

Abstract
Post-synthesis modification of DNA is an important way of functionalizing DNA molecules. Herein we describe a method that first enzymatically incorporates a cyanobenzothiazole (CBT)-modified thymidine. The side chain handle CBT can undergo a rapid and site-specific cyclization reaction with 1,2-aminothiols to afford DNA functionalization in aqueous solution. Another key advantage of this method is the formation of a single stereo/regioisomer in the process, which allows for precise control of DNA modification to yield a single component for aptamer selection work and other applications.
Keywords: post-synthesis modification, DNA labelling, copper free, bioorthogonal reaction, CBT
Introduction
The unique physical and chemical characteristics of DNA as genomic materials also make them very useful for various other applications. For example, DNA has been explored for applications in nanoarchitectures,[1] nanorobots,[2] nanocomputing,[3] reaction encoding,[4] and nanosensing.[5] In addition, DNA can also be used as therapeutic agents[6] and for the in vitro selection of aptamers for various applications.[7] For many applications, DNA must be equipped with specific functionalities, such as fluorescent markers, biotin, thiols, sugars, proteins, positively charged peptides (octaarginines), and boronic acid for endowing different properties.[8] Therefore, there is a critical need to develop chemistry that allows for the ready functionalization of DNA molecules. Nucleic acid functionalization can be conducted through various conjugation methods such as Staudinger ligation, N-hydroxysuccinimide (NHS) ester chemistry and the formation of thiourea, oxime or disulfide linkages.[9] Recent advances in cycloaddition reactions have enabled faster and more efficient conjugation.[8a] Specifically, the labs of Seela[10] and Carell[11] first reported the functionalization of DNA nucleobases through copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction, which was developed by Sharpless[12] and Meldal.[13] This protocol is highly efficient and specific, and results in almost quantitative incorporation of different labels into the DNA. While these seminal reports have allowed easy DNA modification, the CuAAC method has the issue of copper-catalyzed cleavage of DNA.[8a] In addition, Cu(I) also poses stability problems for some other functional groups. For example, boronic acid was reported to be degraded during post-synthetic functionalization of DNA using CuAAC.[14] Copper-free chemistry has thus been used recently by different researchers, including strain-promoted azide-alkyne cycloaddition,[15] Staudinger ligation,[16] Diels-Alder reaction,[17] hydrazone[18] and oxime[19] formation, and native peptide ligation.[20] Although these methods provide alternative ways for copper-free DNA labeling, they still possess some limitations, e.g, low reaction rate, the need of other reagents/organic solvents for functionalization, as well as the possibility of generating stereo/regioisomers.
In our work of preparing modified DNA, we are especially interested in building boronic acid-functionalized DNA libraries for aptamer selection.[8b] We have successfully demonstrated the synthesis and enzymatic incorporation of boronic acid-modified TTPs (B-TTP) into DNA[21] (Route A, Scheme 1), and the chemical phosphoramidite synthesis of DNA with a strained alkyne handle, which allowed introduction of the boronic acid group via copper-free click chemistry (Route B, Scheme 1).[14] Although these paved the way for enzyme-catalyzed functionalization with a boronic acid and the large scale synthesis of single stranded boronic acid-modified DNA for further applications, several limitations still need to be addressed, including the complex synthesis of different B-TTP moieties, Cu(I)-mediated degradation of specific boronic acid functional groups, the potential problems during enzymatic recognition as well as multiple stereoisomers generated due to lack of symmetry. In an attempt to circumvent these restrictions, and most importantly, to develop a novel platform for the rapid and specific post-synthesis modification of DNA that can address some limitations of current methods, we are further interested in the strategy of enzyme-catalyzed (such as PCR) synthesis of DNA with a “general handle”, which can undergo a rapid and specific post-synthesis functionalization. Specifically, we need an approach that allows (1) high enzymatic incorporation efficiency of the “general handle” modified nucleotide; (2) fast, site-specific, and chemoselective post-synthesis modification with its reactive partner in aqueous solution; (3) high yield of a single stereochemically pure product. The last point is especially important and remains a major obstacle for further application in DNA modification, such as aptamer selection. Because even if the yield of the desired isomer is 90% at a single base, the synthesis of a 90-mer DNA would have the possibility of generating multiple isomers with random distribution of regio-/stereo-isomers at different positions. Herein we report our recent endeavor in developing such a method that meets the above-mentioned criteria for post-synthesis modification of DNA.
Scheme 1.
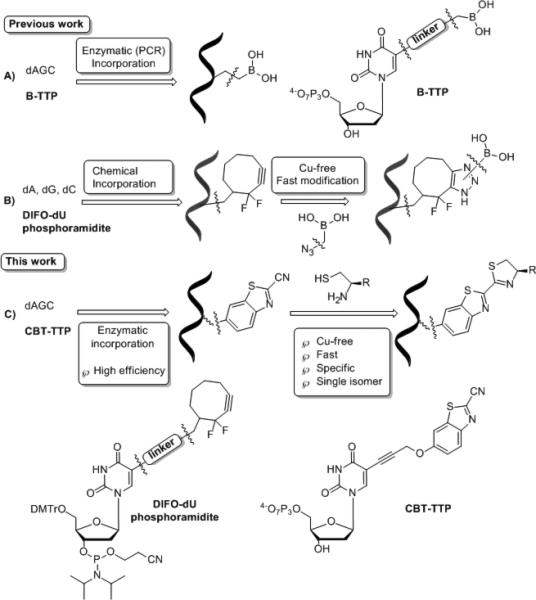
Schematic representations of different boronic acid-functionalized DNA modification strategies. A) Enzymatic incorporation of boronic acid modified nucleotides; B) Chemical incorporation of a nucleotide modified with a reactive group, followed by Cu-free modification with their reaction partners; and C) Enzymatic incorporation of a “general handle” modified nucleotide, followed by Cu-free post-synthesis modification with its partner.
Results and Discussion
To start this project, we first explored the well-developed repertoire of biocompatible reactions to find the appropriate reaction partners for highly reaction selectivity, absence of stereoisomers, and compatibility with biological systems. The condensation of 1,2-aminothiol with 2-cyanobenzothiazole (CBT) was chosen.[22] This reaction, known as the last step of the bio-synthesis of luciferin,[22] has drawn much attentions recently, largely due to the rejuvenation work of Rao[23] and Chin.[24] In employing this reaction for post-synthesis DNA labeling, we have the option of either incorporating a 1,2-aminothiol or CBT into the nucleotide. We selected the pathway of making the CBT-modified nucleotide, since it does not involve protection/deprotection during the synthesis. In addition, the 1,2-aminothiol also renders the click-labeling agent good solubility in aqueous solution. Based on the known fact that 5-position modification of deoxyuridine can be tolerated by polymerases and reverse transcriptases[25] and our own successful experience in developing a series of 5-position boronic-acid functionalized deoxyuridines,[21c] we prepared a CBT-modified thymidine triphosphate in 8 steps. Specifically, starting from commercially available 1,4-benzoquinone (1), ethyl 6-hydroxybenzo[d]thiazole-2-carboxylate (5) was synthesized following literature procedures.[26] Propargylation, followed by ammonolysis and dehydration provided a modified CBT with terminal alkyne moiety 8. Subsequent Sonogashira reaction yielded CBT-T 9. Triphosphorylation was accomplished by the classical one-pot three-step method[27] (Scheme 2) to afford final compound CBT-TTP 10.
Scheme 2.
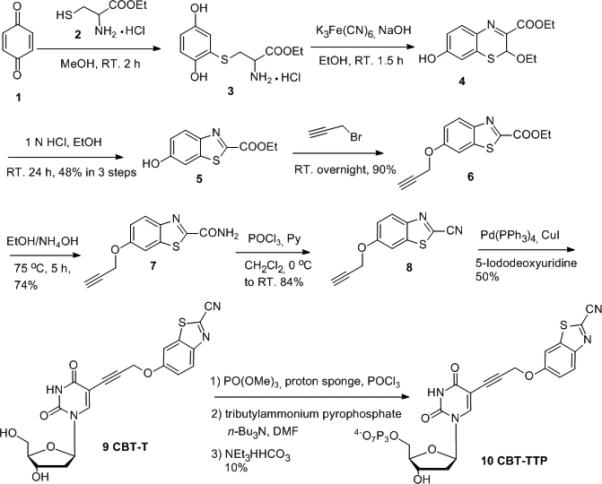
Synthesis of Compound 10 (CBT-TTP)
Before the incorporation study, we first studied the reaction profile. CBT has been reported to react rapidly and specifically with 1, 2-aminothiol with a second-order rate constant of 9 M−1 s−1.[23a] We reasoned that nucleotide-modified CBT would proceed in the same fashion. To confirm this, kinetic study was performed for the reaction between the precursor CBT-T (9, Scheme 2) and 1, 2-aminothiol. The second-order rate constant was determined to be 22 M−1 s−1 (See Figure S1 in Supporting Information) under near physiological conditions (phosphate buffer, pH = 7.4)).
Next, we studied the incorporation of CBT-TTP into DNA by an enzyme-catalyzed reaction. Specifically, primer extension, using CBT-TTP and the Klenow fragment, was conducted using a short sequence of 21-mer oligonucleotide (nt) Template-1 and a 14-mer FAM labeled primer (Figure 1), which have been successfully used in our previous incorporation studies of different functionalized DNA.[21] Klenow fragment (3’-5’ exo-) was used to avoid cleavage of the template. The primer was designed in such a way that the first incorporated base would be a T, so there are two possible scenarios in the extension: either fully extended product or no extension at all. The obtained DNA products were studied using polyacrylamide gel electrophoresis (PAGE). As shown in Figure 1A, negative controls without dTTP (Lane 1, Figure 1A), without Klenow fragment (Lane 2, Figure 1A), and primer plus template only (Lane 3, Figure 1A) showed no full length DNA sequence. Instead, annealing products were observed. Negative control with primer only (Lane 4, Figure 1A) did not show annealing product due to the absence of template. On the other hand, primer extension product using CBT-TTP (Lane 6, Figure 1A) in place of dTTP gave a similar full-length DNA band as that of positive control with Klenow fragment and natural dNTPs only (Lane 5, Figure 1A). Such results indicated that synthesized CBT-TTP could be recognized similarly as dTTP by the Klenow fragment, and incorporated into DNA. After demonstrating the successful incorporation of CBT-TTP, we further explored the feasibility of post-synthesis modification of DNA. Since it is a long-term interest in our group to develop boronic acid-functionalized DNA for further applications, a 1, 2-aminothiol conjugated boronic acid (Cys-BA) probe was synthesized as an example for post-synthesis modification (Figure 1, See Scheme S1 in Supporting information for synthetic route). The same 21-nt template and 14-nt primer without FAM label were used. As shown from Figure 1B, fully extended product was observed using both dNTPs (Lane 1, Figure 1B) and CBT-TTP (Lane 2, Figure 1B). However, after being treated with Cys-BA (1 mM, final concentration (50 equiv.) for 30 min, supplemented with tris(2-carboxyethyl)phosphine (TCEP, 1 mM final concentration)) with the primer extension product, only CBT-DNA21 (CBT incorporated DNA product, 21nt) showed the post-synthesis product as expected (Lane 4, Figure 1B). The reduced mobility of Cys-BA “click”-labeled CBT-DNA21 indicated the reaction, which is presumably due to the interaction between boronic acid and polyacrylamide matrix.[14, 21b] The result was further supported by the band with different mobility after treating the Cys-BA “click” labeled CBT-DNA21 with H2O2 (1 mM final concentration, 1 h) (Lane 5, Figure 1B), which is due to the well-known oxidation reaction of converting phenyl boronic acid functional group into phenol group.[28] As a control, the mobility for the dNTPs-DNA21 (extended DNA product using dNTPs, 21nt) band did not change after treating with Cys-BA (Lane 3, Figure 1B), indicating no reactions as expected.
Figure 1.
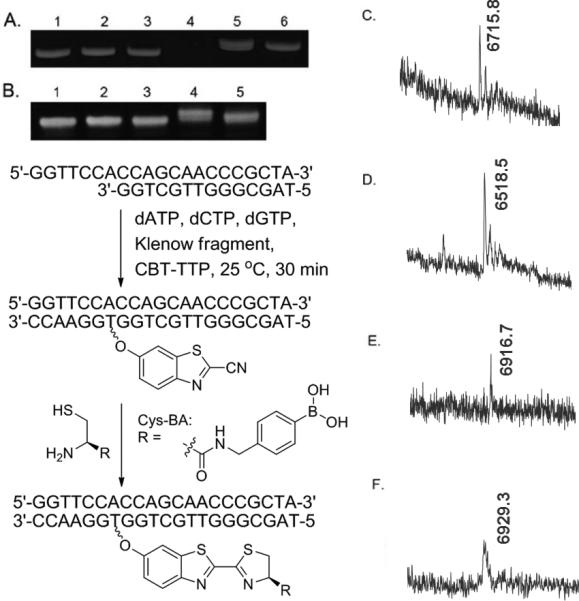
A) Primer extension using CBT-TTP catalyzed by Klenow fragment (3’-5’exo-), 20% PAGE analysis. 1) no dTTP, 2) no enzyme, 3) Template + primer, 4) Primer only, 5) dNTPs, 6) the same as Lane 5 except using CBT-TTP instead of dTTP; B) Post-synthesis labeling using “click” reagent Cys-BA, 20% PAGE analysis. 1) dNTPs-DNA21, 2) CBT-DNA21, 3) dNTPs-DNA21 + Cys-BA, 4) CBT-DNA21 + Cys-BA, 5) Cys-BA “Click” labelled CBT-DNA21 treated with H2O2; C), D), E) and F): MALDI spectra of DNA products corresponding to gel lanes B2, B3, B4, B5, respectively.
Understandably, mobility studies alone would not be enough to prove the post-synthesis modifications. MALDI-MS was used to further examine the DNA products. Specifically, dNTP-DNA21 treated with Cys-BA (Lane 3, Figure 1B) had the same peak with m/z of 6518 (Figure 1D, calculated [M+H]+: 6519) as the original DNA product. Such results indicate that Cys-BA does not interfere/react with dNTP-DNA21, as expected. In contrast, full extension of the primer using CBT-TTP instead of dTTP yielded a CBT-DNA21 (Lane 2, Figure 1B) with m/z of 6716 (Figure 1C, calculated [M+H]+: 6717) in MALDI-MS. When treated with Cys-BA, CBT-DNA21 was converted to a “click” labeled product (Lane 4, Figure 1B) with m/z of 6917 (Figure 1E, calculated [M-2H2O+H]+: 6917), corresponding to the Cys-BA “click” labeled CBT-DNA21 product. After treating the “click” product (Lane 4, Figure 1B) with H2O2, a product (Lane 5, Figure 1B) with m/z of 6929 (Figure 1F, calculated [M+H]+: 6927), corresponding to the boronic acid oxidation product, was observed as expected.[14, 21b] Such results confirmed the intended click-modifications. In addition, the effect of a CBT and boronic acid moiety on the thermostability of a DNA duplex was investigated through thermodenaturation. The results (Figure S8) suggest that the incorporation of one CBT or boronic acid moiety in a 21-bp DNA duplex only slightly decreased its stability (Tm decreased from 73.00 °C to 70.96 °C and 70.24 °C, respectively).
After successful incorporation of one CBT moiety into DNA using Klenow fragment-catalyzed primer extension reaction, we further explored the feasibility of incorporating multiple CBT moieties. Thus, 21-nt Template-2 and Template-3 were designed to incorporate two and three CBT-TTP, respectively. To our surprise, the primer extension reaction catalyzed by Klenow fragment was unsuccessful, as indicated by the presence of multiple incomplete bands, even at elevated temperature or with a longer reaction time (results not shown). Knowing family B polymerases are relatively more tolerant to modified TTP,[29] a family B polymerase (KOD XL) from Thermococcus kodakaraensis, which is a mixture of the natural form and an exo- mutant, was thus chosen for the reaction. As shown in Figure 2A, the electrophoretic mobility of modified DNA21 (lanes 2, 3 and 4) was further decreased with the incorporation of the CBT moiety at one or more positions. This clearly indicated the successful incorporation of multiple CBT-TTP into DNA through enzyme-catalyzed reactions. This was further confirmed by MALDI analysis of the incorporated product using Template-2 and Template-3 (calculated [M+H]+ for Template-2: 6929, found: 6930, Figure S6; calculated [M+H]+ for Template-3 7135, found: 7136, Figure S7). The extension product using Template-3 was further used for post-synthesis modification by Cys-BA. As is shown in Figure 2B, the incorporated product (Lane 2) could be successfully labeled by Cys-BA, showing a slower-moving band (Lane 3), which is consistent with the phenomena observed when using the template with one CBT moiety incorporation.
Figure 2.
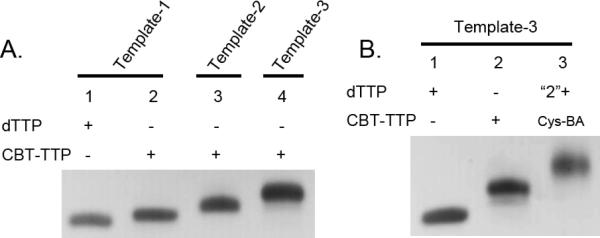
A) Primer extension using CBT-TTP catalyzed by KOD-XL DNA polymerase, 20% PAGE analysis. 1) Template-1 with dTTP, 2) Template-1 with CBT-TTP, 3) Template-2 with CBT-TTP, 4) Template-3 with CBT-TTP; B) Post-synthesis labeling using extension product of Template-3 amd “click” reagent Cys-BA, 20% PAGE analysis, 1) dNTPs-DNA21, 2) CBT-DNA21, 3) CBT-DNA21 + Cys-BA.
Encouraged by the successful incorporation of CBT-TTP into short DNA sequences, we further investigated the PCR amplification of a longer 90-mer DNA strand.[8b] We first studied the incorporation using three commercially available polymerases, including a family A polymerase from Thermus aquaticus (Taq) and family B polymerases from Thermococcus litoralis (Deep VentR exo-) and Thermococcus kodakaraensis (KOD XL), which was used for primer extension experiments. As can be seen from the results summarized in Figure 3A, incorporation of CBT-TTP were inefficient when using Taq (Lane 2, Figure 3A) and Deep Vent (Lane 6, Figure 3A). On the other hand, efficient incorporation was obtained by using KOD XL polymerase, similar to the primer extension experiments for multiple CBT moieties incorporation. Further studies were then conducted using KOD XL as the polymerase. As a negative control, no DNA product (Lane 1, Figure 3B) was observed without using dTTP or CBT-TTP. When the appropriate nucleotides are used, dNTPs-DNA90 (DNA product using dNTPs, 90nt, Lane 2, Figure 3B) and CBT-DNA90 (CBT incorporated DNA product, 90nt, Lane 3, Figure 3B) were obtained. This also indicated that CBT moiety-containing DNA chains could be used as templates for amplification. dNTPs-DNA90 (Lane 2) showed different mobility from that of CBT-DNA90 (Lane 3), presumably because of the added molecular weight of the latter.
Figure 3.
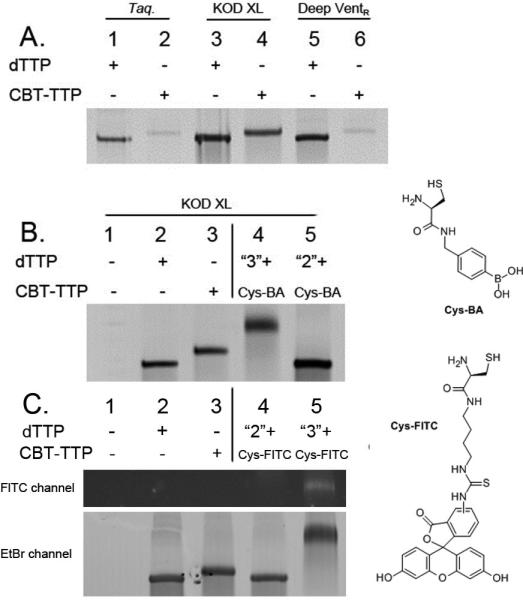
A) Enzyme screening for PCR incorporation, lanes 1, 3, and 5 are using dNTPs, and lanes 2, 4 and 6 are using CBT-TTP instead of dTTP. Lanes 1, 2: Taq polymerase, 3, 4: KOD-XL polymerase, 5, 6: Deep Vent (exo-) Polymerase. B) “Click” labeling of dNTPs/CBT-DNA90 with Cys-BA. 1) no dTTP, 2) dNTPs, 3) CBT-TTP, 4) DNA product after post-synthesis modification of CBT-DNA90 (Lane 3) with Cys-BA for 30 min, 5) DNA yielded after post-synthesis modification of dNTPs-DNA90 (Lane 2) with Cys-BA for 30 min. C) “Click” labeling of dNTPs/CBT-DNA90 with Cys-FITC. 1) no dTTP, 2) dNTP, 3) CBT-TTP, 4) DNA product after post-synthesis modification of CBT-DNA90 (Lane 3) with Cys-FITC 30 min, 5) DNA yielded after post-synthesis modification of dNTPs-DNA90 (Lane 2) with Cys-FITC for 30 min. The FITC channel only detects DNA products with green fluorescence, while EtBr channel detects all DNA products. (15% PAGE; See Supporting Information for the detail protocols).
After post-synthesis modification with Cys-BA (1 mM final concentration for 30 min, supplemented with 1 mM TCEP), a new band with further reduced mobility was observed for the CBT-DNA90 product (Lane 4, Figure 3B). As a control, the same band with the same mobility was observed after the subjection of dNTPs-DNA90 to the same treatment with Cys-BA (Lane 5, Figure 3B). Such results demonstrated the feasibility of post-synthesis click-modification with a longer 90-mer DNA with multiple CBT moieties.
Finally, to further demonstrate the generality of this approach for post-synthesis modification of DNA. 1, 2-aminothiol conjugated FITC (Cys-FITC) (Figure 3) was used instead of Cys-BA. The same phenomenon was observed as with Cys-BA. As can be seen from the EtBr channel in Figure 3C, which stain all DNA products, no DNA product (negative control, Lane 1, Figure 3C) was observed without using dTTP or CBT-TTP. On the other hand, dNTPs-DNA90 (Lane 2, Figure 3C) and CBT-DNA90 (Lane 3, Figure 3C) were observed with different mobilities. After post-synthesis modification with Cys-FITC (1 mM final concentration), supplemented with TCEP (1 mM final concentration) for 30 min, a new band with further reduced mobility was observed (Lane 5, Figure 3C). As a control, dNTPs-DNA90 was also treated with Cys-FITC under the same conditions. No mobility changes was observed after treatment (Lane 4, Figure 3C), as expected. The successful post-synthesis modification of CBT-DNA90 was further confirmed when imaging the same gel through the FITC channel, which only detects green fluorescein signal. Only the band of Cys-FITC labeled CBT-DNA90 (Lane 5) showed FITC signal, with no observable green fluorescein signals for all the other bands as expected.
Experimental Section
1. General procedure for Klenow fragment catalyzed primer extension using dTTP or CBT-TTP
The reaction mixtures of a final volume of 50 μL contained 21-nt template (5’-GGTTCCACCAGCAACCCGCTA-3’, 20 μM), 14-nt primer or 5’-FAM 14-nt primer (5’-(FAM)-TAGCGGGTTGCTGG-3’, 20 μM), 10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, 1 mM Dithiothreitol at pH 7.9, dATP, dCTP, dGTP, dTTP or CBT-TTP at concentrations of 200 μM and 0.5 U μL−1 Klenow fragment. Reactions were performed by incubating the prepared solutions at 25 °C for 30 min. The primer extension products were analyzed by 20% PAGE.
2. General procedure for KOD-XL catalyzed primer extension using dTTP or CBT-TTP
The reaction mixtures of a final volume of 50 μL contained 21-nt template (Template-2: 5’-TCAGTCACCAGCAACCCGCTA-3’, Template-3: 5’-CACGACACCAGCAACCCGCTA-3’, 20 μM), 14-nt primer (5’-TAGCGGGTTGCTGG-3’, 20 μM), 10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, 1 mM Dithiothreitol at pH 7.9, dATP, dCTP, dGTP, dTTP or CBT-TTP at concentrations of 200 μM and 0.5 U μL−1 KOD-XL. Reactions were performed by incubating the prepared solutions at 90 °C for 1 min, 20 °C for 1 min and 66 °C for 20 min. The primer extension products were analyzed by 20% PAGE.
3. Post-synthesis labelling of the primer extension products CBT-DNA21
The primer extension product CBT-DNA21 was purified using Millipore Amicon 3 kDa spin column. 5 μL of 10 mM TCEP in PBS buffer was mixed with 25 μL of 2 mM Cys-BA in 1 × PBS buffer. The reaction was allowed to stand at RT for 10 min. Pre-purified CBT-DNA21 (20 μL) was then added to the mixture, which was further incubated at 37 °C for 1 h. The negative control experiment was performed following the same procedure except using dNTPs-DNA21, the primer extension product using dNTPs and other reagents. The resulting DNA products after post-synthesis modification were purified with Millipore Amicon 3 kDa spin column and analyzed by 20% polyacrylamide gel electrophoresis. For MALDI analysis, the DNA product was further purified on a Sephadex G25 column. Fractions were collected and concentrated using a Millipore Amicon 3 kDa spin column.
4. General procedure for PCR incorporation using dTTP or CBT-TTP
The PCR mixture of a final volume of 50 μL contained DNA 90-nt template (5’-CCTTCGTTGTCTGCCTTCGTGAGCGGAGTCAGACGCACGCTCGTACCTGTGCGCAAGCACTATGACGGACACCCTTCAGAATTCGCACCA-3’, 10 nM), primer 1 (5’-TGGTGCGAATTCTGAAGGGT-3’, 1 μM), primer 2 (5’-CCTTCGTTGTCTGCCTTCGT-3’, 1 μM), dATP, dCTP, dGTP, dTTP or CBT-TTP at a concentration of 200 μM, 0.5 U μL−1 DNA polymerase and 1 × reaction buffer as provided by vender. Ten thermal cycles were conducted with melting at 90 °C for 20 s, annealing at 48 °C for 20 s, and extending at 72 °C for 30 s with initial denaturing at 90 °C for 2 min and final extension at 72 °C for 5 min. The PCR products were then analyzed by 15% PAGE.
5. Post-synthesis labeling of the PCR product CBT-DNA90
Post synthesis labelling of the PCR products was performed using similar procedures as those for primer extension. Specifically, CBT-DNA90 prepared from PCR was purified using Millipore Amicon 10 kDa spin column. 5 μL of 10 m M TCEP was mixed with 25 μL of 2 mM Cys-BA or Cys-FITC and allowed to stand at RT for 10 min. Pre-purified CBT-DNA90 (20 μL) was then added to the mixture, which was further incubated at 37 °C for 30 min. The negative control experiment was performed following the same procedure except using dNTPs-DNA90, the PCR product using dNTPs and other reagents. The resulting DNA product after post-synthesis modification was purified with Millipore Amicon 10 kDa spin column and analyzed by 15% PAGE.
Conclusion
In summary, a novel method for post-synthesis modification of DNA is described thorough the design, synthesis, and successful enzymatic incorporation of a cyanobenzothiazole (CBT)-modified TTP. The CBT-TTP incorporated DNA products can undergo rapid post-synthesis modification via a biocompatible condensation reaction with 1, 2-aminothiol conjugated boronic acid (Cys-BA) or FITC analogue (Cys-FITC). This approach provides a novel way for rapid and site-specific post-synthesis modification of DNA without the issue of regio- or stereo-isomers and the formation of a single isomer makes this approach suitable for DNA-based aptamer selection work and other applications.
Supplementary Material
Acknowledgements
We gratefully acknowledge the financial support of this work by the Molecular Basis of Disease Program at Georgia State University through fellowships to YFC, HJP, and WXC, the GSU University fellowship program through a fellowship to HJP, and the National Institutes of Health (GM086925 and GM084933 to BW).
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Rothemund PWK. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 2.You M, Chen Y, Zhang X, Liu H, Wang R, Wang K, Williams KR, Tan W. Angew. Chem. Int. Ed. 2012;51:2457–2460. doi: 10.1002/anie.201107733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shoshani S, Piran R, Arava Y, Keinan E. Angew. Chem. Int. Ed. 2012;51:2883–2887. doi: 10.1002/anie.201107156. [DOI] [PubMed] [Google Scholar]
- 4.Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metzler R, Ambjornsson T, Hanke A, Zhang YL, Levene S. J. Comput. Theor. Nanos. 2007;4:1–49. [Google Scholar]
- 6.Xu L, Anchordoquy T. J. Pharm. Sci. 2011;100:38–52. doi: 10.1002/jps.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Syed MA, Pervaiz S. Oligonucleotides. 2010;20:215–224. doi: 10.1089/oli.2010.0234. [DOI] [PubMed] [Google Scholar]
- 8.a El-Sagheer AH, Brown T. Chem. Soc. Rev. 2010;39:1388–1405. doi: 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]; b Li M, Lin N, Huang Z, Du L, Altier C, Fang H, Wang B. J. Am. Chem. Soc. 2008;130:12636–12638. doi: 10.1021/ja801510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Singh Y, Murat P, Defrancq E. Chem. Soc. Rev. 2010;39:2054–2070. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]; b Hermanson GT. Bioconjugate Techniques. Academic Press; Rockford: 1996. p. 528. [Google Scholar]
- 10.Seela F, Sirivolu VR. Chem. Biodivers. 2006;3:509–514. doi: 10.1002/cbdv.200690054. [DOI] [PubMed] [Google Scholar]
- 11.Burley GA, Gierlich J, Mofid MR, Nir H, Tal S, Eichen Y, Carell T. J. Am. Chem. Soc. 2006;128:1398–1399. doi: 10.1021/ja055517v. [DOI] [PubMed] [Google Scholar]
- 12.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 14.Dai C, Wang L, Sheng J, Peng H, Draganov AB, Huang Z, Wang B. Chem. Commun. 2011;47:3598–3600. doi: 10.1039/c0cc04546b. [DOI] [PubMed] [Google Scholar]
- 15.Jewett JC, Bertozzi CR. Chem. Soc. Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Gololobov YG, Kasukhin LF. Tetrahedron. 1992;48:1353–1406. [Google Scholar]; b Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 17.a Schoch J, Wiessler M, Jaschke A. J. Am. Chem. Soc. 2010;132:8846–8847. doi: 10.1021/ja102871p. [DOI] [PubMed] [Google Scholar]; b Marchan V, Grandas A. Synthesis of Peptide-oligonucleotide conjugates by Diels-Alder cycloaddition in water. 2007;4 doi: 10.1002/0471142700.nc0432s31. [DOI] [PubMed] [Google Scholar]
- 18.Raindlova V, Pohl R, Klepetarova B, Havran L, Simkova E, Horakova P, Pivonkova H, Fojta M, Hocek M. Chempluschem. 2012;77:652–662. [Google Scholar]
- 19.Karskela M, Helkearo M, Virta P, Lonnberg H. Bioconjugate Chem. 2010;21:748–755. doi: 10.1021/bc900529g. [DOI] [PubMed] [Google Scholar]
- 20.a Singh Y, Murat P, Defrancq E. Chem. Soc. Rev. 2010;39:2054–2070. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]; b Dawson PE, Muir TW, Clarklewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 21.a Yang X, Dai C, Dayan A, Molina C, Wang B. Chem. Commun. 2010;46:1073–1075. doi: 10.1039/b921163b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cheng Y, Dai C, Peng H, Zheng S, Jin S, Wang B. Chem. Asian J. 2011;6:2747–2752. doi: 10.1002/asia.201100229. [DOI] [PubMed] [Google Scholar]; c Lin N, Yan J, Huang Z, Altier C, Li M, Carrasco N, Suyemoto M, Johnston L, Wang S, Wang Q, Fang H, Caton-Williams J, Wang B. Nucleic Acids Res. 2007;35:1222–1229. doi: 10.1093/nar/gkl1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White EH, McCapra F, Field GF, McElroy WD. J. Am. Chem. Soc. 1961;83:2402–2403. [Google Scholar]
- 23.a Ren HJ, Xiao F, Zhan K, Kim YP, Xie HX, Xia ZY, Rao J. Angew. Chem. Int. Ed. 2009;48:9658–9662. doi: 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liang GL, Ren HJ, Rao JH. Nat. Chem. 2010;2:239–239. doi: 10.1038/nchem.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen DP, Elliott T, Holt M, Muir TW, Chin JW. J. Am. Chem. Soc. 2011;133:11418–11421. doi: 10.1021/ja203111c. [DOI] [PubMed] [Google Scholar]
- 25.Eaton BE, Gold L, Hicke BJ, Janjic N, Jucker FM, Sebesta DP, Tarasow TM, Willis MC, Zichi DA. Bioorg. Med. Chem. 1997;5:1087–1096. doi: 10.1016/s0968-0896(97)00044-8. [DOI] [PubMed] [Google Scholar]
- 26.a Meroni G, Ciana P, Maggi A, Santaniello E. Synlett. 2009:2682–2684. [Google Scholar]; b Meroni G, Rajabi M, Ciana P, Maggi A, Santaniello E. Arkivoc. 2010:53–60. [Google Scholar]
- 27.Ludwig J. Acta Biochim. Biophys. Acad. Sci. Hung. 1981;16:131–133. [PubMed] [Google Scholar]
- 28.Kuivila HG. J. Am. Chem. Soc. 1954;76:870–874. [Google Scholar]
- 29.Kuwahara M, Nagashima J, Hasegawa M, Tamura T, Kitagata R, Hanawa K, Hososhima S, Kasamatsu T, Ozaki H, Sawai H. Nucleic Acids Res. 2006;34:5383–5394. doi: 10.1093/nar/gkl637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.