pressures, such as dietary restriction or selection for postponed senescence, exhibit correlated traits indicative of a slowed normal aging program, such as reduced and/or delayed reproduction (Rose, 1984; Chapman and Partridge, 1996), and are valuable for studying the normal aging process (Partridge, 2001). Mounting evidence of changing patterns of gene expression with age from whole genome transcriptional profiles also suggests experimentally extended lifespan may be due to a slower rate of normal aging (Cao et al., 2001; Pletcher et al., 2002; Curtis et al., 2007).
Genome wide expression analysis has been used to identify thousands of genes affecting and affected by the aging process in several model organisms, including S. cerevisiae, C. elegans, D. melanogaster and Mus musculus (Zou et al., 2000; Lund et al., 2002; Pletcher et al., 2002; Jin et al., 2001; Hamatani et al., 2004; Landis et al., 2004; Fabrizio et al., 2005; Curtis et al., 2007; Lai et al., 2007a). However, such studies cannot distinguish causal from co-regulated changes in gene expression, and typically utilize only a single genotype. On the other hand, mapping QTLs affecting naturally occurring variation in lifespan has identified genomic regions affecting variation in longevity in C. elegans (Shook et al., 1996; Ayyadevara et al., 2001), Drosophila (Nuzhdin et al., 1997; Leips and Mackay, 2000; 2002; Vieira et al., 2000; Curtsinger and Khazaeli, 2002; Reiwitch and Nuzhdin, 2002; Forbes et al., 2004; Valenzuela et al., 2004), mice (Jackson et al., 2002; Doria et al., 2004) and humans (Doria et al., 2004). However, QTL regions mapped by linkage to molecular markers typically embrace hundreds of candidate genes. In Drosophila, quantitative complementation tests to deficiencies can greatly refine QTL map positions (Pasyukova et al., 2000); and complementation tests to mutations of positional candidate genes have been successful in identifying genes associated with naturally occurring variation in longevity (De Luca et al., 2003). It is possible that integrating QTL mapping with gene expression analysis will be an even more efficient method for identifying candidate genes affecting variation in lifespan. That is, genes located in regions to which QTLs map, and that exhibit age-related expression differences between lines, are excellent candidates for further study (Wayne and McIntyre, 2002).
Previously (Wilson et al., 2006), we used deficiency complementation mapping to identify 11 QTLs on the third chromosome that affect variation in lifespan between five Drosophila lines selected for postponed senescence via later reproduction (Old, or O lines) and their five unselected controls (Base, or B lines) (Rose, 1984). These QTLs encompass genomic regions ranging in size from 93.3 to 1449.4 kb (4874.8 kb total) and individually contain as few as 12 and as many as 170 genes (598 in total). To investigate each of these genes would be a daunting task, and this is only for one chromosome.
Here, we used Affymetrix microarrays to assess genome wide gene expression in one of the B lines and two O lines, at time points corresponding to the same chronological and physiological ages of the selection lines. We identified genes with statistically significant expression differences between ages averaged over lines, enabling us to identify biomarkers of aging. We also identified genes whose expression differed between B and O lines averaged over ages, enabling us to assess whether the lines selected for postponed senescence exhibit a delayed or different aging program relative to the unselected line. We further tested whether mutations in candidate genes with changes in expression with age affected lifespan, and whether deficiencies uncovering candidate genes with differences in expression between lines exhibited failure to complement the lifespan phenotypes. These analyses have identified novel genes affecting lifespan and candidate genes corresponding to QTLs, and provide insight into the genomic basis of response to selection for postponed senescence.
2. Materials and Methods
2.1 Drosophila stocks
We assessed genome wide gene expression for two lines selected for postponed senescence (O1, O3) and one unselected control line (B3) (Rose, 1984). We maintain B3 on a 14 day generation interval and the O lines on a 70 day generation interval. We tested whether P-element mutations in genes implicated from the gene expression analysis affected lifespan using ten homozygous P-element insertion lines and their co-isogenic controls generated by the Berkeley Drosophila Gene Disruption Project (Bellen et al., 2004): Act5CBG01299, Myo31DFBG01486, Mo25BG01582, Ef2bBG01993, tocBG02065, bin3BG02198, psqBG02327, SdcBG01305, Tis11BG00309 and ThorBG02130. In addition, we performed quantitative complementation tests to seven loss-of-function or hypomorphic mutations (m) in genes implicated from the gene expression analysis that were generated in different backgrounds: Dhc64C4-19, exu1, mbcC1, ple4, Mlc2E38, Hex-CnGB1, grp06034. All flies were reared at 25°C on cornmeal-molasses-agar medium. Flies in aging studies were placed in vials with 5 ml medium.
2.2 Gene expression analysis
We collected six cohorts of flies for aging: 1100 B3 males and 1100 B3 females; and 1200 each of O1 and O3 males and females. Virgin flies for each cohort were collected within a 48-hour period, and placed together in vials with individuals of the opposite sex from the same line, at a density of three males and three females per vial. Flies were transferred to fresh vials every 1-4 days as needed. We sampled ~100 mated flies at two ages for the B3 male and female cohorts, and at three ages for the O1 and O3 male and female cohorts. We divided each sample into three replicates of equal size, and froze the samples at -70°C. The three ages were at day 7 (T1) for all cohorts; when 10% of the B3 population remained alive (T2: 50 and 58 days for females and males, respectively); and when 10% of each O cohort remained alive (T3: 91 and 94 days for O1 females and males, respectively; and 84 and 90 days for O3 females and males, respectively).
We used two of the three replicate samples from each cohort and time point for microarray analysis, for a total of 32 samples (one B line × two sexes × two ages × two replicates + two O lines × two sexes × three ages × two replicates). We extracted RNA from each sample using Qiagen RNAeasy kits. Biotinylated cRNA probes were hybridized to high density oligonucleotide Affymetrix Drosophila GeneChip 1.0 microarrays and visualized with streptavidin-phycoerythrin conjugate, as described in the Affymetrix GeneChip Expression Analysis Technical manual, using internal references for quantification.
We normalized the expression data by scaling overall probe set intensity to 500 on each microarray using standard reference probe sets on each GeneChip for normalization. Every gene on the Affymetrix Drosophila GeneChip 1.0 is represented by a probe set consisting of 14 perfect match (PM) and 14 mismatch (MM) probe pairs. The quantitative estimate of expression of each probe set is the Signal (Sig) metric. Sig is computed using the One-Step Tukey’s Biweight Estimate, which gives the weighted mean of the log (PM-MM) intensities for each probe set (Affymetrix Microarray Suite, Version 4.0). A detection call (present, marginal or absent) is also given for each probe set. We eliminated probe sets from consideration if over one-half were called absent for the same line, age and sex. In practice this retained probe sets with line-, age- and sex-specific expression, and removed those with low and variable Sig values.
We performed two three-way fixed effect analyses of variance (ANOVAs) of the log2 expression values for all probe sets, using the fixed effects factorial model Y = μ + A + L + S + L×A + L×S + L×A×S + A×S + E, where A, L, S are the cross-classified effects of age, line, and sex respectively, and E is the variance between replicate arrays. The first analysis used the T1 and T2 samples from all lines, testing for difference in gene expression between young flies and older flies at the same chronological age. The second analyses used the T1 samples from all lines, but the T2 samples from the B line and the T3 samples from the O lines, testing for differences in gene expression between young flies and old flies at the same physiological age (the last 10% surviving B and O lines). We computed P-values from F-ratio tests of significance for each of the terms in the ANOVA, and used a Q = 0.05 false-discovery rate criterion for significance of any of the terms in the ANOVA model (Storey and Tibshirani, 2003).
2.3 Functional tests of candidate genes
We tested the effects on lifespan of ten P-element insertional mutations in candidate genes with significant changes in gene expression with age, and their corresponding co-isogenic controls. In addition, we performed quantitative complementation tests to seven mutations in candidate genes with significant changes in gene expression among lines. For each quantitative complementation test, we crossed the stock with the mutation (m) and a balancer chromosome (Bal) to the B3, O1 and O3 lines, giving six F1 genotypes: m/O1, m/O3, m/B3, Bal/O1, Bal/O3, and Bal/B3. For both assays, we collected virgin males and females within a 48-hour interval. The sample size for each genotype was N = 60 per sex, with 20 replicate vials and three males and three females in each vial. Flies were transferred to fresh vials every 1-4 days as needed, and scored at 2-day intervals for survival time.
Differences in lifespan between P-element insertion lines and their co-isogenic controls were analyzed using a two-way, mixed model ANOVA, Y = μ + L + S + L×S + R(L×S) + E, where L and S are the fixed cross-classified main effects of line (P-element insertions and control) and sex, R is the random effect of replicate vial, and E is the error variance.
Quantitative complementation tests were analyzed using a three-way factorial, mixed model analysis of variance ANOVA, Y = μ + L + G + S + L×G + L×S + G×S + L×G×S + R(L×G×S) + E, where L, G, and S are the fixed cross-classified main effects of line (O1, O3, B3), genotype (m and Bal), and sex; R is the random effect of replicate vial, and E is the error variance. Reduced ANOVAs were also run for each sex separately. If differences between mutant O and B genetic backgrounds and balancer O and B backgrounds are constant, yielding a non-significant F-statistic (P > 0.05) for the L×G or L×G×S interaction terms, this is interpreted statistically as no interaction and shows quantitative genetic complementation. If differences are not constant and analysis reveals a significant L×G or L×G×S term (P < 0.05), and additionally (m/O − m/B) > (Bal/O − Bal/B), this indicates quantitative failure to complement (Pasyukova et al., 2000). The latter constraint was imposed because significant interactions where the variance among lines was greater in the balancer background than in the mutant background is likely due to epistasis, and is not consistent with an allelic interpretation for failure to complement.
All statistical analyses were conducted using SAS software (SAS Institute).
3. Results and Discussion
3.1 Variation in gene expression
We performed genome wide gene expression analysis for males and females of two Drosophila lines selected for postponed senescence, O1 and O3, and one of the unselected control lines, B3 (Rose, 1984), using Affymetrix high density oligonucleotide microarrays. We collected RNA samples for all lines when all of the flies were seven days old (T1), when 10% of the B flies remained alive (T2) and when 10% of the O flies remained alive (T3) (Fig. 1). We performed three-way factorial ANOVAs (with line, age, and sex as the three cross-classified main effects) for each of the 11,101 probe sets that were expressed in adult flies. We performed two separate analyses. First, we assessed differences associated with young (T1) flies and the second age of collection (T2) for all lines, at which the flies are the same chronological age but have presumably different physiology, given that most of the B flies are dead while the majority of the O flies are alive at T2. Second, we assessed differences at T1 for the B and O lines with T2 for the B line and T3 for O lines; i.e. between young and old flies where the old animals from all lines are near the end of their lifespan and hence at the same physiological age. We used a false discovery rate (FDR) threshold of 0.05 for each term in the analysis to account for multiple tests. P-values and FDRs for each of the terms in the analyses for all expressed probe sets are listed in Supplementary Table S1.
Figure 1.
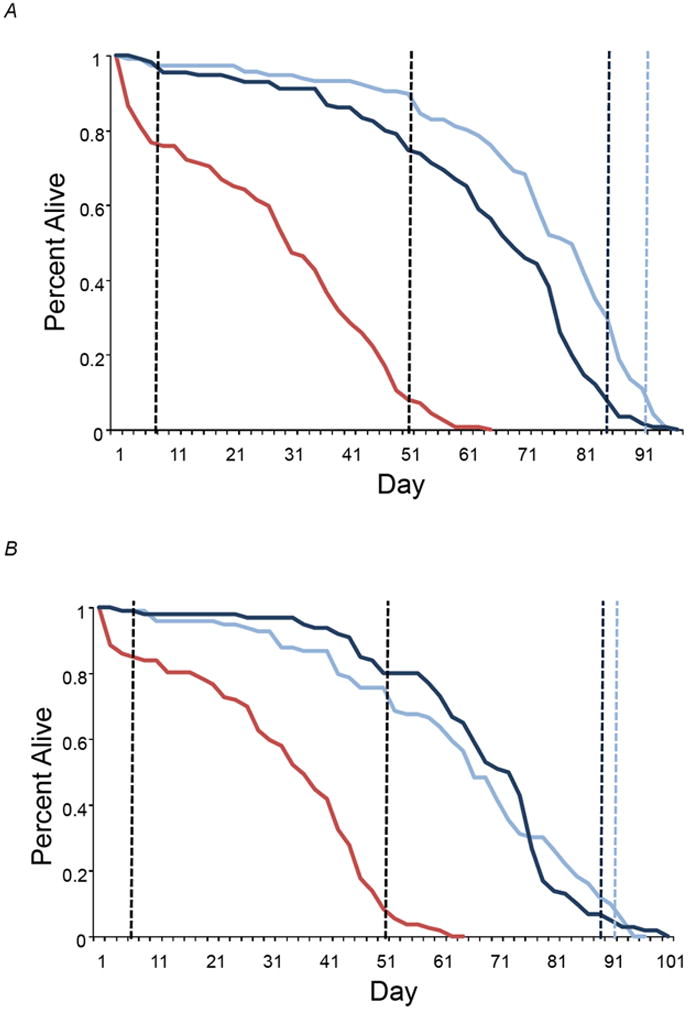
Survival curves for the B3 (red), O1 (light blue) and O3 (dark blue) lines. (A) Females. (B) Males. The vertical dashed lines represent the T1, T2 and T3 time points at which RNA samples were collected for analysis of gene expression. T1 and T2 were the same for all lines. The T3 collection times are given separately for O1 (light blue) and O3 (dark blue).
Table 1 shows the total number of probe sets significant at FDR < 0.05 for each of the terms in the ANOVA for the two analyses. Transcriptional responses to aging, line, and sex are extraordinarily dynamic. In the T1, T2 analyses, the main effect of sex was significant for 8,304 probe sets (75% of the total number expressed), while the main effects of age and line were significant for 834 (7.5%) and 455 (4.1%) probe sets, respectively. The respective numbers of probe sets significant for the sex, age and line terms in the T1, T3 analysis were 8,246 (74%), 2,094 (19%) and 509 (4.6%). 89% of the total number of probe sets significant for the sex effect were in common between the T1, T2 and T1, T3 analyses (Fig. 2). For the main effects of age and line, however, only 35% and 39%, respectively, of the total number of probe sets were in common between the two analyses (Fig. 2). A total of 2,525 probe sets were significant for age, line and/or an interaction with age or line in at least one analysis (Supplementary Table S2).
Table 1.
Number of significant probe sets (FDR < 0.05) for each term in the analyses of variance of gene expression between flies of the same chronological (T1, T2) and physiological (T1, T3) ages
| Term | Comparison | T1, T2 | T1, T3 |
|---|---|---|---|
| Sex (S) | M > F | 3,691 | 3,665 |
| F > M | 4,613 | 4,581 | |
| Age (A) | T1 > T2/T3 | 301 | 863 |
| T2/T3 > T1 | 533 | 1,231 | |
| Line (L) | B3 > O | 253 | 277 |
| O > B3 | 202 | 232 | |
| L×A | 35 | 27 | |
| L×S | 16 | 7 | |
| A×S | 211 | 0 | |
| L×A×S | 9 | 0 |
M and F refer to males and females, respectively. B3 is the short-lived line, and O refers to the average of the two long-lived lines (O1 and O3).
Figure 2.
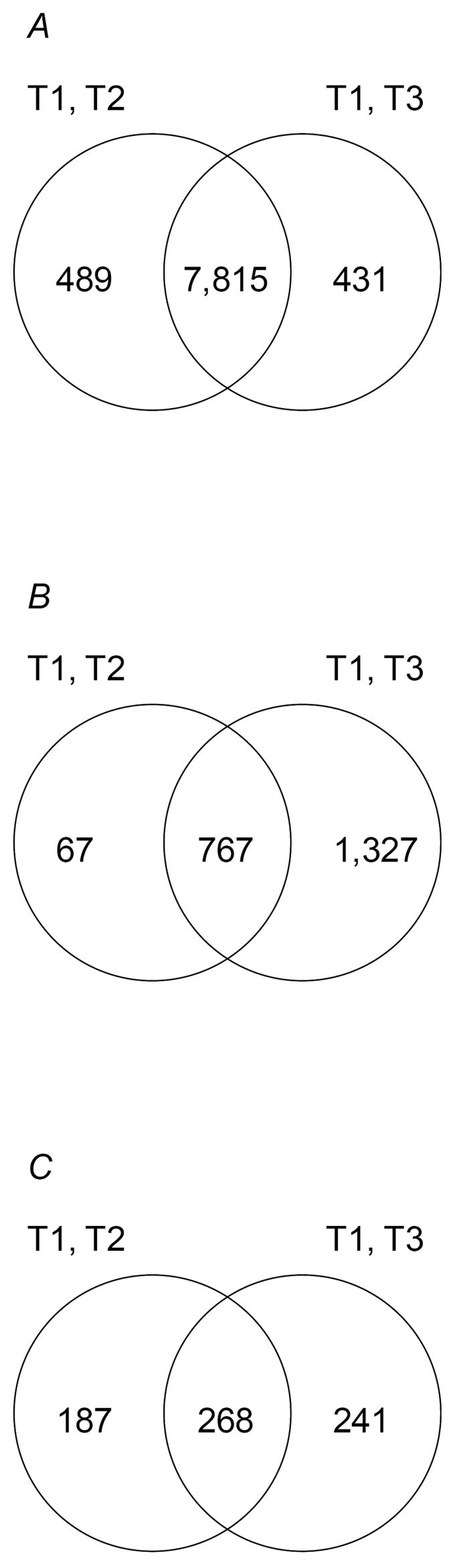
Venn diagrams showing the numbers of overlapping probe sets between the main effects of (A) Sex, (B) Age and (C) Line from analyses of variance of gene expression between flies of the same chronological (T1, T2) and physiological (T1, T3) ages.
3.2 Sex dimorphism in gene expression
Our observation that a large fraction of the transcriptome is sexually dimorphic is consistent with several previous studies documenting widespread sex differences in gene expression (Jin et al., 2001; Rantz et al., 2003; Harbison et al., 2005; Zang et al., 2007; Ayroles et al., 2009). This transcriptional sexual dimorphism is not limited to arthropods, but is also found in C. elegans (Jiang et al., 2001) and mammals (Rinn and Snyder, 2005). We analysed enrichment of Gene Ontology (GO) categories (Huang et al., 2009) for probe sets with higher levels of expression in males, and those with higher levels of expression in females. Male-biased genes were enriched for ontology categories related to the biosynthesis, transport and catabolism of secondary metabolites, and well as those involved in energy production and conversion (Supplementary Table S3). Female-biased genes were enriched for ontology categories related to translation, ribosomal structure and biogenesis; transcription; DNA replication, recombination, and repair; and chromatin structure and dynamics (Supplementary Table S4).
3.3 Transcriptional response to aging
Probe sets that show a significant age (A) and/or age by sex (A×S) interaction term are candidate genes affecting lifespan, and are also biomarkers of aging. We performed GO enrichment analyses (Huang et al., 2009) for the probe sets that were down-regulated with age, and those that were up-regulated with age. The most relevant analysis with respect to age is the T1, T3 comparison, since flies of all lines are the same physiological age. The 863 probe sets that are down-regulated with age in this analysis are strongly enriched for GO categories related to proteolysis, intermediary metabolism, DNA and RNA metabolism, oxidative phosphorylation, and mitochrondrial function (Fig. 3). The 1,231 probe sets that are up-regulated with age are highly enriched for GO categories related to protein synthesis, mitosis, immunity, defense response to bacteria and fungi, detoxification of xenobiotic substances, and the biosynthesis, transport and catabolism of secondary metabolites (Fig. 4). These same GO categories were enriched in previous analyses of changes in transcriptional profiles with age in Drosophila (Zou et al., 2000; Pletcher et al., 2002; Landis et al., 2004; Curtis et al., 2007; Lai et al., 2007a), giving a robust transcriptional signature of senescence. A total of 92% of the probe sets that were differentially expressed with age in the T1, T2 analysis overlapped those that were differentially expressed in the T1, T3 analysis; therefore, the same GO categories were enriched among the probe sets that were down- and up-regulated with age (Supplementary Figs. S1 and S2).
Figure 3.
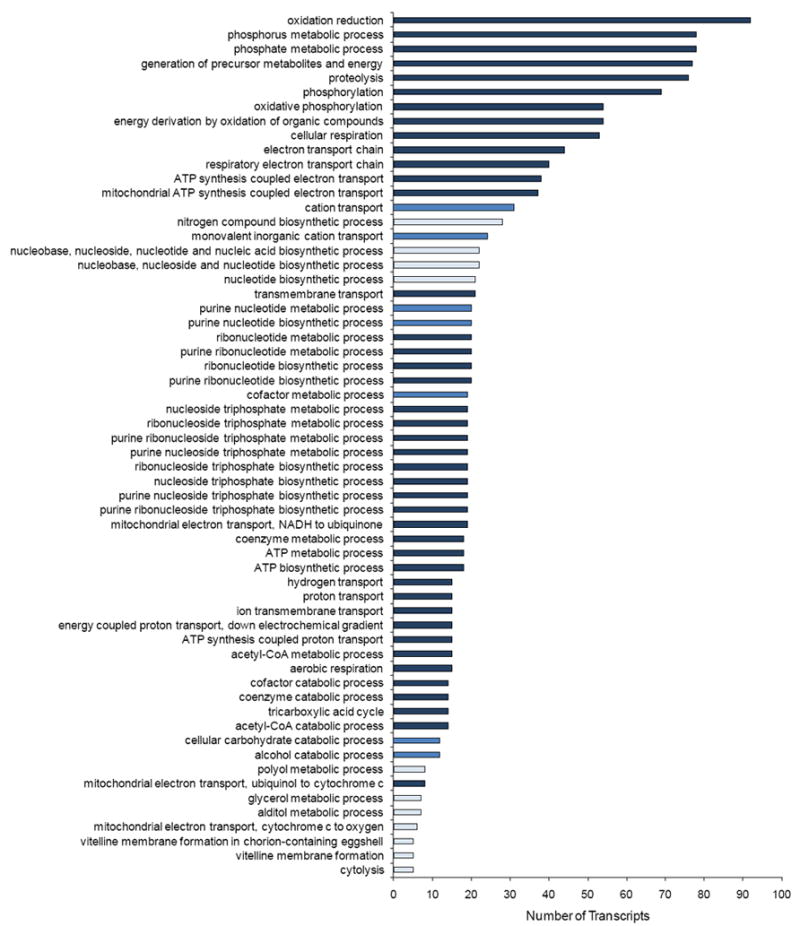
Gene Ontology enrichment analysis for over-represented Biological Process categories for probe sets down-regulated with age in the T1, T3 analysis. The colour of the bars denotes the P-values for significance of enrichment, adjusted for multiple tests using the Benjamini correction. Light blue: P < 0.05; Medium blue: P < 0.01; Dark blue: P < 0.001.
Figure 4.

Gene Ontology enrichment analysis for over-represented Biological Process categories for probe sets up-regulated with age in the T1, T3 analysis. The colour of the bars denotes the P-values for significance of enrichment, adjusted for multiple tests using the Benjamini correction. Light blue: P < 0.05; Medium blue: P < 0.01; Dark blue: P < 0.001.
The large number of probe sets differentially expressed in old age (19% of genes expressed in adult flies) is not surprising as the aging process involves many complex interactions of genetic networks associated with reproduction, metabolism, oxidative stress, sensory perception, serotonin signaling, chromatin silencing, DNA repair, and the heat-shock response (Finch and Ruvkun, 2001; Mackay et al., 2006). Proteolysis is used by cells to break down damaged or misfolded polypeptides, a protective process that could be used during heat shock or oxidative stress (Goldberg, 2003); and alterations in the immune system influence fitness (Hoffmann and Reichart, 2002). Furthermore, the Drosophila immune response to bacterial infection includes the use of lytic peptides to destroy the source of infection (Hoffmann and Reichart, 2002); thus, up- and down- regulation of genes in both categories may be triggered by the same response.
For the 211 probe sets with significant A×S interactions in the T1, T2 analysis (none were significant for this term in the T1, T3 analysis), we performed reduced ANOVAs for males and females separately. We classified the transcript as sex-specific if the expression difference was significant in only one sex, sex-biased if the expression difference was significant in both sexes and in the same direction but different magnitude, and sex antagonistic if the expression difference was significant in both sexes but in opposite directions (Supplementary Table S5). We found 132 (62.6%) male-specific or -biased transcripts, 22 (10.4%) female-specific or -biased transcripts, and 57 (27%) sex-antagonistic transcripts. GO enrichment analyses (data not shown) revealed that the male-specific and -biased transcripts are enriched for GO categories related to the mitotic cell cycle, translation and protein synthesis and ribosome structure. The female-specific and biased transcripts are enriched for starch and sucrose metabolism. The sex-antagonistic transcripts were not significantly enriched for any GO categories. These probe sets do not represent any “typical” sex-specific genes. The 6-fold enrichment of male over female-biased and -specific genes could be simply that males are on average physiologically younger than females.
3.4 Transcriptional response to selection
The transcriptional response to selection for postponed senescence involved ~6% of the probe sets expressed in adults in both the T1, T2 and T1, T3 analyses. This is in contrast to the 21% of the genome with altered differences in transcript abundance between artificial selection lines for increased and decreased mating speed (Mackay et al., 2005) and the ~14% of the genome showing correlated transcriptional responses to divergent artificial selection for aggressive behavior (Edwards et al., 2006), locomotor behavior (Jordan et al., 2007) and alcohol sensitivity (Morozova et al., 2007). There are several possible, non-mutually exclusive, explanations for this difference. The first is that we only examined one of the five B lines and two of the five O lines. In the future, including all selection lines in a similar experiment would increase the power to detect differences among lines, and allow for the identification of effects consistently correlated with increased longevity, as opposed to effects that might be due to chance fixation of alleles in only one line. Second, the distribution of allelic effects for genes affecting life history traits is asymmetrical (Frankham, 1990). That is, if much of the variation for fitness traits is attributable to many loci at which rare deleterious mutations are maintained in the populations by mutation-purifying selection balance (Houle et al., 1996), it will be easier to select for decreased rather than increased fitness. Indeed, the selection response for mating speed (Mackay et al., 2005) was highly asymmetrical, with the greatest response in the direction of increased copulation latency (and presumably decreased fitness). Third, selection for postponed senescence potentially acted on a smaller subset of genes at which intermediate frequency variants were maintained by balancing selection (Wilson et al., 2006), leading to rather fewer transcriptional differences relative to the unselected control.
Transcripts with a significant line (L) term and/or L×A, L×S, or L×A×S interaction terms are potential candidate QTLs for variation in lifespan. In the T1, T2 analysis, genes that are upregulated in the O lines relative to B3 are enriched for proteolysis and detoxification of xenobiotics, while genes upregulated in B3 relative to the O lines are enriched for defense responses to bacteria and fungi and innate immunity, muscle function, and metabolism (Fig. 5). In the T1, T3 analysis, genes that are upregulated in the O lines relative to B3 are also enriched for detoxification of xenobiotics and metabolism; while genes upregulated in B3 relative to the O lines are enriched for the cellular components of lipid particle, contractile fiber, sarcomere and myofibril (data not shown). A total of 27 of these genes co-localized with QTLs on chromosome 3 that are associated with differences in lifespan between the O and B lines (Wilson et al., 2006; Supplementary Table S6).
Figure 5.
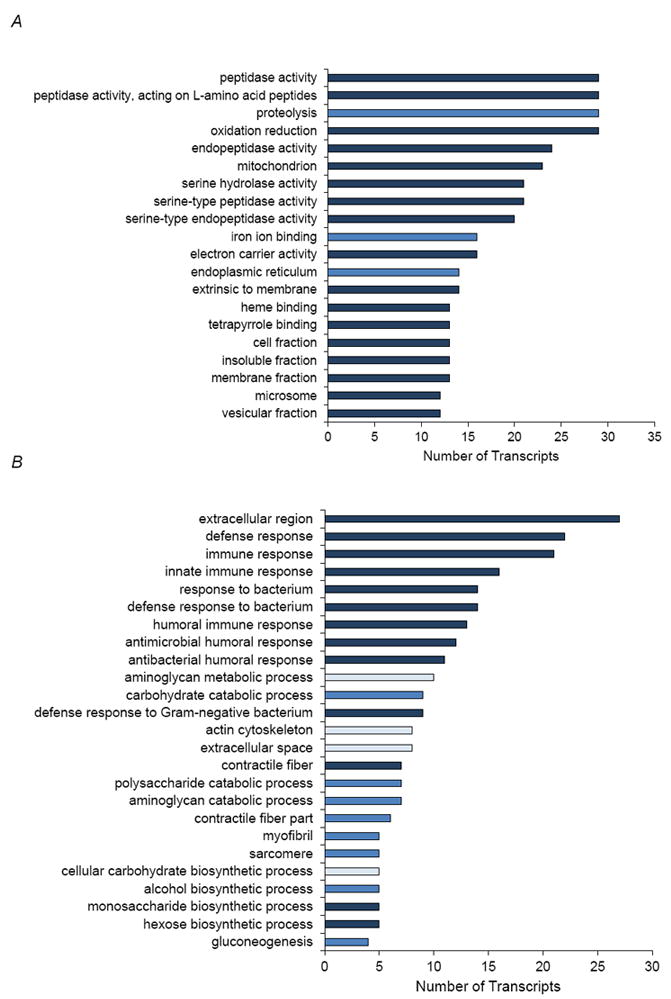
Gene Ontology enrichment analysis for over-represented Biological Process and Cellular Component categories for probe sets upregulated in (A) O lines relative to B3 and (B) B3 relative to O lines in the T1, T2 analysis. The colour of the bars denotes the P-values for significance of enrichment, adjusted for multiple tests using the Benjamini correction. Light blue: P < 0.05; Medium blue: P < 0.01; Dark blue: P < 0.001.
Transcripts with significant L×A effects are particularly interesting as among them are candidate genes for postponed senescence; i.e., transcripts with significant changes in gene expression with age in the B3 line, but not either of the O lines, which remain at the level of young B3 flies. Of the 35 transcripts exhibiting significant L×A effects in the T1, T2, analysis, 21 exhibited this pattern; while three of the 27 transcripts with L×A effects in the T1, T3, analysis showed this signature of postponed senescence (Supplementary Fig. S3). The transcripts with signatures of postponed senescence are implicated in proteolysis, neurogenesis, metabolism, cell death and immune response (Table 2).
Table 2.
Probe sets with signatures of postponed senescence. Raw P-values are given for the Line by Age interaction term (P(L×A)). All FDR values for L×A are < 0.05. Only probe sets that map uniquely to the genome are listed.
| Probe Set | Analysis | P(L×A) | Gene Symbol | Biological Process | Molecular Function |
|---|---|---|---|---|---|
| 141347_at | T1, T2 | 3.5E-05 | CG1806 | not known | not known |
| 141353_at | T1, T2 | 5.5E-05 | Serpin 88Eb | chaeta development | serine-type endopeptidase inhibitor activity |
| 141506_at | T1, T2 | 6.1E-06 | CG7839 | neurogenesis | sequence-specific DNA binding transcription factor activity |
| 142886_at | T1, T2 | 6.1E-06 | CG10165 | not known | not known |
| 142954_at | T1, T2 | 6.6E-05 | CG5773 | not known | not known |
| 144227_at | T1, T2 | 4.4E-05 | CG15293 | not known | not known |
| 145796_at | T1, T2 | 1.5E-05 | Jon25Bii | neurogenesis | serine-type endopeptidase activity |
| 145970_at | T1, T2 | 6.0E-07 | TepII | defense response to Gram-negative bacterium; antibacterial humoral response; phagocytosis, engulfment | peptidase inhibitor activity |
| T1,T3 | 8.9E-05 | ||||
| 147118_at | T1, T2 | 2.6E-06 | CG12374 | proteolysis | metallocarboxypeptidase activity |
| 147469_at | T1, T2 | 1.3E-04 | CG15098 | not known | not known |
| 148228_at | T1, T2 | 2.6E-07 | CG10592 | metabolic process | alkaline phosphatase activity |
| 148332_at | T1, T2 | 6.8E-07 | CG8562 | proteolysis | metallocarboxypeptidase activity |
| 148713_at | T1, T2 | 1.4E-04 | CG14109 | not known | not known |
| T1, T3 | 1.5E-05 | ||||
| 151094_at | T1, T2 | 1.1E-04 | CG3348 | chitin metabolic process | chitin binding |
| 151980_at | T1, T2 | 7.5E-07 | CG5150 | metabolic process | alkaline phosphatase activity |
| 152063_at | T1, T2 | 4.4E-07 | CG4716 | not known | methylenetetrahydrofolate dehydrogenase [NAD(P)+] activity |
| 152369_at | T1, T2 | 9.6E-05 | Bace | proteolysis | aspartic-type endopeptidase activity |
| 152794_at | T1, T2 | 8.8E-06 | CG8560 | proteolysis | zinc ion binding; metallocarboxypeptidase activity |
| 153367_at | T1, T2 | 1.7E-05 | Ect3 | autophagic cell death | beta-galactosidase activity |
| 153743_at | T1, T3 | 6.8E-05 | CG2656 | neurogenesis | purine nucleotide binding |
In addition to formally significant L×A interaction terms, we collated lists of genes that were down-regulated in old age in the T1, T3, analysis but up-regulated in O lines compared to the B line in the T1, T2 analysis; and conversely that were up-regulated in old age in the T1, T3, analysis but down-regulated in O lines compared to the B line in the T1, T2 analysis. These transcripts also exhibit a signature of postponed senescence. The former category of genes was significantly enriched for terms associated with proteolysis and phosphatases, while the latter category was significantly enriched for terms involved in immune/defense response as well as metabolism and catabolism (Supplementary Table S7).
All of these analyses indicate that response to selection for postponed senescence was in part attributable to genes involved with protein breakdown and post-translational modification, immunity and metabolism. These observations are consistent with the increased lipid content and metabolic rate (Service, 1987; Graves et al., 1992) of O lines relative to the B lines. Conspicuously absent from transcripts exhibiting patterns of expression consistent with postponed senescence of O lines and candidate genes affecting lifespan are genes previously associated with Drosophila lifespan. These results highlight the value of interrogating natural variants affecting postponed senescence as a complementary approach to analysis of de novo mutations.
3.5 Functional tests
Transcripts with significant Age effects are candidate longevity genes. Therefore, we tested whether mutations in 10 genes showing significant differences in gene expression with age affected lifespan. We measured longevity of strains containing P-element insertional mutations disrupting each candidate gene, and their co-isogenic wild type controls, separately for males and females. Seven genes - Actin 5C (Act5C), Myosin 31DF (Myo31DF), Mo25, bicoid-interacting protein 3 (bin3), Syndecan (Sdc), Tis11 homolog (Ts11), toucan (toc) - had significant effects on lifespan in analyses pooled across males and females. However, with the exception of Actin5C, the effects on lifespan were sex-specific, as has been found previously for QTLs and mutations (Mackay et al., 2006; Mockett & Sohal, 2006; Tower, 2006; Lai et al., 2007b, Sørensen et al., 2007; Shen et al., 2009; Waskar et al., 2009; Cho et al., 2010; Magwire et al., 2010; Shen & Tower, 2010) affecting Drosophila longevity. All effects were in the direction of reduced lifespan of mutations relative to the wild type control (Fig. 6), suggesting the wild type products of these genes are required for normal lifespan. The high rate of validation (70%) of mutations in genes whose expression changes with age, combined with the large number of these transcripts, is consistent with a large mutational target size and complex genetic control of lifespan. These genes are involved in diverse biological functions, including spermatogenesis (Actin 5C); embryonic (Mo25) and mesoderm (Myo31DF) development; RNA interference (Tis11); mitosis (toc); energy homeostasis (Sdc); nervous system development and function (Act5C, Sdc) and behaviours (Act5C, bin3) (McQuilton et al., 2012).
Figure 6.
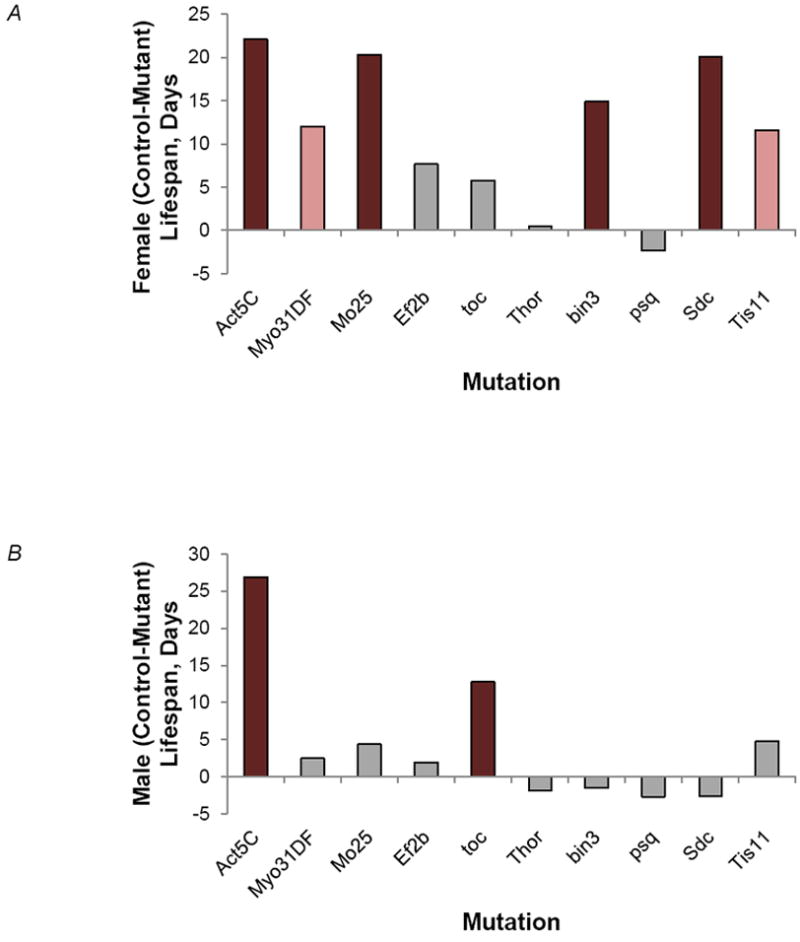
Effects of mutations on lifespan. The bars depict the difference in lifespan between the tested mutation and the control co-isogenic wild type strain. The colour of the bars indicates the significance of the P-value of the line term from the ANOVA. Dark red, P < 0.0001; Light red, P < 0.01; Grey, P > 0.05. (A) Females. (B) Males.
Transcripts with significant Line effects are candidate loci affecting variation in lifespan between the selection lines. Therefore, we performed quantitative complementation tests to mutations in seven genes showing significant differences in gene expression among lines. Five of these genes - Hexokinase C (Hex-C), Dynein heavy chain 64C (Dhc64), myoblast city (mbc), Myosin light chain 2 (Mlc2), exuperantia (exu) - exhibited significant failure to complement consistent with an allelic interaction (i.e., the difference between the wild type and mutant heterozygotes is greater in the O line genetic background than in that of the B line, and the mutant is associated with increased lifespan in the O lines) (Fig. 7). These genes are also involved in biological processes as diverse as embryogenesis (exu), lateral inhibition (Hex-C), muscle development and function (Mlc2, Mbc) and microtubule-based movement (Dhc64). However, as with all complementation tests, we cannot exclude the possibility that epistatic interactions between the targeted mutations and other loci in the selection lines are the cause of the failure to complement (Mackay et al., 2006). In addition, the wild type alleles were on Balancer chromosomes, which contain mutations at other loci, further complicating the interpretation. Nevertheless, the 711 probe sets with significant changes in gene expression involving the Line term (Supplementary Table S8) are attractive candidates for further functional analysis in the future.
Figure 7.
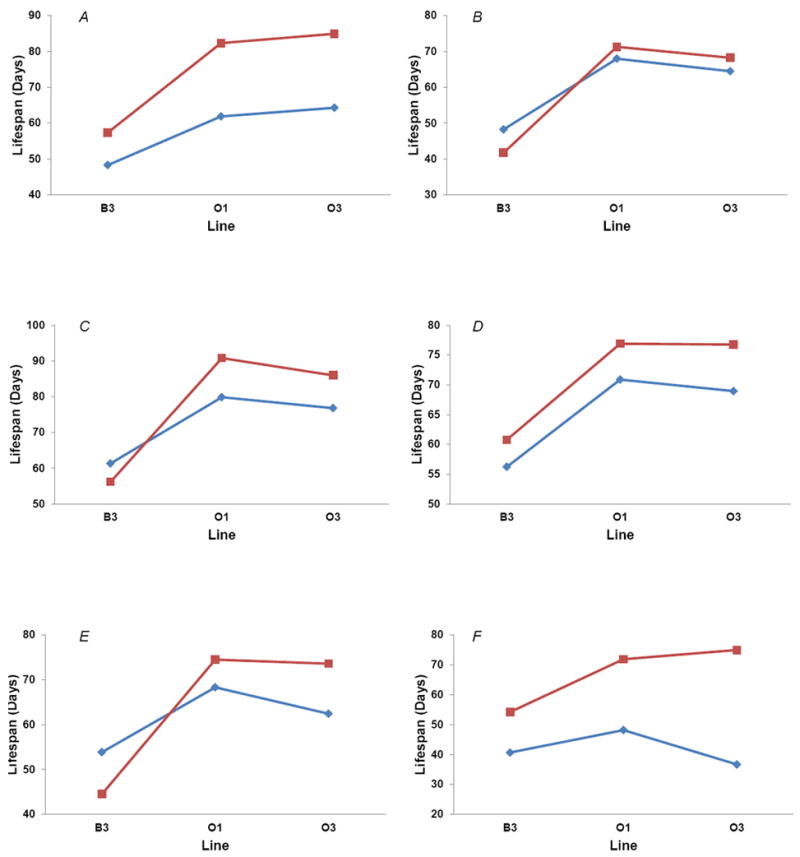
Quantitative complementation tests. Blue lines and diamonds depict F1 heterozygous genotypes between selection lines and a balancer chromosome; red lines and squares depict F1 heterozygous genotypes between selection lines and a target mutation. P-values are given for the significance of the line by genotype interaction term. (A) Hex-CnGB1, pooled across sexes. P < 0.0001. (B) Dhc64C 4-19, pooled across sexes. P = 0.014. (C) mbcC1, pooled across sexes. P < 0.0001. (D) ple4, pooled across sexes. P = 0.72. (E) Mlc2E38, females. P = 0.0043. (F) exu1, females. P < 0.0001.
Supplementary Material
Acknowledgments
We thank Philip Service for the generous gift of the fly stocks used in this study. This work was supported by National Institutes of Health grant R01 GM45146 to T. F. C. M. This is a publication of the W. M. Keck Center for Behavioral Biology.
Footnotes
Disclosure statement: No actual or potential conflicts of interest.
References
- Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, Magwire MM, Rollmann SM, Duncan LH, Lawrence F, Anholt RRH, Mackay TFC. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet. 2009;41:299–307. doi: 10.1038/ng.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S, Ayyadevara R, Hou S, Thaden JJ, Shmookler Reis RJ. Genetic mapping of quantitative trait loci governing longevity of Caenorhabditis elegans in recombinant-inbred progeny of a Bergerac-BO x RC301 interstrain cross. Genetics. 2001;157:655–666. doi: 10.1093/genetics/157.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, Hoskins RA, Spradling AC. The BDGP gene disruption project: Single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SX, Dhahbi JM, Mote PL, Spindler SR. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci USA. 2001;98:10630–10635. doi: 10.1073/pnas.191313598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T, Partridge L. Female fitness in Drosophila melanogaster: an interaction between the effect of nutrition and of encounter rate with males. Proc Roy Soc Lond B. 1996;263:755–759. doi: 10.1098/rspb.1996.0113. [DOI] [PubMed] [Google Scholar]
- Cho I, Horn L, Felix TM, Foster L, Gregory G, Starz-Gaiano M, Chambers MM, De Luca M, Leips J. Age- and diet-specific effects of variation at S6 kinase on life history, metabolic, and immune response traits in Drosophila melanogaster. DNA Cell Biol. 2010;29:473–485. doi: 10.1089/dna.2009.0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Landis GN, Folk D, Wehr NB, Hoe N, Waskar M, Abdueva D, Skvortsov D, Ford D, Luu A, Badrinath A, Levine RL, Bradley TJ, Tavaré S, Tower J. Transcriptional profiling of MnSOD-mediated lifespan extension in Drosophila reveals a species-general network of aging and metabolic genes. Genome Biol. 2007;8:R262. doi: 10.1186/gb-2007-8-12-r262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JW, Khazaeli AA. Lifespan, QTLs, age-specificity, and pleiotropy in Drosophila. Mech Ageing Dev. 2002;123:81–93. doi: 10.1016/s0047-6374(01)00345-1. [DOI] [PubMed] [Google Scholar]
- DeLuca M, Roshina NV, Geiger-Thornsberry GL, Lyman RF, Pasyukova EG, Mackay TFC. Dopa-decarboxylase affects variation in Drosophila longevity. Nat Genet. 2003;34:429–433. doi: 10.1038/ng1218. [DOI] [PubMed] [Google Scholar]
- Doria G, Barattini P, Scarpaci S, Puel A, Guidi L, Frasca D. Role of immune responsiveness and DNA repair capacity genes in ageing. Ageing Res Rev. 2004;3:143–151. doi: 10.1016/j.arr.2003.04.001. [DOI] [PubMed] [Google Scholar]
- Edwards AC, Rollmann SM, Morgan TJ, Mackay TFC. Quantitative genomics of aggressive behavior in Drosophila melanogaster. PLoS Genetics. 2006;2:e154. doi: 10.1371/journal.pgen.0020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Li L, Longo VD. Analysis of gene expression profile in yeast aging chronologically. Mech Ageing Dev. 2005;126:11–6. doi: 10.1016/j.mad.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Finch CE, Ruvkun G. The genetics of aging. Annu Rev Genomics Hum Genetics. 2001;2:435–362. doi: 10.1146/annurev.genom.2.1.435. [DOI] [PubMed] [Google Scholar]
- Forbes SN, Valenzuela RK, Keim P, Service PM. Quantitative trait loci affecting lifespan in replicated populations of Drosophila melanogaster. I. Composite interval mapping. Genetics. 2004;168:301–311. doi: 10.1534/genetics.103.023218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham R. Are responses to artificial selection for reproductive fitness characters consistently asymmetrical? Genet Res. 1990;56:35–42. [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Graves JL, Toolson EC, Jeong C, Vu LN, Rose MR. Desiccation, flight, glycogen, and postponed senescence in Drosophila melanogaster. Physiol Zool. 1992;65:268–286. [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Hamatani T, Falco G, Carter MG, Akutsu H, Stagg CA, Sharov AA, Dudekula DB, VanBuren V, Ko MS. Hum Mol Genet. 2004;13:2263–2278. doi: 10.1093/hmg/ddh241. [DOI] [PubMed] [Google Scholar]
- Harbison ST, Chang S, Kamdar KP, Mackay TFC. Quantitative genomics of starvation stress resistance in Drosophila. Genome Biol. 2005;6:R36. doi: 10.1186/gb-2005-6-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA, Reichhart JM. Drosophila innate immunity: an evolutionary perspective. Nat Immunol. 2002;3:121–126. doi: 10.1038/ni0202-121. [DOI] [PubMed] [Google Scholar]
- Houle D, Morikawa B, Lynch M. Comparing mutational variabilities. Genetics. 1996;143:1467–1483. doi: 10.1093/genetics/143.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jackson AU, Galecki AT, Burke DT, Miller RA. Mouse loci associated with lifespan exhibit sex-specific and epistatic effects. J Gerontol A Bio Sci Med Sci. 2002;57:B9–B15. doi: 10.1093/gerona/57.1.b9. [DOI] [PubMed] [Google Scholar]
- Jiang M, Ryu J, Kiraly M, Duke K, Reinke V, Kim SK. Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2001;98:218–223. doi: 10.1073/pnas.011520898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype, and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Jordan KW, Carbone MA, Yamamoto A, Morgan TJ, Mackay TFC. Quantitative genomics of locomotor behavior in Drosophila melanogaster. Genome Biol. 2007;8:R172. doi: 10.1186/gb-2007-8-8-r172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CQ, Parnell L, Lyman RF, Ordovas JM, Mackay TFC. Candidate genes affecting Drosophila lifespan identified by integrating microarray gene expression analysis and QTL mapping. Mech Ageing and Devel. 2007a;128:237–249. doi: 10.1016/j.mad.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Lai CQ, Leips J, Zou W, Roberts JF, Wollenberg KR, Parnell LD, Zeng ZB, Ordovas JM, Mackay TFC. Speed-mapping quantitative trait loci using microarrays. Nat Methods. 2007b;10:839–841. doi: 10.1038/nmeth1084. [DOI] [PubMed] [Google Scholar]
- Landis GN, Abdueva D, Skvortsov D, Yang J, Rabin BE, Carrick J, Tavaré S, Tower J. Similar gene expression patterns characterize aging and oxidative stress in Drosophila melanogaster. Proc Natl Acad Sci USA. 2004;101:7663–7668. doi: 10.1073/pnas.0307605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leips J, Mackay TFC. Quantitative trait loci for lifespan in Drosophila melanogaster: Interactions with genetic background and larval density. Genetics. 2000;155:1773–1788. doi: 10.1093/genetics/155.4.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leips J, Mackay TFC. The complex genetic architecture of Drosophila lifespan. Exp Aging Res. 2002;28:361–390. doi: 10.1080/03610730290080399. [DOI] [PubMed] [Google Scholar]
- Lund J, Tedesco P, Duke K, Wang J, Kim SK, Johnson TE. Transcriptional profile of aging in C. elegans. Curr Biol. 2002;12:1566–1573. doi: 10.1016/s0960-9822(02)01146-6. [DOI] [PubMed] [Google Scholar]
- Mackay TFC, Heinsohn SL, Lyman RF, Moehring AJ, Morgan TJ, Rollmann SM. Genetics and genomics of Drosophila mating behavior. Proc Natl Acad Sci USA. 2005;102:6622–6629. doi: 10.1073/pnas.0501986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Roshina NV, Leips JW, Pasyukova EG. Complex genetic architecture of Drosophila longevity. In: Masaro EJ, Austad SN, editors. Handbook of the Biology of Aging. Sixth Edition. 2006. pp. 181–216. [Google Scholar]
- Magwire MM, Yamamoto A, Carbone MA, Roshina NV, Pasyukova EG, Morozova TV, Mackay TFC. Quantitative and molecular genetic analyses of mutations increasing Drosophila lifespan. PLoS Genetics. 2010;6:e1001037. doi: 10.1371/journal.pgen.1001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett RJ, Sohal RS. Temperature-dependent trade-offs between longevity and fertility in the Drosophila mutant, methuselah. Exp Gerontol. 2006;41:566–573. doi: 10.1016/j.exger.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Morozova TV, Anholt RRH, Mackay TFC. Phenotypic and transcriptional response to selection for alcohol sensitivity in Drosophila melanogaster. Genome Biol. 2007;8:R231. doi: 10.1186/gb-2007-8-10-r231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P, St Pierre SE, Thurmond J FlyBase Consortium. FlyBase 101 – the basics of navigating FlyBase. Nucl Acids Res. 2012;40:D706–714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV, Pasyukova EG, Dilda C, Mackay TFC. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc Natl Acad Sci USA. 1997;94:9734–9739. doi: 10.1073/pnas.94.18.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge L. Evolutionary theories of ageing applied to long-lived organisms. Exp Gerontol. 2001;36:641–650. doi: 10.1016/s0531-5565(00)00232-1. [DOI] [PubMed] [Google Scholar]
- Pasyukova EG, Vieira C, Mackay TFC. Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics. 2000;156:1129–1146. doi: 10.1093/genetics/156.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher SD, Macdonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, Partridge L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol. 2002;12:712–723. doi: 10.1016/s0960-9822(02)00808-4. [DOI] [PubMed] [Google Scholar]
- Rantz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Reiwitch SG, Nuzhdin SV. Quantitative trait loci for lifespan of mated Drosophila melanogaster affect both sexes. Genet Res. 2002;80:225–230. doi: 10.1017/s0016672302005943. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Snyder M. Sexual dimorphism in mammalian gene expression. Trends Genet. 2005;221:298–305. doi: 10.1016/j.tig.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Rose MR. Laboratory evolution of postponed senescence in Drosophila melanogaster. Evolution. 1984;38:1004–1010. doi: 10.1111/j.1558-5646.1984.tb00370.x. [DOI] [PubMed] [Google Scholar]
- Service PM. Physiological mechanisms of increased stress resistance in Drosophila melanogaster selected for postponed senescence. Physiol Zool. 1987;60:321–326. [Google Scholar]
- Shen J, Ford D, Landis GN, Tower J. Identifying sexual differentiation genes that affect Drosophila life span. BMC Geriatr. 2009;9:56. doi: 10.1186/1471-2318-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tower J. Drosophila foxo acts in males to cause sexual-dimorphism in tissue-specific p53 life span effects. Exp Gerontol. 2010;45:97–105. doi: 10.1016/j.exger.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook DR, Brooks A, Johnson TE. Mapping quantitative trait loci affecting life history traits in the nematode Caenorhabditis elegans. Genetics. 1996;142:801–817. doi: 10.1093/genetics/142.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JG, Kristensen TN, Kristensen KV, Loeschcke V. Sex specific effects of heat induced hormesis in Hsf-deficient Drosophila melanogaster. Exp Gerontol. 2007;42:1123–1129. doi: 10.1016/j.exger.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J. Sex-specific regulation of aging and apoptosis. Mech Ageing Dev. 2006;127:705–718. doi: 10.1016/j.mad.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Valenzuela RK, Forbes SN, Keim P, Service PM. Quantitative trait loci affecting lifespan in replicated populations of Drosophila melanogaster. II. Response to selection. Genetics. 2004;168:313–324. doi: 10.1534/genetics.103.023291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira C, Pasyukova EG, Zeng S, Hackett JB, Lyman RF, Mackay TFC. Genotype-environment interaction for quantitative trait loci affecting lifespan in Drosophila melanogaster. Genetics. 2000;154:213–227. doi: 10.1093/genetics/154.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskar M, Landis GN, Shen J, Curtis C, Tozer K, Abdueva D, Skvortsov D, Tavaré S, Tower J. Drosophila melanogaster p53 has developmental stage-specific and sex-specific effects on adult life span indicative of sexual antagonistic pleiotropy. Aging (Albany NY) 2009;1:903–936. doi: 10.18632/aging.100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne ML, McIntyre LM. Combining mapping and arraying: An approach to candidate gene identification. Proc Natl Acad Sci USA. 2002;99:14903–14906. doi: 10.1073/pnas.222549199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RH, Morgan TJ, Mackay TFC. High resolution mapping of quantitative trait loci affecting Drosophila lifespan. Genetics. 2006;173:1455–1463. doi: 10.1534/genetics.105.055111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–237. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Meadows S, Sharp L, Jan LY, Jan YN. Genome-wide study of aging and oxidative stress response in Drosophila melanogaster. Proc Natl Acad Sci USA. 2000;97:13726–13731. doi: 10.1073/pnas.260496697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.